[level-membership-for-cardiovascular-category]
Chapter 47 Sudden Cardiac Death
Definition
The term sudden cardiac death refers to unexpected natural death from a cardiovascular cause within a short period, generally less than 1 hour from the onset of symptoms.1 The time and mode of death are therefore unexpected.2 By definition, this occurs in individuals who do not have prior conditions that could be fatal in the short term. The use of 1 hour in this definition is arbitrary and therefore subject to different interpretations. It is also important to distinguish a primary arrhythmic cause of death from, for example, an episode of worsening heart failure that culminates in a life-terminating arrhythmia and from other causes of sudden death such as pulmonary embolism, cerebral infarction, and ruptured aneurysms. This distinction is important both for the proper identification of SCD and the study of this phenomenon. More recently, the use of the term sudden cardiac arrest (SCA, synonymous with SCD) has been advocated.
Epidemiology
SCD is a major cause of death in North America and other Western societies, accounting for 10% of all deaths and up to 50% of heart disease–related deaths.3 The incidence of SCD (emergency medical services [EMS]–assessed, out-of-hospital cardiac arrests) in the United States is generally estimated to be 95.7 per 100,000 person years.4 Approximately 60% of out-of-hospital cardiac deaths are treated by EMS personnel.5 Among these EMS-treated, out-of-hospital arrests, 23% have an initial shockable rhythm, and 31% of these receive bystander cardiopulmonary resuscitation (CPR).6 Median survival rate to hospital discharge after EMS-treated, out-of-hospital cardiac arrest with any first recorded rhythm is 7.9% and for ventricular fibrillation (VF) is 21%. However, such median statistics of survival do not reflect regional variations of community-specific survival rates. Of note, the quantification of SCD is so fraught with difficulties that no comprehensive record of all SCDs exists. Surrogate data often are used to estimate its incidence, especially in the out-of-hospital setting. This includes deaths from “coronary heart disease” and “cardiac arrest,” defined as coronary death that occurred within 1 hour of symptom onset in the out-of-hospital setting and without other probable causes of death.7
As medical interventions continue to evolve and improve, the incidence and distribution of risk factors that contribute to SCD continue to change. Although the morbidity and mortality rates of cardiovascular disease have declined in the past 30 years, much less improvement has been observed in the incidence of out-of-hospital cardiac arrests. However, the incidence of cardiac arrest with an initial recorded rhythm of VF specifically has decreased over time, which is a recent trend that reflects the overall decline in cardiac mortality.8,9 The reduction in the incidence of VF is likely multi-factorial in origin and is attributable to improvements in primary and secondary prevention of heart disease. Current epidemiologic data indicate that in developed countries, structural coronary arterial abnormalities and their consequences account for 80% of fatal arrhythmias.10,11 Dilated and hypertrophic cardiomyopathies account for the next largest group of SCD. The remainder are caused by a variety of other cardiac disorders such as congenital heart disease (CHD) and primary electrophysiological disorders.
The epidemiology of SCD in adolescents and young adults (aged 10 to 30 years) is distinctly different from that in the adult population. The incidence of SCD in this population is two orders of magnitude less than in the adult group (1 per 100,000 vs. 1 per 1000 individuals annually).12 Although coronary atherosclerosis accounts for the majority of cases of SCD in individuals older than 40 years, it is an uncommon cause in the younger age group.13 Instead, causes such as hypertrophic cardiomyopathy, myocarditis, right ventricular dysplasia, anomalous coronary arteries, Brugada syndrome, long QT syndrome (LQTS), idiopathic VF, and commotio cordis are the underlying etiologies in this group. Because some of these conditions are genetically determined, a modest age inverse relationship seems to exist in this group, with adolescents having a somewhat higher mortality risk compared with young adults.11 A unique subgroup in this younger population is composed of competitive athletes, among whom the rate of SCD currently is approximately 0.4 to 0.6 per 100,000 person-years.14,15 The large numbers of SCD and the wide range of survival rates have significant potential implications for public health; for example, in the United States and Canada, an additional 7500 lives would be saved if survival from SCD could be increased to 12%.
Mechanisms
From a clinical perspective, the causes of SCD can be divided into two broad categories: (1) ventricular tachyarrhythmias (VA) and (2) pulseless electrical activity (PEA) and asystole. The National Registry of CPR is a prospective, multi-site, in-hospital resuscitation registry sponsored by the American Heart Association. In a study using this database, of the 51,919 index arrests evaluated in 411 centers, pulseless VT was diagnosed in 7%, VF in 17%, PEA in 37%, and asystole in 39%.16 Subsequent ventricular tachycardia (VT) or VF occurred in 26% of patients with an initial documented rhythm of PEA and in 25% of patients with asystole. Thus VT or VF was seen in 44% of all adult in-hospital cardiac arrests. This must be compared with out-of-hospital cardiac arrests, where among cases of EMS-assessed cardiac arrest, the incidence of VT or VF was 13% and PEA and asystole were 33% and 47%, respectively, for cases in which the initial rhythm was unknown, not determined, or not analyzed by EMS.8 A factor that has important implications in out-of-hospital cardiac arrests (as opposed to in-hospital cardiac arrests) is the median time of 7.24 minutes from call to arrival of first advanced life support.8
Pathophysiology
In recent years, significant advances in the understanding of the mechanism of VF have taken place. Controversy exists regarding whether VF is maintained by wandering wavelets with constantly changing re-entrant circuits or by a mother rotor that consists of a sustained and stationary re-entrant circuit that, in turn, gives rise to variable, less-organized daughter wavelets spreading through the rest of the ventricle.17,18 Certain anatomic structures can serve as anchors for rotors, allowing stability within the re-entrant circuit.19 Weiss et al have reported from their studies on porcine hearts that these anatomic sites can be papillary muscles, blood vessels, Purkinje fibers, or locations near the interventricular septum.20 Studies have suggested that Purkinje fibers play an important role in the initiation of VF, whereas others have suggested that they play a role in the maintenance of VF.21,22 Predispositions to SCD may also occur at a cellular level. Ion channelopathies can initiate various electrical disturbances, ranging from torsades de pointes in LQTS to that of idiopathic VF in Brugada syndrome.1,23 Certain mutations are significant enough that their mere presence is associated with a very high risk of SCD. However, other mutations (e.g., the recessive long QT mutation in the HERG gene) may, by themselves, be insufficient to cause SCD but may predispose patients to torsades de pointes in the presence of other factors such as drugs that prolong the Q-T interval.
The role of ischemia as an initiating factor for ventricular arrhythmias has been extensively investigated and is particularly relevant because it is a frequent etiology implicated in SCD. Ischemia causes acute changes at the cellular level that alter local conduction velocity and refractoriness. The resulting dispersion of conduction and repolarization establishes an environment that is ripe for re-entry. In animal experiments, within the first few minutes after coronary occlusion, an arrhythmogenic period that slowly abates after 30 minutes has been observed. These first 30 minutes can be divided broadly into the first 10 minutes, during which the changes are caused by direct ischemic injury, and the second 20 minutes, during which either arrhythmogenicity occurs because of either reperfusion or the evolution of injury in the various layers of the myocardium.24,25 Local changes occur in the ischemic myocardium; these include decrease in local tissue pH to less than 6, increase in interstitial potassium (K+) levels to greater than 15 mmol/L, increases in intracellular calcium (Ca2+), and other neurohormonal changes. All these factors contribute to the altered electrophysiological properties of tissue, including slowed conduction velocity, reduced excitability and prolonged refractoriness, reduced cell-to-cell coupling, and even spontaneous electrical activity.26 In addition to the local micro–re-entrant and macro–re-entrant circuits that may be generated, regional increases in automaticity and triggered activity also occur because of afterdepolarizations.
Disease States Leading to Sudden Cardiac Death
Because the majority of cases of SCD result from CAD, it is not surprising that the risk factors for SCD mirrors those of CAD. The incidence of SCD increases with age and is more common in men than women.27 The incidence of SCD tends to be higher in whites than in other racial groups. Classic coronary risk factors have been noted to be associated with SCD in various studies such as the Framingham Study and the Paris Prospective Study. These risk factors include hypertension, diabetes, high cholesterol levels, smoking, lack of regular exercise, and structural changes such as left ventricular hypertrophy.28–31 These factors are prevalent but are limited by their moderate individual positive predictive value. Left ventricular dysfunction has been shown to be a strong independent predictor of SCD in both ischemic and nonischemic cardiomyopathies. Of note, in patients with severely decreased left ventricular function and advanced heart failure, competing causes of SCD such as electromechanical dissociation and asystole exist.
Observations from population-based studies demonstrate a marked increase in the risk of SCD in first-degree relatives of SCD victims. In a study from Seattle, first-degree relatives of patients with SCD (before age 65 years) had 2.7-fold higher risk of SCD compared with age-matched and gender-matched controls after adjustment for risk factors.32 A Dutch case-control study demonstrated that patients with VF during myocardial infarction (MI) are more likely to have a family history of SCD than those with MI but no VF.33 Polygenic traits leading to SCD and monogenetic SCD syndromes such as LQTS, Brugada syndrome, hypertrophic cardiomyopathy, and arrhythmogenic right ventricular dysplasia contribute to the genetic component of SCD.34 More recently, research has begun to elucidate the role of newly identified genetic variations at the population level.
Coronary Artery Disease
Approximately 80% of patients who experience SCD have coronary atherosclerotic arterial disease as an underlying substrate. In survivors of SCD, critical flow-limiting coronary stenoses are found in approximately 40% to 86% of patients, depending on the age and gender of the population.29,30,35 Although less than 50% of the resuscitated patients have evidence of an acute MI, autopsy studies have revealed that a recent occlusive coronary thrombus can be found in 20% to 95% of victims of SCD.31,36,37 The temporal pattern of SCD closely parallels the patterns of MI and acute ischemic events. In addition, healed infarctions are found in 40% to 75% of hearts of SCD victims at autopsy.36,38–40 The extent to which superimposed acute ischemia plays a role in the pathogenesis of SCD is unclear in patients who do not develop enzymatic or electrocardiographic evidence of an acute MI. However, rapid fibrinolysis of a ruptured plaque could occur such that no visible evidence remains at autopsy. Similarly, cholesterol-laden plaque could conceivably rupture and embolize microscopic debris into distal coronary vessels that could lead to microscopic necrosis and ventricular arrhythmias and yet not be visible at autopsy.41 Because the vast majority of patients die within minutes of onset of symptoms, histologic or enzymatic evidence of ischemia or infarction may be difficult to ascertain. In addition to the most common forms of CAD, nonatherosclerotic CAD includes congenital malformations such as anomalous origin of coronary arteries; inflammatory arteritis also can lead to SCD. These disorders typically manifest early in life but are relatively uncommon.
