CHAPTER 245 Surgery for Malignant Peripheral Nerve Sheath Tumors
Malignant peripheral nerve sheath tumors (MPNSTs) are an uncommon variety of soft tissue sarcoma of ectomesenchymal origin. The World Health Organization (WHO) coined the term MPNST to replace previous heterogeneous and often confusing terminology, such as malignant schwannoma, malignant neurilemoma, neurogenic sarcoma, and neurofibrosarcoma. Although MPNST is now used to identify any malignant tumor arising from a peripheral nerve or its attendant sheath, it does not refer to tumors arising from the epineurium or the vasculature of peripheral nerves.1,2
MPNSTs arise from major or minor peripheral nerve branches3 or sheaths of peripheral nerve fibers4,5 and are derived from Schwann cells or pluripotent cells of neural crest origin.6
Arthur Purdy Stout (1885-1967) played a pivotal role in the development of our current understanding of the pathogenesis of peripheral nerve sheath tumors by identifying the Schwann cell as the major contributor to the formation of benign as well as malignant neoplasms of the nerve sheath.7–9 Although this remains essentially true, the cell of origin of the MPNST remains elusive and has not yet conclusively been identified. Some have suggested these tumors may have multiple cell line origins.
Epidemiology and Risk Factors
It is estimated that from 5% to 10% of the 6000 soft tissue sarcomas diagnosed in the United States per year are malignant nerve sheath tumors, with an incidence of 0.001% in the general population.10 These tumors occur with equal frequency in males and females, although some series have shown a female preponderance.3,4 There is no racial association. Most studies show that the peak incidence of MPNSTs is in the seventh decade of life in the general population but in the third or fourth decade in people with neurofibromatosis type 1 (NF1),11,12 although these tumors may occur at a much younger age in either population.13
Most MPNSTs occur in patients with NF1, with a cumulative lifetime risk of up to 10%. Nonetheless, a surprising number occur as solitary MPNSTs, unassociated with neurofibromatosis or other predilections such as irradiation. Individuals with NF1 and internal plexiform neurofibromas are 18 times more likely to develop MPNSTs than patients without internal plexiform neurofibromas.14 In the general population, plexiform neurofibromas can undergo malignant transformation to an MPNST with an estimated lifetime risk of 3% to 5%, whereas in NF1 patients, it can be as high as 15% to 20%. The dermal neurofibromas seen in NF1, although more numerous and a more troubling cosmetic problem, do not undergo malignant transformation. Only rarely do MPNSTs arise from malignant degeneration of a schwannoma, ganglioneuroma, or pheochromocytoma. Ten percent of these tumors occur in patients who have undergone radiation treatments for other diseases, and they occur on average 15 years after the treatments.15
The effect of radiation on peripheral nerves was described initially in animal experimental work by Bergstrom and Cavanagh.16,17 The incidence of radiation-induced MPNSTs reported in large series ranges from 5.5% to 11%.6,13,18,19 There are also several published reports relating to patients with and without NF1 and radiation treatments.13,20,21 Ducatman and associates13 described 12 patients with postirradiation MPNSTs, 7 of whom had NF1. Two of the 7 had received radiation therapy for optic gliomas 5 and 17 years previously. More recently, Loree and colleagues21 described two of four NF1 patients who developed MPNSTs after head and neck irradiation, whereas Baehring reported such after radiation for Wilms’ tumor and Hodgkin’s disease.6
Diagnosis
The diagnosis of these tumors remains problematic because it is based primarily on clinical suspicion. As with any patient, a history and physical examination are the place to begin the assessment for a peripheral nerve tumor. In the history, special note should be made of when the mass, if palpable, was noticed, and the onset of symptoms such as pain and motor or sensory deficit. Rapid increase in the size of a mass or rapid onset of symptoms should immediately alert the surgeon to the possibility of a malignancy (Fig. 245-1). A patient with a known history of NF1, neurofibromatosis type 2 (NF2), or schwannomatosis who presents with a tumor that shows recent rapid increase in size, a new or progressive neurologic deficit, or pain should alert the examiner to a suspected malignant degeneration. The examiner should question and record the location, quality, and radiation of pain. The location and extent of motor weakness, if present, and the location and extent of sensory deficit should be defined and recorded. A family history of peripheral nerve problems or any other genetic disorders should be closely questioned, and a history of previous radiation treatments should be discussed. Systemic diseases or any preexisting conditions that can contribute to peripheral nerve problems should also be questioned (i.e., diabetes mellitus, cancer). Any recent illnesses, even those as seemingly minor as flu, should be questioned and recorded. Because many prescription medications can cause peripheral neuropathies, a medication history should also be recorded.
