[level-membership-for-pediatrics-category]
Chapter 367 Sudden Infant Death Syndrome
The sudden, unexpected death of an infant that is unexplained by a thorough postmortem examination, which includes a complete autopsy, investigation of the scene of death, and review of the medical history, constitutes sudden infant death syndrome (SIDS). An autopsy is essential to identify possible natural explanations for sudden, unexpected death such as congenital anomalies or infection and to diagnose traumatic child abuse (Tables 367-1, 367-2, 367-3). The autopsy typically cannot distinguish between SIDS and intentional suffocation, but the scene investigation and medical history may be of help if inconsistencies are evident.
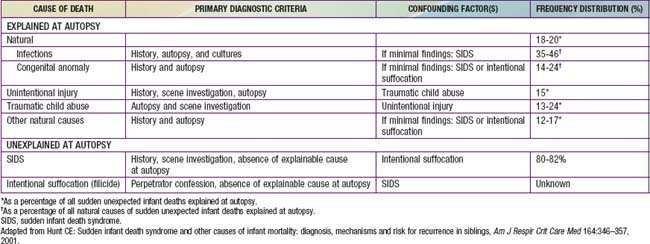
Table 367-2 CONDITIONS THAT CAN CAUSE APPARENT LIFE-THREATENING EVENTS OR SUDDEN DEATH
CENTRAL NERVOUS SYSTEM
CARDIAC
PULMONARY
GASTROINTESTINAL
ENDOCRINE-METABOLIC
INFECTION
TRAUMA
POISONING
From Kliegman RM, Greenbaum LA, Lye PS: Practical strategies in pediatric diagnosis and therapy, ed 2, Philadelphia, 2004, Elsevier Saunders, p 98.
Table 367-3 DIFFERENTIAL DIAGNOSIS OF RECURRENT SUDDEN INFANT DEATH IN A SIBSHIP
IDIOPATHIC
Recurrent true sudden infant death syndrome
CENTRAL NERVOUS SYSTEM
CARDIAC
PULMONARY
Pulmonary hypertension
ENDOCRINE-METABOLIC
See Table 367-2
INFECTION
Disorders of immune host defense
CHILD ABUSE
From Kliegman RM, Greenbaum LA, Lye PS: Practical strategies in pediatric diagnosis and therapy, ed 2, Philadelphia, 2004, Elsevier Saunders, p 101.
Pathology
SIDS victims have several identifiable changes in the lungs and other organs and in brainstem structure and function. Nearly 65% of SIDS victims have structural evidence of pre-existing, chronic low-grade asphyxia, and other studies have identified biochemical markers of asphyxia. SIDS victims have higher levels of vascular endothelial growth factor (VEGF) in the cerebrospinal fluid (CSF). These increases may be related to VEGF polymorphisms (see Genetic Risk Factors) or might indicate recent hypoxemic events, because VEGF is upregulated by hypoxia. Brainstem findings include a persistent increase of dendritic spines and delayed maturation of synapses in the medullary respiratory centers, and decreased tyrosine hydroxylase immunoreactivity and catecholaminergic neurons. Decreases in serotonin 1A (5-HT1A) and 5-HT2A receptor immunoreactivity have been observed in the dorsal nucleus of the vagus, solitary nucleus, and ventrolateral medulla, whereas increases are present in periaqueductal gray matter of the midbrain. The decreased immunoreactivity of receptors is accompanied by brainstem gliosis, and it is therefore unclear whether the decreases are secondary to hypoxia or ischemia or whether they reflect primary alterations in 5-HT metabolism or transport (see Genetic Risk Factors) (Table 367-4).
Table 367-4 IDENTIFIED GENES FOR WHICH THE DISTRIBUTION OF POLYMORPHISMS DIFFERS IN SUDDEN INFANT DEATH SYNDROME INFANTS COMPARED TO CONTROL INFANTS
CARDIA CHANNELOPATHIES (7)
SEROTONIN (5-HT) (3)
GENES PERTINENT TO DEVELOPMENT OF AUTONOMIC NERVOUS SYSTEM (8)
INFECTION AND INFLAMMATION (6)
ENERGY PRODUCTION (1)
Mitochondrial DNA (mtDNA) polymorphisms
Adapted from Hunt CE, Hauck FR: Sudden infant death syndrome: gene-environment interactions. In Brugada R, Brugada J, Brugada P, editors: Clinical care in inherited cardiac syndromes, Guildford, UK, 2009, Springer-Verlag London.
