Chapter 27 Spinal Cord Injury
• Spinal cord injury (SCI) is a problem in both the developed and developing countries and is associated with significant morbidity and long-term human and financial costs.
• Spinal cord injury is a medical emergency. Patients suffering from this injury should be treated in specialized centers with strict adherence to proper clinical examination, imaging, and medical and surgical therapies.
• SCI is a result of primary damage to neural structures and vasculature, followed by secondary damage from physiological insults such as hypotension and hypoxia and resultant cytotoxic swelling and death of neurons and glial cells.
• There have been a multitude of strategies of neuroprotection and repair in SCI and can be found in Box 27.2.
• Spinal shock is represented by depressed or absent spinal reflexes below the injury site. Neurogenic shock after trauma results from disruption of the sympathetic nervous system usually at T6 or higher, which may lead to hypotension and may be difficult to distinguish from hypovolemic shock. The optimal treatment of neurogenic shock is restoring intravascular volume; if symptoms of neurogenic shock persist, vasopressors such as dopamine may be useful.
• Central cord syndrome is often the result of a traumatic spinal cord contusion and affects the central part of the spinal cord. This can cause loss of bilateral pain and temperature sensation and loss of motor function at the level of the lesion and below.
• Early closed reduction of bilateral locked facets in an awake patient, who can participate in a neurological examination, may relieve spinal cord compression and restore normal alignment.
• The use of methylprednisolone as a neuroprotective agent in acute SCI shows moderate benefit in terms of neurological outcome but the benefits of its administration should be considered against the risks associated with medical comorbid conditions in individual patients.
• There are several indications for surgical decompression in SCI. The first is to treat spinal instability caused by fractures and ligamentous injury that may cause further neurological damage. There is some evidence, albeit controversial or evolving, that surgical decompression within 24 hours may improve neurological outcome in some patients with isolated SCI, especially those with cervical SCI who are deteriorating neurologically.
To frame the concepts of SCI—from pathophysiology to translational research to clinical practice—it is important to understand the reasons for undertaking such an endeavor. Spinal cord injury has been studied at a population level in order to understand the incidence, prevalence, and economic costs of this devastating event. These studies tend to come from developed countries that have the financial resources to conduct such research, but this problem certainly does not respect borders. In fact, the frequency of neurotrauma in the developing world is estimated to be higher than in the developed world for reasons such as poor quality of roads, unsafe driving practices, old vehicles without seatbelts or airbags, and a greater proportion of relatively inexpensive transportation such as small motorcycles. Nonetheless, the estimated incidence in the developed world is in the range of 11.5 to 53.4 people per 1 million population.1 This number must be interpreted with caution as some authors point out that the actual incidence may be on the higher side because an unknown number of patients suffering SCI die at the scene of the accident without ever being examined or treated in a hospital.2 Another study estimated that approximately 48% to 79% of individuals die either at the scene of the accident or on arrival to hospital.3 This study was performed in the mid-1970s and most surgeons treating this disease would agree that the number of patients who die is likely less today, mainly as a result of an improved understanding of the cardiorespiratory complications that can arise following injury and advanced intensive care units that are well versed in managing these complications.
One way to comprehend the costs involved in treating a person with SCI is to imagine that a 25-year-old sustains an injury and becomes completely dependent on family and the health care system from this point forward in life. One study estimated that the total cost would be in the range of $3 million (U.S. dollars),4 not to mention the impact of this event on the person’s family and peers. The average age of SCI patients is the late 20s and early 30s and tends to occur more commonly in males. Going back to our example, this event therefore affects people as they are starting their careers and families. A lack of income combined with insurmountable medical costs translates into financial and emotional hardship. Depression and forms of psychological distress are common in this patient group. In well-developed health care systems support networks exist to aid patients and families through the life that they could not have imagined. In third world countries, patients and families are left to their own devices. From either case comes a drive to understand the pathobiology of this devastating condition and hopefully offer treatment strategies. In the pages that follow, we will systematically discuss the events that occur following a traumatic accident—from the shear forces that destroy white matter tracts to the cellular mechanisms that result. With this as a foundation we will discuss different translational research programs that are under way to minimize neurological deficit and hopefully offer neuroregenerative strategies in the near future. Lastly we discuss how a detailed understanding of the pathophysiology of this condition has led to clinical treatment strategies. These strategies are an essential knowledge base for any medical student or resident interested in the treatment of this devastating condition.