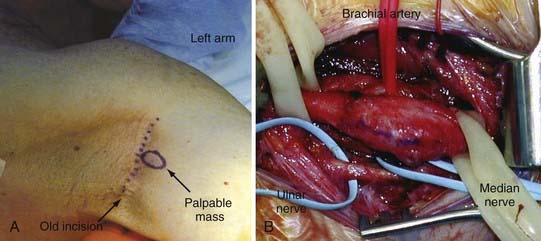
Imaging
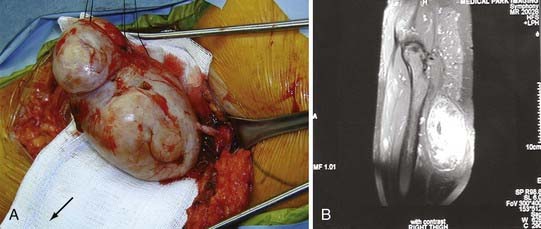
MRI can contribute significant information about the suspected pathology. This information is extremely useful preoperative information. Unfortunately, whether a tumor is benign or malignant cannot be discerned definitively from the image alone (Fig. 245-2). Areas of hemorrhage or necrosis, heterogeneous enhancement, and cystic areas may suggest a malignancy but are no means definitive and can even be seen in benign tumors.
Positron emission tomography (PET) with the glucose analog 18FDG is a dynamic imaging technique that permits the visualization and quantification of glucose metabolism in cells and reflects the increase in metabolism in malignant tumors.22,23 A retrospective study of 18 NF1 patients demonstrated that 18FDG-PET is a potentially useful, noninvasive method for detecting malignant change in plexiform neurofibromas.22 However, the distinction between low-grade MPNSTs and benign plexiform neurofibromas was not clear in all the cases. The new tracer 18F-thymidine, which detects DNA turnover, might be helpful in distinguishing low-grade MPNSTs from active, benign plexiform neurofibromas in future PET-based studies.24
Treatment
When the diagnosis of MPNST is suspected, surgery is the mainstay of treatment of these tumors.24–26 Resectability depends largely on location and ranges from 20% in paraspinal MPNSTs to 95% in tumors of the extremity.2,6,13,18,19,27,28 The ultimate aim of surgery is complete removal of the lesion with tumor-free margins.13,29
Unfortunately, for neurogenic sarcomas that involve brachial or pelvic plexus or the proximal portion of the arm, a wide resection to clean margins is not accomplished without paralysis or even limb loss necessitated by vascular supply sacrifice. Thus, wide local resection appears to work better for neurogenic sarcomas involving the more distal portions of the limb. For more proximal lesions, amputation of the limb may be required, but many patients, given the cosmetic deformity and overall poor prognosis of these tumors, often refuse such recommendations (Fig. 245-3).
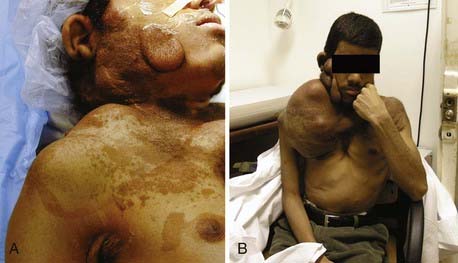
The reported local recurrence rate of MPNST following gross total resection is 32% to 65% after median intervals of 5 to 32.2 months.6,13,18,19,27,28,30 There have been statements that a microscopically non–tumor-free margin may be an indication of a highly aggressive, invasive tumor, rather than being a reflection of inadequate surgical technique, assuming that scrupulous attempts to attain microscopically tumor-free margins have been made.31 Local aggressive resection and control are thought to decrease the risk for systemic metastasis and to contribute to a better overall prognosis.
