Chapter 15 Sudden Unexpected Death in Epilepsy
Definition
All-cause mortality rates in patients with epilepsy are approximately two to three times higher than the general population and are age dependent.1–3 The major contributors of increased mortality are the causes of epilepsy, for example, traumatic brain injury, cerebrovascular disease, brain tumors, and accidents (mostly falls and drowning) or status epilepticus and SUDEP. In contrast, treatment-related mortality, from epilepsy surgery or the adverse effects of antiepileptic medication, is very rare.4
SUDEP is defined as the sudden, unexpected, witnessed or unwitnessed, nontraumatic, and nondrowning death in patients with epilepsy with or without evidence for a seizure and excluding documented status epilepticus, in which postmortem examination does not reveal a structural or toxicologic cause for death.5 Where autopsy is not performed, and for the purpose of epidemiological studies, sudden death occurring in benign circumstances with no known competing cause for death is classified as “probable SUDEP.” Cases classified as “possible SUDEP” are usually not included in epidemiological studies of SUDEP incidence. These are cases in which SUDEP cannot be excluded because either the information regarding circumstances of death is too limited to confirm classification as a probable or definite SUDEP or that adequate or complete documentation is available but there is a plausible competing explanation for the death.3 It must be stressed that this defines a category of sudden death in epilepsy, and not a condition, a concept that is sometimes overlooked. Despite an applicable definition, and clear guidance where there is uncertainty, significant variability in use has hampered efforts to integrate findings from multiple studies on epidemiological and risk factor data and hence establish common relevant factors.6,7
Epidemiology
Sudden unexpected death in the general population is extremely rare in young adults with an incidence of 5 to 10/100, 000 person-years, whereas the rate climbs steeply with advancing age to approximately 300/100, 000 person-years in the elderly.8 The incidence of sudden death in patients with epilepsy is significantly higher and varies markedly with the population studied.9 For example, in population-based studies, the incidence has been reported to be 0.35 and 2.7/1000 person-years, depending on the methodologies employed.10,11 This increases to between 2 and 5.9/1000 person-years in cohorts of patients attending specialist epilepsy clinics,12–14 3.4/1000 person-years in pupils with epilepsy enrolled in a special residential school,15 and up to 9.3/1000 person-years in epilepsy surgery candidates.16,17 The incidence of sudden death in young adults with intractable epilepsy is, therefore, many times that of the general population, with a peak between the ages of 20 and 40 years.18 In older age groups, the relative increased incidence of SUDEP is too small to measure and is confounded by the occurrence of comorbidity such as cardiovascular, respiratory, or cerebrovascular disease. This makes it difficult to ascribe a sudden death to a “pure” SUDEP category but does not exclude the possibility that there is no additive risk from the epilepsy. There is limited data available on the incidence of SUDEP in children. An incidence rate of approximately 0.1 to 0.2/1000 person-years has been estimated from a number of earlier studies.19,20 Significant methodological limitations exist, however, including inadequate follow-up periods, difficulties with case-ascertainment, and assumptions regarding the prevalence of epilepsy in community-based studies. A more recent retrospective study with an 18-year follow-up period confirmed a low incidence rate of approximately 0.4/1000 person-years, despite following a cohort of children with refractory epilepsy and learning disability.21 This is approximately 10 times lower than the incidence rate for similarly affected adults; the rate for children with uncomplicated epilepsy will be lower still.22 The reason for the discrepancy between children and adults is unknown and is unlikely to be entirely due to methodological constraints, but may be as a result of a different cardiorespiratory response from a developing brain compared to a mature brain, a more intensively supervised environment, the inclusion of specific self-limiting pediatric epilepsy syndromes, lack of comorbidity, or possibly, greater autonomic stability.23
Risk Factors
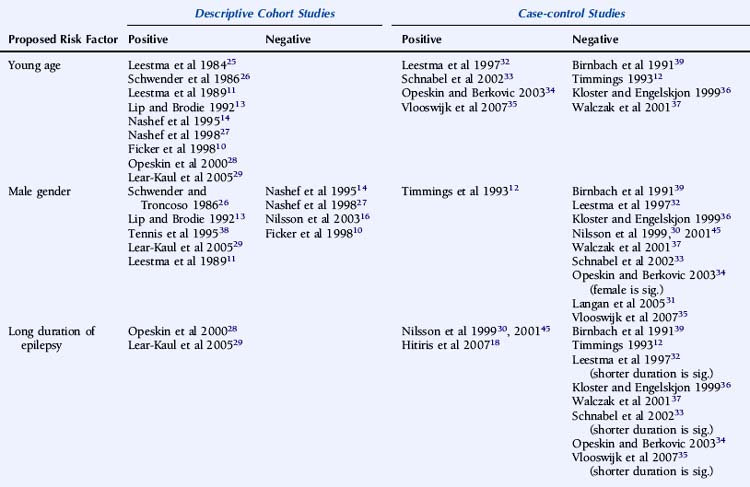
There is significant debate regarding risk factors for SUDEP. A large number of variables that may influence the risk of SUDEP have been proposed, and the significance of each has been discussed at length without clear consensus. Relevant and independent risk factors are difficult to establish given the nonindependence of patient, syndrome, seizures, and treatment characteristics. Multiple logistic regression analyses require large cohorts of patients to achieve statistical significance for each of the variables evaluated, and this is difficult to attain.24 Furthermore, the high variability between studies in terms of patient cohorts, definition, choice of control group, methodology, and overall study quality precludes not only a valid meta-analysis, but even a simple meaningful comparison. In a critical review of the literature regarding risk factors for SUDEP, Tellez-Zenteno noted that a clear definition of SUDEP was stated in only 65% of relevant studies. Furthermore, the low frequency of postmortem data was evident in many studies. This lack of consistency significantly undermines the suitability of such studies to comment on SUDEP causality and risk.7 The preliminary studies of risk factors in SUDEP were descriptive, providing direction for more meaningful case-control studies. The value of these observations, otherwise, is limited, however, because, in general, these studies lack suitable control groups and comprise small numbers of an often highly selected patient population.
A number of studies assessing risk factors used a case-control design (Table 15-1), this often being the only option for the study of relatively uncommon occurrences, such as SUDEP. However, it also has important limitations. For example, case-control studies traditionally evaluate the probability of being exposed to a risk factor, not the risk of developing a condition. Moreover, case-control studies are subject to several sources of bias, such as selection bias of cases and controls and information and recall bias when collecting data on exposure. These biases are particularly important where an objective definition of the condition of interest is not consistently applied, as is the case in the SUDEP literature.
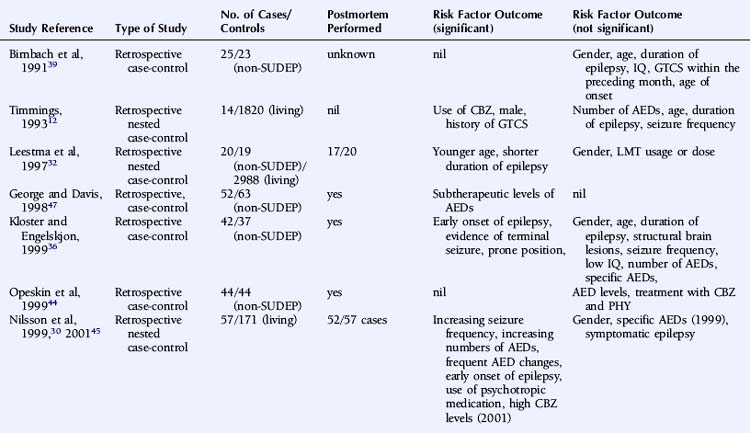

An attempt has been made to resolve the SUDEP risk factor literature and disentangle the often contradictory results on the basis of extensive literature reviews and the implementation of a study-validity scoring system and calculation of relative risk factor ratios.6 Monte and colleagues stratified studies evaluating risk factors on the basis of fulfillment of a number of variables that were considered to be markers of good quality studies. However, the scoring system used was unvalidated and potentially misleading, with, for example, equal credence attributed to controlled and descriptive studies. Papers not achieving an arbitrary threshold were excluded, and the possibility of the same study population being used more than once was not adequately addressed.6 A more simplistic approach was used by Tellez-Zenteno, in which percentages of studies with a positive risk factor were reported. Risk factors that were reported to be significant in more than 50% of studies were a terminal seizure, subtherapeutic antiepileptic drug (AED) levels, high seizure frequency, and a high number of AEDs.7
DEMOGRAPHICS
Descriptive studies have almost universally reported that patients with SUDEP are young adults.10,11,13,14,25–29 A number of biases exist, however, including, as discussed earlier, the exclusion of patients with significant comorbidity associated with increasing age, such as ischemic heart disease or cerebrovascular disease, identified on postmortem examination.13,25,29 Other examples of bias include case identification through self-referral by bereaved relatives, most commonly parents,27 and studies with only small numbers of patients.10,13 Case-control studies are less conclusive. Some studies only included defined age groups and can draw no conclusions regarding other age groups. Nevertheless, it is interesting to note that 70 to 80% of the studied population in a number of case-control studies were less than 45 years old.18,30 Data regarding age, however, is not available from a number of large studies due to age matching of control subjects.18,30,31
Of the remaining studies, the use of a cohort of non-SUDEP deaths as a control group may bias the patient group toward a younger age due to exclusion of comorbid conditions more commonly associated with advancing age, such as atherosclerosis, thromboembolism, and metastatic carcinoma,32–35 although young age as an independent risk factor has not been universally reported.36,37 The likelihood of selection bias is corroborated by finding significantly less comorbidity in the SUDEP group than the non-SUDEP group.35 In studies using living control subjects, younger age was not seen more frequently in the SUDEP group, although numbers of SUDEP patients were small.12
Although a large number of descriptive studies have suggested that male gender is a significant risk factor for SUDEP,11,13,26,29,38 this has not been confirmed by the vast majority of case-control studies.30–33,35–37,39 In addition, a small number of both descriptive and case-control studies have reported a significantly increased standardized mortality rate in female patients, which may be attributable to a lower background rate of death in the female non-SUDEP control group.10,34
EPILEPSY CHARACTERISTICS
