[level-membership-for-neurosurgery-category]
CHAPTER 199 Supratentorial Hemispheric Tumors
Hemispheric neoplasms account for 25% of all primary brain tumors in children beyond infancy.1 This chapter discusses supratentorial hemispheric tumors, which in the pediatric population are generally of a low-grade variety. Hemispheric tumors can be classified by cellular origin: glial, mixed glial and neuronal, and primitive neuroectodermal. Differentiation between these tumors can be accomplished with a careful history and examination and is often fairly apparent before biopsy or resection thanks largely to modern imaging technology. The therapeutic options, prognoses, and molecular genetics of these tumors are unique and frequently vary considerably from such lesions in adults.
Epidemiology
Supratentorial low- and high-grade gliomas each occur annually in less than 1 per 100,000 individuals younger than 20 years, with low-grade being more common.2 In total, glial tumors account for 56% of primary central nervous system (CNS) tumors in children younger than 14 years and for 45% of such tumors in those between the ages of 14 and 19.1 Supratentorial primitive neuroectodermal tumors (SPNETs) occur less frequently, with most children affected when younger than 10 years.3 There has been no demonstration of a higher predilection for gliomas or SPNETs between the sexes. Dysembryoplastic neuroectodermal tumors (DNETs), pleomorphic xanthoastrocytoma (PXA), and low-grade glioneuronal tumors are found in children more so than adults. Oligodendrogliomas and DNETs may occur slightly more frequently in males.4 Oligodendrogliomas are rarer in children than adults and account for less than 1% to 2% of primary pediatric brain tumors.1,5 PXA is less common still.6,7
Familial genetic syndromes increasing the likelihood for the development of primary intracranial tumors include Turcot’s syndrome (colonic polyposis), Li-Fraumeni syndrome, and neurofibromatosis type 1. A history of previous CNS tumor, systemic cancer, or cranial irradiation also increases risk for the development of secondary brain tumors, most notably meningiomas and high-grade gliomas.8 Of note, secondary high-grade gliomas are more commonly supratentorial and may exhibit a distinct genetic profile from their primary counterparts. Once diagnosed, secondary high-grade glioma portends a worse prognosis than primary tumors despite aggressive intervention. The St. Jude’s experience found a cumulative 4% 15-year risk for secondary neoplasms developing after treatment of a primary CNS tumor,9 and in children treated for acute lymphoblastic leukemia, there is a 1.39% cumulative incidence of secondary brain tumors (gliomas and meningiomas) at 20 years, with cranial irradiation being a dose-dependent predisposing factor.
Clinical Findings
Seizures are a frequent symptom of supratentorial lesions in children, particularly low-grade tumors such as fibrillary astrocytoma, PXA, DNET, and ganglioglioma.4,10,11 The likelihood for these tumors to be accompanied by seizures increases with their predilection to occur in the temporal lobe. However, some evidence exists to support the notion that neuronal elements of the tumor itself can act as independent generators of seizures,12 and secondary seizure foci may exist around the tumor.
Glial Tumors
Imaging
The majority of primary supratentorial hemispheric tumors in children are of glial origin. Imaging features that help in predicting the biologic behavior of hemispheric tumors in children include an imaging correlate of cellular density (density or intensity), the degree of mass effect, a cortical location, and any remodeling of the overlying calvaria. Standard evaluation of symptomatic children suspected of harboring an intracranial lesion typically begins with computed tomography (CT). Low-grade tumors are likely to appear hypodense, whereas high-grade gliomas are usually hyperdense. High-grade neoplasms are frequently associated with surrounding hypodense regions representing vasogenic edema. Non–contrast-enhanced CT is sensitive for regions of calcification, usually an indicator of a more indolent lesion, including ganglioglioma or oligodendroglioma.13 Remodeling of the inner table of the skull to conform to the gyral patterns or thinning is commonly seen with low-grade neoplasms. With the administration of intravenous contrast material, high-grade astrocytomas are expected to reveal enhancement attributed to regions of hypervascularity and breakdown of the blood-brain barrier (BBB). Cavitation surrounded by contrast heterogeneity is likely to represent necrotic components, a harbinger of a malignant phenotype. This cavitation is contrasted with the well-circumscribed ring of enhancement that surrounds benign cystic tumors.
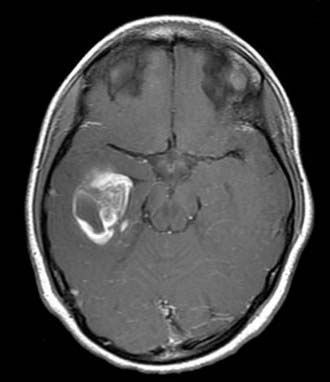
For enhanced anatomic definition and improved surgical planning, magnetic resonance imaging (MRI) has become the standard imaging modality. Low-grade fibrillary astrocytomas are frequently poorly delineated and will typically demonstrate gyral thickening, T1 hypointensity, and T2 hyperintensity, often best appreciated on T2 fluid-attenuated inversion recovery (FLAIR) sequences. PXA is more apt to show enhancing nodularity with frequent cystic components7 (Fig. 199-1).
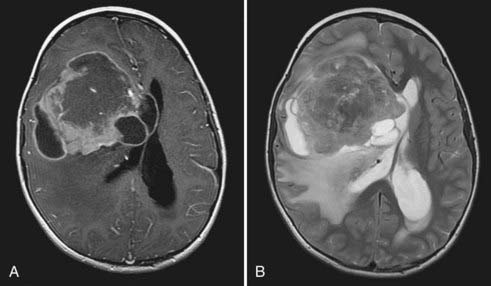
High-grade gliomas have mixed signal intensity, with predominant hypointensity on T1-weighted and hyperintensity on T2-weighted images.13 A broad region of T2 or FLAIR hyperintensity surrounding the tumor represents the highly infiltrative growth characteristic and surrounding edema frequently seen in these aggressive tumors (Fig. 199-2). This signal change most often extends in the direction of anisotropic white matter tracts and may cross the midline along commissural fibers, notably the corpus callosum. Regions of hypercellularity may result in restricted diffusion (bright appearance) within the lesion on diffusion-weighted imaging. Magnetic resonance spectroscopy, which is gaining popularity in narrowing the preoperative diagnosis of intracranial lesions, would be expected in high-grade components to demonstrate elevated peaks of choline, N-acetylaspartate, and lactate and a rise in the choline-to-creatine ratio.14
Pathology
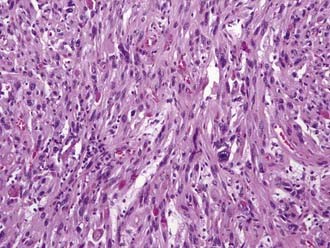
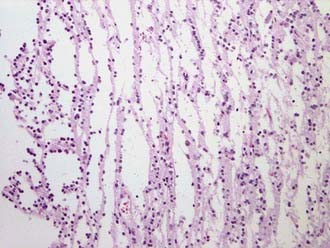
Classification or naming of these tumors is based on their cellular composition. Tumors composed of astrocytes with hypercellularity, infiltration, and a fibrillary matrix are designated fibrillary astrocytoma. The histologic hallmark of oligodendroglioma is perinuclear cytoplasmic shrinkage, which results in a halo appearance surrounding the cells’ nuclei and gives a classic “fried egg” appearance under lower magnification. The microvasculature takes on a “chicken wire” appearance, and calcifications may be seen grossly and under microscopy.15 PXAs have historically been thought to be purely astrocytic in origin, but these tumors may have neuronal progenitors as well. Histologically, these lesions show pleomorphism, lipid, reticulin, and perivascular lymphocytes (Fig. 199-3).
