Parkinson’s disease
Clinical features
Most cases of Parkinson’s disease are idiopathic (meaning that the cause is not known). The main symptoms and signs of idiopathic Parkinson’s disease (IPD) are illustrated in Figure 13.1. The central feature is akinesia or poverty of movement (Greek: a-, without; kinesis, movement) together with marked muscular rigidity. It is therefore classified as an akinetic-rigid syndrome. Another prominent component is bradykinesia, meaning that movements are slow and deliberate (Greek: bradys, slow). In most cases there is also a coarse tremor. Parkinson’s disease is sometimes referred to as an extrapyramidal movement disorder since the pyramidal (primary motor) pathway is unaffected.
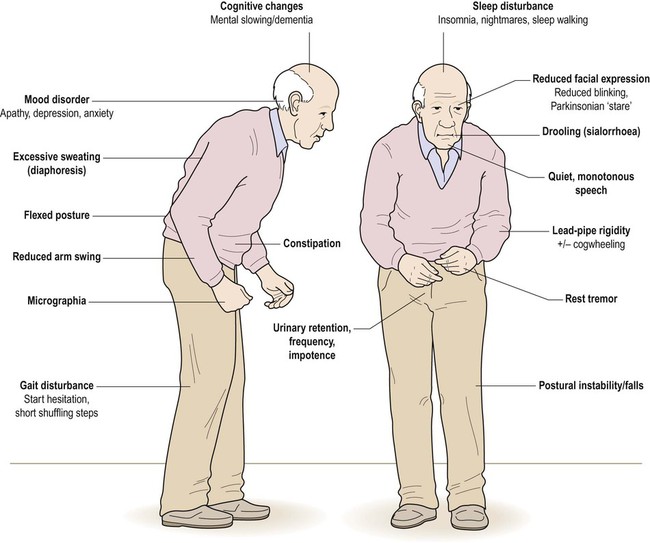
The combination of a flexed posture, slow shuffling gait, mask-like facial expression and unilateral tremor is highly characteristic.
Muscular rigidity
Increased muscle tone (rigidity) may present as stiffness, muscle pain or fatigue. On examination, there is uniform resistance to joint flexion and extension which has been compared to bending a piece of lead pipe. In contrast to spasticity, lead-pipe rigidity is constant (not velocity-dependent) and may be due to over-activity in the long-latency component of the stretch reflex (see Ch. 4).
Rest tremor
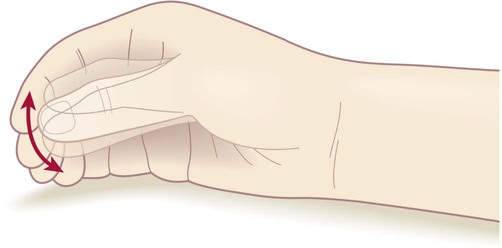
Tremor is a rhythmic ‘back-and-forth’ movement in the limbs, head or jaw and occurs in 75% of patients with Parkinson’s disease. The parkinsonian tremor is usually asymmetric and often begins in one hand or arm. It is classified as a rest tremor because it is much more prominent between movements. It is of large amplitude and low frequency (4–6 Hz) and is not present during sleep. Some patients have a classical ‘pill-rolling’ tremor (Fig. 13.2) which is strongly suggestive of idiopathic Parkinson’s disease. The combination of lead-pipe rigidity and tremor creates a jerky or ‘ratchet-like’ sensation on examination. This is termed cogwheeling and is best appreciated at the wrist.
Other features
Non-motor symptoms in Parkinson’s disease reflect: (i) the role of the basal ganglia in cognition, emotion and behaviour (see Ch. 3); and (ii) the presence of widespread pathological changes in the brain stem, limbic lobe and neocortex. Anxiety, depression or apathy occurs in 40% of patients. There may be sleep disorders including: nocturnal hallucinations, excessive daytime somnolence, vivid dreams, nightmares or sleepwalking. Subtle cognitive changes are common, such as bradyphrenia (generalized slowing of thought) or executive dysfunction (difficulty with organization, planning and decision-making). One in five patients will eventually be diagnosed with dementia (Clinical Box 13.1).
 Clinical Box 13.1: Dementia in Parkinson’s disease
Clinical Box 13.1: Dementia in Parkinson’s disease
Dementia occurs in at least 20% of patients with Parkinson’s disease, usually after a number of years. If cognitive decline appears 12 months or more after the onset of parkinsonian features, then the term Parkinson’s disease with dementia (PDD) is used. If cognitive decline occurs within a year of presentation (or at the same time as the motor features) then a diagnosis of dementia with Lewy bodies (DLB) may be made. Specific features of DLB, in addition to parkinsonism, include fluctuations in cognitive performance and visual hallucinations. It is important to note that the distinction between PDD and DLB is purely clinical, since the pathological changes are identical (see Ch. 12 for further discussion of dementia, including features that distinguish DLB from Alzheimer’s disease).
Diagnosis and course
The diagnosis of Parkinson’s disease is primarily clinical. Routine MRI scans are often normal, but dopamine deficiency in the basal ganglia can be demonstrated using specialized tests (Fig. 13.3). Without treatment there is progressive decline over a 5–10-year period, with gradual deterioration of motor function, worsening postural instability, gait freezing and frequent falls. However, symptoms can usually be controlled for a number of years with dopamine replacement therapy and this is associated with a near-normal life expectancy.
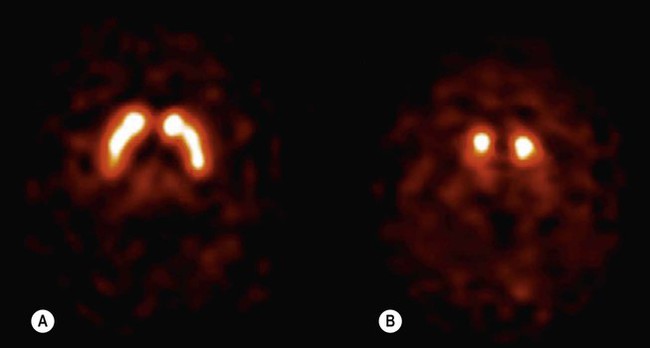
(A) Axial section through the basal ganglia showing the normal pattern of uptake in a healthy control. The characteristic ‘comma’ shape of the striatum (caudate nucleus and putamen) is seen; (B) In a patient with Parkinson’s disease, there is markedly reduced signal in the putamen but not in the head of the caudate nucleus, creating a ‘full stop’ appearance. [Images obtained using single photon emission computed tomography (SPECT) with the radioactively labelled (123I) beta-CIT.] From Seibyl, JP: Single-photon emission computed tomography and positron emission tomography evaluations of patients with central motor disorders. J Semin Nuclear Med (2008) with permission.