Reconstruction of the nerve after surgery for removal of malignant brachial and lumbosacral plexus lesions is not advocated. Because the needed adjuvant radiation and chemotherapy will compromise the ability of the axons to advance and grow down to the target organ, attempts at reconstruction are thought to be a futile exercise. Furthermore, the natural history of MPNST is often not long enough for effective reinnervation.29 Amputation may be indicated for extensive tumors and for MPNSTs that recur after apparently adequate excision.24
Radiotherapy and chemotherapy are unfortunately of only limited value. However, they are routinely applied because our armamentarium against these tumors is so limited. To date, only complete surgical excision before metastasis is likely to give a good prognosis.2,6,13,21,32
Classification and Pathologic Diagnosis and Grading
The NF1 gene on chromosome 17q11.2 was identified by positional cloning, and its protein product, neurofibromin, functions as a tumor suppressor.33–35 One of the functions of neurofibromin is to reduce cell proliferation by accelerating the inactivation of the proto-oncogene p21-ras, which has a pivotal role in mitogenic intracellular signaling pathways.35 Patients with MPNST demonstrate aberrations in 17q11.2-22 and show loss of NF1 gene expression, with resultant increase in ras oncogene expression.
A soft tissue sarcoma is denoted to be of neurogenic origin if it fulfills one the following criteria: (1) macroscopic or microscopic association with a peripheral nerve, (2) malignant transformation of a preexisting neurofibroma, or (3) immunohistochemical or ultrastructural features consistent with peripheral nerve origin.11
Classification of MPNSTs is based on that used for the much more common soft tissue sarcomas, from grade 1 to 3, depending on number of mitotic figures and degree of nuclear and cellular atypia.11
Tumors are also classified based on gross size. Tumor size at surgery is stratified to be greater than or less than 5 cm. This has been based on previous reports in which soft tissue sarcomas larger than 5 cm correlated with a worse prognosis.11,13,36–39 The tumor size influences the treatments and outcomes in several ways. First, patients with neurogenic sarcomas larger than 5 cm present twice as often with neurological motor or sensory deficits compared with patients with neurogenic sarcomas 5 cm or smaller. Second, tumor size correlated with pathologic grade, an important predictor of survival and systemic spread. The most aggressive grade 3 tumors are found in patients with neurogenic sarcomas larger than 5 cm. Third, the size of the presenting neurogenic sarcoma influences the ability to obtain tumor-free margins in the first en bloc resection. Fourth, as with other sarcomas, the presenting size of neurogenic sarcomas appears to negatively influence the survival rate.11
Pathology
Gross inspection of MPNSTs reveals a fusiform, fleshy, tan-white mass with areas of degeneration and secondary hemorrhage. The nerve proximal and distal to the tumor may be thickened owing to spread of the tumor along the epineurium and perineurium. The cell of origin is the Schwann cell, although the mature Schwann cell marker S-100 may be absent in about 50% of cases because of dedifferentiation.29
The minimum histologic examination should comprise sections stained with conventional tinctorial stains including hematoxylin and eosin stain and reticulin stain. At histologic analysis, MPNSTs are nonencapsulated infiltrating tumors composed of spindle cells arranged in a whorling pattern with irregular nuclei, cyst formation, and nuclear palisading.40 Mitotic figures are readily visible, with more than one per high-power field, and 50% to 90% of cases are immunoreactive with S-100 protein staining.41 Necrosis, pseudocystic change, or hemorrhage may also be found. The pathologic criteria for malignancy include invasion of surrounding tissues by tumor cells, vascular invasion, marked nuclear pleomorphism, necrosis, and the presence of mitoses.42
Using the hematoxylin and eosin–stained sections, the tumors are graded 1 to 3, with a scheme for soft tissue sarcomas based on cellularity, nuclear pleomorphism, anaplasia, mitotic rate (mitotic figures in 10 high-power fields), microvascular proliferation, and degree of necrosis and invasion.3,11,43,44 In addition, immunohistochemical stains for S-100 protein, the skeletal muscle markers desmin and myogenin, and a proliferation marker (MIB-1) are required.
