[level-membership-for-neurosurgery-category]
CHAPTER 216 Split Spinal Cord
Descriptions of split cord malformation (SCM) appeared in the medical literature as early as the 17th century.1,2 Historically, the terms diastematomyelia (Greek for “cleft[ed] medulla”) and diplomyelia (Greek for “double[ed] medulla”) were used to describe developmental anomalies now grouped together as split cord malformation.3 The sine qua non of these developmental anomalies is the presence of a longitudinal cleft within the spinal cord across one or more vertebral segments.
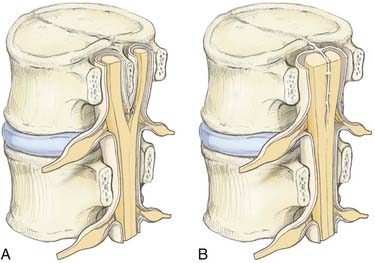
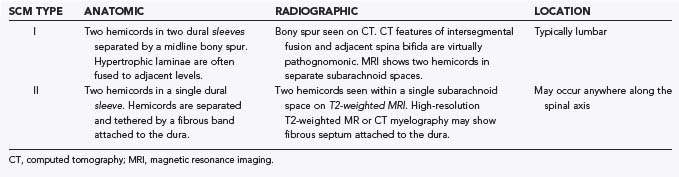
Type I malformations (formerly diastematomyelia) are characterized by a bony septum that cleaves the spinal canal in the sagittal plane and a duplicated thecal sac (Fig. 216-1A).3 Type II malformations (formerly diplomyelia) are characterized by a cleft cord within a single dural sac, often tethered by a fibrous midline septum to the adjacent dura (Fig. 216-1B and Table 216-1). Not all SCMs fit these descriptions precisely, but the classification scheme provides a framework for the surgical approach to these conditions. In 1992, Pang and colleagues proposed a unified theory attributing both type I and type II SCM to a single error in embryogenesis: adhesion between the ectoderm and endoderm leading to a persistent neurenteric canal.3
Not uncommonly, SCMs are accompanied by nonfunctional paramedian nerve roots traveling within a fibrovascular bundle to or beyond the dura. A fibrous tract extending from the epidural space to a small overlying plaque of atretic skin may also be present. Both the intradural bands and the associated skin lesion have been referred to by the term myelomeningocele manqué, which is derived from the French verb manquer (“to miss”) and reflects the outmoded theory that myelomeningocele manqué is a forme fruste of true myelomeningocele.4 Instead, the descriptive nomenclature “dorsal bands” is used here.
Epidemiology of Split Cord Malformations
Although their precise incidence is unknown, SCMs are exceedingly rare and represent 3.8% to 5% of all congenital spinal anomalies.3,5 One recent series estimated the prevalence of SCM to be 1 in 5000 (0.02%) live births.6 Similar to other forms of occult spinal dysraphism, there is a slight female preponderance, approximately 1.3:1.7–10 The peak age at initial medical evaluation is 4 to 7 years, although there is a second peak between 12 and 16 years of age coinciding with the postpubescent growth spurt. A delay in diagnosis until adulthood may be seen, with back pain being the sole complaint7,8,10,11 or pain accompanied by progressive neurological deficits that may be mistaken for degenerative disease. Although estimates of prevalence vary, type I SCMs may occur more frequently than type II lesions.8,10,11
SCMs rarely occur in the absence of associated anomalies. The most common associations (in descending order) are a tethered/low-lying cord (>50%), kyphoscoliosis (44% to 60%), syringomyelia (27.5% to 44%), and spina bifida (11% to 26%).7,8,10 Conversely, SCMs may occur in 5% of patients with congenital scoliosis12 and in up to 15% of patients with myelomeningocele.13,14 When SCM is seen in association with myelomeningocele, it is three to four times more likely to be a type I malformation and tends to occur at or just rostral to the level of the myelomeningocele defect. On occasion, multiple dysraphic lesions may be identified, and occult SCMs can account both for a discrepancy between functional level and anatomic level of the myelomeningocele placode and for late neurological deterioration in patients with myelomeningocele and an undisclosed SCM.11 Dermoids, dermal sinus tracts, and neurenteric cysts may also be found in conjunction with SCMs.
Embryogenesis
SCMs are thought to occur during the third week of embryogenesis, concurrent with gastrulation, as a result of failure of fusion of the notochord in the midline and subsequent persistence of an accessory neurenteric canal.1,3,15,16
On day 17 of human development, cells of Hensen’s node ingress at the embryonic midline to begin formation of the notochord. In a process of cell-to-cell intercalation, bilayers of cells ingress from each side of midline and intermix to form a singular midline notochordal process.17 Oriented cellular division and cell-to-cell intercalation contribute equally to the process of convergent extension by which the notochord elongates. Oriented cellular division refers to the preferential direction of mitosis demonstrated during embryogenesis that allows a mass of cells to extend in a particular plane rather than growing spherically. Intercalation refers to the cellular process by which two groups of cells integrate in a mosaic pattern (Fig. 216-2A). Thus, polar integrated growth of the combined mass occurs in the cranial-caudal direction. Cellular intercalation is partially governed by genes of the wnt family, which are also responsible (along with sonic hedgehog) for dorsal-ventral patterning of the vertebrate nervous system.18 Knockouts of Prickle, another cell polarity molecule that is conserved across species, demonstrate a foreshortened and thickened notochord.