Dilated Cardiomyopathy
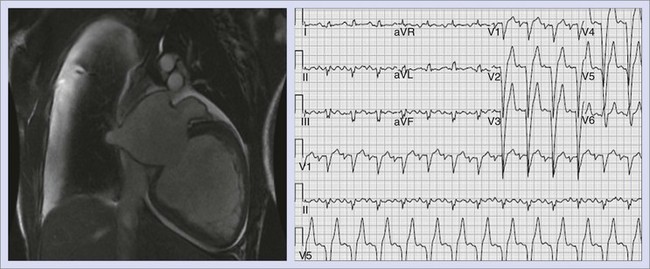
Idiopathic dilated cardiomyopathy accounts for approximately 10% of cases of SCD in the adult population (Figure 47-1). Depending on the severity of the myopathy, the annual incidence can range from 10% to 50%.36 Bundle branch reentrant VT appears to represent an important cause of VAs in this population.37 However, as the myopathy progresses and congestive heart failure worsens, the incidence of VT or VF decreases, and the primary terminal event more frequently becomes electromechanical dissociation or asystole.42 As with ischemic heart disease, the overall left ventricular ejection fraction (LVEF) is an important prognostic factor. Worsening New York Heart Association (NYHA) functional class and the occurrence of syncope are both important clinical prognostic factors for SCD in patients with dilated cardiomyopathy.40
Hypertrophic Cardiomyopathy
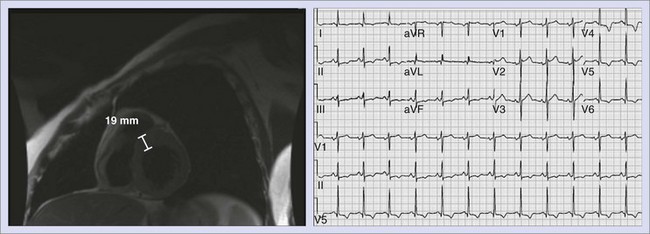
Hypertrophic cardiomyopathy (HCM) is now regarded as the most common cause of SCD in young people, including competitive athletes (Figure 47-2).43–45 The annual mortality rate in patients with HCM in select regional and community-based cohorts is thought to be approximately 1%. The architectural myocardial fiber disorganization, scarring, and the presence of microvascular disease likely account for the proarrhythmic substrate. The conventional risk factors for HCM assume greater weight in patients younger than 50 years and include family history of one or more HCM-related form of SCD, more than one episode of unexplained recent syncope, massive LVH (thickness ≥30 mm), nonsustained VT on 24-hour Holter monitoring, and hypotensive or attenuated blood pressure response to exercise. Although implantable cardioverter defibrillators (ICDs) for primary prevention in this population usually require meeting at least two of these criteria, the need for ICD interventions for VT or VF in patients with single risk factors must be kept in mind when deciding about ICD implantation in this population.46 Of the above criteria, nonsustained VT and blood pressure response have poor positive predictive value.47 It has recently been demonstrated that certain mutations such as nonsarcomeric LAMP2 cardiomyopathy, double sarcomere mutations, and delayed enhancement on cardiac magnetic resonance imaging (MRI) are associated with SCD.48–51 Conversely, the apical variant of hypertrophic cardiomyopathy has been demonstrated to carry a relatively lower risk for SCD.52
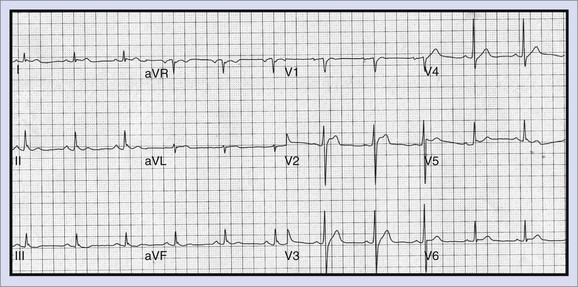
Long QT Syndrome
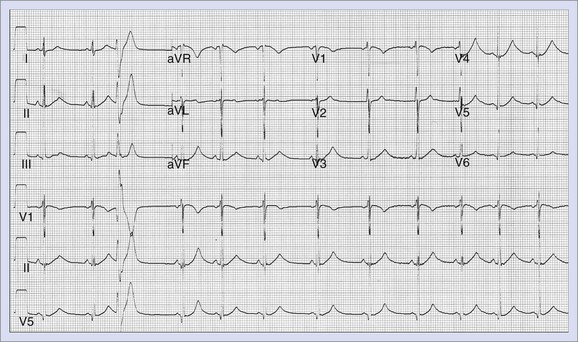
LQTS (Figure 47-3) is a primary cardiac arrhythmogenic disorder typically characterized by prolongation of the Q-T interval corrected for heart rate (QTc) and abnormal T waves. Subjects with LQTS may present with a nearly normal electrocardiogram (ECG) or with a prolonged Q-T interval. The syndrome is associated with a specific ventricular arrhythmia called torsades de pointes and often presents as recurrent syncope. Hundreds of mutations have been identified, and these have been described in up to 12 genes.47 This is most often caused by decreased outward K+ current, IKs (LQT1, LQT5) or IKr (LQT2, LQT6), or by enhanced activity of mutant inward sodium (Na+) current (LQT3). Polymorphic VT associated with a prolonged Q-T interval is believed to be initiated by early afterdepolarizations (EADs) in the Purkinje system and maintained by transmural re-entry in the myocardium. Clinical presentations vary with the specific gene affected and the specific mutation. Some patients with LQTS mutations may not manifest any phenotypic abnormality. In high-risk LQT1 and LQT2, patients should be routinely managed with β-blockers as a first line-therapy and should be referred for primary ICD implantation if they become symptomatic during therapy or when compliance or intolerance to medical therapy is a concern.53 Patients with LQT3, those with frequent and recent syncope, those with excessive Q-T prolongation (>550 ms), and women with LQT2 and a QT interval greater than 500 ms may be at increased risk and may require ICD implantation.
Congenital Short QT Syndrome
Congenital short QT syndrome (SQTS) is a relatively recently described disorder characterized by a very short Q-T interval (<320 ms) and a susceptibility to atrial fibrillation (AF) and VF. At electrophysiology study, short atrial and ventricular refractory periods with easily inducible AF and polymorphic VT have been identified. Gain-of-function mutations in genes encoding K+ channels have been identified, which explains the abbreviated repolarization seen in this condition. The suggested treatment is ICD implantation. The ability of quinidine to prolong the Q-T interval has the potential to be effective pharmacological therapy.54
Arrhythmogenic Right Ventricular Cardiomyopathy
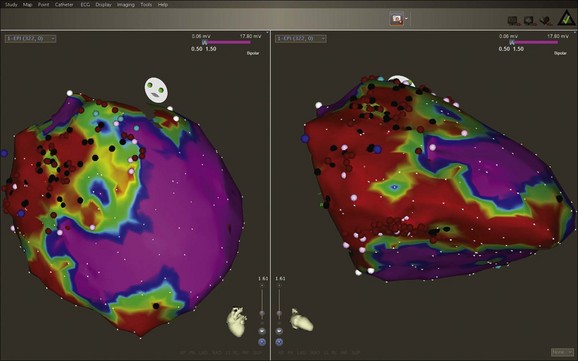
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a progressive, heritable myocardial disorder with a broad phenotypic spectrum that can present with VT, SCD, or both (Figure 47-4). The classic form of the disease has an early predilection for the right ventricle, but left-dominant and biventricular subtypes have been also recognized.55 Desmoplakin and plakophilin-2 are two desmosomal genes that have been implicated in ARVC. A familial form, originally described in people living on the Greek island of Naxos, can present with cardiomyopathy in association with palmo-plantar keratoderma (Naxos disease) and is caused by a defect in the gene for a cellular structural element, plakoglobin.56 ARVC causes progressive fatty replacement of the ventricular wall. Cardiac MRI is a highly sensitive imaging tool to identify the presence and degree of fatty infiltration in the myocardium; however, because this is not specific, formal criteria have been proposed for the diagnosis of ARVD. Phenotypic heterogeneity and the nonspecific nature of its associated features complicate clinical diagnosis, which often requires multiple tests rather than a single test.57 The Revised Task Force criteria for the diagnosis are specific and have helped reduce diagnostic ambiguity, but the sensitivity is low, especially in the “concealed” phase of ARVC.57
Brugada Syndrome
Brugada syndrome is associated with right ventricular conduction delay and ST elevation in the right precordial leads, characterized by syncope and premature SCD caused by VF (Figure 47-5). This syndrome appears to be responsible for a sudden death syndrome seen among Southeast Asian men.58,59 ECG manifestations of the syndrome can often be dynamic. Typical ST-segment changes, if absent at baseline, can be revealed by administration of sodium channel–blocking agents such as ajmaline or procainamide. Mutations in the SCN5A gene cause loss of function in the Na+ current Ina that can lead to accentuation of unopposed Ito currents in the right ventricular epicardium. This leads to loss of the action potential “dome,” resulting in heterogeneity of repolarization, and phase 2 re-entry that precipitates VT or VF. The strategy for risk stratification in Brugada syndrome is controversial with respect to the role of electrophysiology testing in patients who do not present with SCD. ICD is the only treatment with demonstrated efficacy in Brugada syndrome. In general, ICD implantation is recommended for patients with symptoms and for asymptomatic patients with inducible ventricular arrhythmias, especially if they have a spontaneous type I ECG pattern. In asymptomatic patients without a family history of SCD and whose type I ECG pattern is documented only after the administration of sodium channel blockers, periodic follow-up is recommended, but an EPS for risk stratification is not required.60,61 The role of EPS, however, still remains controversial, with some studies not supporting its role in risk stratification.57
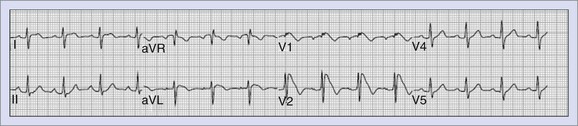
Wolff-Parkinson-White Syndrome
Wolff-Parkinson-White (WPW) syndrome can lead to SCD if the accessory pathway is able to conduct rapidly in the antegrade direction in the presence of rapid atrial arrhythmias such as AF (Figure 47-6). More recently, a long-term follow-up of patients with WPW syndrome reported a mortality rate of 0.02% per year.62 Two additional studies with more than 4000 patient-years of follow-up, estimated mortality rates at approximately 0.05% per year.63,64 However, a study from Italy that prospectively followed up patients with WPW syndrome for 3 years, recorded a much higher event rate (defined as death or potentially lethal arrhythmia recorded on monitoring) at 0.5% per year.65 Predictors for the development of VF include rapid ventricular response during induced AF, with the shortest R-R interval of less than 240 ms and short antegrade pathway refractory periods. High-risk and symptomatic patients are treated with catheter ablation with very high success rates overall.66
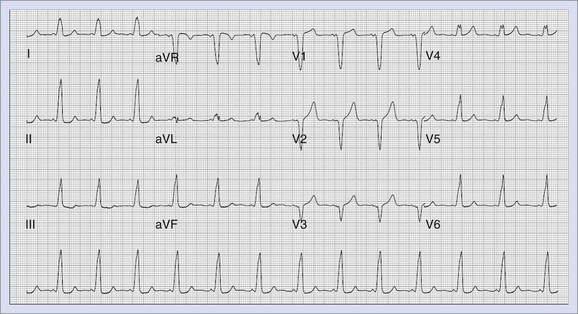
Catecholaminergic Polymorphic Ventricular Tachycardia
Patients with catecholaminergic polymorphic ventricular tachycardia (CPVT) can present with exercise-induced syncope, SCD, or both in the absence of any structural heart disease or prolonged Q-T interval. Inheritance can be autosomal dominant or recessive. These patients often have normal resting ECGs, which makes the diagnosis difficult. Stress-related bi-directional VT has been described classically, but patients can present with polymorphic VT or even frequent ventricular ectopy. Responsible mutations have been shown to reside in the cardiac ryanodine receptor and calsequestrin genes.67–70 Treatment modalities include ICD placement along with β-blocker therapy for symptomatic patients.
Early Repolarization
A multi-center study recently documented an association between idiopathic VF and the presence of early repolarization (ER) abnormalities in inferolateral leads (Figure 47-7).71 Repolarization changes have also frequently been observed in inferolateral precordial leads in patients with Brugada syndrome, mimicking the abnormalities typical of the population presented in the study of Haïssaguerre et al, suggesting a possible overlap of these conditions.72 This subset of idiopathic VF continues to be further studied and, in addition to ICDs, newer therapies such as quinidine and isoproterenol have been noted to play a role in controlling VF storms that can occur in these patients.
Congenital Heart Disease
Patients with CHD represent an anatomically heterogeneous patient group in which risk stratification can be difficult. Current guidelines indicate ICD implantation for survivors of SCD; ICD is considered a reasonable therapy for patients with sustained VT that is not amenable to either ablation or surgery and for those with unexplained syncope and impaired ventricular function. Tetralogy of Fallot (ToF) represents one of the commonly encountered conditions in adult CHD patient populations. Older age at surgery, residual hemodynamic lesions with right heart failure, impaired LVEF, complex ventricular ectopy, inducible ventricular arrhythmias at EPS, and prolongation of the QRS complex represent some of the associations noted in SCD in patients with ToF.73–75
Noncompaction of the left ventricle is a rare congenital cardiomyopathy characterized anatomically by excessive prominent trabeculae and deep intertrabecular recesses in the left ventricle without other major congenital cardiac malfunction. Ventricular arrhythmias and SCD are known to occur, and ICD therapy often is indicated.76–78
Commotio Cordis
The term commotio cordis refers to a blunt, nonpenetrating, and usually innocent-appearing chest blow that can cause VF. The location of the blow to the chest (directly over the heart) and its timing relative to the cardiac cycle (on the upstroke of the T wave, 10 to 20 msec before its peak) are the primary determinants of commotio cordis.79,80
Other Genetic Associations in Patients with Structurally Normal Hearts
SCD likely has a strong genetic component, but only a fraction of the genetic variants that underlie the risk are known; the allelic architecture of SCD thus remains poorly defined.81 Genome-wide association studies have indicated that common variants in NOS1AP are associated with Q-T interval duration and SCD risk in general populations.82 The common polymorphism S1103Y-SCN5A is disproportionately represented in patients with arrhythmia and black patients who have experienced SCD.83 These variants by themselves are unlikely to be sufficient causes of SCD.