Environmental Risk Factors
Declines of 50% or more in rates of SIDS in the USA and around the world have occurred in the past decade, at least in part as a result of national education campaigns directed at reducing risk factors associated with SIDS. The reductions in risk appear to be related primarily to decreases in placing infants prone for sleep and increases in placing them supine. A number of other risk factors also have significant associations with SIDS (Table 367-5); although many are nonmodifiable and most of the modifiable factors have not changed appreciably, self-reported maternal smoking prevalence during pregnancy has decreased by 25% in the past decade.
Table 367-5 ENVIRONMENTAL FACTORS ASSOCIATED WITH INCREASED RISK FOR SUDDEN INFANT DEATH SYNDROME
MATERNAL AND ANTENATAL RISK FACTORS
INFANT RISK FACTORS:
Modifiable Risk Factors
Genetic Risk Factors
As shown in Table 367-4, there are numerous genetic differences identified among healthy infants, infants dying from other causes, and SIDS infants. Polymorphisms occurring at higher incidence in SIDS compared to controls have been identified for 7 cardiac ion channelopathy genes that are proarrhythmic, 3 5-HT genes, 8 autonomic nervous system (ANS) development genes, 6 genes related to infection and inflammation that are pro-inflammatory, and 1 gene related to energy production.
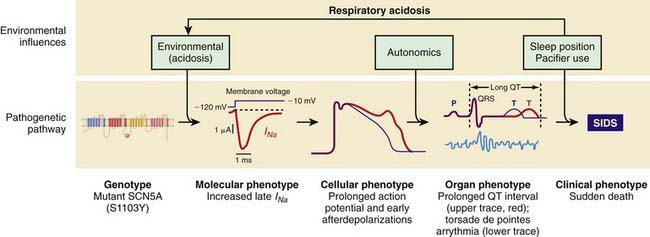
Both LQTS and SQTS are pro-arrhythmic and associated with cardiac arrest and sudden death. However, some of these polymorphisms are associated with concealed phenotypes in which perturbation by a stressor such as acidosis, hypoxemia, or adrenergic stress is required to elicit electrocardiographic evidence of the pro-arrhythmic dysfunction. The other cardiac ion-related channelopathy polymorphisms are also pro-arrhythmic, including Brugada syndrome (BrS1, BrS2), and catecholaminergic paroxysmal ventricular tachycardia (CPVT1). Collectively, these mutations in cardiac ion channels provide a lethal arrhythmogenic substrate in some infants at risk for SIDS (Fig. 367-1) and might account for as many as 10% of SIDS.
Gene-and-Environment Interactions
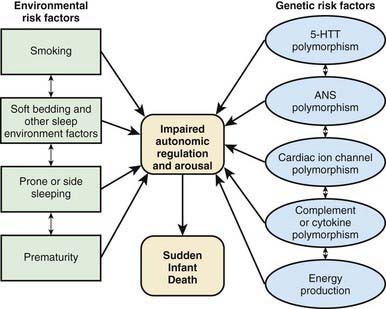
Interactions between genetic and environmental risk factors determine the actual risk for SIDS in individual infants (Fig. 367-2). There appears to be an interaction between prone sleep position and impaired ventilatory and arousal responsiveness. Facedown or nearly facedown sleeping does occasionally occur in prone-sleeping infants and can result in episodes of airway obstruction and asphyxia in healthy full-term infants. Healthy infants arouse before such episodes become life-threatening, but infants with insufficient arousal responsiveness to asphyxia may be at risk for sudden death. There may also be links between modifiable risk factors such as soft bedding, prone sleep position and thermal stress, and links between genetic risk factors such as ventilatory and arousal abnormalities and temperature or metabolic regulation deficits. Cardiorespiratory control deficits could be related to 5-HTT polymorphisms, for example, or to polymorphisms in genes pertinent to ANS development. Affected infants could be at increased risk for sleep-related hypoxemia and hence more susceptible to adverse effects associated with unsafe sleep position or bedding. Infants at increased risk for sleep-related hypoxemia could also be at greater risk for fatal arrhythmias in the presence of a cardiac ion channelopathy polymorphism.