A critical distinction for the pathologist to make is between high- and low-grade lesions. Central review by an expert panel has been used in major studies to fulfill this task.16 Grade I astrocytoma, or juvenile pilocytic astrocytoma (JPA), is uncommonly hemispheric in location. High-grade or malignant gliomas represent World Health Organization (WHO) grades III and IV lesions. Grade III tumors demonstrate cellular pleomorphism, frequent mitoses, and the hypercellularity lacking in grade II lesions. Immunohistochemical testing will reveal positivity for glial fibrillary acidic protein (GFAP) and MIB-1 (anti–Ki-67 antibody). The most common grade III glial neoplasm is anaplastic astrocytoma (AA), but oligodendroglioma and PXA also have the potential to develop into grade III lesions.
The defining presence of necrosis raises the grade of the lesion to WHO IV and a diagnosis of glioblastoma multiforme. Pseudopalisading cellular architecture and significant microvascular proliferation are also seen. Like AA, immunohistochemistry is positive for GFAP. High-grade lesions will demonstrate a high mitotic index, and a high index of proliferation has been well characterized in Children’s Cancer Group (CCG) study 945 to correlate with worsened prognosis.17–19
Treatment
Unlike adult high-grade glioma, this diagnosis in children portends a 5-year survival rate of 30% to 50%, with a higher likelihood of long-term survivors.6,20,21 This prognosis is particularly significant and has relevance to survival from glioblastoma, where the mean survival of adults in contemporary series is less than 2 years.22 Cytoreduction on the order of 90% or greater has been associated with improved outcomes in children with high-grade glioma. The CCG studies 943 and 945 were instrumental in first demonstrating the value of total resection. Although it is difficult to control for adjuvant therapy, which varies from series to series, multiple reports have since corroborated a 5-year progression-free survival benefit of gross total resection on the order of greater than 20%. This holds true for both AA and glioblastoma.6,23–27 CCG 945 included 83 supratentorial high-grade gliomas for review and reported a 5-year progression-free survival rate of 35% for greater than 90% resection of glioblastoma versus 17% for subtotal resection. Furthermore, a 5-year progression-free survival rate of 44% was seen in children after total resection of AAs versus 22% for subtotal resection. These differences were statistically significant.27 The role of a second resection in improving survival for children with recurrent disease has yet to be well defined.
This goal of maximal tumor removal is also enhanced through the incorporation of several operative adjuncts. The principle of discriminating between tumor and parenchyma unifies all past and future improvements aimed at greater removal of tumor. The integration of high-resolution MRI with triplanar navigation is designed to offer the surgeon a reliable method for defining tumor margins with a greater degree of certainty. Errors attributed to brain shifting with partial tumor removal can be overcome with intraoperative modalities, including ultrasound and MRI. Tumor cell fluorescence may offer finer intraoperative resolution of tumor margins. Early results suggest that fluorescence-guided resection of glioblastoma in adults with the use of 5-aminolevulinic acid may improve the degree of tumor removal.28
For tumors associated with primary cortical regions, intraoperative brain mapping is an important safety mechanism. Motor function is assessed most reliably with this method, whereby direct cortical stimulation on the precentral gyrus triggers contralateral movement. The age of the child influences the ability to effectively perform cortical localization because of the variability in electrical impedance of the brain and the need for patient cooperation for language mapping. Older children will typically respond to a stimulus of 4 to 6 mA, whereas young children may require stimuli of 8 to 12 mA.29 Brain immaturity in children younger than 5 years may render direct stimulation ineffectual. In such cases, detection of the phase reversal potential between motor and sensory gyri is most likely to yield useful data. Because language mapping is reliant on intraoperative patient interaction and compliance, cortical localization of primary language representation is difficult in children younger than 10 years.30 Preoperative functional MRI has been shown to predict language dominance in infants and young children.31
Children with high-grade lesions or residual/recurrent low-grade gliomas may benefit from adjuvant radiation therapy. However, because of the well-established cognitive risks associated with cranial irradiation in very young children, radiation therapy is classically reserved for children older than 3 years.32 Cranial irradiation on the order of 50 to 60 Gy in children causes an average 10-point decline in IQ, with younger children being more adversely affected.33 Development of a secondary intracranial tumor is a significant risk in all children undergoing radiation therapy but is typically outweighed by the survival benefit in those with more aggressive malignancies. The risk of a secondary malignancy developing after radiation therapy has been reported to be just under 10%.34 With the development of conformal and fractionated therapy, delivery of radiation has progressed over time to allow higher doses to be given to the tumor while sparing surrounding normal brain.
The benefit of primary radiation therapy for low-grade glioma is dubious at best.16 Noteworthy studies evaluating adjuvant therapy for pediatric high-grade glioma, including the original CCG 943 publication in 1989, have included cranial irradiation as initial management in appropriate children.6,27,35 Most institutions currently use a fairly standard regimen of 50 to 60 Gy delivered to the tumor in roughly 25 fractions over a 5-week period for high-grade glioma. Data from recent studies suggest that the use of stereotactic radiosurgery, whereby a single high dose of radiation is delivered via intersecting beams to a desired tumor volume designated on a radiograph, should be reserved for patients with multiply recurrent low- and high-grade lesions who are not candidates for surgery.36–38
Chemotherapy is a vital component of multimodal treatment of high-grade glioma. The CCG 943 publication in 1989 established the use of chemotherapy as a standard initial adjuvant for pediatric high-grade glioma. That study prospectively randomized 58 children with high-grade glioma, 40 of whom harbored glioblastoma, into two therapeutic arms.25 One group underwent postoperative radiation therapy and combination chemotherapy consisting of lomustine, vincristine, and prednisone, whereas the other group underwent radiation therapy alone. The survival benefit in the chemotherapy group was significant, with a 5-year event-free survival rate of 46% versus 18% for radiation therapy alone.
The next most significant set of results came from CCG 945. Here, an “8 in 1” regimen of chemotherapeutics was administered to an experimental arm, whereas the control arm underwent the chemoradiation protocol in CCG 943. The “8-in-1” regimen consisted of vincristine, carmustine, procarbazine, hydroxyurea, dacarbazine, methylprednisolone, cisplatin, and cytarabine usually given in two cycles before radiation therapy and for up to 1 year afterward. Five-year event-free survival rates were higher in the “8-in-1” than in the standard therapy group (33% versus 26%).21
Oligodendroglioma, especially when anaplastic in nature, is also a good candidate for adjuvant therapy. Procarbazine, lomustine, and vincristine have been evaluated prospectively and been found to improve progression-free survival by almost 1 year in adults when administered in conjunction with surgery and radiation therapy.39,40 Overall survival is also improved in tumors harboring 1p and 19q loss of heterozygosity.
Chemotherapy is an even more important tool for children too young to undergo irradiation. For high-grade glioma, the North American Pediatric Oncology Group found a 3-year event-free survival rate of 43% for paired cyclophosphamide and vincristine and then cisplatin and etoposide without cranial irradiation.23,41 More recently, the French BBSFOP protocol demonstrated a 35% 5-year progression-free survival rate for surgical resection and chemotherapy alone.24 Despite being safer in long-term measures than radiation therapy for young children, the potential for toxicity, myelosuppression, and clinically significant infection is noteworthy and must be considered in patients with poor performance status.
