[level-membership-for-basic-science-category]
Stroke
Types of stroke
Stroke is caused by inadequate tissue perfusion or ischaemia (Greek: isch-, restriction; haema, blood). The brain is particularly sensitive to ischaemia because neurons have a high metabolic rate and can only survive a few minutes without oxygen and glucose. Tissue death as a result of inadequate blood flow (e.g. due to an occluded blood vessel) is called infarction (Latin: infarctus, stuffed). The area of dead tissue is referred to as ‘an infarct’. There are two main types of stroke (Figs 10.1 and 10.2):
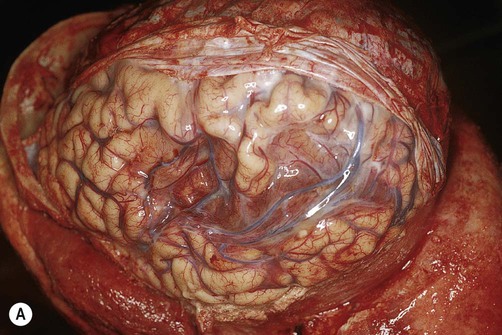
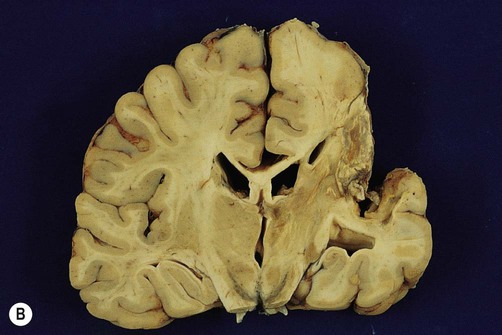
(A) Photograph taken during a post-mortem examination in a patient with an old (‘healed’) left hemisphere ischaemic infarct in the territory of the middle cerebral artery. Extensive resorption of brain tissue (leaving only a fluid-filled cyst cavity) indicates a prolonged period of survival after the stroke; (B) Coronal section of a fixed (preserved) brain slice in a different patient showing an old ischaemic stroke, also in the territory of the middle cerebral artery. From Ellison and Love: Neuropathology 2e (Mosby 2003) with permission.
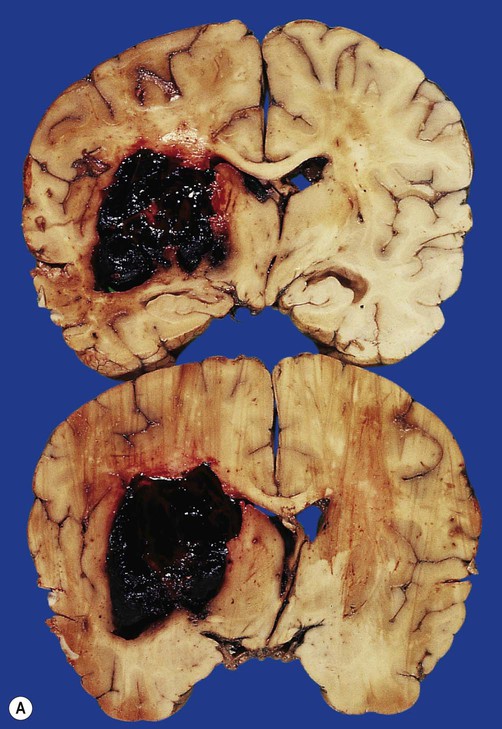
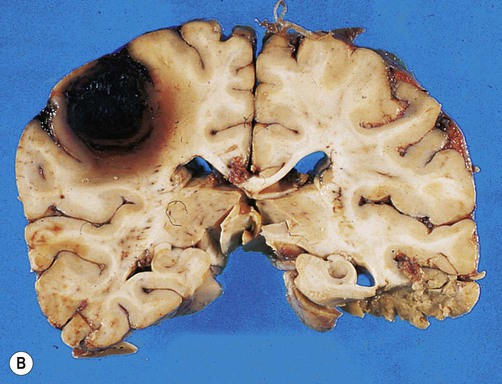
(A) Coronal sections showing a typical hypertension-associated haemorrhagic stroke replacing the basal ganglia; (B) Coronal section showing an atypical (‘lobar’) haemorrhage due to cerebral amyloid angiopathy affecting the cortical and meningeal arteries. Note the superficial (cortical) position of the atypical haemorrhage. From Ellison and Love: Neuropathology 2e (Mosby 2003) with permission.


In both types of stroke the brain tissue normally supplied by the vessel is suddenly deprived of blood and its function is lost. This causes focal neurological deficits that develop very rapidly (e.g. sudden weakness or loss of speech) and abrupt onset is the clinical hallmark of stroke. In haemorrhagic stroke, there may also be mechanical damage, such as tearing and compression of brain tissue, caused by blood escaping under arterial pressure.
Key Points



Ischaemic stroke
Large vessel and cardioembolic stroke
Cerebral blood vessels may be occluded by an embolus (Greek: embolos, wedge or plug). This is a small piece of coagulated blood (or occasionally some other material such as fat) that travels in the circulation and lodges in the vascular tree of the brain. Emboli often originate from the heart (cardioembolic stroke) in association with valve disease or an abnormal heart rhythm (Clinical Box 10.1). Disease in the neck vessels or aortic arch may also give rise to emboli.
 Clinical Box 10.1:
Clinical Box 10.1:Small vessel disease
Both types of pathology cause arteriosclerosis or ‘hardening’ of the arteries (Greek: sklerōs, hard). Sclerotic vessels are unable to dilate in response to reduced flow, leading to lacunar infarcts (see Ch. 12, Figs 12.18 and 12.19). These are small areas of ischaemic tissue damage (often in the basal ganglia or internal capsule) measuring less than 1 cm in diameter (Latin: lacūna, hole or gap). Small vessel disease is also an important cause of vascular cognitive impairment and dementia (Ch. 12).