Other spindle cell tumors may be excluded with appropriate immunohistochemical markers. The diagnosis of a neurogenic sarcoma cannot be reliably made from examination of hematoxylin and eosin–stained sections alone because other soft tissue sarcomas, arising from fibroblasts or smooth muscle cells, may have similar appearances. Three immunohistochemical markers (S-100, Leu-7, and myelin basic protein), although not diagnostic by themselves because of significant false-positive and false-negative rates, are used to facilitate the diagnosis of neurogenic sarcomas.13,45–49
Mitotic rates are evaluated as 0, 1, and 2 depending on the numbers as less than 5, 5 to 10, and more than 10 per high-power field. More than 5 mitoses per 10 high-power fields has been considered to denote a high-grade tumor because a single mitotic figure may be significant in a tumor with hypercellularity and nuclear atypia.24 More than 5% cellular staining of an MIB-1 proliferation marker has been considered to denote a high-grade tumor.50,51
On electron microscopic examination, ultrastructural features suggestive of a neurogenic origin for the tumors recapitulate the features of normal Schwann cells. These features include wavy, buckled, or comma-shaped nuclei arranged in sweeping fascicles with extensive perineural and intraneural spread of the tumor. Also often seen is a proliferation of the tumor in the subendothelial zones of vessels, so much so that neoplastic cells appear to herniate into the lumen.52 Also important is the lack or relative lack of ultrastructural features such as myofibrils, which suggests other origins for soft tissue sarcomas.
These tumors often create diagnostic problems because of their cellular origin and histopathologic similarities with other spindle cell sarcomas such as monophasic synovial sarcoma, leiomyosarcoma, and fibrosarcoma. Another interesting clinical feature of this tumor is multifocality and development of second primary tumors of the same histology.13 However, it is not always possible to demonstrate the origin from a nerve, especially when it arises from a small peripheral branch. This point was exemplified in a series by Nambisan and colleagues, in which nerves could not be identified in 61% of cases of MPNST,26 and in the series by Bilge and coworkers, in which nerve origin could be identified only in 45% to 56% cases.53 Still, there are several distinct features, such as proliferation of tumor in the subendothelial zones of vessels, with neoplastic cell herniation into vessel lumen, and proliferation of small vessels in the walls of the large vessels, which are characteristic features of MPNSTs.54
Radiotherapy
Radiotherapy provides local control and may delay the onset of recurrence but has little effect on long-term survival. However, there are reports of routine postoperative radiotherapy and even radiotherapy as a single modality for MPNST in the literature.55 Currently, postoperative radiotherapy is recommended by the Oncology Consensus Group24 as part of a uniform treatment policy for MPNSTs, much like other high-grade soft tissue sarcomas,2,8 even if clear surgical margins are obtained. Adjuvant radiotherapy should be given whenever possible for intermediate- to high-grade lesions and for low-grade tumors after a marginal excision. Although radiation therapy does provide local control and delay recurrence, it has little effect on long-term survival.
The timing of irradiation (either before or after surgery) and the associated pros and cons are actively being studied by the group at the University of Toronto. Postoperative radiotherapy involves irradiation of the entire operative field, with a 5-cm field margin. Preoperative radiotherapy involves irradiation of the overt tumor alone, again with a 5-cm margin. The usual dose administered is 6000 to 7000 cGy Irradiation before surgery has been recommended if the location, size, and distribution of the tumor make it more technically difficult to provide optimal radiotherapy after excision, dissection is anticipated along a major neurovascular bundle (with the possibility of leaving microscopic disease in critical structures), or remote tissue flaps or skin grafts are required for wound management after resection.11,56,57
Chemotherapy
Like most soft tissue sarcomas, MPNSTs are traditionally chemotherapy insensitive. Chemotherapy for adult soft tissue sarcomas is usually confined to the treatment of metastatic disease. Systemic spread, especially pulmonary metastasis, is the terminal event and, despite limited efficacy, chemotherapy is indicated in this situation.11 Because of the rare incidence of MPNSTs, large trials for effectiveness of chemotherapy in MPNSTs are impossible, and most current data are based on case reports or small case series or on regimens proved successful for other soft tissue sarcomas.
Few drugs have been shown to be effective, and treatment comprises single-agent doxorubicin or a combination of doxorubicin and ifosfamide,58 with a partial response rate of 20% to 25%. With doxorubicin, there was little impact on survival. Dacarbazine was later noted to have activity against these tumors and was combined with doxorubicin in the CYVADIC regimen.59 In the late 1980s, a number of phase II trials demonstrated the superiority of ifosfamide to cyclophosphamide in soft tissue sarcomas. The European Organization for Research and Treatment of Cancer (EORTC) reported a phase III study comparing doxorubicin to doxorubicin plus ifosfamide to CYVADIC and demonstrated an insignificantly increased response rate with combination therapy, at a cost of increased toxicity with doxorubicin plus ifosfamide.58
Chemotherapy is not curative, and therefore its use is controversial. There is a benefit at 10 years for progression-free survival for both local and distant relapse as shown by a meta-analysis.60 However, the magnitude of any overall survival benefit is small (4%, not statistically significant).