Pathophysiology
Acute traumatic injury to the spinal cord can come in the form of a blunt mechanism, such as an acrobat falling onto a hard surface or a person thrown from a car during an automobile collision, or a penetrating injury such as a stab or gunshot wound. The forces involved in each of these events are transmitted to the spinal column and if great enough result in disruption of the bony and ligamentous structures and in damage to the neural elements. Damage to the spinal cord or the exiting nerve roots can result in motor, sensory, or autonomic dysfunction. In an attempt to delineate the precise cause of neurological dysfunction researchers have divided the temporal sequence of destructive events into primary and secondary injury. Primary injury refers to the destructive forces that directly damage the neural structures such as the shear force tearing an axon or direct compressive force occluding a blood vessel, resulting in ischemia. These destructive primary mechanisms not only result in instantaneous damage to neurons and blood vessels but also initiate a cascade of cellular mechanisms that result in ongoing damage to the neural structures, termed secondary injury. In fact, in cases of ongoing primary injury, for example, in the setting of a fracture dislocation where the bony spinal column is displaced and physically pushed up against the spinal cord, these cellular mechanisms are thought to be locked into the “on” position until such physical forces are removed either by closed reduction or surgically. Secondary injury may persist from hours to weeks to years following primary injury. A great deal of work has gone into the detailed understanding of these cellular cascades and along with this work has come an appreciation for the destructive effects of the mechanisms and the role of potential therapeutics to halt these cascades. In the remainder of this section we will highlight the most important secondary mechanisms of injury (Fig. 27.1) and in the section titled “Translational Research” we will discuss some of the therapeutic targets that resulted from these basic science discoveries.
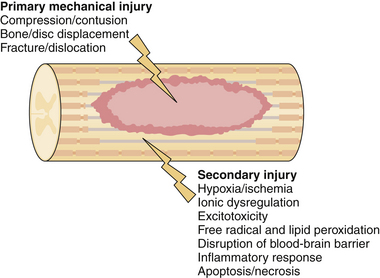
Secondary injury stems from any aberrant physiological or physical force and results in further tissue damage and potential exacerbation of primary injury. Two of the easiest physiological parameters to recognize are hypotension and hypoxia. Both of these conditions are relatively common in trauma patients, who often sustain concomitant injuries and blood loss. Both are also amenable to medical management shortly after the patient arrives at the hospital. Specific treatment options will be discussed later; here we highlight the importance of these two causes of secondary injury and the fact that they further exacerbate secondary cellular cascades. Both global hypotension and hypoxia add to the overall effect of hypoperfusion to the damaged area of the spinal cord. Several other mechanisms add to hypoperfusion including disruption of the microvasculature, loss of normal autoregulatory mechanisms, and increased interstitial pressure.5–7 The effects of this ischemic insult are cytotoxic cell swelling of both neurons and glial cells; as a downstream effect of this, blockade of action potentials has been demonstrated as a result of axonal swelling.8
Ionic dysregulation and excitotoxicity are closely linked events that contribute to tissue damage. Calcium dysregulation in particular has been widely linked to cell death through a number of different processes including calpain activation, mitochondrial dysfunction, and free radical production.9 Whether calcium dysregulation or disruption of other ionic gradients occurs, loss of ionic homeostasis is central to both necrotic and apoptotic cellular death. Excitotoxicity results from activation of glutamate receptors via the influx of both sodium and calcium ions. As cell death occurs, either through the process of necrosis or apoptosis, extracellular glutamate levels rise presumably due to the failure of energy-dependent transporters such as the Na+/K+-ATPase membrane transporter.10 In the next section we will discuss how antagonists of NMDA (N-methyl-D-aspartic acid) receptors are potential targets of translational research in order to minimize cellular death.