Risk Stratification for Sudden Cardiac Death
In SAECG, the terminal part of the QRS complex is evaluated for microvolt potentials that reflect delayed activation in the scarred myocardial substrate, thereby detecting areas of slow conduction.84 In patients with prior MI, the negative predictive value of this test has been demonstrated to be excellent. However, the usefulness of this test has been limited by its low positive predictive value, and its routine use is not common.85–87 However, the presence of an abnormal SAECG continues to have a clinical role in ARVC as a minor criterion for its diagnosis.
Q-T interval dispersion examines the difference between the maximal and minimal Q-T intervals from various standard ECG leads, whereas TWA is defined as microvolt changes in the T-wave amplitude from beat to beat.88,89 Studies evaluating the Q-T interval for prediction of SCD risk in individuals who do not have LQTS have demonstrated mixed results but have generally linked prolonged Q-T interval with increased risk. In recent studies, Q-T interval dispersion has not been shown to be a consistent predictor of SCD.87,90
TWA describes alterations in the amplitude of the T wave at modestly increased heart rates (105 to 110 beats/min) elicited by either exercise or atrial pacing. A number of observational cohort studies have suggested that microvolt TWA may work at least as well as electrophysiological testing to predict SCD or major arrhythmic events in ischemic and nonischemic cardiomyopathy.91–94 The Microvolt T-Wave Alternans Testing for Risk Stratification of Post-Myocardial Infarction Patients (MASTER) study, a prospective study of patients with prior MI and LVEF less than 30%, found that the TWA test results did not influence the frequency of the composite endpoint of arrhythmic death or “appropriate” shock over a 3-year follow-up.93 The recent Alternans Before Cardioverter Defibrillator (ABCD) trial compared the usefulness of EPS versus TWA testing in patients with ischemic cardiomyopathy and LVEF less than 40%.96,97 The results led the authors to conclude that EPS and TWA testing were comparable in terms of predicting risk and were complementary to each other. In contrast, a prospective substudy of the Sudden Cardiac Death in Heart Failure Trial (SCD-HeFT) showed that TWA was not predictive of arrhythmia or death and therefore was not a useful marker in identifying patients who would benefit from an ICD.98,99 These contrasting results have limited the generalized applicability of TWA in the risk stratification for SCD.
Evidence from both clinical and experimental studies supports a role for the autonomic nervous system in the genesis of VT or VF. An association exists between increased sympathetic activity, reduced vagal activity, or both, with a propensity for VF during myocardial ischemia.99 The two major measures of autonomic function that have been tested in clinical studies include heart rate variability (HRV) and baroreflex sensitivity (BRS).40 HRV reflects both the sympathetic and parasympathetic effects on the heart and is measured as the standard deviation of R-R intervals in the heart rate over a 24-hour period. BRS, in contrast, is an indicator of the reflex capacity of the autonomic nervous system and usually is measured by the phenylephrine method.100 Studies suggest that BRS has potential for SCD risk stratification in patients with CAD and that HRV is a predictor of total mortality but may, in fact, be a better marker of nonarrhythmic death.101,102 Further studies are needed to establish the clinical usefulness of these parameters for risk stratification and at this time are of limited utility.
LVEF is a well-established and consistent reported risk factor for overall mortality and SCD. It has been used in the risk stratification schema on many primary prevention clinical trials of SCD. In addition, the more recent Multicenter Autonomic Defibrillator Implantation Trial II (MADIT II) used only LVEF (<30%) for risk stratification for inclusion into the protocol. The results of MADIT II indicate that the use of LVEF alone for risk stratification is associated with significant mortality benefit from prophylactic ICD therapy.103 Impaired EF in certain patient populations, such as in those with early previous MI, have been shown to not be predictive of SCD from trial data.104 Of note, although low LVEF identifies a group with relatively increased risk, most instances of SCD occur in patients with a more preserved LVEF, which highlights the limited sensitivity of this technique.105,106 Heart failure itself also can contribute to arrhythmogenesis in patients with ventricular dysfunction and can increase the mortality rate in patients with dilated cardiomyopathy independent of EF. Qualitative descriptions of functional capacity such as NYHA class, although limited by subjectivity, have been well studied. NYHA classes II and III continue to be accepted as criteria to identify at-risk individuals with impaired left ventricular function for whom ICD is indicated.107–109 The presence of ventricular arrhythmias (premature ventricular contractions [PVCs] and nonsustained ventricular tachycardia [NSVT]) has also been examined in various studies to assess their role in predicting SCD. In one major trial, the positive predictive value of ventricular ectopy after MI for predicting cardiac arrhythmic events or death was limited to 5% to 15%.103 Frequent ectopy or NSVT is insensitive, failing to identify 47% to 94% of those who experience sudden cardiac arrhythmia.110 However, if combined with low LVEF, ventricular ectopy becomes a stronger risk factor for death.
Patients with nonischemic cardiomyopathy frequently have high-grade ventricular ectopy and NSVT. However, the relationship with cardiac arrest is much less clear than is the case of ischemic cardiomyopathy. In general, the incremental use of ambient ventricular arrhythmias is limited and may actually reflect the degree of heart failure rather than providing a specific marker of SCD. NSVT, in particular, has been identified as a risk factor for SCD in patients with HCM. In summary, the role of ambient nonsustained ventricular arrhythmias in predicting SCD appears to be limited. In patients with a history of previous MI, the induction of VT during an EPS has been shown to predict a high risk for recurrent arrhythmias and SCD.111,112 EPS has also been established in a number of other clinical situations, such as in patients with a history of cardiac arrest.113–115 The predictive value of EPS is highest in patients with a history of MI as opposed to those with nonischemic cardiomyopathy.1 Although a positive test result predicts increased risk for SCD, a negative result does not exclude risk, particularly in patients with severe left ventricular dysfunction. In MADIT II, ICD use was associated with a significant survival benefit even in the absence of EPS.103 In the previously reported MADIT I, inducibility in study patients was associated with an increased likelihood of VT; however, the noninducible study subjects had a considerable VT or VF event rate.116 EPS in patients with normal LVEF is of limited clinical benefit. In patients with conditions such as hypertrophic cardiomyopathy, the role of EPS is uncertain, and negative EPS findings do not exclude high risk for SCD. VF is commonly induced in these patient populations and is of uncertain clinical significance.117,118 EPS can discriminate between patients with high risk versus low risk for SCD, but if used in isolation, the sensitivity is inadequate, especially in patients with EF less than 30%. EPS is of more value when used in patients with equivocal results after noninvasive testing than as an initial screening test.119
SCD in competitive athletics generates considerable attention and concern. Although the American Heart Association consensus panel does not endorse mandatory ECG screening for all competitive athletes, it does not discourage screening initiatives by individual organizations.120 In contrast, the European Society of Cardiology recommends that evaluation include electrocardiography, chest radiography, and echocardiography.121
Another risk stratification technique that continues to evolve is that of contrast-enhanced MRI, which, if supported by clinical data, could possibly provide information on susceptibility to VAs and therefore SCD. When assessing risk with algorithms, one should be aware that no single risk factor possesses adequate sensitivity but that the presence of multiple risk factors will inappropriately reduce the number of patients who qualify for ICD therapy. Overcoming such limitations requires balancing the sensitivity and specificity of various risk stratification approaches.103
Interventions Targeting Sudden Cardiac Death
The first experience with pharmacologic prevention of SCD was with β-blocker therapy in the post-infarction population. Although the initial trial designs were not specifically constructed to evaluate SCD, these trials have uniformly demonstrated a clinically important reduction in the incidence of SCD with β-blocker therapy.122,123 The effect is particularly striking in patients with the lowest EFs. The total mortality reduction with these agents is approximately 25% to 40%, with an approximately 32% to 50% reduction in the incidence of SCD.119,125–127 Angiotensin-converting enzyme (ACE) inhibitors have also been established to decrease the overall mortality rate in heart failure. Ramipril has been shown to decrease the incidence of SCD by 30% in patients with previous MI who had heart failure.128 According to a meta-analysis on the use of ACE inhibitors in patients after myocardial infarction, the estimated risk reduction of SCD was approximately 20%.129
A substudy of Multicenter MADIT-II suggested a time-dependent beneficial effect of statin therapy on the incidence of ICD intervention for a first VA or cardiac death in 654 patients with CAD treated with an ICD.130 The cumulative rate of ICD therapy for VT or VF or cardiac death, whichever occurred first, was significantly reduced in those with at least a 90% statin use compared with those with lower statin use (P = .01). With time-dependent statin versus no statin therapy, the hazard ratio (HR) was 0.65 (P < .01) for the endpoint of VT or VF or cardiac death and 0.72 (P = .046) for VT or VF after adjusting for covariates. The Cholesterol Lowering and Arrhythmias Recurrences after Internal Defibrillator Implantation (CLARIDI) study concluded that aggressive treatment with atorvastatin (80 mg daily) in patients with CAD and an ICD resulted in fewer VAs requiring ICD treatment compared with placebo over 1-year follow-up.131 Goldberger et al demonstrated that statin therapy was associated with decreased risk for death from life-threatening arrhythmias (HR, 0.22; 95% confidence interval [CI], 0.09 to 0.55; P = .001) compared with nonstatin therapy in 458 patients with nonischemic dilated cardiomyopathy treated with an ICD in the Defibrillators in Non-Ischemic Cardiomyopathy Treatment Evaluation (DEFINITE) study.132
The role of aldosterone antagonists in preventing SCD was delineated by two landmark trials, the Randomized Aldactone Evaluation Study (RALES) and Eplerenone Post–Acute Myocardial Infarction Heart Failure Efficacy and Survival (EPHESUS) trials. RALES was a double-blind, randomized study of heart failure with impaired LVEF of aldactone versus placebo. SCD occurred in 82 of 822 spironolactone-treated patients versus 110 of 841 placebo-treated patients (relative risk [RR], 0.71; 95% CI, 0.54 to 0.95; P = .02).133 EPHESUS was a double-blind, placebo-controlled study of the eplerenone in patients with previous MI and left ventricular dysfunction.134 SCD occurred in 162 of 3313 in the eplerenone group versus 201 of 3313 in the placebo group (RR, 0.79; 95% CI, 0.64 to 0.97; P = .03).
However, when membrane-active antiarrhythmic drugs were used to treat patients with frequent ventricular ectopy, the incidence of SCD increased.135,136 This was true for both class Ic class III agents such as D-sotalol, as shown in the Cardiac Arrhythmia Suppression Trial (CAST) and the SWORD trials.137–140 The only membrane-active antiarrhythmic medications approved for the treatment of ventricular arrhythmias that do not carry increased mortality risk in appropriately selected patients with ischemic heart disease are D,L-sotalol and amiodarone. However, in two major trials examining the routine use of amiodarone in patients with previous MI (European Myocardial Infarct Amiodarone Trial [EMIAT] and Canadian Amiodarone Myocardial Infarction Arrhythmia Trial [CAMIAT]), amiodarone did not demonstrate a survival benefit.141,142 In a higher risk population of patients (Grupo de Estudio de la Sobrevida en la Insuficiencia Cardiaca en Argentina) [GESICA]: LVEF <35% and either ischemic or nonischemic cardiomyopathy), empiric amiodarone treatment was shown to affect mortality rate favorably.143 These data were not corroborated by another trial examining a population composed primarily of patients with ischemic cardiomyopathy (Congestive Heart Failure Survival Trial of Antiarrhythmic Therapy [CHF-STAT]).144 This trial found no survival benefit with prophylactic amiodarone in patients with congestive heart failure. In the SCD-HeFT study, amiodarone was significantly worse than placebo (HR, 1.44; CI, 1.05 to 1.97; P < .01) in the prespecified NYHA class III patients. However, this trend was not seen in the overall study. A recent large meta-analysis, however, indicated that amiodarone decreases the incidence of SCD but not that of overall mortality.145
Other primary prevention trials have focused on the efficacy of ICD therapy in the prevention of SCD. Two randomized clinical trials have examined the use of ICDs in patients with prior MI, low LVEF, NSVT, and inducible VT at EPS: MADIT I and the Multicenter UnSustained Tachycardia Trial (MUSTT).146,147 Both trials demonstrated a significant mortality benefit in patients who received ICDs compared with those patients treated with or without antiarrhythmic medications. The results of these two trials have been extended by the larger and more reflective of contemporary practice MADIT II, a randomized controlled trial in patients with previous MI and LVEF less than 30%. In this prophylactic ICD trial, NSVT was not a required entry criterion, and all-cause mortality was 20% in the control group and 14.2% in the ICD group (RR, 31%; P = .016).148
Two other ICD trials that deserve special mention include the Coronary Artery Bypass Graft Surgery with/without Simultaneous Epicardial Patch for Automatic Implantable Cardioverter Defibrillator (CABG-PATCH) and Defibrillator IN Acute Myocardial Infarction Trial (DINAMIT) trials. In the CABG-PATCH trial, routine ICD insertion did not improve survival in patients with CAD undergoing CABG who were believed to be at high risk of SCD on the basis of SA ECG and severe left ventricular dysfunction.149 DINAMIT examined the role of ICDs in patients 6 to 40 days after MI with an EF less than 35% and impaired autonomic tone. Prophylactic ICD did not reduce overall mortality in high-risk patients who recently had MI. Although ICD therapy was associated with a reduction in the rate of death from arrhythmia, this was offset by an increase in the rate of death from nonarrhythmic causes.150
Despite the efficacy of ICD therapy in very-high-risk patient subsets, it is important to realize that this subset represents a select minority of the total number at risk for SCD. Protection against SCD by ICD implantation is expensive in high-risk populations and would be significantly more expensive in the larger but lower-risk populations. Substudy analyses of these large trials suggest that much of the benefit of ICD is realized in the sickest patients (i.e., those with the lower LVEFs).151,152 Therefore identifying and improving risk stratification methods to better characterize patients at highest risk for SCD continues to be an area of active research. Other newer implantable therapies such as subcutaneous implantable defibrillators have recently been studied and appear to be a promising alternative in select patients.153 On the invasive front, ablating monomorphic PVCs that repeatedly induce polymorphic VT or VF are increasingly reported. These PVCs are often noted to arise from the Purkinje system. Although the initial case reports were described in patients with idiopathic VF with structurally normal hearts, they have now been described in a wider patient population that has come to include patients with recurrent VF caused by various etiologies such as post-MI syndrome, LQTS, Brugada syndrome, and early repolarization syndrome. Although this is an exciting advance in the treatment of SCD, long-term outcomes in these patients are lacking.154–157
Community-Based Resuscitation
A very important prognostic variable in determining the effectiveness of resuscitation is the time from cardiac arrest to initial defibrillation. “Links in the chain of survival” is a term used to describe such time-sensitive services that influence the likelihood of successful resuscitation. These links include early activation, CPR, defibrillation, and advanced post-resuscitation care.158 One of the most effective EMS in the world is located in Seattle. However, only 40% of cardiac arrest patients in the Seattle experience have VF at initial contact, and 26% of those VF individuals get discharged from the hospital with intact neurologic funtions.159 Thus even in an ideal scenario despite prompt CPR, a good EMS response time, and the use of AEDs, only 10% of all cardiac arrest patients will be satisfactorily discharged from the hospital.160,161 Other communities, however, have been less successful. The reasons for the disappointing success rates are multi-factorial and include various population and community characteristics. Early CPR, usually provided by a layperson before EMS arrival, can improve the likelihood of resuscitation. Efforts by professional organizations to train laypeople in CPR and initiatives such as dispatcher-initiated CPR skills have undoubtedly saved thousands of lives.