A number of case-control studies have suggested that early onset of epilepsy is a significant risk factor for SUDEP.30,33,35,36 For example, Nilsson reported an eightfold higher SUDEP risk in patients with an onset of epilepsy between the ages of 0 and 15 years, compared to patients with seizure onset after 45 years of age.30 However, whereas this may reflect a different etiological basis for the epilepsy, it may also merely be a surrogate marker for an increased cumulative lifetime risk of having seizures for a longer period of time, as suggested by other studies.18,28,29 Conversely, several reports give a shorter duration of epilepsy being associated with an increased risk of SUDEP, although this is most likely as a result of comparison with an older control population.32,33,35 Furthermore, following conditional multiple logistic regression analysis, Walczak showed that a long duration of epilepsy (>30 years) was no longer a risk factor after adjustment for seizure frequency.37
One would expect epilepsy syndrome to be a key factor in defining the risk of SUDEP. Yet, there is only limited evidence to support the association of epilepsy syndrome with an increased risk of SUDEP.30,36 Discordant results from the relatively few case-control studies that assessed this risk factor and low numbers of patients in each group preclude detailed evaluation or definitive conclusions.34 In the study reported by Nilsson, 7 out of 57 (12%) SUDEP cases had primary generalized epilepsy compared to 12 out of 171 (7%) control subjects. Statistical comparison revealed that there was a higher risk of SUDEP in patients with primary generalized epilepsy compared to patients with focal, symptomatic epilepsy, although this was only significant in men.30 Nevertheless, although idiopathic primary generalized epilepsy (IGE) is usually less refractory to treatment, individuals with IGE are well represented in SUDEP cohorts. It is possible that specific epilepsy syndrome subtypes carry an increased risk of sudden death due to phenotypic expression in other cerebral and possibly cardiac structures. For example, Rett syndrome, typically due to mutations in the MECP2 gene, is associated with brain stem immaturity and autonomic, particularly respiratory, instability in association with epilepsy. In addition to respiratory abnormalities, which include apneustic breathing and hyperventilation, patients with Rett syndrome may also present with a prolonged QT interval and reduced heart rate variability.40 The coexistence of cardiac arrhythmia and central apnea may act synergistically in the development of sudden death. Severe myoclonic epilepsy in infancy (SMEI) also appears to be associated with an increased risk of sudden death, although the pathophysiological mechanism remains unclear. A recent report of a pedigree with an SCN1A mutation and two cases of SUDEP is of interest in this respect.42 Although some of these conditions represent the most severely affected group of patients and the high mortality rate may be multifactorial, it is possible that more subtle genetic abnormalities, such as channelopathies in, for example, IGE, may also predispose patients to SUDEP. In this regard, it is important to note that a number of functional cardiac conduction abnormalities, such as long QT syndrome, are also channelopathies, and genetic susceptibility is also well documented in sinus node dysfunction and bradyarrhythmias.43 However, no epidemiologic data indicate a higher incidence of epilepsy among relatives of patients with inherited susceptibility to arrhythmia, and a family history of early sudden cardiac death is not reported in SUDEP series. Furthermore, malignant tachyarrhythmias are relatively uncommon in seizures, and ictal respiratory changes have been documented in the absence of cardiac abnormalities. The proposed cardiorespiratory mechanisms of SUDEP will be discussed in more detail later in the chapter. Clearly, scope exists for potentially useful epidemiologic studies looking at the incidence of epilepsy in families of sudden cardiac death victims; additionally, it may be helpful to study the incidence of syncope in patients with idiopathic epilepsy and their relatives.
Controversy exists on whether high seizure frequency is an independent risk factor for SUDEP. Several descriptive and large case-control studies have reported an increased risk of SUDEP in patients experiencing frequent seizures.28,30,31,33,37,38 This increased risk is most marked for convulsive seizures10–12,27,28,31,37 rather than nonconvulsive episodes, such as complex partial seizures.33 Moreover, on logistic regression analysis, Walczak noted that only the frequency of convulsive seizures was relevant, and not the frequency of all seizures combined.37 Conversely, high seizure frequency was not an independent risk factor in a number of other reports, although a number of methodological issues exist.12,29,34,36 For example, in a retrospective case-control study of 42 patients with SUDEP, reported by Kloster and Engelskjon, there was no reported difference in seizure frequency between the SUDEP and non-SUDEP control group. The study was undertaken at a tertiary referral center with both groups having chronic refractory epilepsy and frequent seizures.36 Other negative studies may have been similarly influenced.34 Intuitively, the severity of convulsive seizures may also be important in SUDEP, but this is more challenging to quantify and hence has not been evaluated as a risk factor.
ANTIEPILEPTIC MEDICATION
The number of antiepileptic medications (AEDs) taken concomitantly have been reported to be an independent risk factor for SUDEP,38 even after correction for seizure frequency.30,37 This is not universally reported, however,12,18,34–36 although small numbers of patients and a high frequency of polytherapy in control subjects may be contributory in these negative studies. Langan found that the risk of SUDEP increased with the number of AEDs previously taken, despite correction for seizure frequency, perhaps a surrogate for epilepsy severity. In addition, risk was also increased in those who had never been on AEDs. This is potentially an important group and needs further clarification. It may include those in whom the epilepsy is considered mild and treatment is not recommended, those who decline treatment, or those with recent onset epilepsy who have not yet been assessed or offered treatment. Risk of SUDEP is also increased in those whose treatment history was unclear, which may reflect the risk associated with the lack of treatment and uncontrolled seizures, although the reason for this was not objectively assessed.31
Despite several descriptive studies suggesting that subtherapeutic levels of AEDs are a risk factor for SUDEP,10,11,25,29 this has not been corroborated by the majority of case-control studies,34,44,45 as it is difficult to study as an independent factor. Of note is that postmortem levels of AEDs may not accurately reflect antemortem levels possibly due to, for example, redistribution and continuing metabolism.46 In a postmortem study reported by George and Davis, however, so-called subtherapeutic drug levels were detected in 69% of the 52 cases of SUDEP, in 75% of the eight cases where a seizure precipitated an accident causing death, and in 34% of the control population.47 This suggests that the increased likelihood of a seizure associated with “subtherapeutic” AED levels, rather than the levels per se, drives the observed elevated mortality rate. Compliance with AED treatment was first proposed as a risk factor for SUDEP in an uncontrolled study by Leestma, who found subtherapeutic AED levels in 68% of SUDEP cases.25 Therapeutic drug monitoring has traditionally been considered a surrogate for medication adherence, although due to the existence of a number of confounding factors, it is clear that the two terms are not interchangeable. For example, in patients with uncontrolled seizures, changes of dose and type of medication are commonplace, and serum levels will not be stable and may frequently be subtherapeutic, despite excellent compliance. More recent studies have attempted to address this by integrating additional clinical information, such as a previous history of noncompliance29 or an arbitrary judgment regarding the degree of compliance made by the treating physician.34 Despite this, conflicting results have been obtained (Table 15-2). Of paramount importance, however, is the understanding that the evaluation of drug levels or noncompliance as independent risk factors for SUDEP must take into account the presence, frequency, and severity of seizures. The issue of variability of AED use was recently addressed in a study by Williams and colleagues comparing hair AED concentration variability in patients with SUDEP, non-SUDEP epilepsy-related deaths, epilepsy outpatients and epilepsy inpatients. The SUDEP group showed greater hair AED concentration variability than either the outpatient or the inpatient groups, reflecting variable AED ingestion over time. However, these variations cannot distinguish prescribed changes from poor compliance or identify consistent noncompliance over time. Second, it does not provide information on drug-taking behavior immediately before death, as it takes about 5 days for a drug sequestrated into the follicle to appear at the scalp; therefore short-term noncompliance immediately before death is not assessed by this study and may have been overlooked.48

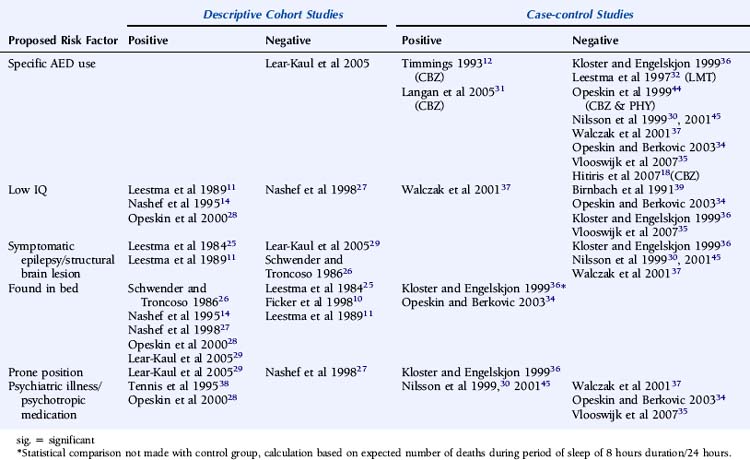
Despite a number of descriptive and controlled studies, no specific AED has been clearly associated with an increased risk of SUDEP,18,29,32,36,37,44,49 although a small number of studies have implicated treatment with carbamazepine as an independent risk factor.31,50,51 For antiepileptic medication in general, proposed mechanisms include perturbed heart rate variability, lengthening of the Q-T interval on the electrocardiogram combined with a mild proarrhythmic effect of epileptic seizure discharges, or excessive postseizure brainstem inhibition producing a blunting or transient abolition of the central hypoxic and hypercarbic respiratory drive, with consequent postictal respiratory arrest.50–52 Elevated serum levels of carbamazepine have been associated with an increased risk of SUDEP, even after adjustments for seizure frequency have been made. Frequent drug changes and multiple concomitant AEDs, conventional markers of severe, and unstable epilepsy increased this risk synergistically.45 On this basis, it is difficult to know whether a high carbamazepine level is an independent risk factor or is merely representative of challenging epilepsy.