Free radical–mediated secondary injury occurs through the process of lipid peroxidation that ultimately results in disruption of axons and death of both neurons and glial cells. Considering the mechanism of free radical damage, peroxynitrite and other reactive oxygen species act through a chain reaction that ultimately destroys cellular membranes and leads to cell lysis, organelle dysfunction. and calcium dysregulation.11 This, in turn, adds to the ionic dysregulation described in the preceding paragraph. Free radicals have been measured in the damaged tissue of experimental models and are found to be elevated for approximately 1 week following injury, returning to preinjury baseline only after 4 to 5 weeks.12 Examples of these free radicals include hydrogen peroxide, hydroxyl radical, nitric oxide, and the superoxide radical. Specific radicals, such as peroxynitrite, have been directly liked to the activation of apoptotic cascades in rat SCI models.13 As with NMDA receptors, free radical oxygen species become a potential target of translational research. If one is theoretically able to mediate the level of these dangerous compounds, could one not prevent the degree of secondary damage that occurs following SCI? This idea will be expanded on in the translational research section below.
Tight junctions between endothelial cells act as an interface between the systemic circulation and the central nervous system. Known as the blood-brain barrier (BBB), it also functions in the spinal cord and acts as a selective barrier that allows passage of solutes such as glucose while maintaining impermeability to macromolecules and infectious agents such as bacteria. Endothelial cells and astrocytes are normally bridged by tight junctions and are interspersed with very selective transmembrane proteins to allow the passage of vital molecules and ions. With the destructive forces that accompany SCI comes the destruction of the BBB. This can result either from direct mechanical destruction, such as damaged blood vessels and spinal cord parenchyma, or from destruction at a cellular level. The latter is best understood as a result of the effects of numerous inflammatory mediators on the endothelial cells that disrupt the normally impermeable barrier. One way researchers have studied this effect is through the use of a compound called horseradish peroxidase. When injected into the peripheral circulation of a normal animal, this compound does not cross the fortress of the BBB and remains in the systemic tissues. When injected into animal models of SCI, the compound was found to permeate into the CNS at a peak time of 24 hours following injury. Such permeability was persistent for up to 2 weeks following injury.14 A number of compounds have been identified that are thought to contribute to this ongoing permeability following injury, each of which is related to the secondary mechanisms of SCI. Inflammatory mediators affecting vascular permeability include tumor necrosis factor-α (TNF-α) and interleukin 1β (IL-1β) and both have been shown to be unregulated following SCI.15,16 Other compounds that play a role include matrix metalloproteinases, histamine, and reactive oxygen species including nitric oxide.12
The inflammatory reaction following SCI has received a renewed interest in recent years largely because of what is thought of as the dual nature of the immune response. On the one hand inflammatory mediators seem to be responsible for the ongoing destruction of tissue, and on the other hand they seem to be clearing cellular debris and optimizing the environment for regenerative growth. Several noncellular mediators are involved in the cascade, the most prominent of which are TNF-α, interferons, and interleukins.17 Cellular mediators following SCI include both resident microglia and peripheral inflammatory cells. A few of the well-characterized examples, mainly from studies in the rodent population, include astrocytes, T cells, neutrophils, and invading monocytes.18 A great example of this dual nature of protection and destruction comes from studies involving TNF-α, which has been demonstrated to be significantly unregulated following SCI.19 Subsequent studies involved the application of neutralizing antibodies to TNF-α following experimental injury, resulting in improved neurological function.20 However, TNF-α-deficient mice have been shown to have higher numbers of apoptotic cells, increased lesion size, and worse function following SCI when compared to wild-type mice.21 This demonstrates the complexity of the inflammatory cascade in SCI and should point the novice reader in the direction of thinking beyond the notion of one type of molecule causing destruction; rather, one must view the many players as acting in a complex pattern, some of which may aid in optimizing function and others that may result in ongoing destruction. The role of research in this field is to tease out these details and provide specific targets for translational research that aim to optimize the potential for recovery.