Haemorrhagic stroke
Spontaneous intracerebral haemorrhage is more common in people with high blood pressure and frequently occurs in the basal ganglia, cerebellum or pons (Fig. 10.2A). However, the incidence of hypertensive intracerebral haemorrhage is declining in the UK and USA and an increasingly important cause is cerebral amyloid angiopathy, particularly in the elderly (see Ch. 12, Clinical Box 12.5). This is caused by deposition of amyloid beta peptide in the walls of cortical and meningeal arteries, leading to atypical (or lobar) haemorrhages that are close to the cortical surface (Fig. 10.2B).
Up to a third of spontaneous intracranial haemorrhages are caused by rupture of an aneurysm (a dilatation in the wall of an artery). Since the cerebral blood vessels travel and branch within the subarachnoid space, this results in subarachnoid haemorrhage (Clinical Box 10.2). Aneurysms that rupture in the substance of the brain may cause devastating brain damage or sudden death.
 Clinical Box 10.2: Subarachnoid haemorrhage
Clinical Box 10.2: Subarachnoid haemorrhage
Bleeding into the subarachnoid space accounts for a third of intracranial haemorrhages. Although this does not necessarily cause a focal neurological deficit, subarachnoid haemorrhage is normally regarded as a type of stroke, accounting for 5% of cases overall. It classically presents with a severe headache (‘the worst headache I have ever had’). The most common cause is a ruptured berry aneurysm, a medical emergency with a high mortality rate (Fig. 10.3). Berry aneurysms are small vascular swellings which develop at congenital weak spots in the arterial wall. They are usually found at points where blood vessels branch, especially in the circle of Willis at the base of the brain. Ischaemic stroke may occur as a late complication. This is due to blood in the subarachnoid space acting as an irritant and triggering vasospasm. Unruptured berry aneurysms are found incidentally in up to 2% of post-mortem examinations.
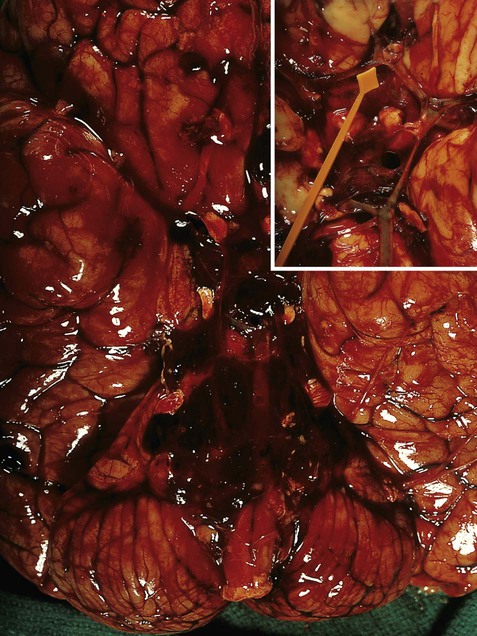
Post-mortem photograph of the brain (ventral aspect) in a patient who collapsed and died after a sudden, severe headache. The subarachnoid space at the base of the brain is filled with fresh blood, obscuring the circle of Willis. Inset: after removal of the blood a ruptured berry aneurysm was found in the region of the anterior communicating artery, indicated by the pointer. From Ellison and Love: Neuropathology 2e (Mosby 2003) with permission.






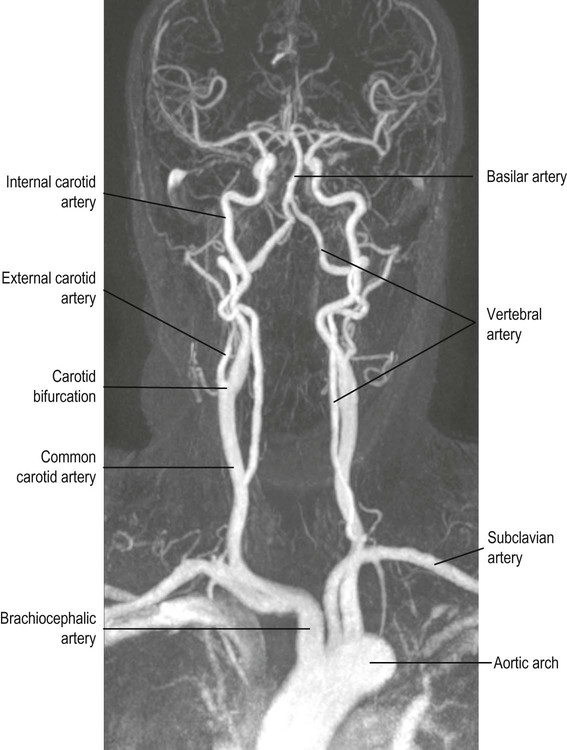
Blood supply to the brain
The brain is supplied by two pairs of arteries that arise from branches of the aortic arch (Fig. 10.4):


The arteries at the base of the brain are linked by communicating vessels to form the circle of Willis. This gives rise to anterior, middle and posterior cerebral arteries on each side, which supply most of the cerebral hemisphere. The cerebral blood supply can be divided into anterior and posterior circulations (Fig. 10.5A).
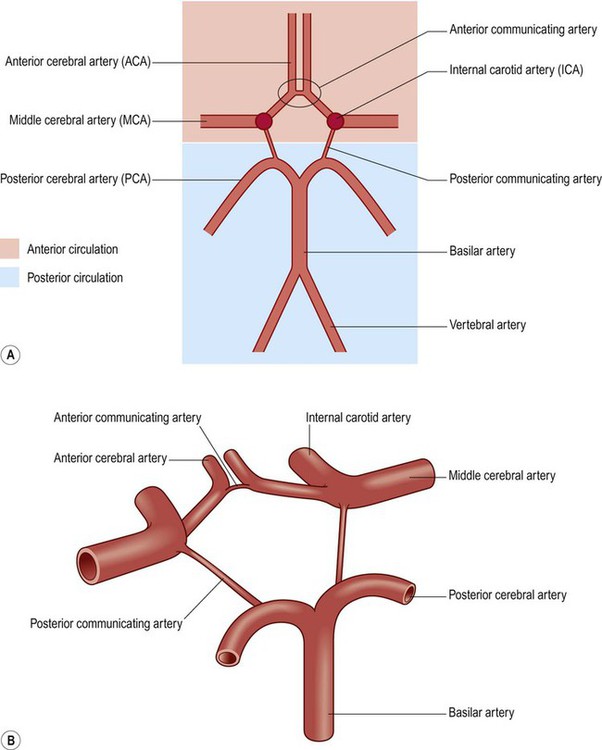
(A) Schematic, two-dimensional, representation of the circle of Willis, showing the anterior (internal carotid) and posterior (vertebrobasilar) circulations; (B) Illustration of the three-dimensional arrangement of the circle of Willis.
