[level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 13
Subacute and Riedel’s Thyroiditis
Subacute Thyroiditis
The term subacute thyroiditis (SAT) describes a self-limited inflammatory disorder and the most common cause of thyroid pain, probably of viral origin.1–5 It was first reported by Mygind6 in 1895, who described 18 cases of “thyroiditis akuta simplex.” The name De Quervain traditionally has been associated with this condition, however, probably because he described the pathology of this disorder thoroughly in 19047 and again in 1936.8 SAT occurs in 5% of patients with clinical thyroid disease9 and frequently follows an upper respiratory tract infection. Its incidence correlates with the peak incidence of enterovirus.10 Other viruses, such as Epstein-Barr virus and cytomegalovirus, also have been reported, but so far clear evidence for a viral cause is still lacking.11 There is a strong preponderance of women over men with this condition.1
SAT has a multiplicity of synonyms, some reflecting misconceptions regarding the etiology or pathology of the condition. These include De Quervain’s thyroiditis, viral thyroiditis, granulomatous thyroiditis, acute or subacute diffuse thyroiditis, acute simple thyroiditis, noninfectious thyroiditis, struma granulomatosa, pseudogranulomatous thyroiditis, giant cell thyroiditis, pseudo–giant cell thyroiditis, migratory “creeping” thyroiditis, and pseudotuberculous thyroiditis. The term subacute thyroiditis connotes a temporal quality that might apply to any inflammatory process of intermediate severity and duration. As the term is generally employed, however, it specifically includes only patients showing a pseudogranulomatous pathologic appearance in the thyroid gland (which is virtually specific for the disease) and a characteristic clinical syndrome in which the painful tender goiter also is associated with considerable malaise, fever, and evidence of thyroid dysfunction (described more fully later).1–5 It generally is distinguishable from a similar disorder, painless or silent thyroiditis, which disturbs thyroid function in a manner similar to SAT but without pain or tenderness and with a different pathologic appearance.
Incidence
Few epidemiologic studies of SAT have been reported.10,14–22 Compared with other thyroid diseases, SAT is uncommon, occurring at the rate of about 1 case per 5 cases of Graves’ disease and 1 case per 15 or 20 cases of Hashimoto’s thyroiditis.19 Although the cause is most likely viral, SAT, similar to all other thyroid conditions, occurs most commonly in women who are 40 to 50 years old. The reported female-to-male ratio is 3 to 6 : 1.19 it has been noted as a rare cause of hyperthyroidism in pregnancy.20 It is rare in children and seems to occur in any season of the year,10,22 with a trend toward more cases in fall and spring.22 Familial or geographic aggregation of cases is seldom noted. SAT has been reported most commonly from the temperate zone, having been observed in North America, Europe, and Japan. Recently, a few cases were reported in Western Saudi Arabia,21 although it is rarely reported from many other parts of the world. Associated autoimmune conditions do not seem more common than autoimmune conditions observed in the general population.22
Although complete recovery is the rule, recurrence after several years has been reported.24–27 In one study, four recurrent episodes of SAT occurred in 3 of 222 patients (1.4%). The recurrent episodes were similar to the first episodes of SAT.27 In a larger study that evaluated data for 3344 patients with SAT between 1970 and 1993, SAT recurred in 48 of 3344 patients (1.4%) (mean 14.5 ± 4.5 years after the first episode). Five patients experienced a third episode (mean 7.6 ± 2.4 years after the second episode).27 Another cohort study showed a 4% recurrence rate after many years.22 Theoretically, late recurrence possibly occurs after the disappearance of immunity to the previous viral infection. During an evaluation of subtypes of hypothyroidism over a 4-year period in Denmark, an incidence of subacute thyroiditis of 1.8% was found in a cohort of 685 patients with hypothyroidism.28
Etiology
In 1952, Fraser and Harrison29 were the first to propose that SAT represents a viral infection of the thyroid gland. Since then, considerable indirect evidence suggests that SAT is most likely the result of a viral infection30–32 that rarely recurs after a complete recovery, possibly because of immunity to the offending virus.
Clinically, the disease has several characteristics typical of viral infections, including a typical prodrome with myalgias, malaise, and fatigue; absence of leukocytosis; and usually a self-limited course.1–5 Additionally, clusters of the disease have been reported during outbreaks of viral infection.1–5,10 It has been described in association with mumps, measles,1 influenza,1 the common cold,1 adenovirus,1 infectious mononucleosis,1,12 coxsackievirus,1 myocarditis,1 cat-scratch fever,1 St. Louis encephalitis,1 hepatitis A,18 parvovirus B19 infection,19 and cytomegalovirus infection.11,13
In an extensive study reported by Volpé and colleagues,33 32 of 71 patients with SAT, who had no evidence of specific viral disease, showed at least fourfold increases in viral antibodies during the thyroid illness. These viral antibodies included antibodies to coxsackievirus, adenovirus, influenza virus, and mumps virus. Coxsackievirus antibodies were found most commonly, and the changes in their titers most closely approximated the course of the disease. In a later study of 10 patients in Singapore, no such antibodies were observed, however.34 It is possible that the presence of these antibodies may not reflect pathogenic significance, but instead may result from an anamnestic response to the inflammatory thyroid lesion. The thyroid responds with the clinical picture of thyroiditis after invasion by a variety of viruses, and a variety of agents may be causative in the syndrome of SAT.
Certain nonviral infections, such as malaria and Q fever, have been associated with a clinical syndrome that at least simulates SAT.1 The significance of these observations remains to be determined. In addition, a case of SAT occurring simultaneously with giant cell arteritis has been reported.35 Several cases of SAT that developed during interferon-α treatment for hepatitis C have been described,36–38 and more recently, a case of SAT that developed after long-term immunosuppression and lithium therapy following an allogeneic bone marrow transplant was reported.39
Several autoimmune phenomena have been described in SAT. Thyroid autoantibodies (antithyroglobulin and antithyroid peroxidase antibodies) have been found in 42% to 64% of patients with SAT.33 In most of these patients, the antibody titer gradually decreased and remained low or disappeared as the disease faded. Thyroid-stimulating hormone (TSH) receptor antibodies also have been reported in patients with SAT,40–42 although changes in antibody titer did not correlate with disease activity.40 Autoantibodies to several novel, uncharacterized thyroid antigenic determinants were found in eight of nine patients with SAT tested.43 These autoantibodies persisted, and their level did not decrease over 39 months after onset of SAT. These antibodies likely arise secondary to the damage caused by viral infection of the thyroid gland because they are typically polyclonal in nature.43
There is evidence that T-cell-mediated immunity against thyroid antigens may play a role in the pathogenesis of SAT. During the initial phase of the disease, the gland is infiltrated by T cells, and sensitization of T cells against thyroid antigens has been shown in such patients.44–46 This sensitization was transitory, however, and likely represented a secondary immune response to the inflammatory release of antigen induced by the viral infection of the gland.1
It has been suggested that thyroid-destructive events in the course of SAT may trigger, under a genetic background, thyroid autoimmune disease of various kinds.47,48 Patients with a previous history of SAT, in about 1% of cases,2 may develop hypothyroidism as a consequence of previous thyroid damage. The occurrence of Graves’ disease after SAT also has been described, although such evidence seems to be extremely rare, with fewer than 20 cases reported in the literature.49–53
Although SAT is shown to be associated with thyroid autoimmune phenomena, after recovery all immunologic phenomena should disappear. This is in contrast to the continuing presence of these abnormalities in autoimmune thyroid disease.54 The transitory immunologic markers observed during the course of SAT seem to be secondary to the release of antigenic material from the thyroid and seem to be a normal, physiologic response to the inflammatory destruction of the gland.55
In light of the previous observations, the lack of any direct evidence, and because it is rare for SAT to progress to either Graves’ disease or Hashimoto’s disease, the corollary is still consistent with the view that antigen-driven events can produce a transient immunologic disturbance, but does not, or is most unlikely to, culminate in chronic autoimmune thyroid disease. It is possible that the illness of SAT might act as a nonspecific stress acting on the immune system to precipitate Graves’ disease in a favorable genetic background.54
An association between SAT and HLABw35 has been noted in all ethnic groups tested.56–59 This haplotype seems to confer an unusual susceptibility to SAT, perhaps because it allows one or more viruses to trigger an immune response directed against thyroid tissue.60 Histocompatibility studies show that 72% of patients with subacute thyroiditis manifest HLA-Bw35.57–60 Familial occurrence of subacute thyroiditis associated with HLA-B35 has been reported.61,62 Another HLABw67 was found in 87% of a Japanese population and correlated with a seasonal appearance and a mild course of disease. Thus the susceptibility to subacute thyroiditis is genetically influenced, and it has also been suggested that subacute thyroiditis might occur by transmission of viral infection in genetically predisposed individuals.13
Clinical Features
Half of patients have a history of an antecedent of upper respiratory infection, followed in days or weeks by the clinical manifestations of SAT itself.1–5,22,63 SAT begins with a prodrome of generalized myalgias, pharyngitis, low-grade fever, and fatigue. The patient notes pain of varying degrees in the region of the thyroid gland. This pain may involve one lobe, part of a lobe, or the whole thyroid, and it typically radiates from the thyroid gland to the angle of the jaw and to the ear of the affected side. If not bilateral initially, the pain and tenderness often spread to the uninvolved side of the thyroid within days or weeks. It also may radiate to the anterior chest or may be centered only over the thyroid. Moving the head, swallowing, or coughing may aggravate it. Transient vocal cord paresis may occur.23
Symptoms of mild to moderate hyperthyroidism occur in the early phase in most patients.22 Fifty percent of patients have symptoms of thyrotoxicosis, and the usual symptoms of nervousness, tremulousness, weight loss, heat intolerance, and tachycardia predominate.1–5,63–66 On physical examination, most patients appear uncomfortable and flushed, with variable fever. The thyroid gland may be only slightly to moderately enlarged, with one lobe larger than the other. The consistency of the involved area is usually firm or hard. With time or treatment, the thyroid tenderness subsides, and the goiter generally disappears within several weeks or months. Signs of mild to moderate hyperthyroidism are present in 50% of cases. About 8% to 16% of patients with this condition are noted to have a preexisting goiter. Cervical lymphadenopathy is rare.
In most patients, SAT lasts 2 to 4 months, although it may last 1 year. When the course is prolonged, the major manifestation is persistent, painful, tender thyroid enlargement, the thyrotoxicosis almost always having subsided earlier. Recurrences after recovery have been reported but are unusual, on the order of 2.3% per year26 or 4% over 21 years after the first episode.22
Sometimes hyperthyroidism may not be apparent clinically but can be detected by biochemical means.64 This situation is due to a disruptive process within the thyroid gland, with continuous leakage of the colloid into the interstitial spaces, where it is broken down into its component parts, liberating thyroid hormones, thyroglobulin, and other iodoamino acids into the circulation.26,64–74 Because the thyroid cells during this phase are virtually incapable of producing new thyroid hormone, the colloid that has been stored within the follicles is depleted within 2 to 3 months, resulting in a phase of transient hypothyroidism in patients in whom the process has persisted over the interval.75 Because disruption of the thyroid parenchyma can continue for months, hypothyroidism may persist for several weeks. As recovery continues, the follicles regenerate, the colloid is repleted, and normal thyroid function is restored. With recovery, the thyroid is reconstituted, repleted with colloid; thyroid function is restored; and a variable amount of interstitial fibrosis persists.1–5,64–75 This transient hypothyroidism may be subclinical or overt and occurs in about two thirds of patients.
SAT rarely progresses to permanent hypothyroidism.1–5,75,76 In these cases, progression may be due to total destruction of the thyroid, with consequent fibrosis. As mentioned before, in rare instances, the disorder may seem to culminate in autoimmune thyroiditis after recovery from SAT.48,51,53
Diagnosis
The typical painful SAT usually is obvious when the patient is first seen and should present no difficulties in diagnosis for the endocrinologist.67,68 In patients who have only a sore throat or ear pain, however, the diagnosis is less obvious, and many patients are initially misdiagnosed with pharyngitis.64 It is important that the thyroid gland be palpated carefully in patients presenting with upper respiratory infections or complaints of sore neck or throat or earache.