Parkinsonism
Vascular pseudoparkinsonism
Patients with cerebrovascular disease may develop an akinetic-rigid syndrome. This is due to microinfarcts (small ischaemic strokes, see Ch. 10) in the basal ganglia or hemispheric white matter. In contrast to idiopathic Parkinson’s disease, symptoms tend to be more severe in the lower limbs, response to dopamine replacement is poor and tremor is usually absent.
Neurodegenerative causes
A number of other neurodegenerative disorders may be confused with Parkinson’s disease. The most important are progressive supranuclear palsy (PSP) and multiple system atrophy (MSA), each with a prevalence of approximately 1 in 20,000. An even rarer form is corticobasal degeneration (CBD), discussed in Clinical Box 13.2.
 Clinical Box 13.2:
Clinical Box 13.2:Progressive supranuclear palsy
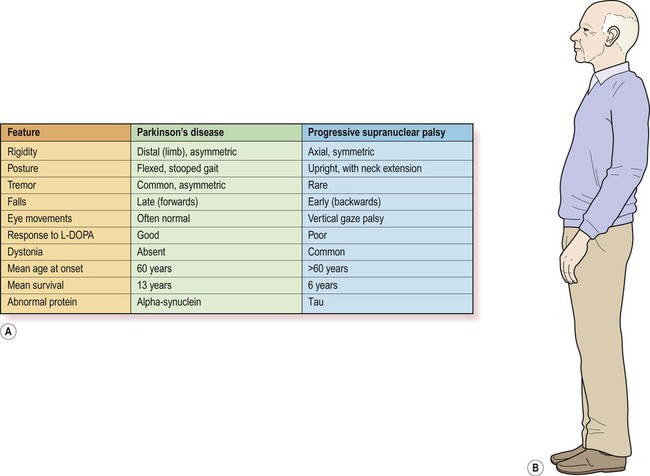
This is the most common neurodegenerative mimic of Parkinson’s disease, accounting for about 5% of people with a parkinsonian syndrome. In more than 50% of cases there is axial rigidity, a hyperextended posture and a characteristic supranuclear gaze palsy with failure in the cortical (‘supranuclear’) control of vertical eye movements. There may also be apathy, cognitive decline and outbursts of inappropriate laughter or tearfulness, termed emotional incontinence. This classical form of PSP is also referred to as Richardson’s syndrome. In up to a third of cases the clinical features closely resemble idiopathic Parkinson’s disease. In this subtype, referred to as PSP-P, the pathological changes are less severe and the clinical course is more favourable. Features of PSP and Parkinson’s disease are compared in Figure 13.4.
Multiple system atrophy
Multiple system atrophy is characterized by parkinsonism, cerebellar ataxia and autonomic dysfunction. There are two patterns. MSA-P is dominated by rigidity, bradykinesia and postural instability and closely resembles idiopathic Parkinson’s disease; whereas MSA-C combines features of cerebellar ataxia with corticospinal tract signs including increased muscle tone and reflexes (see Ch. 4).





