Chapter 5 Spinal Dysraphism and Tethered Spinal Cord
• Spinal dysraphism refers to anomalies of the spine in which the midline structures do not fuse. Myelomeningocele is the most common significant birth defect involving the spine.
• The prevalence of spina bifida in industrialized countries has been decreasing because of the steadily increasing proportion of affected fetuses that are detected prenatally and electively terminated. In addition, there is strong scientific evidence that the use of preconception folate appears to decrease the risk of developing a neural tube defect such as myelomeningocele.
• Embryologically, the abnormality manifests between 3 and 4 weeks of gestation, during the period called neurulation. The abnormality represents the failure of the posterior neuropore to close properly. Patients with myelomeningocele usually have hydrocephalus and a Chiari II malformation. Surgical closure of the dorsal defect is performed shortly after birth.
• In utero closure of myelomeningocele is a promising surgical technique that has been pioneered at several medical centers. The goal is to decrease the incidence of hydrocephalus and hindbrain abnormalities found in this population. A randomized clinical trial demonstrated efficacy of this treatment option in reducing shunt placement and improving motor function.
• Developmental anomalies involving the caudal portion of the neural tube are increasingly important in clinical practice. This is the result of advances in radiological diagnostic techniques and a consequent change in the philosophy of treatment, which includes prophylactic cord untethering to prevent neurological deficits. Greater awareness of the conditions of lipomyelomeningocele, tethered cord, diastematomyelia, and sinus tracts, by pediatricians, orthopedists, pediatric surgeons, and urologists, in concert with the widespread application of magnetic resonance imaging in addition to the clinical examination, have led to earlier recognition of these congenital surgically correctable problems. Recognition of the cutaneous, orthopedic, neurological, and urological stigmata of “occult” spinal anomalies has been helpful for early diagnosis.
• Patients with sacral agenesis, cloacal extrophy, and other caudal regressions syndromes may require magnetic resonance imaging after initial pediatric surgery intervention to identify potential tethered cord anatomy.
Myelomeningocele
Myelomeningocele is the most common significant birth defect involving the spine. Since the early 1980s the prevalence of spina bifida in industrialized countries has been decreasing because of the steadily increasing proportion of affected fetuses that are detected prenatally and electively terminated. The incidence of the condition ranges from less than 1 case per 1000 live births in the United States to almost 9 cases per 1000 in areas of Ireland. The etiology is unknown, but evidence exists for both environmental and genetic influences. A role for genetic risk factors is supported by numerous studies documenting familial aggregation of this condition. In addition, several lines of evidence point to the potential importance of maternal nutritional status as a determinant of the risk for having a child with spina bifida. Indirect evidence for this association is provided by studies indicating that the season of conception, socioeconomic status, and degree of urbanization may be related to the risk of spina bifida. In August 1991, the Centers for Disease Control and Prevention (CDC) advised that women with a history of an affected pregnancy should take 4 mg of folic acid daily, starting at the time they planned to become pregnant, after publication of the Medical Research Council in Britain Vitamin Study Group report.1 This recommendation was based on a randomized, double-blind, multicenter study performed in Europe that clearly showed the protective effect of periconception folate in reducing the recurrence of spina bifida when ingested by the mothers who had previous births of children with spina bifida. A second randomized, double-blind study was performed in Hungary and demonstrated conclusively the beneficial effects of periconception folic acid intake by mothers on decreasing the incidence of first occurrence of spina bifida.2 It was anticipated that these recommendations would have a substantial impact on reducing the risk of neural tube defects in the offspring of such women. Although it is hoped that this benefit will be the case, it should be noted that the vast majority of affected pregnancies (approximately 95%) occur in women with no history of a prior affected fetus or child.3 Currently the recommended daily dose of folic acid is 0.4 mg for all women of childbearing age who are capable of becoming pregnant. Fortification of the food supply may be a more effective strategy at preventing neural tube defects, rather than individual supplementation.
Recent developments in the prenatal diagnosis of fetal anomalies have made antenatal recognition of myelomeningocele commonplace. Families at risk are routinely offered amniocentesis for amniotic alpha fetoprotein and acetylcholinesterase, which are important in separating open lesions from skin-covered masses, such as myelocystocele. Amniocentesis along with ultrasound screening has a combined accuracy of more than 90%. Prenatal magnetic resonance imaging (MRI), using ultrafast T2-weighted sequences, may also be used to characterize the Chiari II malformation and other associated anomalies.4 Furthermore, fetal MRI may augment ultrasound by detecting spinal cord abnormalities underlying bony abnormalities.5 Recent studies indicate that such prenatal imaging studies can help to determine prognosis. Specifically, lesion level determined by prenatal imaging studies appears to predict neurological deficit and ambulatory potential, but not the degree of fetal ventriculomegaly or the extent of hindbrain deformity.6 Families can be professionally counseled regarding the expected prognosis and decisions about abortion or the new option of fetal closure.
The majority of fetuses with spina bifida that are not electively terminated receive no specific treatment until after birth. In the United States, these babies are generally delivered by cesarean section.7 However, the benefit of this approach relative to vaginal delivery has not been clearly demonstrated. Data suggest that if broad-spectrum antibiotics are administered, closure of the myelomeningocele can be safely delayed for up to a week to allow time for discussion with the parents. In most instances, however, the closure is performed within 48 to 72 hours of birth. The parents should be told the infant’s prognosis based on the functional spinal level, and it should be emphasized that closure of the defect is a life-saving measure but will not alter the preexisting neurological deficits. Pending plans for definitive care, the infant is nursed in the prone position with a sterile, saline-soaked gauze dressing loosely applied over the sac or neural placode.
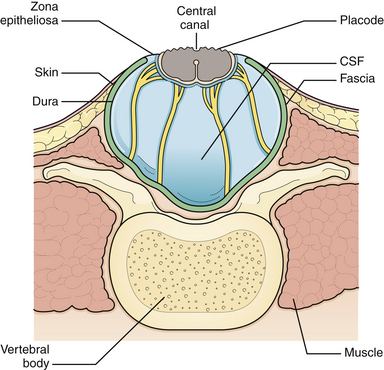
The initial step in managing the newborn with myelomeningocele is a careful physical examination by a pediatrician and neurosurgeon. A thorough evaluation should reveal associated anomalies, including cardiac and renal defects that might contraindicate surgical closure of the spine defect. Approximately 85% of myelomeningocele patients either present with hydrocephalus or develop it during the newborn period.8 A large head or bulging fontanelle suggests active hydrocephalus and indicates the need for a head ultrasound or computed tomography (CT) scan. Stridor, apnea, or bradycardia in the absence of overt intracranial hypertension suggests a symptomatic Chiari II malformation, which carries a poor prognosis. The myelomeningocele is inspected; the red, granular neural placode is surrounded by a pearly “zona epitheliosa” that must be entirely excised to prevent the appearance of a dermoid inclusion cyst. Most myelomeningoceles are slightly oval with the long axis oriented vertically. If the lesion is oriented more horizontally, a horizontal skin closure may be preferable. Neurological examination is difficult in a newborn infant, and it is hard to separate voluntary leg motion from reflex movement. It must be assumed that any leg movement in response to a painful stimulus to that limb is reflexive. Contractures and foot deformity denote paralysis at that segmental level. Virtually all affected neonates have abnormal bladder function, but this is difficult to assess in the newborn. A patulous anus lacking in sensation confirms sacral denervation.