Eventually, patients with Hashimoto’s thyroiditis and a few with silent thyroiditis may present with a painful, tender thyroid enlargement that is indistinguishable from SAT.54 The radioactive iodine uptake is rarely as completely suppressed in Hashimoto’s thyroiditis as it is in SAT, and the titers of thyroid autoantibodies are usually high enough to suggest lymphocytic thyroiditis. Acute suppurative thyroiditis initially may mimic SAT, but with time, the findings of fever, more localized tenderness and swelling, and erythema over the involved area of the thyroid should become obvious. A rapidly growing anaplastic carcinoma77 of the thyroid or hemorrhage into a thyroid nodule can cause thyroid pain and tenderness. In anaplastic carcinomas, the lesion is usually obvious by virtue of its large size, adherence to adjacent structures, lymphadenopathy, and characteristic progressive course. Hemorrhage into a thyroid nodule presents with a localized nature, and the obvious nodule usually leads to the correct diagnosis.
The hallmark of SAT is a markedly elevated erythrocyte sedimentation rate. The serum thyroglobulin and C-reactive protein concentration are similarly elevated.78 The leukocyte count is normal or slightly elevated. Peripheral blood thyroid hormone concentrations are elevated, with ratios of thyroxine (T4) to triiodothyronine (T3) of less than 20, reflecting the proportions of stored hormone within the thyroid,79 and serum concentrations of thyrotropin are low or undetectable. Serum thyroid peroxidase antibody concentrations are usually normal. The 24-hour radioactive iodine uptake is low (<5%) in the toxic phase of SAT, distinguishing this disease from Graves’ disease. Color-flow Doppler ultrasonography also may help to make this distinction; in patients with Graves’ disease, the thyroid gland is hypervascular, whereas in patients with painful SAT, the gland is hypoechogenic and has low to normal vascularity.80
Laboratory and Imaging Findings
Dynamic changes in thyroid function studies occur with the onset of thyroid inflammation (Fig. 13-1). The destruction of the thyroid follicles results in release and breakdown of the colloid into the interstitial tissue and into the circulation of iodinated materials—protein, proteases, peptides, and amino acids. An increase in serum T4, T3, and thyroglobulin and in urinary iodine results.1–5,64,74
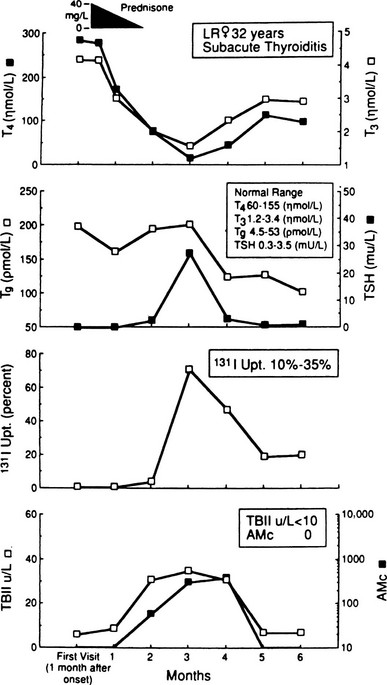
FIGURE 13-1 Salient laboratory features during the course of subacute thyroiditis. AMc, Antimicrosomal (antithyroperoxidase) antibody; T4, thyroxine; T3, triiodothyronine; TBII, thyrotropin-binding inhibitory immunoglobulin; Tg, thyroglobulin.
In addition, iodoproteins, such as thyroglobulin and iodoalbumin, are discharged from the gland into the circulation.69 Plasma thyroglobulin may remain elevated long after all other evidence of the inflammatory process has subsided.71 The decline in plasma T4 is exponential during the first week, and this phase of hyperthyroidism can continue only until the gland is depleted of its preformed colloid.73 TSH is usually undetectable in the hyperthyroid phase,72,73 and the TSH response to thyrotropin-releasing hormone, as expected, is diminished at this time.81,82
At the same time, the damage to the thyroid follicular cells results in impaired iodine transport; the 24-hour radioactive iodine uptake is characteristically suppressed to 0% to 1%, revealing a patchy and irregular distribution of the tracer.1–5,64,69,83,84 Even if only part of the gland is involved, the uptake may be similarly depressed as a result of suppression of pituitary TSH owing to the elevated levels of thyroid hormone.72,73 Increased perfusion is shown in studies with technetium-99m sestamibi during the acute stage of SAT. This increased uptake in the thyroid region suggests the inflammatory phase of this disease.85 SAT is one of the hyperthyroid conditions associated with high levels of thyroid hormones but a low radioactive iodine uptake, and such observations are characteristic in the early phase of this disorder. Under these circumstances, only minimal thyroid hormone biosynthesis is sustained, and what is produced leaks out.69
Evidently, thyroid cell damage reduces the ability of the gland to respond to TSH so that large doses of TSH generally do not cause a rise in the radioactive iodine uptake except when some parts of the gland are uninvolved.82 This lack of response to exogenous TSH administration persists during the first weeks of the disease, reflecting continuing thyroid cell impairment and failure of the iodide-concentrating mechanism. Also, the administration of perchlorate or thiocyanate generally does not cause release of excessive amounts of iodine from the gland.74
The erythrocyte sedimentation rate is characteristically elevated (often >100 mm/h) in SAT.1–5,86 If the test is normal or only slightly elevated, the diagnosis of SAT should be suspected. The leukocyte count is normal in about half of patients and elevated in the remainder1–5,8,86 and has been reported as high as 18 × 109/L. The leukocyte counts correlate with serum concentrations of granulocyte colony-stimulating factor.87 There may be a mild normochromic anemia, and an increase in α2-globulin frequently is seen as a nonspecific inflammatory response.88 Alkaline phosphatase and other hepatic enzymes may be elevated in the early phase.89 It has been suggested that SAT actually represents a multisystem disease also affecting the thyroid gland.90 There also are increases in serum ferritin,94 soluble intercellular adhesion molecule-1,95 selectin,96 and interleukin-697 levels during the inflammatory phase.
Ultrasound examinations show hypoechoic focal areas and can be used for guided fine-needle cytology.84–103 Magnetic resonance imaging of the thyroid also can help distinguish SAT from Graves’ disease during the hyperthyroid phase. The ADC values obtained from the diffusion-weighted images of the patients with Graves’ disease are significantly higher than the values of patients with SAT.104
Recovery Phase
As the process subsides, the serum T4, T3, and thyroglobulin levels decline, but the serum TSH level remains suppressed. The normal concentrations of sex hormone–binding globulin in the hyperthyroid phase probably reflect the short duration of exposure to increased thyroid hormone.105
Later, during the recovery phase, the radioiodine uptake becomes elevated with the resumption of the ability of the thyroid gland to concentrate iodide. The serum T4 concentration may fall below normal; the TSH level may become elevated. Usually, after several weeks or months, all the parameters of thyroid function return to normal. Restoration of iodine stores seems to be much slower and may take more than 1 year after the complete clinical remission.106,107 Ultimate recovery is the general rule. An occasional patient remains permanently hypothyroid.
Tests of thyroid antibodies are positive in a few cases; these develop several weeks after the onset and tend to decline and disappear thereafter.33,108 An antibody against an unpurified thyroid antigen persists for years, however, after clinical features have subsided.43 Also, as mentioned before, antibodies to the TSH receptor, either of the stimulating or of the blocking variety, may appear transiently without relationship to the thyroid functional state.40–42 In about 2% of patients, SAT may trigger autoreactive B cells to produce TSH receptor antibodies, resulting in TSH antibody–associated dysfunction.109
Pathology
From histologic examination, the process may be diffuse or irregular in its involvement, with various stages of the disease sometimes found within the same specimen.110 Initially, there is extensive follicular cell destruction, extravasation of colloid, and infiltration of lymphocytes and histiocytes. The lymphocytes and histiocytes tend to congregate around masses of colloid and coalesce into giant cells. With time, there is a variable degree of fibrosis, and areas of follicular regeneration are seen. After recovery, the thyroid appears normal except for minimal residual fibrosis.
The follicular cells sometimes virtually disappear, leaving a fine follicular lining. The initial phase is characterized by the appearance of neutrophils, followed by large mononuclear cells and lymphocytes (Fig. 13-2). The follicles appear much larger than normal, with disruption of the epithelial lining and hyperplasia of the surviving follicular cells. Histiocytes congregate around masses of colloid within the follicles and in the interstitial tissues, producing “giant cells.” Because often these giant cells actually consist of masses of colloid surrounded by large numbers of individual histiocytes, they should in such cases be termed pseudo–giant cells. True giant cells and granulomas also appear in this disease, however.111 Marked interstitial edema also is present with lymphocytic infiltration.
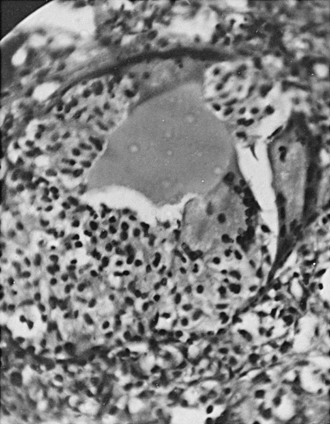
FIGURE 13-2 Pathologic findings in subacute thyroiditis. Note the severe destruction of the thyroid follicle, with the remaining colloid being surrounded by large numbers of histiocytes, giving the appearance of a giant cell (pseudo–giant cell). Marked interstitial edema is noted, with cellular infiltration and considerable destruction of the thyroid parenchyma.
The process often is irregularly distributed in either or both lobes.114 With recovery, the inflammatory reaction recedes, and a variable amount of fibrosis may appear. Areas of follicular regeneration are seen, but there is no caseation, hemorrhage, or calcification. The degree of recovery is generally virtually complete, aside from the residual fibrosis already mentioned. Only in rare instances is there complete destruction of the thyroid parenchyma leading to permanent hypothyroidism.
Thyroid tissue obtained by fine-needle aspiration biopsy (Fig. 13-3) often shows a mixed and polymorphous inflammatory infiltrate of neutrophils, lymphocytes, and large numbers of histiocytes, which can be misinterpreted. The features of SAT—epithelioid granulomas, multinucleated giant cells, and follicular cells with intravacuolar granules—can be identified.87,111–113
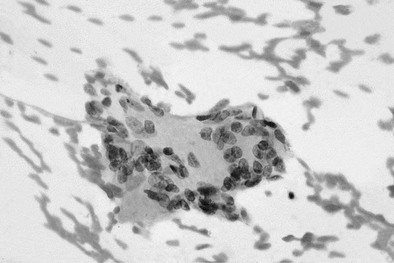
FIGURE 13-3 Fine-needle aspiration findings in subacute thyroiditis. Multinucleated giant cell with lymphocytes.
An immunohistochemical study of six cases showed the cellular composition of SAT.115 The giant cells were CD68 positive, thyroglobulin negative, and cytokeratin negative. Small lymphocytes in the granulomas are CD3-positive, CD8-positive, and CD45RO-positive cytotoxic T cells. In the nongranulomatous lesion, the follicles often were infiltrated by CD8-positive T lymphocytes, plasmacytoid monocytes, and histiocytes, resulting in disrupted basement membrane and rupture of the follicles. These findings suggest an intense cellular immune response in SAT. In addition, apoptotic cells are found in regenerating follicular cells in the area of granuloma and rarely found in sites of fibrosis, which might indicate a healing process.115
Treatment
In some patients, no treatment is required. In mild cases, the relief of symptoms can be achieved with nonsteroidal antiinflammatory drugs or aspirin (2 to 3 g/day).1–4 If this therapy fails, as it often does when the symptoms are severe, prednisone or another similar analogue of cortisol is prescribed.5,116,117 Relief of symptoms occurs often within 24 hours. The basic disease process may not be altered, but the inflammatory response is suppressed, allowing the pathologic process to run its now subclinical course.
Treatment with prednisone generally is begun with a single daily dose of 40 mg. Within 8 to 10 days, symptoms markedly decrease, and the dose can be tapered and completely stopped after 4 weeks. The relief of the tenderness in the neck is so dramatic as to be virtually diagnostic of the problem as being due to SAT. In most instances, exacerbations do not occur, and patients go on to full recovery. Sometimes symptoms flare up again, and the prednisone taper needs to be reversed.1–5 The recurrence rate of SAT after cessation of prednisone therapy is about 20%, but no difference has been found in routine laboratory data between recurrent and nonrecurrent groups of patients.118
There seems to be no significant difference in the incidence of mild thyroid failure between patients receiving corticosteroid therapy and patients receiving nonsteroidal antiinflammatory drugs. In contrast, long-term hypothyroidism requiring T4 therapy is significantly more common in the group receiving corticosteroid therapy, as shown in a more recent report.22 Corticosteroid therapy should be given to improve the symptoms and quality of life without an expectation of reducing long-term thyroid dysfunction.