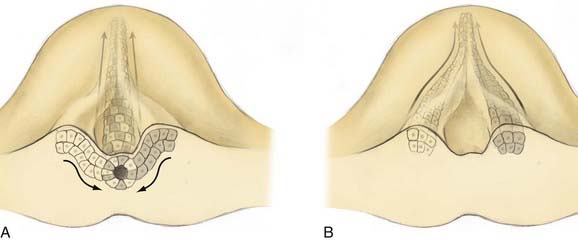
Reciprocal induction of notochordal and paraxial mesodermal cells also plays a key role in this process. Mesodermal somites express protocadherin, which promotes midline intercalation of the notochord. Embryos lacking protocadherin demonstrate normal elongation of the paramedian notochord masses but no integration, reminiscent of the failed integration seen over small spinal segments in human SCM19 (Fig. 216-2B). Once formed, the notochord acts as an organizer by secreting multiple morphogens that are responsible for proper neurulation of the embryo. Experimental transplantation of notochord fragments has demonstrated that presence of the notochord is necessary and sufficient to influence patterning of spinal development20 and that ectopic or redundant notochordal tissue may result in complete duplication of the spinal cord and surrounding tissues.21 Emura and colleagues produced an amphibian model of SCM by microsurgically creating a neurenteric fistula in embryos, which were then incubated to term. They observed SCM in 40% of the embryos that survived the procedure.22
Most theories posit two common prerequisites for the development of SCM.1,3,23,24 The first is persistent adhesion of the ectoderm and endoderm, termed an accessory neurenteric canal by Bremer.24 The second is a cleft notochord, with subsequent induction of a cleft spinal cord. In 1992, Pang proposed a unified theory for the development of both types of SCM in which these two prerequisites occur as the result of a single ontogenic event—failure of midline integration of the anlagen destined to become the notochordal process.3 This theory postulates that the mass of cells derived from the primitive pit does not ingress as a single mass but instead exhibits left-right asymmetry whereby it courses mediolaterally from the primitive pit before reintegrating in the midline (Fig. 216-2B). Failure of reintegration leads to the formation of an accessory neurenteric canal, which Pang labeled the endomesenchymal tract.11 Persistence of the endomesenchymal tract results in a median cleft with two heminotochords, which induces a similarly cleft spinal cord.
This unified theory explains the subsequent occurrence of types I and II SCM based on incorporation of the meninx primitiva into the endomesenchymal tract. Inclusion of meninx primitiva cells, which arise around day 30, results in the formation of two completely separate dural sleeves as seen in a type I SCM. Inasmuch as the meninx primitiva is osteogenic, this also results in the formation of a midline bony spur (see Fig. 216-1A). When the meninx primitiva is not incorporated into the endomesenchymal tract, a single dural sleeve (and single arachnoid space) contains two hemicords separated by a thin fibrous band, as seen in a type II SCM (see Fig. 216-1B). Incorporation of neural crest (other than the meninx primitiva) into the endomesenchymal tract results in the formation of neural elements medial and dorsal to the hemicords that extend to the extradural space and result in the formation of dorsal bands.
The presence of two separate notochordal anlagen during formation of the notochordal process has never been directly observed, but knockout mice lacking genes for intercalation develop two heminotochords that are not fused in the midline.19 The unified theory suggests that caudal failures of midline integration, which occur relatively late during embryonic development, are more likely to coincide with emergence of the meninx primitiva and thus produce type I SCMs. In fact, type I SCM most frequently occurs from L2 to L4 and is rarely seen above the thoracolumbar junction, whereas type II SCM occurs almost exclusively above T8.8
Signs and Symptoms
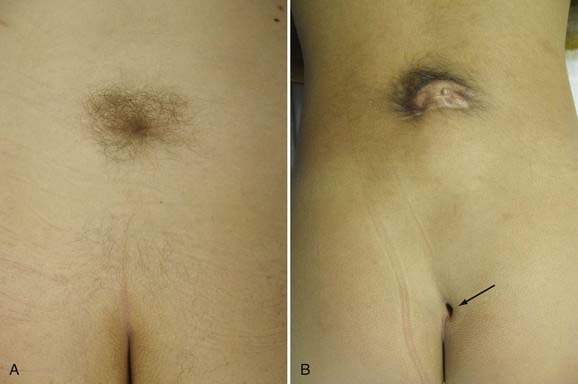
Patients with split cord may be asymptomatic, with only cutaneous stigmata of their spinal dysraphism. In various series, there is some type of cutaneous mark present in up to 92% of patients.7,10,11,14,25 The common stigmata associated with SCM are summarized in Table 216-2.7–11,14,25,26 Because of the rostral migration of neural elements relative to the bony spinal column that normally occurs after the formation of an SCM, cutaneous markers are found somewhat caudal to the level of the neural defect itself. The presence of hypertrichosis, although variable, is highly specific for SCM (Fig. 216-3A). One report identified an odds ratio of 61.6:1 in favor of the occurrence of an SCM if a hairy patch is present.25 One characteristic variation of hypertrichosis, the “faun’s tail,” consists of a patch of unusually coarse, raised hair that is strongly associated with type I SCM.25 There is typically a capillary hemangioma underlying these hairy patches. On occasion (≈7%), hemangiomas alone, without overlying hypertrichosis, may indicate the presence of an SCM. Although sacral dimples, dermal sinuses, and cutaneous lipomas are not specific to SCM per se, they often indicate the presence of an additional underlying dysraphic lesion in a patient with SCM (Fig. 216-3B). True sacral dimples that are suggestive of underlying dysraphism should be distinguished from shallow, symmetrical dimples over the coccygeal midline. The latter is common in the general population and is not indicative of the presence of distal tethering pathology.