The interval from collapse to defibrillation is an exceptionally strong predictor of sudden cardiac arrhythmia survival.162 In a departure from the conventional compression-plus-ventilation approach for bystander CPR, the American Heart Association recommended “hands-only” CPR to be provided by rescuers who are either untrained or not confident in their ability to provide rescue breaths.163 Early defibrillation has been facilitated by the use of the AED, which assesses the rhythm and, when indicated, delivers a potentially lifesaving shock.164 A strategy of broad AED deployment in public places has been implemented to reach out to less-experienced operators. Its success in this setting has prompted the U.S. government to mandate AED placement on all airlines and in airports. In the Home Use of Automated External Defibrillators for Sudden Cardiac Arrest (HAT) trial, placing AEDs in homes, however, did not reduce the mortality rate in patients with prior anterior wall infarctions who did not meet standard indications for ICD compared with a standard response training for cardiac arrest.165 A very low event rate, a high proportion of unwitnessed events, and underuse of AEDs in emergencies, rather than a lack of device efficacy, have been suggested as explanations for these results. Public access defibrillation programs will hopefully improve the outcome from SCD significantly in the future.
Post-Resuscitation Care
Post-resuscitation care involves the care of post-arrest brain and cardiac dysfunctions and the ensuing systemic ischemia or reperfusion response. This is an essential component in the management of SCD because anoxic brain injury is a significant cause of morbidity and mortality in these patients. Evidence from randomized trials support the induction of hypothermia between 32° C and 34° C for 12 to 24 hours in an effort to improve neurologic survival in comatose patients admitted after resuscitation from witnessed VF arrest.166 Such interventions have helped improve outcomes in patients resuscitated after SCD.
Key References
The Antiarrhythmics versus Implantable Defibrillators (AVID) Investigators. A comparison of antiarrhythmic-drug therapy with implantable defibrillators in patients resuscitated from near-fatal ventricular arrhythmias. N Engl J Med. 1997;337:1576.
Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome: Report of the Second Consensus Conference: Endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation. 2005;111:659-670.
Bardy GH, Lee KL, Mark DB, et al. Sudden Cardiac Death in Heart Failure Trial (SCD-HeFT) Investigators: Amiodarone or an implantable cardioverter-defibrillator for congestive heart failure. N Engl J Med. 2005;352:225-237.
Buxton AE. Risk stratification for sudden death in patients with coronary artery disease. Heart Rhythm. 2009;6(6):836-847.
The Cardiac Arrhythmia Suppression Trial II (CAST II) Investigators. Effect of the antiarrhythmic agent moricizine on survival after myocardial infarction. N Engl J Med. 1992;327:227-233.
Haïssaguerre M, Derval N, Sacher F, et al. Sudden cardiac arrest associated with early repolarization. N Engl J Med. 2008;358:2016-2023.
Hohnloser SH, Kuck KH, Dorian P, et al. Prophylactic use of an implantable cardioverter-defibrillator after acute myocardial infarction. N Engl J Med. 2004;351:2481-2488.
Huikuri HV, Castellanos A, Myerburg RJ. Sudden death due to cardiac arrhythmias. N Engl J Med. 2001;345:1473-1482.
Kadish A, Dyer A, Daubert JP, et al. Defibrillators in Non-Ischemic Cardiomyopathy Treatment Evaluation (DEFINITE) Investigators: Prophylactic defibrillator implantation in patients with nonischemic dilated cardiomyopathy. N Engl J Med. 2004;350:2151-2158.
Moss AJ, Hall WJ, Cannom DS, et al. Improved survival with an implanted defibrillator in patients with coronary disease at high risk for ventricular arrhythmia. N Engl J Med. 1996;335:1933-1940.
Moss AJ, Zareba W, Hall WJ, et al. Multicenter Automatic Defibrillator Implantation Trial II Investigators: Prophylactic implantation of a defibrillator in patients with myocardial infarction and reduced ejection fraction. N Engl J Med. 2002;346:877-883.
Nichol G, Thomas E, Callaway CW, et al. Resuscitation Outcomes Consortium Investigators. Regional variation in out-of-hospital cardiac arrest incidence and outcome. JAMA. 2008;300:1423-1431.
Pappone C, Santinelli V, Rosanio S, et al. Usefulness of invasive electrophysiologic testing to stratify the risk of arrhythmic events in asymptomatic patients with Wolff-Parkinson-White pattern: Results from a large prospective long-term follow-up study. J Am Coll Cardiol. 2003;41:239-244.
Pitt B, Remme W, Zannad F, Neaton J, et al. Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study Investigators: Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 2003;48:1309-1321.
Tabereaux PB, Dosdall DJ, Ideker RE. Mechanisms of VF maintenance: Wandering wavelets, mother rotors, or foci. Heart Rhythm. 2009;6(3):405-415.
1 Huikuri HV, Castellanos A, Myerburg RJ. Sudden death due to cardiac arrhythmias. N Engl J Med. 2001;345:1473-1482.
2 Myerburg RJ, Castellanos A. Cardiac arrest and sudden cardiac death. In Zipes DP, Libby P, Bonow RO, Braunwald E, editors: Braunwald’s heart disease: A textbook of cardiovascular medicine, ed 7, Philadelphia: Saunders, 2004.
3 Fox CS, Evans JC, Larson MG, et al. Temporal trends in coronary heart disease mortality and sudden cardiac death from 1950 to 1999: The Framingham Heart Study. Circulation. 2004;110:522-527.
4 Nichol G, Thomas E, Callaway CW, et al. Resuscitation Outcomes Consortium Investigators: Regional variation in out-of-hospital cardiac arrest incidence and outcome. JAMA. 2008;300:1423-1431.
5 Chugh SS, Jui J, Gunson K, et al. Current burden of sudden cardiac death: Multiple source surveillance versus retrospective death certificate–based review in a large U.S. community. J Am Coll Cardiol. 2004;44:1268-1275.
6 American Heart Association. Heart disease and stroke statistics—2010 Update. Dallas, TX: AHA; 2010.
7 Fox CS, Evans JC, Larson MG, et al. A comparison of death certificate out-of-hospital coronary heart disease death with physician-adjudicated sudden cardiac death. Am J Cardiol. 2005;95:856-859.
8 Cobb LA, Fahrenbruch CE, Olsufka M, Copass MK. Changing incidence of out-of-hospital ventricular fibrillation, 1980–2000. JAMA. 2002;288:3008-3013.
9 Rea TD, Pearce RM, Raghunathan TE, et al. Incidence of out-of-hospital cardiac arrest. Am J Cardiol. 2004;93:1455-1460.
10 Myerburg RJ, Interian A, Mitrani RM, et al. Frequency of sudden cardiac death and profiles of risk. Am J Cardiol. 1997;80:10F-19F.
11 Zipes DP, Wellen HJJ. Sudden cardiac death. Circulation. 1998;98:2334-2351.
12 Myerburg RJ. Sudden cardiac death: Exploring the limits of our knowledge. J Cardiovasc Electrophysiol. 2001;12:369-381.
13 Zipes DP, Wellens HJ. Sudden cardiac death. Circulation. 1998;98:2334-2351.
14 Corrado D, Basso C, Pavei A, et al. Trends in sudden cardiovascular death in young competitive athletes after implementation of a preparticipation screening program. JAMA. 2006;296:1593-1601.
15 Maron BJ, Doerer JJ, Haas TS, et al. Sudden deaths in young competitive athletes: Analysis of 1866 deaths in the United States, 1980–2006. Circulation. 2009;119:1085-1092.
16 Meaney PA, Nadkarni VM, et al. Rhythms and outcomes of adult in-hospital cardiac arrest. Crit Care Med. 2010;38(1):101-108.
17 Chen PS, Wu TJ, Ting CT, et al. A tale of two fibrillations. Circulation. 2003;108:2298-2303. Ventricular fibrillation: How do we put the genie back in the bottle? Heart Rhythm, Volume 4, Issue 5, May 2007, Pages 665-674.
18 Tabereaux PB, Dosdall DJ, Ideker RE. Mechanisms of VF maintenance: Wandering wavelets, mother rotors, or foci. Heart Rhythm. 2009;6(3):405-415.
19 Zaitsev AV, Berenfeld O, Mironov SF, et al. Distribution of excitation frequencies on the epicardial and endocardial surfaces of fibrillating ventricular wall of the sheep heart. Circ Res. 2000;86:408-417.
20 Weiss JN, Qu Z, Chen PS, et al. The dynamics of cardiac fibrillation. Circulation. 2005;112:1232-1240.
21 Tabereaux PB, Walcott GP, Rogers JM, et al. Activation patterns of Purkinje fibers during long-duration ventricular fibrillation in an isolated canine heart model. Circulation. 2007;116:1113-1119.
22 Dosdall DJ, Tabereaux PB, Kim JJ, et al. Chemical ablation of the Purkinje system causes early termination and activation rate slowing of long duration ventricular fibrillation in dogs. Am J Physiol Heart Circ Physiol. 2008;295:H883-H889.
23 Zipes DP, Wellen HJJ. Sudden cardiac death. Circulation. 1998;98:2334-2351.
24 Kimura S, Bassett AL, Saoudi NC, et al. Cellular electrophysiologic changes and “arrhythmias” during experimental ischemia and reperfusion in isolated cat ventricular myocardium. J Am Coll Cardiol. 1986;7:833-842.
25 Kimura S, Bassett AL, Kohya T, et al. Simultaneous recording of action potentials from endocardium and epicardium during ischemia in the isolated cat ventricles. Circulation. 1986;64:401-409.
26 Owens LM, Fralix TA, Murphy E, et al. Correlation of ischemia-induced extracellular and intracellular ion changes to cell-to-cell electrical uncoupling in isolated blood-perfused rabbit hearts. Circulation. 1996;94:10-13.
27 Wang JS, Jen CJ, Kung HC, et al. Different effects of strenuous exercise and moderate exercise on platelet function in men. Circulation. 1994;90:2877-2885.
