Chapter 44 Surgical Management of Tumors of the Jugular Foramen
Historical Background
In 1840, Valentin1 observed a small cellular formation near the origin of the tympanic nerve that he identified as ganglionic tissue and termed “ganglionium tympanicum” or “intumescentia gangliosa.” In 1878, Krause2 further demonstrated highly vascular glomus tissue along the tympanic branch of the glossopharyngeal nerve (“glandula tympanica”) that was histologically similar to the carotid body, as reported by Von Lushka in 1862.3 This work received little attention until 1941, when Guild4,5 described glomus tissue as a flattened ovoid body in the adventitia of the dome of the jugular bulb and coined the term “glomus jugularis” to describe paraganglionic tissue composed of capillary or precapillary vessels interspersed with numerous epithelioid cells found along the jugular bulb. In sectioning human temporal bones, Guild found that 50% of this tissue occurred in the jugular bulb, approximately 25% occurred along the course of the tympanic branch of the glossopharyngeal nerve (Jacobson’s nerve), and 25% occurred along the auricular branch of vagus nerve (Arnold’s nerve). This observation explained the existence of “glomus tumors” that occurred both in the middle ear (glomus tympanicum tumors) and in the region of the jugular bulb (glomus jugulare tumors). Rosenwasser6 in 1952, was the first to suggest a possible relationship between the “glomus jugularis” and the “carotid body–like” tumors in the temporal bone. The designation “tumors of the glomus jugulare” was first mentioned by Lattes and Waltner in 1949.7
Management of jugular foramen tumors has evolved over the years. The inaccessibility of the jugular foramen because of its relatively deep location, high vascularity, and proximity to cranial nerves made tumor management extremely challenging; surgery in this region was often associated with a poor outcome. Surgery in the 1930s was primarily performed using a suboccipital approach with removal of bone around the jugular foramen to avoid excessive bleeding.8 A subtotal resection followed by radiation therapy was generally performed.9–12 Most patients had resultant postoperative lower cranial palsies. Mobilization of the facial nerve to better access the jugular foramen was first described by Capps13,14 in 1952 and later by others.15,16 Capps combined this with proximal and distal control of the sigmoid sinus and the jugular vein; however, attempts to remove the jugular bulb were met with excessive bleeding and poor outcomes.
In the 1960s and 1970s, the advent of better surgical technology resulted in better surgical outcomes. These innovations included the operating microscope, microneurosurgery dissection techniques, bipolar electrocautery, safer neuroanesthesia, arteriography,17,18 polytomography,19 retrograde jugular venography,16,20 computed tomography (CT),21 and magnetic resonance imaging (MRI).22 Hearing preservation surgeries were advocated by House and Glasscock23 and Farrior24 (modified endaural postauricular hypotympanotomy). In 1969, McCabe and Fletcher25 proposed that the size and extent of the tumor were the determining factors for selecting the most appropriate surgical approach. Soon after, new classification schemes were proposed by Fisch26 and by Jackson et al.27 based on tumor size, intracranial extension, and surgical operability. The 1970s saw the emergence of multidisciplinary skull base approaches,27 including the combined lateral skull base approach (suboccipital craniectomy with mastoidectomy),28–30 and infratemporal fossa approaches by Fisch31 (modified after Farrior’s hypotympanic approach). Other modifications to access the jugular foramen using skull base approaches were further advocated by Al-Mefty and colleagues,32–34 Bordi et al.,35 Patel et al.,36 and Liu et al.37
Despite the improvements in surgical exposure, extreme vascularity of the tumor was still a major challenge during surgery. The introduction of preoperative superselective transarterial embolization38–41 of jugular foramen tumors significantly reduced the vascularity of the tumor, making surgery safer.
Anatomic Considerations of the Jugular Foramen
The jugular foramen, also called the posterior foramen lacerum, is situated in the posterior fossa lateral to the carotid canal. The walls of the jugular foramen are formed anterolaterally by the petrous bone and posteromedially by the occipital bone.42,43 The foramen is directed in an anterior, lateral, and inferior direction. According to morphometric studies, the jugular foramen can be more accurately described as a triangular canal with an endocranial (~14.5 × 7 mm) and an exocranial opening (~9 × 17 mm). It lies about 23 mm medial to the apex of the mastoid tip, 15 mm medial to the tympanomastoid suture, and 5 mm above the intracranial orifice of the hypoglossal canal.44–47
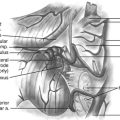
The pars venosa (or pars vascularis) is situated in the posterolateral aspect of the jugular foramen and contains the internal jugular vein (IJV), the jugular bulb, the posterior meningeal branch of the ascending pharyngeal artery, the vagus nerve (cranial nerve X), the auricular branch of the vagus nerve (Arnold’s nerve), and the spinal accessory nerve (cranial nerve XI). A smaller pars nervosa is located in the anteromedial portion of the jugular foramen and contains the glossopharyngeal nerve, the tympanic branch of the glossopharyngeal nerve (Jacobsen’s nerve), and the inferior petrosal sinus. This nomenclature can be somewhat misleading, because both structures contain neural, as well as vascular, structures.44
The important structures surrounding the jugular foramen include the mastoid segment of the facial nerve laterally, the petrous segment of the internal carotid artery (ICA) anteromedially, the vertebral artery inferiorly, and the hypoglossal nerve medially. Access to the jugular foramen from a lateral trajectory is obstructed by the mastoid and styloid processes, the transverse process of C1, and the mandibular ramus.42 The deep potential spaces along the foramen include the middle layer of the deep cervical fascia (buccopharyngeal fascia) anteromedially, the deep layer of the deep cervical fascia (prevertebral fascia) posterolaterally, and the superficial layer of the deep cervical fascia laterally.44 These potential spaces are important in understanding the spread of tumors in this region.
The cranial nerves run anteromedially to the jugular bulb and maintain a multifascicular histoarchitecture, especially cranial nerve X, which is formed by multiple fascicles, in contrast to cranial nerves IX and XI, which are formed of only one or two fascicles. The tympanic branch of the glossopharyngeal nerve (Jacobson’s nerve) and the auricular branch of the vagus nerve (Arnold’s nerve) cross the jugular foramen. A dural septum separates cranial nerve IX from the fascicles of cranial nerves X and XI.45,46,48 The only intradural site at which the glossopharyngeal nerve is consistently distinguishable from the vagus nerve is just proximal to this dural septum. In about 23% to 30% cases,44 a bony partition of the jugular foramen may be seen. The facial nerve exits the stylomastoid foramen approximately 5 mm lateral to the lateral edge of the jugular foramen. The hypoglossal nerve does not traverse the jugular foramen; however, it joins the nerves exiting the jugular foramen just below the skull base and runs with them in the carotid sheath.42 Tumors in this area can cause Vernet syndrome (jugular foramen syndrome),49 which is characterized by paralysis of cranial nerves IX, X, and XI caused by tumor expansion within the jugular foramen.
The muscular relationships encountered during surgery at the jugular foramen include the sternocleidomastoid (SCM), situated superficially in the lateral neck, and the splenius capitis, longissimus capitis, levator scapulae, and scalenus medius muscles in a deeper muscular layer. Anteriorly, the posterior belly of the digastric muscle is encountered. Immediately laterally, the styloid process and its attached musculature (stylopharyngeus, stylohyoid, and styloglossus muscles) appear in a triangular zone bounded by the posterior belly of digastrics muscle, the external auditory canal, and the mandibular ramus. Displacement of the digastric muscles exposes the transverse process of C1 (where the superior and inferior oblique muscles attach). The rectus capitis lateralis is the muscle most intimately related to the jugular formen.42
Specific Pathologic Conditions
Glomus Jugulare Tumors
Glomus jugulare tumors are rare lesions with an annual incidence of approximately 1 in 1.3 million people per year.50,51 Women are affected more often than men (6:1 ratio).52,53 Glomus tumors have also been referred to as paragangliomas, chemodectomas, nonchromaffin tumors, glomerocytomas, and receptomas. Glomus jugulare tumors arise from paraganglia in the adventitia of the jugular vein. In contrast, glomus tympanicum tumors arise from paraganglia associated with Arnold’s and Jacobson’s nerves within the middle ear. Tumors occur most frequently in patients between 40 and 70 years of age, but some have been reported in patients as young as 6 months and as old as 88 years. Although they are histologically benign and only rarely metastatize54 or display malignant features,55 these tumors may locally invade neighboring structures, such as bone, dura, carotid artery, and cranial nerves.48 Jansen et al.56 estimated the doubling time of the size of head and neck paragangliomas to be 13.8 years, with an annual growth rate of 0.79 mm/yr. The incidence of multicentric glomus tumors is 3.7% to 10% in sporadic cases57,58 and up to 55% in familial cases.59 For familial glomus tumors, the mean age of patients at presentation is younger, almost 50% of patients have multicentric tumors, and a higher proportion have bilateral tumors.60,61 Four hereditary paraganglioma syndromes have been described, referred as PGL 1 to 4.62
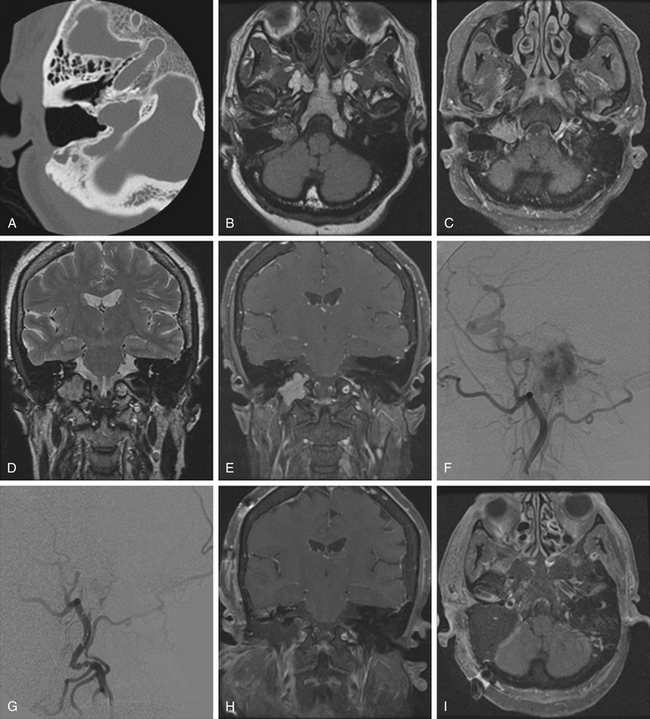
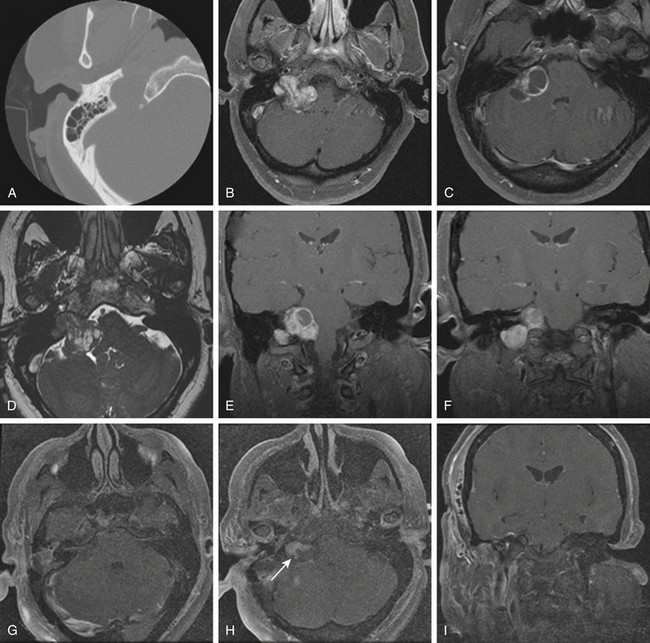
Pathologically, glomus tumors appear as highly vascular nonencapsulated masses (Figs. 44-1 and 44-2). Histologically, they are indistinguishable from glomus tympanicum, glomus vagale, and carotid body tumors. These tumors are characterized by large groups of polyhedral epithelioid cells (chief cells) with centrally located uniformly hyperchromatic nuclei and finely granular cytoplasm.63 There is an extensive thin-walled capillary and reticulin network surrounding the nests of chief cells, creating the characteristic Zellballen appearance.48 Immunohistochemical analysis of chief cells demonstrates positivity for chromogranin, synaptophysin, neuron-specific enolase, and neurofilament. Malignant glomus tumors demonstrate immunoreactivity to MIB-1, p53, Bcl-2, and CD34. The extreme vascularity of these tumors is due to angiogenesis from vascular endothelial growth factor and platelet-derived endothelial cell growth factor.64 Ultrastructurally, secretory granules containing norepinephrine, epinephrine, and dopamine may be seen. Because of the presence of catecholamines and neuropeptides, they are included in the amine precursor uptake and decarboxylase system65 or the diffuse neuroendocrine system.