| SYMPTOMS | PREVALENCE (%) |
|---|---|
| Backache11,26 | 30-50 |
| Progressive neurological deficit11 | 66 |
| Urinary retention/incontinence10,26 | 13-32.5 |
| Leg pain/hyperesthesia10,26 | 7-10 |
| Asymptomatic10 | 13 |
| CUTANEOUS MARKERS | |
| Hypertrichosis9–1125 | 31-61 |
| Dermal sinus8–10 | 7-40 |
| Subcutaneous lipoma8–1014 | 2-25 |
| Sacral dimple8,10 | 5-10 |
| Capillary hemangioma8–1014 | 0.5-7.2 |
| ASSOCIATED FINDINGS ON EXAMINATION | |
| Abnormal urodynamics26 | 74 |
| Left-right motor/sensory asymmetry8 | 21 |
| Left-right asymmetry (limb size/length)10 | 12-15 |
| Talipes equinovarus7,10 | 17-20 |
A majority of patients with SCM have one or more signs and symptoms of neurological deterioration.11 Axial back pain is common and is typically focal and intense around the levels of the split cord. Adults with undiagnosed SCM may have back pain as the only symptom. A radicular component may be present in 10% of both adult and pediatric patients.10,25
Progressive sensory or motor dysfunction (or both) is seen in two thirds of patients. There are two separate, but interrelated causes of asymmetrical motor findings in the lower extremities. Nearly 50% of patients have gross (i.e., structural) asymmetry of the lower extremities, a condition referred to as neuro-orthopedic syndrome. This syndrome is characterized by a triad of limb length discrepancy, muscular atrophy (resulting in secondary weakness), and clubfoot deformity (talipes equinovarus).27–29 The smaller limb is often ipsilateral to a smaller hemicord. Fewer patients (about ≈20%) have an asymmetrical neurological deficit despite structural symmetry of the limbs. This finding is highly associated with hydromyelia of one hemicord.8
Autonomic dysfunction is typically manifested as urologic signs and symptoms. Subjective complaints of enuresis are a poor indicator in comparison to objective urodynamic testing. Although only about one third of patients typically have complaints of urinary dysfunction, urodynamic studies demonstrate abnormal bladder function in nearly three quarters.26 Detailed urologic assessment should be undertaken as part of the initial evaluation and before any surgery.
Surgeons evaluating a patient for suspected SCM should be cognizant of associated anomalies that may require additional surgical intervention. Summarized in Table 216-3,7,8,10 associated anomalies include scoliosis, tethered cord secondary to a thickened filum or terminal lipoma, myelomeningocele (with associated Chiari II malformation), and syringomyelia.
TABLE 216-3 Associated Anomalies
| ANOMALY | PREVALENCE (%) |
|---|---|
| Scoliosis | 44-60 |
| Thickened filum terminale | 22-71 |
| Neuro-orthopedic syndrome | 20-56 |
| Syrinx | 28-44 |
| Myelomeningocele | 11-26 |
| Terminal lipoma | 9-16 |
| Chiari malformation | 6-7 |
Imaging
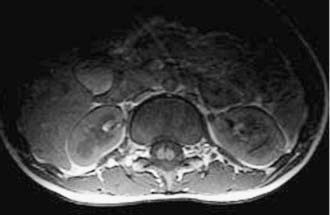
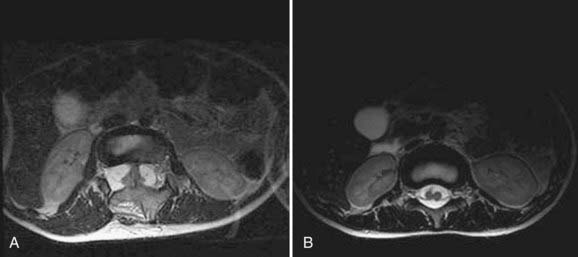
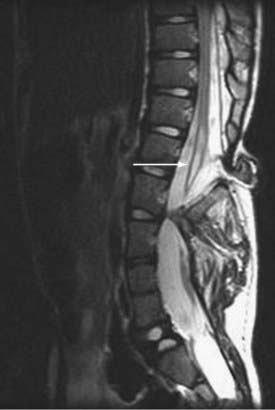
Magnetic resonance imaging (MRI) has led to significant advances in the preoperative diagnosis and planning for surgical management of SCM. The cardinal feature on MRI is the presence of two hemicords, which are best visualized on T1-weighted sequences (Fig. 216-4). The cleft typically begins below T11 and the hemicords most often reunite after two to three vertebral segments.8,30 T1-weighted images are also helpful in demonstrating the presence of an associated fatty filum, which may affect one or both hemicords, a dermal sinus tract, or a terminal lipoma (Fig. 216-5). Associated tethering anomalies are present in approximately half of all patients and in more than 90% of those with a low-lying conus.10,11 T2-weighted MRI typically discriminates between the presence of one or two subdural and subarachnoid spaces, which allows proper categorization of the malformation as type I or type II (Fig. 216-6). T2-weighted axial and coronal images may also demonstrate a hypointense fibrous band in type II SCM. In nearly half of all cases, T2-weighted images also demonstrate syringomyelia proximal to the cleft, which may extend into one or both hemicords30 (Fig. 216-7). Because multiple, often redundant tethering elements may be present at the level of the SCM and elsewhere, careful inspection of the entire spinal intradural space should be undertaken with MRI. Unlike the situation in patients with open dysraphic conditions, brain imaging is not automatically required.