During the initial phase, the patient may be thyrotoxic and need treatment with β-sympathetic blocking agents, such as propranolol. Sodium iopodate has been employed in the management of the hyperthyroidism of SAT.120 The treatment was effective in causing normalization of thyroid function, although the inflammatory state persisted for 6 weeks thereafter. There have been reports that the addition of T4 or T3 after repeated exacerbations can result in amelioration of the condition and prevent further recurrences.1–5 Thyroid hormone administration may be useful in situations in which the patient is not already hyperthyroid due to the release of thyroidal contents into the circulation. It is thought that TSH suppression would reduce the thyroid stimulation, which otherwise might prolong the inflammatory process. Antibiotics are of no value. Thiouracil and TSH have been reported to be beneficial, but such drugs have not found general favor.4 Because recovery is almost certain, thyroidectomy almost never needs to be recommended except in an unusual prolonged course with continuing local distress.3,4,119
Riedel’s Thyroiditis
In 1896, Riedel121–123 described a rare disorder of chronic sclerosing thyroiditis, occurring especially in women, causing pressure symptoms in the neck, which tends to progress inexorably to complete destruction of the thyroid gland. This disorder is characterized pathologically by dense fibrous tissue, which replaces the normal thyroid parenchyma and extends into adjacent tissues, such as muscles, blood vessels, and nerves.1,3,124,125 Synonyms for the term chronic invasive fibrous thyroiditis include Riedel’s struma, struma fibrosa, ligneous struma, chronic fibrous thyroiditis, and chronic productive thyroiditis. The presence of eosinophils has been shown histologically, suggesting a unique autoimmune response to fibrous tissue. It may include sclerosing mediastinitis, retroperitoneal fibrosis, pseudotumors of the orbit, and sclerosis of the biliary tract.126
Incidence
Riedel’s thyroiditis is exceedingly rare. Women are four times more likely to be affected than men, and it occurs most commonly between 30 and 50 years of age. In the Mayo Clinic series, it occurred approximately one fiftieth as frequently as Hashimoto’s thyroiditis.* The operative incidence over 64 years was 0.06%, and the overall incidence in outpatients was 1.06 per 100,000.127–130 Only 37 cases were found on review of histology of 57,000 thyroidectomies.127 In thyroidectomies performed for all disorders, an incidence between 0.03% and 0.98% was reported from a small group of centers.126
Etiology
Although there has been considerable debate as to whether Riedel’s thyroiditis is a primary inflammatory disorder of the thyroid gland, a variant of Hashimoto’s thyroiditis, or even end-stage SAT, current evidence suggests that Riedel’s thyroiditis might be a local manifestation of a systemic fibrosing disease.3,131–133 An autoimmune mechanism is suggested by (1) the presence of mononuclear cell infiltration and vasculitis within the fibrous tissue and the serum antithyroid antibodies present in so many patients134; (2) occasional reports of the coexistence of Riedel’s thyroiditis with autoimmune disorders such as Addison’s disease, type 1 diabetes, pernicious anemia, Graves’ disease, and Hashimoto’s thyroiditis136–139; and (3) the favorable response to glucocorticoid therapy.140
The autoantibodies are thought to be reactive to antigens released from destroyed thyroid tissue.134 The association with other autoimmune diseases is rare and probably coincidental,134–137 and the response to glucocorticoid therapy may be due to decreased production of cytokines with strong fibrogenic properties.140
The absence of other autoantibodies and the presence of a normal serum complement level and a normal lymphocyte subpopulation also are inconsistent with an autoimmune mechanism.141 When followed for many years, patients with Hashimoto’s thyroiditis, a common disease, almost never progress to Riedel’s struma, which is a rare entity. The aforementioned evidence does not confirm an autoimmune basis for the disease, and most cases of Riedel’s struma are clearly unrelated to such autoimmune disease.
It has been suggested that the key event in this disorder might be proliferation by fibroblasts, in turn induced by cytokine production from B or T lymphocytes or both.142 Consistent with this suggestion is the finding by Many and associates142 of histologic modifications similar to modifications observed in Riedel’s thyroiditis in nonobese diabetic (NOD) mice during the development of iodine-induced thyroiditis, in the presence of T-helper 2 cytokines favoring autoantibody production. The observations of Heufelder and colleagues143 of marked tissue eosinophilia and eosinophil degranulation in Riedel’s struma have suggested another possibility—that these elements may represent an important fibrogenic stimulus, possibly via the release of eosinophil-derived products. The nature of such products is as yet unknown.
It now is widely believed that Riedel’s thyroiditis is more likely to be an isolated or local manifestation of a systemic disease called idiopathic multifocal fibrosclerosis. An association between Riedel’s thyroiditis and other fibrosing lesions, including mediastinal fibrosis, was first described by Barret144 in 1958. Since then, many cases of Riedel’s thyroiditis in association with retroperitoneal fibrosis,145–149 mediastinal fibrosis,145,150–153 sclerosing cholangitis,154,155 and pseudotumor of the orbit156,157 have been reported, suggesting they may be variable manifestations of a systemic multifocal fibrosing disorder.
Long-term follow-up of patients with Riedel’s thyroiditis (follow-up time 10 years) has shown that one third develop fibrosing disorders of the retroperitoneal space (often leading to ureteral obstruction), chest, or orbits.138 DeLange and co-workers131 have cited all of the available literature on this point. Two thirds of patients with Riedel’s struma do not develop extracervical fibrosis within the ensuing 10 years, and it is rare for one patient to have extracervical fibrosis in more than one site. Conversely, less than 1% of patients with retroperitoneal fibrosis have Riedel’s struma. It is considered likely that these apparently disparate fibrotic lesions may be different manifestations of the same generalized fibrosing disease; however, the thyroid fibrosis seems common, central, and integral to this disease complex, implying an important role for it in the pathogenesis.
The established association of certain drugs with retroperitoneal fibrosis has not been observed with Riedel’s struma.131 Aside from one example of two brothers, children of consanguineous parents, who developed fibrosclerosis in multiple sites (including Riedel’s struma in one of the brothers),142,150 there does not seem to be a genetic predisposition to this condition.
Clinical Features
The thyroid gland is normal in size or enlarged, usually symmetrically involved, and extremely hard. Occasionally, involvement may be unilateral. Typically, a lobe of the thyroid and the adjacent skeletal muscle, nerves, blood vessels, trachea, and other tissues are extensively replaced by dense, chronically inflamed fibrous tissue. The mass formed is firm to hard, pale gray, and easily mistaken for cancer on clinical examination or by a surgeon at operation. Diagnostic confusion with sarcoma of the thyroid region has been reported.158 The involvement of the thyroid in a sense seems to be incidental to the involvement of the soft tissue of the neck (Table 13-1). It may occur in a multinodular goiter, mimicking thyroid cancer.159 Although the etiology is unknown, the disease may develop in the course of subacute thyroiditis.160
Table 13-1
Clinical Features: Riedel’s versus Hashimoto’s Thyroiditis
| Riedel’s | Hashimoto’s | |
| Age | 23-70 yr (mostly >50 yr) | Any age (mostly >20 yr, gradually increasing with age) |
| Sex (F/M) | 2-4:1 | 4-10:1 |
| Symptoms | Pressure goiter | Often goiter |
| Thyroid involvement | Unilateral or diffuse | Generally diffuse, occasionally goiter quite large |
| Thyroid status | Occasionally hypothyroid, rarely hypoparathyroid | Commonly hypothyroid, but may be euthyroid or hyperthyroid |
| Thyroid antibodies | ≤45% | Almost invariably |
| Follow-up | Often regresses | Usually proceeds to hypothyroidism |
The disease may remain stable over many years, or it may progress slowly and produce hypothyroidism at a prevalence rate of 25% to 29%.124,128,130,131,161,162 Dyspnea, dysphagia, hoarseness, and aphonia are caused by the local pressure, and if there is enough pressure on both recurrent laryngeal nerves, there may be stridor. Sensations of suffocation, cough, and heaviness in the neck are common. Pain is unusual, although the sense of pressure may be out of proportion to the size of the goiter.163–165 The presence or degree of obstruction varies with the extent to which the surrounding structures have been invaded. Occasionally, tetany due to hypoparathyroidism may be observed.161,162
On physical examination, the thyroid gland is rock hard, is densely adherent to adjacent cervical structures (e.g., muscles, blood vessels, and nerves), and may move scarcely on swallowing. It is variable in size, from small to very large.1,124,125 The lesion may be limited to one lobe, may be present in both, or (as mentioned earlier) may involve the entire gland. It has a harder consistency than carcinoma and is only rarely tender. Although adjacent lymph nodes occasionally are enlarged, when they are present and associated with a hard thyroid mass, a diagnosis of carcinoma is often suspected.1,124,125
The clinical importance of Riedel’s thyroiditis lies in its ability to result in local obstructive phenomena; in its potential for being confused with carcinoma, especially lymphoma166 and sarcoma; and in its variable association with fibrosing processes elsewhere in the body.1,124,125,131,161 The local complications of Riedel’s thyroiditis are protean and range from thyroid dysfunction, tracheal and esophageal compression with fibrous mediastinitis,153 bilateral fibrous parotitis,167 occlusive vasculitis148,168 causing an extensive sterile neck abscess,168 superior vena cava syndrome,169,170 and cerebral venous sinus thrombosis,171 obstruction of a ventriculoperitoneal shunt,172 to pituitary failure.173 Spontaneous hypoparathyroidism secondary to Riedel’s thyroiditis seems to be rare. Only nine previous cases of primary hypoparathyroidism secondary to Riedel’s thyroiditis have been reported,132,174–179 and only two of these showed parathyroid recovery.162,179 Parathyroid autoantibodies were tested in some cases and were negative. Vascular compromise and progressive ischemia also may contribute to parathyroid dysfunction. The occurrence of cerebral sinus thrombosis suggests that Riedel’s thyroiditis may cause venous stasis, vascular damage, and possibly hypercoagulability.180 Because of fibrotic lesions elsewhere, the examination must include a careful search for compressive signs.
Laboratory and Imaging Findings
Riedel’s thyroiditis has no characteristic biochemical findings. Most patients are euthyroid, whereas few are hypothyroid.125 This difference is probably due to the extent of the nonfunctioning fibrous infiltration of the gland at the time of diagnosis. In the Cleveland Clinic series, 64% of the patients were found to be euthyroid, 32% hypothyroid, and 4% hyperthyroid.125 Two other large series revealed prevalence rates of hypothyroidism of 25% and 29%.128,131
Antithyroid antibodies are present in 67% of reported cases,129 and a mixed population of B and T cells is present in the thyroid. The occurrence of marked tissue eosinophilia and the extracellular deposition of eosinophil granule major basic protein suggests a role for eosinophils and their products in the development of fibrosis in Riedel’s thyroiditis.143 Fibrosis also may be related to the action of transforming growth factor-β1, as seen in murine thyroiditis.181
Antinuclear factor was once reported as present.161 The white blood cell count may be normal or elevated, and the erythrocyte sedimentation rate is usually moderately elevated.1,124,125 Additionally, fibrosis of the whole gland occasionally can result in hypoparathyroidism, with consequent low serum calcium and high serum phosphorus values.161,162,182 Thyroid and parathyroid function should be measured in all cases.
On thyroid radionuclide imaging, a heterogeneous pattern of isotope uptake or very low uptake is usually seen, similar to other forms of thyroiditis.1,124,125 Fluorine-18 fluorodeoxyglucose positron emission tomography images have shown metabolic activity in an abdominal mass and increased glucose metabolism in the thyroid, probably resulting from active inflammation involving lymphocytes, plasma cells, and fibroblast proliferation.183
Ultrasonography also can be helpful. On ultrasound, Riedel’s thyroiditis is reported to be homogeneously hypoechoic due to fibrosis.161,184 A thyroid hypodense on computed tomography and hypointense on T1-weighted and T2-weighted magnetic resonance images can suggest Riedel’s thyroiditis whether or not invasion to nearby soft tissues is observed. Administration of contrast medium may be helpful in differentiating the normal thyroid parenchyma from the fibrosclerotic mass. Based on current knowledge, no other entity causes diffuse decreased enhancement after gadolinium administration on magnetic resonance imaging or administration of iodinated contrast medium on computed tomography. These radiologic findings may be an important diagnostic tool for Riedel’s thyroiditis.185–187
Pathology
The gland has been described as woody or very hard. On pathologic examination, the thyroid is replaced by dense fibrosis in which are scattered solitary follicular cells and occasional acini with small amounts of colloid (Fig. 13-4).1,124,125,128,130 Extension of the fibrosis beyond the capsule of the thyroid into adjacent structures, such as nerves, blood vessels, and muscles, is a characteristic feature and accounts for the occasional instance in which the parathyroid glands have been obliterated by this fibrosing process. There are no tissue planes, making surgical extirpation virtually impossible (ligneous thyroiditis).1,124,125 An adenoma may occur in the midst of the fibrous mass. Isolated thyroid amyloidosis has been described in one case of Riedel’s struma.124
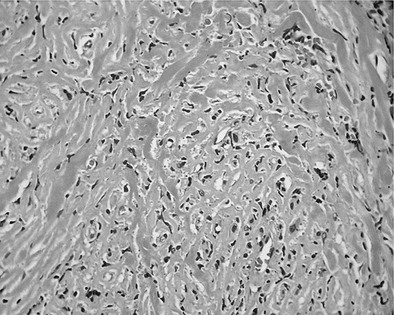
FIGURE 13-4 Pathologic findings of Riedel’s thyroiditis. Extensive follicular cell destruction and replacement of thyroid tissue by a dense fibrous tissue and infiltration of lymphocytes.