Schwannomas
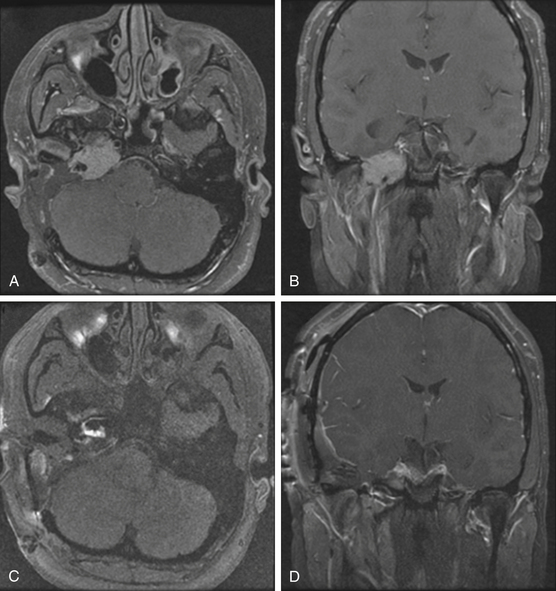
Schwannomas are the second largest group of tumors at the jugular foramen (Figs. 44-3 to 44-5).66 Ninety percent of jugular foramen schwannomas originate from cranial nerve IX or X.67,68 Samii et al.69 classified these tumors into four subtypes based on location. Type A tumors are those that primarily occupy the cerebellopontine angle with minimal enlargement of the jugular foramen. Type B tumors are primarily located in the jugular foramen with intracranial extension. Type C consists of primarily extracranial tumors with extension into the jugular foramen. Type D consists of dumbbell-shaped tumors with both intra- and extracranial components.
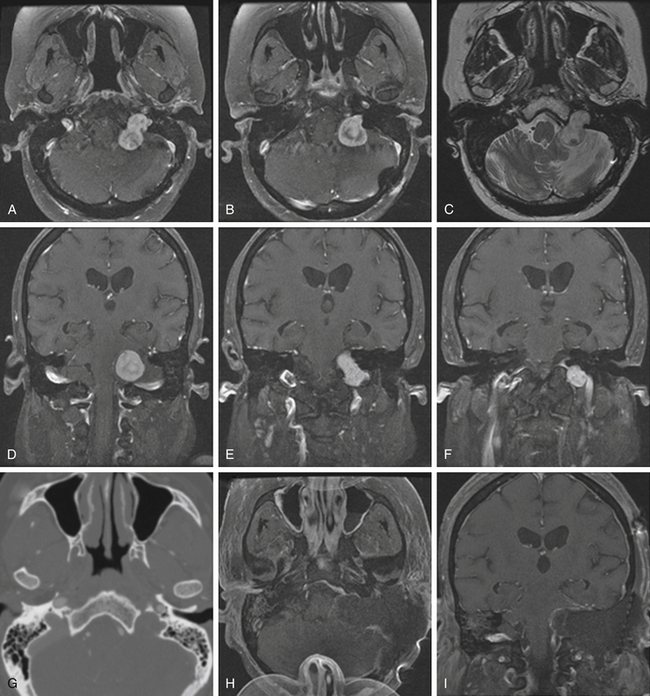
On unenhanced CT scans,70 schwannomas appear isodense to the brain and enhance brightly as well-demarcated tumors after contrast administration. As opposed to glomus tumors, the bony remodeling of the jugular foramen appears smoothly scalloped and well corticated. On T1-weighted MRI,71 schwannomas are isointense to brain and markedly enhance after gadolinium administration. On T2-weighted imaging, they are hyperintense to brain. On angiography, schwannomas do not appear very vascular and often compress the jugular bulb, in contrast to glomus tumors, which are highly vascular and invade the jugular bulb.71
At surgery, jugular foramen schwannomas appear as well-circumscribed, firm or rubbery, tan–white masses. Histologically, they comprise alternating patterns of compact spindle cells called Antoni A areas, interspersed with loose, hypocellular regions called Antoni B areas. Whorling or palisading formation of the nuclei may be seen. The hyperchromatic nucleus is elongated and twisted with indistinct cytoplasmic borders. These tumors often demonstrate retrogressive changes in the form of cystic degeneration, hyalinization, necrosis, calcification, and hemorrhage. Immunohistochemically, they demonstrate a uniformly intense S-100 protein positivity.72 Malignant transformation of these benign lesions is rarely seen.49
Meningiomas
Although basal posterior fossa meningiomas often extend into the jugular fossa, primary jugular foramen meningiomas are rare, with fewer than 100 cases reported in the literature.73–76 The jugular foramen as a location for primary meningiomas accounts for only 4% of all posterior fossa meningiomas.77 Primary jugular foramen meningiomas presumably arise from the arachnoid cap lining the jugular bulb and are more common in women (2:1).78 These meningiomas tend to exhibit an invasive growth pattern with extensive skull base infiltration, differentiating them from the meningiomas that are centered in the posterior fossa and secondarily extend into the jugular foramen.44,79 These lesions infiltrate surrounding temporal bone and neurovascular structures and require wide margins of excision to minimize risk of recurrence. A centrifugal pattern of spread, a permeative-sclerotic appearance of the bone margins of the jugular foramen, the presence of dural tails, and an absence of flow voids are particularly important features that assist in differentiating these from more common jugular foramen tumors. On CT scans, they appear isodense to the brain and show marked enhancement with contrast administration; there is occasional calcification and infiltration of the skull base with a characteristic hyperostosis.75
At surgery, they are solid, well-circumscribed extra-axial masses and are usually sessile with a broad dural base. On MRI studies, they are typically isointense to hypointense on T1-weighted imaging and enhance markedly after gadolinium administration. Moreover, the angiographic blush time is significantly longer than that of a glomus tumor. As compared with surgical intervention for glomus tumors or schwannomas, surgery on primary jugular foramen meningiomas often has a worse cranial nerve outcome76 (approximately 60% for meningiomas, 30% for glomus jugulare tumors, and 15% for schwannomas).80 There is also a higher recurrence rate of up to 25% at 5 years after resection.79
Classification
The main pathways of direct tumor spread are important in understanding the clinical and neurologic manifestations, operative strategies, and risk assessment. Glomus tumors tend to expand within the temporal bone via the pathways of least resistance, that is, air cells, vascular lumens, skull base foramen, and the Eustachian tube.81,82 According to Spector et al.,83 glomus tumors can spread through the following pathways once they have locally invaded into the temporal bone and middle ear: (1) down the Eustachian tube into the nasopharynx and then through the skull base foramina, (2) along the carotid artery into the middle fossa, (3) along the jugular vein or hypoglossal canal toward the posterior fossa, (4) through the tegmen tympani to the middle fossa floor, or (5) through the round window, with extension via the internal auditory canal into the cerebellopontine angle.83,84 Extension within the sigmoid and inferior petrosal sinus may be present as well. Although glomus tumors are usually considered benign and locally invasive, distant metastases to cervical lymph nodes, lung, and bone have been reported in about 4% to 19% of cases.84,85
The two most commonly used classifications of glomus tumors are those proposed by Fisch and colleagues26,86 and Jackson et al.27 (Tables 44-1 and 44-2). These systems are based primarily on tumor location and size. In 2002, Al-Mefty and Teixeira32 defined a subgroup of complex glomus jugulare tumors as those having one or more of the following criteria: giant size, multiple paragangliomas (bilateral or ipsilateral), malignancy, catecholamine secretion, association with other lesions such as dural arteriovenous malformations or adrenal tumors, or previous treatment with adverse outcome (prior surgery, radiation, or embolization).