28 McLenachan JM, Henderson E, Morris KI, Dargie HJ. Ventricular arrhythmias in patients with hypertensive left ventricular hypertrophy. N Engl J Med. 1987;317:787-792.
29 El Fawal MA, Berg GA, Whealey DJ, et al. Sudden coronary death in Glasgow: Nature and frequency of acute coronary lesions. Br Heart J. 1987;57:329-335.
30 Davies M, Bland J, Hangartner J, et al. Factors influencing the presence or absence of acute coronary artery thrombi in sudden ischemic death. Eur Heart J. 1989;10:203-208.
31 Poole JE, Bardy GH. Sudden cardiac death. In: Zipes DP, Jalife F, editors. Cardiac electrophysiology: From cell to bedside. Philadelphia: Saunders, 2000.
32 Friedlander Y, Siscovick DS, Arbogast P, et al. Sudden death and myocardial infarction in first degree relatives as predictors of primary cardiac arrest. Atherosclerosis. 2002;162:211-216.
33 Dekker LR, Bezzina CR, Henriques JP, et al. Familial sudden death is an important risk factor for primary ventricular fibrillation: A case-control study in acute myocardial infarction patients. Circulation. 2006;114:1140-1145.
34 Noseworthy PA, Newton-Cheh C. Genetic determinants of sudden cardiac death [review of 109 references]. Circulation. 2008;118(18):1854-1863.
35 Davies MJ, Thomas A. Thrombosis and acute coronary artery lesions in sudden cardiac ischemic death. N Engl J Med. 1984;310:1137-1140.
36 Tamburro P, Wilber D. Sudden death in idiopathic dilated cardiomyopathy. Am Heart J. 1992;124:1035-1045.
37 Blanck Z, Dhala A, Deshpande S, et al. Bundle branch reentrant ventricular tachycardia: Cumulative experience in 48 patients. J Cardiovasc Electrophysiol. 1993;4:253-262.
38 Brooks SC, Schmicker RH, Rea TD, et al. ROC Investigators: Out-of-hospital cardiac arrest frequency and survival: Evidence for temporal variability resuscitation. Resuscitation. 2010;81(2):175-181.
39 Reichenbach DD, Moss NS, Meyer E. Pathology of the heart in sudden cardiac death. Am J Cardiol. 1977;39:865.
40 Gomes JA, Alexopoulos D, Winters SL, et al. The role of silent ischemia, the arrhythmic substrate and the short-long sequence in the genesis of sudden cardiac death. J Am Coll Cardiol. 1989;14:1618-1625.
41 Farb A, Tang AL, Burke AP, et al. Sudden coronary death: Frequency of active coronary lesions, inactive coronary lesions, and myocardial infarction. Circulation. 1995;92:1701-1709.
42 Stevenson WG, Stevenson LW, Middlekauff HR, Saxon LA. Sudden death prevention in patients with advanced ventricular dysfunction. Circulation. 1993;88:2953-2961.
43 Spirito P, Seidman CE, McKenna WJ, Maron BJ. The management of hypertrophic cardiomyopathy. N Engl J Med. 1997;336:775-785.
44 Maron BJ. Sudden death in young athletes. N Engl J Med. 2003;349:1064-1075.
45 Maron BJ, McKenna WJ, Danielson GK, et al. for the Task Force on Clinical Expert Consensus Documents, American College of Cardiology; Committee for Practice Guidelines, European Society of Cardiology: American College of Cardiology/European Society of Cardiology clinical expert consensus document on hypertrophic cardiomyopathy: A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. J Am Coll Cardiol. 2003;42:1687-1713.
46 Maron BJ. Contemporary insights and strategies for risk stratification and prevention of sudden death in hypertrophic cardiomyopathy. Circulation. 2010;121(3):445-456. Review
47 Goldenberg I, Moss AJ. Long QT syndrome. J Am Coll Cardiol. 2008;51:2291-2300.
48 Kelly M, Semsarian C. Multiple mutations in genetic cardiovascular disease: A marker of disease severity? Circ Cardiovasc Genet. 2009;2:182-190.
49 Maron BJ, Roberts WC, Arad M, et al. Clinical outcome and phenotypic expression in LAMP2 cardiomyopathy. JAMA. 2009;301:1253-1259.
50 Adabag AS, Maron BJ, Appelbaum E, et al. Occurrence and frequency of arrhythmias in hypertrophic cardiomyopathy in relation to delayed enhancement on cardiovascular magnetic resonance. J Am Coll Cardiol. 2008;51(14):1369-1374.
51 Kwon DH, Setser RM, Popovic ZB, et al. Association of myocardial fibrosis, electrocardiography and ventricular tachyarrhythmia in hypertrophic cardiomyopathy: A delayed contrast enhanced MRI study. Int J Cardiovasc Imaging. 2008;24:617-625.
52 Eriksson MJ, Sonnenberg B, Woo A, et al. Long-term outcome in patients with apical hypertrophic cardiomyopathy. J Am Coll Cardiol. 2002;39:638-645.
53 Goldenberg I, Bradley J, Moss A, et al. on behalf of the international LQTS registry investigators: Beta-blocker efficacy in high-risk patients with the congenital long-QT syndrome types 1 and 2: Implications for patient management. J Cardiovasc Electrophysiol. 2010;21(8):893-901.
54 Patel U, Pavri BB. Short QT syndrome: A review. Cardiol Rev. 2009;17(6):300-303.
55 Sen-Chowdhry S, Morgan RD, Chambers JC, McKenna WJ. Arrhythmogenic cardiomyopathy: Etiology, diagnosis, and treatment. Annu Rev Med. 2010;61:233-253.
56 McKoy G, Protonotarios N, Crosby A, et al. Identification of a deletion in plakoglobin arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and wooly hair (Naxos disease). Lancet. 2000;355:2119-2124.
57 Probst V, Veltmann C, Eckardt L, et al. Long-term prognosis of patients diagnosed with Brugada syndrome: Results from the FINGER Brugada Syndrome Registry. Circulation. 2010;121(5):635-643.
58 Nademanee K, Veerakul G, Nimmannit S, et al. Arrhythmogenic marker for the sudden unexplained death syndrome in Thai men. Circulation. 1997;96:2595-2600.
59 Gussak I, Antzelevitch C, Bjerregaard P, et al. The Brugada syndrome: Clinical, electrophysiologic and genetic aspects. J Am Coll Cardiol. 1999;33:5-15.
60 Benito B, Brugada J, Brugada R, Brugada P. Brugada syndrome. Rev Esp Cardiol. 2009;62(11):1297-1315.
61 Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome: Report of the Second Consensus Conference: Endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation. 2005;111:659-670.
62 Fitzsimmons PJ, McWhirter PD, Peterson DW, et al. The natural history of Wolff-Parkinson-White syndrome in 228 military aviators: A long-term follow-up of 22 years. Am Heart J. 2001;142:530-536.
63 Goudevenos JA, Katsouras CS, Graekas G, et al. Ventricular pre-excitation in the general population: A study on the mode of presentation and clinical course. Heart. 2000;83:29-34.
64 Pietersen AH, Andersen ED, Sandoe E. Atrial fibrillation in the Wolff-Parkinson-White syndrome. Am J Cardiol. 1992;70:38A-43A.
65 Pappone C, Santinelli V, Rosanio S, et al. Usefulness of invasive electrophysiologic testing to stratify the risk of arrhythmic events in asymptomatic patients with Wolff-Parkinson-White pattern: Results from a large prospective long-term follow-up study. J Am Coll Cardiol. 2003;41:239-244.
66 Klein GJ, Bashore TM, Sellers TD, et al. Ventricular fibrillation in the Wolff-Parkinson-White syndrome. N Engl J Med. 1979;301:1080-1085.
67 Laitinen PJ, Brown KM, Piippo K, et al. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation. 2001;103:485-490.
68 Priori SG, Napolitano C, Tiso N, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103:196-200.
69 Lahat H, Pras E, Olender T, et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am J Hum Genet. 2001;69:1378-1384.
70 Postma AV, Denjoy I, Hoorntje TM, et al. Absence of calsequestrin 2 causes severe forms of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2002;91:e21-e26.
71 Haïssaguerre M, Derval N, Sacher F, et al. Sudden cardiac arrest associated with early repolarization. N Engl J Med. 2008;358:2016-2023.
72 Sarkozy A, Chierchia GB, Paparella G, et al. Inferior and lateral electrocardiographic repolarization abnormalities in Brugada syndrome. Circ Arrhythm Electrophysiol. 2009;2:154-161.
73 Kavey RE, Thomas FD, Byrum CJ, et al. Ventricular arrhythmias and biventricular dysfunction after repair of tetralogy of Fallot. J Am Coll Cardiol. 1984;4:126-131.
74 Gatzoulis MA, Balaji S, Webber SA, et al. Risk factors for arrhythmia and sudden cardiac death late after repair of tetralogy of Fallot: A multicentre study. Lancet. 2000;356:975-981.
75 Ghai A, Silversides C, Harris L, et al. Left ventricular dysfunction is a risk factor for sudden cardiac death in adults late after repair of tetralogy of Fallot. J Am Coll Cardiol. 2002;40(9):1675-1680.
76 Kobza R, Steffel J, Erne P, et al. Implantable cardioverter defibrillator and cardiac resynchronization therapy in patients with left ventricular noncompaction. Heart Rhythm. 2010;7(11):1545-1549.
77 Oechslin EN, Attenhofer Jost CH, Rojas JR, et al. Long-term follow-up of 34 adults with isolated left ventricular noncompaction: A distinct cardiomyopathy with poor prognosis. J Am Coll Cardiol. 2000;36:493-500.
78 Priori SG, Aliot E, Blomstrom-Lundqvist C, et al. Task Force on Sudden Cardiac Death of the European Society of Cardiology. Eur Heart J. 2001;22:1374-1450.
79 Link MS, Maron BJ, VanderBrink BA, et al. Impact directly over the cardiac silhouette is necessary to produce ventricular fibrillation in an experimental model of commotio cordis. J Am Coll Cardiol. 2001;37:649-654.
80 Link MS, Wang PJ, Pandian NG, et al. An experimental model of sudden death due to low-energy chest-wall impact (commotio cordis). N Engl J Med. 1998;338:1805-1811.
81 Newton-Cheh C, Shah R. Genetic determinants of QT interval variation and sudden cardiac death. Curr Opin Genet Dev. 2007;17:213-221.
82 Kao WH, Arking DE, Post W, et al. Genetic variations in nitric oxide synthase 1 adaptor protein are associated with sudden cardiac death in US white community-based populations. Circulation. 2009;119:940-951.
83 Splawski I, Timothy KW, Tateyama M, et al. Variant of SCN5A sodium channel implicated in risk of cardiac arrhythmia. Science. 2002;297:1333-1336.
84 El-Sherif N, Denes P, Katz R, et al. Definition of the best prediction criteria of the time-domain signal-averaged electrocardiogram for serious arrhythmic events in the postinfarction period. J Am Coll Cardiol. 1995;25:908-914.
85 McClements BM, Adgey AAJ. Value of signal-averaged electrocardiography, radionuclide ventriculography, Holter monitoring and clinical variables for prediction of arrhythmic events in survivors of acute myocardial infarction in the thrombolytic era. J Am Coll Cardiol. 1993;21:1419-1427.
86 Gomes JA, Winter SL, Stewart D, et al. A new noninvasive index to predict sustained ventricular tachycardia and sudden death in the first year after myocardial infarction: Based on signal averaged electrocardiogram, radionuclide ejection fraction and Holter monitoring. J Am Coll Cardiol. 1987;10:349-357.
87 Glancy JM, Garratt CJ, Woods KL, de Bono DP. QT dispersion and mortality after myocardial infarction. Lancet. 1995;345:945-948.
88 Day CP, McComb JM, Campbell RW. QT dispersion: An indication of arrhythmia risk in patients with long QT intervals. Br Heart J. 1990;63:342-344.
89 Smith JM, Clancy EA, Valeri CR, et al. Electrical alternans and cardiac electrical instability. Circulation. 1988;77:110-121.
90 Zabel M, Klingenheben T, Franz MR, Hohnloser SH. Assessment of QT dispersion for prediction of mortality or arrhythmic events after myocardial infarction: Results of a prospective, long-term follow-up study. Circulation. 1998;97:2543-2550.
91 Hohnloser SH, Klingenheben T, Bloomfield D, et al. Usefulness of microvolt T-wave alternans for prediction of ventricular tachyarrhythmic events in patients with dilated cardiomyopathy: Results from a prospective observational study. J Am Coll Cardiol. 2003;41:2220-2224.