PERIMORTEM FEATURES
There is evidence from both descriptive and controlled studies that a terminal convulsive seizure,11,14,25,27,29,34,36,53 being found alone in bed,14,26–28,34,36 and being in the prone position29,36 are independent risk factors for SUDEP. Whereas a small number of descriptive studies have not found an association (Table 15-2), all case-control studies that have evaluated these factors have found a positive relationship with the risk of SUDEP. For example, the descriptive study reported by Ficker failed to find an association between being in bed and SUDEP or evidence for a terminal seizure, although only nine patients were evaluated.10 In a report by Nashef, following interviews with bereaved relatives, evidence for a terminal seizure was found in 24 of 26 cases, but it is of interest that only two were witnessed. The observation that, in most studies, unwitnessed cases far outnumber those witnessed suggests that enhanced surveillance of patients with epilepsy may be protective.27 This is corroborated by a study of young patients with epilepsy at a special residential school. All sudden deaths that occurred during the period of the study were when the pupils were not under the close supervision of the school, and most were unwitnessed.15 Similar findings of a protective effect of enhanced supervision at night were also found in a large controlled study, where supervision was defined as the presence in the bedroom of an individual of normal intelligence and at least 10 years old or the use of special precautions, such as checks throughout the night or the use of a listening device.31
In some cases when a prone position was not observed, other factors that might compromise breathing were identified. For example, Nashef noted that only five of 26 people were found facedown in the pillow and a sixth with the head in carpet pile. In total, however, in 11 of 26 cases, an extrinsic or intrinsic positional obstruction to breathing amenable to intervention may have contributed.27 Moreover, it is possible that this may be an underestimate, as obstructive apnea can occur in an apparently benign position.54
OTHER FEATURES
There is limited evidence for an independent relationship between learning disability and an increased risk of SUDEP. Early descriptive and population-based studies, in which learning disability was determined by observer impressions rather than by formal IQ examination, provided only weak support for this association.11,55 Most recent studies have found no clear correlation.27,34–36,39 However, Walczak identified an IQ of less than 70 to be a risk factor for SUDEP, even after accounting for seizure frequency.37 Observational studies are likely to be biased. Nashef found that five of 11 patients with SUDEP had a low IQ, but the study was undertaken at a tertiary referral center with a higher-than-average background incidence of learning disability.14 Similar bias was introduced in an observational study by Opeskin, where all patients in the study region who died in institutions for the disabled were mandatory coroner’s cases, thus artificially elevating SUDEP cases with learning disability.28 It has been postulated that patients with learning disability are more susceptible to central apnea and positional asphyxia that may cause SUDEP as a result of prolonged postictal encephalopathy,56 decreased postictal respiratory drive, and impaired movement and righting reflexes.37 This is likely to reflect associated pathology, rather than the learning difficulty per se. Despite early reports of an increased incidence of structural lesions in patients with SUDEP,11,25,57 this has not been confirmed by more recent controlled studies.30,36,37 Although there is evidence that psychotropic medication can influence the risk of sudden death in general, there is no convincing evidence of this being particularly relevant in SUDEP. Logistic regression analysis on 18 patients with SUDEP suggested an independent association of the risk of SUDEP with the number of concomitant psychotropic medications; however, the study was observational and uncontrolled, and the principal inclusion criteria for the study included any patient prescribed regular antiepileptic medication. Thus, the study population excluded those with epilepsy on no medication and included patients without epilepsy who were prescribed AEDs for an unrelated condition, for example, migraine, neuropathic pain, or psychiatric disorders.38 A single case-control study has also reported an increased risk of SUDEP in women on neuroleptic medication and in men prescribed anxiolytic medication. The explanation for this is unclear and most likely represents an effect of confounding factors.30 A number of more recent case-control studies have failed to identify a similar association.34,35,37
Pathophysiology of SUDEP
CEREBROGENIC AUTONOMIC CONTROL
The components of the central autonomic network involved in the functional relationships between cortical, subcortical, and somatic regions have been elucidated from experimental and human stimulation and lesional studies. The network comprises the insular cortex, central nucleus of the amygdala, and hypothalamus with interconnections with the mesial temporal and frontal areas.58 It has been demonstrated that limbic structures, especially the amygdala and pyriform cortex, modulate hypothalamic function, and stimulation of these foci can elicit both sympathetic and parasympathetic visceromotor autonomic responses.59 Both experimental and ictal electrical stimulation of the cingulate gyrus and orbitofrontal cortex also produce changes in respiration and heart rate.60–64 Intraoperative stimulation of the left insular cortex produces bradycardia and hypotension, whereas the converse is true of right-sided stimulation.65 Unilateral hemispheric inactivation with an intracarotid amobarbital infusion produces bradycardia and reduced sympathetic tone when performed on the right side and tachycardia and reduced parasympathetic tone when applied on the left side.66,67 Similar lateralization findings have been reported in patients undergoing unilateral electroconvulsive therapy.68
Other than visual inspection of a standard 12-lead electrocardiogram (ECG), more sophisticated methods to interrogate the cardiac autonomic system have been developed, for example, measures of heart rate variability (HRV). In its simplest form, this is measured in a time domain analysis as the standard deviation of R-R wave intervals.69,70 Frequency domain analysis permits the calculation of high-frequency (HF) and low-frequency (LF) components that assess the relative contribution of parasympathetic and sympathetic autonomic activity.71 The HF component is attributed to vagal mechanisms, and the LF component reflects sympathetic activity with parasympathetic modulation.72 However, measures of HRV may be more complex than purely sympathovagal balance. For example, baroreflex sensitivity modulation may be an important influence.73 More advanced, nonlinear measures of “regularity,” such as entropy, have recently been used that facilitate further analyses of autonomic function.74 The balance between parasympathetic and sympathetic regulation is dynamic, and in healthy subjects, cyclic variation is commonly observed during the ventilatory cycle and in sleep.75 Reduced HRV has been reported in infants with aborted sudden infant death syndrome,76 in heart transplant patients,77 and as an independent risk factor for sudden arrhythmic death after myocardial infarction.78
CARDIAC MECHANISMS
Structural Cardiac Pathology
The exclusion of cardiac pathology as a contributing factor in SUDEP is challenging due to the presence of, for example, subtle abnormalities that only a detailed microscopic examination of cardiac tissue can elucidate, such as conducting system fibrosis or cardiomyopathy,79 tissue decomposition precluding the acquisition of suitable material for evaluation, lack of an appropriate control group for comparison, and the possibility of a functional rather than a structural disorder, such as ion channelopathies or preexcitation syndromes, with normal macroscopic and microscopic examinations being implicated.43 Furthermore, inconsistencies in the methodology and reporting of cardiac abnormalities in SUDEP postmortem reports prevent meaningful comparison of results.11,30,36,80
Increased cardiac weight has been observed in male SUDEP cases compared to control subjects,11 although it is likely that this is due to demographic and methodological confounding factors, such as a disproportionate prevalence of excess alcohol consumption in the study population and the derivation of expected cardiac weight from a regression equation based on body height, which has been demonstrated to be less accurate than using body weight.81 Recent studies, using more convincing methodology, have failed to replicate this earlier finding, and cardiac weight is not considered to differ between SUDEP and non-SUDEP cases.80,82,83 Minor, nonspecific pathological change presumed to be nonfatal, such as atherosclerosis, conducting system fibrosis, and diffuse myocardial fibrosis, was identified in 33% (14 out of 42) of SUDEP cases, compared to 16% (six out of 37) of control cases, in an unblinded study reported by Kloster and Engelskjon in 1999.36 This study corroborated earlier findings by Falconer, who demonstrated arterial, interstitial, and subendocardial fibrosis with myofibrillar degeneration in SUDEP cases,84 and Natelson, who identified perivascular and interstitial fibrosis and subendocardial myocyte vacuolization in five of seven SUDEP cases. In the latter study, these changes were not seen in the control group, which comprised 13 cases of hanging or drug overdose in otherwise healthy, similarly aged individuals,82 although in this unblinded study the pathologist was aware of the relevant clinical information. Interestingly, myocyte vacuolization has also been observed in rats who developed asystole following stimulation of the insular cortex.82 Myocyte vacuolization is considered a reversible pathological entity, occurring in the context of subendocardial ischemia.85 However, the patients universally had normal appearing coronary arteries. It has been postulated that neurogenic coronary vasospasm may be implicated, and that if recurrent, it may eventually progress to perivascular and interstitial fibrosis.86 This may, in turn, predispose the heart to arrhythmogenesis, particularly in the setting of considerable autonomic imbalance during seizures.87,88 The occurrence and significance of these pathological changes in SUDEP is not universally agreed, however. Opeskin failed to identify similar fibrotic changes in SUDEP cases but did demonstrate cardiac conduction system pathology and myocyte vacuolization, although these abnormalities were no more common in the SUDEP group than in the control subjects.80 Nevertheless, the pathological changes may still be important in sudden death, but not specific for SUDEP. It is acknowledged that the identification of subtle interstitial and perivascular fibrosis is subjective, and quantitative histopathological evaluations were not performed. Furthermore, the results were not corrected for seizure frequency. More recently, qualitative and quantitative histopathological assessments of myocardial fibrosis were undertaken in SUDEP and age- and gender-matched non-SUDEP cases, and although visual assessment showed significantly more fibrosis in the SUDEP cases, this was not verified on quantitation, further reinforcing the view that a purely qualitative evaluation is subjective and possibly unreliable. Furthermore, no abnormalities of the cardiac conducting system were demonstrated.89
Interictal
At the simplest level, interictal cardiac function can be evaluated by visually assessing a standard 12-lead ECG, primarily for evidence of conduction abnormalities, although these are frequently normal90–92 or show only minor, nonsignificant changes.93 However, a recent preliminary study of 128 patients with severe refractory epilepsy and learning disability revealed interictal ECG abnormalities in approximately 60% of patients, including first degree atrioventricular block and poor R-wave progression.94 Potentially, a number of important confounding factors exist, though, including whether this study population is adequately representative, whether account was made of the presence of systemic comorbidity or use of psychotropic medication, and whether the conduction abnormalities are clinically significant.