Cellular death via the apoptotic pathway is thought to play little role in the fate of neurons22 but a considerable role in the fate of oligodendrocytes23 following SCI. Although it may be difficult to demonstrate the apoptotic mechanism of neuronal death in human SCI, there is some evidence for its role in animal models.24 In contrast, the apoptotic mechanism of oligodendrocyte death has been convincingly demonstrated and its mechanism at least partially elucidated. Following SCI, microglia are activated within the first 2 to 48 hours and express the Fas ligand.25 This ligand has a receptor largely expressed on the oligodendrocyte, and communication occurs via the p75 neurotrophin receptor.26 The interaction of the Fas ligand and its receptor initiate apoptosis by way of the caspase cascade that ultimately results in proteolysis, DNA cleavage, and cellular death. This pathway has been the target of translational research strategies and will be expanded on in the next section.
In contrast, cellular death via the necrotic pathway has been demonstrated in both the neuronal and non-neuronal cell populations. Several of the secondary mechanisms of injury previously discussed, such as hypoperfusion and excitotoxicity, culminate in the necrotic death of neurons. Oligodendrocytes are also susceptible to this death pathway and the loss of this cell type has the consequence of axonal demyelination. This demyelination, in combination with other insults on the neuronal processes such as lipid peroxidation and ischemic swelling, results in death of the associated neuronal cell bodies.27 Animal studies have provided evidence that preserved axons represent a critical therapeutic target in order to regain neurological function.28 In stark contrast to this, postmortem human studies have failed to identify demyelinated axons29 and may represent an important disconnect between animal and human pathology following SCI.
Translational Research
As previously stated, an in-depth understanding of secondary mechanisms of SCI has led to identification of numerous possible targets for prevention of this secondary damage and possible regeneration of partially damaged neural circuits. Here we will review a number of agents that are currently under investigation. Broad categories include neuroprotective agents (minocycline and riluzole), myelin-associated inhibitors of neural regeneration (ATI355 and Cethrin), and cellular transplantation strategies (activated autologous macrophages, bone marrow stromal cells, and human embryonic stem cells). This list is by no means exhaustive. Numerous other strategies have been attempted in the past and many lessons have been learned—both positive and negative. Readers interested in a recent history of translational research in the field of SCI are referred to a recently published open source review.30 Alongside developments in neurobiology have come advances in clinical management and operative strategies. In a similar fashion to the scrutiny of drug delivery trials, these clinical and surgical options have also been the focus of clinical trials and extensive review to determine if their effects provide better outcomes for patients. Research that has gone into blood pressure management, methylprednisolone therapy, closed reduction of bilateral locked facets, and early surgical decompression will be discussed in the following section on clinical management.
Neuroprotective Agents
Several promising neuroprotective strategies are under investigation (Box 27.1); two of these are reviewed in greater detail here. Minocycline is a tetracycline derivative that has been of interest to neuroscience for quite some time. It has been shown to have neuroprotective properties in a diversity of animal models, including those that aim to study stroke, Parkinson’s disease, Huntington disease, amyotrophic lateral sclerosis (ALS), and multiple sclerosis.31 As a result of its promise in these conditions, its potential application was carried over to the realm of SCI. Initially studied in animal models, its benefit became apparent in improved neurological outcomes32 and its mechanism of action was thought to be a result of decreased microglia activation and antiapoptotic pathways mediated by the inhibition of cytochrome c release. Because of its extensive use in other neurological conditions, and promise in animal models of SCI, at least two clinical trials are under way to test the efficacy of minocycline in human SCI.