92 Narayan SM. T-wave alternans and the susceptibility to ventricular arrhythmias. J Am Coll Cardiol. 2006;47:269-281.
93 Chow T, Kereiakes DJ, Onufer J, et al. MASTER Trial Investigators: Does microvolt T-wave alternans testing predict ventricular tachyarrhythmias in patients with ischemic cardiomyopathy and prophylactic defibrillators? The MASTER (Microvolt T Wave Alternans Testing for Risk Stratification of Post-Myocardial Infarction Patients) trial. J Am Coll Cardiol. 2008;52:1607-1615.
94 Gold MR, Ip JH, Costantini O, et al. Role of microvolt T-wave alternans in assessment of arrhythmia vulnerability among patients with heart failure and systolic dysfunction: Primary results from the T-wave alternans sudden cardiac death in heart failure trial substudy. Circulation. 2008;118:2022-2028.
95 Reference deleted in proofs
96 Costantini O, Hohnloser SH, Kirk MM, et al. ABCD Trial Investigators: The ABCD (Alternans Before Cardioverter Defibrillator) Trial: Strategies using T-wave alternans to improve efficiency of sudden cardiac death prevention. J Am Coll Cardiol. 2009;53(6):471-479.
97 Amit G, Rosenbaum DS, Super DM, Costantini O. Microvolt T-wave alternans and electrophysiologic testing predict distinct arrhythmia substrates: Implications for identifying patients at risk for sudden cardiac death. Heart Rhythm. 2010;7(6):763-768.
98 Costantini O, Hohnloser SH, Kirk MM, et al. ABCD Trial Investigators: The ABCD (Alternans Before Cardioverter Defibrillator) trial: Strategies using T-wave alternans to improve efficiency of sudden cardiac death prevention. J Am Coll Cardiol. 2009;53:471-479.
99 Schwartz PJ, La Rovere MT, Vanoli E. Autonomic nervous system and sudden cardiac death: Experimental basis and clinical observations for post-myocardial infarction risk stratification. Circulation. 1992;85(Suppl I):I77-I91.
100 Eckberg DL, Sleight P. Human baroreflexes in health and disease. Oxford, UK: Clarendon Press; 1992.
101 La Rovere MT, Bigger JTJr, Marcus FI, et al. ATRAMI (Autonomic Tone and Reflexes After Myocardial Infarction) Investigators: Baroreflex sensitivity and heart-rate variability in prediction of total cardiac mortality after myocardial infarction. Lancet. 1998;351:478-484.
102 Camm AJ, Pratt CM, Schwartz PJ, et al. AzimiLide post Infarct surVival Evaluation (ALIVE) Investigators: Mortality in patients after a recent myocardial infarction: A randomized, placebo-controlled trial of azimilide using heart rate variability for risk stratification. Circulation. 2004;109:990-996.
103 Maggioni AP, Zuanetti G, Franzosi MG, et al. Prevalence and prognostic significance of ventricular arrhythmias after acute myocardial infarction in the fibrinolytic era. GISSI-2 results. Circulation. 1993;87:312-322.
104 Hohnloser SH, Kuck KH, Dorian P, et al. Prophylactic use of an implantable cardioverter-defibrillator after acute myocardial infarction. N Engl J Med. 2004;351:2481-2488.
105 Stecker EC, Vickers C, Waltz J, et al. Population-based analysis of sudden cardiac death with and without left ventricular systolic dysfunction. Two-year findings from the Oregon Sudden Unexpected Death Study. J Am Coll Cardiol. 2006;47:1161-1166.
106 Gorgels APM, Gijsbers C, de Vreede-Swagemakers J, et al. Out-of-hospital cardiac arrest—the relevance of heart failure. The Maastricht Circulatory Arrest Registry. Eur Heart J. 2003;24:1204-1209.
107 Kadish A, Dyer A, Daubert JP, et al. Defibrillators in Non-Ischemic Cardiomyopathy Treatment Evaluation (DEFINITE) Investigators: Prophylactic defibrillator implantation in patients with nonischemic dilated cardiomyopathy. N Engl J Med. 2004;350:2151-2158.
108 Moss AJ, Zareba W, Hall WJ, et al. Multicenter Automatic Defibrillator Implantation Trial II Investigators: Prophylactic implantation of a defibrillator defibrillator in patients with myocardial infarction and reduced ejection fraction. N Engl J Med. 2002;346:877-883.
109 Bardy GH, Lee KL, Mark DB, et al. Sudden Cardiac Death in Heart Failure Trial (SCD-HeFT) Investigators: Amiodarone or an implantable cardioverter-defibrillator for congestive heart failure. N Engl J Med. 2005;352:225-237.
110 Buxton AE. Risk stratification for sudden death in patients with coronary artery disease. Heart Rhythm. 2009;6(6):836-847.
111 Buxton AE, Lee KL, DiCarlo L, et al. Electrophysiologic testing to identify patients with coronary artery disease who are at risk for sudden death. Multicenter Unsustained Tachycardia Trial Investigators. N Engl J Med. 2000;342:1937-1945.
112 Moss AJ, Hall WJ, Cannom DS, et al. Improved survival with an implanted defibrillator in patients with coronary disease at high risk for ventricular arrhythmia. N Engl J Med. 1996:3351933-3351940.
113 Kuck KH, Cappato R, Siebels J, Ruppel R. Randomized comparison of antiarrhythmic drug therapy with implantable defibrillators in patients resuscitated from cardiac arrest: The Cardiac Arrest Study Hamburg (CASH). Circulation. 2000;102:748-754.
114 Connolly SJ, Gent M, Roberts RS, et al. Canadian Implantable Defibrillator Study (CIDS): Randomized trial of the implantable cardioverter defibrillator against amiodarone. Circulation. 2000;101:1287-1302.
115 The Antiarrhythmics versus Implantable Defibrillators (AVID) Investigators. A comparison of antiarrhythmic-drug therapy with implantable defibrillators in patients resuscitated from near-fatal ventricular arrhythmias. N Engl J Med. 1997;337:1576.
116 Daubert JP, Zareba W, Hall WJ, et al. MADIT II Study Investigators: Predictive value of ventricular arrhythmia inducibility for subsequent ventricular tachycardia or ventricular fibrillation in Multicenter Automatic Defibrillator Implantation Trial (MADIT) II patients. J Am Coll Cardiol. 2006:4798-4807.
117 Poole JE, Bardy GH. Sudden cardiac death. In: Zipes DP, Jalife F, editors. Cardiac electrophysiology: From cell to bedside. Philadelphia: Saunders, 2000.
118 Zipes DP, Wellen HJJ. Sudden cardiac death. Circulation. 1998;98:2334-2351.
119 Dargie HJ. Effect of carvedilol on outcome after myocardial infarction in patients with left-ventricular dysfunction: The CAPRICORN randomised trial. Lancet. 2001;357(9266):1385-1390.
120 Maron BJ, Thompson PD, Ackerman MJ, et al. American Heart Association Council on Nutrition, Physical Activity, and Metabolism: Recommendations and considerations related to preparticipation screening for cardiovascular abnormalities in competitive athletes: 2007 update: A scientific statement from the American Heart Association Council on Nutrition, Physical Activity, and Metabolism: Endorsed by the American College of Cardiology Foundation. Circulation. 2007;115:1643-1655.
121 Pelliccia A, Fagard R, Bjørnstad HH, et al. Study Group of Sports Cardiology of the Working Group of Cardiac Rehabilitation and Exercise Physiology; Working Group of Myocardial and Pericardial Diseases of the European Society of Cardiology: Recommendations for competitive sports participation in athletes with cardiovascular disease: A consensus document from the Study Group of Sports Cardiology of the Working Group of Cardiac Rehabilitation and Exercise Physiology and the Working Group of Myocardial and Pericardial Diseases of the European Society of Cardiology. Eur Heart J. 2005;26(14):1422-1445.
122 Gottlieb SS, McCarter RJ, Vogel RA. Effect of beta-blockade on mortality among high-risk and low-risk patients after myocardial infarction. N Engl J Med. 1998;3339:489-497.
123 Yusuf S, Peto R, Lewis J, et al. Beta blockade during and after myocardial infarction: An overview of the randomized trials. Prog Cardiovasc Dis. 1985;27:335-371.
124 Reference deleted in proofs
125 Cucherat M, Boissel JP, Leizorovicz A. The APSI investigators: Persistent reduction of mortality for five years after one year of acebutolol treatment initiated during acute myocardial infarction. Am J Cardiol. 1997;79(5):587-589.
126 A randomized trial of propranolol in patients with acute myocardial infarction. I. Mortality results. JAMA. 1982;247(12):1707-1714.
127 Timolol-induced reduction in mortality and reinfarction in patients surviving acute myocardial infarction. N Engl J Med. 1981;304(14):801-807.
128 Cleland JG, Erhardt L, Murray G, et al. Effect of ramipril on morbidity and mode of death among survivors of acute myocardial infarction with clinical evidence of heart failure. A report from the AIRE Study Investigators. Eur Heart J. 1997;18(1):41-51.
129 Domanski MJ, Exner DV, Borkowf CB, et al. Effect of angiotensin converting enzyme inhibition on sudden cardiac death in patients following acute myocardial infarction. A meta-analysis of randomized clinical trials. J Am Coll Cardiol. 1999;33:598-604.
130 Vyas AK, Guo H, Moss AJ, et al. MADIT-II Research Group: Reduction in ventricular tachyarrhythmias with statins in the Multicenter Automatic Defibrillator Implantation Trial (MADIT)-II. J Am Coll Cardiol. 2006;47:769-773.
131 De Sutter J, Tavernier R, De Bacquer D, et al. Coronary risk factors and inflammation in patients with coronary artery disease and internal cardioverter defibrillator implants. Int J Cardiol. 2006;112(1):72-79.
132 Goldberger JJ, Subacius H, Schaechter A, et al. DEFINITE Investigators: Effects of statin therapy on arrhythmic events and survival in patients with nonischemic dilated cardiomyopathy. J Am Coll Cardiol. 2006;48(6):1228-1233.
133 Pitt B, Zannad F, Remme WJ, et al. Randomized Aldactone Evaluation Study Investigators: The effect of spironolactone on morbidity and mortality in patients with severe heart failure. N Engl J Med. 1999;341:709-717.
134 Pitt B, Remme W, Zannad F, et al. Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study Investigators: Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 2003;48:1309-1321.
135 The Cardiac Arrhythmia Suppression Trial II (CAST II) Investigators. Effect of the antiarrhythmic agent moricizine on survival after myocardial infarction. N Engl J Med. 1992;327:227-233.
136 The Cardiac Arrhythmia Suppression Trial (CAST) Investigators. Preliminary report: Effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N Engl J Med. 1989;321:406-412.
137 Stanton MS, Prystowsky EN, Fineberg NS, et al. Arrhythmogenic effects of antiarrhythmic drugs: A study of 506 patients treated for ventricular tachycardia or fibrillation. J Am Coll Cardiol. 1989;14:209-215.
138 IMPACT Research Group. International mexiletine and placebo antiarrhythmic coronary trial: Report on arrhythmia and other findings. J Am Coll Cardiol. 1984;4:1148-1163.
139 Coplen SE, Antman EM, Berlin JA, et al. Efficacy and safety of quinidine therapy for maintenance of sinus rhythm after cardioversion: A meta-analysis of randomized control trials. Circulation. 1990;82:1106-1116.
140 Waldo AL, Camm AJ, deRuyter H, et al. Effect of d-sotalol on mortality in patients with left ventricular dysfunction after recent and remote myocardial infarction (SWORD). Lancet. 1996;348:7-12.
141 Cairns JA, Connolly SJ, Roberts R, Gent M. Randomized trial of outcome after myocardial in patients with frequent or repetitive ventricular premature depolarizations: CAMIAT. Lancet. 1997;349:675-682.
142 Julian DG, Camm AJ, Frangin G, et al. Randomized trial of effect of amiodarone on mortality in patients with left ventricular dysfunction after recent myocardial infarction: EMIAT. Lancet. 1997;349:667-684.
143 Doval HC, Nul DR, Grancelli HO, et al. GESICA-GEMA Investigators: Nonsustained ventricular tachycardia in severe heart failure: Independent marker of increased mortality due to sudden death. Circulation. 1996;94:3198-3203.
144 Massie BM, Fisher SG, Radford M, et al. Effect of amiodarone on clinical status and left ventricular function in patients with congestive heart failure. Circulation. 1996;93:2128-2134.
145 Piccini JP, Berger JS, O’Conner CM. Amiodarone for the prevention of sudden cardiac death: A meta-analysis of randomized controlled trials. Eur Heart J. 2009;30(10):1245-1253.
146 Buxton AE, Lee KL, Fisher JD, et al. A randomized study of the prevention of sudden death in patients with coronary artery disease. N Engl J Med. 1999;341:1882-1890.