Early experimental studies demonstrated that interictal epileptiform activity was associated with sympathetic and parasympathetic autonomic dysfunction in a time-locked synchronized pattern.95,96 In the first clinical reports, analysis of interictal heart rate variability in 19 patients with refractory temporal lobe epilepsy revealed frequent, high-amplitude fluctuations in heart rate that were most pronounced in poor surgical candidates.97 Reduced sympathetic tone, demonstrated by decreased LF power, has been seen in both focal and, albeit less markedly, primary generalized epilepsy.69,92,97,98 These findings have been corroborated by more recent studies,74,99,100 although the pattern of interictal autonomic disturbance in patients with epilepsy is contentious. For example, Evrengul reported an increase in LF power and a reduction of HF values consistent with an increase in the sympathetic control of the heart rate in patients with untreated generalized tonic-clonic seizures.71 It is possible that the disparity between the studies may be, at least partly, due to antiepileptic medication. Tomson showed that patients on carbamazepine had a significantly lower standard deviation of RR-intervals, LF power, and a LF/HF power ratio than matched healthy control subjects. In patients on sodium valproate, only the ratio of LF/HF power was lower.69 Interictal autonomic dysfunction associated with carbamazepine use has also been implicated in a number of similar studies. Isojärvi evaluated 84 patients with a variety of epilepsies and observed autonomic dysfunction in only those patients taking carbamazepine.101 Confusingly, rapid withdrawal of carbamazepine has been associated with both increased sympathetic tone during sleep as measured by the LH/HF ratio51 and a significant reduction in heart-rate variability in both the time and frequency domains with, in particular, a significant reduction in LF power and sympathetic tone.102 The reason for the divergence between these studies is not clear, but it is important to note that both were small series studies and may have lacked sufficient power to convincingly demonstrate a clear effect. Furthermore, there may have been methodological differences. To address the possible modulatory effect of antiepileptic medication, patients with untreated epilepsy have been studied and shown to have both increased LF power, and thus augmented sympathetic tone71 and, in a similar study, normal HRV parameters.103 Again, it is difficult to satisfactorily explain the variance in the results, although important differences in the study population may be at least partly responsible.103 In an interesting study of patients with chronic temporal lobe epilepsy (TLE), postganglionic cardiac sympathetic innervation was quantified using (123)I-metaiodobenzylguanidine-single photon computed tomography (MIBG-SPECT). Cardiac MIBG uptake was significantly less in the TLE patients than in the controls, but did not differ between subgroups with and without carbamazepine treatment. The findings are consistent with either postganglionic transsynaptic degeneration resulting from a prolonged increase in central sympathetic interictal discharges or competitive inhibition of MIBG uptake due to continuously enhanced sympathetic activity. The authors concluded that this may translate into an increased risk of cardiac instability and arrhythmias.100
Of recent interest in the pursuit for pathophysiological mechanisms of SUDEP is the potential association between cardiac and epileptic ion channelopathies.43 For example, it is well known that long-QT syndrome, a defect of cardiac repolarization typically due to potassium or sodium channel mutations, produces prolongation of the action potential, propensity to malignant tachyarrhythmias, and sudden death.104 Ion channelopathies are also implicated in familial, monogenic idiopathic epilepsies, such as generalized epilepsy with febrile seizures plus.105 Direct evidence linking these cardiac inherited gene determinants and SUDEP is lacking at present; however, there is emerging evidence of genetic susceptibility in sinus node dysfunction and bradyarrhythmias, and this more complex pattern of inheritance may have more relevance to SUDEP than monogenic disorders (see review by Nashef et al, 200743). Future work in this area should include much larger studies of ECG changes and QT interval in patients with epilepsy and epidemiological studies exploring the association between syncope, inherited arrhythmias, and epilepsy.
Ictal
There is extensive literature on the presence of ECG changes, for example T-wave inversion, ST segment elevation, and a prolonged QT interval, in patients with intracranial pathology, such as subarachnoid hemorrhage and cerebrovascular accident, despite normal cardiac examination at autopsy (see Samuels, 2007 for review106). It is understood that these changes are a manifestation of massive catecholamine release and autonomic dysregulation resulting in ventricular wall motion abnormalities, vasospasm, and subsequent cardiac contraction-band necrosis, rather than being due to established structural cardiac pathology, such as atheroma.107,108 Predominantly neurogenic, rather than humorally driven, autonomic dysfunction has been postulated as the cause of ECG abnormalities during convulsive seizures.
Arrhythmias, conduction block, and repolarization ECG abnormalities, such as atrial fibrillation, marked sinus arrhythmia, supraventricular tachycardia, atrial and ventricular premature depolarization, bundle-branch block, high-grade atrioventricular conduction block, ST segment depression, and T-wave inversion have been reported in up to 56% of seizures. Abnormalities appear to be more common in nocturnal, prolonged, and generalized seizures than in focal seizures or those occurring during wakefulness.59,109–112 Tavernor reported minor prolongation of the ictal, compared to interictal, corrected QT interval (QTc) in a group analysis of 11 patients who subsequently died of SUDEP. The same QTc prolongation was not seen in living age- and sex-matched control subjects who also had refractory epilepsy. The authors concluded that prolongation of the QT interval may provoke malignant ventricular tachyarrhythmias, which may be a potential cause of SUDEP.113 However, although ictal sinus tachycardia is a frequent occurrence, atrial or ventricular tachyarrhythmias are only rarely seen.91,109,114
Sinus rate change is the most common cardiac accompaniment to ictal discharge. Sinus tachycardia has been reported in 50 to 100% of seizures and is dependent on the definition used and population studied.90–92,112,114–119 Although the heart rate in ictal tachycardia is typically 100 to 120 beats per minute,90 there are reports of rates exceeding 170 beats per minute, even during simple partial seizures.91,115 Ictal tachycardia is most commonly seen in the early ictal phase, soon after seizure onset,114,115,118,119 or rarely before clear evidence of electroclinical onset.112 This contrasts with ictal bradycardia, which is seen during the late ictal phase or in the immediate postictal period.120,121 Some evidence exists for right-sided lateralization and temporal lobe localization in patients with ictal tachycardia,114,116,119 corroborating the reports of early experimental and clinical stimulation studies,65,68,122 although it is important to note that most temporal lobe seizures are associated with ictal tachycardia, irrespective of lateralization. In contrast, in patients with unilateral temporal lobe epilepsy being evaluated with extensive intracranial electroencephalogram (EEG) electrodes, irrespective of lateralization of ictal onset, heart rate was seen to increase incrementally as new cortical regions anywhere in the brain were recruited.123
The first report of ictal asystole was by Russell in 1906, who noted the disappearance of a young male patient’s pulse during a seizure.124 The published literature since that time is, unsurprisingly, mostly case reports or small-series studies, which significantly limit the number and confidence of any conclusions extracted from the data. Ictal bradycardia is observed in <5% of recorded seizures,91,114,118,125 but may occur in a higher percentage of patients because a consistent cardiac response to each apparently electroclinically identical seizure is not seen.91
A recent literature review by Britton revealed that of 65 cases of ictal bradycardia with sufficient EEG and ECG data, seizure onset was localized to the temporal lobe in 55%, the frontal lobe in 20%, the frontotemporal region in 23%, and the occipital lobe in 2% of cases. Information regarding seizure-onset lateralization was available in 56 cases. Seizure onset was lateralized to the left hemisphere in 63%, the right in 34%, and bilaterally in 4%. Interestingly, of 22 cases with EEG data available at the onset of the bradycardia, 12 showed bilateral hemispheric ictal activity, whereas six showed left, and four showed right-sided activity.121 No control group data is available, however. Nevertheless, it appears that there is a trend toward the left temporal lobe being implicated in ictal bradycardia, but this is not sufficiently specific to be valuable localizing semiological information.121,125 Of greater interest is the frequent observation of bilateral ictal activity during bradycardia.121,126,127 This may cause a more significant imbalance of parasympathetic and sympathetic dysfunction than unilateral stimulation via either corticocardiac pathways or through connections with subcortical and brainstem regions, which are frequently activated in seizures and that may potentially contribute to a bradycardic response.128,129
Ictal asystole, lasting between 4 and 60 seconds, is reported, albeit rarely, in patients with refractory epilepsy.54,91,112,126,130,131 In addition, experimental data suggests that ictal bradyarrhythmias can lead to complete heart block.96 It is possible, however, that bradycardia and asystole are conditions with distinct pathophysiological bases rather than two points on a continuum of cerebrogenic arrhythmias. However, the characteristics of the epilepsy with respect to localization, lateralization, seizure types, and population demographics are identical to those of patients with ictal bradycardia, suggesting that the two entities are possibly linked. Short periods of EEG/ECG monitoring may underestimate the prevalence of ictal asystole. For example, Schuele searched a database of 6825 patients undergoing inpatient video-EEG monitoring and found ictal asystole in only 0.27% of all patients with epilepsy. In contrast, Rugg-Gunn reported on 19 patients with refractory focal epilepsy who were implanted with an ECG loop recorder for up to 18 months. Over 220,000 patient hours of ECG recording were monitored, during which time 3377 seizures (1897 complex partial or secondarily generalized tonic-clonic seizures and 1480 simple partial seizures) were reported by patients. Cardiac rhythm was captured on the implantable loop recorders in 377 seizures. Ictal bradycardia, defined as a rate of less than 40 beats per minute, was seen in 0.24% of all seizures over the study period and 2.1% of the recorded seizures. One patient developed supraventricular tachycardia (rate 120 bpm) lasting approximately 30 seconds, during a complex partial seizure. Seven of the 19 patients experienced ictal bradycardia. Four of these had severe bradycardia or periods of asystole, which led to the insertion of a permanent pacemaker. The small number of patients involved precluded statistical analysis of localization and lateralization data. There was no clear correlation between cardiac events and specific AEDs. Notably, only a small proportion of seizures for every patient were associated with significant cardiac events, despite identical seizure characteristics.91 The wider significance of these findings remains to be established but may be addressed by a larger UK-based multicenter study that is currently underway. For example, although this study showed asystole in patients with refractory focal seizures, a similar intractable group of patients with generalized epilepsy has not been studied. Nevertheless, the identification and targeting of patients at risk of ictal asystole, preferably with an interictal surrogate marker, is an important goal.