Riluzole is a benzothiazole anticonvulsant that has been in clinical use for greater than a decade. Used primarily in patients with ALS, it has been shown to prolong the lives of persons by 2 to 3 months.33 Relevant to our earlier discussion of ionic dysregulation as a secondary means of injury, riluzole is thought to act by blocking voltage-sensitive sodium channels, whose overactivity after trauma has been associated with neural tissue destruction. In addition, riluzole has been shown to block presynaptic calcium-dependent glutamate release, whose deleterious effects have been discussed. Multicenter clinical trials are under way to study this agent. Its dosing will be similar to that for ALS patients and the duration will be 10 days. This duration was decided as a direct effect of studies into secondary injury cascades, where results indicate that sodium- and glutamate-mediated damage persists for that time period—an excellent example of how translational research is the direct result of thorough and comprehensive basic science research.
A nonpharmacological means of neuroprotection currently under study in the SCI population is hypothermia. Cooling the human body to slow metabolism and enzymatic processes is certainly not new in clinical medicine. However, its application for SCI has received recent attention likely due to new methods that allow for easy and faster cooling via a femoral sheath catheter. A recent retrospectively designed study34 demonstrated the safety of this method and gathered preliminary data with regard to cooling temperature, time to target temperature, duration of cooling, and any adverse events. The long-term follow-up of these patients compared to control subjects will be necessary to demonstrate any functional benefit.
Myelin-Associated Inhibitors of Neural Regeneration
Many regenerative strategies for the treatment of SCI are under investigation; the most prominent molecular and pharmaceutical strategies are listed in Box 27.2 and selected cellular and bioengineered strategies are listed in Box 27.3. Here, we further discuss the evolution of this field along with two therapeutic agents. The notion that the central nervous system cannot regenerate axons after injury was convincingly disproved in the 1980s35 and with numerous other studies after this. Axons, however, do not typically regrow after injury for the many reasons discussed in the secondary injury cascade. Of the innate mechanisms that stunt this growth are the myelin-associated proteins whose activity has been directly linked to a lack of regenerative capacity. Many inhibitors of myelin-associated proteins have been researched in cell culture and animal models and two have been the focus of investigation in clinical trials: ATI335 and Cethrin.
ATI335 has a rich history ranging from basic laboratory science through animal models and into the realm of translational research. CNS myelin, at this time known to be an inhibitor of axonal growth, was biochemically separated into component proteins.36 Antibodies to these proteins were then developed and applied to in vitro models that demonstrated their ability to diminish the inhibitory effects of myelin. Subsequent animal models demonstrated improved neurological outcomes with administration of these antibodies.37 After a flurry of excitement in the animal world, Nogo was characterized as the target antigen and human antibodies were subsequently developed. These antibodies were demonstrated to promote axonal growth and functional recovery in primate models of SCI. Clinical trials began shortly thereafter and are currently under way in both Europe and Canada.
Myelin inhibitors of axonal growth are known to signal through the Rho cascade. Briefly, Rho is a guanosine triphosphatase that, when activated, binds to Rho kinase (ROCK). ROCK, in turn, is a key regulator of axonal growth cone dynamics and cellular apoptosis. Disruption of this cascade has been shown to facilitate axon growth and functional recovery in mice.38 Initial studies made use of a toxin produced by Clostridium botulinum termed C3 transferase, a specific inhibitor of Rho. Eventually, a recombinant protein was created, commercialized, and brought to clinical trial. Cethrin is a combination of this recombinant protein and fibrin glue—the mixture is applied directly to the dura at the time of surgical decompression of SCI. Initial results of a multicenter phase I/IIa trial are becoming available and appear promising in terms of neurological recovery. A phase II study is being planned.
Cellular Transplantation Strategies
Activated Autologous Macrophages
It has been realized for about 2 decades that macrophages play a vital role in the regeneration of peripheral nervous function and that this role does not exist in the central nervous system. Following injury to a nerve in the peripheral nervous system, macrophages are recruited to the site of injury and are responsible for clearing myelin debris and optimizing the local environment for regeneration. In order to capitalize on this finding in the central nervous system, researchers attempted to take autologous macrophages from SCI patients, activate them with peripheral myelin, and inject them into the damaged area of the spinal cord shortly after injury. The initial results were promising, although only a small number of individuals received treatment. Owing to financial circumstances, the trial was never expanded beyond the initial study. This strategy once again highlights a bridge between understanding the secondary mechanisms of damage, in this case how central myelin acts to inhibit neural regeneration and repair, and the development of potential therapy. Hopefully this promising strategy will be revitalized and studied in a larger population.