| Fisch Grade | Extent of Tumor |
|---|---|
| A | Middle ear cleft (glomus tympanicum) |
| B | Tympanomastoid area with no infralabyrinthine compartment involvement |
| C | Infralabyrinthine compartment of the temporal bone and extending into the petrous apex |
| C1 | Limited involvement of the vertical portion of the carotid canal |
| C2 | Invasion of the vertical portion of the carotid canal |
| C3 | Invasion of the horizontal portion of the carotid canal |
| D1 | Intracranial extension <2 cm in diameter |
| D2 | Intracranial extension >2 cm in diameter |
TABLE 44-2 Glasscock-Jackson Classification
| Glasscock-Jackson Grade | Extent of Tumor |
|---|---|
| I | Involves the jugular bulb, middle ear, and mastoid; generally small in size |
| II | Extends under the internal auditory canal; intracranial canal extension possible |
| III | Extends into the petrous apex; intracranial canal extension possible |
| IV | Extends beyond the petrous apex into the clivus or intratemporal fossa; intracranial canal extension possible |
Clinical Presentation and Diagnosis
Patients with jugular foramen tumors often present with initial symptoms of hearing loss and swallowing difficulty, as well as pulsatile tinnitus with glomus tumors. Invasion of the middle ear results in conductive hearing loss. The glomus tumor may erode through the floor of the hypotympanum, presenting as a middle ear mass; if there is erosion of the tympanic membrane, it may present as an aural polyp. Extension of the tumor through the facial recess results in facial nerve encasement and a deficit of the facial nerve.87,88
Glomus tumors have been associated with multiple endocrine neoplasia type 2, neurofibromatosis type 1, and Von Hippel–Lindau syndrome.87 Familial paragangliomas, in contrast to sporadic cases, are commonly multiple and bilateral, present at an earlier age, and are inherited almost exclusively via the paternal allele.53,89 Genetic mapping has identified loci 11q13 and q23 as sites likely responsible for tumorogenesis.90
Although the incidence of catecholamine secretion is approximately 4%,52,91 complications of blood pressure and pulse fluctuations intraoperatively can occur during surgical manipulation.32 Therefore, preoperative assessment for catecholamine secretion should be performed, including urinary vanillylmandelic acid and metanephrines.92,93 An abdominal CT or MRI should be obtained in all such patients to rule out a pheochromocytoma as an adrenal tumor source of catecholamine secretion.32 In patients with catecholamine-secreting tumors, pretreatment with alpha- or beta-blockers is warranted. Epinephrine release during surgical manipulation can provoke a hypertensive crisis, whereas histamine and bradykinin release can cause severe hypotension.94
Radiologic Imaging
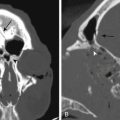
On thin-section axial CT, glomus jugulare tumors produce a characteristic “moth-eaten” pattern of destruction of the temporal bone and adjacent structures, particularly the jugular spine and the carotid crest (the bone separating the petrous carotid artery from the jugular bulb). These tumors can also cause dehiscence of the floor of the tympanic cavity, with extension into the middle ear and destruction of the bony labyrinth. The tendency of glomus jugulare tumors to erode bone helps distinguish them from glomus tympanicum tumors, which are generally smaller and arise from the cochlear promontory, enveloping but not usually destroying the ossicular chain.44 In some cases, however, the differentiation cannot be made between the two tumors; therefore, they are called glomus jugulotympanicum tumors. On T1-weighted MRI, glomus tumors demonstrate a characteristic “salt and pepper” pattern caused by flow voids within the highly vascular tumor.95 They enhance heterogeneously after gadolinium administration.96 More recently, 111indium octreotide, a radiologic somatostatin analogue, has been used to selectively identify paragangliomas, especially for detecting multicentricity, recurrence, and metastatic disease.97
Jugular foramen schwannomas, on the other hand, enlarge the jugular foramen with widened smooth, scalloped, sclerotic margins seen on CT scans. Unlike meningiomas, there is a lack bony invasion in schwannomas.95,98 Meningiomas frequently invade bone, particularly the jugular spine and jugular tubercle, resulting in hyperostosis.96 Schwannomas are usually solid and well circumscribed but occasionally demonstrate cystic degeneration. On MRI, they appear hypointense on T1-weighted images and hyperintense on T2-weighted images; they also brightly enhance, though the enhancement can be heterogeneous if there is associated necrosis or cystic degeneration.95 When there are intra- and extracranial components, a pathognomonic dumbbell-shaped tumor can be seen best on coronal or sagittal views. Meningiomas typically appear isointense to hypointense on T1-weighted images and enhance considerably after gadolinium administration. A pathognomonic “dural tail” is usually seen.
On diagnostic cerebral angiography, glomus jugulare tumors demonstrate an intense tumor blush because of their hypervascularity.95 Large feeding vessels and early draining veins are commonly associated with glomus tumors. The main blood supply of glomus tumors comes from the external carotid artery via the inferior tympanic branch of the ascending pharyngeal artery.89 Large tumors may also parasitize their blood supply from meningeal branches of the occipital artery, the posterior auricular artery, caroticotympanic branches from the petrous ICA, ascending cervical branches from the thyrocervical trunk, the posterior inferior cerebellar artery, and the vertebral artery.87,96 Narrowing and irregularities of the ICA may indicate tumor infiltration of the carotid wall. In comparison with glomus tumors, meningiomas and schwannomas are only mild to moderately vascular. Meningiomas of the jugular foramen do not necessarily demonstrate the early, prominent, and prolonged tumor blush that is frequently seen with supratentorial meningiomas.96 Cerebral angiography is also useful in demonstrating the patency of the sigmoid sinus and jugular vein to assess the venous outflow of the tumor during preoperative planning.
Treatment
In 1934, Seiffert performed the first reported exploration of the jugular bulb.9 Since then, several techniques and modalities have been tried to find the optimal approach to facilitate complete removal of paragangliomas. Early treatment options were extremely limited because of the inaccessibility and the relatively deep location of the jugular foramen,99 as well as the proximity to and involvement of the cranial nerves and the very high vascularity.100 With the advancements in the field of neuroimaging, endovascular techniques, anesthesia, microsurgical techniques, and radiosurgery, the treatment of even large jugular foramen tumors has become safer with good outcome.
For catecholamine-secreting tumors, pretreatment is required. Alpha- and beta-blockers are used for catecholamine-secreting tumors and are given 2 to 3 weeks before surgery or embolization to avoid potentially lethal intraoperative blood pressure lability and arrhythmias. Beta-blockers should never be started before full alpha-blockade, because unopposed alpha-agonism in the setting of beta-blockade can lead to severe vasoconstriction and a hypertensive crisis; the beta-blocked heart may not be able to compensate for the increased systemic vascular resistance, leading to myocardial ischemia, infarction, and heart failure.94 The recommended regimen includes phenoxybenzamine administered 1 to 2 weeks before elective surgery (initial dosage of 10 mg twice daily gradually increasing to final dosages of 40 to 100 mg/day). This drug blocks postsynaptic alpha1-receptor and presynaptic alpha2-receptors, stimulation of which causes increased catecholamine secretion and symptoms thereof. In emergent cases, 3 days of treatment is adequate. Prazosin, a selective alpha2-blocker, can also be used (1 mg three times a day gradually increasing to a final dosage of 8 to 12 mg/day). This has the advantage of causing less tachycardia and is shorter acting, therefore resulting in less prolonged postoperative hypotension. Labetelol (combined alpha- and beta-adrenergic blocker) can also be used. For serotonin-producing tumors, a combination of somatostatin and octreotide is used.94
Preoperative Considerations
Patient Considerations
Small, asymptomatic tumors in patients without a family history can be monitored radiographically with serial imaging. The growth rate of glomus tumors has been estimated at 0.79 mm/yr, with a doubling time of 13.8 years.56 Surgical removal should be considered in younger patients with symptomatic tumors, in patients with intracranial extension, in those with neural compression, and in patients with tumors demonstrating progressive growth.101 In patients with bilateral glomus tumors, the larger and more symptomatic side should be treated first. If the resection does not result in significant cranial nerve palsies, then surgery on the remaining contralateral side can be considered. If the patient already has significant postoperative cranial neuropathies, then radiation therapy should be considered on the contralateral side.32,61 Radiation therapy should also be considered in patients of advanced age with significant medical comorbidities who cannot tolerate the surgical risk, although other studies have shown that the patient’s age is not correlated with the postoperative surgical outcome.69
Endocrine Secretion
Paragangliomas are capable of secreting catecholamines and may produce symptoms similar to those of pheochromocytomas. Although the incidence of catecholamine secretion is approximately 4%,52,91 complications of wide blood pressure and pulse fluctuations intraoperatively can occur during surgical manipulation.32 Therefore, preoperative assessment for catecholamine secretion is done in all patients.92,93 This includes measuring plasma catecholamines, metanephrines/normetanephrines and urinary metanephrines/normetanephrines, and/or vanillylmandelic acid. Preoperative hypertension, arrhythmias, palpitations, headaches, or nausea are indicative of a norepinephrine-secreting tumor.89 An abdominal CT or MRI scan should be obtained in all patients with elevated levels of catecholamines to rule out an adrenal source of secretion.32
The endocrine secretion capabilities of paragangliomas are not limited to norepinephrine. Paragangliomas can also secrete 5-hydroxytryptamine (serotonin), kallikrein, and 5-hydroxytryptophan, which are precursors of serotonin and histamine, producing a carcinoid-like syndrome.94 Clinical symptoms of bronchoconstriction, tricuspid regurgitation, pulmonary stenosis, abdominal pain, explosive diarrhea, violent headaches, cutaneous flushing, hypertension, hepatomegaly, and hyperglycemia are all consistent with carcinoid syndrome. Serotonin-secreting tumors are even less common than norepinephrine-secreting tumors; therefore, preoperative screening is needed only if there are clinical symptoms of concern.
Preoperative Endovascular Treatment
Extreme vascularity of the tumor is another preoperative consideration. Preoperative embolization of the paragangliomas is performed for highly vascular lesions and is useful to reduce bleeding, surgical time, and intraoperative blood transfusion.41 The various endovascular embolization agents described are Gelfoam, polyvinyl alcohol particles, cyanoacrylate glue,102 and Onyx.103 Not all paragangliomas need preoperative embolization (e.g., tympanic paragangliomas confined to the middle ear cavity do not). Balloon test occlusion (BTO) can be helpful in assessing the degree of collateral flow if the ICA were to be sacrificed. If there is poor collateral flow, a superficial temporal artery–middle cerebral artery104 or saphenous vein reconstruction and high-flow bypass of the carotid artery may be necessary.105,106 The risk of permanent carotid artery occlusion is high even in patients who pass a preoperative BTO test,33,107 which carries an inherent risk of 3.7%.108 Sanna et al.104 advocate ICA stent placement preoperatively in cases with extensive ICA involvement. They suggest that this maneuver also decreases the intraoperative risk of carotid artery injury; however, if a stent is placed, the patient must be continued on prolonged antiplatelet therapy until the stent has endothelialized.