Pathology of Parkinson’s disease
The key pathological change in Parkinson’s disease is loss of dopaminergic neurons in the substantia nigra of the midbrain (Fig. 13.5). This is associated with degeneration of the nigrostriatal tract, leading to a profound reduction of dopamine in the basal ganglia (typically below 20% of normal at presentation). Surviving nigral neurons contain cytoplasmic inclusions called Lewy bodies, which can be identified by antibody labelling for the major component, alpha-synuclein protein. This reveals widespread pathological changes throughout the brain stem, limbic lobe and neocortex.
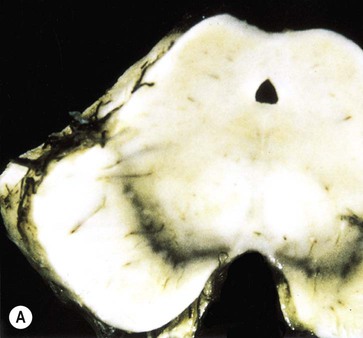
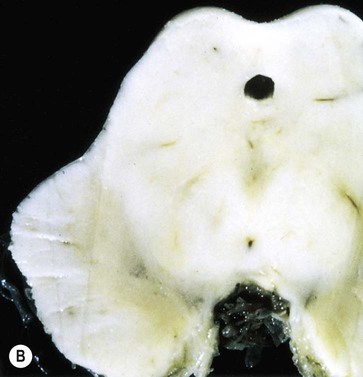
(A) Normal midbrain (in cross section) with a deeply pigmented substantia nigra; (B) Pallor of the substantia nigra in a case of Parkinson’s disease. From Kumar et al: Robbins and Cotran’s Pathologic Basis of Disease 7e (Saunders 2004) with permission.
Neuronal loss
The substantia nigra is a large midbrain nucleus that can be divided into compact and reticular parts. The pars compacta contains the cell bodies of dopaminergic neurons contributing to the nigrostriatal tract, whereas the pars reticulata consists of GABAergic neurons and is analogous to the globus pallidus. The substantia nigra is almost black in the adult brain (Latin: nigra, black) due to the accumulation of neuromelanin as a by-product of dopamine synthesis (see Ch. 7). Loss of dopaminergic neurons in Parkinson’s disease causes pallor of the substantia nigra which can be seen at post-mortem examination. The lateral part of the substantia nigra (which projects to the putamen or ‘motor striatum’) is more severely affected than the medial portion (which projects to the caudate nucleus).
Neuronal loss is also seen in other parts of the nervous system in patients with Parkinson’s disease. These include the noradrenergic locus coeruleus of the pons (see Ch. 1). Post-mortem examination of the brain in Parkinson’s disease may therefore show pallor of the loci coerulei as well as the substantia nigra. Despite normal age-related degeneration of the substantia nigra, most people have sufficient reserve capacity so that striatal dopamine levels never fall below 20% of normal.
Lewy bodies
The pathological hallmark of Parkinson’s disease is the Lewy body (Fig. 13.6). This is a type of pathological inclusion (abnormal protein aggregate) found in the cytoplasm of surviving neurons. Lewy bodies are spherical structures, measuring 5–30 µm in diameter. They are pink on standard histological preparations (because they take up the red tissue dye eosin) and are surrounded by a pale halo.
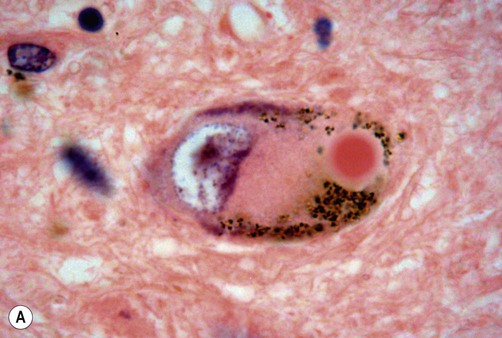
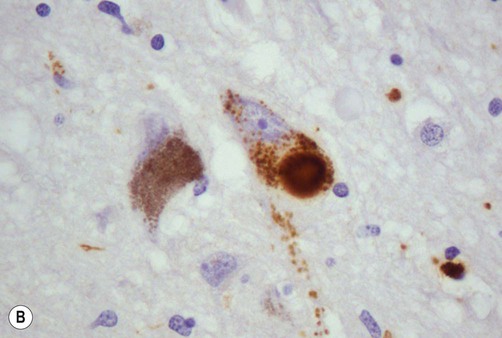
(A) On routine haematoxylin and eosin (H&E)-stained sections Lewy bodies appear as bright pink structures in the neuronal cytoplasm, surrounded by a pale halo; (B) They can also be demonstrated by immunohistochemistry for alpha-synuclein protein, which is the main constituent. Courtesy of Professor Steve Gentleman.
Progression of Lewy body pathology
Lewy body pathology begins in the medulla and olfactory bulbs, spreading progressively through six Braak stages to involve the pons, midbrain, limbic lobe, amygdala and neocortex (Fig. 13.7). Cortical Lewy bodies are similar to those encountered in the brain stem, but do not have a halo and are present even in cases without dementia. Pathological inclusions are also found in the autonomic nervous system, including the enteric nervous system in the gastrointestinal tract.
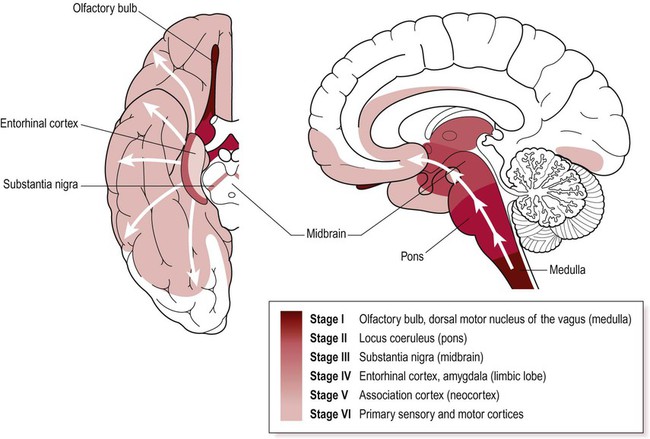
Lewy bodies first appear in the medulla and olfactory bulb and then progressively spread to the pons, midbrain, limbic lobe and neocortex. Key anatomical structures involved in each of the six Braak stages are indicated and disease progression is colour-coded on the inferior and midsagittal views of the cerebral hemisphere. Modified from Braak H et al: Neurobiology of Aging 24 (2003) with permission.
Alpha-synuclein
The main constituent of Lewy bodies is alpha-synuclein. This is a synaptic protein that is present in presynaptic terminals in association with synaptic vesicles. It seems to be involved in neurotransmitter release and synaptic plasticity (which is critical for learning and memory; see Ch. 7). It may also take part in the regulation of dopamine storage and synaptic vesicle recycling.
Accumulation of alpha-synuclein (within neurons and glia) occurs in several other parkinsonian syndromes including Parkinson’s disease with dementia, dementia with Lewy bodies (DLB) and MSA (Fig. 13.8) which are all classified as synucleinopathies. In other forms of parkinsonism such as PSP and CBD there is accumulation of the microtubule-associated protein tau and these disorders are therefore classified as tauopathies. The molecular classification of neurodegenerative diseases is discussed in Ch. 8.
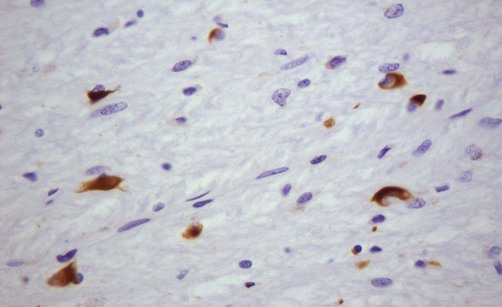
This microscopic image shows glial cytoplasmic inclusions (GCIs) within oligodendrocytes in a case of MSA, highlighted by immunohistochemistry (antibody labelling). Courtesy of Professor Steve Gentleman.