With the addition in recent years of temozolomide to the armamentarium, the standards of treatment in adult neuro-oncology have decidedly changed. However, this generally well tolerated oral DNA-methylating agent has yet to show similar activity against pediatric high-grade glioma. This may in part be attributable to the differential response of methylguanine methyltransferase or the mismatch repair system in children, both of which potentially confer resistance. The Children’s Oncology Group recently published a large series of children undergoing temozolomide therapy for recurrent CNS tumors, 23 of which were high-grade gliomas. One child with high-grade glioma showed a partial response.42 With mechanisms of resistance in mind, the Pediatric Brain Tumor Consortium has recently reported phase I results for temozolomide given with the methylguanine methyltransferase inactivator O6-benzylguanine for recurrent high-grade glioma. Only four of eight patients completed therapy, but three did show a partial radiographic response to treatment.43
When high-grade gliomas arise as secondary tumors after treatment of a previous malignancy, the results are even more dismal. Responses to radiation therapy and chemotherapy are blunted, perhaps as a result of previous administration, and survival averages less than 1 year from the time of diagnosis.44
Mixed Glial/Neuronal Tumors
Tumors with mixed glial and neuronal components include DNET, ganglioglioma, and low-grade glioneuronal tumors. On MRI, gangliogliomas and DNETs are generally better circumscribed, occur most commonly in the frontotemporal cortex, and also show T1 hypointensity and T2 hyperintensity. Calcification should raise suspicion for ganglioglioma. Both ganglioglioma and DNET may show contrast enhancement in heterogeneous or ring-like patterns4,10,11 (Fig. 199-4). Characteristically, DNET has disproportionately less mass effect than would be predicted by the tumor size because of its “dysplastic” nature.
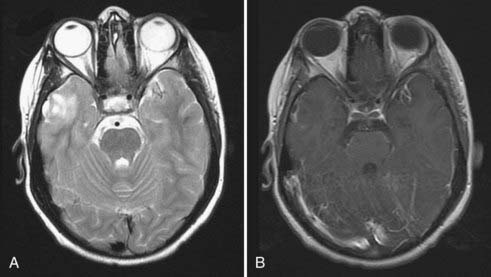
Gangliogliomas are a low-grade neoplasm in which the astrocytic components are distinct from the neuronal components. Non-neoplastic cysts and microcystic degeneration may be seen, along with hypercellularity, vascular proliferation, and calcification. Binucleate heterotopic neurons are a common finding.45 DNETs show dysplastic cortical neuronal elements with astrocytic features in a perpendicular columnar orientation with reference to the cortical surface45–47 (Fig. 199-5). Other less common primary tumors with mixed components include astroblastoma, a cystic lesion that histologically mimics ependymoma with the presence of pseudorosettes, and low-grade glioneuronal tumors. Glioneuronal tumors can be identified by the presence of a neurocytic component in the neuropil matrix that will stain positive for the neuronal marker synaptophysin, as well as an astrocytic component immunoreactive to GFAP.48
DNETs generally do not recur or progress after partial or total resection. Seizures are expected to improve in more than 80% of patients after surgery, with potentially greater than 70% being seizure free.4,49 Recurrent DNET has recently been recognized and should raise awareness that long-term follow-up is mandatory.50 Patients with ganglioglioma classically experience greater than a 90% 5-year progression-free survival rate after gross total resection.7,16,18
Primitive Neuroectodermal Tumors
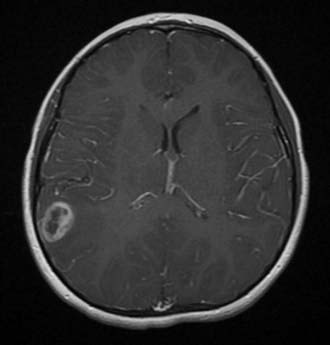
Along with high-grade glioma, SPNETs are aggressive childhood hemispheric malignancies and offer a significant challenge to treatment. On imaging, SPNETs predictably exhibit heterogeneous regions of contrast enhancement with possible surrounding edema.51 SPNETs may show similar mixed intensity and heterogeneous enhancement as a high-grade glioma, but they are usually better demarcated from surrounding brain (Fig. 199-6).
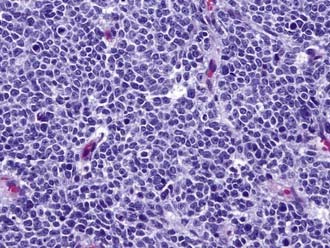
Unlike invasive glial tumors, SPNETs are delineated from normal brain. Hemispheric SPNETs fall under the broader pathologic classification of neuroectodermal tumors, which include medulloblastoma, retinoblastoma, and pineoblastoma. They are histologically represented as a classic small blue cell tumor with dense, hypercellular blue cells that have scant nuclei and frequent mitoses52 (Fig. 199-7). The cells are typically synaptophysin and GFAP positive, and hypervascularity is also common. Intratumoral hemorrhage and calcification may be seen.
Like children with high-grade glioma, the treatment of children with SPNETs is dependent on multimodal therapy consisting of surgery, radiation therapy, and chemotherapy. Aggressive resection has been shown to improve survival in children with SPNETs. Albright’s Pittsburgh series reported a 4-year survival rate of 40% in patients with less than a 1.5-cm2 postoperative residual versus 13% in those for whom this extent of resection was not met.3 Similarly, 5-year progression-free survival rates of up to 30% for children with no or minimal residual disease after surgery have been reported.53,54
Radiation therapy is a critical component in the management of newly diagnosed SPNETs. The Prospective German Brain Tumor Trials HIT 87, 88, 89, 91, and 92 studied 63 children with SPNETs, with 29 children being younger than 37 months. They found implementation of postsurgical radiotherapy to be the only significant predictor for progression-free survival in both children and infants.55,56 McBride and coauthors reported 15 patients treated with either radiation therapy and chemotherapy or chemotherapy alone after surgery. Only patients given radiation therapy remained progression free.57 Unlike glioma, the higher likelihood of primitive neuroectodermal tumor (PNET) to disseminate along the neuraxis supports prophylactic craniospinal irradiation with an involved boost to the primary site.56
Although no study has evaluated the efficacy of chemotherapy for SPNETs in a fashion analogous to the CCG study 943 for gliomas, it has become standard over the past 20 years in the initial management of SPNETs at large institutions.3,51,53 The Prospective German Brain Tumor Trials administered chemotherapy before and after irradiation. Agents used included ifosfamide, vincristine, methotrexate, cisplatin, and lomustine. This study found a 49.3% 3-year progression-free survival rate with optimal surgery followed by chemoradiation.55 The “8-in-1” regimen found efficacious in treating high-grade glioma has not produced similar results in children with SPNETs.58 Finally, the use of temozolomide has yet to meet significant success. Of the Children’s Oncology Group’s recent series of recurrent CNS tumors, 1 of 25 children with PNETs/medulloblastoma experienced a partial response.42
Interestingly, comparison of survival rates in patients with hemispheric PNET and pineoblastoma consistently reveals a difference in outcome. Unlike hemispheric PNET, aggressive multimodal therapy for pineoblastoma has yielded 3-year progression-free survival rates as high as 60% in some series.59,60 Greater ability to perform complete resection or increased permeability of the BBB in the pineal region may partly account for this survival difference, although the literature has yet to fully substantiate this theory.