In >50% of SIDS victims, recent febrile illnesses, often related to upper respiratory infection, have been documented (see Table 367-5). Otherwise benign infections might increase risk for SIDS if interacting with genetically determined impaired immune responses including those due to partial deletions in the complement C4 gene or to interleukin polymorphisms (see Table 367-4). Deficient inflammatory responsiveness can also occur due to mast cell degranulation, which has been reported in SIDS infants; this is consistent with an anaphylactic reaction to a bacterial toxin, and some family members of SIDS infants also have mast cell hyper-releasability and degranulation, suggesting that increased susceptibility to an anaphylactic reaction is another genetic factor influencing fatal outcomes to otherwise minor infections in infants. Interactions between upper respiratory infections or other minor illnesses and other factors such as prone sleeping might also play a role in the pathogenesis of SIDS.
Infant Groups at Increased Risk for Sids
Subsequent Siblings of a SIDS Victim
The next-born siblings of first-born infants dying of any noninfectious natural cause are at significantly increased risk for infant death from the same cause, including SIDS. The relative risk is 9.1 for the same cause of recurrent death vs 1.6 for a different cause of death. The relative risk for recurrent SIDS (range, 5.4-5.8) is similar to the relative risk for non-SIDS causes of recurrent death (range, 4.6-12.5). The risk for recurrent infant mortality from the same cause as in the index sibling thus appears to be increased to a similar degree in subsequent siblings for both explained causes and for SIDS. This increased risk in SIDS families is consistent with genetic risk factors interacting with environmental risk factors (Table 367-5 and Fig. 367-2).
Physiologic Studies
Physiologic studies have been performed on healthy infants in early infancy, a few of whom later died of SIDS. Physiologic studies have also been performed on infant groups at increased risk for SIDS, especially those with ALTE and subsequent siblings of SIDS. In the aggregate, these studies indicate brainstem abnormalities in neuroregulation of cardiorespiratory control or other autonomic functions and are consistent with the autopsy findings and genetic studies in SIDS victims (see the “Pathology” and “Genetic Risk Factors” sections).
The ability to shorten the QT interval as heart rate increases appears to be impaired in some SIDS victims, suggesting that such infants may be predisposed to ventricular arrhythmia. This is consistent with the observations of cardiac ion channel gene polymorphisms in other SIDS victims (Table 367-4), but there are no antemortem QT interval data in the SIDS infants having postmortem channelopathy polymorphisms. Infants studied physiologically and dying of SIDS a few weeks later have higher heart rates in all sleep-wake states, diminished heart rate variability during wakefulness, and significantly lower heart rate variability at the respiratory frequency across all sleep-wake cycles. Also, these SIDS infants have longer QT intervals than control infants in both REM and non-REM sleep, especially in the late hours of the night when most SIDS likely occurs. In only 1 of these SIDS infants, however, did the QT interval exceed 440 ms.
Clinical Strategies
Home Monitoring
SIDS cannot be prevented in individual infants because it is not possible to identify prospective SIDS infants, and no effective intervention has been established even if SIDS infants could be prospectively identified. Studies of cardiorespiratory pattern or other autonomic abnormalities do not have sufficient sensitivity and specificity to be clinically useful as screening tests. Home electronic surveillance using existing technology does not reduce the risk of SIDS. Although a prolonged QT interval in an infant may be treated if diagnosed, neither the role of routine postnatal electrocardiographic (ECG) screening nor the cost-effectiveness of diagnosis and treatment and safety of treatment has been established (Chapter 429.5). Parental ECG screening is not helpful because spontaneous mutations are common.