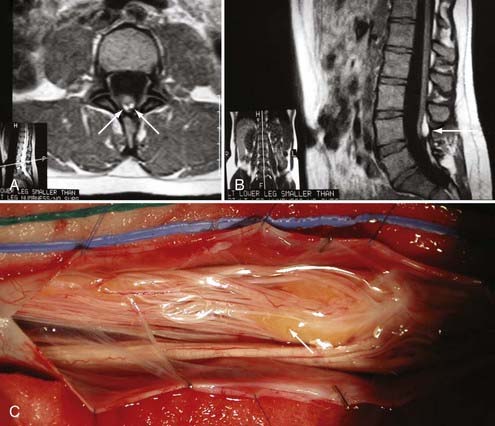
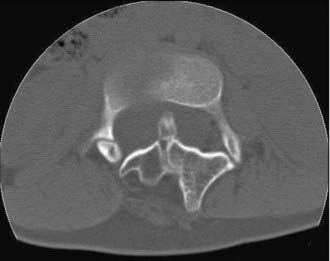
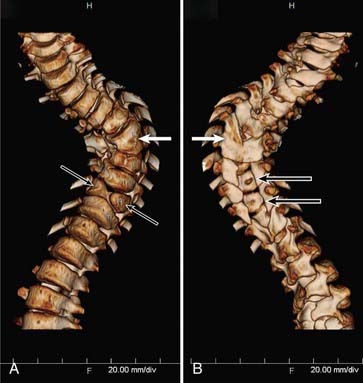
In type I malformations, computed tomography (CT) best delineates the configuration of the osseous spur. Most spurs occur as a midline bony outgrowth of the posterior aspect of the vertebral body, although spurs with oblique orientation and spurs arising from the neural arch have been reported5 (Fig. 216-8). SCM is associated with bony anomalies in more than 85% of patients.5 The more common bony segmentation defects include bifid vertebrae, bifid laminae, Klippel-Feil malformation, butterfly vertebrae, and hemivertebrae. SCM is associated with scoliosis in 50% to 60% of patients, and SCM is present in 5% of patients with congenital scoliosis.2,31 The apex of the major curvature generally coincides with the level of the SCM because of a strong association of segmentation and vertebral body defects at that level.8 Both tethering and congenital vertebral anomalies contribute to the formation and progression of scoliosis. The scoliotic component may best be appreciated on coronal images from MRI, CT, or standing plain films. Based on published series, CT myelography is superior to MRI in detecting the bony septum of type I SCM (100% versus 82%), bifid vertebrae (100% versus 65%), and hypertrophic neural arches (100% versus 54%).8 Complex, hypertrophic neural arches are seen most often with type I malformations, presumably because of the presence of additional osteogenic meninx primitiva during development. Hypertrophic arches are often fused to the lamina of an adjacent segment, a condition referred to as intersegmental laminar fusion. When associated with bony spina bifida (60% of cases), this finding is virtually pathognomonic for type I SCM31 (Fig. 216-9).
Surgical Management
The mere presence of a type I SCM is often taken as indication for operative intervention. A unique, but small long-term natural history study in patients with type I SCM from the early 1970s observed that 85% of patients without intervention suffer from a progressive neurological deficit versus only 4.5% after surgical treatment.27 Pang observed a similarly high rate of progressive neurological deterioration in patients with type II as with type I SCM and therefore advocated “prophylactic” surgical exploration of both types of malformation.8 By contrast, many surgeons elect to serially observe neurologically and urologically asymptomatic patients with type II SCM discovered incidentally or as a result of the presence of a cutaneous marker only. Unfortunately, data on the long-term natural history of type II SCM and contemporary long-term data from the MRI era regarding type I SCM are not available.
Care must be taken during midline exposure to avoid durotomy and neural injury given the high incidence of concomitant neural arch defects. The complex bony anatomy associated with the SCM may be correlated with preoperative and intraoperative bone imaging. Exposure generally extends one to two levels above and below the SCM to allow dissection to extend from normal to abnormal anatomy. At this point, operations for types I and II SCM differ significantly. For type I SCM, the initial laminectomies should be limited to adjacent levels while initially avoiding exposure of dura at the level of the midline bony spur. Rongeurs or a high-speed drill (or both) should then be used to perform bilateral paramedian laminectomies while preserving the midline lamina and spinous process and thus preventing any torque or lateral force from disrupting the bony spur prematurely.11 Once the spur is isolated and exposed, blunt subperiosteal dissection may be performed to separate the dural sleeves from the spur bilaterally. Dissection typically occurs in a rostral to caudal fashion—from the least adherent to the most adherent dura. Once freed, the spur may be resected carefully with a small rongeur or high-speed drill. Significant bleeding may occur from medullary vessels within the spur itself and can often be controlled with bone wax. Bone removal is complete when the spur is flush with the posterior wall of the vertebral body.
Any associated tethering lesion (sinus tract, fatty filum, or terminal lipoma) should also be addressed. Most authors advocate sectioning of a normal filum in all patients with a low-lying conus.7 Distal tethering of the conus is nearly always present in patients with lower thoracic and lumbar SCMs and, although less frequent, cannot be excluded even in those with more cranially located malformations.8 Dorsal bands are seen with equal frequency in type I and type II malformations and represent an additional tethering lesion.7 Depending on the location of the SCM, distal untethering and treatment of the SCM itself may be undertaken through a single or through two separate incisions. Any additional tethering bands should also be transected so that both hemicords and the conus can move freely within the spinal canal. Successful untethering often indirectly treats syringomyelia effectively. Large, progressive, or recurrent syringes, however, may require direct surgical management. Dorsally, the dura is closed in watertight fashion, if necessary, with a dural patch. One potential advantage of expansion duraplasty is that it discourages retethering. Anterior dural defects are rarely the source of a cerebrospinal fluid (CSF) leak and are not typically closed.