On histologic examination, early lesions show an intense infiltration of lymphocytes, plasma cells, neutrophils, and eosinophils. Subsequently, dense fibrous bands divide the thyroid into progressively smaller lobules. Eventually, dense hyalinized fibrous tissue with a few lymphocytes, plasma cells, and eosinophils replaces the thyroid parenchyma.128,130 Similar features also are observed in the extracervical fibrosclerotic lesions in the retroperitoneal or mediastinal regions, in the orbit or lacrimal glands, or in cholangitis. An associated arteritis and phlebitis with intimal proliferation, medial destruction, adventitial inflammation, and frequent thrombosis also may occur.1,124,125,128,130
Diagnosis and Treatment
The criteria most commonly used to diagnose Riedel’s thyroiditis, as reported by Woolner and colleagues128 and modified further by Schwaegerle and associates,125 are (1) gross description of a visible fibroinflammatory process involving all or a portion of the thyroid gland, (2) gross or histologic evidence of extension into adjacent structures, (3) absence of granulomatous reaction, and (4) absence of neoplasm.
Although there is no specific therapy for Riedel’s thyroiditis, several management strategies are available depending on the clinical features of the disease in the individual patient. Hypothyroidism should be treated with levothyroxine, although it rarely has an effect on goiter size or the progressive spread of fibrosclerosis. In addition, calcium and vitamin D therapy are indicated in cases with associated hypoparathyroidism.161 Surgical intervention is indicated on two grounds: (1) to exclude malignancy and (2) to relieve tracheal or esophageal compression. In this case, operation should be limited to relief of obstruction, for example, by wedge excision of the thyroid isthmus. Thyroidectomy or neck dissection is not the rule, because the lack of resection planes and the risk of injury to adjacent structures make surgery quite hazardous.129 Recurrence after surgery also has been reported.134,135
Because of the rarity of Riedel’s thyroiditis, there have been no extensive clinical trials of the efficacy of medical therapy. Nevertheless, small studies and case reports consistently show a good response to glucocorticoids.134,162,182,188 These drugs often are used as first-line therapy because of the progressive perithyroidal infiltration and fibrosis with potentially life-threatening destruction of local structures.
In some instances, dramatic responses to glucocorticoids have been described and lasting benefit even after withdrawal.141,189 In other instances, relapses have occurred when these drugs were stopped.141,182 The reasons for this variation are unclear, but inflammatory activity and duration of disease may be relevant factors. Glucocorticoids are considered to be more effective when given early in the disease.182 Initial doses of 100 mg/day of prednisone have been used, but sustained improvement has been reported with lower doses of 15 to 60 mg/day.166
In patients who fail to respond to steroid therapy or relapse after withdrawal, tamoxifen therapy should be tried. Tamoxifen, an antiestrogen drug with inhibitory properties for connective tissue proliferation, has been used successfully in the treatment of Riedel’s thyroiditis. In one series of four patients with Riedel’s thyroiditis who had progressive symptomatic disease despite glucocorticoid therapy and surgery, tamoxifen resulted in subjective and objective improvements.190 Each of the four patients had a decrease in goiter size of 50% or more, and one had total resolution of the disease. Although not fully understood, tamoxifen’s effectiveness in treating disorders such as Riedel’s thyroiditis could have been related to the stimulation of transforming growth factor β production. Transforming growth factor β is a known potent growth inhibitor of immature fibroblasts and epithelial cells.191 Until more effective drugs are found, tamoxifen may be the drug of choice for managing intractable Riedel’s thyroiditis. Side effects of tamoxifen include hot flashes and menstrual irregularity. Men report decreased libido.
Prognosis
The mortality rate has been reported to range from 6% to 10%, with deaths usually attributed to asphyxia secondary to tracheal compression or laryngospasm.1,124,125 The mortality rates mentioned are derived from the older literature, however, and may not reflect the (presumably lower) current rates. The course of the lesion may be slowly progressive, may stabilize, or may remit. After surgery, the disease sometimes subsides or takes a benign, self-limiting course. Spontaneous remissions without surgery may occur, and secondary surgery is only rarely required.1,124,125
Acknowledgments
The author expresses her gratitude and appreciation to Dr. Inês V. Castro and Dr. Paulo Carneiro for the preparation of Figs. 13-3 and 13-4. The author also thanks Dr. Robert Volpé for permission to use Figs. 13-1 and 13-2.
References
1. Volpé, R. Subacute and sclerosing thyroiditis. In: DeGroot LG, ed. Endocrinology. 3rd ed. Philadelphia: WB Saunders; 1995:742–751.
2. Bastenie, PA, Ermans, AM. Thyroiditis and thyroid function: clinical, morphological and physiological studies. International Series of Monographs in Pure and Applied Biology. Modern Trends in Physiological Sciences, vol. 36. New York: Pergamon Press, 1972.
3. LiVolsi VA, LoGerfo P, eds. Thyroiditis. CRC Press: Boca Raton, FL, 1981:21–42.
4. Greene, JN. Subacute thyroiditis. Am J Med. 1971;51:97–108.
5. Steinberg, FU. Subacute granulomatous thyroiditis: a review. Ann Intern Med. 1960;52:1014–1025.
6. Mygind, H. Thyroiditis akuta simplex. J Laryngol. 1895;91:181–193.
7. De Quervain, F. Die akute nicht eiterige Thyreoiditis und die Beteiligung der Schilddrüse und akuten Intoxikationen und Infectionen überhaupt. Mitt Grenzgebieiten Med Chir. 1904;2(Suppl):1–165.
8. De Quervain, F, Giordandengo, G. Die akute und subakute nicht eiterige thyroiditis. Mitt Grenzgeheiten Med Chir. 1936;44:538–590.
9. Volpe, R, Row, VV, Ezrin, C. Circulating viral and thyroid antibodies in subacute thyroiditis. J Clin Endocrinol Metab. 1967;27:1275–1284.
10. Martino, E, Buratti, L, Bartalena, L, et al. High prevalence of subacute thyroiditis during summer season in Italy. J Endocrinol Invest. 1987;10:321–323.
11. Kawano, C, Muroi, K, Akioka, T, et al. Cytomegalovirus pneumonitis, activated prothrombin time prolongation and subacute thyroiditis after unrelated allogeneic bone marrow transplantation. Bone Marrow Transplant. 2000;26:1347–1349.
12. Volta, C, Carano, N, Street, ME, et al. Atypical subacute thyroiditis caused by Epstein-Barr virus infection in a three-year-old girl. Thyroid. 2005;15:1189–1191.
13. Al Maawali, A, Al Yaarubi, S, Al Futaisi, A. An infant with cytomegalovirus-induced subacute thyroiditis. J Pediatr Endocrinol Metab. 2008;21:191–193B.
14. Dulipsingh, L, Ikram, Z, Malchoff, CD, et al. A cluster of cases of subacute and silent thyroiditis in the northern Connecticut, Greater Hartford area. Conn Med. 1998;62:395–397.
15. Oksa, H, Jarvenpaa, P, Metsahonkala, L, et al. No seasonal distribution in subacute de Quervain’s thyroiditis in Finland. J Endocrinol Invest. 1989;12:495.
16. Cordray, JP, Nys, P, Merceron, RE, et al. Frequency of hypothyroidism after de Quervain’s thyroiditis and contribution of ultrasonographic thyroid volume measurement [French]. Ann Med Interne (Paris). 2001;152:84–88.
17. de Bruin, TW, Riekhoff, FP, de Boer, JJ. An outbreak of thyrotoxicosis due to atypical subacute thyroiditis. J Clin Endocrinol Metab. 1990;70:396–402.
18. Nordyke, RA, Gilbert, FI, Jr., Lew, C. Painful subacute thyroiditis in Hawaii. West J Med. 1991;155:61–63.
19. Nikolai, TF. Silent thyroiditis and subacute thyroiditis. In: Braverman LE, Utiger R, eds. Werner and Ingbar’s The Thyroid: A Fundamental and Clinical Text. ed 6. Philadelphia: JB Lippincott; 1991:720–727.
20. B Hiraiwa, T, Kubota, S, Imagawa, A, et al. B Two cases of subacute thyroiditis presenting in pregnancy. B J Endocrinol Invest. 2006;29:924–927.
21. Qari, FA, Maimani, AA. Subacute thyroiditis in Western Saudi Arabia. Saudi Med J. 2005;26:630–633.
22. Fatourechi, V, Aniszewski, J, Fatourechi, G, et al. Clinical features and outcome of subacute thyroiditis in an incidence cohort: Olmsted County, Minnesota study. J Clin Endocrinol Metab. 2003;88:2100.
23. B Dedivitis, RA, Coelho, LS. Vocal fold paralysis in subacute thyroiditis. B Rev Bras Otorrinolaringol. 2007;73:138.
24. Tauveron, I, Thieblot, P, Marcheix, JC. Recurrence after 12 years of de Quervain-Crile subacute thyroiditis [French]. Rev Med Interne. 1991;12:396.
25. Bauman, A, Friedman, A. Recurrent subacute thyroiditis: a report of three cases. N Y State J Med. 1983;83:987–988.
26. Iitaka, M, Momotani, N, Ishii, J, et al. Incidence of subacute thyroiditis recurrences after a prolonged latency: 24 year survey. J Clin Endocrinol Metab. 1996;81:466–469.
27. Yamamoto, M, Saito, S, Sakurada, T, et al. Recurrence of subacute thyroiditis over 10 years after the first attack in three cases. Endocrinol Jpn. 1988;35:833–839.
28. Carle, A, Laurberg, P, Pedersen, IB, et al. Epidemiology of subtypes of hypothyroidism in Denmark. Eur J Endocrinol. 2006;154:21–28.
29. Fraser, R, Harrison, RJ. Subacute thyroiditis. Lancet. 1952;1:382–386.
30. Vejlgaard, TB, Nielsen, OB. Subacute thyroiditis in parvovirus B19 infection. Ugeskr Laeger. 1994;156:6039–6040.
31. Brouqui, P, Raoult, D, Conte-Devolx, B. Coxsackie thyroiditis. Ann Intern Med. 1991;114:1063–1064.
32. Sato, M. Virus-like particles in the follicular epithelium of the thyroid from a patient with subacute thyroiditis (de Quervain). Acta Pathol Jpn. 1975;25:499–501.
33. Volpé, R, Row, VV, Ezrin, C. Circulating viral and thyroid antibodies in subacute thyroiditis. J Clin Endocrinol Metab. 1967;27:1275–1284.
34. Yeo, PPB, Rauff, A, Chan, SW, et al. Subacute (de Quervain’s) thyroiditis in the tropics. In: Stockigt JR, Nagataki S, eds. Thyroid Research VIII. Canberra: Australian Academy of Science; 1980:570–574.
35. Arend, SM, Westedt, ML. Simultaneous onset of giant cell arteritis and subacute thyroiditis. Ann Rheum Dis. 1993;52:839–840.
36. Falaschi, P, Martocchia, A, D’Urso, R, et al. Subacute thyroiditis during interferon-alpha therapy for chronic hepatitis C. J Endocrinol Invest. 1997;20:24–28.
37. Parana, R, Cruz, M, Lyra, L, et al. Subacute thyroiditis during treatment with combination therapy (interferon plus ribavirin) for hepatitis C virus. J Viral Hepatol. 2000;7:393–395.
38. B Moser, C, Furrer, J, Ruggieri, F. Neck pain and fever after peginterferon alpha-2a. B Schweiz Rundsch Med Pradx. 2007;96:205–207.
39. Obuobie, K, Al-Sabah, A, Lazarus, JH. Subacute thyroiditis in an immunosuppressed patient. J Endocrinol Invest. 2002;25:169–171.