Anterior circulation
The internal carotid artery divides at the base of the brain, giving rise to the middle cerebral artery (MCA) and the anterior cerebral artery (ACA). The MCA is the larger of the two and receives 80% of the internal carotid blood flow. For this reason, cardiogenic emboli are much more likely to enter the MCA than the ACA. The middle cerebral artery continues laterally between the frontal and temporal lobes and emerges from the lateral sulcus to supply most of the hemispheric convexity (Figs 10.6A and 10.7).
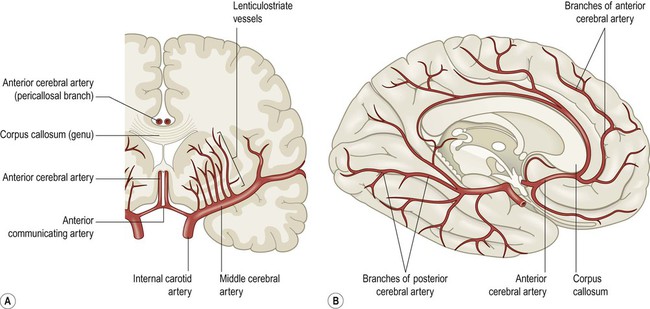
(A) Coronal view of the cerebral hemisphere showing the anterior and middle cerebral arteries. The lenticulostriate (deep perforating) vessels can be seen arising at right-angles from the middle cerebral artery; (B) Midsagittal view of the cerebral hemisphere showing the anterior and posterior cerebral arteries.
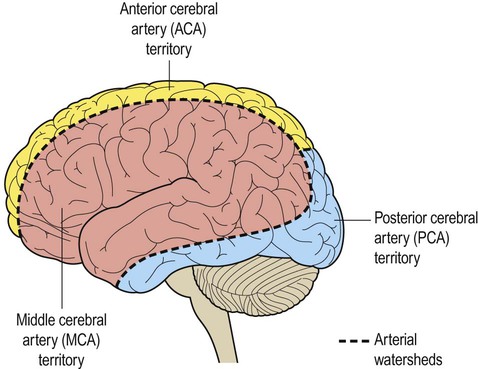
The dashed line indicates the arterial watershed zone between the main vascular territories.
The anterior cerebral artery passes forward and medially to meet its partner between the cerebral hemispheres. It winds around the corpus callosum and supplies the medial aspect of the hemisphere as far back as the parieto-occipital sulcus (Figs 10.6 and 10.8). Posterior to this point the posterior cerebral artery takes over to supply the occipital lobe. Perforating branches of the anterior circulation supply the optic radiations, so that anterior circulation strokes may cause a contralateral visual field defect.
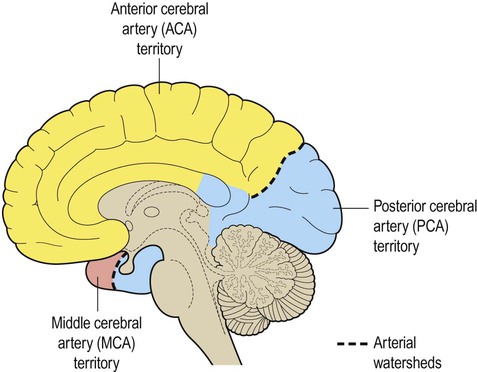
Posterior circulation
The posterior circulation supplies the remainder of the cerebral hemisphere and the posterior fossa contents (brain stem and cerebellum). The two vertebral arteries unite in front of the brain stem, at the junction between pons and medulla, to become the basilar artery. For this reason, the posterior circulation is also known as the vertebrobasilar circulation. The basilar artery ascends along the basal pons to reach the midbrain where it splits into the two posterior cerebral arteries (PCA). These vessels wind around the midbrain and pass posteriorly, above the tentorium cerebelli, to supply the occipital lobe and the inferior surface of the temporal lobe (Figs 10.6B, 10.7 and 10.8).
Circle of Willis
The circle of Willis is a polygonal arrangement of blood vessels surrounding the optic chiasm and pituitary stalk. It connects the anterior and posterior circulations via the single anterior communicating artery and the paired posterior communicating arteries (Figs 10.5B and 10.9). The circle of Willis shows considerable anatomical variation and is incomplete in 50% of people. It is an example of a collateral circulation, an arrangement of interconnected vascular channels permitting blood flow via an alternative route in the event of an obstruction. Interconnections between vessels exist elsewhere (e.g. between the distal territories of the three main cerebral blood vessels) but are not always effective, particularly in the elderly. Some structures do not have a collateral blood supply (e.g. the internal capsule and basal ganglia) and are therefore more vulnerable to stroke.
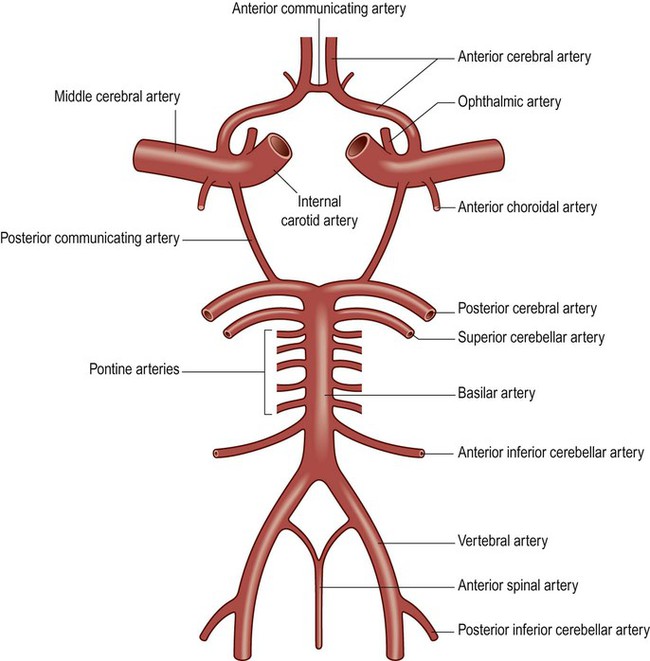
The ophthalmic artery is the first branch of the intracranial portion of the internal carotid artery and supplies the orbital contents. The anterior choroidal artery is the final branch of the internal carotid artery (or an early branch of the MCA); despite being a small vessel, it supplies portions of the central visual pathways, medial temporal lobe (e.g. hippocampus, amygdala) and part of the basal ganglia and internal capsule. The anterior spinal artery supplies the basal portion of the medulla and the anterior two thirds of the spinal cord.