Patients should be kept flat postoperatively for 1 to 2 days to allow a preliminary seal to form along the dural suture line. Some surgeons prefer to maintain patients in the prone position during this initial recovery to discourage adhesion of the spinal cord to the dural closure site. The Foley catheter is removed at the time of mobilization, with monitoring of postvoid residuals. Transient urinary retention affects up to 20% of patients and may require intermittent straight catheterization or discharge with a Foley catheter temporarily in place.32
Outcomes
Surgical repair of SCMs results in improvement or stabilization of progressive neurological deficits in more than 90% of patients.7–1133 Functional outcomes are summarized in Table 216-4.7–10 Axial back pain is improved in the majority of patients and otherwise generally decreases to a tolerable level.8 The remainder of symptoms can be conveniently divided among motor, sensory, and autonomic (bladder) dysfunction. Progressive sensory and motor deficits show the most pronounced improvement after surgical intervention.9 Up to one third of patients will regain urinary function postoperatively.7 The extent of neurological and urologic recovery is related in part to the timing of intervention. Outcomes are most favorable in asymptomatic patients identified by cutaneous stigmata—with most series describing complete preservation of neurological function in patients in whom prophylactic repair is undertaken.7,8 A stable neurological state may in some cases persist for decades, and perhaps indefinitely, after surgical SCM repair.34
TABLE 216-4 Outcomes After Surgical Intervention for Split Cord Malformation
| IMPROVED (%) | UNCHANGED/STABLE (%) | |
|---|---|---|
| Pain | 93 | 0-6 |
| Sensory deficit | 41-58 | 35 |
| Motor deficit | 38-41 | 54-59 |
| Bowel/bladder dysfunction | 21-30 | 64-68 |
| Scoliosis | 0 | 85 |
Complications associated with repair of SCM include CSF leak, wound infection, and new neurological deficits (Table 216-5).7–1132 Secondary repair of an SCM in a patient with a previously closed myelomeningocele at the same level carries a risk for failed wound healing as high as 10%.7,14 Transient urinary retention is the most common new neurological deficit and is seen in up to 25% of patients.32 New sensorimotor deficits may develop in 7% of patients. The majority of these deficits are transient; however, there is a 3% incidence of new permanent deficits.7–11
TABLE 216-5 Complications After Surgical Intervention for Split Cord Malformation
| COMPLICATION | INCIDENCE (%) |
|---|---|
| Transient urinary retention | 20-25 |
| Cerebrospinal fluid leak | 5.1-9.9 |
| Wound infection | 2.5-3 |
| New sensory/motor deficit | 7 |
| Transient | 4.2 |
| Permanent | 1.4-3 |
Bademci G, Saygun M, Batay F, et al. Prevalence of primary tethered cord syndrome associated with occult spinal dysraphism in primary school children in Turkey. Pediatr Neurosurg. 2006;42:4.
Barkovich AJ. Congenital anomalies of the spine. In: Barkovich AJ, editor. Pediatric Neuroimaging. 3rd ed. Philadelphia: Lippincott Williams & Wilkins; 1990:658.
Beardmore HE, Wiglesworth FW. Vertebral anomalies and alimentary duplications; clinical and embryological aspects. Pediatr Clin North Am. 1958:457.
Bremer JL. Dorsal intestinal fistula; accessory neurenteric canal; diastematomyelia. AMA Arch Pathol. 1952;54:132.
Dias MS. Normal and abnormal development of the spine. Neurosurg Clin N Am. 2007;18:415.
Dias MS, Pang D. Split cord malformations. Neurosurg Clin N Am. 1995;6:339.
Dias MS, Walker ML. The embryogenesis of complex dysraphic malformations: a disorder of gastrulation? Pediatr Neurosurg. 1992;18:229.
Gower DJ, Del Curling O, Kelly DLJr, et al. Diastematomyelia—a 40-year experience. Pediatr Neurosci. 1988;14:90.
Guthkelch AN. Diastematomyelia with median septum. Brain. 1974;97:729.
Iskandar BJ, McLaughlin C, Oakes WJ. Split cord malformations in myelomeningocele patients. Br J Neurosurg. 2000;14:200.
James CC, Lassman LP. Diastematomyelia and the tight filum terminale. J Neurol Sci. 1970;10:193.
Kumar R, Bansal KK, Chhabra DK. Occurrence of split cord malformation in meningomyelocele: complex spina bifida. Pediatr Neurosurg. 2002;36:119.
Mahapatra AK, Gupta DK. Split cord malformations: a clinical study of 254 patients and a proposal for a new clinical-imaging classification. J Neurosurg. 2005;103:531.
Pang D. Split cord malformations: theory and practice. In: Batjer HH, Loftus CM, editors. Textbook of Neurological Surgery, vol 1. Philadelphia: Lippincott Williams & Wilkins; 2002:916.
Pang D. Split cord malformation: part II: clinical syndrome. Neurosurgery. 1992;31:481.
Pang D, Dias MS, Ahab-Barmada M. Split cord malformation: part I: a unified theory of embryogenesis for double spinal cord malformations. Neurosurgery. 1992;31:451.
Proctor MR, Bauer SB, Scott RM. The effect of surgery for split spinal cord malformation on neurologic and urologic function. Pediatr Neurosurg. 2000;32:13.
Proctor MR, Scott RM. Long-term outcome for patients with split cord malformation. Neurosurg Focus. 2001;10(1):e5.