Familial Parkinson’s disease
Five to ten percent of Parkinson’s disease is familial. Around a dozen genes have been identified and the six best understood are shown in Figure 13.9. Some genes have one name connected with the protein encoded and another that is based on the order of discovery (PARK1, PARK2, etc.). The names can be confusing (for instance, it turns out that PARK1 and PARK4 are the same gene).
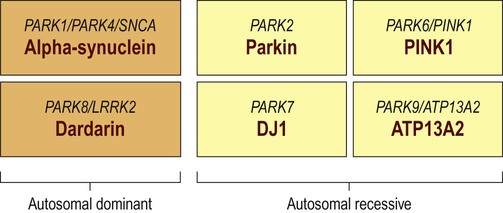
Around a dozen genes have been identified in association with familial Parkinson’s disease, but the six best understood are illustrated. In each case the different names for the genes are shown above the protein encoded.
Autosomal recessive PD
With Parkin gene (PARK2) mutations, disease onset is usually below the age of 40 years and these mutations account for 50% of autosomal recessive juvenile parkinsonism (ARJP). Parkin is a ubiquitin-ligase which is involved in ubiquitination and targeting of proteins for degradation by the proteasome (discussed below; see also Ch. 8). Most Parkin gene mutations reduce the ability to form protein aggregates and Lewy bodies are therefore absent.




Treatment of Parkinson’s disease
The core features of Parkinson’s disease can be treated by replacing striatal dopamine, enhancing transmission at dopaminergic synapses or by stimulating dopamine receptors (see Fig. 13.10; and discussion in following sections).
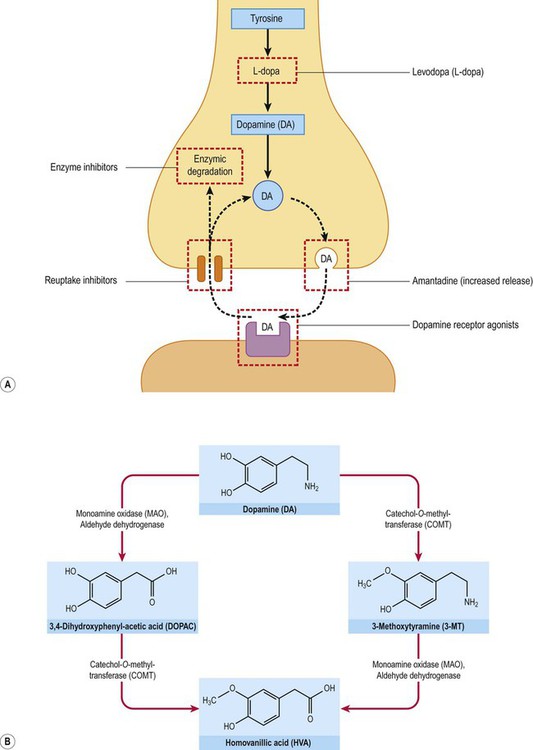
(A) Several agents used in the symptomatic treatment of Parkinson’s disease act at the dopaminergic synapse, affecting dopamine synthesis, release, reuptake or metabolism. A number of dopamine receptor agonists are also available which act directly at post-synaptic dopamine receptors in the striatum; (B) The two main pathways for enzymic degradation of dopamine are shown, via monoamine oxidase (MAO) and catechol-ortho-methyltransferase (COMT). MAO/COMT inhibitors are also used in Parkinson’s disease to increase availability of dopamine.
Dopamine replacement
Levodopa is a prodrug because it has to be taken up by neurons and glia where it is converted to the active drug, dopamine. Peripheral activation would release dopamine into the bloodstream, causing hypotension and nausea (Clinical Box 13.3). Uptake by sympathetic neurons and conversion to noradrenaline would also interfere with autonomic control of the cardiovascular system. These side effects are avoided by co-administration of a dopa-decarboxylase inhibitor that is unable to cross the blood–brain barrier, increasing availability to the brain and significantly reducing the oral dose. Two commonly used peripheral decarboxylase inhibitors are carbidopa and benserazide (contained in combined preparations: co-careldopa and co-beneldopa).
 Clinical Box 13.3:
Clinical Box 13.3:Problems with levodopa therapy
Side-effects of levodopa
Excessive dopaminergic stimulation may also cause psychotic features which may be difficult to manage as most antipsychotic agents block central dopamine receptors and therefore exacerbate parkinsonism. Some patients respond to ‘atypical’ neuroleptics (e.g. clozapine) which also antagonize serotonin receptors. Other side effects include confusion and behavioural changes (Clinical Box 13.4).
 Clinical Box 13.4: Dopamine dysregulation syndrome
Clinical Box 13.4: Dopamine dysregulation syndrome
A small percentage of patients with Parkinson’s disease become ‘addicted’ to their medication and use much higher doses than they need. This is associated with impulse control problems (e.g. hypersexuality, pathological gambling, over-eating, over-spending). Another feature is punding, the compulsive repetition of purposeless actions (e.g. lining up objects in rows). Dopamine dysregulation syndrome is thought to be caused by over-stimulation of dopamine receptors in the ventral striatum, part of the basal ganglia that is known to be involved in reward-based learning and addiction (see Ch. 3).
Other agents
Other drugs used to treat the symptoms of Parkinson’s disease include dopamine receptor agonists, enzyme inhibitors (see Fig. 13.10) and anticholinergic agents.
Amantadine
This was originally used as an antiviral agent to treat influenza, but provides mild symptomatic benefit in early Parkinson’s disease. It can be used as a dopamine-sparing agent, delaying the onset of levodopa-associated side effects and loss of efficacy. Amantadine increases dopamine release and reduces its reuptake. It is also a weak antagonist at the NMDA glutamate receptor (see Ch. 7). The beneficial effect of amantadine is modest overall and tends to wear off after a few weeks. Side effects include ankle oedema, skin rash, confusion and hallucinations.