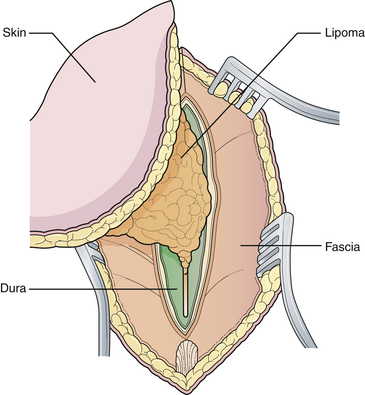
Generally, the back is closed first, and a CSF shunting procedure is deferred unless necessary. In cases with overt hydrocephalus, the back closure and the shunt can be performed at the same time to protect the back closure from CSF leakage. The goal of back closure is to seal the spinal cord with multiple tissue layers to inhibit the entrance of bacteria from the skin and to prevent CSF leakage while preserving neurological function and preventing tethering of the spinal cord. Accomplishing this goal requires a thorough understanding of the three-dimensional anatomy of the tissue layers involved (Fig. 5.1).
Surgical Technique
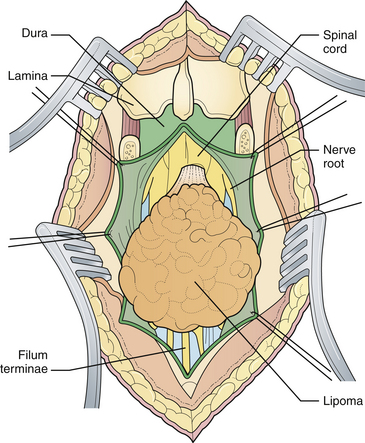
General anesthesia is used, and the patient is placed in the prone position, with rolls under the chest and hips to allow the abdomen to hang freely (Fig. 5.2). If the sac remains intact, fluid is aspirated and sent for culture. The surgeon gently attempts to approximate the base of the sac or defect vertically, then horizontally, to determine which direction of closure will produce the smallest skin defect. An elliptical incision is made, oriented along that axis, just outside the junction of the normal, full-thickness skin and the thin, pearly zona epitheliosa. Full-thickness skin forming the base of the sac is viable and should not be excised. The incision is carried through the subcutaneous tissue until the glistening layer of everted dura or fascia is encountered. The base of the sac is mobilized medially until it is seen to enter the fascial defect (Fig. 5.3A). The sac is entered by radially incising the cuff of skin surrounding the neural placode. This skin is sharply excised circumferentially around the placode with scissors and discarded (Fig. 5.3B). It is important to excise all of the zona epitheliosa to prevent later formation of an epidermoid cyst. At this point, the neural placode is lying freely above the everted dura (Fig. 5.4).
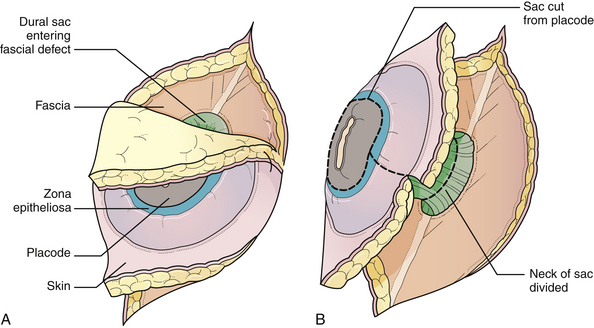
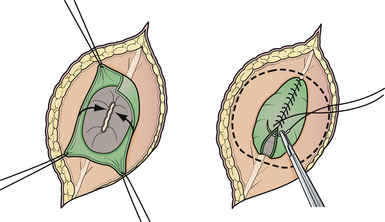
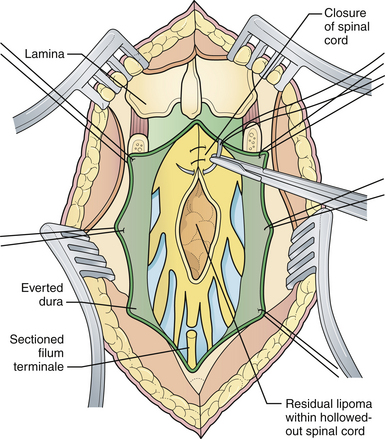
Attention is then directed to the dura, which is everted and loosely attached to the underlying fascia. The dura is undermined bluntly and reflected medially on each side until enough has been mobilized to enable a closure (see Fig. 5.4). The dura is closed in a watertight fashion using a running suture of 4-0 neurilon. If possible, the fascia is closed as a separate layer by incising it laterally in a semicircle on both sides, elevating it from the underlying muscle, and reflecting it medially. Like the dura, the fascia is closed with a continuous stitch of 4-0 suture material (Fig. 5.5). The fascia is poor at the caudal end of a lumbar myelomeningocele as well as with most sacral lesions; thus, closure at this level may be incomplete.
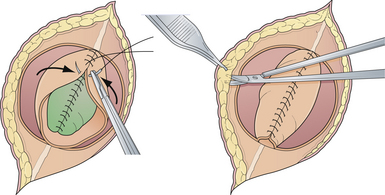
The skin is mobilized by blunt dissection with dissecting scissors or a finger. It may be necessary to free up the skin ventrally all the way to the abdomen (see Fig. 5.5). In most instances, midsagittal (vertical) plane closure is easiest, but occasionally horizontal closure results in less tension. A two-layer closure using vertical interrupted mattress skin sutures is preferred.
Very large lesions require special techniques. Various types of “Z-plasties” and relaxation incisions have been described and may be necessary in very large or difficult lesions. Large circular defects can be closed using a simple rotation flap (Fig. 5.6). Alternative techniques such as allogeneic skin grafts and tissue expansion may be used in rare circumstances.9,10
Care of the child with a myelomeningocele is life-long; it only begins with the surgical closure. Any deterioration in neurological function signals a progressive process such as shunt malfunction, hydromyelia, tethered cord, or symptomatic Chiari II malformation. Significant advancements have been made in the treatment of these children over the past two decades, particularly in the widespread use of multidisciplinary teams of specialists to manage their urological, orthopedic, and other needs. Among those who undergo early back closure, 92% will survive to 1 year.11 From prospective outcome cohort data, it is known that the survival rate until age 17 is 78%,12 but it drops to 46% by the fourth decade of life.13 Death is the result of problems associated with the Chiari II malformation, restrictive lung disease secondary to chest deformity, shunt malfunction, or urinary sepsis. A sensory level higher than T11 is associated with increased risk of mortality,13 likely due to increased risk of urosepsis.14 Approximately 75% of children with myelomeningocele are ambulatory, although most require braces and crutches. Approximately 75% of surviving infants will have normal intelligence (defined as IQ > 80), although only 60% of those requiring shunts for hydrocephalus will have normal intelligence.15 Normal intelligence drops to 70% in surviving adults.16
In a few centers, the fetus with spina bifida may be a candidate for in utero treatment, because this condition is routinely detected before 20 weeks of gestation. There is evidence that neurological deterioration occurs during gestation.17 Normal lower extremity movement can be seen on sonograms of affected fetuses before 17 to 20 weeks of gestation, but most late-gestation fetuses and newborns have some degree of deformity and paralysis. Such deterioration could be the result of exposure of neural tissue to amniotic fluid and meconium or direct trauma as the exposed neural placode impacts against the uterine wall. In theory, such deterioration could be reduced or eliminated by in utero closure of the lesion. Animal studies (in which a model for spina bifida is created by laminectomy and exposure of the fetal spinal cord to the amniotic fluid) have demonstrated improved leg function if the lesion is closed before birth.18 There is also evidence that the Chiari II malformation, which occurs in the vast majority of individuals with spina bifida, is acquired and could potentially be prevented by in utero closure.19
The first cases of in utero spina bifida repair were performed in 1994 using an endoscopic technique. This technique proved unsatisfactory and was quickly abandoned. In 1997, in utero repair of spina bifida was performed by hysterotomy at Vanderbilt University and at the Children’s Hospital of Philadelphia.20,21 Fetuses treated in utero are delivered by cesarean section because the forces of labor are likely to produce a uterine dehiscence. The early experience at both institutions suggested that relative to babies treated postnatally, those treated in utero had a decreased incidence of hindbrain herniation, and possibly a decreased need for shunting.22,23 The combined experience at the Children’s Hospital of Philadelphia and Vanderbilt, indicates that the incidence of hydrocephalus requiring shunting in patients treated in utero is less than in historical control subjects stratified by spinal level who received standard postnatal care.24,25 It is hypothesized that fetal closure of the spinal lesion reduces the need for shunting by eliminating the leakage of spinal fluid which puts back-pressure on the hindbrain. This allows reduction of the hindbrain hernia and relieves the obstruction of the outflow from the fourth ventricle.26
In utero spina bifida closure appears to be generally well tolerated by the expectant mothers. Approximately 5% of fetuses have died from complications associated with uncontrollable labor and premature birth. An analysis of leg function in children treated prenatally revealed no significant difference from a set of historical control subjects who were treated with conventional postnatal repair.27 However, many of the children evaluated in this series had lower limb paralysis at the time of the surgery, which may have diluted any possible benefit. In contrast, a series from the Children’s Hospital of Philadelphia suggested potentially improved leg function in patients with prenatally confirmed intact leg movement on ultrasound prior to fetal surgery.28 Problems with delayed development of dermoid inclusion cysts and tethered cord may adversely affect outcome in the long term.29 The preliminary experience suggests that children treated in utero have the same urodynamic abnormalities that are seen in conventionally treated children with spina bifida.30,31 The incidence of the Chiari II malformation, and the need for shunting may be decreased,23 but there are currently no long-term data.