Perforating vessels
The circle of Willis gives rise to a number of central perforating vessels which supply the internal substance of the brain. Perforating vessels from the anterior circulation enter the brain just lateral to the optic chiasm on each side, via the anterior perforated area. A second group arises from the posterior circulation and enters the brain at the interpeduncular fossa of the midbrain, via the posterior perforated area. The anterior group includes the lenticulostriate vessels (or ‘arteries of stroke’) which supply the basal ganglia and internal capsule (Fig. 10.6A). They are an important site of spontaneous intracerebral haemorrhage and lacunar infarcts (both caused by small-vessel disease).
The arterial territories of the three main cerebral vessels and deep perforating vessels are illustrated in coronal section in Figure 10.10. The regions between territories are the watershed zones and are particularly vulnerable to a sudden reduction in arterial blood pressure. This may lead to a characteristic pattern of watershed infarction at the arterial border zones following profound arterial hypotension (e.g. after cardiorespiratory arrest, anaphylactic shock or major blood loss).
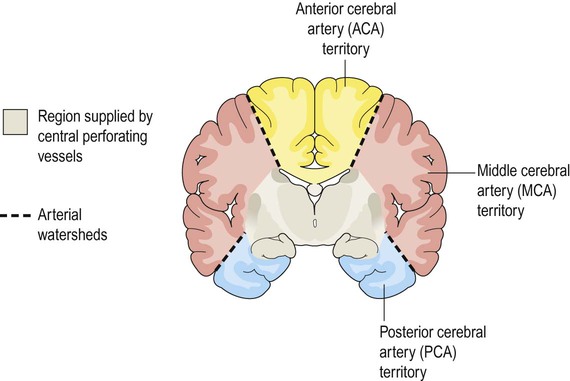
The grey zone is supplied by deep (perforating) branches of the circle of Willis.
Blood supply to the cerebellum
The cerebellum is supplied by three long circumferential vessels that arise from different parts of the posterior circulation (Figs 10.9 and 10.11):
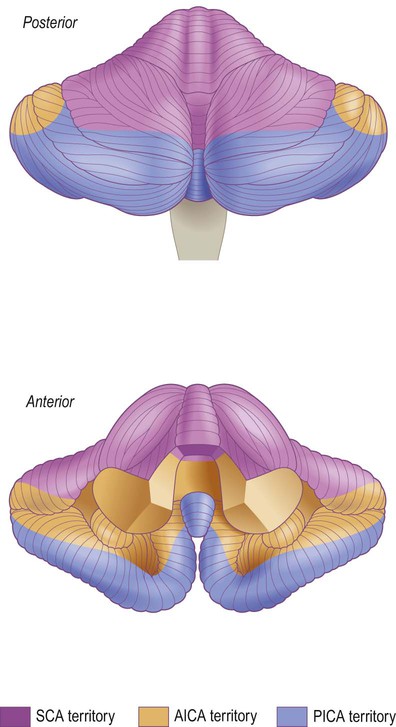
The vascular territories of the three main blood vessels supplying the cerebellum are illustrated. The superior cerebellar artery (SCA) and anterior inferior cerebellar artery (AICA) both arise from the basilar artery; the posterior inferior cerebellar artery (PICA) usually arises from the vertebral artery.
 The superior cerebellar artery (SCA) arises from the upper part of the basilar artery, immediately below the origin of the posterior cerebral artery.
The superior cerebellar artery (SCA) arises from the upper part of the basilar artery, immediately below the origin of the posterior cerebral artery.
 The anterior inferior cerebellar artery (AICA) is a caudal (inferior) branch of the basilar artery.
The anterior inferior cerebellar artery (AICA) is a caudal (inferior) branch of the basilar artery.
 The posterior inferior cerebellar artery (PICA) usually arises from the vertebral artery.
The posterior inferior cerebellar artery (PICA) usually arises from the vertebral artery.
The posterior inferior cerebellar artery also supplies the lateral medulla. Occlusion of this vessel is associated with the lateral medullary syndrome (Clinical Box 10.3).
 Clinical Box 10.3: Lateral medullary syndrome
Clinical Box 10.3: Lateral medullary syndrome
Occlusion of the posterior inferior cerebellar artery (PICA), supplying the lateral part of the medulla, causes: (i) ipsilateral loss of pain and temperature sensation in the face; and (ii) contralateral loss of pain and temperature sensationin in the trunk and limbs. This results from damage to the trigeminal sensory nucleus and spinothalamic tract respectively (see Ch. 4). Sympathetic fibres passing from the hypothalamus to the spinal cord may also be affected, leading to loss of sympathetic innervation to the ipsilateral face. This is one cause of Horner’s syndrome, characterized by ptosis (drooping eyelid), miosis (small, constricted pupil) and facial anhidrosis (loss of sweating). Other features of the lateral medullary syndrome reflect damage to local brain stem structures and connections with the cerebellum: difficulty swallowing, double vision, poor coordination, vertigo and nausea.