Intraoperative Neurophysiologic Monitoring
Real-time intraoperative neurophysiologic monitoring is critical not only in preventing inadvertent intraoperative injury to neural elements but also in mitigating further nerve injury after injury is first detected intraoperatively.109 Baseline curves for brain stem auditory evoked potentials, somatosensory evoked potentials, motor evoked potentials, and facial nerve monitoring are obtained before starting surgery, monitoring is continued intraoperatively, and final runs are obtained at the conclusion of surgery. Electrodes are placed into the SCM muscle and tongue for monitoring cranial nerves XI and XII, and an electromyographic endotracheal tube (Medtronic Xomed Inc., Jacksonville, FL) is used for cranial nerve X monitoring. Cranial nerves at risk during surgery at the jugular foramen include cranial nerves IX, X, XI, and XII (and V, VI, VII, and VIII in the case of large tumors).37
Surgical Procedure
Choice of Approach
The location, size, and presence of vascular encasement dictate the choice of approach. Access to the jugular foramen is blocked laterally by mastoid and styloid processes, the transverse process of the atlas, and the mandibular ramus.42 Tumors of the jugular foramen can be typically accessed through a transjugular posterior infratemporal fossa approach. This generally involves mastoid–neck approach that includes a retrolabyrinthine mastoidectomy, upper cervical neck exposure, and skeletonization of the sigmoid sinus and jugular bulb with or without anterior translocation of the facial nerve.110 For resection of large paragangliomas (Fisch types C and D), several lateral infratemporal fossa approaches have been described by Fisch.111 These involve transection and blind sac closure of the external ear canal with permanent routing of the facial nerve and anterior displacement of the mandible to expose the vertical segment of the ICA (C7). This procedure has inherent limitations, including conductive hearing loss, facial nerve palsy, and problems with mastication because of the displacement of mandible. For Fisch type D2 (greater than 2-cm intracranial extension), a combined two-stage resection has also been described.112–115
We previously described a one-stage transjugular posterior infratemporal fossa approach that allows radical resection of tumors that are located around the jugular foramen, lower clivus, and high cervical region from an anterolateral direction.37 Transection of the external ear canal, permanent rerouting of the facial nerve, and mandibular displacement are not required as in the Fisch approach. Instead, slight anterior transposition of the facial nerve, in select cases, allows exposure of the vertical C7 segment of the petrous ICA. Both intracranial and extracranial components of tumor can be removed in one stage with total exposure of the jugular foramen. Additional exposure of the lower clivus can be achieved by translocating the vertical infratemporal ICA anteriorly and translocating the lower cranial nerves in the pars nervosa inferiorly. If, however, the patient has nonserviceable hearing and the tumor extends into the middle ear (as in glomus jugulotympanicum tumors), transection of the external ear canal with blind sac closure may be necessary to achieve better tumor exposure and removal.
One-Stage Transjugular Posterior Infratemporal Fossa Approach
The one-stage transjugular posterior infratemporal fossa approach is a combination of the transmastoid, retro- and infralabyrinthine, transjugular, extreme lateral infrajugular–transcondylar–transtubercular, and high-cervical approaches.116 Multidirectional working corridors to the jugular foramen can be achieved including suprajugular/infralabyrinthine, transjugular, and infrajugular (retrosigmoid–transcondylar) exposures. The main steps of this approach are: (1) postauricular infratemporal incision; (2) retrolabyrinthine mastoidectomy; (3) high cervical exposure; (4) skeletonization and anterior translocation of the facial nerve; (5) lateral suboccipital craniotomy and transcondylar–transtubercular exposure; (6) removal of the IJV, jugular bulb, and sigmoid sinus; and (7) intradural exposure. This approach is versatile and can be used to remove a variety of jugular foramen tumors, including glomus tumors, schwannomas, meningiomas, chordomas, and chondrosarcomas.
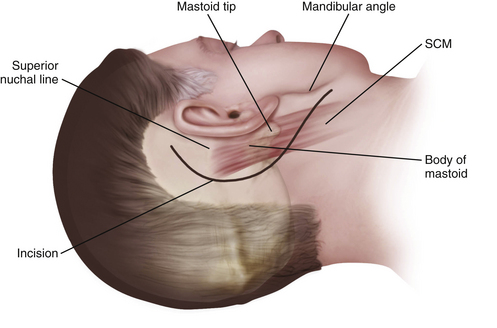
Postauricular Infratemporal Incision
A curvilinear C-shaped combined retroauricular–high cervical skin incision is made starting 2 to 3 cm posterior to the upper border of the ear and continuing inferiorly toward the neck over the anterior border of the SCM muscle (Fig. 44-6). After the galeal layer/temporoparietal fascia is undermined, the skin flap is reflected anteriorly and the posterior auricular muscle is identified behind the external ear canal. At this point, we must identify and preserve the greater auricular nerve, which runs obliquely 2 to 3 cm below the mastoid across the anterior border of the SCM. If facial nerve reconstruction is planned, the nerve is harvested now. As noted earlier, the sural nerve is another alternative for reconstruction. The muscular attachments at the mastoid tip are then carefully dissected in layers. First the superficial layer (including the SCM and splenius capitis) and then the middle layer (longissimus capitis and semispinalis capitis) are reflected posteriorly, exposing the deep layer muscles of the suboccipital triangle. Next, the deep layer, including the rectus capitis posterior major and obliquus capitis superior and obliquus capitis inferior (suboccipital triangle), is dissected. The styloid diaphragm, a membranous structure covering the posterior belly of the digastrics muscle, is also identified. The occipital artery runs under the styloid diaphragm and the posterior belly of the digastric muscle.
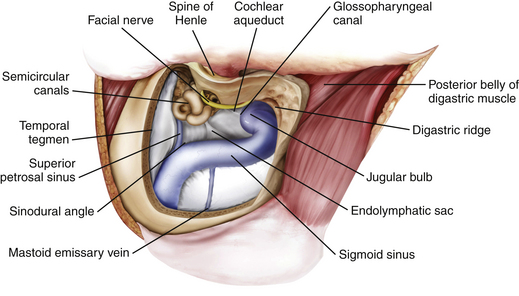
Retrolabyrinthine Mastoidectomy
A retrolabyrinthine mastoidectomy with skeletonization of the sigmoid sinus and jugular bulb is performed, and the mastoid air cells are totally removed until the presigmoid dura, the superior petrosal sinus, the middle fossa dura, the sinodural angle, and the retrosigmoid dura are identified (Fig. 44-7). The digastric ridge, which marks the exit of cranial nerve VII from the fallopian canal through the stylomastoid foramen, is then identified. The fallopian canal is located 12 to 15 mm deep to the outer cortical surface of the mastoid and 1 to 3 mm anterior and parallel to the posterior semicircular canal. The facial nerve is carefully skeletonized using a high-speed drill under constant copious irrigation to prevent thermal injury to the facial nerve. The retrofacial air cells are removed to further skeletonize the jugular bulb.
High Cervical Exposure
The goal of the high cervical exposure part of the procedure is identification of the extracranial portions of the lower cranial nerves, the ICA, and the IJV (Fig. 44-8).117 The anterior limit of the approach is defined by the posterior border of the angle of the mandible, and the posterior limit is defined by the mastoid tip. The subcutaneous tissue and the platysma are divided, and blunt dissection is used to demarcate the posterior angle of the mandible, along with the anterior border of the SCM. The anterior part of the SCM is retracted posteriorly to achieve adequate exposure of the posterior belly of the digastric muscle. Next, the stylomastoid diaphragm is removed and the underlying occipital artery coagulated. The posterior belly of the digastric muscle is reflected superoanteriorly to cover and protect the facial nerve. For identification of the accessory nerve, the transverse process of C1 is palpated.118 The lateral point located 3 to 15 mm inferolaterally to the anterior edge of the C1 transverse process is an important landmark for identifying the accessory nerve, which runs in a posteroinferior direction between the posterior belly of digastric and the IJV. The hypoglossal nerve is identified running over the IJV on the lateral surface of the carotid sheath. After the carotid sheath is opened, the vagus nerve is identified in the dorsal aspect of the ICA. The stylohyoid muscle is identified lying anterior to the ICA and attached posteriorly to the styloid process at the cranial base. The stylopharyngeus and styloglossus are also attached to the styloid process, which can be subsequently removed from its insertion at the skull base to increase the exposure of the ICA where it enters the cranial base. The glossopharyngeal nerve can be seen crossing the ICA 1 to 2 cm inferior to this point, and posterior retraction of the IJV helps expose the carotid branch of the glossopharyngeal nerve (the carotid sinus nerve). The pharyngeal branch of the vagus nerve is identified inferior to the glossopharyngeal nerve.
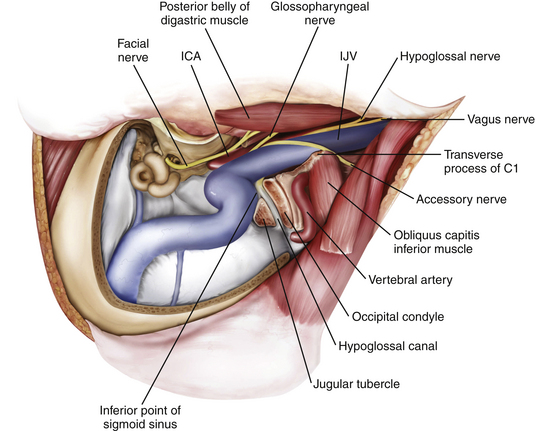
Retrosigmoid and Transcondylar–Transtubercular Exposure
A retrosigmoid craniectomy is performed and carried down to include the lip of the foramen magnum. Extradural reduction of the occipital condyle and jugular tubercle allows exposure of the hypoglossal canal and the posterior condyle and opens up the posterior rim of the jugular foramen (Fig. 44-9). Care is taken to identify and protect the vertebral artery, which lies in the C1 vertebral sulcus on the arch of C1. A safe way to identify the vertebral sulcus is to follow the lamina of C1 toward the transverse process. A J-shaped groove can be identified where the vertebral artery, which is encased by a venous plexus, courses from the foramen transversarium of C1 behind the lateral mass of C1 in the vertebral sulcus and then turns medially to pierce the atlanto-occipital membrane and dura.
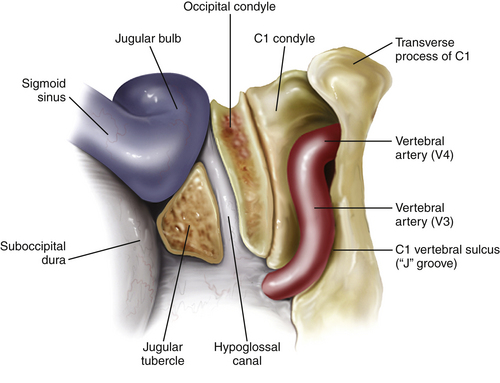
The posteromedial aspect of the occipital condyle is removed with a high-speed diamond drill while protecting the vertebral artery. Venous bleeding from the condylar emissary vein can be controlled with bone wax and Surgicel. Further drilling exposes the extradural hypoglossal canal, which is situated superior to the occipital condyle and inferior to the jugular tubercle. The canal contains the hypoglossal nerve, a meningeal branch of the ascending pharyngeal artery and the venous plexus of the hypoglossal canal, which communicates the basilar venous plexus with the marginal sinus that encircles the foramen magnum. Identification of the medial aspect of the hypoglossal canal usually indicates that approximately one third of the posterior condyle has been removed. Because the hypoglossal canal is directed anteriorly and laterally at a 45-degree angle with the sagittal plane, further skeletonization of the canal to its lateral extent usually results in approximate removal of the lateral aspect of the posterior two thirds of the condyle. Skeletonization of the entire hypoglossal canal should not result in occipitocervical instability as long as care is taken not to drill the occipital condyle inferior to the hypoglossal canal and violate the articulation to the lateral mass of C1. Further drilling of the jugular tubercle can facilitate an unobstructed view of the basal cistern and clivus anterior to the lower cranial nerves.