Prior to the Management of Myelomeningocele Study (MOMS) trial,32 outcomes for spina bifida babies treated in utero were assessed relative to outcomes in conventionally treated, historical control subjects.8 Such comparisons are, however, prone to substantial biases because fetuses that undergo in utero closures represent a highly selected subset of cases. In addition, the medical management of spina bifida is continuously improving, making comparisons with historical control subjects particularly problematic.
A consortium of three institutions (Children’s Hospital of Philadelphia, Vanderbilt, and University of California San Francisco) performed an unblinded, randomized, controlled trial of in utero treatment of spina bifida ([MOMS] to obtain definitive answers regarding the benefits of fetal myelomeningocele closure.32). Pregnant women who receive a prenatal diagnosis of spina bifida between 16 and 25 weeks of gestation were randomized to either in utero repair at 19 to 25 weeks’ gestation or cesarean delivery after demonstration of lung maturity. The primary study end points were the need for a shunt procedure at 12 months, and fetal/infant death. Secondary end points included neurological function, cognitive outcome, and maternal morbidity. The intent to treat analysis demonstrated a significant risk reduction with regard to the primary endpoint, and the study was closed early due to efficacy of prenatal surgery. The prenatal surgery group benefited from decreased shunt requirement (40% versus 82%) and a higher proportion of normal hindbrain anatomy, and it was more likely to ambulate independently at 30 months compared to the postnatal group. There were no maternal deaths, and adverse neonatal outcomes were similar between groups; however, prenatal surgery was associated with more pregnancy complications, increased frequency of pre-term delivery, and a higher rate of respiratory distress syndrome in the neonate. To date, this is the only randomized study that demonstrates clear benefits of in utero treatment of spina bifida. These benefits were realized at experienced centers with strict inclusion criteria and must be carefully weighted against the higher rates of prematurity and maternal morbidity. Longer follow up is required to determine the longevity of these benefits as well as the effect on urinary function.
Occult Spina Bifida and the Tethered Cord Syndrome
The term occult spinal dysraphism actually encompasses several separate, possibly coexisting, entities. Most of these entities are localized to the lower spine segments and hidden by full-thickness skin. Embryologically, they arise from abnormal retrogressive differentiation of the caudal cell mass, a process by which the previously formed tail structures undergo a precise, ordered necrosis, leaving only the filum terminale, the coccygeal ligament, and the terminal ventricle of the conus as remnants by 11 weeks of gestation. Failure of regression presumably gives rise to the hypertrophied filum terminale; abnormal and incomplete regression result in lipomyelomeningocele. The embryology of diastematomyelia remains poorly understood,33 but it may involve persistence of the fetal neurenteric canal between the yolk sac and the amniotic cavity, allowing herniation of endodermal elements through a split notochord, and causing migrating mesenchymal elements to form the bony “spike.”
To recognize occult dysraphic states, one must appreciate the significance of the various syndromes that occur in association with the various entities (Table 5.1). The cutaneous syndrome refers to any midline skin anomaly overlying the lower spine. This anomaly often signals a dysraphic state, and its recognition is especially important in the infant, in whom urological or orthopedic complaints are not yet manifest. Dimples may be significant if they are at the level of the upper sacral or lumbar spine above the gluteal fold, but the common coccygeal pit overlying the lowest point of the coccyx in or below the gluteal fold has no particular significance.34,35 The cutaneous abnormality may include the striking “faun’s tail” of hair (Fig. 5.7), dermal sinus tract, hemangioma (Fig. 5.8), or skin-covered fatty mass (Fig. 5.9). The orthopedic syndrome is apparent at birth or develops progressively in childhood. Common components include high arched feet, claw toes, unequal leg length, and scoliosis. The urological syndrome should be considered in any infant or small child who has an abnormal voiding pattern, a child with a new onset of incontinence after toilet training, or with urinary tract infection in a child of any age. The neurological syndrome presents as leg muscle atrophy or weakness, numbness of the feet, or radicular lower extremity pain and can occur at any age. In summary, patients may present with any of the abovementioned syndromes, but in general, infants primarily present with skin manifestations, older children present with urological, neurological, or orthopedic syndromes, and adults often complain of pain (see Table 5.1).36
TABLE 5.1 Presenting Symptoms and Signs of Occult Spinal Dysraphism
| Symptoms/Signs | Frequency |
|---|---|
| Foot deformity | 39% |
| Scoliosis | 14% |
| Gait abnormality | 16% |
| Leg weakness | 48% |
| Sensory abnormality | 32% |
| Urinary incontinence | 36% |
| Recurrent urinary tract infections | 20% |
| Fecal incontinence | 32% |
| Cutaneous abnormality | 48% |
Adapted from Pang D: Sacral agenesis and caudal spinal cord malformations. Neurosurgery 1993;32:755-758.
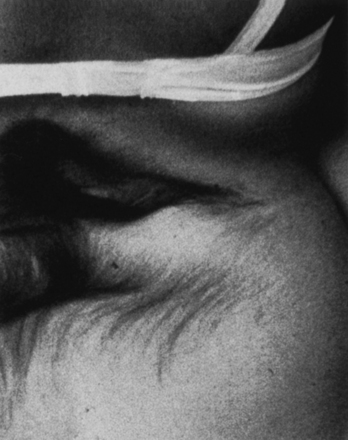
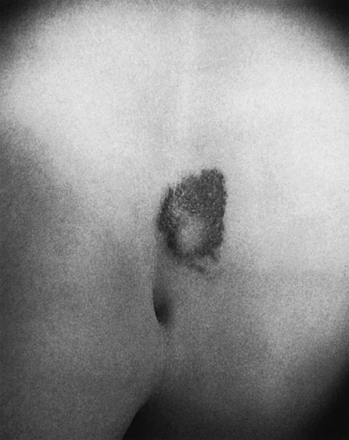
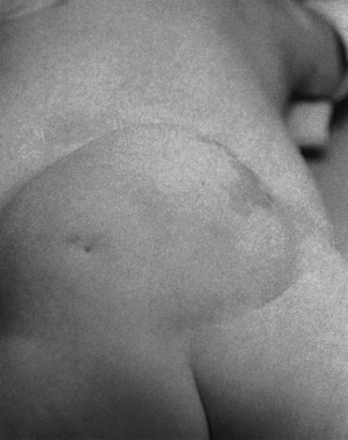
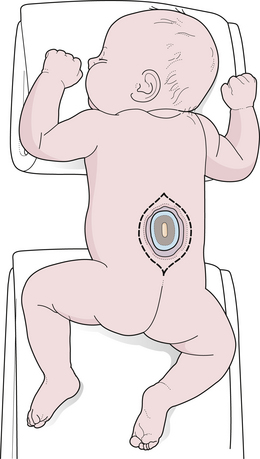
FIGURE 5.9 Lipomyelomeningocele in an infant. The skin-covered fatty mass in the lumbosacral region is typical.