Surgery in Parkinson’s disease
Neurosurgery may be an option in longstanding Parkinson’s disease that is no longer responsive to levodopa, particularly in patients with severe dyskinesias and no evidence of dementia. Destructive lesions in the thalamus or pallidum (thalamotomy and pallidotomy) can be effective and a small number of patients have received experimental transplants from the substantia nigra of fetuses (Clinical Box 13.5). However, the most successful surgical approach involves the placement of deep brain electrodes which are used to stimulate various parts of the basal ganglia.
 Clinical Box 13.5:
Clinical Box 13.5:Deep brain stimulation (DBS)
Using a stereotactic frame and MRI guidance (Fig. 13.11) it is possible to implant electrodes at precise subcortical targets, deep within the brain. Specific nuclei can then be stimulated via a subcutaneous pacemaker in the chest. The main surgical risks are intracerebral haemorrhage and infection, but serious complications occur in less than 2% of cases in specialist centres.
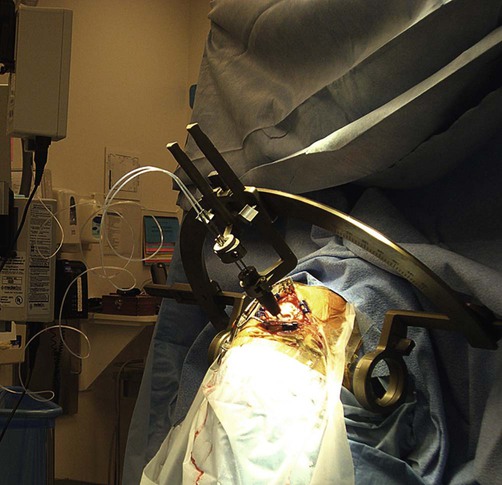
Intraoperative photograph of a patient wearing a stereotactic frame undergoing functional neurosurgery for drug-resistant Parkinson’s disease. From Larson, PS: Neurosurg Clin North Am 2010; 21(4): 691–698, with permission.
The most effective approach is bilateral stimulation of the subthalamic nucleus (Fig. 13.12) which provides excellent relief of akinesia and bradykinesia. It also allows the oral levodopa dose to be reduced and improves dopa-induced dyskinesias. Other DBS targets include the thalamus (particularly in people with severe, drug-resistant tremor) and, much less commonly, the pedunculopontine nucleus at the junction of the midbrain and pons (a small brain stem structure that is involved in gait initiation and is known to degenerate in Parkinson’s disease).
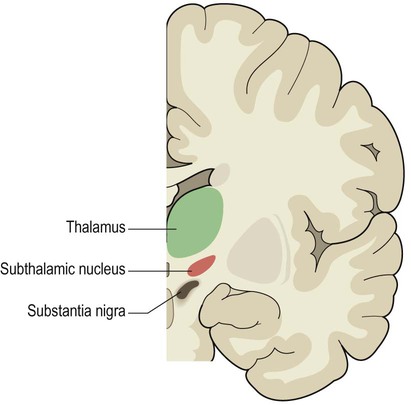
This small, lens-shaped nucleus belongs to the basal ganglia. It lies just below the thalamus, close to the substantia nigra and posterior limb of the internal capsule.




Pathophysiology
The anatomy of the corpus striatum, its functional divisions and the concept of basal ganglia loops (including their non-motor roles in cognition, emotion and behaviour) have been introduced in Chapter 3. This section will explore the contribution of the basal ganglia to voluntary movement and how this is disturbed in Parkinson’s disease.
The voluntary motor loop
Initiation of voluntary actions involves a basal ganglia loop that originates and terminates in the supplementary motor area (SMA) (Fig. 13.13; see also Ch. 3). Activity in the SMA and voluntary motor loop is facilitated by dopamine, which lowers the threshold for movement initiation. This helps to determine whether an intention to act is translated into an actual movement. Reduced activity in the SMA (due to striatal dopamine deficiency) is responsible for the akinesia (poverty of movement) in Parkinson’s disease.
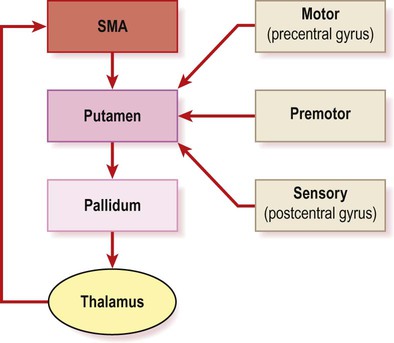
The central, ‘closed loop’ component originates and terminates in the supplementary motor area (SMA) in the medial frontal lobe (see Ch. 3, Fig. 3.4b). Other sensory and motor areas form ‘open-loop’ components.
The SMA is involved in self-initiated actions (e.g. throwing a ball, rising from a chair) rather than movements that occur in response to an external stimulus or trigger (e.g. catching a ball, stepping over a piece of chalk). This has been exploited with the creation of virtual reality glasses that provide artificial visual cues for parkinsonian patients (projections of horizontal lines to ‘step over’). This leads to improvement in gait initiation, stride length and pace, with fewer falls. In some cases, powerful emotions can overcome akinesia (Clinical Box 13.6).
 Clinical Box 13.6: Paradoxical kinesis
Clinical Box 13.6: Paradoxical kinesis
A strong sense of urgency or fear can sometimes overcome akinesia in patients with Parkinson’s disease. The classic example is the profoundly akinetic patient who is suddenly able to run freely from a burning building, only to ‘freeze’ again once he reaches a place of safety. This is referred to as paradoxical kinesis and may be mediated by projections from the amygdala to the ventral striatum which is the ‘limbic’ (emotion-related) portion of the basal ganglia (see Ch. 3).