Molecular Biology
Understanding abnormal gene amplification and expression continues to gain importance in oncology. Molecular profiling is clinically relevant in understanding tumorigenesis and resistance to chemotherapy, formulating prognoses, and developing tumor-specific therapy. This is perhaps most apparent in the study of high-grade glioma. Mutations known to occur in adult high-grade glioma and implications in terms of prognosis do not hold for children, even though these tumors appear to be histologically identical. Perhaps the best example of this is epidermal growth factor receptor (EGFR) gene amplification and overexpression. Well known to occur in adult high-grade glioma, EGFR gene amplification occurs in less than 5% of pediatric high-grade gliomas. Interestingly, a majority of pediatric high-grade gliomas do overexpress the EGFR protein.61 However, there is no clear evidence that EGFR gene amplification or overexpression bears any effect in terms of prognosis in adults or children.62–64
The TP53 tumor suppressor gene is perhaps the best described genetic mutation present in childhood high-grade glioma. CCG study 945, the most significant large genetic database of pediatric gliomas to date, reported that 26 of 77 lesions harbored TP53 mutations.65,66 Sung and colleagues studied the molecular genetics of 29 pediatric high-grade gliomas and found all but 1 to demonstrate preferential inactivation of the p53 tumor suppressor pathway.63 Overexpression of p53 correlates with overexpression of the PTEN (phosphatase and tensin homolog) protein and is associated with significantly poorer outcomes in children.67 This mutation is similarly seen in adult high-grade glioma in roughly 30% to 50% of cases. However, in adults there has been no revelation of prognostic correlation.68 The platelet-derived growth factor (PDGF) receptor has also been found to be overexpressed.69
Investigation of medulloblastoma, which may bear relevance in understanding the molecular pathology in SPNETs, have revealed abnormalities in activation of the Wnt, Hedgehog, and Notch pathways.70,71 Also in the realm of small blue cell tumors, retinoblastoma is known to develop as a result of mutations in the Rb tumor suppressor gene. Molecular characterization of PXA has been recently undertaken. A recent investigation found reduced transcription of the tumor-suppressor gene TSC1 in 50 samples. Other interesting observations included losses on chromosome 9 in half the tumors, as well as impaired p53 and pRb1 pathways secondary to CDKN2A and CDKN2B gene mutations. These findings were consistent with low-grade PXA, as well as those with anaplastic features.72 A proportion of oligodendrogliomas have been described to harbor losses in chromosomes 1p and 19q. This has clinical significance in predicting enhanced chemosensitivity.39,40,73 The results of such research will probably prove most beneficial to clinicians in assessing prognosis and developing novel therapy for such tumors posing risk for relapse.
Experimental Therapy
Alternative modes of therapy and potentiation have been preliminarily evaluated with mixed benefit. High-dose chemotherapy with autologous stem cell rescue was recently shown to provide minimal benefit and confer significant toxicity in children with recurrent high-grade glioma. For PNETs, however, such aggressive therapy has been shown to be of greater benefit and to provide the potential for complete response in children with recurrent disease.74 High-dose methotrexate may have potential benefit when given at the time of diagnosis with concomitant chemoradiation therapy.26
Bypass of the BBB remains a significant obstacle for systemically delivered agents to achieve therapeutic concentrations in brain tumors. The Children’s Oncology Group recently reported a failed phase II effort to augment carboplatin with lobradimil, which improves BBB permeability, in the treatment of pediatric high-grade glioma.75 A phase I study has been successfully completed for the treatment of recurrent high-grade glioma with imatinib, an antibody that binds the PDGF receptor.76 Molecular research continues to describe possible targets for therapy.
Bypass of the BBB altogether may be achieved by interstitial infusion (convection-enhanced delivery, intracerebral clysis), whereby small catheters placed in solid tumor or resection cavities are used as a vehicle for direct delivery of agents to the parenchyma.77–81 Use of a pressure gradient has been demonstrated to improve distribution and uniformity of the therapeutic agent while limiting systemic exposure and potential toxicity. This technique has widened the scope of agents potentially available to treat intracranial lesions, including tumor-targeting immunotoxins, antibodies, and viral vectors unlikely to be effective when administered intravenously. In adults, phase I and II efforts have been completed,82,83 as well as a phase III trial using the tumor-specific recombinant immunotoxin interleukin-13–PE38 for the treatment of recurrent adult high-grade glioma.82 However, with further study of nontraditional therapeutic agents, interstitial infusion could hold major importance for their delivery in hemispheric malignancies, in particular those not amenable to complete resection.
Albright AL, Wisoff JH, Zeltzer P, et al. Prognostic factors in children with supratentorial (nonpineal) primitive neuroectodermal tumors. A neurosurgical perspective from the Children’s Cancer Group. Pediatr Neurosurg. 1995;22:1-7.
Bobo RH, Laske DW, Akbasak A, et al. Convection-enhanced delivery to macromolecules in the brain. Proc Nat Acad Sci U S A. 1994;91:2076-2080.
Bredel M, Pollack IF, Hamilton RL, et al. Epidermal growth factor receptor expression and gene amplification in high-grade non-brainstem gliomas of childhood. Clin Cancer Res. 1999;5:1786-1792.
Cohen BH, Zeltzer PM, Boyett JM, et al. Prognostic factors and treatment results for supratentorial primitive neuroectodermal tumors in children using radiation and chemotherapy: a Children’s Cancer Group randomized trial. J Clin Oncol. 1995;13:1687-1696.
Daumas-Duport C, Scheithauer BW, Chodkiewicz JP, et al. Dysembryoplastic neuroepithelial tumor: a surgically curable tumor of young patients with intractable partial seizures. Report of thirty-nine cases. Neurosurgery. 1988;23:545-556.
Finlay JL, Boyett JM, Yates AJ, et al. Randomized phase III trial in childhood high-grade astrocytoma comparing vincristine, lomustine, and prednisone with the eight-drugs-in-1-day regimen. Children’s Cancer Group. J Clin Oncol. 1995;13:112-123.
Giulioni M, Galassi E, Zucchelli M, et al. Seizure outcome of lesionectomy in glioneuronal tumors associated with epilepsy in children. J Neurosurg. 2005;102:288-293.
Khajavi K, Comair YG, Prayson RA, et al. Childhood ganglioglioma and medically intractable epilepsy. A clinicopathological study of 15 patients and a review of the literature. Pediatr Neurosurg. 1995;22:181-188.
McBride S, Daganzo S, Banerjee A, et al. Radiation is an important component of multimodality therapy for pediatric non-pineal supratentorial primitive neuroectodermal tumors. Int J Radiat Oncol Biol Phys. 2008;72:1319-1323.
Pollack IF. Brain tumors in children. N Engl J Med. 1994;331:1500-1507.
Pollack IF, Finkelstein SD, Burnham J, et al. Age and TP53 mutation frequency in childhood malignant gliomas: results in a multi-institutional cohort. Cancer Res. 2001;61:7404-7407.
Pollack IF, Finkelstein SD, Woods J, et al. Expression of p53 and prognosis in children with malignant gliomas. N Engl J Med. 2002;346:420-427.
Raffel C, Frederick L, O’Fallon JR, et al. Analysis of oncogene and tumor suppressor gene alterations in pediatric malignant astrocytomas reveals reduced survival for patients with PTEN mutations. Clin Cancer Res. 1999;5:4085-4090.
Reddy AT, Janss AJ, Phillips PC, et al. Outcome for children with supratentorial primitive neuroectodermal tumors treated with surgery, radiation, and chemotherapy. Cancer. 2000;88:2189-2193.
Sampson JH, Akabani G, Friedman AH, et al. Comparison of intratumoral bolus injection and convection-enhanced delivery of radiolabeled antitenascin monoclonal antibodies. Neurosurg Focus. 2006;20(4):E14.
Sandberg DI, Edgar MA, Souweidane MM. Convection-enhanced delivery into the rat brain stem. J Neurosurg. 2002;96:885-891.
Sposto R, Ertel IJ, Jenkin RD, et al. The effectiveness of chemotherapy for treatment of high grade astrocytoma in children: results of a randomized trial. A report from the Children’s Cancer Study Group. J Neurooncol. 1989;7:165-177.
Timmermann B, Kortmann RD, Kuhl J, et al. Role of radiotherapy in the treatment of supratentorial primitive neuroectodermal tumors in childhood: results of the prospective German brain tumor trials HIT 88/89 and 91. J Clin Oncol. 2002;20:842-849.
Wisoff JH, Boyett JM, Berger MS, et al. Current neurosurgical management and the impact of the extent of resection in the treatment of malignant gliomas of childhood: a report of the Children’s Cancer Group trial no. CCG-945. J Neurosurg. 1998;89:52-59.