The current method of choice for a suspected occult spinal dysraphic lesion is MRI scanning, which is usually definitive. In newborn infants the image quality can be suboptimal because of their small size, and if the clinical suspicion is high, a repeat scan at 6 months is indicated. The scan is examined for the level of the conus, which should not be below the L2-L3 interspace, and for the presence of fatty masses, a split cord, or a thickened filum. A large distended urinary bladder suggests sacral root dysfunction. In some cases of hypertrophied filum terminale the MRI scan may be equivocal, and if the clinical suspicion is high, surgical exploration may be warranted.37,38 Fat in the filum is a frequent incidental MRI finding, and if the conus is at a normal level and there are no clinical indications of a tethered cord, surgery is usually not indicated. Fat that occurs near the conus may represent a different clinical situation and may be more likely to cause tethering.39
Lipomyelomeningocele
Lipomyelomeningocele is one of the more common forms of occult spinal dysraphism seen in pediatric neurosurgical practice. The term is actually a misnomer, because it suggests herniation of neural elements through a spina bifida defect into a meningeal sac, which is not the case. In fact, the lipomatous tissue inserts into the conus, and it is fat that herniates through the bony defect dorsally to attach to a subcutaneous mass. Nonetheless, the term has gained wide acceptance and is likely to stay. The distinction between lipomyelomeningoceles that insert caudally into the conus and those that attach to the dorsal surface of the spinal cord is of considerable value in planning the operative approach.40 If the lipoma inserts into the dorsal surface of the conus there is usually a substantial subcutaneous mass (Fig. 5.10). Along the lateral interface of the attachment of the lipoma to the spinal cord, the dura and pia are also fused. Sensory roots emerge just anterior to this “lateral line of fusion,” and as a result, neither the sensory nor the motor roots lie within the actual substance of the lipoma. Alternatively, the lipoma joins the conus at its caudal end, almost as a continuation of the cord itself. The remaining lipomatous mass then lies entirely within the spinal canal or extends dorsally through a spina bifida defect. The fatty tumor either replaces the filum terminale, or a separate filum lies anteriorly. The nerve roots usually lie anterior to the lipoma, although they can lie within the fibrous anterior portion of the mass itself (Fig. 5.11). A third, least common, type is the chaotic lipomyelomeningocele, which has a prominent ventral component, as described by Pang and associates.41 Transitional forms of these types may occur, but this schema is extremely useful in planning surgery.
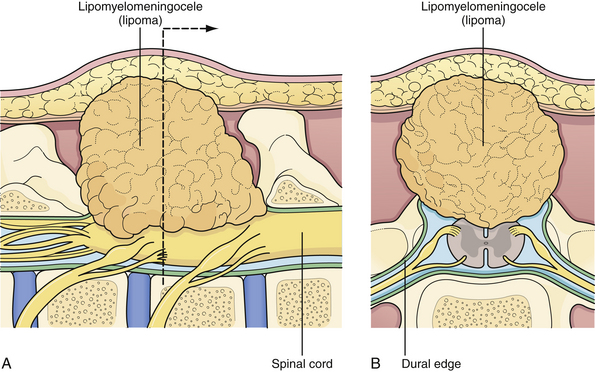
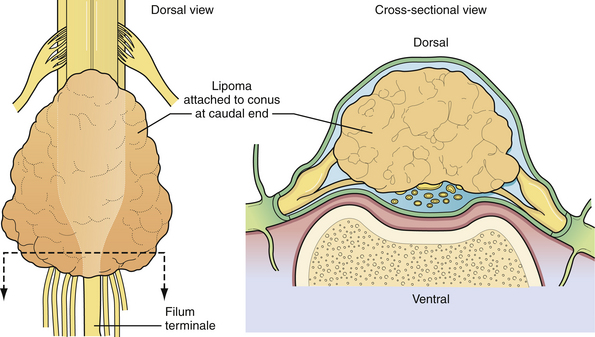
The surgical indications for lipomyelomeningocele have changed over the past 20 years. Although some neurosurgeons have questioned the value of prophylactic surgery,42 almost all modern authorities strongly favor it, preferably in the first 6 months of life.43–45 The rationale is that once a significant neurological deficit occurs, due to natural history in a lipomyelomeningocele patient, the chance of reversing this deficit is not uniformly assured. The risks of creating a new neurological deficit in an experienced neurosurgeon’s hands are low albeit not negligible. Thus, the goals of prophylactic surgery are to untether the spinal cord, remove as much of the lipomatous mass as possible, and reconstruct the dura to avoid leakage of CSF and to discourage retethering. This seems to provide a better outcome than the natural history of this condition in which neurological deficits can occur during periods of rapid growth and activity in which the spinal cord can become stretched and compromised. Surgical planning begins with review of the MRI scan. The lipoma can usually be determined to fit either the dorsal group (Fig. 5.12) or the caudal group (Fig. 5.13).
Surgical Technique
General anesthesia is used, and the patient is positioned prone with rolls under the hips and chest so the abdomen hangs free. If electrophysiological monitoring with electromyography (EMG) or continuous motor evoked potentials is to be used, muscle relaxants must be discontinued before the dura is exposed. An elliptical skin incision surrounding the subcutaneous mass is made along a vertical axis. The subcutaneous tissue is then incised circumferentially down to the lumbodorsal fascia (Fig. 5.14). The lipoma is undermined and separated bluntly from the underlying fascia, until it can be seen to enter the fascial defect medially. A self-retaining retractor is inserted, and the lowest intact laminar arch is palpated. The fascia overlying this spinous process and lamina is opened in the midline, and a laminectomy of this segment is performed, exposing the underlying normal dura. At this point it can help to amputate the large fatty mass with its island of skin attached at the level of its stalk.
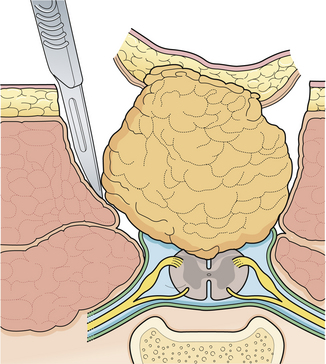
Starting at the level of normal dura cephalad to the mass, the epidural fat is melted with a bipolar cautery until the dural defect with fatty tissue extruding through it is encountered. A midline dural opening is made above the defect, exposing the spinal cord. As the dural opening is carried inferiorly toward the defect, a transverse band of thick, fibrous tissue, which kinks the spinal cord, is noted at the rostral end of the lipoma stalk. This is opened widely along with the dura. The dural opening is extended caudally on either side of the exiting lipoma circumferentially (Fig. 5.15).
At this point, the lipoma will usually be found to correspond to one of the two types previously described. Lipomas that insert into the conus dorsally can be removed from the dorsal aspect of the cord in a plane superficial to the lateral lines of fusion, with the nerve roots emerging anteriorly (Fig. 5.16). These lines of fusion are divided laterally, first on one side and then the other, with a bipolar cautery and microscissors or a knife blade over a dissector. A CO2 laser may be of help to shave down the mass of the lipoma. The filum is identified and divided. After the lipoma has been largely removed from the spinal cord, it is sometimes possible to reapproximate the pial edges of the cord to reconstitute the normal tubular configuration (Fig. 5.17), which discourages retethering.