RESPIRATORY MECHANISMS
It is likely that primary respiratory dysfunction is involved in an important proportion of SUDEP.54,132–138 Central and obstructive apnea, excess bronchial and oral secretions, pulmonary edema, and hypoxia during seizures are all well documented.54,136,138–140 Central chemoreceptive areas responsible for sensitivity to increases in carbon dioxide have been shown to be located in several areas of the brainstem, including the nucleus tractus solitarii, locus coeruleus, medullary raphe, and ventrolateral medulla, which comprises an extensive network of respiratory neurons, known collectively as the ventral respiratory group, involving the nucleus ambiguous (NA) and Bötzinger and pre-Bötzinger complexes.141 Changes in the arterial partial pressure of CO2 evoke rapid alterations in respiration.142 This brainstem network receives supranuclear afferents from cortical areas, such as the rolandic region, or via cranial nerves, such as the trigeminal and glossopharyngeal nerves.60 Central apnea can therefore occur secondary to the ictal discharge, acting at either the cortical or medullary level or possibly as a result of secondary endogenous opioid release influencing the brainstem respiratory nuclei directly. During postictal impairment of consciousness, hypercapnia and hypoxia may be less potent respiratory stimuli.
In a study of 17 patients with epilepsy who underwent polysomnography with cardiorespiratory monitoring in a supervised environment, ictal apnea of greater than 10 seconds in duration was demonstrated in 20 of 47 seizures. Oxyhemoglobin saturation decreased to less than 85% in 10 seizures. Central apnea, which may evolve during a focal or generalized seizure, was seen more frequently than obstructive apnea; however, the study was in a controlled environment, and assistance may have minimized the likelihood of obstructive apnea being observed.54 Interictal obstructive sleep apnea has been reported to be more frequent in patients with epilepsy than in the general population, although the reasons for this are unclear.143 Interestingly, transient bradycardia or sinus arrest has been seen in association with ictal apnea, suggesting that the reported seizure-related arrhythmias may be consecutive to ictal apnea.54 Similar findings have been reported in children.138 Additional reports of ictal apnea are typically case studies recorded incidentally during video-EEG telemetry.134–136 In a study of 135 SUDEP cases, 15 of which were witnessed, observers described respiratory difficulties, such as apnea and obvious respiratory obstruction, in 12 patients, although the conclusions that may be drawn are significantly limited by the quality of the retrieved information and lack of additional relevant cardiorespiratory parameters.137 Witnesses have reported a delay between the seizure and time of death, which is more consistent with primary respiratory inhibition followed by respiratory arrest and the development of hypoxia and pulmonary edema, than “primary” ictal cardiac asystole.29 Analogous with reports of patients with ictal bradycardia and asystole, the majority of published cases of ictal central apnea had temporal lobe epilepsy, although this is likely to represent selection bias. No clear lateralizing information is available from the published literature.
Neurogenic pulmonary edema, which may in itself be insufficient to be fatal, has been implicated in theories regarding respiratory dysfunction and SUDEP following a number of postmortem reports and case studies.11,29,133,140 In a sheep model of ictal sudden death, animals that died had a greater increase in pulmonary vascular pressure and hypoventilation. When airway obstruction was excluded by tracheostomy, central apnea and hypoventilation were observed in all, causing or contributing to death in two, whereas a third animal developed heart failure with significant pathologic cardiac ischemic changes.132,144 The apparent protective effect of supervision favors an important primary role for respiratory factors,31 as these can be influenced by relatively unskilled intervention, such as airway protection, repositioning, or stimulation. It is unknown what proportion of SUDEP cases may be prevented by such intervention.
SUPPRESSION OF CEREBRAL ACTIVITY
The possibility of progressive suppression and eventually cessation of cerebral activity as a cause of SUDEP, despite normal cardiac function, was introduced with the publication of a case report of an intracranially monitored patient who died of SUDEP. Bird described a seizure starting in sleep in one hemisphere and spreading to the other for several minutes. The EEG pattern on the original side then changed to burst-suppression with spindling spike discharges, followed by complete cessation of activity. The other hemisphere continued to show spike discharges until ceasing suddenly a few seconds later. A pulse artifact on the EEG continued for an additional 2 minutes; there was no recording of respiratory activity. Postmortem examination showed mild congestion of the lungs. It was postulated that the loss of EEG activity was not preceded by anoxia, as both hemispheres were not simultaneously affected.145
Excessive postictal brainstem inhibition due to seizure-induced release of GABA and other neuroinhibitory peptides may contribute to death in some patients. This may be compounded by antiepileptic medication.50 This endogenous seizure-terminating mechanism could result in blunting of the central hypoxic and hypercarbic respiratory drive, resulting in postictal respiratory arrest, subsequent exacerbation of hypoxia, further cardiac destabilization, and death due to hypoxia and secondary cardiac arrhythmia. This is consistent with the observation that SUDEP occurs after a seizure and could be a consequence of failed reestablishment of respiration in the postictal phase.
SUDEP and Epilepsy Surgery
Compelling evidence shows that patients with poorly controlled, predominantly generalized tonic-clonic seizures are at greatest risk of SUDEP, and a seizure is frequently seen as the terminal event. Intuitively, therefore, good seizure control should translate into reduced risk of SUDEP. Sperling evaluated the mortality rates of 393 patients who underwent epilepsy surgery. The standardized mortality ratio (SMR) for patients with recurrent seizures postoperatively was 4.69, with a SUDEP incidence of 7.5/1000 patient-years, whereas in patients who became seizure free, there was no difference in mortality rate compared with an age- and sex-matched population.146 This compares with the results from Hennessy, who identified an overall postoperative SMR of 4.5 and SUDEP incidence of 2.2/1000 patient-years, although the results were not stratified for postoperative seizure outcome,147 and Salanova, who found a SMR of 1.8 in those with a good postoperative outcome versus 7.4 in those who failed surgery.148 In a large, population-based epilepsy surgery cohort, Nilsson failed to demonstrate an association between mortality rates and seizure outcomes, although there was a clear difference between patients who underwent surgery (SUDEP incidence 2.4 per 1000 patient-years) and those who failed presurgical assessment (SUDEP incidence 6.3 per 1000 patient-years).16 There has been recent interest in the tenet that a common factor predisposes to surgical failure and an increased risk of SUDEP so that patients who respond poorly to surgery also carry an increased risk of SUDEP and that, overall, surgery does not alter the risk of SUDEP.149 Proposed common factors include TLE, which extends beyond the temporal lobe into the insula, frontal orbital, or frontal operculum region, which may favor ictal arrhythmias, central apnea, and secondary generalization. This, in turn, would increase the risk of SUDEP, and the wide epileptogenic field would translate into a poor postoperative seizure outcome.149 This is supported by a study by Persson, who reported on 21 patients undergoing temporal lobe surgery and found that postoperative measures of HRV did not differ from preoperative HRV. Patients with a poor outcome after surgery had significantly lower pre- and postoperative HRV than patients with a good outcome, who had similar HRV measures to healthy control subjects.70,150 In contrast, Hilz found a postoperative reduction in sympathetic cardiovascular modulation and baroreflex sensitivity in patients undergoing temporal lobe resection, consistent with stabilization of cardiac autonomic control.151
Mortality studies performed in patients with vagal nerve stimulators have shown that excess mortality associated with refractory epilepsy reduced as a function of duration of use. The rate of SUDEP was 5.5 per 1000 patient-years in the first 24 months and 1.7 per 1000 patient-years thereafter, possibly reflecting gradual increase in efficacy over time. Stabilization of measures of heart rate variability post-VNS (vagal nerve stimulation) implantation152–154 have paralleled the improved mortality rates, although these findings are not universal.155,156
Implications for Management
Given the disturbance in cardiac autonomic control in patients with epilepsy, there has been speculation as to whether cardiotropic medication, such as beta-antagonists, may have a protective effect, although no studies have been performed in this regard.111 Experimental studies in rats with audiogenic seizures and ictal apnea have shown that selective serotonin reuptake inhibitors have a protective effect,157 although relevant confirmatory clinical studies are lacking. Of interest, however, is the recent finding of neuropathological evidence of involvement of the medullary serotonergic network in sudden infant death syndrome cases with a significantly lower density of serotonin receptor binding sites, particularly in male SIDS cases compared to controls.158 Whether pharmacological modulation of the brainstem serotonergic network or cardiac autonomic function results in a protective effect remains to be seen.
Supervision of patients with epilepsy has emerged as the only clinically important protective factor, independent of seizure control. The basis for this remains unclear but may relate to body positioning and alleviation of obstructive apnea or possibly brainstem arousal mechanisms.15,27,31,137 Strategies to adequately monitor patients with epilepsy at night, evaluating either cardiac, respiratory, or body movement parameters, have been developed, but issues with, for example, high false-positive rates render the devices less user friendly. As a result, there is an urgent need to develop more sophisticated, unobtrusive, reliable, and affordable monitoring equipment.
In the U.K. the NICE guidelines state that tailored information and discussion between the individual, family, and/or caregivers and health care professional should take account of the small but definite risk of SUDEP.159 Several studies have suggested that the relatives of people who died from SUDEP wished they had known of the risk of sudden death.27,160 Whereas it is not certain that knowledge of the risks of epilepsy would necessarily prevent death, some evidence suggests that observation, positioning, and where necessary, stimulation after a seizure may protect against death. It is also likely that patients who know of the risks of epilepsy might be more adherent to AED regimens and the avoidance of trigger factors, thus reducing the frequency of seizures. In contrast, a cohort-controlled study of SUDEP from Australia concluded that because no clear risk factors were modifiable by practical intervention, disclosure to the patient of the possibility of SUDEP was inappropriate.161
1. Hauser WA, Annegers JF, Elveback LR. Mortality in patients with epilepsy. Epilepsia. 1980;21:399-412.
2. Cockerell OC, Johnson AL, Sander JW, Hart YM, Goodridge DM, Shorvon SD. Mortality from epilepsy: results from a prospective population-based study. Lancet. 1994;344:918-921.