Stroke syndromes
For practical purposes, ischaemic strokes can be classified into one of four major clinical categories based on the maximum deficit following a single stroke, illustrated in Figure 10.12:
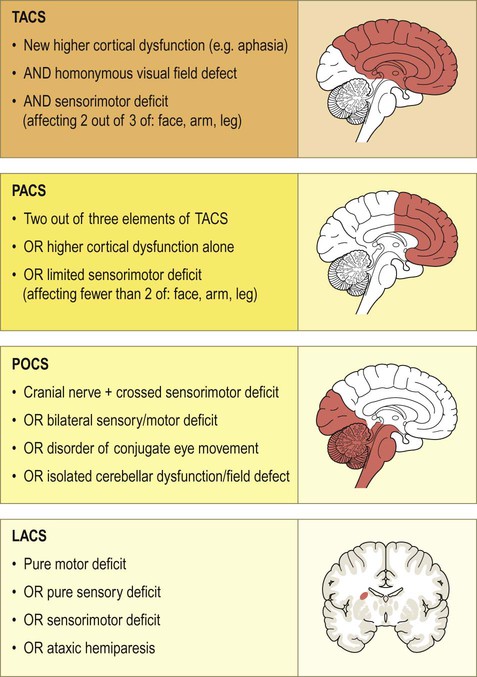
Ischaemic strokes can be classified as a total anterior circulation syndrome (TACS), partial anterior circulation syndrome (PACS), posterior circulation syndrome (POCS) or lacunar syndrome (LACS). The classification is based on the maximum observed deficit following a single stroke. For the lacunar syndrome, the sensorimotor deficit must affect at least two out of three areas (i.e. leg, arm and face).




This system is easy to use and provides useful prognostic information. For example, the proportion of patients dying within the first year in each type is approximately 60% (TACS), 20% (PACS or POCS) and 10% (LACS). In the first week after a stroke, the most common cause of death is raised intracranial pressure due to brain swelling (cerebral oedema); this causes herniation of the cerebellar tonsils through the skull base, compressing the brain stem (which is referred to as ‘coning’, see Ch. 9).





Atherosclerosis
Atheromatous plaques
The characteristic lesion in atherosclerosis is called an atheroma (from the Latin word for porridge). This is a deposit of lipid-rich material in the vessel wall that develops over many years. The process begins in the innermost (intimal) layer of large arteries and slowly expands. Atheromatous deposits therefore cause progressive stenosis (narrowing) of the vessel (Fig. 10.13). This gradually impinges on the lumen, but may remain asymptomatic for many years. The precursor lesions in atherosclerosis are known as fatty streaks. These are pale areas beneath the arterial endothelium that can be identified in the arteries of children and consist of lipid-laden macrophages.
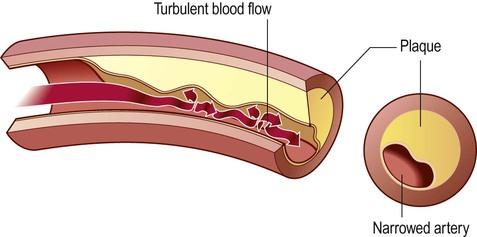
In a normal artery, the blood flow is smooth and laminar, but development of atheromatous plaques causes progressive stenosis (narrowing) of the arterial lumen and leads to abnormal, turbulent blood flow.
The distribution of atheromatous plaques is not random. Areas exposed to rapid, turbulent blood flow are particularly susceptible, especially arterial branch-points. These include the aortic arch and large arteries of the neck, both of which may be a source of emboli to the brain (Clinical Box 10.4). Atherosclerosis is also common in the intracranial portion of the internal carotid artery and at the origin and major branch-points of the middle cerebral artery.
 Clinical Box 10.4:
Clinical Box 10.4:
Plaque formation
Plaque progression
Inflammatory mediators released by activated platelets recruit monocytes from the bloodstream. These migrate into the vessel wall and differentiate into macrophages where they take up cholesterol and lipids to become foam cells. Growth factors and cytokines released by foam cells initiate a cascade of pathological events in which there is further accumulation of cholesterol and proliferation of intimal smooth muscle. This process is accelerated if the serum cholesterol is high (Clinical Box 10.5). Smooth muscle cells secrete extracellular matrix proteins including collagen, which generates a tough fibrous cap over the soft plaque core, creating a relatively stable fibrous plaque (Fig. 10.14A). If the fibrous cap ruptures or the overlying endothelial layer becomes ulcerated, the plaque is referred to as complicated (Fig. 10.14B). This may trigger thrombosis, leading to vessel occlusion and stroke.
 Clinical Box 10.5:
Clinical Box 10.5: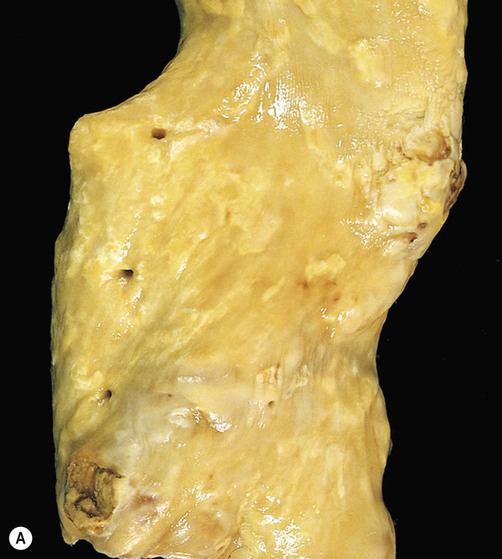
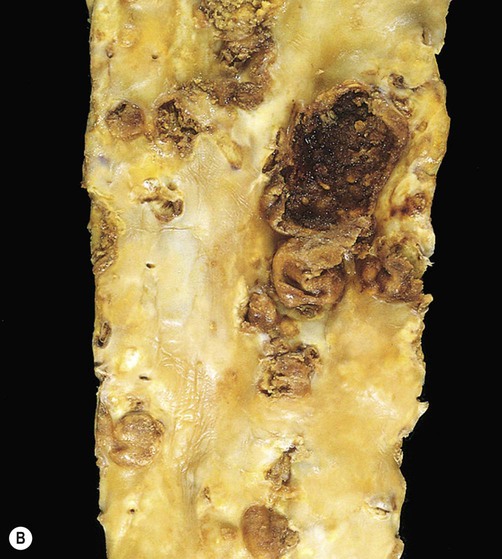
(A) This image shows numerous yellow-white fibrous plaques in the wall of the aorta. These lesions have a cholesterol-rich core covered by a collagenous cap and they often remain stable (without causing symptoms) for many years; (B) This example (in a different patient) shows complicated atheromatous plaques, in which there has been rupture/haemorrhage of the collagenous cap or ulceration of the overlying endothelium – in this case adherent thrombus is clearly visible on some of the plaques. From Kumar et al: Robbins Basic Pathology 8e (Saunders 2007) with permission.