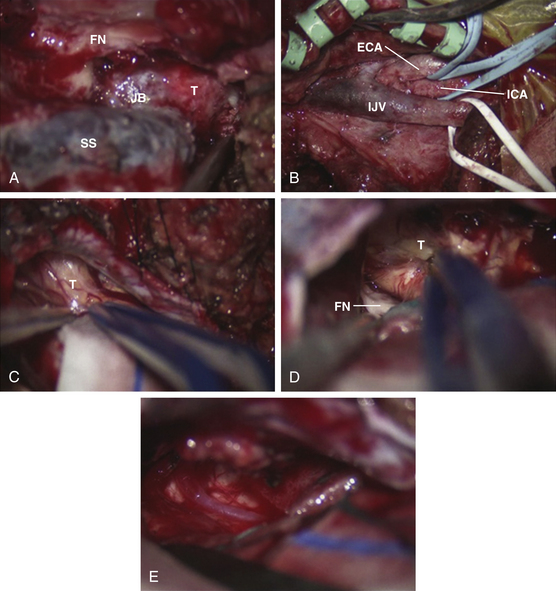
Removal of IJV, Jugular Bulb, and Sigmoid Sinus
In glomus jugulare tumors, the tumor situated within the IJV, jugular bulb, and sigmoid sinus can now be palpated. The jugular bulb is the connective link between the sigmoid sinus and the IJV and varies in both height and position. The average bulb has a width of approximately 15 mm and a height of about 20 mm.32 The hypervascularity of the tumor contributes to the prominence of the vasa vasorum of the IJV. All arterial feeders to the tumor are then meticulously coagulated. The tumor mass is isolated by ligating the IJV inferior to the tumor mass and ligating the sigmoid sinus superior to the tumor mass (Figs. 44-10 and 44-11). The lateral wall of the IJV is then opened with a No. 11 blade, and the tumor is removed from within. Brisk venous bleeding arising from the inferior petrosal sinus can be readily controlled with Surgiflo (Ethicon Inc.), followed by gentle packing with cottonoid patties. The tumor is coagulated with a bipolar cautery and carefully dissected from the pars nervosa while preserving the plane of dissection between the tumor and the medial wall of the jugular bulb. By preserving the medial wall of the jugular bulb, the lower cranial nerves at the jugular foramen are protected and are at less risk for injury. This technique can only be performed as long as the tumor has not invaded the medial wall of the jugular bulb or infiltrated the lower cranial nerves. The lower cranial nerves in the pars nervosa can be translocated inferiorly, and the vertical C7 segment of the ICA can be translocated anteriorly to establish an operative corridor to the lower clivus if necessary. For functional preservation of the lower cranial nerves, we must preserve the dural sleeve covering of the nerves. For schwannomas and meningiomas extending through the jugular foramen, ligation of the IJV and sigmoid sinus and opening of the IJV and jugular bulb can be useful in removing these tumors situated at the jugular bulb.
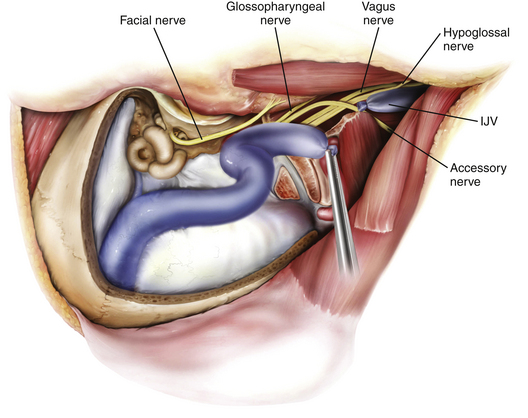
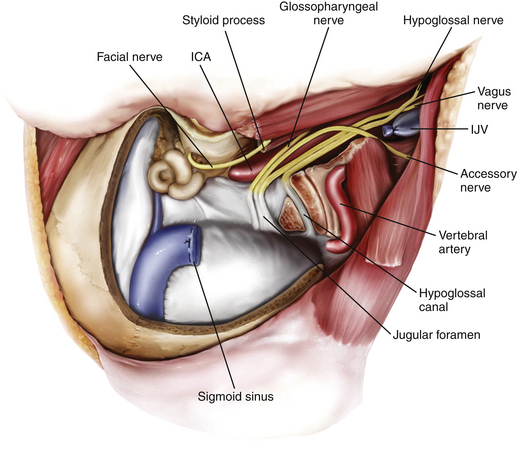
Retrosigmoid Intradural Exposure
Intradural exposure provides access to the ventral craniovertebral junction, including the inferior part of the cerebellopontine angle and the cerebellomedullary angle, and allows removal of the intradural part of the tumor, including careful dissection of the tumor off the lower cranial nerves (Figs. 44-12 and 44-13). A curvilinear incision of the dura is made 3 to 4 mm posterior to the sigmoid sinus all the way inferiorly to meet the point where the vertebral artery enters the dura. Additional exposure of the cerebellopontine angle is achieved by extending this incision superiorly to the transverse–sigmoid junction. An adequate reduction of the occipital condyle and jugular tubercle, as explained in the previous section, is key for providing a good surgical corridor to the ventral craniovertebral junction and the inferior part of the cerebellopontine angle, including the cerebellomedullary angle. This gives an excellent view of cranial nerves V through XII, the vertebral artery, the vertebrobasilar junction, the basilar artery, the posterior inferior cerebellar artery, and the anterior inferior cerebellar artery. The next step is inspection of the intradural part of the jugular foramen for any tumor invasion and dissecting the tumor carefully off the lower cranial nerves.
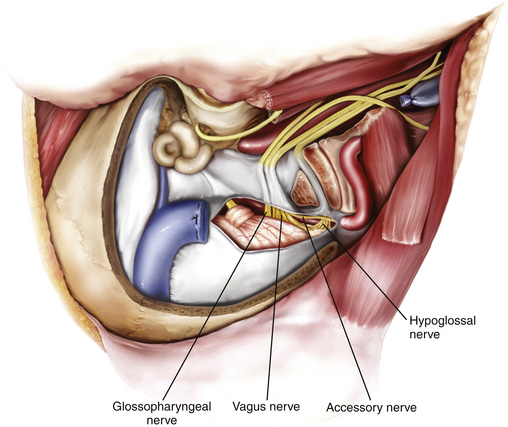
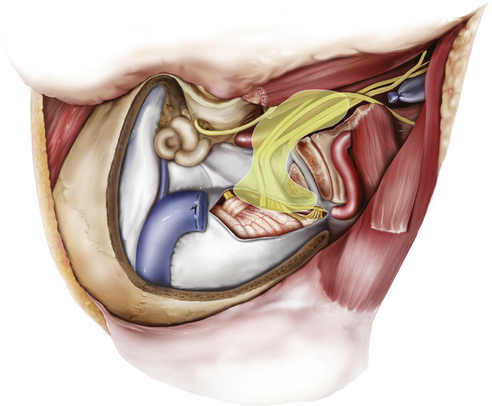
Reconstruction of Skull Base after Tumor Removal
Cranial base reconstruction and excellent watertight dural closure are key to prevention of cerebrospinal fluid (CSF) leakage. A watertight dural closure should be the goal if possible. Autologous fascia lata or a dural substitute can be used to close dural defects primarily. If a primary closure is not attainable, then an autologous fat graft is used to plug the remaining dural defect. For larger defects, an autologous fascial graft or pericranial flap can be used followed by fibrin glue. Care is taken to seal off any entrance to the middle ear with autologous muscle, bone wax, or bone cement to prevent CSF fistula to the middle ear. Additional fat is used to plug the mastoid defect and any remaining anatomic dead space. The mastoid and suboccipital cranial defect can be repaired with a sheet of malleable titanium embedded in polyethylene (Medpor Titan) that is easy to shape and secure to the skull with titanium screws. Multilayer soft tissue wound closure is performed. Temporary lumbar drainage can be used to facilitate healing of the dural closure.
Complication Avoidance
Complications from surgical removal of jugular foramen tumors include new cranial nerve deficits, vascular injury, CSF leakage, and meningitis. Postoperative morbidity can be considerable, largely from permanent cranial nerve palsies.119 Although patients with larger tumors can present with preexisting cranial nerve palsies, it is important not to incur new and permanent cranial nerve palsies. Anatomic preservation of cranial nerves and intraoperative cranial nerve monitoring are useful in minimizing these complications. Anterior translocation of the facial nerve should be used judiciously, because this can increase the risk of transient facial nerve palsy. Preservation of the medial wall of the jugular bulb is helpful in preserving lower cranial nerve integrity and function, as long as there is no tumor invasion of the medial wall or cranial nerves. In a report by Green et al.,57 29% of patients had postoperative dysphagia, 29% had persistent hoarseness, 19% were treated for vocal cord paralysis (medialization laryngoplasty), 8% required primary thyroplasty, 8% required prolonged nasogastric feedings, and 2% to 6% experienced additional complications, including pneumonia, pulmonary embolism, wound infection, aspiration, and meningitis. Patients with facial nerve palsy and incomplete eye closure may require a tarsorrhaphy or upper lid gold weight to protect the eye until facial nerve function improves. In cases of intraoperative carotid injury, attempts should be made at repairing the ICA primarily. Alternatively, ICA sacrifice with or without a bypass can be performed, depending on the results of the preoperative BTO study.111,120,121
Treatment of Residual and Recurrent Lesions
Although gross total resection is the goal of surgery, residual tumor may be left behind in some cases to preserve lower cranial nerve function. Treatment options of residual tumor include conservative management with periodic reimaging (every 3 to 6 months), reoperation, stereotactic radiosurgery, or conventional fractionated external beam radiation.122 Repeat surgery for tumor recurrence, sometimes even in cases of failed radiation, is technically more difficult because of tissue fibrosis, loss of surgical planes, and previous irradiation.
Radiosurgery
The use of stereotactic radiosurgery as a primary and a secondary treatment modality for jugular foramen tumors has been increasing in frequency.123 Radiosurgery is effective in controlling growth while minimizing permanent disability in patients with jugular foramen tumors.124 It involves a shorter treatment time than conventional fractionated external beam radiation and offers precise stereotactic localization. Some have recommended radiosurgery for tumors of less than 2 to 3 cm, with documentation of growth on serial MRI and for patients with other relative contraindications to surgery, such as patient age, significant comorbidities, or simply patient preference.125 The main goal of radiosurgery is tumor control. The main indications of primary radiosurgery are (1) age greater than 60 years; (2) medical or surgical contraindications to surgery; (3) unresectable or bilaterally large tumors; (4) major vascular risk, including failed BTO; (5) patient preference; and (6) absence of neurologic deficits.126–128 Radiosurgery should not be considered primary therapy in patients with large tumors resulting in mass effect and brain stem compression.