Lipomas that insert caudally into the conus must be sectioned distally to the take-off of any functional roots. It is unnecessary to remove all of the gross lipoma; attempting this can cause damage to the conus and roots. Simple sectioning of the lipoma releases the point of tethering and typically fulfills the goal of the operation. However, Pang and colleagues have reported that a more aggressive sharp excision of the lipoma can decrease the rate of retethering.46
Surgery is relatively safe. The major problems are postoperative CSF leaks and pseudomeningoceles, which require re-exploration. Most series report a small number of patients whose condition is made worse by the procedure; however, when the outcome of the procedure is compared with that of untreated cases, which are characterized by progressive worsening and disability, the benefits outweigh the risks. The major late problem is retethering, which is suggested by clinical deterioration, and requires re-exploration.47
Diastematomyelia and the Split Cord Malformations
The term diastematomyelia, which derives from the Greek word diastema, meaning cleft, refers to a congenital splitting of the spinal cord. The term is used to describe the split, not the bony spike that often accompanies the abnormality. Clinically, it presents as tethered cord syndrome.48 It occurs predominantly in females and most often in the lower thoracic or upper lumbar spine. Most patients have a midline cutaneous abnormality, but it does not necessarily correspond to the level of the cleft. The most common finding is a hairy patch, but a variety of other cutaneous abnormalities are seen. The spinal deformity (kyphoscoliosis), which eventually develops in virtually all patients, is thought to be primarily the result of the bony structure abnormalities, rather than of neurological involvement.
Pang has suggested a useful classification scheme.33,48 It is proposed that the term diastematomyelia be replaced by the more general term “split cord malformation” (SCM), which may occur as one of two types: The type I SCM consists of two hemicords separated by a bony or cartilaginous median septum, with each housed in its own dural sheath. The type II SCM consists of two hemicords enveloped in the same dural sheath, and separated by a fibrous septum. Both are associated with tethering.
The classic appearance of the SCM on plain spine roentgenograph is a fusiform interpedicular widening of the spinal canal on the anteroposterior view with a midline oval bony mass projecting posteriorly from the vertebral body. The spur is usually not visible on lateral views. CT myelography will clarify the diagnosis, but MRI is currently the primary diagnostic test. The coronal study shows the split nicely, but severe kyphoscoliosis can make the study difficult to interpret. Newer imaging sequences, in which the scan is obtained along the curve of the spine, will probably solve this problem. It is important to evaluate the entire spine so that secondary lesions such as lipomas or hypertrophied fila are not missed.
Clear indications for surgery include progressive neurological deficit and scoliosis. When performing surgery to untether the cord for scoliosis, it is usually advisable to operate on the SCM first as a separate procedure; removal of the bony spike most often results in the temporary loss of evoked potential signals, which can reduce the safety of the orthopedic procedure. In select cases, however, the two procedures can be performed together.49 The management of the asymptomatic patient remains controversial. Historically, some authors have favored a more conservative approach because of the potential risks of surgery and the significant number of patients who remain asymptomatic (or with stable deficits) throughout growth.50 However, more recently, other authors have advocated for prophylactic surgery within the first 2 years in asymptomatic children.51,52 Risk of postsurgical neurological decline is likely highest in SCM subtypes in which the bony septum maximally overlaps the region of cord split.52
Surgical Technique
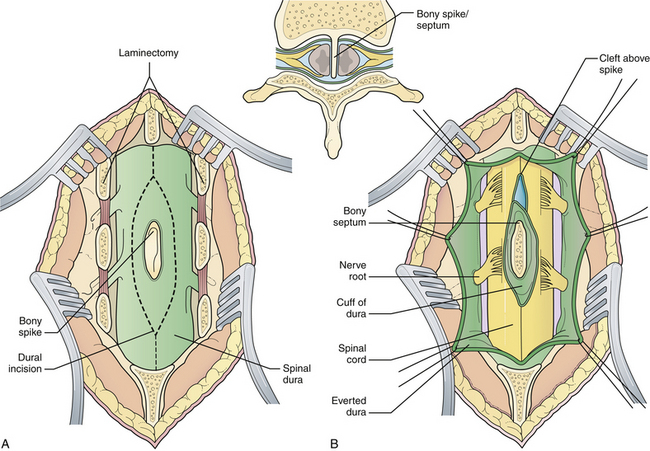
The patient is positioned as for a standard laminectomy. The paraspinal muscles on either side of the midline are freed and retracted laterally as in any standard laminectomy, but vigorous blunt dissection with a periosteal elevator and sponges is avoided, because spina bifida can coexist with the bony septum. The laminectomy is initiated at least one full segment above and below the septum, and it is carried out around the bony spike itself, exposing the dural cleft (Fig. 5.18A). The cleft will usually extend cephalad to the spur but hug it tightly caudally, which indicates tethering. A septal elevator frees the septum from the surrounding dura. The superficial portion of the septum is removed by a rongeur or a high-speed drill that has a diamond burr within the investing dural sheath, which protects the spinal cord. Once the cleft is decompressed, the dura is opened around the cleft, and all intradural adhesions at the cleft are divided (Fig. 5.18B). The dural cuff and the deeper portions of the septum are removed to the level of the anterior spinal canal. It is not necessary or appropriate to close the anterior dura. The posterior dura is closed in a watertight fashion, using a graft if necessary. If an associated hypertrophied filum is suspected, it is divided, using a separate laminectomy if needed.
Anterior Sacral Meningocele
Aspiration of the cyst through the rectum or vagina may result in meningitis and should not be performed. If the meningocele is discovered at laparotomy for other reasons, the operation should be terminated and further workup carried out. Surgical treatment via laparotomy has been described historically and recently.53,54 Nevertheless, most authors prefer the sacral laminectomy approach because it allows visualization of the intraspinal contents of the cyst, resection of adhesions, and sectioning of the filum terminale.55 The goal of surgery is to untether the spinal cord, decompress the pelvic mass, and obliterate the CSF fistula.
Surgical Technique
The surgical technique has been reviewed.56 Antibiotic coverage and a bowel preparation are begun 48 hours prior to surgery in case bowel perforation occurs. Under general anesthesia the patient is positioned for laminectomy. A lumbosacral laminectomy is performed from L5 to S4, and the posterior dura is opened longitudinally (Fig. 5.19). Nerve roots within the dural canal are carefully retracted, and the filum terminale is divided to expose the dural ostium leading to the pelvic sac. If no roots enter the sac and the neck is narrow, the anterior dura is simply oversewn (Fig. 5.20). If the sac arises as a caudal extension of the dural sac, and the sacral roots have exited above, the dural sleeve may simply be ligated. If the anterior defect is wide and cannot be mobilized into the field sufficiently for primary closure, digital collapse of the sac through the rectum can be helpful or a fascial graft can be sewn to the edges of the defect. If roots exit through the defect, the dura or graft will have to be plicated around the roots as they exit. The posterior dura is closed. Postoperatively, stool softeners are given to prevent straining. In difficult cases, a second pelvic procedure is required.