147 Moss AJ, Hall WJ, Cannom DS, et al. Multicenter automatic defibrillator implantation trial investigators: Improved survival with an implanted defibrillator in patients with coronary disease at high risk for ventricular arrhythmia. N Engl J Med. 1996;335:1933-1940.
148 Moss AJ, Zareba W, Hall WJ, et al. Prophylactic implantation of a defibrillator in patients with myocardial infarction and reduced ejection fraction. N Engl J Med. 2002;346:877-883.
149 Bigger JT. Prophylactic use of implantable cardiac defibrillators in patients at high risk for ventricular arrhythmias after coronary artery bypass graft surgery. N Engl J Med. 1997;335:1569-1575.
150 Hohnloser SH, Kuck KH, Dorian P, et al. DINAMIT Investigators: Prophylactic use of an implantable cardioverter-defibrillator after acute myocardial infarction. N Engl J Med. 2004;351(24):2481-2488.
151 Domanski MJ, Saksena S, Epstein AE, et al. Relative effectiveness of the implantable cardioverter-defibrillator and antiarrhythmic drugs in patients with varying degrees of left ventricular dysfunction who have survived malignant ventricular arrhythmias. J Am Coll Cardiol. 1999;34:1090-1095.
152 Moss AJ. Implantable cardioverter defibrillator: The sickest patients benefit the most. Circulation. 2000;101:1638-1640.
153 Bardy GH, Smith WM, Hood MA, et al. An entirely subcutaneous implantable cardioverter-defibrillator. N Engl J Med. 2010;363(1):36-44.
154 Bogun F, Good E, Riech S, et al. Role of Purkinje fibers in post-infarction ventricular tachycardia. J Am Coll Cardiol. 2006;48:2500-2507.
155 Nogami A, Sugiyasu A, Kubota S, Kato K. Mapping and ablation of idiopathic ventricular fibrillation from the Purkinje system. Heart Rhythm. 2005;2:646-649.
156 Bansch D, Oyang F, Antz M, et al. Successful catheter ablation of electrical storm after myocardial infarction. Circulation. 2003;108:3011-3016.
157 Haïssaguerre M, Extraminana F, Hocini M, et al: Mapping and ablation of ventricular fibrillation associated with long QT and Brugada syndromes, Circulation 48:2500–2507, 2003
158 Rea TD, Page RL. Community approaches to improve resuscitation after out-of-hospital sudden cardiac arrest. Circulation. 2010;121(9):1134-1140.
159 Weaver WE, Hill D, Fahrenbruch CD, et al. Use of the automatic external defibrillator in the management of out-of-hospital cardiac arrest. N Engl J Med. 1988;319:661-666.
160 Cobb LA, Weaver WD, Fahrenbruch CE, et al. Community-based interventions for sudden cardiac death: Impact, limitations, and changes. Circulation. 1992;85:I98-I102.
161 Weaver WD, Copass MK, Hill DL, et al. Cardiac arrest treated with a new automatic external defibrillator by out-of-hospital first responders. Am J Cardiol. 1986;57:1017-1021.
162 Valenzuela TD, Roe DJ, Cretin S, et al. Estimating effectiveness of cardiac arrest interventions: A logistic regression survival model. Circulation. 1997;96:3308-3313.
163 Sayre MR, Berg RA, Cave DM, et al. American Heart Association Emergency Cardiovascular Care Committee): Hands-only (compression-only) cardiopulmonary resuscitation: A call to action for bystander response to adults who experience out-of-hospital sudden cardiac arrest: A science advisory for the public from the American Heart Association Emergency Cardiovascular Care Committee. Circulation. 2008;117:2162-2167.
164 Rho RW, Page RL. The automated external defibrillator. J Cardiovasc Electrophysiol. 2007;18:896-899.
165 Bardy GH, Lee KL, Mark DB, et al. HAT Investigators: Home use of automated external defibrillators for sudden cardiac arrest. N Engl J Med. 2008;358(17):1793-1804.
166 Bernard SA, Gray TW, Buist MD, et al. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med. 2002;346:557-563.
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]
Chapter 47 Sudden Cardiac Death
Definition
The term sudden cardiac death refers to unexpected natural death from a cardiovascular cause within a short period, generally less than 1 hour from the onset of symptoms.1 The time and mode of death are therefore unexpected.2 By definition, this occurs in individuals who do not have prior conditions that could be fatal in the short term. The use of 1 hour in this definition is arbitrary and therefore subject to different interpretations. It is also important to distinguish a primary arrhythmic cause of death from, for example, an episode of worsening heart failure that culminates in a life-terminating arrhythmia and from other causes of sudden death such as pulmonary embolism, cerebral infarction, and ruptured aneurysms. This distinction is important both for the proper identification of SCD and the study of this phenomenon. More recently, the use of the term sudden cardiac arrest (SCA, synonymous with SCD) has been advocated.
Epidemiology
SCD is a major cause of death in North America and other Western societies, accounting for 10% of all deaths and up to 50% of heart disease–related deaths.3 The incidence of SCD (emergency medical services [EMS]–assessed, out-of-hospital cardiac arrests) in the United States is generally estimated to be 95.7 per 100,000 person years.4 Approximately 60% of out-of-hospital cardiac deaths are treated by EMS personnel.5 Among these EMS-treated, out-of-hospital arrests, 23% have an initial shockable rhythm, and 31% of these receive bystander cardiopulmonary resuscitation (CPR).6 Median survival rate to hospital discharge after EMS-treated, out-of-hospital cardiac arrest with any first recorded rhythm is 7.9% and for ventricular fibrillation (VF) is 21%. However, such median statistics of survival do not reflect regional variations of community-specific survival rates. Of note, the quantification of SCD is so fraught with difficulties that no comprehensive record of all SCDs exists. Surrogate data often are used to estimate its incidence, especially in the out-of-hospital setting. This includes deaths from “coronary heart disease” and “cardiac arrest,” defined as coronary death that occurred within 1 hour of symptom onset in the out-of-hospital setting and without other probable causes of death.7
As medical interventions continue to evolve and improve, the incidence and distribution of risk factors that contribute to SCD continue to change. Although the morbidity and mortality rates of cardiovascular disease have declined in the past 30 years, much less improvement has been observed in the incidence of out-of-hospital cardiac arrests. However, the incidence of cardiac arrest with an initial recorded rhythm of VF specifically has decreased over time, which is a recent trend that reflects the overall decline in cardiac mortality.8,9 The reduction in the incidence of VF is likely multi-factorial in origin and is attributable to improvements in primary and secondary prevention of heart disease. Current epidemiologic data indicate that in developed countries, structural coronary arterial abnormalities and their consequences account for 80% of fatal arrhythmias.10,11 Dilated and hypertrophic cardiomyopathies account for the next largest group of SCD. The remainder are caused by a variety of other cardiac disorders such as congenital heart disease (CHD) and primary electrophysiological disorders.
The epidemiology of SCD in adolescents and young adults (aged 10 to 30 years) is distinctly different from that in the adult population. The incidence of SCD in this population is two orders of magnitude less than in the adult group (1 per 100,000 vs. 1 per 1000 individuals annually).12 Although coronary atherosclerosis accounts for the majority of cases of SCD in individuals older than 40 years, it is an uncommon cause in the younger age group.13 Instead, causes such as hypertrophic cardiomyopathy, myocarditis, right ventricular dysplasia, anomalous coronary arteries, Brugada syndrome, long QT syndrome (LQTS), idiopathic VF, and commotio cordis are the underlying etiologies in this group. Because some of these conditions are genetically determined, a modest age inverse relationship seems to exist in this group, with adolescents having a somewhat higher mortality risk compared with young adults.11 A unique subgroup in this younger population is composed of competitive athletes, among whom the rate of SCD currently is approximately 0.4 to 0.6 per 100,000 person-years.14,15 The large numbers of SCD and the wide range of survival rates have significant potential implications for public health; for example, in the United States and Canada, an additional 7500 lives would be saved if survival from SCD could be increased to 12%.
Mechanisms
From a clinical perspective, the causes of SCD can be divided into two broad categories: (1) ventricular tachyarrhythmias (VA) and (2) pulseless electrical activity (PEA) and asystole. The National Registry of CPR is a prospective, multi-site, in-hospital resuscitation registry sponsored by the American Heart Association. In a study using this database, of the 51,919 index arrests evaluated in 411 centers, pulseless VT was diagnosed in 7%, VF in 17%, PEA in 37%, and asystole in 39%.16 Subsequent ventricular tachycardia (VT) or VF occurred in 26% of patients with an initial documented rhythm of PEA and in 25% of patients with asystole. Thus VT or VF was seen in 44% of all adult in-hospital cardiac arrests. This must be compared with out-of-hospital cardiac arrests, where among cases of EMS-assessed cardiac arrest, the incidence of VT or VF was 13% and PEA and asystole were 33% and 47%, respectively, for cases in which the initial rhythm was unknown, not determined, or not analyzed by EMS.8 A factor that has important implications in out-of-hospital cardiac arrests (as opposed to in-hospital cardiac arrests) is the median time of 7.24 minutes from call to arrival of first advanced life support.8
Pathophysiology
In recent years, significant advances in the understanding of the mechanism of VF have taken place. Controversy exists regarding whether VF is maintained by wandering wavelets with constantly changing re-entrant circuits or by a mother rotor that consists of a sustained and stationary re-entrant circuit that, in turn, gives rise to variable, less-organized daughter wavelets spreading through the rest of the ventricle.17,18 Certain anatomic structures can serve as anchors for rotors, allowing stability within the re-entrant circuit.19 Weiss et al have reported from their studies on porcine hearts that these anatomic sites can be papillary muscles, blood vessels, Purkinje fibers, or locations near the interventricular septum.20 Studies have suggested that Purkinje fibers play an important role in the initiation of VF, whereas others have suggested that they play a role in the maintenance of VF.21,22 Predispositions to SCD may also occur at a cellular level. Ion channelopathies can initiate various electrical disturbances, ranging from torsades de pointes in LQTS to that of idiopathic VF in Brugada syndrome.1,23 Certain mutations are significant enough that their mere presence is associated with a very high risk of SCD. However, other mutations (e.g., the recessive long QT mutation in the HERG gene) may, by themselves, be insufficient to cause SCD but may predispose patients to torsades de pointes in the presence of other factors such as drugs that prolong the Q-T interval.
The role of ischemia as an initiating factor for ventricular arrhythmias has been extensively investigated and is particularly relevant because it is a frequent etiology implicated in SCD. Ischemia causes acute changes at the cellular level that alter local conduction velocity and refractoriness. The resulting dispersion of conduction and repolarization establishes an environment that is ripe for re-entry. In animal experiments, within the first few minutes after coronary occlusion, an arrhythmogenic period that slowly abates after 30 minutes has been observed. These first 30 minutes can be divided broadly into the first 10 minutes, during which the changes are caused by direct ischemic injury, and the second 20 minutes, during which either arrhythmogenicity occurs because of either reperfusion or the evolution of injury in the various layers of the myocardium.24,25 Local changes occur in the ischemic myocardium; these include decrease in local tissue pH to less than 6, increase in interstitial potassium (K+) levels to greater than 15 mmol/L, increases in intracellular calcium (Ca2+), and other neurohormonal changes. All these factors contribute to the altered electrophysiological properties of tissue, including slowed conduction velocity, reduced excitability and prolonged refractoriness, reduced cell-to-cell coupling, and even spontaneous electrical activity.26 In addition to the local micro–re-entrant and macro–re-entrant circuits that may be generated, regional increases in automaticity and triggered activity also occur because of afterdepolarizations.
Disease States Leading to Sudden Cardiac Death
Because the majority of cases of SCD result from CAD, it is not surprising that the risk factors for SCD mirrors those of CAD. The incidence of SCD increases with age and is more common in men than women.27 The incidence of SCD tends to be higher in whites than in other racial groups. Classic coronary risk factors have been noted to be associated with SCD in various studies such as the Framingham Study and the Paris Prospective Study. These risk factors include hypertension, diabetes, high cholesterol levels, smoking, lack of regular exercise, and structural changes such as left ventricular hypertrophy.28–31 These factors are prevalent but are limited by their moderate individual positive predictive value. Left ventricular dysfunction has been shown to be a strong independent predictor of SCD in both ischemic and nonischemic cardiomyopathies. Of note, in patients with severely decreased left ventricular function and advanced heart failure, competing causes of SCD such as electromechanical dissociation and asystole exist.