Reducing the Risk of SIDS
AAP Task Force on Infant Sleep Position and Sudden Infant Death Syndrome. The changing concept of sudden infant death syndrome: diagnostic coding shifts, controversies regarding the sleeping environment, and new variables to consider in reducing risk. Pediatrics. 2005;116:1245-1255.
AL-Kindy HA, Gelinas JF, Hatzakis G, et al. Risk factors for extreme events in infants hospitalized for apparent life-threatening events. J Pediatr. 2009;154:332-337.
Audero E, Gross C. Could serotonin play a role in sudden infant death? Commentary. Pediatr Res. 2009;65:131.
Bacon CJ, Hall DBM, Stephenson TJ, et al. How common is repeat sudden infant death syndrome? Arch Dis Child. 2008;93:323-326.
Blair PS, Sidebotham P, Evason-Coombe C, et al. Hazardous cosleeping environments and risk factors amendable to change: case-control study of SIDS in south west England. BMJ. 2009;339:911.
Bonkowsky JL, Guenther E, Srivastava R, et al. Seizures in children following an apparent life-threatening event. J Child Neurol. 2009;24:709-713.
Broadbelt KG, Barger MA, Paterson DS, et al. Serotinin-related FEV gene variant on the sudden infant death syndrome is a common polymorphism in the African-American population. Pediatr Res. 2009;66:631-635.
Campbell MJ, Hall D, Stephenson T, et al. Recurrence rates for sudden infant death syndrome (SIDS): the importance of risk stratification. Arch Dis Child. 2008;93:936-939.
Centers for Disease Control and Prevention. Release of sudden, unexplained infant death investigation reporting form. MMWR. 2006;55:212-213.
Coleman-Phox K, Odouli R, Li DK. Use of a fan during sleep and the risk of sudden infant death syndrome. Arch Pediatr Adolesc Med. 2008;162:963-968.
Darnall RA. ALTEs: Still a puzzle after all these years. J Pediatr. 2009;154:317-319.
Duncan JR, Paterson DS, Hoffman JM, et al. Brainstem serotonergic deficiency in sudden infant death syndrome. JAMA. 2010;303:430-437.
Esani N, Hodgman JE, Ehsani N, et al. Apparent life-threatening events and sudden infant death syndrome: comparison of risk factors. J Pediatr. 2008;152:365-370.
Franco P, Groswasser J, Scaillet S, et al. QT prolongation in future SIDS victims: a polysomnographic study. Sleep. 2008;31:1691-1699.
Hauck FR, Omojokun OO, Siadaty MS. Do pacifiers reduce the risk of sudden infant death syndrome? A meta-analysis. Pediatrics. 2005;116:e716-e723.
Hauck FR, Tanabe KO. International trends in sudden infant death syndrome. stabilization of rates requires further action. Pediatrics. 2008;122:660-666.
Heron M, Hoyert DL. Deaths: final data for 2006. Natl Vital Stat Rep. 2009;57:1-136.
Kiehne N, Kauferstein S. Mutations in the SCN5A gene: evidence for a link between long QT syndrome and sudden death? Forensic Sci Int: Genetics. 2007;1:170-174.
Kinney HC, Thach BT. The sudden infant death syndrome. N Engl J Med. 2009;361:795-804.
Krous HF, Ferandos C, Masoumi H, et al. Myocardial inflammation, cellular death, and viral detection on sudden infant death caused by SIDS, suffocation, or myocarditis. Pediatr Res. 2009;66:17-21.
Kuntschar M, Reichenpfader B, Saternus K-S. A functional polymorphism in the tyrosine hydroxylase gene indicates a role of noradrenalinergic signaling in sudden infant death syndrome. J Pediatr. 2008;153:190-193.
Lavezzi AM, Casale V, Oneda R, et al. Sudden infant death syndrome and sudden intrauterine unexplained death: correlation between hypoplasia of raphe nuclei and serotonin transporter gene promotor polymorphism. Pediatr Res. 2009;66:22-27.
Li DK, Willinger M, Petitti DB, et al. Use of a dummy (pacifier) during sleep and risk of sudden infant death syndrome (SIDS): Population based case-control study. BMJ. 2006;332:18-21.