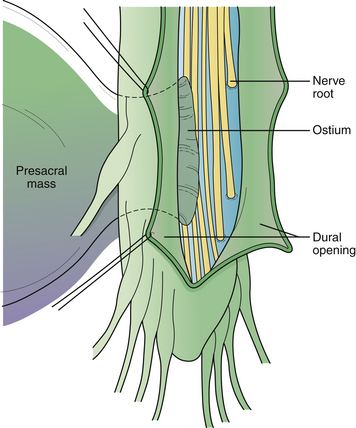
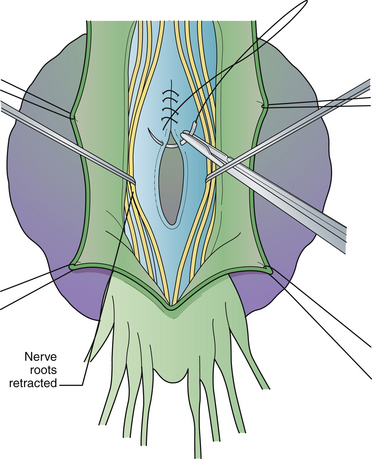
The results of surgery described in the literature have been generally good.54 Complications include meningitis, CSF leak, and neurological problems when nerve roots enter the meningocele sac.
Congenital Dermal Sinus and Hypertrophied Filum Terminale
The term congenital dermal sinus refers to a group of congenital malformations in which a tubular tract lined with squamous epithelium extends from the skin overlying the spine inward to varying depths. The sinus terminates in the subcutaneous tissue, bone, dura, subarachnoid space, filum terminale, or within an intradural dermoid cyst or neuroglial mass within the spinal cord itself. They occur at all levels within the spine but are most commonly seen in the lower lumbosacral area, where they are frequently confused with simple pilonidal sinuses or coccygeal pits. Pilonidal sinuses are acquired lesions in adults, believed to be secondary to trauma or chronic inflammation, that have no connection with the subarachnoid space or neural elements. Coccygeal pits represent a minor embryonic defect in which the sacroccygeal ligament produces a dimple in the overlying skin as the spine begins to elongate. There is no connection with the spinal canal, and the coccyx can be palpated directly beneath. In contrast, a congenital dermal sinus is a significant lesion because it enables skin flora to enter the spinal fluid pathways, resulting in repeated bouts of meningitis. It also causes spinal cord tethering, which leads to progressive neurological problems. The hallmark of the lesion is a midline cutaneous dimple overlying the lumbosacral spine above the gluteal fold. There can be other associated cutaneous abnormalities (see Fig. 5.10), such as hemangiomas or hairy patches.
A somewhat related condition is the so-called meningocele manqué, which is believed to represent an incomplete form of open dysraphism in which bands of meninges, fibrous tissue, and some neural tissue tether the cord to a small area of atretic skin on the back. The radiographic hallmark is a tract extending from the low-lying conus to the cutaneous abnormality. However, it is now recognized that the spinal cord may be tethered even with the conus at a normal level.57 This condition may be suspected only if there are clear clinical symptoms.
Prophylactic surgery is performed as early as possible, even in newborns, to excise the entire tract.58,59 When there is clinical evidence of spinal cord compression, MRI scanning is indicated to determine the extent of abscesses or dermoid cysts. In asymptomatic cases, the lesions can simply be explored and the tract followed to its termination. The surgeon undertaking such an operation must be prepared to carry out an extensive intradural dissection, because the tract can extend for a considerable distance. In typical cases, the sinus tract begins at the skin dimple and proceeds cephalad through the soft tissues overlying the spine to traverse the dorsal dura. Once intradural, the tract often becomes continuous with the filum terminale, which is thickened and may contain dermoid elements (Fig. 5.21).
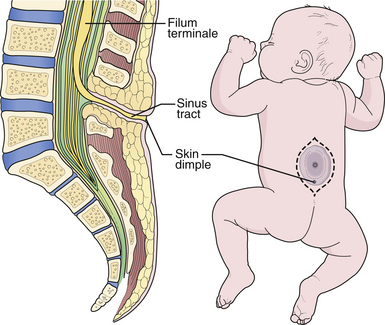
Surgical Technique
The operation begins with an elliptical skin incision that surrounds the sinus opening and encompasses any abnormal skin surrounding it. The tract is sharply dissected and followed through the defect in the fascia. If the tract appears to continue through the dura, a laminectomy is performed above the level of the tract. If the tract attaches to the dura, the dura is opened above the attachment in the midline and incised inferiorly around the point of entry. Any intradural tract must be followed to its termination, even if this involves an extensive laminectomy, because remaining tissue has the capacity to grow into a dermoid inclusion cyst. Intradural dermoids are completely removed, if possible without violating the capsule. If the cyst has ruptured or has been infected, a dense arachnoiditis with scarred nerve roots will prevent complete excision. In this case, judicious intracapsular removal of purulent material and dermoid material is performed, and no attempt is made to remove the scarred capsular wall from the nerve roots. A watertight dural closure is accomplished, except when closure would compress residual infected dermoid cyst material, in which case the dura is left open and the muscle and fascia are closed.
Sacral Agenesis, Myelocystocele, and Cloacal Extrophy
A number of complex anomalies involving the caudal spine have been described, some of which may come to the attention of the neurosurgeon.61,62 These may involve multiple organ systems, and include imperforate anus, the VACTERL syndrome (vertebral anomalies, anal atresia, cardiac anomalies, tracheoesophageal fistula, esophageal atresia, renal and limb anomalies), the OEIS complex (omphalocele, cloacal extrophy, imperforate anus, spinal deformities), and sacral agenesis. Any of these conditions may be associated with the tethered cord syndrome.
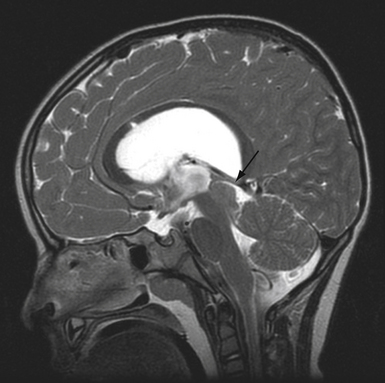
Some consider these caudal regression syndromes to be a continuum, while others feel that they are distinct entities. The severity of the anomaly determines the likely spinal pathology and the management. Simple imperforate anus is associated with hypertrophy of the filum terminale, and there may be no cutaneous stigmata of this condition. Screening with MRI is often recommended. Sacral agenesis is suspected by flattening of the buttocks, shortening of the intergluteal cleft, and prominence of the iliac crest. The newborn with an omphalocele, ambiguous genitalia, and cloacal extrophy may have an associated skin-covered lumbosacral mass, which may represent a myelocystocele or a lipomyelomeningocele. Myelocystocele may occur in association with these complex syndromes, or in isolation. It is generally considered to be an extreme form of the common ventriculus terminalis, which is a normal anatomical variant seen often on MRI scans performed for unrelated indications. The central canal of the terminal spinal cord is massively dilated to form a huge cystic structure, which presents as a skin-covered lumbosacral mass. Fetal ultrasound may confuse a myelocystocele with a cystic myelomeningocele. The spinal cord is invariably tethered. Sacral agenesis is associated with maternal diabetes. Pang has divided these cases into five types, based on the appearance of the sacrum.61 As a practical matter, one can divide these anomalies into those with a high conus and those with a low conus. High symmetrical sacral agenesis is correlated with a truncated, club-shaped conus ending around T11 or T12. Tethering is not present, although dural canal stenosis has been reported as the cause of delayed deterioration. The lower asymmetrical forms of sacral dysgenesis are more likely to have a low-lying tethered cord. Altogether, tethering is reported in 24% of children with anorectal malformations and as frequently as 43% in those with complex malformations.63 Mechanisms of tethering include myelocystocele, lipomyelomeningocele, or simple hypertrophy of the filum.