Observations from population-based studies demonstrate a marked increase in the risk of SCD in first-degree relatives of SCD victims. In a study from Seattle, first-degree relatives of patients with SCD (before age 65 years) had 2.7-fold higher risk of SCD compared with age-matched and gender-matched controls after adjustment for risk factors.32 A Dutch case-control study demonstrated that patients with VF during myocardial infarction (MI) are more likely to have a family history of SCD than those with MI but no VF.33 Polygenic traits leading to SCD and monogenetic SCD syndromes such as LQTS, Brugada syndrome, hypertrophic cardiomyopathy, and arrhythmogenic right ventricular dysplasia contribute to the genetic component of SCD.34 More recently, research has begun to elucidate the role of newly identified genetic variations at the population level.
Coronary Artery Disease
Approximately 80% of patients who experience SCD have coronary atherosclerotic arterial disease as an underlying substrate. In survivors of SCD, critical flow-limiting coronary stenoses are found in approximately 40% to 86% of patients, depending on the age and gender of the population.29,30,35 Although less than 50% of the resuscitated patients have evidence of an acute MI, autopsy studies have revealed that a recent occlusive coronary thrombus can be found in 20% to 95% of victims of SCD.31,36,37 The temporal pattern of SCD closely parallels the patterns of MI and acute ischemic events. In addition, healed infarctions are found in 40% to 75% of hearts of SCD victims at autopsy.36,38–40 The extent to which superimposed acute ischemia plays a role in the pathogenesis of SCD is unclear in patients who do not develop enzymatic or electrocardiographic evidence of an acute MI. However, rapid fibrinolysis of a ruptured plaque could occur such that no visible evidence remains at autopsy. Similarly, cholesterol-laden plaque could conceivably rupture and embolize microscopic debris into distal coronary vessels that could lead to microscopic necrosis and ventricular arrhythmias and yet not be visible at autopsy.41 Because the vast majority of patients die within minutes of onset of symptoms, histologic or enzymatic evidence of ischemia or infarction may be difficult to ascertain. In addition to the most common forms of CAD, nonatherosclerotic CAD includes congenital malformations such as anomalous origin of coronary arteries; inflammatory arteritis also can lead to SCD. These disorders typically manifest early in life but are relatively uncommon.
Dilated Cardiomyopathy
Idiopathic dilated cardiomyopathy accounts for approximately 10% of cases of SCD in the adult population (Figure 47-1). Depending on the severity of the myopathy, the annual incidence can range from 10% to 50%.36 Bundle branch reentrant VT appears to represent an important cause of VAs in this population.37 However, as the myopathy progresses and congestive heart failure worsens, the incidence of VT or VF decreases, and the primary terminal event more frequently becomes electromechanical dissociation or asystole.42 As with ischemic heart disease, the overall left ventricular ejection fraction (LVEF) is an important prognostic factor. Worsening New York Heart Association (NYHA) functional class and the occurrence of syncope are both important clinical prognostic factors for SCD in patients with dilated cardiomyopathy.40
Hypertrophic Cardiomyopathy
Hypertrophic cardiomyopathy (HCM) is now regarded as the most common cause of SCD in young people, including competitive athletes (Figure 47-2).43–45 The annual mortality rate in patients with HCM in select regional and community-based cohorts is thought to be approximately 1%. The architectural myocardial fiber disorganization, scarring, and the presence of microvascular disease likely account for the proarrhythmic substrate. The conventional risk factors for HCM assume greater weight in patients younger than 50 years and include family history of one or more HCM-related form of SCD, more than one episode of unexplained recent syncope, massive LVH (thickness ≥30 mm), nonsustained VT on 24-hour Holter monitoring, and hypotensive or attenuated blood pressure response to exercise. Although implantable cardioverter defibrillators (ICDs) for primary prevention in this population usually require meeting at least two of these criteria, the need for ICD interventions for VT or VF in patients with single risk factors must be kept in mind when deciding about ICD implantation in this population.46 Of the above criteria, nonsustained VT and blood pressure response have poor positive predictive value.47 It has recently been demonstrated that certain mutations such as nonsarcomeric LAMP2 cardiomyopathy, double sarcomere mutations, and delayed enhancement on cardiac magnetic resonance imaging (MRI) are associated with SCD.48–51 Conversely, the apical variant of hypertrophic cardiomyopathy has been demonstrated to carry a relatively lower risk for SCD.52
Long QT Syndrome
LQTS (Figure 47-3) is a primary cardiac arrhythmogenic disorder typically characterized by prolongation of the Q-T interval corrected for heart rate (QTc) and abnormal T waves. Subjects with LQTS may present with a nearly normal electrocardiogram (ECG) or with a prolonged Q-T interval. The syndrome is associated with a specific ventricular arrhythmia called torsades de pointes and often presents as recurrent syncope. Hundreds of mutations have been identified, and these have been described in up to 12 genes.47 This is most often caused by decreased outward K+ current, IKs (LQT1, LQT5) or IKr (LQT2, LQT6), or by enhanced activity of mutant inward sodium (Na+) current (LQT3). Polymorphic VT associated with a prolonged Q-T interval is believed to be initiated by early afterdepolarizations (EADs) in the Purkinje system and maintained by transmural re-entry in the myocardium. Clinical presentations vary with the specific gene affected and the specific mutation. Some patients with LQTS mutations may not manifest any phenotypic abnormality. In high-risk LQT1 and LQT2, patients should be routinely managed with β-blockers as a first line-therapy and should be referred for primary ICD implantation if they become symptomatic during therapy or when compliance or intolerance to medical therapy is a concern.53 Patients with LQT3, those with frequent and recent syncope, those with excessive Q-T prolongation (>550 ms), and women with LQT2 and a QT interval greater than 500 ms may be at increased risk and may require ICD implantation.
Congenital Short QT Syndrome
Congenital short QT syndrome (SQTS) is a relatively recently described disorder characterized by a very short Q-T interval (<320 ms) and a susceptibility to atrial fibrillation (AF) and VF. At electrophysiology study, short atrial and ventricular refractory periods with easily inducible AF and polymorphic VT have been identified. Gain-of-function mutations in genes encoding K+ channels have been identified, which explains the abbreviated repolarization seen in this condition. The suggested treatment is ICD implantation. The ability of quinidine to prolong the Q-T interval has the potential to be effective pharmacological therapy.54
Arrhythmogenic Right Ventricular Cardiomyopathy
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a progressive, heritable myocardial disorder with a broad phenotypic spectrum that can present with VT, SCD, or both (Figure 47-4). The classic form of the disease has an early predilection for the right ventricle, but left-dominant and biventricular subtypes have been also recognized.55 Desmoplakin and plakophilin-2 are two desmosomal genes that have been implicated in ARVC. A familial form, originally described in people living on the Greek island of Naxos, can present with cardiomyopathy in association with palmo-plantar keratoderma (Naxos disease) and is caused by a defect in the gene for a cellular structural element, plakoglobin.56 ARVC causes progressive fatty replacement of the ventricular wall. Cardiac MRI is a highly sensitive imaging tool to identify the presence and degree of fatty infiltration in the myocardium; however, because this is not specific, formal criteria have been proposed for the diagnosis of ARVD. Phenotypic heterogeneity and the nonspecific nature of its associated features complicate clinical diagnosis, which often requires multiple tests rather than a single test.57 The Revised Task Force criteria for the diagnosis are specific and have helped reduce diagnostic ambiguity, but the sensitivity is low, especially in the “concealed” phase of ARVC.57
Brugada Syndrome
Brugada syndrome is associated with right ventricular conduction delay and ST elevation in the right precordial leads, characterized by syncope and premature SCD caused by VF (Figure 47-5). This syndrome appears to be responsible for a sudden death syndrome seen among Southeast Asian men.58,59 ECG manifestations of the syndrome can often be dynamic. Typical ST-segment changes, if absent at baseline, can be revealed by administration of sodium channel–blocking agents such as ajmaline or procainamide. Mutations in the SCN5A gene cause loss of function in the Na+ current Ina that can lead to accentuation of unopposed Ito currents in the right ventricular epicardium. This leads to loss of the action potential “dome,” resulting in heterogeneity of repolarization, and phase 2 re-entry that precipitates VT or VF. The strategy for risk stratification in Brugada syndrome is controversial with respect to the role of electrophysiology testing in patients who do not present with SCD. ICD is the only treatment with demonstrated efficacy in Brugada syndrome. In general, ICD implantation is recommended for patients with symptoms and for asymptomatic patients with inducible ventricular arrhythmias, especially if they have a spontaneous type I ECG pattern. In asymptomatic patients without a family history of SCD and whose type I ECG pattern is documented only after the administration of sodium channel blockers, periodic follow-up is recommended, but an EPS for risk stratification is not required.60,61 The role of EPS, however, still remains controversial, with some studies not supporting its role in risk stratification.57
Wolff-Parkinson-White Syndrome
Wolff-Parkinson-White (WPW) syndrome can lead to SCD if the accessory pathway is able to conduct rapidly in the antegrade direction in the presence of rapid atrial arrhythmias such as AF (Figure 47-6). More recently, a long-term follow-up of patients with WPW syndrome reported a mortality rate of 0.02% per year.62 Two additional studies with more than 4000 patient-years of follow-up, estimated mortality rates at approximately 0.05% per year.63,64 However, a study from Italy that prospectively followed up patients with WPW syndrome for 3 years, recorded a much higher event rate (defined as death or potentially lethal arrhythmia recorded on monitoring) at 0.5% per year.65 Predictors for the development of VF include rapid ventricular response during induced AF, with the shortest R-R interval of less than 240 ms and short antegrade pathway refractory periods. High-risk and symptomatic patients are treated with catheter ablation with very high success rates overall.66
Catecholaminergic Polymorphic Ventricular Tachycardia
Patients with catecholaminergic polymorphic ventricular tachycardia (CPVT) can present with exercise-induced syncope, SCD, or both in the absence of any structural heart disease or prolonged Q-T interval. Inheritance can be autosomal dominant or recessive. These patients often have normal resting ECGs, which makes the diagnosis difficult. Stress-related bi-directional VT has been described classically, but patients can present with polymorphic VT or even frequent ventricular ectopy. Responsible mutations have been shown to reside in the cardiac ryanodine receptor and calsequestrin genes.67–70 Treatment modalities include ICD placement along with β-blocker therapy for symptomatic patients.
Early Repolarization
A multi-center study recently documented an association between idiopathic VF and the presence of early repolarization (ER) abnormalities in inferolateral leads (Figure 47-7).71 Repolarization changes have also frequently been observed in inferolateral precordial leads in patients with Brugada syndrome, mimicking the abnormalities typical of the population presented in the study of Haïssaguerre et al, suggesting a possible overlap of these conditions.72 This subset of idiopathic VF continues to be further studied and, in addition to ICDs, newer therapies such as quinidine and isoproterenol have been noted to play a role in controlling VF storms that can occur in these patients.
Congenital Heart Disease
Patients with CHD represent an anatomically heterogeneous patient group in which risk stratification can be difficult. Current guidelines indicate ICD implantation for survivors of SCD; ICD is considered a reasonable therapy for patients with sustained VT that is not amenable to either ablation or surgery and for those with unexplained syncope and impaired ventricular function. Tetralogy of Fallot (ToF) represents one of the commonly encountered conditions in adult CHD patient populations. Older age at surgery, residual hemodynamic lesions with right heart failure, impaired LVEF, complex ventricular ectopy, inducible ventricular arrhythmias at EPS, and prolongation of the QRS complex represent some of the associations noted in SCD in patients with ToF.73–75
Noncompaction of the left ventricle is a rare congenital cardiomyopathy characterized anatomically by excessive prominent trabeculae and deep intertrabecular recesses in the left ventricle without other major congenital cardiac malfunction. Ventricular arrhythmias and SCD are known to occur, and ICD therapy often is indicated.76–78
Commotio Cordis
The term commotio cordis refers to a blunt, nonpenetrating, and usually innocent-appearing chest blow that can cause VF. The location of the blow to the chest (directly over the heart) and its timing relative to the cardiac cycle (on the upstroke of the T wave, 10 to 20 msec before its peak) are the primary determinants of commotio cordis.79,80
Other Genetic Associations in Patients with Structurally Normal Hearts
SCD likely has a strong genetic component, but only a fraction of the genetic variants that underlie the risk are known; the allelic architecture of SCD thus remains poorly defined.81 Genome-wide association studies have indicated that common variants in NOS1AP are associated with Q-T interval duration and SCD risk in general populations.82 The common polymorphism S1103Y-SCN5A is disproportionately represented in patients with arrhythmia and black patients who have experienced SCD.83 These variants by themselves are unlikely to be sufficient causes of SCD.
Risk Stratification for Sudden Cardiac Death
In SAECG, the terminal part of the QRS complex is evaluated for microvolt potentials that reflect delayed activation in the scarred myocardial substrate, thereby detecting areas of slow conduction.84
[/not-level-membership-for-cardiovascular-category]