Bone Marrow Stromal Cells
The use of bone marrow stromal cells aims to take advantage of a relatively accessible multipotent stem cell that has the potential to differentiate and integrate into existing spinal circuits and result in neural recovery. A number of groups around the world are reporting the use of these cells including scientists from Korea, China, Russia, the Czech Republic, and Brazil. To date, the trials have been small and are often not blinded, prompting cautious interpretation of the results. Nonetheless, researchers report significant neurological recovery after direct injection of this cell type in the damaged area of the spinal cord. Researchers in Prague, Czech Republic, did have blind assessors evaluate patients at follow-up in terms of both the ASIA (American Spinal Injury Association) scale and electrophysiologically. Significant improvement was noted in patients who received therapy within 3 to 4 weeks of injury but not in those who were in the chronic stages of injury.39
Human Embryonic Stem Cells
Although human embryonic stem cells are theorized as one of the more promising strategies in cell replacement in the spinal cord, there are a number of hurdles that must be overcome. This cell type is first cultured in vitro and the purity of these cultures is paramount and somewhat challenging. In addition, viral contamination via delivery vectors and the acquisition of membrane polysaccharides that may react with the host immune system are all concerns that are being addressed. Significant advances have been made in this field, mainly out of the University of California at Irvine,40,41 and are currently being evaluated by the Food and Drug Administration (FDA) in the United States. The goal of this therapy is to achieve differentiation of these stem cells into oligodendrocytes that would aid in remyelination of spared but demyelinated axons. This represents a promising avenue of research in the decades to come.
Clinical Management
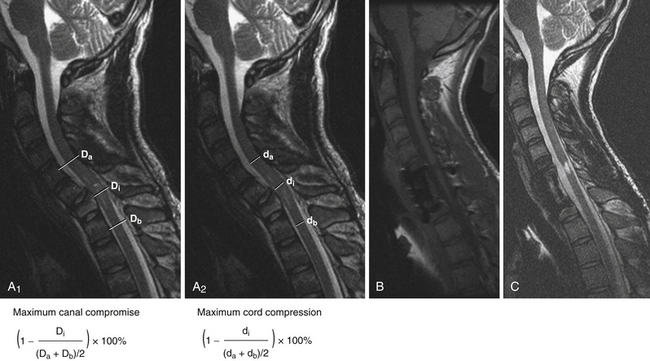
A patient who sustains an SCI has often sustained concomitant injuries and may be medically unstable; in fact, the treating physician may not recognize the presence of SCI immediately. Adherence to Advanced Trauma Life Support (ATLS) protocol is essential. Airway, breathing, and circulation (ABCs) are of paramount importance, followed by the treatment of any immediate life-threatening condition. If there is concern that any one of these domains is unstable, it should be revisited before moving on. Hypotension, in particular, must be observed for an ongoing secondary SCI can result. Following stabilization of the patient, the treating physician should then proceed with the neurological examination. This comes in the form of testing for motor power, sensory impairment, and reflexes including rectal tone. This examination should be documented on the standard ASIA forms.42 Following a detailed examination, and assuming the finding of a deficit, imaging of the spinal column with computed tomography (CT) scan and imaging of the spinal cord with magnetic resonance imaging (MRI) should follow in short order. To frame the concepts of SCI, refer to Figure 27.2, which displays the MRI of an 18-year-old male subject who sustained an SCI. Also shown in this figure is the method for calculating the maximal spinal canal compromise and the maximal cord compression—two measurements that are useful for quantifying the degree of injury.43 In the remainder of this chapter we will review the following important concepts that are essential to understand in order to properly care for the SCI patient: spinal shock, neurogenic shock, spinal cord syndromes, evidence for early closed reduction of bilateral locked facets, evidence for methylprednisolone therapy, and evidence for early surgical decompression.