The primary effects of radiation therapy occur from direct injury to the cells by damaging cellular deoxyribonucleic acid and by radiation-induced vascular injury.129 There is no association between radiation dose and clinical outcome,130 tumor control,130,131 or toxicity.132,133 Although paragangliomas treated primarily with radiation have shown excellent tumor control,134 there is no effect of radiation on the catecholamine secretion of the tumor.125,135
Stereotactic radiosurgery can be used in the treatment of jugular foramen tumors as a primary therapy,136 in conjunction with surgery137 for unresectable or subtotally resected tumors, or when the patient may not be a candidate for surgery based on age,138 medical problems, tumor size, or prior treatment failure. A multicenter study139 involving 66 patients with paragangliomas treated with gamma knife radiosurgery demonstrated that 95% had clinical improvement or remained stable, 60% had no change in tumor size on follow-up CT or MRI, and 40% had reduction in the size of their tumor. For jugular foramen schwannomas, stereotactic radiosurgery showed 5- and 10-year control rates of 97% and 94%, respectively.140
Radiation-induced neoplasms,136,141 hearing loss, alopecia, facial weakness, imbalance, and vocal cord dysfunction125,129,141 have all been described after radiotherapy for benign paragangliomas. Acute complications of radiosurgery include mucositis and nausea. Late complications include hair loss, xerostomia,142 otitis media, vertigo, secondary tumor development, radiation necrosis of brain, bone, and muscle,133 radiation-induced cranial neuropathies,143 ICA thrombosis, and poor wound healing. Although excellent tumor control rates have been reported over relatively short periods, only a few studies have reported long-term tumor control rates.133,143,144
Al-Mefty O., Fox J.L., Rifai A., et al. A combined infratemporal and posterior fossa approach for the removal of giant glomus tumors and chondrosarcomas. Surg Neurol. 1987;28(6):423-431.
Al-Mefty O., Teixeira A. Complex tumors of the glomus jugulare: criteria, treatment, and outcome. J Neurosurg. 2002;97(6):1356-1366.
Arnautovic K.I., Al-Mefty O. Primary meningiomas of the jugular fossa. J Neurosurg. 2002;97(1):12-20.
Brackmann D.E., Fayad J.N., Owens R.M. Paragangliomas and schwannomas of the jugular foramen. In: Sekhar L.N., Fessler R.G. Atlas of Neurosurgical Techniques: Brain. New York: Thieme; 2006:752-758.
Fayad J.N., Schwartz M.S., Brackmann D.E. Treatment of recurrent and residual glomus jugulare tumors. Skull Base. 2009;19(1):92-98.
Fisch U. Infratemporal fossa approach for glomus tumors of the temporal bone. Ann Otol Rhinol Laryngol. 1982;91(5 Pt 1):474-479.
Fisch U. Infratemporal fossa approach to tumours of the temporal bone and base of the skull. J Laryngol Otol. 1978;92(11):949-967.
Gardner G., Cocke E.W.Jr., Robertson J.T., et al. Combined approach surgery for removal of glomus jugulare tumors. Laryngoscope. 1977;87(5 Pt 1):665-688.
Gottfried O.N., Liu J.K., Couldwell W.T. Comparison of radiosurgery and conventional surgery for the treatment of glomus jugulare tumors. Neurosurg Focus. 2004;17(2):E4.
Graham M.D., Larouere M.J., Kartush J.M. Jugular foramen schwannomas: diagnosis and suggestions for surgical management. Skull Base Surg. 1991;1(1):34-38.
Green J.D.Jr., Brackmann D.E., Nguyen C.D., et al. Surgical management of previously untreated glomus jugulare tumors. Laryngoscope. 1994;104(8 Pt 1):917-921.
Jackson C.G., Glasscock M.E.3rd, Harris P.F. Glomus Tumors. Diagnosis, classification, and management of large lesions. Arch Otolaryngol. 1982;108(7):401-410.
Katsuta T., Rhoton A.L.Jr., Matsushima T. The jugular foramen: microsurgical anatomy and operative approaches. Neurosurgery. 1997;41(1):149-201. discussion 201-202
Liu J.K., Sameshima T., Gottfried O.N., et al. The combined transmastoid retro- and infralabyrinthine transjugular transcondylar transtubercular high cervical approach for resection of glomus jugulare tumors. Neurosurgery. 2006;59(1 suppl 1):ONS115-ONS125.
Patel S.J., Sekhar L.N., Cass S.P., et al. Combined approaches for resection of extensive glomus jugulare tumors. A review of 12 cases. J Neurosurg. 1994;80(6):1026-1038.
Ramina R., Maniglia J.J., Fernandes Y.B., et al. Tumors of the jugular foramen: diagnosis and management. Neurosurgery. 2005;57(suppl 1):59-68.
Ramina R., Neto M.C., Fernandes Y.B., et al. Meningiomas of the jugular foramen. Neurosurg Rev. 2006;29(1):55-60.
Samii M., Babu R., Tatgiba M., et al. Surgical treatment of jugular foramen schwannomas. J Neurosurg. 1995;82:924-932.
Sheehan J., Kondziolka D., Flickinger J., et al. Gamma knife surgery for glomus jugulare tumors: an intermediate report on efficacy and safety. J Neurosurg. 2005;102(suppl):241-246.
Varma A., Nathoo N., Neyman G., et al. Gamma knife radiosurgery for glomus jugulare tumors: volumetric analysis in 17 patients. Neurosurgery. 2006;59(5):1030-1036. discussion 1036
1. Valentin G. Über eine gangliose Anschwellung in der Jacobson’schen Anastomose des Menschen. Arch Anat Physiol. 1840:287-290.
2. Krause W. Die Glandula tympanica des Menschen. Zbl Med Wiss. 1878;16:737-739.
3. Jackson C.G., Welling D.B., Chironis P., et al. Glomus tympanicum tumors: contemporary concepts in conservation surgery. Laryngoscope. 1989;99(9):875-884.
4. Guild S. A hitherto unrecognized structure: the glomus jugularis in man. Anat Rec. 1941;79:28.
5. Guild S. Glomus jugulare in man. Ann Otol Rhinol Laryngol. 1953;62:1045.
6. Rosenwasser H. Glomus jugularis tumor of the middle ear; carotid body tumor, tympanic body tumor, nonchromaffin paraganglioma. Trans Am Laryngol Rhinol Otol Soc. 1952;3(56th Meeting):94-106.
7. Lattes R., Waltner J.G. Nonchromaffin paraganglioma of the middle ear; carotid-body-like tumor; glomus-jugulare tumor. Cancer. 1949;2(3):447-468.
8. Weille F.L., Lane C.S.Jr. Surgical problems involved in the removal of glomus-jugulare tumors. Laryngoscope. 1951;61(5):448-459.
9. Lundgren N. Tympanic body tumours in the middle ear; tumours of carotid body type. Acta Otolaryngol. 1949;37(4):367-379. illust
10. Alexander E.Jr., Beamer P.R., Williams J.O. Tumor of the glomus jugulare with extension into the middle ear; nonchromaffin paraganglioma or carotid-body-type tumor. J Neurosurg. 1951;8(5):515-523.
11. Capps F.C. Carotid body tumors and their surgical management. Proc R Soc Med. 1964;57:954-955.
12. Thoms O.J., Shaw D.T., Trowbridge W.V. Glomus jugulare tumor: report of a case with surgical removal. J Neurosurg. 1960;17:500-504.
13. Michael L.M.2nd, Robertson J.H. Glomus jugulare tumors: historical overview of the management of this disease. Neurosurg Focus. 2004;17(2):E1.
14. Capps F.C. Glomus jugulare tumours of the middle ear. J Laryngol Otol. 1952;66(7):302-314.
15. Shapiro M.J., Neues D.K. Technique for removal of glomus jugulare tumors. Arch Otolaryngol. 1964;79:219-224.
16. Gejrot T. Surgical Treatment of glomus jugulare tumours with special reference to the diagnostic value of retrograde jugularography. Acta Otolaryngol. 1965;60:150-168.
17. Riemenschneider P.A., Hoople G.D., Brewer D., et al. Roentgenographic diagnosis of tumors of the glomus jugularis. Am J Roentgenol Radium Ther Nucl Med. 1953;69(1):59-65.
18. Hoople G.D. Personal experiences with glomus jugulare tumors. Ann Otol Rhinol Laryngol. 1964;73:778-790.
19. Valvassori G.E. Laminagraphy of the ear. Normal roentgenographic anatomy. Am J Roentgenol Radium Ther Nucl Med. 1963;89:1155-1167.
20. Gejrot T. Retrograde jugularography in the diagnosis of abnormalities of the superior bulb of the internal jugular vein. Acta Otolaryngol. 1964;57:177-180.
21. Mafee M.F., Valvassori G.E., Shugar M.A., et al. High resolution and dynamic sequential computed tomography. Use in the evaluation of glomus complex tumors. Arch Otolaryngol. 1983;109(10):691-696.
22. Valvassori G.E., Garcia Morales F., Palacios E., et al. MR of the normal and abnormal internal auditory canal. AJNR Am J Neuroradiol. 1988;9(1):115-119.
23. House W.F., Glasscock M.E.3rd. Glomus tympanicum tumors. Arch Otolaryngol. 1968;87(5):550-554.
24. Farrior J.B. Glomus tumors. Postauricular hypotympanotomy and hypotympanoplasty. Arch Otolaryngol. 1967;86(4):367-373.
25. McCabe B.F., Fletcher M. Selection of therapy of glomus jugulare tumors. Arch Otolaryngol. 1969;89(1):156-159.
26. Fisch U. Infratemporal fossa approach to tumours of the temporal bone and base of the skull. J Laryngol Otol. 1978;92(11):949-967.
27. Jackson C.G., Glasscock M.E.3rd, Harris P.F. Glomus tumors. Diagnosis, classification, and management of large lesions. Arch Otolaryngol. 1982;108(7):401-410.
28. Gardner G., Cocke E.W.Jr., Robertson J.T., et al. Combined approach surgery for removal of glomus jugulare tumors. Laryngoscope. 1977;87(5 Pt 1):665-688.
29. Gardner G., Cocke E.W.Jr., Robertson J.T., et al. Glomus jugulare tumours—combined treatment: part II. J Laryngol Otol. 1981;95(6):567-580.
30. Kempe L.G., VanderArk G.D., Smith D.R. The neurosurgical treatment of glomus jugulare tumors. J Neurosurg. 1971;35(1):59-64.