Prognosis
Patients (and parents of pediatric patients) with NF1 should be made aware of the relatively high risk for MPNST associated with the disease. They should be instructed to contact their physician sooner rather than later should rapid enlargement, pain, or neurologic deficit occur.12 MPNSTs have a poor prognosis, and in most studies, the prognosis appears to be worse in patients with NF1.13,30 MPNSTs in broad terms can be considered fatal, with the worst outcomes related consistently to incomplete tumor resection.13,19 Sarcomatous cells spread extensively within the fascial planes, resulting in high recurrence rates and ultimately systemic spread.11,61
MPNSTs have a poor prognosis because metastases to the lung, liver, brain, soft tissue, bone, regional lymph nodes, skin, and retroperitoneum are common.13 Adverse prognostic factors include large size (>5 cm), higher tumor grade, advanced histology, non–tumor-free surgical margin, and associated NF1.11
Basso-Ricci demonstrated a 56% disease-free survival rate using combined surgery and radiation therapy for MPNST.62 MPNSTs have the highest recurrence rate of any sarcomas,63 and adequate initial treatment gives the best chance for survival.64 Even with aggressive therapy, local recurrence of tumor is seen in 50% of patients.65 Angelov and coworkers recommend that sacrifice of major neural structures such as the sciatic nerve should be performed, without exception, to achieve resection with tumor-free margins.11 Other studies using preoperative radiotherapy or interstitial radiotherapy demonstrated that only a minority of patients with non–tumor-free margins ultimately develop locally recurrent or distant metastatic disease.66–68 Hematogenous metastatic spread occurs most commonly to the lungs. The 5-year survival rates in large series have been reported to range between 16% and 52%.13,18,19,69 The reported 5-year survival rate for patients with MPNSTs without NF1 is as high as 50%. It drops to as low as 10% for MPNSTs in patients with NF1.30
Angelov L, Davis A, O’Sullivan B, et al. Neurogenic sarcomas: experience at the University of Toronto. Neurosurgery. 1998;43:56-64.
Baehring J, Betensky R, Batchelor T. Malignant peripheral nerve sheath tumor: the clinical spectrum and outcome of treatment. Neurology. 2003;61:696-698.
Bhattacharyya AK, Perrin R, Guha A. Peripheral nerve tumors: management strategies and molecular insights. J Neurooncol. 2004;69:335-349.
Bilge B, Ates LE, Demiryont M, et al. Malignant peripheral nerve sheath tumors associated with neurofibromatosis type 1. Pathol Oncol Res. 2000;9:201-205.
Cashen DV, Parisien RC, Raskin K, et al. Survival data for patients with malignant schwannoma. Clin Orthop Relat Res. 2004;426:69-73.
Enziger F, Weiss S. Benign Tumors of Peripheral Nerves: Malignant Tumors of the Peripheral Nerves, 4th ed. St. Louis: Mosby; 2001.
Evans DG, Baser ME, McGaughran J, et al. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet. 2002;39:311-314.
Ferner RE, Gutmann DH. International consensus statement on malignant peripheral nerve sheath tumors in neurofibromatosis. Cancer Res. 2002;62:1573-1577.
Loree TR, North JHJr, Werness BA, et al. Malignant peripheral nerve sheath tumors of the head and neck: analysis of prognostic factors. Otolaryngol Head Neck Surg. 2000;122:667-672.
Pisters P, Leung D, Woodruff J, et al. Analysis of prognostic factors in 1,041 patients with localized soft tissue sarcomas of the extremities. J Clin Oncol. 1996;14:1679-1689.
Stout A. The peripheral manifestation of the specific nerve sheath tumor (neurilemoma). Am Cancer. 1935;24:751-796.
Suh JS, Abenoza P, Galloway HR, et al. Peripheral (extracranial) nerve tumors: correlation of MR imaging and histologic findings. Radiology. 1992;183:341-346.
Tucker T, Wolkenstein P, Revuz J, et al. Association between benign and malignant peripheral nerve sheath tumors in NF1. Neurology. 2005;65:205-211.
Wong WW, Hirose T, Scheithauer BW, et al. Malignant peripheral nerve sheath tumor: analysis of treatment outcome. Int J Radiat Oncol Biol Phys. 1998;42:351-360.