Initial management of infants with multisystem anomalies is usually non-neurosurgical, and consists of colostomy, closure of an omphalocele, urinary diversion, and reconstruction for tracheoesophageal fistula. An MRI of the spine is obtained electively, when the infant is stable (Fig. 5.22). Fetal MRI can also make the diagnosis.64 When the conus is at a normal level, neurosurgical intervention is usually not required. Those cases in which the conus is low-lying should undergo tethered cord release at about 3 months of age, or when the systemic condition permits. It is useful to perform the tethered cord release prior to reversing the colostomy, because the wound is protected from fecal contamination. Even infants with high motor levels should undergo prophylactic untethering, because improvement is possible.
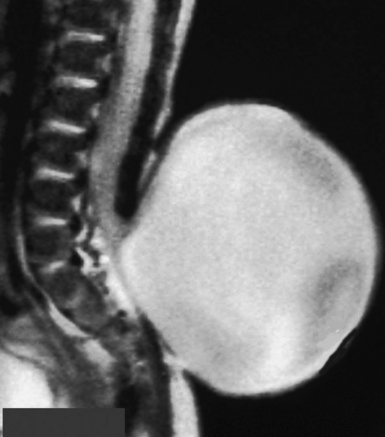
Surgery is similar to that for other tethered cord syndromes. Surgery for myelocystocele consists of defining normal anatomy above the sac, and amputating the sac and all of the tissue below the level of the last intact nerve roots with untethering (Fig. 5.23). The dural reconstruction may require a graft.
Conclusions
The treatment of spinal dysraphism is still evolving. Little in the way of randomized controlled studies is available to support current practice (Table 5.2), However, the recently completed MOMS trial may provide the impetus for further work in this area.
| Statement | Reference∗ | LOE |
|---|---|---|
| Periconceptional folate results in a 72% relative risk reduction in the recurrence of spina bifida when taken by mothers with previous birth of a child with spina bifida. | 1 | I |
| Periconceptional folic acid intake results in a 42% relative risk reduction in the incidence of first occurrence of spina bifida. | 2 | I |
| In utero spina bifida repair is associated with a 52% relative risk reduction in the need for shunt placement at 12 months and twice the likelihood of independent ambulation at 30 months. (Reference 32, LOE I.) For the fetus with uncomplicated myelomeningocele, cesarean delivery before the onset of labor results in an average postnatal motor functional level 2.2 segments lower (better) than that noted for vaginal delivery. | 7 | II |
| In patients with lumbosacral dimples, ultrasound examination is more cost-effective than MRI in screening for occult spinal dysraphism. MRI becomes more effective for higher-risk patients, such as those with anorectal malformations. | 35 | II/III |
| Patients requiring revision spinal lipoma surgery are 2.2 times more likely to have an enlarged neural tube–to–canal ratio than those initially presenting for spinal lipoma surgery. | 41 | III |
| Patients with symptomatic retethering after lipomyelomeningocele repair are 6.6 times more likely to have a transitional lipomyelomeningocele than those who do not experience retethering. | 47 | III |
| Patients with skin stigmata of occult spinal dysraphism who present with neurological deficit are 11 times more likely to be older than 1 year of age than those presenting without neurological deficit. | 58 | III |
LOE, level of evidence.
Adzick N.S., Thom E.A., Spong C.Y., et al. A randomized trial of prenatal versus postnatal repair of myelomeningocele. N Engl J Med. 2011;364:993-1004.
Chapman P. Congenital intraspinal lipomas. Anatomic considerations and surgical treatment. Childs Brain. 1982;9:37-47.
Pang D., Dias M., Ahab-Barmada M. Split cord malformation: Part I: a unified theory of embryogenesis for double cord malformations. Neurosurgery. 1992;31(3):451-480.
Pang D. Split cord malformation: Part II: clinical syndrome. Neurosurgery. 1992;31(3):481-500.
Warder D., Oakes W. Tethered cord syndrome and the conus in a normal position. Neurosurgery. 1993;33(3):374-378.
Please go to expertconsult.com to view the complete list of references.
1. Group M.R.C.V.R.S. Prevention of neural tube defects: results of the Medical Research Council Vitamin Study. Lancet. 1991;338(8760):131-137.
2. Czeizel A.E., Dudas I. Prevention of the first occurrence of neural-tube defects by periconceptional vitamin supplementation. N Engl J Med. 1992;327(26):1832-1835.
3. Botto L., Moore C., Khoury M., Erickson J. Neural tube defects. N Engl J Med. 1999;341(20):1509-1519.
4. Simon E., Goldstein R., Coakley F., et al. Fast MR imaging of fetal CNS anomalies in utero. Am J Neuroradiol. 2000;21(9):1688-1698.
5. von Koch C.S., Glenn O.A., Goldstein R.B., Barkovich A.J. Fetal magnetic resonance imaging enhances detection of spinal cord anomalies in patients with sonographically detected bony anomalies of the spine. J Ultrasound Med. 2005;24(6):781-789.
6. Cochrane D., Wilson R., Steinbok P., et al. Prenatal spinal evaluation and functional outcome of patients born with myelomeningocele: information for improved prenatal counselling and outcome prediction. Fetal Diagn Ther. 1996;11(3):159-168.
7. Luthy D., Wardinsky T., Shurtleff D. Cesarean section before the onset of labor and subsequent motor function in infants with myelomeningocele diagnosed antenatally. N Engl J Med. 1991;324(10):662-666.
8. Rintoul N., Sutton L., Hubbard A., et al. A new look at myelomeningoceles: functional level, vertebral level, shunting, and the implications for fetal intervention. Pediatrics. 2002;109(3):409-413.
9. Danish S.F., Samdani A.F., Storm P.B., Sutton L. Use of allogeneic skin graft for the closure of large meningomyeloceles: technical case report. Neurosurgery. 2006;58(4 Suppl. 2):ONS-E376. discussion ONS-E376
10. Mowatt D.J., Thomson D.N., Dunaway D.J. Tissue expansion for the delayed closure of large myelomeningoceles. J Neurosurg. 2005;103(Suppl 6):544-548.
11. Bol K.A., Collins J.S., Kirby R.S. Survival of infants with neural tube defects in the presence of folic acid fortification. Pediatrics. 2006;117(3):803-813.
12. Hunt G. Open spina bifida: outcome for a complete cohort treated unselectively and followed into adulthood. Dev Med Child Neurol. 1990;32(2):108-188.
13. Hunt G.M., Oakeshott P. Outcome in people with open spina bifida at age 35: prospective community based cohort study. BMJ. 2003;326(7403):1365-1366.
14. Oakeshott P., Hunt G.M., Whitaker R.H., Kerry S. Perineal sensation: an important predictor of long-term outcome in open spina bifida. Arch Dis Child. 2007;92(1):67-70.
15. Sutton L., Charney E., Bruce D. Myelomeningocele—the question of selection. Clin Neurosurg. 1986;33:371-382.
16. Oakeshott P., Hunt G.M. Long-term outcome in open spina bifida. Br J Gen Pract. 2003;53(493):632-636.
17. Stiefel D., Meuli M. Scanning electron microscopy of fetal murine myelomeningocele reveals growth and development of the spinal cord in early gestation and neural tissue destruction around birth. J Pediatr Surg. 2007;42(9):1561-1565.
18. Meuli M., Meuli-Simmen C., Yingling C., et al. Creation of myelomeningocele in utero: a model of functional damage from spinal cord exposure in fetal sheep. J Pediatr Surg. 1995;30(7):1028-1032.
19. Osaka K., Tanimura T., Hirayama A., Matsumoto S. Myelomeningocele before birth. J Neurosurgery. 1978;49(5):711-724.
20. Adzick N., Sutton L., Crombleholme T., Flake A. Successful fetal surgery for spina bifida. Lancet. 1998;352(9141):1675-1676.
21. Tulipan N., Bruner J. Myelomeningocele repair in utero: A report of three cases. Pediatr Neurosurg. 1998;28(4):177-180.