31. Fisch U. Infratemporal fossa approach for glomus tumors of the temporal bone. Ann Otol Rhinol Laryngol. 1982;91(5 Pt 1):474-479.
32. Al-Mefty O., Teixeira A. Complex tumors of the glomus jugulare: criteria, treatment, and outcome. J Neurosurg. 2002;97(6):1356-1366.
33. Anand V.K., Leonetti J.P., Al-Mefty O. Neurovascular considerations in surgery of glomus tumors with intracranial extensions. Laryngoscope. 1993;103(7):722-728.
34. Al-Mefty O., Fox J.L., Rifai A., et al. A combined infratemporal and posterior fossa approach for the removal of giant glomus tumors and chondrosarcomas. Surg Neurol. 1987;28(6):423-431.
35. Bordi L.T., Cheesman A.D., Symon L. The surgical management of glomus jugulare tumours—description of a single-staged posterolateral combined otoneurosurgical approach. Br J Neurosurg. 1989;3(1):21-30.
36. Patel S.J., Sekhar L.N., Cass S.P., et al. Combined approaches for resection of extensive glomus jugulare tumors. A review of 12 cases. J Neurosurg. 1994;80(6):1026-1038.
37. Liu J.K., Sameshima T., Gottfried O.N., et al. The combined transmastoid retro- and infralabyrinthine transjugular transcondylar transtubercular high cervical approach for resection of glomus jugulare tumors. Neurosurgery. 2006;59(1 suppl 1):ONS115-ONS125.
38. Hilal S.K., Michelsen J.W. Therapeutic percutaneous embolization for extra-axial vascular lesions of the head, neck, and spine. J Neurosurg. 1975;43(3):275-287.
39. Brismar J., Cronqvist S. Therapeutic embolization in the external carotid artery region. Acta Radiol Diagn (Stockh). 1978;19(5):715-731.
40. Simpson G.T.2nd, Konrad H.R., Takahashi M., et al. Immediate postembolization excision of glomus jugulare tumors: advantages of new combined techniques. Arch Otolaryngol. 1979;105(11):639-643.
41. Murphy T.P., Brackmann D.E. Effects of preoperative embolization on glomus jugulare tumors. Laryngoscope. 1989;99(12):1244-1247.
42. Rhoton A.L.Jr. Jugular foramen. Neurosurgery. 2000;47(suppl 3):S267-S285.
43. Katsuta T., Rhoton A.L.Jr., Matsushima T. The jugular foramen: microsurgical anatomy and operative approaches. Neurosurgery. 1997;41(1):149-201. discussion 201-202
44. Vogl T.J., Bisdas S. Differential diagnosis of jugular foramen lesions. Skull Base. 2009;19(1):3-16.
45. Tekdemir I., Tuccar E., Aslan A., et al. Comprehensive microsurgical anatomy of the jugular foramen and review of terminology. J Clin Neurosci. 2001;8(4):351-356.
46. Tekdemir I., Tuccar E., Aslan A., et al. The jugular foramen: a comparative radioanatomic study. Surg Neurol. 1998;50(6):557-562.
47. Schwaber M.K., Netterville J.L., Maciunas R. Microsurgical anatomy of the lower skullbase—a morphometric analysis. Am J Otol. 1990;11(6):401-405.
48. Fujimoto K., Yoshino H., Saito I.. Conformation dependent DNA photoligation via sensitizer tethered 5-carboxyvinyluracil. Nucleic Acids Res Suppl, 2002;2:155-156
49. Maniglia A.J., Chandler J.R., Goodwin W.J.Jr., et al. Schwannomas of the parapharyngeal space and jugular foramen. Laryngoscope. 1979;89(9 Pt 1):1405-1414.
50. Moffat D.A., Hardy D.G. Surgical management of large glomus jugulare tumours: infra- and trans-temporal approach. J Laryngol Otol. 1989;103(12):1167-1180.
51. Havekes B., van der Klaauw A.A., Hoftijzer H.C., et al. Reduced quality of life in patients with head-and-neck paragangliomas. Eur J Endocrinol. 2008;158(2):247-253.
52. Brown J.S. Glomus jugulare tumors revisited: a ten-year statistical follow-up of 231 cases. Laryngoscope. 1985;95(3):284-288.
53. Heth J. The basic science of glomus jugulare tumors. Neurosurg Focus. 2004;17(2):E2.
54. Brewis C., Bottrill I.D., Wharton S.B., et al. Glomus jugulare tumour with metastases to cervical lymph nodes. J Laryngol Otol. 2000;114(1):67-69.
55. Johnston F., Symon L. Malignant paraganglioma of the glomus jugulare: a case report. Br J Neurosurg. 1992;6(3):255-259.
56. Jansen J.C., van den Berg R., Kuiper A., et al. Estimation of growth rate in patients with head and neck paragangliomas influences the treatment proposal. Cancer. 2000;88(12):2811-2816.
57. Green J.D.Jr., Brackmann D.E., Nguyen C.D., et al. Surgical management of previously untreated glomus jugulare tumors. Laryngoscope. 1994;104(8 Pt 1):917-921.
58. Woods C.I., Strasnick B., Jackson C.G. Surgery for glomus tumors: the Otology Group experience. Laryngoscope. 1993;103(11 Pt 2 suppl 60):65-70.
59. van Baars F., van den Broek P., Cremers C., et al. Familial non-chromaffinic paragangliomas (glomus tumors): clinical aspects. Laryngoscope. 1981;91(6):988-996.
60. Sugarbaker E.V., Chretien P.B., Jacobs J.B. Bilateral familial carotid body tumors: report of a patient with an occult contralateral tumor and postoperative hypertension. Ann Surg. 1971;174(2):242-247.
61. van der Borden J. Bilateral non-chromaffin tympano-jugular paraganglioma. J Laryngol Otol. 1967;81(4):445-448.
62. Boedeker C.C., Neumann H.P., Offergeld C., et al. Clinical features of paraganglioma syndromes. Skull Base. 2009;19(1):17-25.
63. Rao A.B., Koeller K.K., Adair C.F. From the archives of the AFIP. Paragangliomas of the head and neck: radiologic–pathologic correlation. Armed Forces Institute of Pathology. Radiographics. 1999;19(6):1605-1632.
64. Jyung R.W., LeClair E.E., Bernat R.A., et al. Expression of angiogenic growth factors in paragangliomas. Laryngoscope. 2000;110(1):161-167.
65. Farrior J.B.3rd, Hyams V.J., Benke R.H., et al. Carcinoid apudoma arising in a glomus jugulare tumor: review of endocrine activity in glomus jugulare tumors. Laryngoscope. 1980;90(1):110-119.
66. Bakar B. The jugular foramen schwannomas: review of the large surgical series. J Korean Neurosurg Soc. 2008;44(5):285-294.
67. Graham M.D., Larouere M.J., Kartush J.M. Jugular foramen schwannomas: diagnosis and suggestions for surgical management. Skull Base Surg. 1991;1(1):34-38.
68. Song M.H., Lee H.Y., Jeon J.S., et al. Jugular foramen schwannoma: analysis on its origin and location. Otol Neurotol. 2008;29(3):387-391.
69. Samii M., Babu R., Tatgiba M., et al. Surgical treatment of jugular foramen schwannomas. J Neurosurg. 1995;82:924-932.
70. Uchino A., Hasuo K., Fukui M., et al. Computed tomography of jugular foramen neurinomas—report of four cases. Neurol Med Chir (Tokyo). 1987;27(7):628-632.
71. Eldevik O.P., Gabrielsen T.O., Jacobsen E.A. Imaging findings in schwannomas of the jugular foramen. AJNR Am J Neuroradiol. 2000;21(6):1139-1144.
72. Brackmann D.E., Fayad J.N., Owens R.M. Paragangliomas and schwannomas of the jugular foramen. In: Sekhar L.N., Fessler R.G. Atlas of Neurosurgical Techniques: Brain. New York: Thieme; 2006:752-758.
73. Arnautovic K.I., Al-Mefty O. Primary meningiomas of the jugular fossa. J Neurosurg. 2002;97(1):12-20.
74. Ramina R., Neto M.C., Fernandes Y.B., et al. Meningiomas of the jugular foramen. Neurosurg Rev. 2006;29(1):55-60.
75. Macdonald A.J., Salzman K.L., Harnsberger H.R., et al. Primary jugular foramen meningioma: imaging appearance and differentiating features. AJR Am J Roentgenol. 2004;182(2):373-377.
76. Gilbert M.E., Shelton C., McDonald A., et al. Meningioma of the jugular foramen: glomus jugulare mimic and surgical challenge. Laryngoscope. 2004;114(1):25-32.
77. Roberti F., Sekhar L.N., Kalavakonda C., et al. Posterior fossa meningiomas: surgical experience in 161 cases. Surg Neurol. 2001;56(1):8-20. discussion 20-21
78. Toyama C., Santiago Gebrim E.M., Brito R., et al. Primary jugular foramen meningioma. Otol Neurotol. 2008;29(3):417-418.
79. Molony T.B., Brackmann D.E., Lo W.W. Meningiomas of the jugular foramen. Otolaryngol Head Neck Surg. 1992;106(2):128-136.
80. Lustig L.R., Jackler R.K. The variable relationship between the lower cranial nerves and jugular foramen tumors: implications for neural preservation. Am J Otol. 1996;17(4):658-668.
81. Gulya A.J. The glomus tumor and its biology. Laryngoscope. 1993;103(11 Pt 2 suppl 60):7-15.
82. Dickens W.J., Million R.R., Cassisi N.J., et al. Chemodectomas arising in temporal bone structures. Laryngoscope. 1982;92(2):188-191.
83. Spector G.J., Druck N.S., Gado M. Neurologic manifestations of glomus tumors in the head and neck. Arch Neurol. 1976;33(4):270-274.
84. Druck N.S., Spector G.J., Ciralsky R.H., et al. Malignant glomus vagale: report of a case and review of the literature. Arch Otolaryngol. 1976;102(10):534-536.
85. Pluta R.M., Ram Z., Patronas N.J., et al. Long-term effects of radiation therapy for a catecholamine-producing glomus jugulare tumor. Case report. J Neurosurg. 1994;80(6):1091-1094.
86. Jenkins H.A., Fisch U. Glomus tumors of the temporal region. Technique of surgical resection. Arch Otolaryngol. 1981;107(4):209-214.
87. Semaan M., Megerian C. Current assessment and management of glomus tumors. Curr Opin Otolaryngol Head Neck Surg.. 2008;16:420-426.
88. Ramina R., Maniglia J.J., Fernandes Y.B., et al. Tumors of the jugular foramen: diagnosis and management. Neurosurgery. 2005;57(suppl 1):59-68.