Rajpal S, Salamat MS, Tubbs RS, et al. Tethering tracts in spina bifida occulta: revisiting an established nomenclature. J Neurosurg Spine. 2007;7:315.
Schijman E. Split spinal cord malformations: report of 22 cases and review of the literature. Childs Nerv Syst. 2003;19:96.
Schropp C, Sorensen N, Collmann H, et al. Cutaneous lesions in occult spinal dysraphism—correlation with intraspinal findings. Childs Nerv Syst. 2006;22:125.
Sinha S, Agarwal D, Mahapatra AK. Split cord malformations: an experience of 203 cases. Childs Nerv Syst. 2006;22:3.
Tortori-Donati PT, Rossi A, Biancheri R, et al. Congenital malformations of the spine and spinal cord. In: Tortori-Donati PT, Rossi A, editors. Pediatric Neuroradiology: Brain, Head, Neck and Spine. New York: Springer-Verlag; 2005:1587.
Tubbs RS, Wellons JC3rd, Oakes WJ. Lumbar split cord malformation and Klippel-Feil syndrome. Pediatr Neurosurg. 2003;39:305.
Warder DE. Tethered cord syndrome and occult spinal dysraphism. Neurosurg Focus. 2001;10(1):e1.
1 Dias MS, Walker ML. The embryogenesis of complex dysraphic malformations: a disorder of gastrulation? Pediatr Neurosurg. 1992;18:229.
2 Tubbs RS, Wellons JC3rd, Oakes WJ. Lumbar split cord malformation and Klippel-Feil syndrome. Pediatr Neurosurg. 2003;39:305.
3 Pang D, Dias MS, Ahab-Barmada M. Split cord malformation: part I: a unified theory of embryogenesis for double spinal cord malformations. Neurosurgery. 1992;31:451.
4 Rajpal S, Salamat MS, Tubbs RS, et al. Tethering tracts in spina bifida occulta: revisiting an established nomenclature. J Neurosurg Spine. 2007;7:315.
5 Tortori-Donati PT, Rossi A, Biancheri R, et al. Congenital malformations of the spine and spinal cord. In: Tortori-Donati PT, Rossi A, editors. Pediatric Neuroradiology: Brain, Head, Neck and Spine. New York: Springer-Verlag; 2005:1587.
6 Bademci G, Saygun M, Batay F, et al. Prevalence of primary tethered cord syndrome associated with occult spinal dysraphism in primary school children in Turkey. Pediatr Neurosurg. 2006;42:4.
7 Mahapatra AK, Gupta DK. Split cord malformations: a clinical study of 254 patients and a proposal for a new clinical-imaging classification. J Neurosurg. 2005;103:531.
8 Pang D. Split cord malformation: part II: clinical syndrome. Neurosurgery. 1992;31:481.
9 Proctor MR, Scott RM. Long-term outcome for patients with split cord malformation. Neurosurg Focus. 2001;10(1):e5.
10 Sinha S, Agarwal D, Mahapatra AK. Split cord malformations: an experience of 203 cases. Childs Nerv Syst. 2006;22:3.
11 Pang D. Split cord malformations: theory and practice. In: Batjer HH, Loftus CM, editors. Textbook of Neurological Surgery, vol 1. Philadelphia: Lippincott Williams & Wilkins; 2002:916.
12 Mohanty S, Kumar N. Patterns of presentation of congenital scoliosis. J Orthop Surg (Hong Kong). 2000;8:33.
13 Iskandar BJ, McLaughlin C, Oakes WJ. Split cord malformations in myelomeningocele patients. Br J Neurosurg. 2000;14:200.
14 Kumar R, Bansal KK, Chhabra DK. Occurrence of split cord malformation in meningomyelocele: complex spina bifida. Pediatr Neurosurg. 2002;36:119.
15 Dias MS. Normal and abnormal development of the spine. Neurosurg Clin N Am. 2007;18:415.
16 Dias MS, Pang D. Split cord malformations. Neurosurg Clin N Am. 1995;6:339.
17 Schoenwolf GC, Larsen WJ. Larsen’s Human Embryology, 4th ed. Philadelphia: Churchill Livingstone; 2009.
18 Chuai M, Weijer CJ. The mechanisms underlying primitive streak formation in the chick embryo. Curr Top Dev Biol. 2008;81:135.
19 Tada M. Notochord morphogenesis: a prickly subject for ascidians. Curr Biol. 2005;15:R14.
20 Echelard Y, Epstein DJ, St-Jacques B, et al. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell. 1993;75:1417.
21 Cogliatti SB. Diplomyelia: caudal duplication of the neural tube in mice. Teratology. 1986;34:343.
22 Emura T, Asashima M, Furue M, et al. Experimental split cord malformations. Pediatr Neurosurg. 2002;36:229.
23 Beardmore HE, Wiglesworth FW. Vertebral anomalies and alimentary duplications; clinical and embryological aspects. Pediatr Clin North Am. 1958:457.
24 Bremer JL. Dorsal intestinal fistula; accessory neurenteric canal; diastematomyelia. AMA Arch Pathol. 1952;54:132.
25 Schropp C, Sorensen N, Collmann H, et al. Cutaneous lesions in occult spinal dysraphism—correlation with intraspinal findings. Childs Nerv Syst. 2006;22:125.
26 Proctor MR, Bauer SB, Scott RM. The effect of surgery for split spinal cord malformation on neurologic and urologic function. Pediatr Neurosurg. 2000;32:13.
27 Guthkelch AN. Diastematomyelia with median septum. Brain. 1974;97:729.
28 James CC, Lassman LP. Diastematomyelia and the tight filum terminale. J Neurol Sci. 1970;10:193.
29 Warder DE. Tethered cord syndrome and occult spinal dysraphism. Neurosurg Focus. 2001;10(1):e1.
30 Moore KR. III-6: Diastematomyelia. In: Barkovich AJ, editor. Diagnostic Imaging— Pediatric Neuroradiology. Salt Lake City, UT: Amirsys; 2007:40.
31 Barkovich AJ. Congenital anomalies of the spine. In: Barkovich AJ, editor. Pediatric Neuroimaging. 3rd ed. Philadelphia: Lippincott Williams & Wilkins; 1990:658.
32 Schijman E. Split spinal cord malformations: report of 22 cases and review of the literature. Childs Nerv Syst. 2003;19:96.
33 Gan YC, Sgouros S, Walsh AR, et al. Diastematomyelia in children: treatment outcome and natural history of associated syringomyelia. Childs Nerv Syst. 2007;23:515.
34 Gower DJ, Del Curling O, Kelly DLJr, et al. Diastematomyelia—a 40-year experience. Pediatr Neurosci. 1988;14:90.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
CHAPTER 216 Split Spinal Cord
Descriptions of split cord malformation (SCM) appeared in the medical literature as early as the 17th century.1,2 Historically, the terms diastematomyelia (Greek for “cleft[ed] medulla”) and diplomyelia (Greek for “double[ed] medulla”) were used to describe developmental anomalies now grouped together as split cord malformation.3 The sine qua non of these developmental anomalies is the presence of a longitudinal cleft within the spinal cord across one or more vertebral segments.
Type I malformations (formerly diastematomyelia) are characterized by a bony septum that cleaves the spinal canal in the sagittal plane and a duplicated thecal sac (Fig. 216-1A).3 Type II malformations (formerly diplomyelia) are characterized by a cleft cord within a single dural sac, often tethered by a fibrous midline septum to the adjacent dura (Fig. 216-1B and Table 216-1). Not all SCMs fit these descriptions precisely, but the classification scheme provides a framework for the surgical approach to these conditions. In 1992, Pang and colleagues proposed a unified theory attributing both type I and type II SCM to a single error in embryogenesis: adhesion between the ectoderm and endoderm leading to a persistent neurenteric canal.3
Not uncommonly, SCMs are accompanied by nonfunctional paramedian nerve roots traveling within a fibrovascular bundle to or beyond the dura. A fibrous tract extending from the epidural space to a small overlying plaque of atretic skin may also be present. Both the intradural bands and the associated skin lesion have been referred to by the term myelomeningocele manqué, which is derived from the French verb manquer (“to miss”) and reflects the outmoded theory that myelomeningocele manqué is a forme fruste of true myelomeningocele.4 Instead, the descriptive nomenclature “dorsal bands” is used here.
Epidemiology of Split Cord Malformations
Although their precise incidence is unknown, SCMs are exceedingly rare and represent 3.8% to 5% of all congenital spinal anomalies.3,5 One recent series estimated the prevalence of SCM to be 1 in 5000 (0.02%) live births.6 Similar to other forms of occult spinal dysraphism, there is a slight female preponderance, approximately 1.3:1.7–10 The peak age at initial medical evaluation is 4 to 7 years, although there is a second peak between 12 and 16 years of age coinciding with the postpubescent growth spurt. A delay in diagnosis until adulthood may be seen, with back pain being the sole complaint7,8,10,11 or pain accompanied by progressive neurological deficits that may be mistaken for degenerative disease. Although estimates of prevalence vary, type I SCMs may occur more frequently than type II lesions.8,10,11
SCMs rarely occur in the absence of associated anomalies. The most common associations (in descending order) are a tethered/low-lying cord (>50%), kyphoscoliosis (44% to 60%), syringomyelia (27.5% to 44%), and spina bifida (11% to 26%).7,8,10 Conversely, SCMs may occur in 5% of patients with congenital scoliosis12 and in up to 15% of patients with myelomeningocele.13,14 When SCM is seen in association with myelomeningocele, it is three to four times more likely to be a type I malformation and tends to occur at or just rostral to the level of the myelomeningocele defect. On occasion, multiple dysraphic lesions may be identified, and occult SCMs can account both for a discrepancy between functional level and anatomic level of the myelomeningocele placode and for late neurological deterioration in patients with myelomeningocele and an undisclosed SCM.11 Dermoids, dermal sinus tracts, and neurenteric cysts may also be found in conjunction with SCMs.
Embryogenesis
SCMs are thought to occur during the third week of embryogenesis, concurrent with gastrulation, as a result of failure of fusion of the notochord in the midline and subsequent persistence of an accessory neurenteric canal.1,3,15,16
On day 17 of human development, cells of Hensen’s node ingress at the embryonic midline to begin formation of the notochord. In a process of cell-to-cell intercalation, bilayers of cells ingress from each side of midline and intermix to form a singular midline notochordal process.17 Oriented cellular division and cell-to-cell intercalation contribute equally to the process of convergent extension by which the notochord elongates. Oriented cellular division refers to the preferential direction of mitosis demonstrated during embryogenesis that allows a mass of cells to extend in a particular plane rather than growing spherically. Intercalation refers to the cellular process by which two groups of cells integrate in a mosaic pattern (Fig. 216-2A). Thus, polar integrated growth of the combined mass occurs in the cranial-caudal direction. Cellular intercalation is partially governed by genes of the wnt family, which are also responsible (along with sonic hedgehog) for dorsal-ventral patterning of the vertebrate nervous system.18 Knockouts of Prickle, another cell polarity molecule that is conserved across species, demonstrate a foreshortened and thickened notochord.
Reciprocal induction of notochordal and paraxial mesodermal cells also plays a key role in this process. Mesodermal somites express protocadherin, which promotes midline intercalation of the notochord. Embryos lacking protocadherin demonstrate normal elongation of the paramedian notochord masses but no integration, reminiscent of the failed integration seen over small spinal segments in human SCM19 (Fig. 216-2B). Once formed, the notochord acts as an organizer by secreting multiple morphogens that are responsible for proper neurulation of the embryo. Experimental transplantation of notochord fragments has demonstrated that presence of the notochord is necessary and sufficient to influence patterning of spinal development20 and that ectopic or redundant notochordal tissue may result in complete duplication of the spinal cord and surrounding tissues.21 Emura and colleagues produced an amphibian model of SCM by microsurgically creating a neurenteric fistula in embryos, which were then incubated to term. They observed SCM in 40% of the embryos that survived the procedure.22
Most theories posit two common prerequisites for the development of SCM.1,3,23,24 The first is persistent adhesion of the ectoderm and endoderm, termed an accessory neurenteric canal by Bremer.24 The second is a cleft notochord, with subsequent induction of a cleft spinal cord. In 1992, Pang proposed a unified theory for the development of both types of SCM in which these two prerequisites occur as the result of a single ontogenic event—failure of midline integration of the anlagen destined to become the notochordal process.3 This theory postulates that the mass of cells derived from the primitive pit does not ingress as a single mass but instead exhibits left-right asymmetry whereby it courses mediolaterally from the primitive pit before reintegrating in the midline (Fig. 216-2B). Failure of reintegration leads to the formation of an accessory neurenteric canal, which Pang labeled the endomesenchymal tract.11 Persistence of the endomesenchymal tract results in a median cleft with two heminotochords, which induces a similarly cleft spinal cord.
This unified theory explains the subsequent occurrence of types I and II SCM based on incorporation of the meninx primitiva into the endomesenchymal tract. Inclusion of meninx primitiva cells, which arise around day 30, results in the formation of two completely separate dural sleeves as seen in a type I SCM. Inasmuch as the meninx primitiva is osteogenic, this also results in the formation of a midline bony spur (see Fig. 216-1A). When the meninx primitiva is not incorporated into the endomesenchymal tract, a single dural sleeve (and single arachnoid space) contains two hemicords separated by a thin fibrous band, as seen in a type II SCM (see Fig. 216-1B). Incorporation of neural crest (other than the meninx primitiva) into the endomesenchymal tract results in the formation of neural elements medial and dorsal to the hemicords that extend to the extradural space and result in the formation of dorsal bands.
The presence of two separate notochordal anlagen during formation of the notochordal process has never been directly observed, but knockout mice lacking genes for intercalation develop two heminotochords that are not fused in the midline.19 The unified theory suggests that caudal failures of midline integration, which occur relatively late during embryonic development, are more likely to coincide with emergence of the meninx primitiva and thus produce type I SCMs. In fact, type I SCM most frequently occurs from L2 to L4 and is rarely seen above the thoracolumbar junction, whereas type II SCM occurs almost exclusively above T8.8
Signs and Symptoms
Patients with split cord may be asymptomatic, with only cutaneous stigmata of their spinal dysraphism. In various series, there is some type of cutaneous mark present in up to 92% of patients.7,10,11,14,25 The common stigmata associated with SCM are summarized in Table 216-2.7–11,14,25,26 Because of the rostral migration of neural elements relative to the bony spinal column that normally occurs after the formation of an SCM, cutaneous markers are found somewhat caudal to the level of the neural defect itself. The presence of hypertrichosis, although variable, is highly specific for SCM (Fig. 216-3A). One report identified an odds ratio of 61.6:1 in favor of the occurrence of an SCM if a hairy patch is present.25 One characteristic variation of hypertrichosis, the “faun’s tail,” consists of a patch of unusually coarse, raised hair that is strongly associated with type I SCM.25 There is typically a capillary hemangioma underlying these hairy patches. On occasion (≈7%), hemangiomas alone, without overlying hypertrichosis, may indicate the presence of an SCM. Although sacral dimples, dermal sinuses, and cutaneous lipomas are not specific to SCM per se, they often indicate the presence of an additional underlying dysraphic lesion in a patient with SCM (Fig. 216-3B). True sacral dimples that are suggestive of underlying dysraphism should be distinguished from shallow, symmetrical dimples over the coccygeal midline. The latter is common in the general population and is not indicative of the presence of distal tethering pathology.
| SYMPTOMS | PREVALENCE (%) |
|---|---|
| Backache11,26 | 30-50 |
| Progressive neurological deficit11 | 66 |
| Urinary retention/incontinence10,26 | 13-32.5 |
| Leg pain/hyperesthesia10,26 | 7-10 |
| Asymptomatic10 | 13 |
| CUTANEOUS MARKERS | |
| Hypertrichosis9–1125 | 31-61 |
| Dermal sinus8–10 | 7-40 |
| Subcutaneous lipoma8–1014 | 2-25 |
| Sacral dimple8,10 | 5-10 |
| Capillary hemangioma8–1014 | 0.5-7.2 |
| ASSOCIATED FINDINGS ON EXAMINATION | |
| Abnormal urodynamics26 | 74 |
| Left-right motor/sensory asymmetry8 | 21 |
| Left-right asymmetry (limb size/length)10 | 12-15 |
| Talipes equinovarus7,10 | 17-20 |
[/not-level-membership-for-neurosurgery-category]