3. Annegers JF, Coan SP. SUDEP: overview of definitions and review of incidence data. Seizure. 1999;8:347-352.
4. Shorvon S. The treatment of chronic epilepsy: a review of recent studies of clinical efficacy and side effects. Curr Opin Neurol. 2007;20:159-163.
5. Nashef L. Sudden unexpected death in epilepsy: terminology and definitions. Epilepsia. 1997;38:S6-S8.
6. Monte CP, Arends JB, Tan IY, Aldenkamp AP, Limburg M, De Krom MC. Sudden unexpected death in epilepsy patients: risk factors. A systematic review. Seizure. 2007;16:1-7.
7. Tellez-Zenteno JF, Ronquillo LH, Wiebe S. Sudden unexpected death in epilepsy: evidence-based analysis of incidence and risk factors. Epilepsy Res. 2005;65:101-115.
8. Elveback LR, Connolly DC, Kurland LT. Coronary heart disease in residents of Rochester, Minnesota. II. Mortality, incidence, and survivorship, 1950-1975. Mayo Clin Proc. 1981;56:665-672.
9. Tomson T, Walczak T, Sillanpaa M, Sander JW. Sudden unexpected death in epilepsy: a review of incidence and risk factors. Epilepsia. 2005;46(Suppl 11):54-61.
10. Ficker DM, So EL, Shen WK, et al. Population-based study of the incidence of sudden unexplained death in epilepsy. Neurology. 1998;51:1270-1274.
11. Leestma JE, Walczak T, Hughes JR, Kalelkar MB, Teas SS. A prospective study on sudden unexpected death in epilepsy. Ann Neurol. 1989;26:195-203.
12. Timmings PL. Sudden unexpected death in epilepsy: a local audit. Seizure. 1993;2:287-290.
13. Lip GY, Brodie MJ. Sudden death in epilepsy: an avoidable outcome? J R Soc Med. 1992;85:609-611.
14. Nashef L, Fish DR, Sander JW, Shorvon SD. Incidence of sudden unexpected death in an adult outpatient cohort with epilepsy at a tertiary referral centre. J Neurol Neurosurg Psychiatry. 1995;58:462-464.
15. Nashef L, Fish DR, Garner S, Sander JW, Shorvon SD. Sudden death in epilepsy: a study of incidence in a young cohort with epilepsy and learning difficulty. Epilepsia. 1995;36:1187-1194.
16. Nilsson L, Ahlbom A, Farahmand BY, Tomson T. Mortality in a population-based cohort of epilepsy surgery patients. Epilepsia. 2003;44:575-581.
17. Dasheiff RM. Sudden unexpected death in epilepsy: a series from an epilepsy surgery program and speculation on the relationship to sudden cardiac death. J Clin Neurophysiol. 1991;8:216-222.
18. Hitiris N, Suratman S, Kelly K, Stephen LJ, Sills GJ, Brodie MJ. Sudden unexpected death in epilepsy: a search for risk factors. Epilepsy Behav. 2007;10:138-141.
19. Donner EJ, Smith CR, Snead OCIII. Sudden unexplained death in children with epilepsy. Neurology. 2001;57:430-434.
20. Camfield CS, Camfield CP. Good news—a population based study indicates that SUDEP is very unusual in childhood onset epilepsy. Epilepsia. 1999;40(Suppl 7):159.
21. Weber P, Bubl R, Blauenstein U, Tillmann BU, Lutschg J. Sudden unexplained death in children with epilepsy: a cohort study with an eighteen-year follow-up. Acta Paediatr. 2005;94:564-567.
22. Camfield P, Camfield C. Sudden unexpected death in people with epilepsy: a pediatric perspective. Semin Pediatr Neurol. 2005;12:10-14.
23. Sillanpää M, Jalava M, Kaleva O, Shinnar S. Long-term prognosis of seizures with onset in childhood. N Engl J Med. 1998;338:1715-1722.
24. Schraeder PL, Delin K, McClelland RL, So EL. Coroner and medical examiner documentation of sudden unexplained deaths in epilepsy. Epilepsy Res. 2006;68:137-143.
25. Leestma JE, Kalelkar MB, Teas SS, Jay GW, Hughes JR. Sudden unexpected death associated with seizures: analysis of 66 cases. Epilepsia. 1984;25:84-88.
26. Schwender LA, Troncoso JC. Evaluation of sudden death in epilepsy. Am J Forensic Med Pathol. 1986;7:283-287.
27. Nashef L, Garner S, Sander JW, Fish DR, Shorvon SD. Circumstances of death in sudden death in epilepsy: interviews of bereaved relatives. J Neurol Neurosurg Psychiatry. 1998;64:349-352.
28. Opeskin K, Harvey AS, Cordner SM, Berkovic SF. Sudden unexpected death in epilepsy in Victoria. J Clin Neurosci. 2000;7:34-37.
29. Lear-Kaul KC, Coughlin L, Dobersen MJ. Sudden unexpected death in epilepsy: a retrospective study. Am J Forensic Med Pathol. 2005;26:11-17.
30. Nilsson L, Farahmand BY, Persson PG, Thiblin I, Tomson T. Risk factors for sudden unexpected death in epilepsy: a case-control study. Lancet. 1999;353:888-893.
31. Langan Y, Nashef L, Sander JW. Case-control study of SUDEP. Neurology. 2005;64:1131-1133.
32. Leestma JE, Annegers JF, Brodie MJ, et al. Sudden unexplained death in epilepsy: observations from a large clinical development program. Epilepsia. 1997;38:47-55.
33. Schnabel R, Beblo M, May TW, Burmester L. Is sudden unexplained death in adult epileptic patients associated with geomagnetic disturbances at the day of death or the 4 days before? Neurosci Lett. 2002;329:261-264.
34. Opeskin K, Berkovic SF. Risk factors for sudden unexpected death in epilepsy: a controlled prospective study based on coroners cases. Seizure. 2003;12:456-464.
35. Vlooswijk MC, Majoie HJ, De Krom MC, Tan IY, Aldenkamp AP. SUDEP in the Netherlands: a retrospective study in a tertiary referral center. Seizure. 2007;16:153-159.
36. Kloster R, Engelskjon T. Sudden unexpected death in epilepsy (SUDEP): a clinical perspective and a search for risk factors. J Neurol Neurosurg Psychiatry. 1999;67:439-444.
37. Walczak TS, Leppik IE, D’Amelio M, et al. Incidence and risk factors in sudden unexpected death in epilepsy: a prospective cohort study. Neurology. 2001;56:519-525.
38. Tennis P, Cole TB, Annegers JF, Leestma JE, McNutt M, Rajput A. Cohort study of incidence of sudden unexplained death in persons with seizure disorder treated with antiepileptic drugs in Saskatchewan, Canada. Epilepsia. 1995;36:29-36.
39. Birnbach CD, Wilensky AJ, Dodrill CB. Predictors of early mortality and sudden death in epilepsy: A multidisciplinary approach. J Epilepsy. 1991;4:11-17.
40. Sekul EA, Moak JP, Schultz RJ, Glaze DG, Dunn JK, Percy AK. Electrocardiographic findings in Rett syndrome: an explanation for sudden death? J Pediatr. 1994;125:80-82.
41. Nabbout R. Can SCN1A mutations account for SUDEP? Commentary on Hindocha et al. Epilepsia. 2008;49:367-368.
42. Hindocha N, Nashef L, Elmslie F, et al. Two cases of sudden unexpected death in epilepsy in a GEFS+ family with an SCN1A mutation. Epilepsia. 2008;49:360-365.
43. Nashef L, Hindocha N, Makoff A. Risk factors in sudden death in epilepsy (SUDEP): the quest for mechanisms. Epilepsia. 2007;48:859-871.
44. Opeskin K, Burke MP, Cordner SM, Berkovic SF. Comparison of antiepileptic drug levels in sudden unexpected deaths in epilepsy with deaths from other causes. Epilepsia. 1999;40:1795-1798.
45. Nilsson L, Bergman U, Diwan V, Farahmand BY, Persson PG, Tomson T. Antiepileptic drug therapy and its management in sudden unexpected death in epilepsy: a case-control study. Epilepsia. 2001;42:667-673.
46. Tomson T, Skold AC, Holmgen P, Nilsson L, Danielsson B. Postmortem changes in blood concentrations of phenytoin and carbamazepine: an experimental study. Ther Drug Monit. 1998;20:309-312.
47. George JR, Davis GG. Comparison of anti-epileptic drug levels in different cases of sudden death. J Forensic Sci. 1998;43:598-603.
48. Williams J, Lawthom C, Dunstan FD, et al. Variability of antiepileptic medication taking behaviour in sudden unexplained death in epilepsy: hair analysis at autopsy. J Neurol Neurosurg Psychiatry. 2006;77:481-484.
49. Walczak T. Do antiepileptic drugs play a role in sudden unexpected death in epilepsy? Drug Saf. 2003;26:673-683.
50. Timmings PL. Sudden unexpected death in epilepsy: is carbamazepine implicated? Seizure. 1998;7:289-291.
51. Hennessy MJ, Tighe MG, Binnie CD, Nashef L. Sudden withdrawal of carbamazepine increases cardiac sympathetic activity in sleep. Neurology. 2001;57:1650-1654.
52. Tomson T, Kenneback G. Arrhythmia, heart rate variability and anti-epileptic drugs. Epilepsia. 1997;38:S48-S51.
53. Thom M, Seetah S, Sisodiya S, Koepp M, Scaravilli F. Sudden and unexpected death in epilepsy (SUDEP): evidence of acute neuronal injury using HSP-70 and c-Jun immunohistochemistry. Neuropathol Appl Neurobiol. 2003;29:132-143.
54. Nashef L, Walker F, Allen P, Sander JW, Shorvon SD, Fish DR. Apnoea and bradycardia during epileptic seizures: relation to sudden death in epilepsy. J Neurol Neurosurg Psychiatry. 1996;60:297-300.
55. Hirsch CS, Martin DL. Unexpected death in young epileptics. Neurology. 1971;21:682-690.
56. Biton V, Gates JR, dePadua SL. Prolonged postictal encephalopathy. Neurology. 1990;40:963-966.
57. Annegers JF, Hauser WA, Shirts SB. Heart disease mortality and morbidity in patients with epilepsy. Epilepsia. 1984;25:699-704.