Afferent and efferent connections
Projections into the basal ganglia
All basal ganglia loops arise and terminate in the frontal lobe. The frontal cortex projects to the striatum (caudate-putamen) which is therefore the afferent (or ‘input’) part of the basal ganglia. Cortical afferents terminate on medium spiny neurons, which make up 95% of basal ganglia cells. They are so-named because they have medium-sized cell bodies and numerous dendritic spines (Fig. 13.14). The head of each spine receives a single afferent projection from the frontal cortex, whereas the shaft receives a dopaminergic projection from the nigrostriatal pathway (which has a modulating effect). It is important to note that the vast majority of intrinsic basal ganglia cells are GABAergic and that the outflow of the basal ganglia is entirely inhibitory.
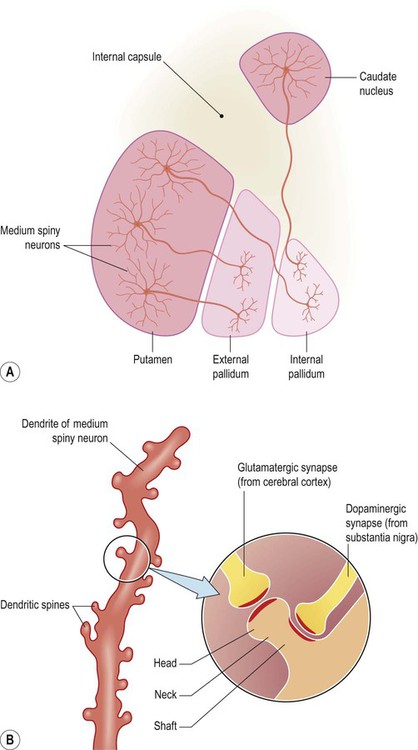
(A) Approximately 95% of basal ganglia neurons are classified as ‘medium spiny’ cells. Several medium spiny striatal neurons are illustrated in the figure: some projecting to the external pallidum, others projecting to the internal pallidum. These two types of neuron belong to the ‘indirect’ and ‘direct’ pathways, respectively [see text for further discussion]; (B) Medium spiny neurons in the striatum (caudate nucleus/putamen) receive excitatory projections from the frontal lobe. These excitatory cortico-striatal projections terminate on the heads of dendritic spines. In contrast, dopaminergic projections (from the substantia nigra) synapse on the shafts of dendritic spines, facilitating direct pathway neurons but inhibiting those belonging to the indirect pathway.
Outflow of the basal ganglia
By default, the internal pallidum inhibits thalamocortical neurons that take part in basal ganglia loops. The ‘default action’ of the basal ganglia is thus to prevent unwanted movements, thoughts and behaviours (Fig. 13.15). Although tonically active, the internal pallidum is also constantly stimulated by the subthalamic nucleus, which is excitatory and glutamatergic. This reinforces the ‘default state’ of pallidal inhibition and explains why destruction of the subthalamic nucleus causes involuntary movements (Clinical Box 13.7).
 Clinical Box 13.7: Hemiballismus
Clinical Box 13.7: Hemiballismus
Hemiballismus is a rare condition characterized by violent flinging movements on one side of the body (Greek: hemi, half; ballismos, jumping). It is usually caused by a small stroke (Ch. 10) affecting the subthalamic nucleus, which normally helps to block unwanted movements by exciting the internal pallidum (which is the inhibitory outflow nucleus or ‘brake’ of the basal ganglia). In the absence of excitation from the subthalamic nucleus, the internal pallidum no longer suppresses unwanted movements. It is as though the ‘foot’ has been taken off the ‘brake’.
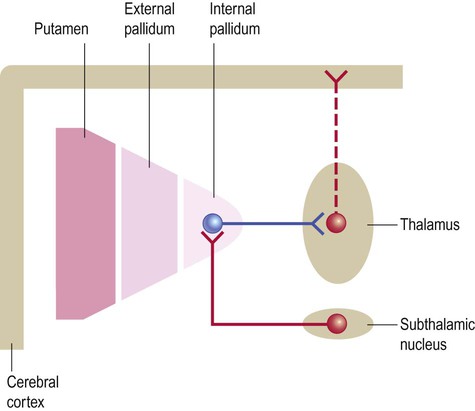
This simplified diagram illustrates the ‘default’ state of the basal ganglia. The internal pallidum is tonically active and powerfully inhibits nuclei in the ventral thalamus, blocking activity in thalamocortical projections that take part in basal ganglia loops. The subthalamic nucleus (which belongs to the basal ganglia) is constantly active and excites the internal pallidum, reinforcing its restraining action on the thalamus. (If the internal pallidum is a ‘brake’, then the subthalamic nucleus is a ‘foot’ on the brake pedal.)