40. Strakosch, CR, Joyner, D, Wall, JR. Thyroid stimulating antibodies in subacute thyroiditis. J Clin Endocrinol Metab. 1978;46:345–348.
41. Wall, JR, Strakosch, CR, Brandy, P, et al. Nature of thyrotropin displacement activity in subacute thyroiditis. J Clin Endocrinol Metab. 1982;54:349–353.
42. Tamai, H, Nozaki, T, Mukuta, T, et al. The incidence of thyroid-stimulating blocking antibodies during the hypothyroid phase in patients with subacute thyroiditis. J Clin Endocrinol Metab. 1991;73:245–250.
43. Weetman, AP, Smallridge, RC, Nutman, TB, et al. Persistent thyroid autoimmunity after subacute thyroiditis. J Lab Clin Immunol. 1987;23:1–6.
44. Wall, JR, Fang, SL, Ingbar, SH, et al. Lymphocyte transformation in response to human thyroid extract in patients with subacute thyroiditis. J Clin Endocrinol Metab. 1976;43:587–590.
45. Totterman, TH, Gordin, A, Hayry, P, et al. Accumulation of thyroid antigen-reactive T lymphocytes in the gland of patients with subacute thyroiditis. Clin Exp Immunol. 1978;32:153–158.
46. Galluzzo, A, Giordano, C, Andronico, F, et al. Leucocyte migration test in subacute thyroiditis: hypothetical role of cell mediated immunity. J Clin Endocrinol Metab. 1980;50:1038–1041.
47. Fukata, S, Matsuzuka, F, Kobayashi, A, et al. Development of Graves’ disease after subacute thyroiditis: two unusual cases. Acta Endocrinol (Copenh). 1992;126:495–496.
48. Bartalena, L, Bogazzi, F, Pecori, F, et al. Graves’ disease occurring after subacute thyroiditis: report of a case and review of the literature. Thyroid. 1996;6:345–348.
49. Sheets, RF. The sequential occurrence of acute thyroiditis and thyrotoxicosis. JAMA. 1955;157:139–140.
50. Perloff, WH. Thyrotoxicosis following acute thyroiditis: a report of 5 cases. J Clin Endocrinol Metab. 1956;16:542–546.
51. Werner, SC. Graves’ disease following acute (subacute) thyroiditis. Arch Intern Med. 1979;139:1313–1315.
52. Sartani, A, Feigl, D, Zaidel, L, et al. Painless thyroiditis followed by autoimmune disorders of the thyroid: a case report with biopsy. J Endocrinol Invest. 1980;3:169–172.
53. Wartofsky, L, Schaaf, M. Graves’ disease with thyrotoxicosis following subacute thyroiditis. Am J Med. 1987;83:761–763.
54. Volpé, R. Autoimmune thyroiditis. In: Braverman LE, Utiger R, eds. Werner and Ingbar’s The Thyroid: A Fundamental and Clinical Text. ed 6. Philadelphia: JB Lippincott; 1991:921–933.
55. Volpé, R. Immunology of the thyroid. In: Volpé R, ed. Autoimmune Diseases of the Endocrine System. Boca Raton, FL: CRC Press; 1990:73–240.
56. Bech, K, Nerup, J, Thomsen, M, et al. Subacute thyroiditis de Quervain: a disease associated with HLA-B antigen. Acta Endocrinol. 1977;86:504–509.
57. Nyulassy, S, Hnilica, P, Buc, M, et al. Subacute (de Quervain’s) thyroiditis: association with HLA-Bw35 antigen, and abnormalities of the complement system, immunoglobulins and other serum proteins. J Clin Endocrinol Metab. 1977;45:270–274.
58. Tamai, H, Goto, H, Uno, H, et al. HLA in Japanese patients with subacute (de Quervain’s) thyroiditis. Tissue Antigens. 1984;24:58–59.
59. Yeo, PPB, Chan, SH, Aw, TC, et al. HLA and Chinese patients with subacute (de Quervain’s) thyroiditis. Tissue Antigens. 1981;17:249–250.
60. Ohsako, N, Tamai, H, Sudo, T, et al. Clinical characteristics of subacute thyroiditis classified according to HLA typing. J Clin Endocrinol Metab. 1995;80:3653–3656.
61. Zein, EF, Karaa, SE, Megarbane, A. Familial occurrence of painful subacute thyroiditis associated with human leukocyte antigen-B35. Presse Med. 2007;36:808–809.
62. Hsiao, Jy, Hsin, SC, Hsieh, MC, et al. Subacute thyroiditis following influenza vaccine (Vaxigrip) in a young female. Kaohsiung J Med Sci. 2006;22:297–300.
63. Volpé, R, Johnston, MW. Subacute thyroiditis: A disease commonly mistaken for pharyngitis. Can Med Assoc J. 1957;77:297–307.
64. Volpé, R, Johnston, MW, Huber, N. Thyroid function in subacute thyroiditis. J Clin Endocrinol Metab. 1958;18:65–78.
65. Alper, AT, Hasdemir, H, Akyol, A, et al. Incessant ventricular tachycardia due to subacute thyroiditis. B Int J Cardiol. 2007;116:E22–E24.
66. Swinburne, JL, Kreisman, SH. A rare case of subacute thyroiditis causing thyroid storm. Thyroid. 2007;17:73–76.
67. Nishihara, E, Ohye, H, Amino, N, et al. Clinical characteristics of 852 patients with subacute thyroiditis before treatment. Intern Med. 2008;47:725–729.
68. Benbassat, CA, Olchovsky, D, Tsvetov, G, et al. Subacute thyroiditis: clinical characteristics and treatment outcome in fifty-six consecutive patients diagnosed between 1999 and 2005. J Endocrinol Invest. 2007;30:631–635.
69. Ingbar, SH, Freinkel, N. Thyroid function and metabolism of iodine in patients with subacute thyroiditis. Arch Intern Med. 1958;101:339–346.
70. Dorta, T, Beraud, T. New investigations on subacute thyroiditis. Helv Med Acta. 1961;28:19–41.
71. Izumi, M, Larsen, PR. Correlation of sequential change in serum thyroglobulin, triiodothyronine, and thyroxine in patients with Graves’ disease and subacute thyroiditis. Metabolism. 1978;27:449–460.
72. Larsen, PR. Serum triiodothyronine and thyrotropin during hyperthyroid and recovery phases of subacute non-suppurative thyroiditis. Metabolism. 1974;23:467–471.
73. Weihl, AC, Daniels, GH, Ridgeway, ED, et al. Thyroid function during the early days of subacute thyroiditis. J Clin Endocrinol Metab. 1977;44:1107–1114.
74. Glinoer, D, Puttemans, N, Van Herle, AJ, et al. Sequential study of the impairment of thyroid function in the early stage of subacute thyroiditis. Acta Endocrinol. 1974;77:26–39.
75. Lio, S, Pontecorvi, A, Caruso, M, et al. Transitory subclinical and permanent hypothyroidism in the course of subacute thyroiditis (de Quervain). Acta Endocrinol. 1984;106:67–70.
76. Jay, HK. Permanent myxedema: an unusual complication of granulomatous thyroiditis. J Clin Endocrinol Metab. 1961;21:1384–1387.
77. Rosen, F, Row, VV, Volpé, R, et al. Anaplastic carcinoma of thyroid with abnormal circulating iodoprotein: a case simulating subacute thyroiditis. Can Med Assoc J. 1966;95:1039–1041.
78. Pearce, EN, Martino, E, Bogazzi, F, et al. The prevalence of elevated serum C-reactive protein levels in inflammatory and noninflammatory thyroid disease. Thyroid. 2003;13:643–648.
79. Amino, N, Yabu, Y, Miki, T, et al. Serum ratio of triiodothyronine to thyroxine, and thyroxine-binding globulin and calcitonin concentrations in Graves’ disease and destruction-induced thyrotoxicosis. J Clin Endocrinol Metab. 1981;53:113–116.
80. Hiromatsu, Y, Ishibashi, M, Miyake, I, et al. Color Doppler ultrasonography in patients with subacute thyroiditis. Thyroid. 1999;9:1189–1193.
81. Demeester-Mirkine, N, Brauman, H, Corvilain, J. Delayed adjustment of the pituitary response in circulating thyroid hormones in a case of subacute thyroiditis. Clin Endocrinol. 1976;5:9–14.
82. Staub, JJ. The TRH test in subacute thyroiditis. Lancet. 1975;1:868–870.
83. Lewitus, W, Rechnic, J, Lubin, E. Sequential scanning of the thyroid as an aid to the diagnosis of subacute thyroiditis. Isr J Med Sci. 1967;3:847–854.
84. Hamburger, JL, Kadian, G, Rossin, HW. Subacute thyroiditis—evaluations depicted by serial 131I scintigrams. J Nucl Med. 1965;6:560–565.
85. Hiromatsu, Y, Ishibashi, M, Nishida, H, et al. Technetium-99m sestamibi imaging in patients with subacute thyroiditis. Endocr J. 2003;50:239–244.
86. Nicklaus Muller, E, Mullhaupt, B, Perschak, H. Steroid therapy and course of blood sedimentation rate in de Quervain’s thyroiditis. Schweiz Rundsch Med Prax. 1994;83:95–100.
87. Sakane, S, Murakami, Y, Sasaki, M, et al. Serum concentrations of granulocyte colony-stimulating factor (G-CSF) determined by a highly-sensitive chemiluminescent immunoassay during the clinical course of subacute thyroiditis. Endocr J. 1995;42:391–396.
88. Skillern, PG, Lewis, LA. Fractional plasma protein values in subacute thyroiditis. J Clin Invest. 1957;36:780–783.
89. Kubota, S, Matsumoto, Y, Amino, N, et al. Serial changes in liver function tests in patients with subacute thyroiditis. Thyroid. 2008;18:815.
90. Hamada, S, Yagura, T, Ishii, H, et al. Subacute thyroiditis as a systemic multisystem disease. In: Nagataki S, Torizuka K, eds. The Thyroid 1988. Excerpta Medica International Congress Series 796. Amsterdam: Elsevier; 1988:521–525.
91. Kunz, A, Blank, W, Braun, B. De Quervain’s subacute thyroiditis by colour Doppler sonography findings. Ultraschall Med. 2005;26:102–106.
92. Park, SY, Kim, EK, Kim, MJ, et al. Ultrasonographic characteristics of subacute granulomatous thyroiditis. Korean J Radiol. 2006;7:229–234.
93. Omori, N, OmoriK, Takano, K. Association of the ultrasonographic findings of subacute thyroiditis with thyroid pain and laboratory findings. Endocr J. 2008;55:583–588.
94. Sakata, S, Nagai, K, Maekawa, H, et al. Serum ferritin concentrations in subacute thyroiditis. Metabolism. 1991;40:682–688.
95. Ozata, M, Bolu, E, Sengul, A, et al. Soluble intercellular adhesion molecule-1 concentrations in patients with subacute thyroiditis and in patients with Graves’ disease with or without ophthalmopathy. Endocr J. 1996;43:517–525.
96. Hara, H, Sugita, E, Sato, R, et al. Plasma selectin levels in patients with Graves’ disease. Endocr J. 1996;43:709–713.
97. Yamada, T, Sato, A, Aizawa, T. Dissociation between serum interleukin 6 rise and other parameters of disease activity in subacute thyroiditis during treatment with corticosteroid. J Clin Endocrinol Metab. 1996;81:577–579.
98. Vulpoi, C, Zbranca, E, Preda, C, et al. Contribution of ultrasonography in the evaluation of subacute thyroiditis. Rev Med Chir Soc Med Nat Iasi. 2001;105:749–755.
99. Lu, CP, Chang, TC, Wang, CY, et al. Serial changes in ultrasound-guided fine needle aspiration cytology in subacute thyroiditis. Acta Cytol. 1997;41:238–243.
100. Bennedbaek, FN, Gram, J, Hegedus, L. The transition of subacute thyroiditis to Graves’ disease as evidenced by diagnostic imaging. Thyroid. 1996;6:457–459.
101. Bennedbaek, FN, Hegedus, L. The value of ultrasonography in the diagnosis and follow-up of subacute thyroiditis. Thyroid. 1997;7:45–50.
102. Benker, G, Olbricht, TH, Windeck, R, et al. The sonographic and functional sequelae of de Quervain’s subacute thyroiditis. Acta Endocrinol. 1988;117:435–441.
103. Tokuda, Y, Kasagi, K, Iida, Y, et al. Sonography of subacute thyroiditis: changes in the findings during the course of the disease. J Clin Ultrasound. 1990;18:21–26.