Neurogenic Shock
Neurogenic shock is a potentially life-threatening condition and must be managed as such. Without a clear understanding of this condition inappropriate management of a trauma patient, who often suffers concomitant hemodynamic instability, could be fatal. Neurogenic shock is defined as disruption of the sympathetic nervous system with preserved parasympathetic activity. This typically occurs with patients suffering a severe SCI at the level of T6 or higher. Disruption of the sympathetic division of the autonomic nervous system affects three areas of the cardiovascular system: coronary blood flow, cardiac contractility, and heart rate. With preserved parasympathetic activity this translates clinically into bradycardia (and possibly other cardiac arrhythmias) in the setting of profound hypotension. A prudent clinician must look for these characteristics in combination, as many trauma patients are hypotensive as a result of blood loss or intravascular hypovolemia but will mount an appropriate tachycardic response.
Spinal Cord Syndromes
Paresis is a term used to describe weakness or partial paralysis. For example, hemiparesis describes weakness on one side of the body. In contrast, both paralysis and the suffix –plegia refer to no movement. For example, hemiplegia refers to no movement on one side of the body. Keeping these terms in mind, we turn our attention to spinal cord syndromes that occur when a select region of the spinal cord is damaged, resulting in a predictable pattern of neurological deficit. These syndromes are expanded on in the paragraphs that follow and are depicted in Figure 27.3, along with a description of the ASIA impairment scale.
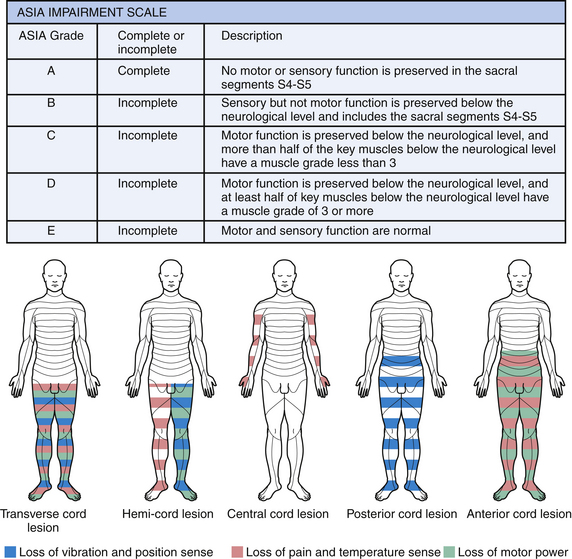
Hemisection of the spinal cord, commonly referred to as Brown-Sequard syndrome, is characterized by damage to one half of the spinal cord and all motor and sensory pathways at that level and neurological deficits at and below that level. This results in ipsilateral upper motor neuron weakness and ipsilateral loss of vibration and position sense at and below the level of lesion. There is contralateral loss of pain and temperature sensation below the level of the lesion. There may also be ipsilateral loss of pain and temperature sensation at the level of the lesion for one or two spinal segments if the lesion has damaged posterior horn cells before fibers have crossed to the other side.
Anterior cord syndrome results in loss of pain and temperature sensation below the level of the lesion (spinothalamic pathways), lower motor neuron signs at the level of the lesion (anterior horn cell damage), and upper motor neuron signs below the level of the lesion (lateral corticospinal tracts). In addition, urinary incontinence is common because of the ventral location of the descending pathways controlling sphincter function. Common causes of this syndrome include trauma and anterior spinal artery infarct.