Stroke management




The collective impact of these simple measures is a substantial reduction in long-term neurological impairment, with a 25% increase in the number of people returning to work. A small proportion of patients with ischaemic stroke may be suitable for thrombolysis (Clinical Box 10.6).
 Clinical Box 10.6:
Clinical Box 10.6:Stroke prevention
Risk factors for stroke
Arterial hypertension is the most important factor and clinical trials show that a 5 mmHg reduction in diastolic blood pressure cuts stroke risk by around a third, even in patients with normal blood pressure. Decreasing serum cholesterol is also highly beneficial, and even people with normal levels may benefit from cholesterol-lowering agents such as statins (inhibitors of the cholesterol-synthesizing liver enzyme hydroxy-methyl-glutaryl-CoA reductase). Reduced consumption of saturated animal fats is also helpful, together with increased intake of polyunsaturated vegetable fats and fish oils (containing omega-3 fatty acids). In patients with diabetes, who are particularly predisposed to arterial disease, good control of blood sugar is essential. Daily low-dose aspirin and other antiplatelet agents may also of value (Clinical Box 10.7).
 Clinical Box 10.7:
Clinical Box 10.7:Other factors
Oral contraceptives with a high oestrogen content increase relative stroke risk by enhancing the tendency of blood to coagulate, but since the background risk is tiny in young females, the absolute risk remains extremely low. Elevation of the amino acid homocysteine in the serum, as a result of folate or vitamin B deficiency or in the rare genetic condition homocysteineuria, is also associated with premature vascular disease, but it is not clear if reducing homocysteine levels is beneficial in preventing stroke. Modest alcohol intake may reduce stroke risk (perhaps in part by increasing plasma HDL concentration) but consuming more than two units per day (i) begins to reverse the benefits, (ii) has other negative effects on health and (iii) is an important cause of haemorrhagic stroke in younger people (Fig. 10.15).
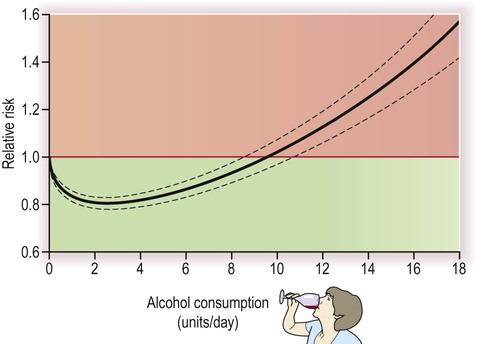
With increasing levels of consumption, the negative effects quickly outweigh the benefits. This is described as a J-shaped relationship.





Pathophysiology of stroke
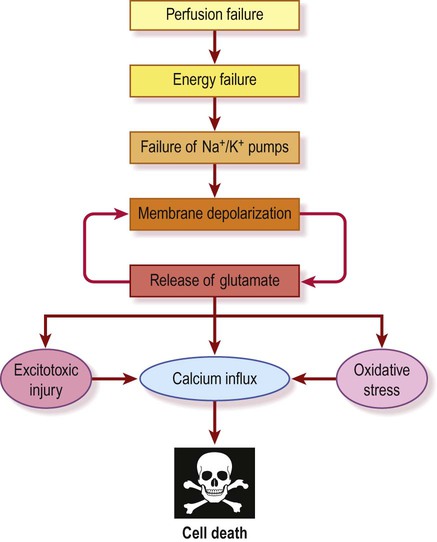
The ischaemic cascade (Fig. 10.16)
Anaerobic respiration
In the absence of oxygen, cellular metabolism switches from aerobic respiration to anaerobic glycolysis. This extracts much less energy from the limited glucose available and generates lactate as a by-product, exacerbating the developing acidosis. The mitochondrial Na+/H+ pump, which expels hydrogen ions from the cell, is retarded as the pH falls below 6.0. This compromises cellular respiration further and a vicious cycle develops. Under these conditions harmful reactive oxygen species such as superoxide anion (O2−) and hydroxyl radical (OH−) are liberated in large quantities, creating oxidative stress and further tissue damage (see Ch. 8).
Failure of the sodium pump
The sodium pump (Na+/K+-ATPase) is responsible for two thirds of the basal energy expenditure of the brain. It works constantly in the background to maintain the intracellular and extracellular sodium and potassium ion concentrations and is therefore essential for electrical activity in nerve cells (see Ch. 6). As the ATP supply falls to critical levels, the sodium pump fails. As a result, the sodium and potassium gradients dissipate and neuronal membranes depolarize. A critical point is reached when cerebral blood flow falls to less than 20% of normal (below 10 mL per 100 g per minute). Depolarized neurons reach their ‘firing thresholds’ and there is large-scale release of neurotransmitters, including the excitatory amino acid glutamate which is toxic at high concentrations.
Calcium and excitotoxicity
As a result of these events the extracellular glutamate concentration is increased to ten times its normal value. At these levels glutamate behaves as an excitotoxin by allowing excessive calcium influx through NMDA channels (which have been liberated from their normal magnesium block) together with contributions from voltage-gated calcium channels and calcium-permeable AMPA receptors (see Chs 7 and 8). Elevation of intracellular calcium is a critical event in neuronal cell death since calcium activates a host of harmful enzymes, including calpains, proteases, phospholipases, endonucleases and nitric oxide synthase (see Ch. 8). The final ‘point of no return’ occurs as plasma membrane integrity is lost and water enters the cell. This is called cytotoxic oedema which causes cell rupture and death, followed by an inflammatory response.






The ischaemic penumbra
Within an hour of stroke onset a necrotic core lesion is established in which the blood flow is below 20% of normal. This is surrounded by a poorly perfused border zone known as the ischaemic penumbra in which blood flow is between 20–40% of normal (Fig. 10.17). Penumbral neurons may remain viable for up to 24 hours and can potentially be salvaged, but if perfusion is not restored the core lesion will gradually expand.