Machaalani R, Meichien S, Waters KA. Serotonergic receptor 1A in the sudden infant death syndrome brainstem medulla and associations with clinical risk factors. Acta Neuropathol. 2009;117:257-265.
Marzano FN, Maldini M, Filonzi L, et al. Genes regulating the serotonin metabolic pathway in the brain stem and their role in the etiopathogenesis of the sudden infant death syndrome. Genomics. 2008;91:485-491.
McGovern MC, Smith MBH. Causes of apparent life threatening events in infants: a systematic review. Arch Dis Child. 2004;89:1043-1048.
Mitchell EA. Risk factors for SIDS. BMJ. 2009;339:873-874.
Mitchell EA, Thompson JMD, Becroft DMO, et al. Head covering and the risk for SIDS: findings from the New Zealand and German SIDS case-control studies. Pediatrics. 2008;121:e1478-e1483.
Moon RY, Horne RSC, Hauck FR. Sudden infant death syndrome. Lancet. 2007;370:1578-1587.
Morris JA, Harrison LM. Sudden unexpected death in infancy: evidence of infection. Lancet. 2008;371:1815-1816.
National Infant Sleep Position and Collaborative Home Infant Monitoring Evaluation. Public access. (website) http://slone-web2.bu.edu/ChimeNisp/ Accessed June 19, 2010
O’Connor NR, Tanabe KO, Siadaty MS, et al. Pacifiers and breastfeeding. A systematic review. Arch Pediatr Adolesc Med. 2009;163:378-382.
Otagiri T, Kijima K, Osawa M, et al. Cardiac ion channel gene mutations in sudden infant death syndrome. Pediatr Res. 2008;64:482-487.
Paterson DS, Trachtenberg FL, Thompson EG, et al. Multiple serotonergic brainstem abnormalities in sudden infant death syndrome. JAMA. 2006;296:2124-2132.
Pitetti RD, Whitman E, Zaylor A. Accidental and nonaccidental poisonings as a cause of apparent life-threatening events in infants. Pediatrics. 2008;122:e359-e362.
Poetsch M, Nottebaum BJ, Wingenfeld L, et al. Impact of sodium/proton exchanger 3 gene variants on sudden infant death syndrome. J Pediatr. 2010;156:44-48.
Ramanathan R, Corwin MJ, Hunt CE, et al. Cardiorespiratory events recorded on home monitors: comparison of healthy infants with those at increased risk for SIDS. Collaborative Home Infant Monitoring Evaluation (CHIME) Study Group. JAMA. 2001;285:2199-2207.
Tester DJ, Ackerman MJ. Cardiomyopathic and channelopathic causes of sudden unexplained death in infants and children. Ann Rev Med. 2009;60:69-84.
Thach BT. Does swaddling decrease or increase the risk of sudden infant death syndrome? J Pediatr. 2009;155:461-462.
Van Norstrandt DW, Ackerman MJ. Sudden infant death syndrome: do ion channels play a role? Heart Rhythm. 2009;6:272-278.
Van Wouwe J, HiraSing RA. Prevention of sudden unexpected infant death. Lancet. 2006;367:277-278.
Vennemann MMT, Bajanowski T, Brinkmann B, et al. Sleep environment risk factors for sudden infant death syndrome: the German sudden infant death syndrome study. J Pediatr. 2009;123:1162-1170.
Vennemann MMT, Höffgen M, Bajanowski T, et al. Do immunizations reduce the risk of SIDS? A meta-analysis. Vaccines. 2007;25:4875-4879.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 367 Sudden Infant Death Syndrome
The sudden, unexpected death of an infant that is unexplained by a thorough postmortem examination, which includes a complete autopsy, investigation of the scene of death, and review of the medical history, constitutes sudden infant death syndrome (SIDS). An autopsy is essential to identify possible natural explanations for sudden, unexpected death such as congenital anomalies or infection and to diagnose traumatic child abuse (Tables 367-1, 367-2, 367-3). The autopsy typically cannot distinguish between SIDS and intentional suffocation, but the scene investigation and medical history may be of help if inconsistencies are evident.