1 CBTRUS. Statistical Report: Primary Brain Tumors in the United States, 2000-2004. Central Brain Tumor Registry of the United States; 2008.
2 Pollack IF. Brain tumors in children. N Engl J Med. 1994;331:1500-1507.
3 Albright AL, Wisoff JH, Zeltzer P, et al. Prognostic factors in children with supratentorial (nonpineal) primitive neuroectodermal tumors. A neurosurgical perspective from the Children’s Cancer Group. Pediatr Neurosurg. 1995;22:1-7.
4 Daumas-Duport C, Scheithauer BW, Chodkiewicz JP, et al. Dysembryoplastic neuroepithelial tumor: a surgically curable tumor of young patients with intractable partial seizures. Report of thirty-nine cases. Neurosurgery. 1988;23:545-556.
5 Favier J, Pizzolato GP, Berney J. Oligodendroglial tumors in childhood. Childs Nerv Syst. 1985;1:33-38.
6 Campbell JW, Pollack IF, Martinez AJ, et al. High-grade astrocytomas in children: radiologically complete resection is associated with an excellent long-term prognosis. Neurosurgery. 1996;38:258-264.
7 Pahapill PA, Ramsay DA, Del Maestro RF. Pleomorphic xanthoastrocytoma: case report and analysis of the literature concerning the efficacy of resection and the significance of necrosis. Neurosurgery. 1996;38:822-828.
8 Romeike BF, Kim YJ, Steudel WI, et al. Diffuse high-grade gliomas as second malignant neoplasms after radio-chemotherapy for pediatric malignancies. Childs Nerv Syst. 2007;23:185-193.
9 Broniscer A, Ke W, Fuller CE, et al. Second neoplasms in pediatric patients with primary central nervous system tumors: the St. Jude Children’s Research Hospital experience. Cancer. 2004;100:2246-2252.
10 Khajavi K, Comair YG, Prayson RA, et al. Childhood ganglioglioma and medically intractable epilepsy. A clinicopathological study of 15 patients and a review of the literature. Pediatr Neurosurg. 1995;22:181-188.
11 Raymond AA, Fish DR, Sisodiya SM, et al. Abnormalities of gyration, heterotopias, tuberous sclerosis, focal cortical dysplasia, microdysgenesis, dysembryoplastic neuroepithelial tumour and dysgenesis of the archicortex in epilepsy. Clinical, EEG and neuroimaging features in 100 adult patients. Brain. 1995;118:629-660.
12 Giulioni M, Galassi E, Zucchelli M, et al. Seizure outcome of lesionectomy in glioneuronal tumors associated with epilepsy in children. J Neurosurg. 2005;102:288-293.
13 Osborn A. Diagnostic Imaging: Brain. Salt Lake City, UT: Amirsys; 2004.
14 Girard N, Wang ZJ, Erbetta A, et al. Prognostic value of proton MR spectroscopy of cerebral hemisphere tumors in children. Neuroradiology. 1998;40:121-125.
15 Pollack I. Supratentorial high-grade gliomas. In: Berger M, Prados M, editors. Textbook of Neuro-Oncology. Philadelphia: Elsevier; 2005:605-611.
16 Fouladi M, Hunt DL, Pollack IF, et al. Outcome of children with centrally reviewed low-grade gliomas treated with chemotherapy with or without radiotherapy on Children’s Cancer Group high-grade glioma study CCG-945. Cancer. 2003;98:1243-1252.
17 Kepes JJ, Rubinstein LJ, Ansbacher L, et al. Histopathological features of recurrent pleomorphic xanthoastrocytomas: further corroboration of the glial nature of this neoplasm. A study of 3 cases. Acta Neuropathol. 1989;78:585-593.
18 Macaulay RJ, Jay V, Hoffman HJ, et al. Increased mitotic activity as a negative prognostic indicator in pleomorphic xanthoastrocytoma. Case report. J Neurosurg. 1993;79:761-768.
19 Pollack IF, Hamilton RL, Burnham J, et al. Impact of proliferation index on outcome in childhood malignant gliomas: results in a multi-institutional cohort. Neurosurgery. 2002;50:1238-1244.
20 Broniscer A, Gajjar A. Supratentorial high-grade astrocytoma and diffuse brainstem glioma: two challenges for the pediatric oncologist. Oncologist. 2004;9:197-206.
21 Finlay JL, Boyett JM, Yates AJ, et al. Randomized phase III trial in childhood high-grade astrocytoma comparing vincristine, lomustine, and prednisone with the eight-drugs-in-1-day regimen. Children’s Cancer Group. J Clin Oncol. 1995;13:112-123.
22 Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987-996.
23 Duffner PK, Horowitz ME, Krischer JP, et al. The treatment of malignant brain tumors in infants and very young children: an update of the Pediatric Oncology Group experience. Neuro Oncol. 1999;1:152-161.
24 Dufour C, Grill J, Lellouch-Tubiana A, et al. High-grade glioma in children under 5 years of age: a chemotherapy only approach with the BBSFOP protocol. Eur J Cancer. 2006;42:2939-2945.
25 Sposto R, Ertel IJ, Jenkin RD, et al. The effectiveness of chemotherapy for treatment of high grade astrocytoma in children: results of a randomized trial. A report from the Children’s Cancer Study Group. J Neurooncol. 1989;7:165-177.
26 Wagner S, Reinert C, Schmid HJ, et al. High-dose methotrexate prior to simultaneous radiochemotherapy in children with malignant high-grade gliomas. Anticancer Res. 2005;25:2583-2587.
27 Wisoff JH, Boyett JM, Berger MS, et al. Current neurosurgical management and the impact of the extent of resection in the treatment of malignant gliomas of childhood: a report of the Children’s Cancer Group trial no. CCG-945. J Neurosurg. 1998;89:52-59.
28 Stummer W, Pichlmeier U, Meinel T, et al. Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: a randomised controlled multicentre phase III trial. Lancet Oncol. 2006;7:392-401.
29 Chitoku S, Otsubo H, Harada Y, et al. Extraoperative cortical stimulation of motor function in children. Pediatr Neurol. 2001;24:344-350.
30 Gupta N, Berger MS. Brain mapping for hemispheric tumors in children. Pediatr Neurosurg. 2003;38:302-306.
31 Souweidane MM, Kim KH, McDowall R, et al. Brain mapping in sedated infants and young children with passive-functional magnetic resonance imaging. Pediatr Neurosurg. 1999;30:86-92.
32 Silber JH, Radcliffe J, Peckham V, et al. Whole-brain irradiation and decline in intelligence: the influence of dose and age on IQ score. J Clin Oncol. 1992;10:1390-1396.
33 Radcliffe J, Packer RJ, Atkins TE, et al. Three- and four-year cognitive outcome in children with noncortical brain tumors treated with whole-brain radiotherapy. Ann Neurol. 1992;32:551-554.
34 Gold DG, Neglia JP, Dusenbery KE. Second neoplasms after megavoltage radiation for pediatric tumors. Cancer. 2003;97:2588-2596.
35 Phuphanich S, Edwards MS, Levin VA, et al. Supratentorial malignant gliomas of childhood. Results of treatment with radiation therapy and chemotherapy. J Neurosurg. 1984;60:495-499.
36 Hadjipanayis CG, Kondziolka D, Gardner P, et al. Stereotactic radiosurgery for pilocytic astrocytomas when multimodal therapy is necessary. J Neurosurg. 2002;97:56-64.
37 Hodgson DC, Goumnerova LC, Loeffler JS, et al. Radiosurgery in the management of pediatric brain tumors. Int J Radiat Oncol Biol Phys. 2001;50:929-935.
38 Marcus KJ, Goumnerova L, Billett AL, et al. Stereotactic radiotherapy for localized low-grade gliomas in children: final results of a prospective trial. Int J Radiat Oncol Biol Phys. 2005;61:374-379.
39 Intergroup Radiation Therapy Oncology Group Trial 9402Cairncross G, Berkey B, Shaw E, et al. Phase III trial of chemotherapy plus radiotherapy compared with radiotherapy alone for pure and mixed anaplastic oligodendroglioma: Intergroup Radiation Therapy Oncology Group Trial 9402. J Clin Oncol. 2006;24:2707-2714.
40 van den Bent MJ, Carpentier AF, Brandes AA, et al. Adjuvant procarbazine, lomustine, and vincristine improves progression-free survival but not overall survival in newly diagnosed anaplastic oligodendrogliomas and oligoastrocytomas: a randomized European Organisation for Research and Treatment of Cancer phase III trial. J Clin Oncol. 2006;24:2715-2722.
41 Duffner PK, Horowitz ME, Krischer JP, et al. Postoperative chemotherapy and delayed radiation in children less than three years of age with malignant brain tumors. N Engl J Med. 1993;328:1725-1731.
42 Nicholson HS, Kretschmar CS, Krailo M, et al. Phase 2 study of temozolomide in children and adolescents with recurrent central nervous system tumors: a report from the Children’s Oncology Group. Cancer. 2007;110:1542-1550.
43 Broniscer A, Gururangan S, MacDonald TJ, et al. Phase I trial of single-dose temozolomide and continuous administration of O6-benzylguanine in children with brain tumors: a pediatric brain tumor consortium report. Clin Cancer Res. 2007;13:6712-6718.
44 Carret AS, Tabori U, Crooks B, et al. Outcome of secondary high-grade glioma in children previously treated for a malignant condition: a study of the Canadian Pediatric Brain Tumour Consortium. Radiother Oncol. 2006;81:33-38.
45 Daumas-Duport C, Varlet P, Bacha S, et al. Dysembryoplastic neuroepithelial tumors: nonspecific histological forms—a study of 40 cases. J Neurooncol. 1999;41:267-280.
46 Leung SY, Gwi E, Ng HK, et al. Dysembryoplastic neuroepithelial tumor. A tumor with small neuronal cells resembling oligodendroglioma. Am J Surg Pathol. 1994;18:604-614.
47 Taylor M, Mainprize T, Hawkins C. Dysembryoplastic neuroepithelial tumor. In: Berger M, Prados M, editors. Textbook of Neuro-Oncology. Philadelphia: Elsevier; 2005:682-686.
48 Kuo YH, Edgar MA, Luther N, et al. Novel low-grade glioneuronal neoplasm presenting in an octogenarian: case report and review of the literature. Clin Neurol Neurosurgy. 2006;108:426-432.
49 Burneo J, Tellez-Zenteno J, Steven D, et al. Adult-onset epilepsy associated with dysembryoplastic neuroepithelial tumor. Seizure. 2008;17:498-504.
50 Maher C, White J, Scheithauer B, et al. Recurrence of dysembryoplastic neuroepithelial tumor following resection. Pediatr Neurosurg. 2008;44:333-336.
51 Cohen BH, Zeltzer PM, Boyett JM, et al. Prognostic factors and treatment results for supratentorial primitive neuroectodermal tumors in children using radiation and chemotherapy: a Children’s Cancer Group randomized trial. J Clin Oncol. 1995;13:1687-1696.
52 Rorke LB, Trojanowski JQ, Lee VM, et al. Primitive neuroectodermal tumors of the central nervous system. Brain Pathol. 1997;7:765-784.
53 Reddy AT, Janss AJ, Phillips PC, et al. Outcome for children with supratentorial primitive neuroectodermal tumors treated with surgery, radiation, and chemotherapy. Cancer. 2000;88:2189-2193.
54 Tomita T, McLone DG, Yasue M. Cerebral primitive neuroectodermal tumors in childhood. J Neurooncol. 1988;6:233-243.
55 Timmermann B, Kortmann RD, Kuhl J, et al. Role of radiotherapy in the treatment of supratentorial primitive neuroectodermal tumors in childhood: results of the prospective German brain tumor trials HIT 88/89 and 91. J Clin Oncol. 2002;20:842-849.
56 Timmermann B, Kortmann RD, Kuhl J, et al. Role of radiotherapy in supratentorial primitive neuroectodermal tumor in young children: results of the German HIT-SKK87 and HIT-SKK92 trials. J Clin Oncol. 2006;24:1554-1560.
57 McBride S, Daganzo S, Banerjee A, et al. Radiation is an important component of multimodality therapy for pediatric non-pineal supratentorial primitive neuroectodermal tumors. Int J Radiat Oncol Biol Phys. 2008;72:1319-1323.
58 Geyer JR, Zeltzer PM, Boyett JM, et al. Survival of infants with primitive neuroectodermal tumors or malignant ependymomas of the CNS treated with eight drugs in 1 day: a report from the Children’s Cancer Group. J Clin Oncol. 1994;12:1607-1615.
59 Gilheeney SW, Saad A, Chi S, et al. Outcome of pediatric pineoblastoma after surgery, radiation and chemotherapy. J Neurooncol. 2008;89:89-95.
60 Jakacki RI, Zeltzer PM, Boyett JM, et al. Survival and prognostic factors following radiation and/or chemotherapy for primitive neuroectodermal tumors of the pineal region in infants and children: a report of the Children’s Cancer Group. J Clin Oncol. 1995;13:1377-1383.
61 Liang ML, Ma J, Ho M, et al. Tyrosine kinase expression in pediatric high grade astrocytoma. J Neurooncol. 2008;87:247-253.
62 Bredel M, Pollack IF, Hamilton RL, et al. Epidermal growth factor receptor expression and gene amplification in high-grade non-brainstem gliomas of childhood. Clin Cancer Res. 1999;5:1786-1792.
63 Sung T, Miller DC, Hayes RL, et al. Preferential inactivation of the p53 tumor suppressor pathway and lack of EGFR amplification distinguish de novo high grade pediatric astrocytomas from de novo adult astrocytomas. Brain Pathol. 2000;10:249-259.
64 Zhou YH, Tan F, Hess KR, et al. The expression of PAX6, PTEN, vascular endothelial growth factor, and epidermal growth factor receptor in gliomas: relationship to tumor grade and survival. Clin Cancer Res. 2003;9:3369-3375.
65 Pollack IF, Finkelstein SD, Burnham J, et al. Age and TP53 mutation frequency in childhood malignant gliomas: results in a multi-institutional cohort. Cancer Res. 2001;61:7404-7407.
66 Pollack IF, Finkelstein SD, Woods J, et al. Expression of p53 and prognosis in children with malignant gliomas. N Engl J Med. 2002;346:420-427.
67 Raffel C, Frederick L, O’Fallon JR, et al. Analysis of oncogene and tumor suppressor gene alterations in pediatric malignant astrocytomas reveals reduced survival for patients with PTEN mutations. Clin Cancer Res. 1999;5:4085-4090.
68 Shiraishi S, Tada K, Nakamura H, et al. Influence of p53 mutations on prognosis of patients with glioblastoma. Cancer. 2002;95:249-257.
69 Rood BR, MacDonald TJ. Pediatric high-grade glioma: molecular genetic clues for innovative therapeutic approaches. J Neurooncol. 2005;75:267-272.
70 Clifford SC, Lusher ME, Lindsey JC, et al. Wnt/Wingless pathway activation and chromosome 6 loss characterize a distinct molecular sub-group of medulloblastomas associated with a favorable prognosis. Cell Cycle. 2006;5:2666-2670.
71 Thompson MC, Fuller C, Hogg TL, et al. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol. 2006;24:1924-1931.
72 Weber RG, Hoischen A, Ehrler M, et al. Frequent loss of chromosome 9, homozygous CDKN2A/p14(ARF)/CDKN2B deletion and low TSC1 mRNA expression in pleomorphic xanthoastrocytomas. Oncogene. 2007;26:1088-1097.
73 Pollack IF, Finkelstein SD, Burnham J, et al. Association between chromosome 1p and 19q loss and outcome in pediatric malignant gliomas: results from the CCG-945 cohort. Pediatr Neurosurg. 2003;39:114-121.
74 Shih CS, Hale GA, Gronewold L, et al. High-dose chemotherapy with autologous stem cell rescue for children with recurrent malignant brain tumors. Cancer. 2008;112:1345-1353.
75 Warren K, Jakacki R, Widemann B, et al. Phase II trial of intravenous lobradimil and carboplatin in childhood brain tumors: a report from the Children’s Oncology Group. Cancer Chemother Pharmacol. 2006;58:343-347.
76 Pollack IF, Jakacki RI, Blaney SM, et al. Phase I trial of imatinib in children with newly diagnosed brainstem and recurrent malignant gliomas: a Pediatric Brain Tumor Consortium report. Neuro Oncol. 2007;9:145-160.
77 Luther N, Cheung NK, Dunkel IJ, et al. Intraparenchymal and intratumoral interstitial infusion of anti-glioma monoclonal antibody 8H9. Neurosurgery. 2008;63:1166-1174.
78 Bobo RH, Laske DW, Akbasak A, et al. Convection-enhanced delivery to macromolecules in the brain. Proc Nat Acad Sci U S A. 1994;91:2076-2080.
79 Lonser RR, Walbridge S, Garmestani K, et al. Successful and safe perfusion of the primate brainstem: in vivo magnetic resonance imaging of macromolecular distribution during infusion. J Neurosurg. 2002;97:905-913.
80 Sampson JH, Akabani G, Friedman AH, et al. Comparison of intratumoral bolus injection and convection-enhanced delivery of radiolabeled antitenascin monoclonal antibodies. Neurosurg Focus. 2006;20(4):E14.
81 Sandberg DI, Edgar MA, Souweidane MM. Convection-enhanced delivery into the rat brain stem. J Neurosurg. 2002;96:885-891.
82 Kunwar S, Prados MD, Chang SM, et al. Direct intracerebral delivery of cintredekin besudotox (IL13-PE38QQR) in recurrent malignant glioma: a report by the Cintredekin Besudotox Intraparenchymal Study Group. J Clin Oncol. 2007;25:837-844.
83 Lidar Z, Mardor Y, Jonas T, et al. Convection-enhanced delivery of paclitaxel for the treatment of recurrent malignant glioma: a phase I/II clinical study. J Neurosurg. 2004;100:472-479.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
CHAPTER 199 Supratentorial Hemispheric Tumors
Hemispheric neoplasms account for 25% of all primary brain tumors in children beyond infancy.1 This chapter discusses supratentorial hemispheric tumors, which in the pediatric population are generally of a low-grade variety. Hemispheric tumors can be classified by cellular origin: glial, mixed glial and neuronal, and primitive neuroectodermal. Differentiation between these tumors can be accomplished with a careful history and examination and is often fairly apparent before biopsy or resection thanks largely to modern imaging technology. The therapeutic options, prognoses, and molecular genetics of these tumors are unique and frequently vary considerably from such lesions in adults.
Epidemiology
Supratentorial low- and high-grade gliomas each occur annually in less than 1 per 100,000 individuals younger than 20 years, with low-grade being more common.2 In total, glial tumors account for 56% of primary central nervous system (CNS) tumors in children younger than 14 years and for 45% of such tumors in those between the ages of 14 and 19.1 Supratentorial primitive neuroectodermal tumors (SPNETs) occur less frequently, with most children affected when younger than 10 years.3 There has been no demonstration of a higher predilection for gliomas or SPNETs between the sexes. Dysembryoplastic neuroectodermal tumors (DNETs), pleomorphic xanthoastrocytoma (PXA), and low-grade glioneuronal tumors are found in children more so than adults. Oligodendrogliomas and DNETs may occur slightly more frequently in males.4 Oligodendrogliomas are rarer in children than adults and account for less than 1% to 2% of primary pediatric brain tumors.1,5 PXA is less common still.6,7
Familial genetic syndromes increasing the likelihood for the development of primary intracranial tumors include Turcot’s syndrome (colonic polyposis), Li-Fraumeni syndrome, and neurofibromatosis type 1. A history of previous CNS tumor, systemic cancer, or cranial irradiation also increases risk for the development of secondary brain tumors, most notably meningiomas and high-grade gliomas.8 Of note, secondary high-grade gliomas are more commonly supratentorial and may exhibit a distinct genetic profile from their primary counterparts. Once diagnosed, secondary high-grade glioma portends a worse prognosis than primary tumors despite aggressive intervention. The St. Jude’s experience found a cumulative 4% 15-year risk for secondary neoplasms developing after treatment of a primary CNS tumor,9 and in children treated for acute lymphoblastic leukemia, there is a 1.39% cumulative incidence of secondary brain tumors (gliomas and meningiomas) at 20 years, with cranial irradiation being a dose-dependent predisposing factor.
Clinical Findings
Seizures are a frequent symptom of supratentorial lesions in children, particularly low-grade tumors such as fibrillary astrocytoma, PXA, DNET, and ganglioglioma.4,10,11 The likelihood for these tumors to be accompanied by seizures increases with their predilection to occur in the temporal lobe. However, some evidence exists to support the notion that neuronal elements of the tumor itself can act as independent generators of seizures,12 and secondary seizure foci may exist around the tumor.
Glial Tumors
Imaging
The majority of primary supratentorial hemispheric tumors in children are of glial origin. Imaging features that help in predicting the biologic behavior of hemispheric tumors in children include an imaging correlate of cellular density (density or intensity), the degree of mass effect, a cortical location, and any remodeling of the overlying calvaria. Standard evaluation of symptomatic children suspected of harboring an intracranial lesion typically begins with computed tomography (CT). Low-grade tumors are likely to appear hypodense, whereas high-grade gliomas are usually hyperdense. High-grade neoplasms are frequently associated with surrounding hypodense regions representing vasogenic edema. Non–contrast-enhanced CT is sensitive for regions of calcification, usually an indicator of a more indolent lesion, including ganglioglioma or oligodendroglioma.13 Remodeling of the inner table of the skull to conform to the gyral patterns or thinning is commonly seen with low-grade neoplasms. With the administration of intravenous contrast material, high-grade astrocytomas are expected to reveal enhancement attributed to regions of hypervascularity and breakdown of the blood-brain barrier (BBB). Cavitation surrounded by contrast heterogeneity is likely to represent necrotic components, a harbinger of a malignant phenotype. This cavitation is contrasted with the well-circumscribed ring of enhancement that surrounds benign cystic tumors.
For enhanced anatomic definition and improved surgical planning, magnetic resonance imaging (MRI) has become the standard imaging modality. Low-grade fibrillary astrocytomas are frequently poorly delineated and will typically demonstrate gyral thickening, T1 hypointensity, and T2 hyperintensity, often best appreciated on T2 fluid-attenuated inversion recovery (FLAIR) sequences. PXA is more apt to show enhancing nodularity with frequent cystic components7 (Fig. 199-1).