89. Ramina R., Maniglia J.J., Fernandes Y.B., et al. Jugular foramen tumors: diagnosis and treatment. Neurosurg Focus. 2004;17(2):E5.
90. Bikhazi P.H., Roeder E., Attaie A., et al. Familial paragangliomas: the emerging impact of molecular genetics on evaluation and management. Am J Otol. 1999;20(5):639-643.
91. Jackson C.G., Harris P.F., Glasscock M.E.3rd, et al. Diagnosis and management of paragangliomas of the skull base. Am J Surg. 1990;159(4):389-393.
92. Nelson M.D., Kendall B.E. Intracranial catecholamine secreting paragangliomas. Neuroradiology. 1987;29(3):277-282.
93. Schwaber M.K., Glasscock M.E., Nissen A.J., et al. Diagnosis and management of catecholamine secreting glomus tumors. Laryngoscope. 1984;94(8):1008-1015.
94. Jensen N.F. Glomus tumors of the head and neck: anesthetic considerations. Anesth Analg. 1994;78(1):112-119.
95. Lowenheim H., Koerbel A., Ebner F., et al. Differentiating imaging findings in primary and secondary tumors of the jugular foramen. Neurosurg Rev. 2006;29:1-11.
96. Caldemeyer K., Mathews V., Azzarelli B., et al. The jugular foramen: a review of anatomy, masses and imaging characteristics. RadioGraphics. 1997;17:1123-1139.
97. Kwekkeboom D.J., van Urk H., Pauw B.K., et al. Octreotide scintigraphy for the detection of paragangliomas. J Nucl Med. 1993;34(6):873-878.
98. Larson T.C.3rd, Reese D.F., Baker H.L.Jr., et al. Glomus tympanicum chemodectomas: radiographic and clinical characteristics. Radiology. 1987;163(3):801-806.
99. Dichiro G., Fisher R.L., Nelson K.B. The jugular foramen. J Neurosurg. 1964;21:447-460.
100. Simko T.G., Griffin T.W., Gerdes A.J., et al. The role of radiation therapy in the treatment of glomus jugulare tumors. Cancer. 1978;42(1):104-106.
101. Tran Ba Huy P., Kania R., Duet M., et al. Evolving concepts in the management of jugular paraganglioma: a comparison of radiotherapy and surgery in 88 cases. Skull Base. 2009;19(1):83-91.
102. Persky M.S., Setton A., Niimi Y., et al. Combined endovascular and surgical treatment of head and neck paragangliomas—a team approach. Head Neck. 2002;24(5):423-431.
103. Rimbot A., Mounayer C., Loureiro C., et al. [Preoperative mixed embolization of a paraganglioma using Onyx.]. J Neuroradiol. 2007;34(5):334-339.
104. Sanna M., Piazza P., De Donato G., et al. Combined endovascular–surgical management of the internal carotid artery in complex tympanojugular paragangliomas. Skull Base. 2009;19(1):26-42.
105. Miyazaki S., Fukushima T., Fujimaki T. Resection of high-cervical paraganglioma with cervical-to-petrous internal carotid artery saphenous vein bypass. Report of two cases. J Neurosurg. 1990;73(1):141-146.
106. Sekhar L.N., Kalavakonda C. Cerebral revascularization for aneurysms and tumors. Neurosurgery. 2002;50(2):321-331.
107. Origitano T.C., Al-Mefty O., Leonetti J.P., et al. Vascular considerations and complications in cranial base surgery. Neurosurgery. 1994;35(3):351-362. discussion 362-363
108. Tarr R.W., Jungreis C.A., Horton J.A., et al. Complications of preoperative balloon test occlusion of the internal carotid arteries: experience in 300 cases. Skull Base Surg. 1991;1(4):240-244.
109. Shils J., Martin C., Deletis V. Intraoperative monitoring for surgeries at the jugular foramen. Operative Techniques in Neurosurgery. 2005;8(1):42-49.
110. Hirsch B., Shekhar L., Kamerer D. Transtemporal and infratemporal approach for benign tumors of the jugular foramen and temporal bone. In: Sekhar L., Jacecka I. Surgery of Cranial Base Tumors. New York: Raven, 1993.
111. Fisch U. Infratemporal fossa approach for lesions in the temporal bone and base of the skull. Adv Otorhinolaryngol. 1984;34:254-266.
112. Cheek J.C. Posterior fossa intraoperative monitoring. J Clin Neurophysiol. 1993;10(4):412-424.
113. Sekiya T., Hatayama T., Akasaka K., et al. [Intraoperative monitoring of the lower cranial nerves during skull base surgery: evoked electromyographic technique.]. No Shinkei Geka. 1995;23(6):491-496.
114. Maurer J., Pelster H., Amedee R.G., et al. Intraoperative monitoring of motor cranial nerves in skull base surgery. Skull Base Surg. 1995;5(3):169-175.
115. Sekhar L.N., Bejjani G., Nora P., et al. Neurophysiologic monitoring during cranial base surgery: is it necessary? Clin Neurosurg. 1995;42:180-202.
116. Erol F.S., Kaplan M., Kavakli A., et al. Jugular foramen syndrome caused by choleastatoma. Clin Neurol Neurosurg. 2005;107(4):342-346.
117. Sheen T.S., Yen K.L., Ko J.Y., et al. Usefulness of the C1 transverse process as a reference guide in the dissection of the upper lateral neck. Otolaryngol Head Neck Surg. 2000;122(2):284-289.
118. Salas E., Ziyal I.M., Bank W.O., et al. Extradural origin of the posteroinferior cerebellar artery: an anatomic study with histological and radiographic correlation. Neurosurgery. 1998;42(6):1326-1331.
119. Makek M., Franklin D.J., Zhao J.C., et al. Neural infiltration of glomus temporale tumors. Am J Otol. 1990;11(1):1-5.
120. van der Mey A.G., Frijns J.H., Cornelisse C.J., et al. Does intervention improve the natural course of glomus tumors? A series of 108 patients seen in a 32-year period. Ann Otol Rhinol Laryngol. 1992;101(8):635-642.
121. Andrews J.C., Valavanis A., Fisch U. Management of the internal carotid artery in surgery of the skull base. Laryngoscope. 1989;99(12):1224-1229.
122. Krych A.J., Foote R.L., Brown P.D., et al. Long-term results of irradiation for paraganglioma. Int J Radiat Oncol Biol Phys. 2006;65(4):1063-1066.
123. Feigenberg S.J., Mendenhall W.M., Hinerman R.W., et al. Radiosurgery for paraganglioma of the temporal bone. Head Neck. 2002;24(4):384-389.
124. Huy P.T., Kania R., Duet M., et al. Evolving concepts in the management of jugular paraganglioma: a comparison of radiotherapy and surgery in 88 cases. Skull Base. 2009;19(1):83-91.
125. Sheehan J., Kondziolka D., Flickinger J., et al. Gamma knife surgery for glomus jugulare tumors: an intermediate report on efficacy and safety. J Neurosurg. 2005;102(suppl):241-246.
126. Hawthorne M.R., Makek M.S., Harris J.P., et al. The histopathological and clinical features of irradiated and nonirradiated temporal paragangliomas. Laryngoscope. 1988;98(3):325-331.
127. Brackmann D.E., House W.F., Terry R., et al. Glomus jugulare tumors: effect of irradiation. Trans Am Acad Ophthalmol Otolaryngol. 1972;76(6):1423-1431.
128. Fayad J.N., Schwartz M.S., Brackmann D.E. Treatment of recurrent and residual glomus jugulare tumors. Skull Base. 2009;19(1):92-98.
129. Poznanovic S.A., Cass S.P., Kavanagh B.D. Short-term tumor control and acute toxicity after stereotactic radiosurgery for glomus jugulare tumors. Otolaryngol Head Neck Surg. 2006;134(3):437-442.
130. Varma A., Nathoo N., Neyman G., et al. Gamma knife radiosurgery for glomus jugulare tumors: volumetric analysis in 17 patients. Neurosurgery. 2006;59(5):1030-1036. discussion 1036
131. Henzel M., Hamm K., Gross M.W., et al. Fractionated stereotactic radiotherapy of glomus jugulare tumors. Local control, toxicity, symptomatology, and quality of life. Strahlenther Onkol. 2007;183(10):557-562.
132. Lim M., Bower R., Nangiana J.S., et al. Radiosurgery for glomus jugulare tumors. Technol Cancer Res Treat. 2007;6(5):419-423.
133. Saringer W., Khayal H., Ertl A., et al. Efficiency of gamma knife radiosurgery in the treatment of glomus jugulare tumors. Minim Invasive Neurosurg. 2001;44(3):141-146.
134. Schwaber M.K., Gussack G.S., Kirkpatrick W. The role of radiation therapy in the management of catecholamine-secreting glomus tumors. Otolaryngol Head Neck Surg. 1988;98(2):150-154.
135. Eustacchio S., Leber K., Trummer M., et al. Gamma knife radiosurgery for glomus jugulare tumours. Acta Neurochir (Wien). 1999;141(8):811-818.
136. Foote R.L., Pollock B.E., Gorman D.A., et al. Glomus jugulare tumor: tumor control and complications after stereotactic radiosurgery. Head Neck. 2002;24(4):332-338. discussion 338-339
137. Jordan J.A., Roland P.S., McManus C., et al. Stereotastic radiosurgery for glomus jugulare tumors. Laryngoscope. 2000;110(1):35-38.
138. Martin J.J., Kondziolka D., Flickinger J.C., et al. Cranial nerve preservation and outcomes after stereotactic radiosurgery for jugular foramen schwannomas. Neurosurgery. 2007;61(1):76-81. discussion 81
139. Liscak R., Vladyka V., Wowra B., et al. Gamma knife radiosurgery of the glomus jugulare tumour—early multicentre experience. Acta Neurochir (Wien). 1999;141(11):1141-1146.
140. Lalwani A.K., Jackler R.K., Gutin P.H. Lethal fibrosarcoma complicating radiation therapy for benign glomus jugulare tumor. Am J Otol. 1993;14(4):398-402.
141. Pollock B.E. Stereotactic radiosurgery in patients with glomus jugulare tumors. Neurosurg Focus. 2004;17(2):E10.
142. Redvanly R.D., Hudgins P.A., Gussack G.S., et al. CT of muscle necrosis following radiation therapy in a patient with head and neck malignancy. AJNR Am J Neuroradiol. 1992;13(1):220-222.
143. Gottfried O.N., Liu J.K., Couldwell W.T. Comparison of radiosurgery and conventional surgery for the treatment of glomus jugulare tumors. Neurosurg Focus. 2004;17(2):E4.
144. Foote R.L., Coffey R.J., Gorman D.A., et al. Stereotactic radiosurgery for glomus jugulare tumors: a preliminary report. Int J Radiat Oncol Biol Phys. 1997;38(3):491-495.