Yamaguchi U, Hasegawa T, Hirose T, et al. Low grade malignant peripheral nerve sheath tumour: varied cytological and histological patterns. J Clin Pathol. 2003;56:826-830.
1 Dasgupta TK, Choudhuri PK. Tumors of Soft Tissue, 2nd ed. Norwalk, Conn: Appleton and Lange; 1998.
2 Wanebo JE, Malik JM, VandenBerg SR, et al. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 28 cases. Cancer. 1993;71:1247-1253.
3 D’Agostino AN, Soule EH, Miller RH. Sarcomas of the peripheral nerves and somatic soft tissues associated with multiple neurofibromatosis (von Recklinghausen’s disease). Cancer. 1963;16:1015-1027.
4 Cashen DV, Parisien RC, Raskin K, et al. Survival data for patients with malignant schwannoma. Clin Orthop Relat Res. 2004;426:69-73.
5 Hirose T, Scheithauer BW, Sano T. Perineurial malignant peripheral nerve sheath tumor (MPNST): a clinicopathologic, immunohistochemical, and ultrastructural study of seven cases. Am J Surg Pathol. 1998;22:1368-1378.
6 Baehring J, Betensky R, Batchelor T. Malignant peripheral nerve sheath tumor: the clinical spectrum and outcome of treatment. Neurology. 2003;61:696-698.
7 Stout A. The peripheral manifestation of the specific nerve sheath tumor (neurilemoma). Am Cancer. 1935;24:751-796.
8 Stout A. Tumors of Peripheral Nervous System. Washington, DC: Armed Forces Institute of Pathology; 1949. Vol Sect. 2, fasc. 6
9 Woodruff JM. Arthur Purdy Stout and the evolution of modern concepts regarding peripheral nerve sheath tumors. Am J Surg Pathol. 1986;10(suppl 1):63-67.
10 Hadju S. Peripheral nerve sheath tumors. Histogenesis, classification, and prognosis [comment]. Cancer. 1993;72:3549-3552.
11 Angelov L, Davis A, O’Sullivan B, et al. Neurogenic sarcomas: experience at the University of Toronto. Neurosurgery. 1998;43:56-64.
12 Evans DG, Baser ME, McGaughran J, et al. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet. 2002;39:311-314.
13 Ducatman BS, Scheithauer BW, Piepgras DG, et al. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer. 1986;57:2006-2021.
14 Tucker T, Wolkenstein P, Revuz J, et al. Association between benign and malignant peripheral nerve sheath tumors in NF1. Neurology. 2005;65:205-211.
15 Amin A, Saifuddin A, Flanagan A, et al. Radiotherapy-induced malignant peripheral nerve sheath tumor of the cauda equina. Spine. 2004;29:E506-509.
16 Bergstrom R. Morphological studies on peripheral nerves exposed to a beam of high energy protons. Acta Pathol Microbiol Scand Suppl. 1962;154:80-82.
17 Cavanagh JB. Effects of x-irradiation on the proliferation of cells in peripheral nerve during Wallerian degeneration in the rat. Br J Radiol. 1968;41:275-281.
18 Kourea HP, Bilsky MH, Leung DH, et al. Subdiaphragmatic and intrathoracic paraspinal malignant peripheral nerve sheath tumors: a clinicopathologic study of 25 patients and 26 tumors. Cancer. 1998;82:2191-2203.
19 Wong WW, Hirose T, Scheithauer BW, et al. Malignant peripheral nerve sheath tumor: analysis of treatment outcome. Int J Radiat Oncol Biol Phys. 1998;42:351-360.
20 D’Agostino AN, Soule EH, Miller RH. Primary malignant neoplasms of nerves (malignant neurilemomas) in patients without manifestations of multiple neurofibromatosis (von Recklinghausen’s disease). Cancer. 1963;16:1003-1014.
21 Loree TR, North JHJr, Werness BA, et al. Malignant peripheral nerve sheath tumors of the head and neck: analysis of prognostic factors. Otolaryngol Head Neck Surg. 2000;122:667-672.
22 Ferner RE, Lucas JD, O’Doherty MJ, et al. Evaluation of (18)fluorodeoxyglucose positron emission tomography ((18)FDG PET) in the detection of malignant peripheral nerve sheath tumours arising from within plexiform neurofibromas in neurofibromatosis 1. J Neurol Neurosurg Psychiatry. 2000;68:353-357.