22. Tulipan N., Hernanz-Schulman M., Bruner J. Reduced hindbrain herniation after intrauterine myelomeningocele repair: a report of four cases. Pediatr Neurosurg. 1998;29(5):274-278.
23. Sutton L., Adzick N., Bilaniuk L., et al. Improvement in hindbrain herniation demonstrated by serial fetal magnetic resonance imaging following fetal surgery for myelomeningocele. JAMA. 1999;282(19):1826-1831.
24. Tulipan N., Sutton L., Bruner J., et al. The effect of intrauterine myelomeningocele repair on the incidence of shunt-dependent hydrocephalus. Pediatr Neurosurg. 2003;38(1):27-33.
25. Johnson M.P., Gerdes M., Rintoul N., et al. Maternal-fetal surgery for myelomeningocele: neurodevelopmental outcomes at 2 years of age. Am J Obstet Gynecol. 2006;194(4):1145-1150. discussion 1150–1142
26. Sutton L., Sun P., Adzick N. Fetal neurosurgery. Neurosurgery. 2001;48(1):124-142.
27. Tulipan N., Bruner J.P., Hernanz-Schulman M., et al. Effect of intrauterine myelomeningocele repair on central nervous system structure and function. Pediatr Neurosurg. 1999;31(4):183-188.
28. Johnson M., Sutton L., Rintoul N., et al. Fetal myelomeningocele repair: short term clinical outcomes. Am J Obstet Gynecol. 2003;189(2):482-487.
29. Mazzola C., Albright A., Sutton L., et al. Dermoid inclusion cysts and early spinal cord tethering after fetal surgery for myelomeningocele. N Engl J Med. 2002;347(4):256-259.
30. Holzbeierlein J., Pope J.I., Adams M.C., et al. The urodynamic profile of myelodysplasia in childhood with spinal closure during gestation. J Urol. 2000;164(4):1336-1339.
31. Holmes N.M., Nguyen H.T., Harrison M.R., et al. Fetal intervention for myelomeningocele: effect on postnatal bladder function. J Urol. 2001;166(6):2383-2386.
32. Adzick N.S., Thom E.A., Spong C.Y., et al. A randomized trial of prenatal versus postnatal repair of myelomeningocele. N Engl J Med. 2011;364:993-1004.
33. Pang D., Dias M., Ahab-Barmada M. Split cord malformation: Part I: a unified theory of embryogenesis for double cord malformations. Neurosurgery. 1992;31(3):451-480.
34. Gibson P., Britton J., Hall D., Hill C. Lumbosacral skin markers and identification of occult spinal dysraphism in neonates. Acta Paediatr. 1995;84(2):208-209.
35. Medina L.S., Crone K., Kuntz K.M. Newborns with suspected occult spinal dysraphism: a cost-effectiveness analysis of diagnostic strategies. Pediatrics. 2001;108(6):E101.
36. James H.E., Walsh J.W. Spinal dysraphism. Curr Probl Pediatr. 1981;11(8):6-25.
37. Warder D., Oakes W. Tethered cord syndrome and the conus in a normal position. Neurosurgery. 1993;33(3):374-378.
38. Warder D.E. Tethered cord syndrome and occult spinal dysraphism. Neurosurg Focus. 2001;10(1):e1.
39. Bulsara K., Zomorodi A., Enterline D., George T. The value of magnetic resonance imaging in the evaluation of fatty filum terminale. Neurosurgery. 2004;54:375-379.
40. Chapman P. Congenital intraspinal lipomas. Anatomic considerations and surgical treatment. Childs Brain. 1982;9:37-47.
41. Pang D., Zovickian J., Oviedo A. Long-term outcome of total and near-total resection of spinal cord lipomas and radical reconstruction of the neural placode: Part I: surgical technique. Neurosurgery. 2009;65(3):511-528. discussion 528–519
42. Kulkarni A., Pierre-Kahn A., Zerah M. Conservative management of asymptomatic spinal lipomas of the conus. Neurosurgery. 2004;54(2):868-875.
43. Kanev P., Bierbrauer K. Reflections on the natural history of lipomyelomeningocele. J Neurosurg. 1995;22(3):137-140.
44. Wu H., Kogan B., Baskin L., Edwards M. Long-term benefits of early neurosurgery for lipomyelomeningocele. J Urol. 1998;160(2):511-514.
45. Xenos C., Sgouros S., Walsh R., Hockley A. Spinal lipomas in children. Pediatr Neurosurg. 2000;32(6):295-307.
46. Pang D., Zovickian J., Oviedo A. Long-term outcome of total and near total resection of spinal cord lipomas and radical reconstruction of the neural placode. Part II: outcome analysis and preoperative profiling. Neurosurgery. 2010;66(2):253-272.
47. Colak A., Pillack I., Albright A. Recurrent tethering: a common long-term problem after lipomyelomeningocele repair. Pediatr Neurosurg. 1998;29(4):184-190.
48. Pang D. Split cord malformation: Part II: clinical syndrome. Neurosurgery. 1992;31(3):481-500.
49. Samdani A.F., Asghar J., Pahys J., et al. Concurrent spinal cord untethering and scoliosis correction: case report. Spine. 2007;32(26):E832-836.
50. James C., Lassman L, Diastematomyelia L. A critical survey of 24 cases submitted to laminectomy. Arch. Dis Child. 1964;39:125-130.
51. Dias M., Pand D. Split cord malformations. Neurosurg Clin North Am. 1995;6:339-358.
52. Mahapatra A.K., Gupta D.K. Split cord malformations: a clinical study of 254 patients and a proposal for a new clinical-imaging classification. J Neurosurg. 2005;103(6 Suppl):531-536.
53. Amacher A., Drake C., McLaughlin A. Anterior sacral meningocele. Surg Gynecol Obstet. 1968;126:986-994.
54. Ashley W.W.Jr., Wright N.M. Resection of a giant anterior sacral meningocele via an anterior approach: case report and review of literature. Surg Neurol. 2006;66(1):89-93. discussion 93
55. Lee S.C., Chun Y.S., Jung S.E., et al. Currarino triad: anorectal malformation, sacral bony abnormality, and presacral mass—a review of 11 cases. J Pediatr Surg. 1997;32(1):58-61.
56. Mapstone T.B., White R.J., Takaoka Y. Anterior sacral meningocele. Surg Neurol. 1981;16(1):44-47.
57. Warder D.E., Oakes W.J. Tethered cord syndrome: the low-lying and normally positioned conus. Neurosurgery. 1994;34(4):597-600. discussion 600
58. Ackerman L.L., Menezes A.H. Spinal congenital dermal sinuses: a 30-year experience. Pediatrics. 2003;112(3):641-647.
59. Ackerman L.L., Menezes A.H., Follett K.A. Cervical and thoracic dermal sinus tracts. A case series and review of the literature. Pediatr Neurosurg. 2002;37(3):137-147.
60. van Aalst J., Beuls E.A., Cornips E.M., et al. Anatomy and surgery of the infected dermal sinus of the lower spine. Childs Nerv Syst. 2006;22(10):1307-1315.
61. Pang D. Sacral agenesis and caudal spinal cord malformations. Neurosurgery. 1993;32(5):755-778.
62. Estin D., Cohen A. Caudal agenesis and associated caudal spinal cord malformations. Neurosurg Clin North Am. 1995;6:377-391.
63. Levitt M., Patel M., Rodriguez G., et al. The tethered spinal cord in patients with anorectal malformation. J Pediatr Surg. 1997;32(3):462-468.
64. Midrio P., Silberstein H., Bilaniuk L., et al. Prenatal diagnosis of terminal myelocystocele in the fetal surgery era: case report. Neurosurgery. 2002;50(5):1152-1155.