Direct and indirect pathways
The striatum gives rise to two sets of basal ganglia connections: the direct and indirect pathways, which are both stimulated by afferent (corticostriatal) projections. Dopamine stimulates striatal neurons belonging to the direct pathway, but at the same time inhibits those of the indirect pathway. This is because the two types of neuron express different dopamine receptors. There are five main types of dopamine receptor, arranged in two groups: D1-like and D2-like (Fig. 13.16). Direct pathway neurons are excited by dopamine since they express D1-like receptors; indirect pathway neurons are inhibited by dopamine because they express D2-like receptors (Fig. 13.17). The action of dopamine is therefore to shift the balance in favour of the direct pathway and this leads to increased activity in the SMA/motor loop.
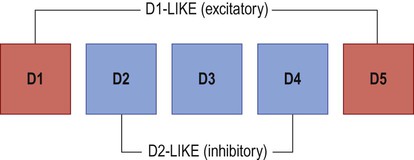
There are two major subtypes of dopamine receptor. D1-like receptors (D1, D5) are excitatory, whereas D2-like receptors (D2, D3, D4) are inhibitory.
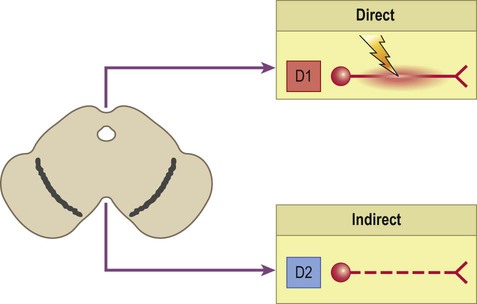
Neurons that are part of the direct pathway express D1-like dopamine receptors and are excited by dopamine. Neurons that are part of the indirect pathway express D2-like dopamine receptors and are inhibited by dopamine. Dopamine therefore shifts the balance of activity in favour of the direct pathway.
Direct pathway
The internal connections of the basal ganglia work by disinhibition (release of inhibition). This happens when two inhibitory neurons are arranged in series, so that the first one inhibits the braking action of the second (illustrated in Fig. 13.18).
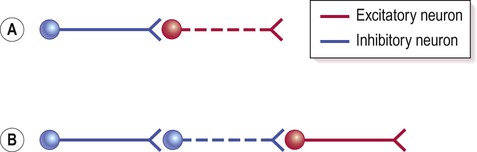
(A) Simple illustration of an inhibitory interaction between two nerve cells. Activity in the excitatory neuron (red) is inhibited by an inhibitory interneuron (blue); (B) Disinhibition (release of inhibition) occurs when two inhibitory neurons occur in sequence. In this case the excitatory neuron (red) has been released from inhibition (or ‘disinhibited’) as the neuron that was previously restraining its action has itself been inhibited. In functional terms, disinhibition is equivalent to excitation.
The arrangement of connections in the direct pathway is shown in Figure 13.19A in the context of the voluntary motor loop. Striatal neurons belonging to the direct pathway project directly to the internal pallidum and inhibit it, thereby releasing thalamocortical neurons from their normal state of inhibition. This promotes activity in the motor loop and SMA, facilitating voluntary movement.
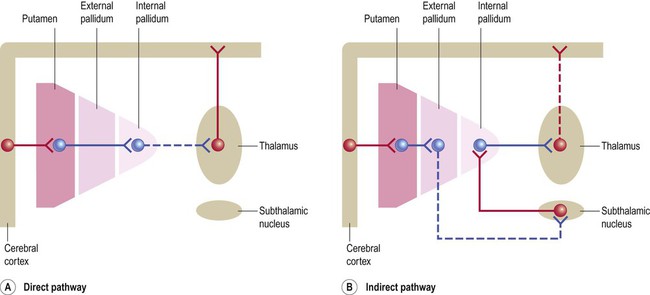
(A) Striatal neurons belonging to the direct pathway project directly to the internal pallidum and inhibit it, thereby releasing thalamocortical neurons from inhibition. In the voluntary motor loop, the direct pathway therefore facilitates movement; (B) Striatal neurons belonging to the indirect pathway project to the external pallidum, where they inhibit a group of neurons that would normally reduce the firing rate of the subthalamic nucleus. This means that the indirect pathway disinhibits the subthalamic nucleus and accentuates its excitation of the internal pallidum. The indirect pathway therefore reinforces the default (inhibitory) outflow of the basal ganglia and prevents unwanted movements.
Indirect pathway
The indirect pathway is illustrated in Figure 13.19B. Striatal neurons belonging to the indirect pathway project to the external pallidum, where they inhibit a group of cells that would normally reduce the firing rate of the subthalamic nucleus. This means that the indirect pathway disinhibits the subthalamic nucleus and accentuates its excitation of the internal pallidum. The indirect pathway therefore reinforces the ‘default’ (inhibitory) outflow of the basal ganglia. Selective loss of indirect pathway neurons leads to involuntary movements in Huntington’s disease (Clinical Box 13.8).
 Clinical Box 13.8: Huntington’s disease
Clinical Box 13.8: Huntington’s disease
This is an autosomal dominant disorder with variable penetrance caused by expansion of a CAG (trinucleotide) repeat within the huntingtin gene on chromosome 4. It is therefore classified as a trinucleotide repeat expansion disorder and the number of abnormal repeats determines disease severity and age at onset. Huntington’s disease is characterized by involuntary movements with a ‘jerky’ or dance-like quality, termed chorea (Greek: khoreia, a choral dance). This is caused by selective loss of indirect pathway neurons which normally inhibit movement. Accompanying degeneration of the caudate nucleus and cerebral cortex is associated with dementia and psychiatric features (Fig. 13.20).
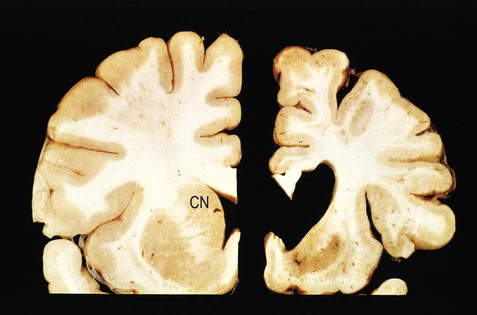
Post-mortem examination of the brain in an individual with Huntington’s disease [right] compared to a normal control brain [left] shows marked cortical thinning, extensive loss of subcortical white matter and dilation of the lateral ventricle. There is also striking atrophy of the caudate nucleus (labelled “CN” in the normal control brain) which takes part in basal ganglia loops passing through the prefrontal cortex that are concerned with cognition and behaviour. From Kumar et al.: Robbins and Cotran’s Pathologic Basis of Disease 7e (Saunders 2004) with permission.





Basal ganglia oscillations



In Parkinson’s disease there is excessive beta activity, which is normalized by dopamine replacement. The effect of surgical intervention (whether by stimulation or destruction) may be to interfere with pathological oscillations, which might enable other parts of the brain to compensate. This is summarized in the idea that ‘silence is better than noise’.