23 Strauss LG, Conti PS. The applications of PET in clinical oncology. J Nucl Med. 1991;32:623-648.
24 Ferner RE, Gutmann DH. International consensus statement on malignant peripheral nerve sheath tumors in neurofibromatosis. Cancer Res. 2002;62:1573-1577.
25 Ducatman BS, Scheithauer BWP, Reiman DG, et al. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer. 1986;57:2006-2021.
26 Nambisan RN, Rao U, Moore R, Karakousis CP. Malignant soft tissue tumors of nerve sheath origin. J Surg Oncol. 1984;25:268-272.
27 Hruban RH, Shiu MH, Senie RT, Woodruff JM. Malignant peripheral nerve sheath tumors of the buttock and lower extremity. A study of 43 cases. Cancer. 1990;66:1253-1265.
28 Vege DS, Chinoy RF, Ganesh B, Parikh DM. Malignant peripheral nerve sheath tumors of the head and neck: a clinicopathological study. J Surg Oncol. 1994;55:100-103.
29 Bhattacharyya AK, Perrin R, Guha A. Peripheral nerve tumors: management strategies and molecular insights. J Neurooncol. 2004;69:335-349.
30 Doorn PF, Molenaar WM, Buter J, Hoekstra HJ. Malignant peripheral nerve sheath tumors in patients with and without neurofibromatosis. Eur J Surg Oncol. 1995;21:78-82.
31 Brennan MF. The enigma of local recurrence. The Society of Surgical Oncology. Ann Surg Oncol. 1997;4:1-12.
32 Greager JA, Reichard KW, Campana JP, DasGupta TK. Malignant schwannoma of the head and neck. Am J Surg. 1992;163:440-442.
33 Viskochil D, Buchberg AM, Xu G, et al. Deletions and a translocation interrupt a cloned gene at the neurofibromatosis type 1 locus. Cell. 1990;62:187-192.
34 Wallace MR, Marchuk DA, Andersen LB, et al. Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients. Science. 1990;249:181-186.
35 Xu GF, O’Connell P, Viskochil D, et al. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell. 1990;62:599-608.
36 LeVay J, O’Sullivan B, Catton C, et al. Outcome and prognostic factors in soft tissue sarcomas in the adult. Int J Radiat Oncol Biol Phys. 1993;27:1091-1099.
37 Pisters P, Leung D, Woodruff J, et al. Analysis of prognostic factors in 1,041 patients with localized soft tissue sarcomas of the extremities. J Clin Oncol. 1996;14:1679-1689.
38 Saddegh MK, Lindholm J, Lundberg A, et al. Staging of soft-tissue sarcomas. Prognostic analysis of clinical and pathological features. J Bone Joint Surg Br. 1992;74:495-500.
39 Singer S, Corson JM, Gonin R, et al. Prognostic factors predictive of survival and local recurrence for extremity soft tissue sarcoma. Ann Surg. 1994;219:165-173.
40 Cotran R, Kumar V, Robbins S. Robbins Pathologic Basis of Disease. Philadelphia: Saunders; 1994.
41 Johnson MD, Glick AD, Davis BW. Immunohistochemical evaluation of Leu-7, myelin basic-protein, S100-protein, glial-fibrillary acidic-protein, and LN3 immunoreactivity in nerve sheath tumors and sarcomas. Arch Pathol Lab Med. 1988;112:155-160.
42 Enziger F, Weiss S. Benign Tumors of Peripheral Nerves: Malignant Tumors of the Peripheral Nerves, 4th ed. St. Louis: Mosby; 2001.
43 Trojanowski JQ, Kleinman GM, Proppe KH. Malignant tumors of nerve sheath origin. Cancer. 1980;46:1202-1212.
44 Wilson AN, Davis A, Bell RS, et al. Local control of soft tissue sarcoma of the extremity: the experience of a multidisciplinary sarcoma group with definitive surgery and radiotherapy. Eur J Cancer. 1994;30A:746-751.
45 Hirose T, Hasegawa T, Kudo E, et al. Malignant peripheral nerve sheath tumors: an immunohistochemical study in relation to ultrastructural features. Hum Pathol. 1992;23:865-870.
46 Kikuchi A, Akiyama M, Han-Yaku H, et al. Solitary cutaneous malignant schwannoma. Immunohistochemical and ultrastructural studies. Am J Dermatopathol. 1993;15:15-19.