High-grade gliomas have mixed signal intensity, with predominant hypointensity on T1-weighted and hyperintensity on T2-weighted images.13 A broad region of T2 or FLAIR hyperintensity surrounding the tumor represents the highly infiltrative growth characteristic and surrounding edema frequently seen in these aggressive tumors (Fig. 199-2). This signal change most often extends in the direction of anisotropic white matter tracts and may cross the midline along commissural fibers, notably the corpus callosum. Regions of hypercellularity may result in restricted diffusion (bright appearance) within the lesion on diffusion-weighted imaging. Magnetic resonance spectroscopy, which is gaining popularity in narrowing the preoperative diagnosis of intracranial lesions, would be expected in high-grade components to demonstrate elevated peaks of choline, N-acetylaspartate, and lactate and a rise in the choline-to-creatine ratio.14
Pathology
Classification or naming of these tumors is based on their cellular composition. Tumors composed of astrocytes with hypercellularity, infiltration, and a fibrillary matrix are designated fibrillary astrocytoma. The histologic hallmark of oligodendroglioma is perinuclear cytoplasmic shrinkage, which results in a halo appearance surrounding the cells’ nuclei and gives a classic “fried egg” appearance under lower magnification. The microvasculature takes on a “chicken wire” appearance, and calcifications may be seen grossly and under microscopy.15 PXAs have historically been thought to be purely astrocytic in origin, but these tumors may have neuronal progenitors as well. Histologically, these lesions show pleomorphism, lipid, reticulin, and perivascular lymphocytes (Fig. 199-3).
A critical distinction for the pathologist to make is between high- and low-grade lesions. Central review by an expert panel has been used in major studies to fulfill this task.16 Grade I astrocytoma, or juvenile pilocytic astrocytoma (JPA), is uncommonly hemispheric in location. High-grade or malignant gliomas represent World Health Organization (WHO) grades III and IV lesions. Grade III tumors demonstrate cellular pleomorphism, frequent mitoses, and the hypercellularity lacking in grade II lesions. Immunohistochemical testing will reveal positivity for glial fibrillary acidic protein (GFAP) and MIB-1 (anti–Ki-67 antibody). The most common grade III glial neoplasm is anaplastic astrocytoma (AA), but oligodendroglioma and PXA also have the potential to develop into grade III lesions.
The defining presence of necrosis raises the grade of the lesion to WHO IV and a diagnosis of glioblastoma multiforme. Pseudopalisading cellular architecture and significant microvascular proliferation are also seen. Like AA, immunohistochemistry is positive for GFAP. High-grade lesions will demonstrate a high mitotic index, and a high index of proliferation has been well characterized in Children’s Cancer Group (CCG) study 945 to correlate with worsened prognosis.17–19
Treatment
Unlike adult high-grade glioma, this diagnosis in children portends a 5-year survival rate of 30% to 50%, with a higher likelihood of long-term survivors.6,20,21 This prognosis is particularly significant and has relevance to survival from glioblastoma, where the mean survival of adults in contemporary series is less than 2 years.22 Cytoreduction on the order of 90% or greater has been associated with improved outcomes in children with high-grade glioma. The CCG studies 943 and 945 were instrumental in first demonstrating the value of total resection. Although it is difficult to control for adjuvant therapy, which varies from series to series, multiple reports have since corroborated a 5-year progression-free survival benefit of gross total resection on the order of greater than 20%. This holds true for both AA and glioblastoma.6,23–27 CCG 945 included 83 supratentorial high-grade gliomas for review and reported a 5-year progression-free survival rate of 35% for greater than 90% resection of glioblastoma versus 17% for subtotal resection. Furthermore, a 5-year progression-free survival rate of 44% was seen in children after total resection of AAs versus 22% for subtotal resection. These differences were statistically significant.27 The role of a second resection in improving survival for children with recurrent disease has yet to be well defined.
This goal of maximal tumor removal is also enhanced through the incorporation of several operative adjuncts. The principle of discriminating between tumor and parenchyma unifies all past and future improvements aimed at greater removal of tumor. The integration of high-resolution MRI with triplanar navigation is designed to offer the surgeon a reliable method for defining tumor margins with a greater degree of certainty. Errors attributed to brain shifting with partial tumor removal can be overcome with intraoperative modalities, including ultrasound and MRI. Tumor cell fluorescence may offer finer intraoperative resolution of tumor margins. Early results suggest that fluorescence-guided resection of glioblastoma in adults with the use of 5-aminolevulinic acid may improve the degree of tumor removal.28
For tumors associated with primary cortical regions, intraoperative brain mapping is an important safety mechanism. Motor function is assessed most reliably with this method, whereby direct cortical stimulation on the precentral gyrus triggers contralateral movement. The age of the child influences the ability to effectively perform cortical localization because of the variability in electrical impedance of the brain and the need for patient cooperation for language mapping. Older children will typically respond to a stimulus of 4 to 6 mA, whereas young children may require stimuli of 8 to 12 mA.29 Brain immaturity in children younger than 5 years may render direct stimulation ineffectual. In such cases, detection of the phase reversal potential between motor and sensory gyri is most likely to yield useful data. Because language mapping is reliant on intraoperative patient interaction and compliance, cortical localization of primary language representation is difficult in children younger than 10 years.30 Preoperative functional MRI has been shown to predict language dominance in infants and young children.31
Children with high-grade lesions or residual/recurrent low-grade gliomas may benefit from adjuvant radiation therapy. However, because of the well-established cognitive risks associated with cranial irradiation in very young children, radiation therapy is classically reserved for children older than 3 years.32 Cranial irradiation on the order of 50 to 60 Gy in children causes an average 10-point decline in IQ, with younger children being more adversely affected.33 Development of a secondary intracranial tumor is a significant risk in all children undergoing radiation therapy but is typically outweighed by the survival benefit in those with more aggressive malignancies. The risk of a secondary malignancy developing after radiation therapy has been reported to be just under 10%.34 With the development of conformal and fractionated therapy, delivery of radiation has progressed over time to allow higher doses to be given to the tumor while sparing surrounding normal brain.
The benefit of primary radiation therapy for low-grade glioma is dubious at best.16 Noteworthy studies evaluating adjuvant therapy for pediatric high-grade glioma, including the original CCG 943 publication in 1989, have included cranial irradiation as initial management in appropriate children.6,27,35 Most institutions currently use a fairly standard regimen of 50 to 60 Gy delivered to the tumor in roughly 25 fractions over a 5-week period for high-grade glioma. Data from recent studies suggest that the use of stereotactic radiosurgery, whereby a single high dose of radiation is delivered via intersecting beams to a desired tumor volume designated on a radiograph, should be reserved for patients with multiply recurrent low- and high-grade lesions who are not candidates for surgery.36–38
Chemotherapy is a vital component of multimodal treatment of high-grade glioma. The CCG 943 publication in 1989 established the use of chemotherapy as a standard initial adjuvant for pediatric high-grade glioma. That study prospectively randomized 58 children with high-grade glioma, 40 of whom harbored glioblastoma, into two therapeutic arms.25 One group underwent postoperative radiation therapy and combination chemotherapy consisting of lomustine, vincristine, and prednisone, whereas the other group underwent radiation therapy alone. The survival benefit in the chemotherapy group was significant, with a 5-year event-free survival rate of 46% versus 18% for radiation therapy alone.
The next most significant set of results came from CCG 945. Here, an “8 in 1” regimen of chemotherapeutics was administered to an experimental arm, whereas the control arm underwent the chemoradiation protocol in CCG 943. The “8-in-1” regimen consisted of vincristine, carmustine, procarbazine, hydroxyurea, dacarbazine, methylprednisolone, cisplatin, and cytarabine usually given in two cycles before radiation therapy and for up to 1 year afterward. Five-year event-free survival rates were higher in the “8-in-1” than in the standard therapy group (33% versus 26%).21
Oligodendroglioma, especially when anaplastic in nature, is also a good candidate for adjuvant therapy. Procarbazine, lomustine, and vincristine have been evaluated prospectively and been found to improve progression-free survival by almost 1 year in adults when administered in conjunction with surgery and radiation therapy.39,40 Overall survival is also improved in tumors harboring 1p and 19q loss of heterozygosity.
[/not-level-membership-for-neurosurgery-category]