Aetiology and pathogenesis
Environmental factors
There are several examples of acquired akinetic-rigid syndromes that have features in common with idiopathic Parkinson’s disease. These are caused by various environmental, infectious or toxic agents (e.g. carbon monoxide, manganese and carbon disulphide). Parkinsonian syndromes have also been described following viral or bacterial infections (see Clinical Boxes 13.9 and 13.10).
 Clinical Box 13.9: Encephalitis lethargica
Clinical Box 13.9: Encephalitis lethargica
In the early 20th century a viral pandemic swept across central Europe. Features included fever, headache and marked somnolence or coma caused by inflammation of the brain: encephalitis lethargica (the ‘sleepy sickness’). It affected children and young adults and carried a mortality of 40%. More than half of the survivors developed a syndrome of post-encephalitic parkinsonism and these individuals were among the first recipients of levodopa therapy in the 1960s. Post-mortem examination of the brain showed degeneration of the substantia nigra and accumulation of neurofibrillary tangles in surviving neurons, similar to those found in Alzheimer’s disease (Ch. 12).
 Clinical Box 13.10: Autoimmune basal ganglia damage
Clinical Box 13.10: Autoimmune basal ganglia damage
The extrapyramidal movement disorder Sydenham’s chorea is characterized by ‘jerky’ involuntary limb movements (‘St Vitus’s dance’). It is now rare, but classically occurred a few weeks after recovery from rheumatic fever (caused by a streptococcal infection that affects the heart and joints). There is also a group of conditions referred to as PANDAS (paediatric autoimmune neuropsychiatric disorders associated with streptococcal infection). Affected children may develop tics, features of obsessive-compulsive disorder (see Ch. 3, Clinical Box 3.10) or Tourette’s syndrome. The underlying cause in both disorders is autoimmune CNS damage due to ‘cross-reaction’ with a basal ganglia surface antigen that is similar to one present on the streptococcal bacterium. Interestingly, adults with OCD and related disorders are significantly more likely to show serological evidence of a previous streptococcal infection than controls.



MPTP in animal models
Mechanism of MPTP toxicity
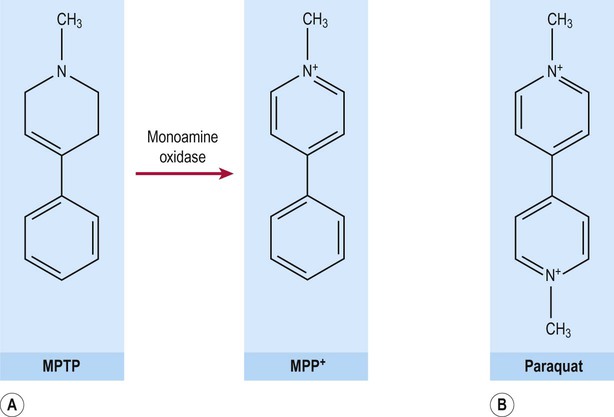
MPTP crosses the blood–brain barrier where it is taken up by neurons and astrocytes and metabolized by monoamine oxidase (MAO) to form MPP+ (1-methyl-4-phenylpyridinium) (Fig. 13.21A). This molecule is a highly reactive free radical species with an unpaired electron and is similar to the neurotoxic herbicide paraquat (Fig. 13.21B). MPP+ is taken up by dopaminergic neurons (via a specific monoamine transporter) where it binds to neuromelanin and becomes concentrated. MPP+ has been shown to inhibit complex I of the mitochondrial respiratory chain, leading to oxidative stress in dopaminergic neurons.
Mitochondria and oxidative stress
Inhibition of complex I of the mitochondrial electron transport chain (NADH dehydrogenase) is a key event in the pathogenesis of Parkinson’s disease. This reduces ATP production and impairs energy-dependent cellular processes. It also leads to the generation of free radicals, causing additional oxidative stress and lowering the threshold for apoptosis (programmed cell death; see Ch. 8).




Protein aggregation
Neurotoxicity of alpha-synuclein
It is not clear whether or not Lewy bodies are neurotoxic and it may be that an intermediate oligomeric species (formed during their synthesis) is responsible for damaging the cell. It has been shown that monomers of alpha-synuclein associate with cell membranes and form ring-like oligomeric assemblies with a central pore that can perforate the cell membrane. This is similar to the membrane attack complex of the complement cascade and enables free calcium to enter the cell (a final common pathway in neuronal cell death; see Ch. 8). A similar mechanism has been postulated for amyloid beta toxicity in Alzheimer’s disease (Ch. 12).
Dysfunction of the ubiquitin-proteasome system
The ubiquitin-proteasome system (UPS) is an important cellular mechanism for the disposal of abnormal or misfolded proteins, particularly when attempts to deal with them have failed (e.g. the unfolded protein response or upregulation of molecular chaperones, see Ch. 8).




Interaction of pathogenetic mechanisms
Sporadic Parkinson’s disease is thought to be caused by a combination of environmental and constitutional factors. There are three main elements (illustrated in Figure 13.22):
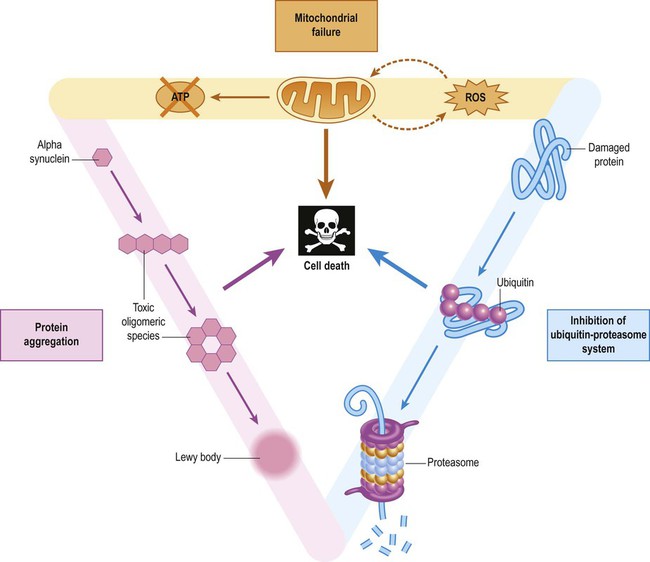
Interaction between the three main factors creates a vicious cycle in dopaminergic neurons (ROS = reactive oxygen species).



Exposure to a relevant environmental or toxic factor in a genetically predisposed individual (e.g. with reduced capacity to deal with oxidative stress, mitochondrial dysfunction or to handle misfolded proteins) is thought to trigger a set of events that culminates in a vicious cycle incorporating these three key pathogenetic elements. This occurs on a background of variable nigral reserve capacity and normal age-related degeneration (in some cases with additional contributory factors, such as head injury).