104. Tezuka, M, Murata, Y, Ishida, R, et al. MR imaging of the thyroid: correlation between apparent diffusion coefficient and thyroid gland scintigraphy. J Magn Reson Imaging. 2003;17:163–169.
105. Vierhapper, H, Bieglmayer, CH, Nowotny, P, et al. Normal serum concentrations of sex hormone-binding globulin in patients with hyperthyroidism due to subacute thyroiditis. Thyroid. 1998;8:1107–1111.
106. Fragu, P, Rougier, P, Schlumberger, M, et al. Evolution of thyroid 127I stores measured by x-ray fluorescence in subacute thyroiditis. J Clin Endocrinol Metab. 1982;54:162–166.
107. Rapoport, B, Block, MB, Hoffer, PB, et al. Depletion of thyroid iodine during subacute thyroiditis. J Clin Endocrinol Metab. 1973;36:610–611.
108. Bech, K, Feldt-Rasmussen, U, Bliddal, H, et al. Persistence of autoimmune reactions during recovery of subacute thyroiditis. In: Pinchera A, Ingbar SH, McKenzie JM, Fenzi GF, eds. Thyroid Autoimmunity. New York: Plenum Press; 1987:623–625.
109. Itaka, M, Momotani, N, Hisaoka, T, et al. TSH receptor antibody-associated thyroid dysfunction following subacute thyroiditis. Clin Endocrinol. 1998;48:445–453.
110. Volpé, R. Subacute and sclerosing thyroiditis. In: DeGroot LG, ed. Endocrinology. ed 4. Philadelphia: WB Saunders; 1999:742–751.
111. Solano, JC, Bascunana, AG, Perez, JS, et al. Fine-needle aspiration of subacute granulomatous thyroiditis (de Quervain’s thyroiditis): a clinico-cytologic review of 36 cases. Diagn Cytopathol. 1997;16:214–220.
112. Shabb, NS, Salti, I. Subacute thyroiditis: fine-needle aspiration cytology of 14 cases presenting with thyroid nodules. Diagn Cytopathol. 2006;34:18–23.
113. Liel, Y. The survivor: association of an autonomously functioning thyroid nodule and subacute thyroiditis. Thyroid. 2007;17:183–184.
114. Sari, O, Erbas, B, Erbas, T. Subacute thyroiditis in a single lobe. Clin Nucl Med. 2001;26:400–401.
115. Koga, M, Hiromatsu, Y, Jimi, A, et al. Immunohistochemical analysis of Bcl-2, Bax, and Bak expression in thyroid glands from patients with subacute thyroiditis. J Clin Endocrinol Metab. 2002;84:2221–2225.
116. Singer, PA. Thyroiditis: acute, subacute, and chronic. Med Clin North Am. 1991;75:61–77.
117. Volpe, R. The management of subacute (de Quervain’s) thyroiditis. Thyroid. 1993;5:253–255.
118. Mizukoshi, T, Noguchi, S, Murakami, T, et al. Evaluation of recurrence in 36 subacute thyroiditis patients managed with prednisolone. Intern Med. 2001;40:292–295.
119. Bogazzi, F, Dell’Unto, E, Tanda, ML, et al. Long-term outcome of thyroid function after amiodarone-induced thyrotoxicosis, as compared to subacute thyroiditis. J Endocrinol Invest. 2006;29:694–698.
120. Chopra, IJ, Van Herle, AJ, Korenman, SG, et al. Use of sodium iodate in management of hyperthyroidism in subacute thyroiditis. J Clin Endocrinol Metab. 1995;80:2178–2180.
121. Riedel, BM. Ueber Verlauf and Ausgang der chronischer Strumitis. Munch Med Wochenschr. 1910;57:1946–1947.
122. Riedel, BM. Vorstellung eines Kranken mit chronischer Strumitis. Verh Ges Chir. 1896;26:127–129.
123. Riedel, BM. Die chronische zur Bildung eisenharter Tumoren führende Entzündung der Schilddrüse. Verh Ges Chir. 1896;25:101–105.
124. Bastenie PA: Invasive fibrous thyroiditis (Riedel), In Bastenie PA, Ermans AM, editors: Thyroiditis and Thyroid Function. International Series of Monographs in Pure and Applied Biology, Modern Trends in Physiological Sciences, vol. 36, Oxford, 1972, Pergamon Press, pp 99–108.
125. Schwaegerle, SM, Bauer, TW, Esselstyn, CB. Riedel’s thyroiditis. Am J Clin Pathol. 1988;90:715–722.
126. DeCourcey, JL. A new theory concerning the etiology of Riedel’s struma. Surgery. 1942;12:754–762.
127. Hay, ID. Thyroiditis: a clinical update. Mayo Clin Proc. 1985;60:836–843.
128. Woolner, LB, McConahey, WM, Beahrs, O. Invasive fibrous thyroiditis (Riedel’s struma). J Clin Endocrinol Metab. 1957;17:201–220.
129. Hay, ID, McConahey, WM, Carney, JA, et al. Invasive fibrous thyroiditis (Riedel’s struma) and associated extracervical fibrosclerosis: Bowlby’s disease revisited [abstract]. Ann Endocrinol. 1982;43:29A.
130. Woolner, LB. Thyroiditis: classification and clinicopathologic correlations. In: Hazard JB, Smith DE, eds. The Thyroid. Baltimore: Williams & Wilkins; 1964:123–142.
131. DeLange, WE, Freling, NJM, Molenaar, WM, et al. Invasive fibrous thyroiditis (Riedel’s struma): a case report with review of the literature. QJM. 1989;268:709–717.
132. Volpé, R. Suppurative thyroiditis. In: Werner SC, Ingbar SH, eds. The Thyroid: A Fundamental and Clinical Text. ed 4. New York: Harper & Row; 1978:983–985.
133. Degroot, LJ, Stanbury, JB. The Thyroid and Its Diseases, ed 4. New York: Wiley & Sons; 1975.
134. Zimmermann-Belsing, T, Feldt-Rasmussen, U. Riedel’s thyroiditis: an autoimmune or primary fibrotic disease? J Intern Med. 1994;235:271–274.
135. Lorenz, K, Gimm, O, Holzhausen, HJ, et al. Riedel’s thyroiditis: impact and strategy of a challenging surgery. Langenbecks Arch Surg. 2007;392:405–412.
136. Vaidya, B, Harris, PE, Barrett, P, et al. Corticosteroid therapy in Riedel’s thyroiditis. Postgrad Med J. 1997;73:817.
137. Drury, ME, Sweeney, EC, Heffernan, SJ. Invasive fibrous thyroiditis. Ir Med J. 1974;67:388–390.
138. Merrington, WR. Chronic thyroiditis: a case showing features of both Riedel’s and Hashimoto’s thyroiditis. Br J Surg. 1948;35:423–426.
139. Rose, E, Rayster, HP. Invasive fibrous thyroiditis (Riedel’s struma). JAMA. 1961;176:224–226.
140. Bendtzen, K. Clinical significance of cytokines: Natural and therapeutic regulation. Semin Clin Immunol. 1991;8:5–18.
141. Katsikas, D, Shorthouse, AJ, Taylor, S. Riedel’s thyroiditis. Br J Surg. 1976;63:929–931.
142. Many, MC, Carpentier, S, Eggermont, J, et al. Towards an experimental model for Riedel’s fibrous thyroiditis [abstract]. J Endocrinol Invest. 1998;4(Suppl):92.
143. Heufelder, AE, Goellner, JR, Bahn, RS, et al. Tissue eosinophilia and eosinophil degranulation in Riedel’s invasive fibrous thyroiditis. J Clin Endocrinol Metab. 1996;81:977–984.
144. Barret, NR. Idiopathic mediastinal fibrosis. Br J Surg. 1958;46:207–218.
145. Rao, CR, Ferguson, GC, Kyle, VN. Retroperitoneal fibrosis associated with Riedel’s struma. Can Med Assoc J. 1973;108:1019–1021.
146. Turner-Warwich, R, Nabarro, JD, Doniach, D. Riedel’s thyroiditis and retroperitoneal fibrosis. Proc R Soc Med. 1966;59:596–598.
147. Gleeson, MH, Taylor, S, Dowling, RH. Multifocal fibrosclerosis. Proc R Soc Med. 1970;63:1309–1311.
148. Meijer, S, Hoitsma, HF, Scholtmeijer, R. Idiopathic retroperitoneal fibrosis in multifocal fibrosclerosis. Eur Urol. 1976;2:258–260.
149. Nielson, HK. Multifocal idiopathic fibrosclerosis: two cases with simultaneous occurrence of retroperitoneal fibrosis and Riedel’s thyroiditis. Acta Med Scand. 1980;208:119–123.
150. Comings, DS, Skubi, KB, Van Eyes, J, et al. Familial multifocal fibrosclerosis. Ann Intern Med. 1967;66:884–892.
151. Husband, P, Knudsen, A. Idiopathic cervical and retroperitoneal fibrosis: report of a case treated with steroids. Postgrad Med J. 1976;52:788–793.
152. Raphael, HA, Beahrs, OH, Woolner, LB, et al. Riedel’s struma associated with fibrous mediastinitis: report of a case. Mayo Clin Proc. 1966;41:375–382.
153. Wold, LE, Weiland, LH. Tumefactive fibro-inflammatory lesions of the head and neck. Am J Surg Pathol. 1983;7:477–482.
154. Hache, L, Utz, DC, Woolner, LB. Idiopathic fibrous retroperitonitis. Surg Gynecol Obstet. 1962;115:737–744.
155. Bartholemew, LG, Cain, JC, Woolner, LB, et al. Sclerosing cholangitis: its possible association with Riedel’s struma and fibrous retroperitonitis. N Engl J Med. 1963;269:8–12.
156. Andersen, SR, Seedorf, HH, Halberg, P. Thyroiditis with myxedema and orbital pseudotumor. Acta Ophthalmol (Rbh). 1963;41:120–125.
157. Arnott, EJ, Greaves, DP. Orbital involvement in Riedel’s thyroiditis. Br J Ophthalmol. 1965;49:1–5.
158. Torres-Montaner, A, Beltran, M, Romero de la Osa, A, et al. Sarcoma of the thyroid region mimicking Riedel’s thyroiditis. J Clin Pathol. 2001;54:570–572.
159. Annaert, M, Thijs, M, Sciot, R, et al. Riedel’s thyroiditis occurring in a multinodular goiter, mimicking thyroid cancer. J Clin Endocrinol Metab. 2007;92:2005–2006.
160. Cho, MH, Kim, CS, Park, JS, et al. Riedel’s thyroiditis in a patient with recurrent subacute thyroiditis: a case report and review of the literature. Endocr J. 2007;54:559–562.
161. Best, TB, Munro, RE, Burwell, S, et al. Riedel’s thyroiditis associated with Hashimoto’s thyroiditis, hypoparathyroidism, and retroperitoneal fibrosis. J Endocrinol Invest. 1991;14:767–772.
162. Chopra, D, Wool, MS, Crosson, A, et al. Riedel’s struma associated with subacute thyroiditis, hypothyroidism and hypoparathyroidism. J Clin Endocrinol Metab. 1978;46:869–871.
163. Heufelder, AE, Hay, ID, Carney, JA, et al. Coexistence of Graves’ disease and Riedel’s (invasive fibrous) thyroiditis: further evidence of a link between Riedel’s thyroiditis and organ-specific autoimmunity. Clin Invest. 1994;72:788–793.
164. Heufelder, AE, Hay, ID. Further evidence for autoimmune mechanisms in the pathogenesis of Riedel’s invasive thyroiditis. J Intern Med. 1995;238:85–86.
165. Shaw, AFB, Smith, RP. Riedel’s chronic thyroiditis: with a report of six cases and a contribution to the pathology. Br J Surg. 1925;13:93–108.
166. Vigouroux, C, Escourolle, H, Mosnier-Pudar, H, et al. Riedel’s thyroiditis and lymphoma: Diagnostic difficulties. Presse Med. 1996;25:28–30.
167. Hines, RC, Scheuermann, HA, Royster, HP, et al. Invasive fibrous (Riedel’s) thyroiditis with bilateral fibrous parotitis. JAMA. 1970;213:869–871.
168. Geissler, B, Wagner, T, Dorn, R, et al. Extensive sterile abscess in an invasive fibrous thyroiditis (Riedel’s thyroiditis) caused by an occlusive vasculitis. J Endocrinol Invest. 2001;24:111–115.
169. Abet, D, Francisci, MP, Sevestre, H, et al. Superior vena cava syndrome and Riedel’s thyroiditis: Report of a case: Review of the literature. J Mal Vasc. 1991;16:298–300.