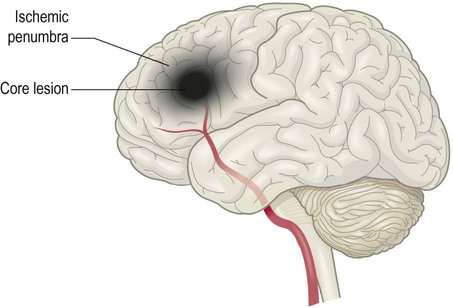
Following an ischaemic stroke there is often a core lesion of irreversible cell death, surrounded by a poorly perfused penumbral region in which neurons are functionally compromised but may still be salvaged.



Neuroprotection
Animal models of stroke
Organotypic cultures
This method employs intact brain slices (e.g. rodent hippocampus) that are maintained in an incubated culture medium. Preparations of this kind can remain viable for as long as six weeks, maintaining normal connectivity and activity (Fig. 10.18A). Cerebral ischaemia can be simulated by combined oxygen–glucose deprivation, producing reproducible lesions that compare well with their in vivo counterparts (Fig. 10.18B). Slice cultures therefore combine many of the positive characteristics of whole animal studies without the major drawbacks of dissociated cell cultures.
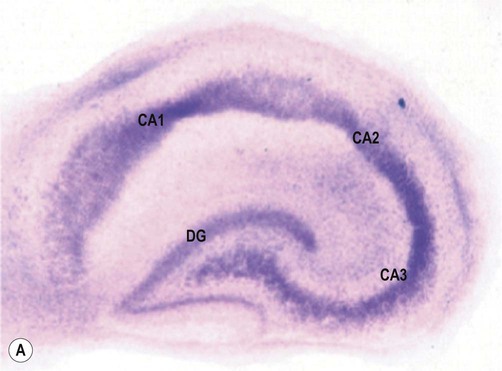

(A) A slice culture preparation of rodent hippocampus after two weeks in culture showing normal hippocampal anatomy. The tissue slice has been stained with thionine to demonstrate neuronal cell bodies [DG= dentate gyrus, CA1-3 = subfields of Ammon’s horn/cornu Ammonis]; (B) Rodent hippocampal slice 24 hours after a 60-minute period of combined oxygen-glucose deprivation (simulated ischaemia). There is marked neuronal loss in the CA1 subfield of the hippocampus [visualized with fluorescent tissue dye propidium iodide, which is only able to enter damaged cells with impaired membrane integrity]. The CA1 subfield (sometimes referred to as Sommer’s sector) is also particularly susceptible to hypoxia and ischaemia in the human brain. From Sundstrom et al: Drug Discovery Today 2005;10(14), with permission.
Neuroprotective agents
Failure of drug delivery
Agents travelling in the bloodstream may not be able to reach the targeted area of the brain, either due to poor perfusion of ischaemic tissues or failure to cross the blood–brain barrier (see Ch. 5), but this is not relevant in many animal models. Furthermore, side effects of certain drugs can prevent administration of effective doses that are equivalent to those used in preclinical studies.






[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Stroke
Types of stroke
Stroke is caused by inadequate tissue perfusion or ischaemia (Greek: isch-, restriction; haema, blood). The brain is particularly sensitive to ischaemia because neurons have a high metabolic rate and can only survive a few minutes without oxygen and glucose. Tissue death as a result of inadequate blood flow (e.g. due to an occluded blood vessel) is called infarction (Latin: infarctus, stuffed). The area of dead tissue is referred to as ‘an infarct’. There are two main types of stroke (Figs 10.1 and 10.2):
(A) Photograph taken during a post-mortem examination in a patient with an old (‘healed’) left hemisphere ischaemic infarct in the territory of the middle cerebral artery. Extensive resorption of brain tissue (leaving only a fluid-filled cyst cavity) indicates a prolonged period of survival after the stroke; (B) Coronal section of a fixed (preserved) brain slice in a different patient showing an old ischaemic stroke, also in the territory of the middle cerebral artery. From Ellison and Love: Neuropathology 2e (Mosby 2003) with permission.
(A) Coronal sections showing a typical hypertension-associated haemorrhagic stroke replacing the basal ganglia; (B) Coronal section showing an atypical (‘lobar’) haemorrhage due to cerebral amyloid angiopathy affecting the cortical and meningeal arteries. Note the superficial (cortical) position of the atypical haemorrhage. From Ellison and Love: Neuropathology 2e (Mosby 2003) with permission.
In both types of stroke the brain tissue normally supplied by the vessel is suddenly deprived of blood and its function is lost. This causes focal neurological deficits that develop very rapidly (e.g. sudden weakness or loss of speech) and abrupt onset is the clinical hallmark of stroke. In haemorrhagic stroke, there may also be mechanical damage, such as tearing and compression of brain tissue, caused by blood escaping under arterial pressure.
Key Points
Ischaemic stroke
Large vessel and cardioembolic stroke
Cerebral blood vessels may be occluded by an embolus (Greek: embolos, wedge or plug). This is a small piece of coagulated blood (or occasionally some other material such as fat) that travels in the circulation and lodges in the vascular tree of the brain. Emboli often originate from the heart (cardioembolic stroke) in association with valve disease or an abnormal heart rhythm (Clinical Box 10.1). Disease in the neck vessels or aortic arch may also give rise to emboli.
Small vessel disease
Both types of pathology cause arteriosclerosis or ‘hardening’ of the arteries (Greek: sklerōs, hard). Sclerotic vessels are unable to dilate in response to reduced flow, leading to lacunar infarcts (see Ch. 12, Figs 12.18 and 12.19). These are small areas of ischaemic tissue damage (often in the basal ganglia or internal capsule) measuring less than 1 cm in diameter (Latin: lacūna, hole or gap). Small vessel disease is also an important cause of vascular cognitive impairment and dementia (Ch. 12).