47 Matus A, Mughal S. Immunohistochemical localization of S-100 protein in the brain. Nature. 1975;258:746-748.
48 Moore BW, McGregor D. Chromatographic and electrophoretic fractionation of soluble proteins of brain and liver. J Biol Chem. 1965;240:1647-1653.
49 Wick MR, Swanson PE, Scheithauer BW, Manivel JC. Malignant peripheral nerve sheath tumor. An immunohistochemical study of 62 cases. Am J Clin Pathol. 1987;87:425-433.
50 Yamaguchi U, Hasegawa T, Hirose T, et al. Low grade malignant peripheral nerve sheath tumour: varied cytological and histological patterns. J Clin Pathol. 2003;56:826-830.
51 Zhou H, Coffin CM, Perkins SL, et al. Malignant peripheral nerve sheath tumor: a comparison of grade, immunophenotype, and cell cycle/growth activation marker expression in sporadic and neurofibromatosis 1-related lesions. Am J Surg Pathol. 2003;27:1337-1345.
52 Feldkamp MM, Lau N, Provias JP, et al. Acute presentation of a neurogenic sarcoma in a patient with neurofibromatosis type 1: a pathological and molecular explanation. Case report. J Neurosurg. 1996;84:867-873.
53 Bilge B, Ates LE, Demiryont M, et al. Malignant peripheral nerve sheath tumors associated with neurofibromatosis type 1. Pathol Oncol Res. 2000;9:201-205.
54 Weiss SJr. Soft Tissue Tumors, 4th ed. St. Louis: Mosby; 2001.
55 Suh JS, Abenoza P, Galloway HR, et al. Peripheral (extracranial) nerve tumors: correlation of MR imaging and histologic findings. Radiology. 1992;183:341-346.
56 Neilson O, Cummings B, O’Sullivan B, et al. Comparison of treatment volumes in preoperative and postoperative irradiation for soft tissue sarcomas. Int J Radiat Oncol Biol Phys. 1991;21:1595-1599.
57 Peat B, Bell R, Davis A, et al. Wound healing complications after soft tissue sarcomas surgery. Plast Reconstr Surg. 1994;93:980-987.
58 Santoro A, Tursz T, Mouridsen H, et al. Doxorubicin versus CYVADIC versus doxorubicin plus ifosfamide in first-line treatment of advanced soft tissue sarcomas: a randomized study of the European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. J Clin Oncol. 1995;13:1537-1545.
59 Bramwell V, Rouesse J, Steward W, et al. Adjuvant CYVADIC chemotherapy for adult soft tissue sarcoma—reduced local recurrence but no improvement in survival: a study of the European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. J Clin Oncol. 1994;12:1137-1149.
60 Collaboration SM. Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Sarcoma Meta-analysis Collaboration. Lancet. 1997;350:1647-1654.
61 Simon M, Enneking W. The management of soft tissue sarcomas of the extremities. J Bone Joint Surg Am. 1976;58:317-327.
62 Basso-Ricci S. Therapy of malignant schwannomas: usefulness of an integrated radiologic-surgical therapy. J Neurosurg Sci. 1989;33:253-257.
63 Collin C, Godbold J, Hajdu S, Brennan M. Localized extremity soft tissue sarcoma: an analysis of factors affecting survival. J Clin Oncol. 1987;5:601-612.
64 Ghosh BC, Ghosh L, Huvos AG, Fortner JG. Malignant schwannoma. A clinicopathologic study. Cancer. 1973;31:184-190.
65 Murphey M, Smith W, Smith S, et al. Imaging of musculoskeletal neurogenic tumors: radiologic-pathologic correlation. Radiographics. 1999;19:1253-1280.
66 Pollock R. Neurogenic sarcomas: experience at the University of Toronto [comment]. Neurosurgery. 1998;43:65.
67 Tanabe KK, Pollock RE, Ellis LM, et al. Influence of surgical margins on outcome in patients with preoperatively irradiated extremity soft tissue sarcomas. Cancer. 1994;73:1652-1659.
68 Zelefsky M, Nori D, Shiu M, Brennan M. Limb salvage in soft tissue sarcomas involving neurovascular structures using combined surgical resection and brachytherapy. Int J Radiat Oncol Biol. 1990;19:913-918.
69 Sordillo PP, Helson L, Hajdu SI, et al. Malignant schwannoma—clinical characteristics, survival, and response to therapy. Cancer. 1981;47:2503-2509.