170. Yasmeen, T, Khan, S, Gonsh, F, et al. Riedel’s thyroiditis: report of a case complicated by spontaneous hypoparathyroidism, recurrent laryngeal nerve injury, and Horner’s syndrome. J Clin Endocrinol Metab. 2002;87:3543–3547.
171. Vaidya, B, Coulthard, A, Goonetilleke, A, et al. Cerebral venous sinus thrombosis: a late sequel of invasive fibrous thyroiditis. Thyroid. 1998;8:787–790.
172. Natt, N, Heufelder, AE, Hay, ID, et al. Extracervical fibrosclerosis causing obstruction of a ventriculo-peritoneal shunt in a patient with hydrocephalus and invasive fibrous thyroiditis (Riedel’s struma). Clin Endocrinol (Oxf). 1997;47:107–111.
173. Kelly, WF, Mashiter, K, Taylor, S, et al. Riedel’s thyroiditis leading to severe but reversible pituitary failure. Postgrad Med J. 1979;55:194–198.
174. Lo, JC, Loh, KC, Rubin, AL, et al. Riedel’s thyroiditis presenting with hypothyroidism and hypoparathyroidism: dramatic response to glucocorticoid and thyroxine therapy. Clin Endocrinol (Oxf). 1998;48:815–818.
175. Crile, C, Jr. Thyroiditis. Ann Surg. 1948;127:640–654.
176. Austoni, M, Conte, N, Zaccaria, M, et al. Tiroidite di Riedel associata a ipoparatiroidismo. Folia Endocrinol. 1972;25(Suppl 6):495–501.
177. Marin, F, Araujo, R, Paramo, C, et al. Riedel’s thyroiditis associated with hypothyroidism and hypoparathyroidism. Postgrad Med J. 1989;268:709–717.
178. McRorie, ER, Chalmers, J, Campbell, IW. Riedel’s thyroiditis complicated by hypoparathyroidism and hypothyroidism. Scott Med J. 1993;38:27–28.
179. Casoli, P, Tumiati, B. Hypoparathyroidism secondary to Riedel’s thyroiditis: A case report and review of the literature. Ann Ital Med Int. 1999;14:54–57.
180. Vaiydya, B, Coulthard, A, Goonetilleke, A, et al. Cerebral venous sinus thrombosis: a late sequel of invasive fibrous thyroiditis. Thyroid. 1998;8:787–790.
181. Chen, K, Wei, Y, Sharp, GC, et al. Characterization of thyroid fibrosis in a murine model of granulomatous experimental autoimmune thyroiditis. J Leukoc Biol. 2000;68:828–835.
182. Thomson, JA, Jackson, IMD, Duguid, WP. The effect of steroid therapy on Riedel’s thyroiditis. Scott Med J. 1968;13:13–16.
183. Drieskens, O, Blockmans, D, Van den Bruel, A, et al. Riedel’s thyroiditis and retroperitoneal fibrosis in multifocal fibrosclerosis: positron emission tomographic findings. Clin Nucl Med. 2002;27:413–415.
184. Perez Fontan, FJ, Cordido Carballido, F, Pombo Felipe, F, et al. Riedel thyroiditis: US, CT, and MR evaluation. J Comput Assist Tomogr. 1993;17:324–325.
185. Ozgen, A, Cila, A. Riedel’s thyroiditis in multifocal fibrosclerosis: CT and MR imaging findings. Am J Neuroradiol. 2000;21:320–321.
186. Takahashi, N, Okamoto, K, Sakai, K, et al. MR Findings with dynamic evaluation in Riedel’s thyroiditis. Clin Imaging. 2002;26:89–91.
187. Papi, G, Corrado, S, Cesinaro, AM, et al. Riedel’s thyroiditis: clinical, pathological and imaging features. Int J Clin Pract. 2002;56:65–67.
188. Rodriguez, I, Ayala, E, Caballero, C, et al. Solitary fibrous tumor of the thyroid gland: report of seven cases. Am J Surg Pathol. 2001;25:1424–1428.
189. Owen, K, Lane, H, Jones, MK. Multifocal fibrosclerosis: a case of thyroiditis and bilateral lacrimal gland involvement. Thyroid. 2001:1187–1190.
190. Few, J, Thompson, NW, Angelos, P, et al. Riedel’s thyroiditis: treatment with tamoxifen. Surgery. 1996;120:993–999.
191. Arteaga, CL, Tandon, AK, Von Hoff, DD, et al. Transforming growth factor β: potential autocrine growth inhibitor of estrogen receptor-negative human breast cancer cell. Cancer Res. 1988;48:3808–3904.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 13
Subacute and Riedel’s Thyroiditis
Subacute Thyroiditis
The term subacute thyroiditis (SAT) describes a self-limited inflammatory disorder and the most common cause of thyroid pain, probably of viral origin.1–5 It was first reported by Mygind6 in 1895, who described 18 cases of “thyroiditis akuta simplex.” The name De Quervain traditionally has been associated with this condition, however, probably because he described the pathology of this disorder thoroughly in 19047 and again in 1936.8 SAT occurs in 5% of patients with clinical thyroid disease9 and frequently follows an upper respiratory tract infection. Its incidence correlates with the peak incidence of enterovirus.10 Other viruses, such as Epstein-Barr virus and cytomegalovirus, also have been reported, but so far clear evidence for a viral cause is still lacking.11 There is a strong preponderance of women over men with this condition.1
SAT has a multiplicity of synonyms, some reflecting misconceptions regarding the etiology or pathology of the condition. These include De Quervain’s thyroiditis, viral thyroiditis, granulomatous thyroiditis, acute or subacute diffuse thyroiditis, acute simple thyroiditis, noninfectious thyroiditis, struma granulomatosa, pseudogranulomatous thyroiditis, giant cell thyroiditis, pseudo–giant cell thyroiditis, migratory “creeping” thyroiditis, and pseudotuberculous thyroiditis. The term subacute thyroiditis connotes a temporal quality that might apply to any inflammatory process of intermediate severity and duration. As the term is generally employed, however, it specifically includes only patients showing a pseudogranulomatous pathologic appearance in the thyroid gland (which is virtually specific for the disease) and a characteristic clinical syndrome in which the painful tender goiter also is associated with considerable malaise, fever, and evidence of thyroid dysfunction (described more fully later).1–5 It generally is distinguishable from a similar disorder, painless or silent thyroiditis, which disturbs thyroid function in a manner similar to SAT but without pain or tenderness and with a different pathologic appearance.
Incidence
Few epidemiologic studies of SAT have been reported.10,14–22 Compared with other thyroid diseases, SAT is uncommon, occurring at the rate of about 1 case per 5 cases of Graves’ disease and 1 case per 15 or 20 cases of Hashimoto’s thyroiditis.19 Although the cause is most likely viral, SAT, similar to all other thyroid conditions, occurs most commonly in women who are 40 to 50 years old. The reported female-to-male ratio is 3 to 6 : 1.19 it has been noted as a rare cause of hyperthyroidism in pregnancy.20 It is rare in children and seems to occur in any season of the year,10,22 with a trend toward more cases in fall and spring.22 Familial or geographic aggregation of cases is seldom noted. SAT has been reported most commonly from the temperate zone, having been observed in North America, Europe, and Japan. Recently, a few cases were reported in Western Saudi Arabia,21 although it is rarely reported from many other parts of the world. Associated autoimmune conditions do not seem more common than autoimmune conditions observed in the general population.22
Although complete recovery is the rule, recurrence after several years has been reported.24–27 In one study, four recurrent episodes of SAT occurred in 3 of 222 patients (1.4%). The recurrent episodes were similar to the first episodes of SAT.27 In a larger study that evaluated data for 3344 patients with SAT between 1970 and 1993, SAT recurred in 48 of 3344 patients (1.4%) (mean 14.5 ± 4.5 years after the first episode). Five patients experienced a third episode (mean 7.6 ± 2.4 years after the second episode).27 Another cohort study showed a 4% recurrence rate after many years.22 Theoretically, late recurrence possibly occurs after the disappearance of immunity to the previous viral infection. During an evaluation of subtypes of hypothyroidism over a 4-year period in Denmark, an incidence of subacute thyroiditis of 1.8% was found in a cohort of 685 patients with hypothyroidism.28
Etiology
In 1952, Fraser and Harrison29 were the first to propose that SAT represents a viral infection of the thyroid gland. Since then, considerable indirect evidence suggests that SAT is most likely the result of a viral infection30–32 that rarely recurs after a complete recovery, possibly because of immunity to the offending virus.
Clinically, the disease has several characteristics typical of viral infections, including a typical prodrome with myalgias, malaise, and fatigue; absence of leukocytosis; and usually a self-limited course.1–5 Additionally, clusters of the disease have been reported during outbreaks of viral infection.1–5,10 It has been described in association with mumps, measles,1 influenza,1 the common cold,1 adenovirus,1 infectious mononucleosis,1,12 coxsackievirus,1 myocarditis,1 cat-scratch fever,1 St. Louis encephalitis,1 hepatitis A,18 parvovirus B19 infection,19 and cytomegalovirus infection.11,13
In an extensive study reported by Volpé and colleagues,33 32 of 71 patients with SAT, who had no evidence of specific viral disease, showed at least fourfold increases in viral antibodies during the thyroid illness. These viral antibodies included antibodies to coxsackievirus, adenovirus, influenza virus, and mumps virus. Coxsackievirus antibodies were found most commonly, and the changes in their titers most closely approximated the course of the disease. In a later study of 10 patients in Singapore, no such antibodies were observed, however.34 It is possible that the presence of these antibodies may not reflect pathogenic significance, but instead may result from an anamnestic response to the inflammatory thyroid lesion. The thyroid responds with the clinical picture of thyroiditis after invasion by a variety of viruses, and a variety of agents may be causative in the syndrome of SAT.
Certain nonviral infections, such as malaria and Q fever, have been associated with a clinical syndrome that at least simulates SAT.1 The significance of these observations remains to be determined. In addition, a case of SAT occurring simultaneously with giant cell arteritis has been reported.35 Several cases of SAT that developed during interferon-α treatment for hepatitis C have been described,36–38 and more recently, a case of SAT that developed after long-term immunosuppression and lithium therapy following an allogeneic bone marrow transplant was reported.39
Several autoimmune phenomena have been described in SAT. Thyroid autoantibodies (antithyroglobulin and antithyroid peroxidase antibodies) have been found in 42% to 64% of patients with SAT.33 In most of these patients, the antibody titer gradually decreased and remained low or disappeared as the disease faded. Thyroid-stimulating hormone (TSH) receptor antibodies also have been reported in patients with SAT,40–42 although changes in antibody titer did not correlate with disease activity.40 Autoantibodies to several novel, uncharacterized thyroid antigenic determinants were found in eight of nine patients with SAT tested.43 These autoantibodies persisted, and their level did not decrease over 39 months after onset of SAT. These antibodies likely arise secondary to the damage caused by viral infection of the thyroid gland because they are typically polyclonal in nature.43
There is evidence that T-cell-mediated immunity against thyroid antigens may play a role in the pathogenesis of SAT. During the initial phase of the disease, the gland is infiltrated by T cells, and sensitization of T cells against thyroid antigens has been shown in such patients.44–46 This sensitization was transitory, however, and likely represented a secondary immune response to the inflammatory release of antigen induced by the viral infection of the gland.1
It has been suggested that thyroid-destructive events in the course of SAT may trigger, under a genetic background, thyroid autoimmune disease of various kinds.47,48 Patients with a previous history of SAT, in about 1% of cases,2 may develop hypothyroidism as a consequence of previous thyroid damage. The occurrence of Graves’ disease after SAT also has been described, although such evidence seems to be extremely rare, with fewer than 20 cases reported in the literature.49–53
Although SAT is shown to be associated with thyroid autoimmune phenomena, after recovery all immunologic phenomena should disappear. This is in contrast to the continuing presence of these abnormalities in autoimmune thyroid disease.54 The transitory immunologic markers observed during the course of SAT seem to be secondary to the release of antigenic material from the thyroid and seem to be a normal, physiologic response to the inflammatory destruction of the gland.55
In light of the previous observations, the lack of any direct evidence, and because it is rare for SAT to progress to either Graves’ disease or Hashimoto’s disease, the corollary is still consistent with the view that antigen-driven events can produce a transient immunologic disturbance, but does not, or is most unlikely to, culminate in chronic autoimmune thyroid disease. It is possible that the illness of SAT might act as a nonspecific stress acting on the immune system to precipitate Graves’ disease in a favorable genetic background.54
An association between SAT and HLABw35 has been noted in all ethnic groups tested.56–59 This haplotype seems to confer an unusual susceptibility to SAT, perhaps because it allows one or more viruses to trigger an immune response directed against thyroid tissue.60 Histocompatibility studies show that 72% of patients with subacute thyroiditis manifest HLA-Bw35.57–60 Familial occurrence of subacute thyroiditis associated with HLA-B35 has been reported.61,62 Another HLABw67 was found in 87% of a Japanese population and correlated with a seasonal appearance and a mild course of disease. Thus the susceptibility to subacute thyroiditis is genetically influenced, and it has also been suggested that subacute thyroiditis might occur by transmission of viral infection in genetically predisposed individuals.13
Clinical Features
Half of patients have a history of an antecedent of upper respiratory infection, followed in days or weeks by the clinical manifestations of SAT itself.1–5,22,63 SAT begins with a prodrome of generalized myalgias, pharyngitis, low-grade fever, and fatigue. The patient notes pain of varying degrees in the region of the thyroid gland. This pain may involve one lobe, part of a lobe, or the whole thyroid, and it typically radiates from the thyroid gland to the angle of the jaw and to the ear of the affected side. If not bilateral initially, the pain and tenderness often spread to the uninvolved side of the thyroid within days or weeks. It also may radiate to the anterior chest or may be centered only over the thyroid. Moving the head, swallowing, or coughing may aggravate it. Transient vocal cord paresis may occur.23
Symptoms of mild to moderate hyperthyroidism occur in the early phase in most patients.22 Fifty percent of patients have symptoms of thyrotoxicosis, and the usual symptoms of nervousness, tremulousness, weight loss, heat intolerance, and tachycardia predominate.1–5,63–66 On physical examination, most patients appear uncomfortable and flushed, with variable fever. The thyroid gland may be only slightly to moderately enlarged, with one lobe larger than the other. The consistency of the involved area is usually firm or hard. With time or treatment, the thyroid tenderness subsides, and the goiter generally disappears within several weeks or months. Signs of mild to moderate hyperthyroidism are present in 50% of cases. About 8% to 16% of patients with this condition are noted to have a preexisting goiter. Cervical lymphadenopathy is rare.
In most patients, SAT lasts 2 to 4 months, although it may last 1 year. When the course is prolonged, the major manifestation is persistent, painful, tender thyroid enlargement, the thyrotoxicosis almost always having subsided earlier. Recurrences after recovery have been reported but are unusual, on the order of 2.3% per year26 or 4% over 21 years after the first episode.22
Sometimes hyperthyroidism may not be apparent clinically but can be detected by biochemical means.64 This situation is due to a disruptive process within the thyroid gland, with continuous leakage of the colloid into the interstitial spaces, where it is broken down into its component parts, liberating thyroid hormones, thyroglobulin, and other iodoamino acids into the circulation.26,64–74 Because the thyroid cells during this phase are virtually incapable of producing new thyroid hormone, the colloid that has been stored within the follicles is depleted within 2 to 3 months, resulting in a phase of transient hypothyroidism in patients in whom the process has persisted over the interval.75 Because disruption of the thyroid parenchyma can continue for months, hypothyroidism may persist for several weeks. As recovery continues, the follicles regenerate, the colloid is repleted, and normal thyroid function is restored. With recovery, the thyroid is reconstituted, repleted with colloid; thyroid function is restored; and a variable amount of interstitial fibrosis persists.1–5,64–75 This transient hypothyroidism may be subclinical or overt and occurs in about two thirds of patients.
SAT rarely progresses to permanent hypothyroidism.1–5,75,76 In these cases, progression may be due to total destruction of the thyroid, with consequent fibrosis. As mentioned before, in rare instances, the disorder may seem to culminate in autoimmune thyroiditis after recovery from SAT.48,51,53
Diagnosis
The typical painful SAT usually is obvious when the patient is first seen and should present no difficulties in diagnosis for the endocrinologist.67,68 In patients who have only a sore throat or ear pain, however, the diagnosis is less obvious, and many patients are initially misdiagnosed with pharyngitis.64 It is important that the thyroid gland be palpated carefully in patients presenting with upper respiratory infections or complaints of sore neck or throat or earache.
Eventually, patients with Hashimoto’s thyroiditis and a few with silent thyroiditis may present with a painful, tender thyroid enlargement that is indistinguishable from SAT.54 The radioactive iodine uptake is rarely as completely suppressed in Hashimoto’s thyroiditis as it is in SAT, and the titers of thyroid autoantibodies are usually high enough to suggest lymphocytic thyroiditis. Acute suppurative thyroiditis initially may mimic SAT, but with time, the findings of fever, more localized tenderness and swelling, and erythema over the involved area of the thyroid should become obvious. A rapidly growing anaplastic carcinoma77 of the thyroid or hemorrhage into a thyroid nodule can cause thyroid pain and tenderness. In anaplastic carcinomas, the lesion is usually obvious by virtue of its large size, adherence to adjacent structures, lymphadenopathy, and characteristic progressive course. Hemorrhage into a thyroid nodule presents with a localized nature, and the obvious nodule usually leads to the correct diagnosis.
The hallmark of SAT is a markedly elevated erythrocyte sedimentation rate. The serum thyroglobulin and C-reactive protein concentration are similarly elevated.78 The leukocyte count is normal or slightly elevated. Peripheral blood thyroid hormone concentrations are elevated, with ratios of thyroxine (T4) to triiodothyronine (T3) of less than 20, reflecting the proportions of stored hormone within the thyroid,79 and serum concentrations of thyrotropin are low or undetectable. Serum thyroid peroxidase antibody concentrations are usually normal. The 24-hour radioactive iodine uptake is low (<5%) in the toxic phase of SAT, distinguishing this disease from Graves’ disease. Color-flow Doppler ultrasonography also may help to make this distinction; in patients with Graves’ disease, the thyroid gland is hypervascular, whereas in patients with painful SAT, the gland is hypoechogenic and has low to normal vascularity.80
Laboratory and Imaging Findings
Dynamic changes in thyroid function studies occur with the onset of thyroid inflammation (Fig. 13-1). The destruction of the thyroid follicles results in release and breakdown of the colloid into the interstitial tissue and into the circulation of iodinated materials—protein, proteases, peptides, and amino acids. An increase in serum T4, T3, and thyroglobulin and in urinary iodine results.1–5,64,74
FIGURE 13-1 Salient laboratory features during the course of subacute thyroiditis. AMc, Antimicrosomal (antithyroperoxidase) antibody; T4, thyroxine; T3, triiodothyronine; TBII, thyrotropin-binding inhibitory immunoglobulin; Tg, thyroglobulin.
In addition, iodoproteins, such as thyroglobulin and iodoalbumin, are discharged from the gland into the circulation.69 Plasma thyroglobulin may remain elevated long after all other evidence of the inflammatory process has subsided.71 The decline in plasma T4 is exponential during the first week, and this phase of hyperthyroidism can continue only until the gland is depleted of its preformed colloid.73 TSH is usually undetectable in the hyperthyroid phase,72,73 and the TSH response to thyrotropin-releasing hormone, as expected, is diminished at this time.81,82
At the same time, the damage to the thyroid follicular cells results in impaired iodine transport; the 24-hour radioactive iodine uptake is characteristically suppressed to 0% to 1%, revealing a patchy and irregular distribution of the tracer.1–5,64,69,83,84 Even if only part of the gland is involved, the uptake may be similarly depressed as a result of suppression of pituitary TSH owing to the elevated levels of thyroid hormone.72,73 Increased perfusion is shown in studies with technetium-99m sestamibi during the acute stage of SAT. This increased uptake in the thyroid region suggests the inflammatory phase of this disease.85 SAT is one of the hyperthyroid conditions associated with high levels of thyroid hormones but a low radioactive iodine uptake, and such observations are characteristic in the early phase of this disorder. Under these circumstances, only minimal thyroid hormone biosynthesis is sustained, and what is produced leaks out.69
Evidently, thyroid cell damage reduces the ability of the gland to respond to TSH so that large doses of TSH generally do not cause a rise in the radioactive iodine uptake except when some parts of the gland are uninvolved.82 This lack of response to exogenous TSH administration persists during the first weeks of the disease, reflecting continuing thyroid cell impairment and failure of the iodide-concentrating mechanism. Also, the administration of perchlorate or thiocyanate generally does not cause release of excessive amounts of iodine from the gland.74
The erythrocyte sedimentation rate is characteristically elevated (often >100 mm/h) in SAT.1–5,86 If the test is normal or only slightly elevated, the diagnosis of SAT should be suspected. The leukocyte count is normal in about half of patients and elevated in the remainder1–5,8,86 and has been reported as high as 18 × 109/L. The leukocyte counts correlate with serum concentrations of granulocyte colony-stimulating factor.87 There may be a mild normochromic anemia, and an increase in α2-globulin frequently is seen as a nonspecific inflammatory response.88 Alkaline phosphatase and other hepatic enzymes may be elevated in the early phase.89 It has been suggested that SAT actually represents a multisystem disease also affecting the thyroid gland.90 There also are increases in serum ferritin,94 soluble intercellular adhesion molecule-1,95 selectin,96 and interleukin-697 levels during the inflammatory phase.
Ultrasound examinations show hypoechoic focal areas and can be used for guided fine-needle cytology.84–103 Magnetic resonance imaging of the thyroid also can help distinguish SAT from Graves’ disease during the hyperthyroid phase. The ADC values obtained from the diffusion-weighted images of the patients with Graves’ disease are significantly higher than the values of patients with SAT.104
Recovery Phase
As the process subsides, the serum T4, T3, and thyroglobulin levels decline, but the serum TSH level remains suppressed. The normal concentrations of sex hormone–binding globulin in the hyperthyroid phase probably reflect the short duration of exposure to increased thyroid hormone.105
Later, during the recovery phase, the radioiodine uptake becomes elevated with the resumption of the ability of the thyroid gland to concentrate iodide. The serum T4 concentration may fall below normal; the TSH level may become elevated. Usually, after several weeks or months, all the parameters of thyroid function return to normal. Restoration of iodine stores seems to be much slower and may take more than 1 year after the complete clinical remission.106,107 Ultimate recovery is the general rule. An occasional patient remains permanently hypothyroid.
Tests of thyroid antibodies are positive in a few cases; these develop several weeks after the onset and tend to decline and disappear thereafter.33,108 An antibody against an unpurified thyroid antigen persists for years, however, after clinical features have subsided.43 Also, as mentioned before, antibodies to the TSH receptor, either of the stimulating or of the blocking variety, may appear transiently without relationship to the thyroid functional state.40–42 In about 2% of patients, SAT may trigger autoreactive B cells to produce TSH receptor antibodies, resulting in TSH antibody–associated dysfunction.109
Pathology
From histologic examination, the process may be diffuse or irregular in its involvement, with various stages of the disease sometimes found within the same specimen.110 Initially, there is extensive follicular cell destruction, extravasation of colloid, and infiltration of lymphocytes and histiocytes. The lymphocytes and histiocytes tend to congregate around masses of colloid and coalesce into giant cells. With time, there is a variable degree of fibrosis, and areas of follicular regeneration are seen. After recovery, the thyroid appears normal except for minimal residual fibrosis.
The follicular cells sometimes virtually disappear, leaving a fine follicular lining. The initial phase is characterized by the appearance of neutrophils, followed by large mononuclear cells and lymphocytes (Fig. 13-2). The follicles appear much larger than normal, with disruption of the epithelial lining and hyperplasia of the surviving follicular cells. Histiocytes congregate around masses of colloid within the follicles and in the interstitial tissues, producing “giant cells.” Because often these giant cells actually consist of masses of colloid surrounded by large numbers of individual histiocytes, they should in such cases be termed pseudo–giant cells. True giant cells and granulomas also appear in this disease, however.111 Marked interstitial edema also is present with lymphocytic infiltration.
FIGURE 13-2 Pathologic findings in subacute thyroiditis. Note the severe destruction of the thyroid follicle, with the remaining colloid being surrounded by large numbers of histiocytes, giving the appearance of a giant cell (pseudo–giant cell). Marked interstitial edema is noted, with cellular infiltration and considerable destruction of the thyroid parenchyma.
The process often is irregularly distributed in either or both lobes.114 With recovery, the inflammatory reaction recedes, and a variable amount of fibrosis may appear. Areas of follicular regeneration are seen, but there is no caseation, hemorrhage, or calcification. The degree of recovery is generally virtually complete, aside from the residual fibrosis already mentioned. Only in rare instances is there complete destruction of the thyroid parenchyma leading to permanent hypothyroidism.
Thyroid tissue obtained by fine-needle aspiration biopsy (Fig. 13-3
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