Haemorrhagic stroke
Spontaneous intracerebral haemorrhage is more common in people with high blood pressure and frequently occurs in the basal ganglia, cerebellum or pons (Fig. 10.2A). However, the incidence of hypertensive intracerebral haemorrhage is declining in the UK and USA and an increasingly important cause is cerebral amyloid angiopathy, particularly in the elderly (see Ch. 12, Clinical Box 12.5). This is caused by deposition of amyloid beta peptide in the walls of cortical and meningeal arteries, leading to atypical (or lobar) haemorrhages that are close to the cortical surface (Fig. 10.2B).
Up to a third of spontaneous intracranial haemorrhages are caused by rupture of an aneurysm (a dilatation in the wall of an artery). Since the cerebral blood vessels travel and branch within the subarachnoid space, this results in subarachnoid haemorrhage (Clinical Box 10.2). Aneurysms that rupture in the substance of the brain may cause devastating brain damage or sudden death.
Clinical Box 10.2: Subarachnoid haemorrhage
Bleeding into the subarachnoid space accounts for a third of intracranial haemorrhages. Although this does not necessarily cause a focal neurological deficit, subarachnoid haemorrhage is normally regarded as a type of stroke, accounting for 5% of cases overall. It classically presents with a severe headache (‘the worst headache I have ever had’). The most common cause is a ruptured berry aneurysm, a medical emergency with a high mortality rate (Fig. 10.3). Berry aneurysms are small vascular swellings which develop at congenital weak spots in the arterial wall. They are usually found at points where blood vessels branch, especially in the circle of Willis at the base of the brain. Ischaemic stroke may occur as a late complication. This is due to blood in the subarachnoid space acting as an irritant and triggering vasospasm. Unruptured berry aneurysms are found incidentally in up to 2% of post-mortem examinations.
Post-mortem photograph of the brain (ventral aspect) in a patient who collapsed and died after a sudden, severe headache. The subarachnoid space at the base of the brain is filled with fresh blood, obscuring the circle of Willis. Inset: after removal of the blood a ruptured berry aneurysm was found in the region of the anterior communicating artery, indicated by the pointer. From Ellison and Love: Neuropathology 2e (Mosby 2003) with permission.
Blood supply to the brain
The brain is supplied by two pairs of arteries that arise from branches of the aortic arch (Fig. 10.4):
The arteries at the base of the brain are linked by communicating vessels to form the circle of Willis. This gives rise to anterior, middle and posterior cerebral arteries on each side, which supply most of the cerebral hemisphere. The cerebral blood supply can be divided into anterior and posterior circulations (Fig. 10.5A).
(A) Schematic, two-dimensional, representation of the circle of Willis, showing the anterior (internal carotid) and posterior (vertebrobasilar) circulations; (B) Illustration of the three-dimensional arrangement of the circle of Willis.
Anterior circulation
The internal carotid artery divides at the base of the brain, giving rise to the middle cerebral artery (MCA) and the anterior cerebral artery (ACA). The MCA is the larger of the two and receives 80% of the internal carotid blood flow. For this reason, cardiogenic emboli are much more likely to enter the MCA than the ACA. The middle cerebral artery continues laterally between the frontal and temporal lobes and emerges from the lateral sulcus to supply most of the hemispheric convexity (Figs 10.6A and 10.7).
(A) Coronal view of the cerebral hemisphere showing the anterior and middle cerebral arteries. The lenticulostriate (deep perforating) vessels can be seen arising at right-angles from the middle cerebral artery; (B) Midsagittal view of the cerebral hemisphere showing the anterior and posterior cerebral arteries.
The dashed line indicates the arterial watershed zone between the main vascular territories.
The anterior cerebral artery passes forward and medially to meet its partner between the cerebral hemispheres. It winds around the corpus callosum and supplies the medial aspect of the hemisphere as far back as the parieto-occipital sulcus (Figs 10.6 and 10.8). Posterior to this point the posterior cerebral artery takes over to supply the occipital lobe. Perforating branches of the anterior circulation supply the optic radiations, so that anterior circulation strokes may cause a contralateral visual field defect.
Posterior circulation
The posterior circulation supplies the remainder of the cerebral hemisphere and the posterior fossa contents (brain stem and cerebellum). The two vertebral arteries unite in front of the brain stem, at the junction between pons and medulla, to become the basilar artery. For this reason, the posterior circulation is also known as the vertebrobasilar circulation. The basilar artery ascends along the basal pons to reach the midbrain where it splits into the two posterior cerebral arteries (PCA). These vessels wind around the midbrain and pass posteriorly, above the tentorium cerebelli, to supply the occipital lobe and the inferior surface of the temporal lobe (Figs 10.6B, 10.7 and 10.8).
Circle of Willis
The circle of Willis is a polygonal arrangement of blood vessels surrounding the optic chiasm and pituitary stalk. It connects the anterior and posterior circulations via the single anterior communicating artery and the paired posterior communicating arteries (Figs 10.5B and 10.9). The circle of Willis shows considerable anatomical variation and is incomplete in 50% of people. It is an example of a collateral circulation, an arrangement of interconnected vascular channels permitting blood flow via an alternative route in the event of an obstruction. Interconnections between vessels exist elsewhere (e.g. between the distal territories of the three main cerebral blood vessels) but are not always effective, particularly in the elderly. Some structures do not have a collateral blood supply (e.g. the internal capsule and basal ganglia) and are therefore more vulnerable to stroke.
The ophthalmic artery is the first branch of the intracranial portion of the internal carotid artery and supplies the orbital contents. The anterior choroidal artery is the final branch of the internal carotid artery (or an early branch of the MCA); despite being a small vessel, it supplies portions of the central visual pathways, medial temporal lobe (e.g. hippocampus, amygdala) and part of the basal ganglia and internal capsule. The anterior spinal artery supplies the basal portion of the medulla and the anterior two thirds of the spinal cord.
Key Points
 The anterior (internal carotid) and posterior (vertebrobasilar) circulations are united at the base of the brain to form the circle of Willis. This is an example of a collateral circulation.
The anterior (internal carotid) and posterior (vertebrobasilar) circulations are united at the base of the brain to form the circle of Willis. This is an example of a collateral circulation.
[/not-level-membership-for-basic-science-category]