58. Benarroch EE. Overview of the organisation of the central autonomic network. In: Benarroch EE, editor. Central Autonomic Network. New York: Futura Publishing; 1997:3.
59. Altenmuller DM, Zehender M, Schulze-Bonhage A. High-grade atrioventricular block triggered by spontaneous and stimulation-induced epileptic activity in the left temporal lobe. Epilepsia. 2004;45:1640-1644.
60. Penfield W, Jasper H. Summary of clinical analysis and seizure patterns. In: Epilepsy and the Functional Anatomy of the Human Brain. Boston: Little Brown; 1954:818-844.
61. van Buren J. Sensory, motor and autonomic effects of mesial temporal stimulation in man. J Neurosurg. 1961;18:273-288.
62. Oppenheimer SM, Cechetto DF, Hachinski VC. Cerebrogenic cardiac arrhythmias. Cerebral electrocardiographic influences and their role in sudden death. Arch Neurol. 1990;47:513-519.
63. Leung H, Schindler K, Kwan P, Elger C. Asystole induced by electrical stimulation of the left cingulate gyrus. Epileptic Disord. 2007;9:77-81.
64. Mascia A, Quarato PP, Sparano A, et al. Cardiac asystole during right frontal lobe seizures: a case report. Neurol Sci. 2005;26:340-343.
65. Oppenheimer SM, Gelb A, Girvin JP, Hachinski VC. Cardiovascular effects of human insular cortex stimulation. Neurology. 1992;42:1727-1732.
66. Zamrini EY, Meador KJ, Loring DW, et al. Unilateral cerebral inactivation produces differential left/right heart rate responses. Neurology. 1990;40:1408-1411.
67. Yoon BW, Morillo CA, Cechetto DF, Hachinski V. Cerebral hemispheric lateralization in cardiac autonomic control. Arch Neurol. 1997;54:741-744.
68. Swartz CM, Abrams R, Lane RD, DuBois MA, Srinivasaraghavan J. Heart rate differences between right and left unilateral electroconvulsive therapy. J Neurol Neurosurg Psychiatry. 1994;57:97-99.
69. Tomson T, Ericson M, Ihrman C, Lindblad LE. Heart rate variability in patients with epilepsy. Epilepsy Res. 1998;30:77-83.
70. Persson H, Kumlien E, Ericson M, Tomson T. Preoperative heart rate variability in relation to surgery outcome in refractory epilepsy. Neurology. 2005;65:1021-1025.
71. Evrengul H, Tanriverdi H, Dursunoglu D, et al. Time and frequency domain analyses of heart rate variability in patients with epilepsy. Epilepsy Res. 2005;63:131-139.
72. Task Force of the European Society of Cardiology and the North American Society of Pacing and Electrophysiology. Heart rate variability: standards of measurement, physiological interpretation, and clinical use. Circulation. 1996;93:1043-1065.
73. Sleight P, La Rovere MT, Mortara A, et al. Physiology and pathophysiology of heart rate and blood pressure variability in humans: is power spectral analysis largely an index of baroreflex gain? Clin Sci (Lond). 1995;88:103-109.
74. Ansakorpi H, Korpelainen JT, Huikuri HV, Tolonen U, Myllyla VV, Isojärvi JI. Heart rate dynamics in refractory and well controlled temporal lobe epilepsy. J Neurol Neurosurg Psychiatry. 2002;72:26-30.
75. Baharav A, Kotagal S, Gibbons V, et al. Fluctuations in autonomic nervous activity during sleep displayed by power spectrum analysis of heart rate variability. Neurology. 1995;45:1183-1187.
76. Pincus SM, Cummins TR, Haddad GG. Heart rate control in normal and aborted-SIDS infants. Am J Physiol. 1993;264:R638-R646.
77. Guzzetti S, Signorini MG, Cogliati C, et al. Non-linear dynamics and chaotic indices in heart rate variability of normal subjects and heart-transplanted patients. Cardiovasc Res. 1996;31:441-446.
78. Bigger JTJr, Steinman RC, Rolnitzky LM, Fleiss JL, Albrecht P, Cohen RJ. Power law behavior of RR-interval variability in healthy middle-aged persons, patients with recent acute myocardial infarction, and patients with heart transplants. Circulation. 1996;93:2142-2151.
79. Corrado D, Basso C, Thiene G. Sudden cardiac death in young people with apparently normal heart. Cardiovasc Res. 2001;50:399-408.
80. Opeskin K, Thomas A, Berkovic SF. Does cardiac conduction pathology contribute to sudden unexpected death in epilepsy? Epilepsy Res. 2000;40:17-24.
81. Hangartner JR, Marley NJ, Whitehead A, Thomas AC, Davies MJ. The assessment of cardiac hypertrophy at autopsy. Histopathology. 1985;9:1295-1306.
82. Natelson BH, Suarez RV, Terrence CF, Turizo R. Patients with epilepsy who die suddenly have cardiac disease. Arch Neurol. 1998;55:857-860.
83. Davis GG, McGwin GJr. Comparison of heart mass in seizure patients dying of sudden unexplained death in epilepsy to sudden death due to some other cause. Am J Forensic Med Pathol. 2004;25:23-28.
84. Falconer B, Rajs J. Post-mortem findings of cardiac lesions in epileptics: a preliminary report. Forensic Sci. 1976;8:63-71.
85. Pirolo JS, Hutchins GM, Moore GW. Myocyte vacuolization in infarct border zones is reversible. Am J Pathol. 1985;121:444-450.
86. Cordero DL, Cagin NA, Natelson BH. Neurocardiology update: role of the nervous system in coronary vasomotion. Cardiovasc Res. 1995;29:319-328.
87. Natelson BH, Chang Q. Sudden death. A neurocardiologic phenomenon. Neurol Clin. 1993;11:293-308.
88. Kawara T, Derksen R, de Groot JR, et al. Activation delay after premature stimulation in chronically diseased human myocardium relates to the architecture of interstitial fibrosis. Circulation. 2001;104:3069-3075.
89. P-Codrea Tigaran S, Dalager-Pedersen S, Baandrup U, Dam M, Vesterby-Charles A. Sudden unexpected death in epilepsy: is death by seizures a cardiac disease? Am J Forensic Med Pathol. 2005;26:99-105.
90. Blumhardt LD, Smith PE, Owen L. Electrocardiographic accompaniments of temporal lobe epileptic seizures. Lancet. 1986;1:1051-1056.
91. Rugg-Gunn FJ, Simister RJ, Squirrell M, Holdright DR, Duncan JS. Cardiac arrhythmias in focal epilepsy: a prospective long-term study. Lancet. 2004;364:2212-2219.
92. Massetani R, Strata G, Galli R, et al. Alteration of cardiac function in patients with temporal lobe epilepsy: different roles of EEG-ECG monitoring and spectral analysis of RR variability. Epilepsia. 1997;38:363-369.
93. Drake ME, Reider CR, Kay A. Electrocardiography in epilepsy patients without cardiac symptoms. Seizure. 1993;2:63-65.
94. Petkar S, Cooper P, Fitzpatrick A. High incidence of ECG abnormalities in Severe epilepsy. World Congress of Cardiology. 2006:1335.
95. Lathers CM, Schraeder PL, Weiner FL. Synchronization of cardiac autonomic neural discharge with epileptogenic activity: the lockstep phenomenon. Electroencephalogr Clin Neurophysiol. 1987;67:247-259.
96. Lathers CM, Schraeder PL. Autonomic dysfunction in epilepsy: characterization of autonomic cardiac neural discharge associated with pentylenetetrazol-induced epileptogenic activity. Epilepsia. 1982;23:633-647.
97. Frysinger RC, Engel J, Harper RM. Interictal heart rate patterns in partial seizure disorders. Neurology. 1993;43:2136-2139.
98. Toichi M, Murai T, Sengoku A, Miyoshi K. Interictal change in cardiac autonomic function associated with EEG abnormalities and clinical symptoms: a longitudinal study following acute deterioration in two patients with temporal lobe epilepsy. Psychiatry Clin Neurosci. 1998;52:499-505.
99. Ansakorpi H, Korpelainen JT, Suominen K, Tolonen U, Myllyla VV, Isojärvi JI. Interictal cardiovascular autonomic responses in patients with temporal lobe epilepsy. Epilepsia. 2000;41:42-47.
100. Druschky A, Hilz MJ, Hopp P, et al. Interictal cardiac autonomic dysfunction in temporal lobe epilepsy demonstrated by [(123)I]metaiodobenzylguanidine-SPECT. Brain. 2001;124:2372-2382.
101. Isojärvi JI, Ansakorpi H, Suominen K, Tolonen U, Repo M, Myllyla VV. Interictal cardiovascular autonomic responses in patients with epilepsy. Epilepsia. 1998;39:420-426.
102. Kenneback G, Ericson M, Tomson T, Bergfeldt L. Changes in arrhythmia profile and heart rate variability during abrupt withdrawal of antiepileptic drugs. Implications for sudden death. Seizure. 1997;6:369-376.
103. Persson H, Ericson M, Tomson T. Heart rate variability in patients with untreated epilepsy. Seizure. 2007;16:504-508.
104. Kass RS, Moss AJ. Long QT syndrome: novel insights into the mechanisms of cardiac arrhythmias. J Clin Invest. 2003;112:810-815.
105. Escayg A, MacDonald BT, Meisler MH, et al. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet. 2000;24:343-345.
106. Samuels MA. The brain-heart connection. Circulation. 2007;116:77-84.
107. Mayer SA, LiMandri G, Sherman D, et al. Electrocardiographic markers of abnormal left ventricular wall motion in acute subarachnoid hemorrhage. J Neurosurg. 1995;83:889-896.
108. Samuels MA. Neurogenic heart disease: a unifying hypothesis. Am J Cardiol. 1987;60:15J-19J.
109. Nei M, Ho RT, Abou-Khalil BW, et al. EEG and ECG in sudden unexplained death in epilepsy. Epilepsia. 2004;45:338-345.