Table 367-2 CONDITIONS THAT CAN CAUSE APPARENT LIFE-THREATENING EVENTS OR SUDDEN DEATH
CENTRAL NERVOUS SYSTEM
CARDIAC
PULMONARY
GASTROINTESTINAL
ENDOCRINE-METABOLIC
INFECTION
TRAUMA
POISONING
From Kliegman RM, Greenbaum LA, Lye PS: Practical strategies in pediatric diagnosis and therapy, ed 2, Philadelphia, 2004, Elsevier Saunders, p 98.
Table 367-3 DIFFERENTIAL DIAGNOSIS OF RECURRENT SUDDEN INFANT DEATH IN A SIBSHIP
IDIOPATHIC
Recurrent true sudden infant death syndrome
CENTRAL NERVOUS SYSTEM
CARDIAC
PULMONARY
Pulmonary hypertension
ENDOCRINE-METABOLIC
See Table 367-2
INFECTION
Disorders of immune host defense
CHILD ABUSE
From Kliegman RM, Greenbaum LA, Lye PS: Practical strategies in pediatric diagnosis and therapy, ed 2, Philadelphia, 2004, Elsevier Saunders, p 101.
Pathology
SIDS victims have several identifiable changes in the lungs and other organs and in brainstem structure and function. Nearly 65% of SIDS victims have structural evidence of pre-existing, chronic low-grade asphyxia, and other studies have identified biochemical markers of asphyxia. SIDS victims have higher levels of vascular endothelial growth factor (VEGF) in the cerebrospinal fluid (CSF). These increases may be related to VEGF polymorphisms (see Genetic Risk Factors) or might indicate recent hypoxemic events, because VEGF is upregulated by hypoxia. Brainstem findings include a persistent increase of dendritic spines and delayed maturation of synapses in the medullary respiratory centers, and decreased tyrosine hydroxylase immunoreactivity and catecholaminergic neurons. Decreases in serotonin 1A (5-HT1A) and 5-HT2A receptor immunoreactivity have been observed in the dorsal nucleus of the vagus, solitary nucleus, and ventrolateral medulla, whereas increases are present in periaqueductal gray matter of the midbrain. The decreased immunoreactivity of receptors is accompanied by brainstem gliosis, and it is therefore unclear whether the decreases are secondary to hypoxia or ischemia or whether they reflect primary alterations in 5-HT metabolism or transport (see Genetic Risk Factors) (Table 367-4).
Table 367-4 IDENTIFIED GENES FOR WHICH THE DISTRIBUTION OF POLYMORPHISMS DIFFERS IN SUDDEN INFANT DEATH SYNDROME INFANTS COMPARED TO CONTROL INFANTS
CARDIA CHANNELOPATHIES (7)
SEROTONIN (5-HT) (3)
GENES PERTINENT TO DEVELOPMENT OF AUTONOMIC NERVOUS SYSTEM (8)
INFECTION AND INFLAMMATION (6)
ENERGY PRODUCTION (1)
Mitochondrial DNA (mtDNA) polymorphisms
Adapted from Hunt CE, Hauck FR: Sudden infant death syndrome: gene-environment interactions. In Brugada R, Brugada J, Brugada P, editors: Clinical care in inherited cardiac syndromes, Guildford, UK, 2009, Springer-Verlag London.
Environmental Risk Factors
Declines of 50% or more in rates of SIDS in the USA and around the world have occurred in the past decade, at least in part as a result of national education campaigns directed at reducing risk factors associated with SIDS. The reductions in risk appear to be related primarily to decreases in placing infants prone for sleep and increases in placing them supine. A number of other risk factors also have significant associations with SIDS (Table 367-5); although many are nonmodifiable and most of the modifiable factors have not changed appreciably, self-reported maternal smoking prevalence during pregnancy has decreased by 25% in the past decade.
Table 367-5 ENVIRONMENTAL FACTORS ASSOCIATED WITH INCREASED RISK FOR SUDDEN INFANT DEATH SYNDROME
MATERNAL AND ANTENATAL RISK FACTORS
INFANT RISK FACTORS:
