[level-membership-for-pediatrics-category]Chapter 45
Spinal Tumor and Tumorlike Conditions
Spinal Neoplasms
Overview: Spinal tumors occur predominantly in young or middle-aged adults and tend to be less common in children. The most common presentation is 3 to 6 months of pain, gait disturbance, change in spinal curvature, motor weakness, and bowel and bladder dysfunction. In the setting of acute trauma, peritumoral edema may cause paresis or paralysis. In this chapter, general concepts regarding spinal neoplasms will be discussed, followed by specific anatomic categories of lesions. Categories will include neoplasms located in the intramedullary, intradural extramedullary, or extradural compartments.1
Imaging: The imaging features of spinal neoplasms are often nonspecific, and findings overlap. Familiarity with age at diagnosis, key imaging findings, and associations can narrow the differential diagnosis, as summarized in Table 45-1.
Intraoperative ultrasound may be used to determine tumor location and borders during exposure and resection. Baseline postoperative MRI is deferred for at least 12 weeks because surgical changes make early postoperative scans difficult to interpret.2
Treatment: The goal with benign, noninfiltrative spinal neoplasms is complete excision of both the solid tumor and associated syrinx cavities. If any portion of the neoplasm infiltrates the cord, surgical success decreases. Adjuvant radiation therapy and chemotherapy may be used, but the prognosis with an infiltrative, malignant lesion is dismal.3
Typically, a laminectomy is performed to gain access to the spinal cord lesion. Myelotomies are performed in the midline between the posterior columns or along the dorsal root entry zone. The tumor must be centrally debulked. Electrophysiologic monitoring techniques can be helpful as the periphery of the tumor is approached to prevent damage to the lateral columns of the spinal cord. The wound is closed when the tumor has been removed to its interface with normal-appearing spinal cord and the associated syrinx cavity has been carefully inspected for additional tumor deposits.3
Prognosis: Preoperative neurologic deficits may persist postoperatively, but most tumors can be removed without new long-term disabilities. If complete tumor excision is not possible with histologically benign pediatric intramedullary gliomas, adjuvant radiation therapy is preferentially avoided because of adverse effects on the immature spinal cord and spinal column. Residual and recurrent tumor deposits often remain static for years or grow very slowly. Symptomatic recurrences can be managed by operating again.
Surgical management is considered with extradural tumors to establish a pathologic diagnosis with biopsy, to decompress the spinal cord in the setting of progressive myelopathy, for reconstruction in cases of spinal instability, and with an attempted radical resection for cure. Most childhood spinal epidural lesions are responsive to radiation or chemotherapy.4
Intramedullary Neoplasms
Overview: Pediatric intramedullary tumors occur most commonly in the cervicothoracic cord.5 Nonspecific symptoms often lead to a delay in diagnosis and vary with the age of the child. Younger children may present with spinal pain (dull and aching) or root pain, rigidity, persistent unexplained torticollis, and muscle spasm. Older children may present with gait disturbance and/or progressive scoliosis. Extremity weakness and paresthesias are common as well.6 A subset of patients may present with symptoms of increased intracranial pressure (ICP) and hydrocephalus.
Overview and Origin: Up to 60% of intramedullary tumors in children are astrocytomas, and the cervical cord is most commonly involved.3 Spinal astrocytomas usually occur in children around 10 years of age, with an equal sex predilection. Spinal astrocytomas are rarely seen in neonates and infants and may present with irritability, torticollis, and loss or absence of developmental milestones. Symptoms often are protracted.6 Astrocytomas arise from astrocytes and range from benign (grade I) to malignant (grade IV).7 Spongioblastomas and pilocytic astrocytomas are at the benign end of the spectrum, and high-grade astrocytomas and glioblastoma multiforme tumors are at the malignant end of the spectrum.
Spinal astrocytomas may be cystic, mixed cystic and solid, solid, or contain necrotic components. Malignant astrocytomas can mimic spinal vascular malformations as a result of hypervascularity with possible intratumoral hemorrhage. Lesion size varies from focal to involvement of the entire spinal cord (a holocord tumor), which usually is seen during the first year of life.8
Imaging: Contrast-enhanced spinal MRI is critical to the identification of small tumors with associated syringohydromyelia and to detect subarachnoid seeding. Key features of astrocytomas include cord expansion, eccentric location, hypointense to isointense appearance on T1-weighted imaging, heterogeneous hyperintensity on T2-weighted imaging, and variable enhancement, sometimes of a mural nodule, on postcontrast images (Figs. 45-1 and 45-2). T2 heterogeneity depends on the presence of solid, cystic, and necrotic components.5
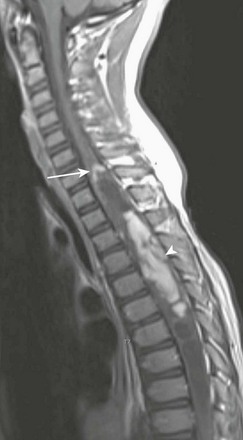
Figure 45-1 An astrocytoma in an 8-year-old boy with back pain.
A sagittal T1-weighted contrast-enhanced image demonstrates an intramedullary cystic mass with dorsal nodular (arrowhead) and peripheral cranial (arrow) enhancement.
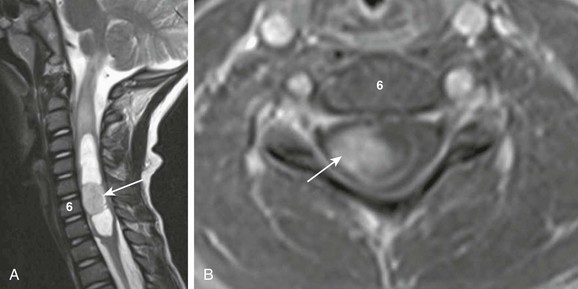
Figure 45-2 An astrocytoma in a 6-year-old boy with pain and decreased use of his right arm.
A, A sagittal T2-weighted image shows an intramedullary cystic cervical cord mass with an enhancing mural nodule (arrow). Note T2 bright cord edema above the mass. B, An axial T1-weighted image with contrast through the enhancing nodule shows that the lesion is eccentric.
Ependymoma
Overview and Origin: Up to 30% of intramedullary tumors in children are ependymomas. Ependymomas present in an older age group than do astrocytomas (at age 13 or 14 years), with a slight female predilection.3 Ependymomas originate from ependymal cells within the central spinal cord or filum terminale, and they frequently span multiple segments. Holocord involvement, as with astrocytomas, is possible.9 A variety of histologic subtypes of ependymomas exist, with cellular ependymoma the most common. Myxopapillary ependymoma occurs exclusively in the lower cord and filum terminale. Surgical management of the myxopapillary subtype is difficult because of varied physical consistency of the lesion and relationship to surrounding structures. When occurring in the cauda equina, this subtype may be associated with subarachnoid hemorrhage, back pain, lower extremity weakness, numbness, pain, and even bowel and bladder incontinence.10
Imaging: Ependymomas are distinguished by the findings of central cord location, cord expansion, heterogeneous signal intensity on all sequences, and a T2 hypointense hemosiderin cap along the cranial or caudal aspects of the tumor (Figs. 45-3 and 45-4). Contrast-enhanced spinal MRI is useful in the evaluation of small drop metastasis, a common feature of ependymomas.11,12 The presence of metastatic spread to the brain with resultant hydrocephalus can be evaluated with cranial MRI. Multiple ependymomas should prompt consideration of neurofibromatosis type 2 (NF2).13
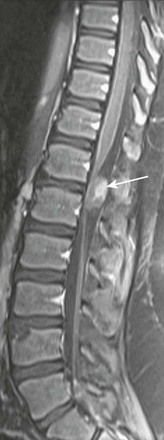
Figure 45-3 An ependymoma in an 11-year-old boy with unexplained altered mental status.
A sagittal T1-weighted image with fat suppression demonstrates an extramedullary heterogeneously enhancing mass (arrow) at T12-L1 displacing the nerve roots ventrally.
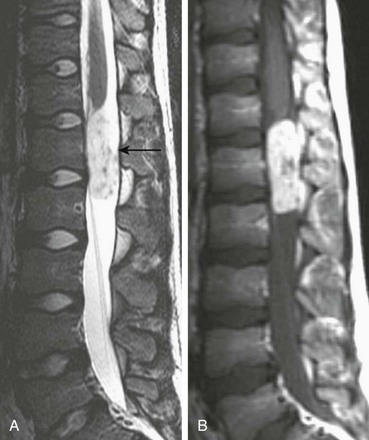
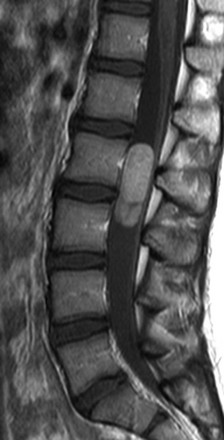
Figure 45-4 A myxopapillary ependymoma in an 11-year-old girl with back pain.
A, Sagittal T2-weighted imaging demonstrates a hyperintense, well-defined mass with internal flow voids arising just below the conus medullaris (arrow). B, A sagittal contrast-enhanced T1-weighted image confirms a mass and shows avid enhancement.
Ganglioglioma
Overview and Origin: Gangliogliomas contain both astrocytic and neuronal components. They present in the first three decades with an average presentation age of 12 years.5,14
Imaging: Gangliogliomas may be solid, cystic, calcified, or hemorrhagic. They demonstrate heterogeneous signal intensity on all sequences.15 Notable imaging features include a lack of peritumoral edema, even when large in size, and associated adjacent osseous erosion (Fig. 45-5). Given this lesion’s similar appearance to an ependymoma on MRI and its association with patients who have NF2, it should be included in the differential diagnosis when ependymoma is a diagnostic consideration.14
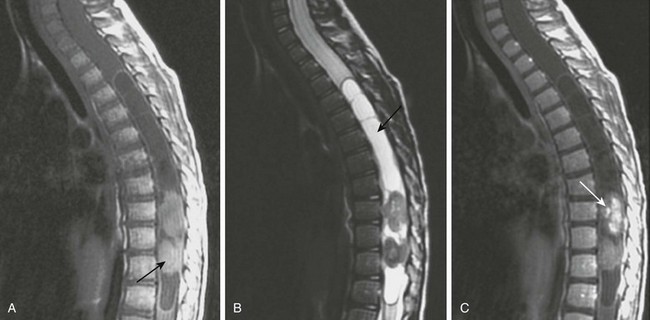
Figure 45-5 Ganglioglioma.
A, A sagittal T1-weighted image shows a holocord tumor. Rounded hyperintense elements seen at the thoracolumbar junction were found at the time of surgery to represent hemorrhagic components of the tumor (arrow). B, A sagittal fat-suppressed T2-weighted magnetic resonance (MR) image shows the hemorrhagic components of the lesion as hypointense masses. Note the tumor cyst in the midthoracic cord (arrow). C, A sagittal postenhanced T1-weighted MR image shows enhancement of the caudal tumor (arrow).
Hemangioblastoma
Overview and Origin: Hemangioblastomas are most commonly intramedullary, but they can be pial, subpial, or combined intramedullary-extramedullary.16 The presence of this lesion should prompt consideration of von Hippel–Lindau disease, and an appropriate workup should be initiated. Treatment includes chemotherapy and surgical resection.17,18
Imaging: Hemangioblastomas frequently are cystic masses with variable T1 signal intensity depending on the proteinaceous content of the cyst fluid, T2 hyperintensity, variable enhancement of mural nodules, and a syrinx in up to 50% of patients (Fig. 45-6). Flow voids are produced by the arterial supply and draining veins of the lesion. Associated hemorrhage and peritumoral edema are common.19
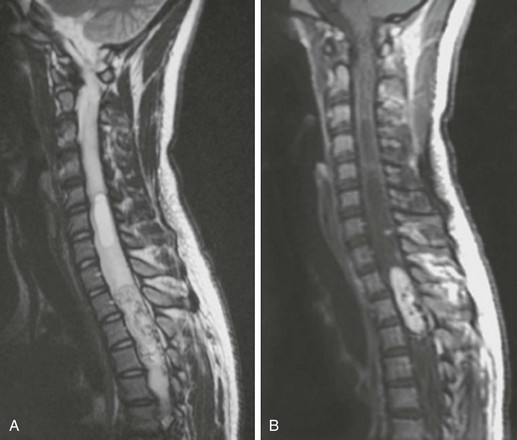
Figure 45-6 A hemangioblastoma in a 14-year-old child with von Hippel–Lindau disease.
A, A sagittal T2-weighted image shows a cystic component superior to the heterogeneous lesion within the spinal cord. The heterogeneous lesion has flow voids. B, A sagittal contrast-enhanced T1 sequence shows marked enhancement of the tumor and multiple small, bright foci of other hemangioblastomas in the cystic portion of the cord mass. A cystic component with a low signal again is seen.
Intradural-Extramedullary Neoplasms
Overview and Origin: The two main spinal nerve sheath tumors are neurofibromas and schwannomas, with the focus in this chapter on neurofibromas.20 Neurofibromas may be derived from an intermediate cell or from a combination of Schwann cells and perineural fibroblasts. Most spinal neurofibromas are intradural-extramedullary lesions. The two morphologic types are fusiform (arising from a single nerve fascicle) and plexiform (involving multiple nerve branches). The lesions may be single, multiple, or diffuse. Neurofibromas usually are encountered in children with neurofibromatosis type 1 (NF1), but they may occur as an isolated lesion.21
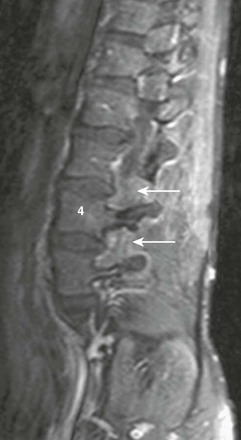
Imaging: Neurofibromas have a variable T1 signal, are T2 hyperintense, and enhance after administration of contrast material. Diffuse neurofibromas may extend along the paraspinal region. Key imaging features of neurofibromas include remodeling and expansion of the intervertebral foramina, which is indicative of slow indolent growth, and a target appearance (central low signal with peripheral high signal) on T2-weighted images (Fig. 45-7). Rib erosion may be seen, and when it is severe over a long length of the rib, it may cause “ribbon ribs.” Malignant degeneration into a neurofibrosarcoma is uncommon but should be considered in large lesions with bone destruction or necrosis.
Meningioma
Overview and Imaging: Meningiomas of the spinal cord are rare in childhood.22 When they are seen in patients with NF2, they may occur intracranially and intraspinally. Meningiomas are T1 hypointense to isointense, T2 hyperintense, and uniformly enhance after administration of contrast material (Fig. 45-8). Areas of hypointensity may be due to calcification.23
Leptomeningeal Metastases
Overview and Origin: Metastatic spread to the spinal canal most commonly occurs via the cerebrospinal fluid (CSF) as a result of drop metastases from primary supratentorial and posterior fossa brain neoplasms, such as medulloblastoma, ependymoma, and atypical teratoid-rhabdoid tumors.24
Imaging: CSF metastases may be solitary, multiple, or diffuse, coating the spinal cord and nerve roots. Metastatic nodules may range in size from 1 to 2 mm to 1 to 2 cm and may block CSF flow (Fig. 45-9). The most common site is the distal thecal sac in the lumbosacral area, followed by the thoracic, and finally the cervical spine.22 To avoid false-positive MRI examinations, the spinal cord should be scanned before surgery for newly diagnosed brain tumors with a propensity to spread via the CSF. If preoperative MRI is not feasible, MRI should be delayed for 4 to 6 weeks after surgery to avoid confusion and possible false positve results.11
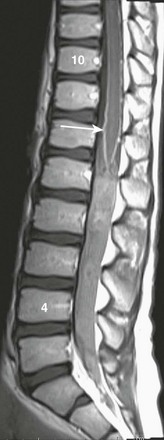
Figure 45-9 Leptomeningeal metastases in a 9-year-old girl with an atypical rhabdoid teratoid tumor who has undergone radiation.
A sagittal contrast-enhanced T1-weighted image demonstrates leptomeningeal enhancement (arrow) of the spinal cord from the lower thoracic cord to the conus medullaris. Extensive solid tissue enhancement from L1-L5 fills the distal thecal sac.
Extradural Tumors
Overview: Most childhood extradural neoplasms arise in the vertebral bodies or the paravertebral soft tissues and invade the spinal canal via the intervertebral foramina, such as neuroblastomas and sacrococcygeal teratomas. Primary osseous lesions, such as aneurysmal bone cysts, lymphoma, and Ewing sarcoma, as well as metastatic lesions, also may involve the vertebra and, secondarily, the spinal canal. Imaging is used to determine the extent of spinal involvement, the degree of neural element compression, and the degree of destabilization of the spinal column by destruction of osseous elements.12,25
Overview and Origin: Tumors of neural crest cell origin include neuroblastomas, ganglioneuroblastomas, and ganglioneuromas. Neuroblastoma is by far the most common extracranial solid neoplasm, usually presenting between 1 and 5 years of age. Neuroblastomas are classified as primitive neuroectodermal tumors (PNETs) when they involve the central nervous system.
Imaging: Neuroblastomas are paraspinal soft tissue masses, often with intraspinal extension. The lesion is hypointense to isointense on T1-weighted imaging compared with the spinal cord, demonstrates subtle hyperintensity on T2-weighted imaging, and has variable enhancement after administration of contrast material (Fig. 45-10). Hypointense areas on T1- and T2-weighted imaging may indicate calcifications. Fat-suppressed T2-weighted imaging is important to determine the extent of disease. Imaging to evaluate for extension across the midline is important in staging.1
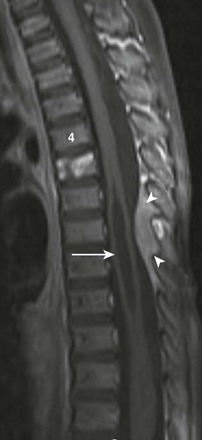
Figure 45-10 A metastatic neuroblastoma in a 2-year-old boy with leg weakness, vomiting, and diarrhea.
A sagittal T1-weighted, fat-suppressed image demonstrates an enhancing metastatic lesion in the T5 vertebral body, a syrinx from T5-T11 (arrow), and a hyperintense, enhancing dorsal epidural lesion (arrowheads). A primary right adrenal mass was found but is not shown.
Sacrococcygeal Teratomas
Overview and Origin: Sacrococcygeal teratomas (SCTs) are neoplasms derived from all three germinal layers. These neoplasms may contain neural elements, squamous and intestinal epithelium, skin appendages, teeth, and calcium. Four types of SCTs are found; type 1 is external, type 2 is predominantly external with a small intrapelvic portion, type 3 is predominantly intrapelvic with a small external component, and type 4 is completely intrapelvic. All four types may present as an extradural mass and may be associated with sacrococcygeal bony erosions. SCTs can be cystic, mixed cystic-solid, or solid lesions. Lesions are more likely to have a favorable prognosis in younger patients, in female patients, when they are mostly cystic masses, when they are less heterogeneous masses, when more calcifications are present, and when they are type 1 (external). SCTs may be benign or malignant.
Imaging: CT is used to demonstrate osseous destruction of the sacrum and coccyx, as well as calcification within the lesion. Cystic components of SCT are T1 hypointense and T2 hyperintense. The MR signal of hemorrhagic components will depend on the age of the hemorrhage. Solid components may enhance after administration of contrast material (Fig. 45-11).
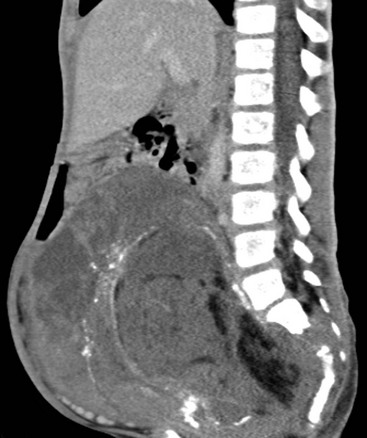
The location of the SCT in relationship to the gluteal fold helps to differentiate this lesion from back masses associated with spinal dysraphic conditions. Specifically, SCTs typically extend below the gluteal fold, and masses associated with spinal dysraphism typically extend above the gluteal fold.26
Lymphoma and Leukemia
Overview and Origin: Lymphoma involves the spine less commonly than it does other reticuloendothelial system structures, such as lymph nodes. Single or multiple vertebral bodies may be involved, especially with non-Hodgkin type lymphoma (Fig. 45-12). A diffuse dural infiltration, a localized dural-based mass, a primary osseous mass with spinal canal extension, or spinal cord masses may be present. Single or multiple vertebral levels may be involved. Epidural deposits from leukemia more commonly produce clinical symptoms in children than in adults, with myelogenous leukemia more likely presenting as a focal mass (Fig. 45-13).27
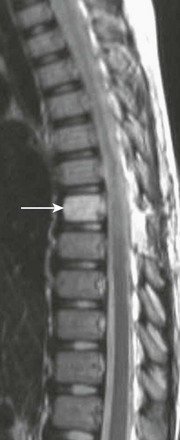
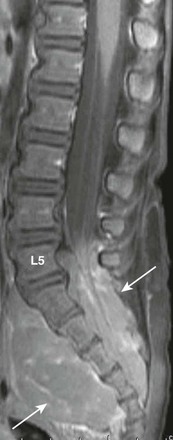
Imaging: Epidural involvement in patients with lymphoma and leukemia is usually isointense on noncontrast T1-weighted imaging. Homogeneous enhancement is seen after administration of contrast material. A key imaging feature suggesting lymphoma or a myelogenous leukemic mass is slightly low signal on T2-weighted images, along with lack of vertebral compression or other osseous destruction.28
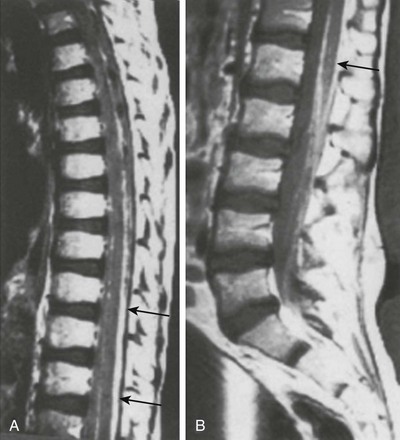
Overview: Neurocutaneous melanosis is a rare, nonfamilial phakomatosis caused by melanocyte proliferation within the epidermis and leptomeninges. It is a nonfamilial disease characterized by large cutaneous pigmented nevi and melanosis of the leptomeninges. A forme fruste may occur in which only leptomeningeal involvement is present without cutaneous manifestations. In the pediatric population, leptomeningeal involvement may present with signs of raised ICP and hydrocephalus.
Imaging: The most common appearance is diffuse leptomeningeal thickening and enhancement after administration of contrast material, as can be seen with leptomeningeal metastases. Although leptomeningeal metastatic disease is more common than leptomeningeal melanosis, the presence of melanin, which is seen as hyperintense signal on noncontrast T1-weighted images, should prompt consideration of this disease (Fig. 45-14). T1 hyperintense melanin deposits most commonly involve the hippocampus and brainstem.29
Extradural Bony Lesions
Overview: Numerous vertebral osseous abnormalities may extend outside of the spine and produce extradural mass effect. Lesions in the spine tend to arise either from the posterior elements or the vertebral body. Common posterior element lesions include osteoid osteomas, aneurysmal bone cysts, and osteoblastomas. Common primary pediatric vertebral masses include Ewing sarcoma, lymphoma, Langerhans cell histiocytosis, and metastatic disease such as neuroblastoma, leukemia, and PNET.
Imaging: The imaging appearance of extradural bony masses varies depending on the specific type of lesion. Unlike with most spinal tumors, CT plays a greater role in the imaging of osseous masses and neoplasms and is complimentary to MRI. Posterior element lesions, which include osteoid osteomas, have a focal sclerotic nidus surrounded by a lucent halo on CT and show a “double density sign” on nuclear bone scintigraphy. On MRI, these lesions show impressive surrounding edema. Aneurysmal bone cysts classically appear as an expansile, multilocular lesion with fluid-fluid levels due to internal hemorrhage and debris (Fig. 45-15). Osteoblastomas are expansile solid masses of the posterior elements and rarely have internal fluid levels (Fig. 45-16). Focal vertebral body lesions, as may be seen with Ewing sarcoma and lymphoma, tend to cause T1 hypointense, T2 hyperintense marrow signal with enhancement after administration of contrast material in a single vertebra with little loss of vertebral body height. Similar features are less often seen with multiple vertebral body involvement.30
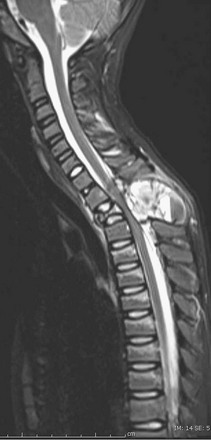
Figure 45-15 An aneurysmal bone cyst in a 10-year-old girl with back pain.
A sagittal T2-weighted image demonstrates a multiloculated, expansile lesion with fluid levels in the T3 posterior elements. Compression of T3 causes effacement of the adjacent spinal canal.
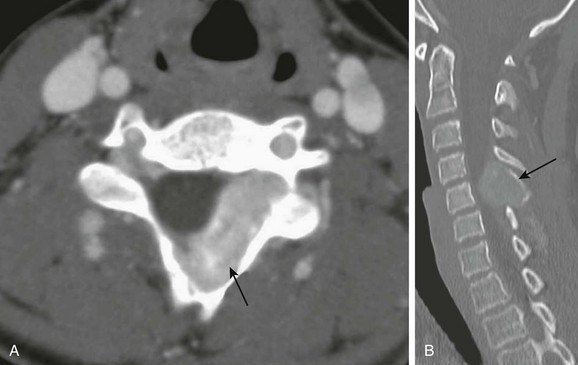
Figure 45-16 Osteoblastoma in 14-year-old boy with left neck pain and paresthesias.
A, Axial contrast-enhanced computed tomography (CT) soft tissue windows demonstrate an enhancing epidural lesion (arrow) with rightward cord displacement. B, A sagittal cervical spine CT image demonstrates an expansile mass arising from the posterior element of C5 (arrow).
Crawford, JR, Zaninovic, A, Santi, M, et al. Primary spinal cord tumors of childhood: effects of clinical presentation, radiographic features, and pathology on survival. J Neurooncol. 2009;95:259–269.
Jallo, GI, Freed, D, Epstein, F. Intramedullary spinal cord tumors in children. Childs Nerv Syst. 2003;19:641–649.
Rossi, A, Gandolfo, C, Morana, G, et al. Tumors of the spine in children. Neuroimaging Clin N Am. 2007;17(1):17–35.
References
1. Dietrich, RB, Kangarloo, H. Retroperitoneal mass with intradural extension: value of magnetic resonance imaging in neuroblastoma. AJR Am J Roentgenol. 1986;146(2):251–254.
2. Keiper, MD, Zimmerman, RA, Bilaniuk, LT. MRI in the assessment of the supportive soft tissues of the cervical spine in acute trauma in children. Neuroradiology. 1998;40(6):359–363.
3. Houten, JK, Weiner, HL. Pediatric intramedullary spinal cord tumors: special considerations. J Neurooncol. 2000;47(3):225–230.
4. Sakai, Y, Matsuyama, Y, Katayama, Y, et al. Spinal myxopapillary ependymoma: neurological deterioration in patients treated with surgery. Spine (Phila Pa 1976). 2009;34(15):1619–1624.
5. Huisman, TA. Pediatric tumors of the spine. Cancer Imaging. 2009;9(Spec No A):S45–S48.
6. Young Poussaint, T, Yousuf, N, Barnes, PD, et al. Cervicomedullary astrocytomas of childhood: clinical and imaging follow-up. Pediatr Radiol. 1999;29(9):662–668.
7. Roonprapunt, C, Houten, JK. Spinal cord astrocytomas: presentation, management, and outcome. Neurosurg Clin N Am. 2006;17(1):29–36.
8. Schittenhelm, J, Ebner, FH, Tatagiba, M, et al. Holocord pilocytic astrocytoma—case report and review of the literature. Clin Neurol Neurosurg. 2009;111(2):203–207.
9. Sarikaya, S, Acikgoz, B, Tekkok, IH, et al. Conus ependymoma with holocord syringohydromyelia and syringobulbia. J Clin Neurosci. 2007;14(9):901–904.
10. Wippold, FJ, 2nd., Smirniotopoulos, JG, Moran, CJ, et al. MR imaging of myxopapillary ependymoma: findings and value to determine extent of tumor and its relation to intraspinal structures. AJR Am J Roentgenol. 1995;165(5):1263–1267.
11. Khan, SN, Donthineni, R. Surgical management of metastatic spine tumors. Orthop Clin North Am. 2006;37(1):99–104.
12. Gebauer, GP, Farjoodi, P, Sciubba, DM, et al. Magnetic resonance imaging of spine tumors: classification, differential diagnosis, and spectrum of disease. J Bone Joint Surg Am. 2008;90(suppl 4):146–162.
13. Aguilera, DG, Mazewski, C, Schniederjan, MJ, et al. Neurofibromatosis-2 and spinal cord ependymomas: report of two cases and review of the literature. Childs Nerv Syst. 2011;27(5):757–764.
14. Patel, U, Pinto, RS, Miller, DC, et al. MR of spinal cord ganglioglioma. AJNR Am J Neuroradiol. 1998;19(5):879–887.
15. Jallo, GI, Freed, D, Epstein, FJ. Spinal cord gangliogliomas: a review of 56 patients. J Neurooncol. 2004;68(1):71–77.
16. Bostrom, A, Hans, FJ, Reinacher, PC, et al. Intramedullary hemangioblastomas: timing of surgery, microsurgical technique and follow-up in 23 patients. Eur Spine J. 2008;17(6):882–886.
17. Sardi, I, Sanzo, M, Giordano, F, et al. Monotherapy with thalidomide for treatment of spinal cord hemangioblastomas in a patient with von Hippel-Lindau disease. Pediatr Blood Cancer. 2009;53(3):464–467.
18. Vougioukas, VI, Glasker, S, Hubbe, U, et al. Surgical treatment of hemangioblastomas of the central nervous system in pediatric patients. Childs Nerv Syst. 2006;22(9):1149–1153.
19. Murota, T, Symon, L. Surgical management of hemangioblastoma of the spinal cord: a report of 18 cases. Neurosurgery. 1989;25(5):699–707. [discussion 708].
20. Kim, NR, Suh, YL, Shin, HJ. Thoracic pediatric intramedullary schwannoma: report of a case. Pediatr Neurosurg. 2009;45(5):396–401.
21. Jinnai, T, Koyama, T. Clinical characteristics of spinal nerve sheath tumors: analysis of 149 cases. Neurosurgery. 2005;56(3):510–515. [discussion 510-515].
22. Liu, PI, Liu, GC, Tsai, KB, et al. Intraspinal clear-cell meningioma: case report and review of literature. Surg Neurol. 2005;63(3):285–288. [discussion 288-289].
23. Solero, CL, Fornari, M, Giombini, S, et al. Spinal meningiomas: review of 174 operated cases. Neurosurgery. 1989;25(2):153–160.
24. Parmar, H, Hawkins, C, Bouffet, E, et al. Imaging findings in primary intracranial atypical teratoid/rhabdoid tumors. Pediatr Radiol. 2006;36(2):126–132.
25. Sciubba, DM, Hsieh, P, McLoughlin, GS, et al. Pediatric tumors involving the spinal column. Neurosurg Clin N Am. 2008;19(1):81–92.
26. Fadler, KM, Askin, DF. Sacrococcygeal teratoma in the newborn: a case study of prenatal management and clinical intervention. Neonatal Netw. 2008;27(3):185–191.
27. Vazquez, E, Lucaya, J, Castellote, A, et al. Neuroimaging in pediatric leukemia and lymphoma: differential diagnosis. Radiographics. 2002;22(6):1411–1428.
28. Murphey, MD, Andrews, CL, Flemming, DJ, et al. From the archives of the AFIP: primary tumors of the spine: radiologic pathologic correlation. Radiographics. 1996;16(5):1131–1158.
29. Hayashi, M, Maeda, M, Maji, T, et al. Diffuse leptomeningeal hyperintensity on fluid-attenuated inversion recovery MR images in neurocutaneous melanosis. AJNR Am J Neuroradiol. 2004;25(1):138–141.
30. Beltran, J, Noto, AM, Chakeres, DW, et al. Tumors of the osseous spine: staging with MR imaging versus CT. Radiology. 1987;162(2):565–569.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]Chapter 45
Spinal Tumor and Tumorlike Conditions
Spinal Neoplasms
Overview: Spinal tumors occur predominantly in young or middle-aged adults and tend to be less common in children. The most common presentation is 3 to 6 months of pain, gait disturbance, change in spinal curvature, motor weakness, and bowel and bladder dysfunction. In the setting of acute trauma, peritumoral edema may cause paresis or paralysis. In this chapter, general concepts regarding spinal neoplasms will be discussed, followed by specific anatomic categories of lesions. Categories will include neoplasms located in the intramedullary, intradural extramedullary, or extradural compartments.1
Imaging: The imaging features of spinal neoplasms are often nonspecific, and findings overlap. Familiarity with age at diagnosis, key imaging findings, and associations can narrow the differential diagnosis, as summarized in Table 45-1.
Intraoperative ultrasound may be used to determine tumor location and borders during exposure and resection. Baseline postoperative MRI is deferred for at least 12 weeks because surgical changes make early postoperative scans difficult to interpret.2
Treatment: The goal with benign, noninfiltrative spinal neoplasms is complete excision of both the solid tumor and associated syrinx cavities. If any portion of the neoplasm infiltrates the cord, surgical success decreases. Adjuvant radiation therapy and chemotherapy may be used, but the prognosis with an infiltrative, malignant lesion is dismal.3
Typically, a laminectomy is performed to gain access to the spinal cord lesion. Myelotomies are performed in the midline between the posterior columns or along the dorsal root entry zone. The tumor must be centrally debulked. Electrophysiologic monitoring techniques can be helpful as the periphery of the tumor is approached to prevent damage to the lateral columns of the spinal cord. The wound is closed when the tumor has been removed to its interface with normal-appearing spinal cord and the associated syrinx cavity has been carefully inspected for additional tumor deposits.3
Prognosis: Preoperative neurologic deficits may persist postoperatively, but most tumors can be removed without new long-term disabilities. If complete tumor excision is not possible with histologically benign pediatric intramedullary gliomas, adjuvant radiation therapy is preferentially avoided because of adverse effects on the immature spinal cord and spinal column. Residual and recurrent tumor deposits often remain static for years or grow very slowly. Symptomatic recurrences can be managed by operating again.
Surgical management is considered with extradural tumors to establish a pathologic diagnosis with biopsy, to decompress the spinal cord in the setting of progressive myelopathy, for reconstruction in cases of spinal instability, and with an attempted radical resection for cure. Most childhood spinal epidural lesions are responsive to radiation or chemotherapy.4
Intramedullary Neoplasms
Overview: Pediatric intramedullary tumors occur most commonly in the cervicothoracic cord.5 Nonspecific symptoms often lead to a delay in diagnosis and vary with the age of the child. Younger children may present with spinal pain (dull and aching) or root pain, rigidity, persistent unexplained torticollis, and muscle spasm. Older children may present with gait disturbance and/or progressive scoliosis. Extremity weakness and paresthesias are common as well.6 A subset of patients may present with symptoms of increased intracranial pressure (ICP) and hydrocephalus.
Overview and Origin: Up to 60% of intramedullary tumors in children are astrocytomas, and the cervical cord is most commonly involved.3 Spinal astrocytomas usually occur in children around 10 years of age, with an equal sex predilection. Spinal astrocytomas are rarely seen in neonates and infants and may present with irritability, torticollis, and loss or absence of developmental milestones. Symptoms often are protracted.6 Astrocytomas arise from astrocytes and range from benign (grade I) to malignant (grade IV).7 Spongioblastomas and pilocytic astrocytomas are at the benign end of the spectrum, and high-grade astrocytomas and glioblastoma multiforme tumors are at the malignant end of the spectrum.
Spinal astrocytomas may be cystic, mixed cystic and solid, solid, or contain necrotic components. Malignant astrocytomas can mimic spinal vascular malformations as a result of hypervascularity with possible intratumoral hemorrhage. Lesion size varies from focal to involvement of the entire spinal cord (a holocord tumor), which usually is seen during the first year of life.8
Imaging: Contrast-enhanced spinal MRI is critical to the identification of small tumors with associated syringohydromyelia and to detect subarachnoid seeding. Key features of astrocytomas include cord expansion, eccentric location, hypointense to isointense appearance on T1-weighted imaging, heterogeneous hyperintensity on T2-weighted imaging, and variable enhancement, sometimes of a mural nodule, on postcontrast images (Figs. 45-1 and 45-2). T2 heterogeneity depends on the presence of solid, cystic, and necrotic components.5
Figure 45-1 An astrocytoma in an 8-year-old boy with back pain.
A sagittal T1-weighted contrast-enhanced image demonstrates an intramedullary cystic mass with dorsal nodular (arrowhead) and peripheral cranial (arrow) enhancement.
Figure 45-2 An astrocytoma in a 6-year-old boy with pain and decreased use of his right arm.
A, A sagittal T2-weighted image shows an intramedullary cystic cervical cord mass with an enhancing mural nodule (arrow). Note T2 bright cord edema above the mass. B, An axial T1-weighted image with contrast through the enhancing nodule shows that the lesion is eccentric.
Ependymoma
Overview and Origin: Up to 30% of intramedullary tumors in children are ependymomas. Ependymomas present in an older age group than do astrocytomas (at age 13 or 14 years), with a slight female predilection.3 Ependymomas originate from ependymal cells within the central spinal cord or filum terminale, and they frequently span multiple segments. Holocord involvement, as with astrocytomas, is possible.9 A variety of histologic subtypes of ependymomas exist, with cellular ependymoma the most common. Myxopapillary ependymoma occurs exclusively in the lower cord and filum terminale. Surgical management of the myxopapillary subtype is difficult because of varied physical consistency of the lesion and relationship to surrounding structures. When occurring in the cauda equina, this subtype may be associated with subarachnoid hemorrhage, back pain, lower extremity weakness, numbness, pain, and even bowel and bladder incontinence.10
Imaging: Ependymomas are distinguished by the findings of central cord location, cord expansion, heterogeneous signal intensity on all sequences, and a T2 hypointense hemosiderin cap along the cranial or caudal aspects of the tumor (Figs. 45-3 and 45-4). Contrast-enhanced spinal MRI is useful in the evaluation of small drop metastasis, a common feature of ependymomas.11,12 The presence of metastatic spread to the brain with resultant hydrocephalus can be evaluated with cranial MRI. Multiple ependymomas should prompt consideration of neurofibromatosis type 2 (NF2).13
Figure 45-3 An ependymoma in an 11-year-old boy with unexplained altered mental status.
A sagittal T1-weighted image with fat suppression demonstrates an extramedullary heterogeneously enhancing mass (arrow) at T12-L1 displacing the nerve roots ventrally.
Figure 45-4 A myxopapillary ependymoma in an 11-year-old girl with back pain.
A, Sagittal T2-weighted imaging demonstrates a hyperintense, well-defined mass with internal flow voids arising just below the conus medullaris (arrow). B, A sagittal contrast-enhanced T1-weighted image confirms a mass and shows avid enhancement.
Ganglioglioma
Overview and Origin: Gangliogliomas contain both astrocytic and neuronal components. They present in the first three decades with an average presentation age of 12 years.5,14
Imaging: Gangliogliomas may be solid, cystic, calcified, or hemorrhagic. They demonstrate heterogeneous signal intensity on all sequences.15 Notable imaging features include a lack of peritumoral edema, even when large in size, and associated adjacent osseous erosion (Fig. 45-5). Given this lesion’s similar appearance to an ependymoma on MRI and its association with patients who have NF2, it should be included in the differential diagnosis when ependymoma is a diagnostic consideration.14
Figure 45-5 Ganglioglioma.
A, A sagittal T1-weighted image shows a holocord tumor. Rounded hyperintense elements seen at the thoracolumbar junction were found at the time of surgery to represent hemorrhagic components of the tumor (arrow). B, A sagittal fat-suppressed T2-weighted magnetic resonance (MR) image shows the hemorrhagic components of the lesion as hypointense masses. Note the tumor cyst in the midthoracic cord (arrow). C, A sagittal postenhanced T1-weighted MR image shows enhancement of the caudal tumor (arrow).