110. Nei M, Ho RT, Sperling MR. EKG abnormalities during partial seizures in refractory epilepsy. Epilepsia. 2000;41:542-548.
111. Opherk C, Coromilas J, Hirsch LJ. Heart rate and EKG changes in 102 seizures: analysis of influencing factors. Epilepsy Res. 2002;52:117-127.
112. Zijlmans M, Flanagan D, Gotman J. Heart rate changes and ECG abnormalities during epileptic seizures: prevalence and definition of an objective clinical sign. Epilepsia. 2002;43:847-854.
113. Tavernor SJ, Brown SW, Tavernor RM, Gifford C. Electrocardiograph QT lengthening associated with epileptiform EEG discharges–a role in sudden unexplained death in epilepsy? Seizure. 1996;5:79-83.
114. Leutmezer F, Schernthaner C, Lurger S, Potzelberger K, Baumgartner C. Electrocardiographic changes at the onset of epileptic seizures. Epilepsia. 2003;44:348-354.
115. Galimberti CA, Marchioni E, Barzizza F, Manni R, Sartori I, Tartara A. Partial epileptic seizures of different origin variably affect cardiac rhythm. Epilepsia. 1996;37:742-747.
116. Kirchner A, Pauli E, Hilz MJ, Neundorfer B, Stefan H. Sex differences and lateral asymmetry in heart rate modulation in patients with temporal lobe epilepsy. J Neurol Neurosurg Psychiatry. 2002;73:73-75.
117. Opherk C, Coromilas J, Hirsch LJ. Heart rate and EKG changes in 102 seizures: analysis of influencing factors. Epilepsy Res. 2002;52:117-127.
118. Smith PE, Howell SJ, Owen L, Blumhardt LD. Profiles of instant heart rate during partial seizures. Electroencephalogr Clin Neurophysiol. 1989;72:207-217.
119. Mayer H, Benninger F, Urak L, Plattner B, Geldner J, Feucht M. EKG abnormalities in children and adolescents with symptomatic temporal lobe epilepsy. Neurology. 2004;63:324-328.
120. Schuele SU, Bermeo AC, Alexopoulos AV, et al. Video-electrographic and clinical features in patients with ictal asystole. Neurology. 2007;69:434-441.
121. Britton JW, Ghearing GR, Benarroch EE, Cascino GD. The ictal bradycardia syndrome: localization and lateralization. Epilepsia. 2006;47:737-744.
122. Oppenheimer SM, Cechetto DF. Cardiac chronotropic organization of the rat insular cortex. Brain Res. 1990;533:66-72.
123. Epstein MA, Sperling MR, O’Connor MJ. Cardiac rhythm during temporal lobe seizures. Neurology. 1992;42:50-53.
124. Russell AE. Cessation of the pulse during the onset of epileptic fits. Lancet. 1906;2:152-154.
125. Tinuper P, Bisulli F, Cerullo A, et al. Ictal bradycardia in partial epileptic seizures: Autonomic investigation in three cases and literature review. Brain. 2001;124:2361-2371.
126. Rossetti AO, Dworetzky BA, Madsen JR, Golub O, Beckman JA, Bromfield EB. Ictal asystole with convulsive syncope mimicking secondary generalisation: a depth electrode study. J Neurol Neurosurg Psychiatry. 2005;76:885-887.
127. Devinsky O, Pacia S, Tatambhotla G. Bradycardia and asystole induced by partial seizures: a case report and literature review. Neurology. 1997;48:1712-1714.
128. Oppenheimer SM, Saleh T, Cechetto DF. Lateral hypothalamic area neurotransmission and neuromodulation of the specific cardiac effects of insular cortex stimulation. Brain Res. 1992;581:133-142.
129. Lee KH, Meador KJ, Park YD, et al. Pathophysiology of altered consciousness during seizures: Subtraction SPECT study. Neurology. 2002;59:841-846.
130. Li LM, Roche J, Sander JW. Ictal ECG changes in temporal lobe epilepsy. Arq Neuropsiquiatr. 1995;53:619-624.
131. Rocamora R, Kurthen M, Lickfett L, Von OJ, Elger CE. Cardiac asystole in epilepsy: clinical and neurophysiologic features. Epilepsia. 2003;44:179-185.
132. Johnston SC, Siedenberg R, Min JK, Jerome EH, Laxer KD. Central apnea and acute cardiac ischemia in a sheep model of epileptic sudden death. Ann Neurol. 1997;42:588-594.
133. Terrence CF, Rao GR, Perper JA. Neurogenic pulmonary edema in unexpected, unexplained death of epileptic patients. Ann Neurol. 1981;9:458-464.
134. Coulter DL. Partial seizures with apnea and bradycardia. Arch Neurol. 1984;41:173-174.
135. Singh B, al Shahwan A, al Deeb SM. Partial seizures presenting as life-threatening apnea. Epilepsia. 1993;34:901-903.
136. So EL, Sam MC, Lagerlund TL. Postictal central apnea as a cause of SUDEP: evidence from near-SUDEP incident. Epilepsia. 2000;41:1494-1497.
137. Langan Y, Nashef L, Sander JW. Sudden unexpected death in epilepsy: a series of witnessed deaths. J Neurol Neurosurg Psychiatry. 2000;68:211-213.
138. O’Regan ME, Brown JK. Abnormalities in cardiac and respiratory function observed during seizures in childhood. Dev Med Child Neurol. 2005;47:4-9.
139. Blum AS, Ives JR, Goldberger AL, et al. Oxygen desaturations triggered by partial seizures: implications for cardiopulmonary instability in epilepsy. Epilepsia. 2000;41:536-541.
140. Swallow RA, Hillier CE, Smith PE. Sudden unexplained death in epilepsy (SUDEP) following previous seizure-related pulmonary oedema: case report and review of possible preventative treatment. Seizure. 2002;11:446-448.
141. Benarroch EE. Brainstem respiratory chemosensitivity: new insights and clinical implications. Neurology. 2007;68:2140-2143.
142. Thomas T, Ralevic V, Gadd CA, Spyer KM. Central CO2 chemoreception: a mechanism involving P2 purinoceptors localized in the ventrolateral medulla of the anaesthetized rat. J Physiol. 1999;517(Pt 3):899-905.
143. Malow BA, Levy K, Maturen K, Bowes R. Obstructive sleep apnea is common in medically refractory epilepsy patients. Neurology. 2000;55:1002-1007.
144. Johnston SC, Horn JK, Valente J, Simon RP. The role of hypoventilation in a sheep model of epileptic sudden death. Ann Neurol. 1995;37:531-537.
145. Bird JM, Dembny KAT, Sandeman D. Sudden unexplained death in epilepsy: an intracranially monitored case. Epilepsia. 1997;38(Suppl. 11):S52-S56.
146. Sperling MR, Feldman H, Kinman J, Liporace JD, O’Connor MJ. Seizure control and mortality in epilepsy. Ann Neurol. 1999;46:45-50.
147. Hennessy MJ, Langan Y, Elwes RD, Binnie CD, Polkey CE, Nashef L. A study of mortality after temporal lobe epilepsy surgery. Neurology. 1999;53:1276-1283.
148. Salanova V, Markand O, Worth R. Temporal lobe epilepsy surgery: outcome, complications, and late mortality rate in 215 patients. Epilepsia. 2002;43:170-174.
149. Ryvlin P, Kahane P. Does epilepsy surgery lower the mortality of drug-resistant epilepsy? Epilepsy Res. 2003;56:105-120.
150. Persson H, Kumlien E, Ericson M, Tomson T. No apparent effect of surgery for temporal lobe epilepsy on heart rate variability. Epilepsy Res. 2006;70:127-132.
151. Hilz MJ, Devinsky O, Doyle W, Mauerer A, Dutsch M. Decrease of sympathetic cardiovascular modulation after temporal lobe epilepsy surgery. Brain. 2002;125:985-995.
152. Galli R, Limbruno U, Pizzanelli C, et al. Analysis of RR variability in drug-resistant epilepsy patients chronically treated with vagus nerve stimulation. Auton Neurosci. 2003;107:52-59.
153. Kamath MV, Upton AR, Talalla A, Fallen EL. Effect of vagal nerve electrostimulation on the power spectrum of heart rate variability in man. Pacing Clin Electrophysiol. 1992;15:235-243.
154. Kamath MV, Upton AR, Talalla A, Fallen EL. Neurocardiac responses to vagoafferent electrostimulation in humans. Pacing Clin Electrophysiol. 1992;15:1581-1587.
155. Ronkainen E, Korpelainen JT, Heikkinen E, Myllyla VV, Huikuri HV, Isojärvi JI. Cardiac autonomic control in patients with refractory epilepsy before and during vagus nerve stimulation treatment: a one-year follow-up study. Epilepsia. 2006;47:556-562.
156. Setty AB, Vaughn BV, Quint SR, Robertson KR, Messenheimer JA. Heart period variability during vagal nerve stimulation. Seizure. 1998;7:213-217.
157. Tupal S, Faingold CL. Evidence supporting a role of serotonin in modulation of sudden death induced by seizures in DBA/2 mice. Epilepsia. 2006;47:21-26.
158. Paterson DS, Trachtenberg FL, Thompson EG, et al. Multiple serotonergic brainstem abnormalities in sudden infant death syndrome. JAMA. 2006;296:2124-2132.
159. National Institute for Clinical Excellence. The epilepsies: the diagnosis and management of the epilepsies in adults and children in primary and secondary care. Clinical Guideline. 2004:20.
160. Kennelly C, Riesel J. Sudden Death and Epilepsy. The Views and Experiences of Bereaved Relatives and Carers. Oxon, UK: Epilepsy Bereaved, 2002.
161. Beran RG, Weber S, Sungaran R, Venn N, Hung A. Review of the legal obligations of the doctor to discuss Sudden Unexplained Death in Epilepsy (SUDEP)—a cohort controlled comparative cross-matched study in an outpatient epilepsy clinic. Seizure. 2004;13:523-528.
162. Opeskin K, Clarke I, Berkovic SF. Prolactin levels in sudden unexpected death in epilepsy. Epilepsia. 2000;41:48-51.