Evidence for Early Closed Reduction of Bilateral Locked Facets
Treatment of this injury usually begins with closed reduction of the facet dislocation; whether or not one investigates the cervical spine with MRI prior to this maneuver is a controversial issue. An important qualifying factor in this situation is that the patient must be awake, alert, and able to participate in repeated neurological examinations. This notion rests in the dictum of “do no harm to your patients.” Early closed reduction of bilateral locked facets aims to relieve severe spinal cord compression in the setting of significant ligamentous damage by restoring normal bony alignment. The technique should be performed in a monitored setting and involves applying either Gardner-Wells tongs or a halo crown as a rigid fixation device to the skull and applying a traction force. Increasing weight is added over time while neurological status is monitored for stability and lateral radiographs are used to follow the bony anatomy. The goal is to obtain reduction of the cervical facets. A postreduction MRI should be obtained to rule out disk herniation. This maneuver should only be performed in specialty centers with surgeons experienced in its application. Under no circumstances should this maneuver delay operative management. The evidence for this technique was recently reviewed and consisted of a number of class II and III studies, with retrospective and prospective designs.44 The results of these studies varied from neurological deterioration to no neurological change to neurological improvement. In order to further distill the controversy, several authors reported that the method is safe and that neurological decline is often transient and improves with removal of added weight. Furthermore, a number of studies report neurological improvement that led to the recommendation of early closed reduction as a clinical guideline in patients with bilateral locked facets and an incomplete tetraplegia or in those with deteriorating neurological status.
Evidence for Methylprednisolone Therapy
The use of methylprednisolone as a neuroprotective agent to mitigate the deleterious effects of secondary injury is still controversial. It has been shown to offer modest benefit in terms of neurological outcome following SCI.45 The National Acute Spinal Cord Injury Study (NASCIS) aimed to investigate the question of whether or not patients would benefit from its administration. In the paragraphs that follow we will review the results of this study along with the method and dose by which physicians administered the drug in each trial.
NASCIS was designed as a multicenter trial and was completed as three separate trials: NASCIS I, NASCIS II, and NASCIS III. The first study compared low- and high-dose methylprednisolone (no placebo), the second trial was randomized and controlled with a placebo, and the third trial compared the effects of 24- versus 48-hour treatment. NASCIS I randomized SCI patients into two cohorts: the first group received a 100-mg loading dose of methylprednisolone followed by 25 mg every 6 hours for 10 days, and the second group received a 1000-mg loading dose of methylprednisolone followed by 250 mg every 6 hours for 10 days.46 There were no differences in neurological outcome between the two groups at 1 year after injury. NASCIS II investigated a 30-mg/kg bolus of methylprednisolone over 1 hour followed by 5.4 mg/kg/hour over the following 23 hours and included a placebo group and naloxone administration group. The reason for the higher doses (in comparison to NASCIS I) involved the analysis of animal research that suggested a therapeutic benefit only above a certain threshold. Results from the overall group showed no significant differences in neurological outcome at 6 months; a subgroup analysis demonstrated improved motor and sensory outcomes in patients receiving methylprednisolone within 8 hours of injury.47 NASCIS III examined outcomes in acute SCI patients receiving a 30-mg/kg bolus of methypredniosolone followed by 5.4 mg/kg/hour for either 23 hours or 47 hours. Patients treated with methylprednisolone for 48 hours had better neurological outcomes in comparison to the other treatment groups if therapy was initiated within 3 to 8 hours of injury; however, the 48-hour regimen was associated with increased risk of sepsis and pneumonia.48 Following the completion of each of these studies, Bracken conducted a reviewed along with two other independent trials and concluded that high-dose therapy was safe and afforded a modest benefit in terms of neurological outcome.49
Clearly the results were not overwhelmingly in favor of methylprednisolone administration and there is still considerable debate as to whether or not it should be used. Furthermore, if a practicing physician decides to use this therapy, there is debate as to which administration protocol should be used. The senior author of this chapter has published his protocol for administering methylprednisolone following acute SCI based on the timing of administration.45 Patients with acute nonpenetrating SCI should receive methylprednisolone as per the NASCIS II protocol if started less than 3 hours after injury. If started between 3 and 8 hours after injury, then the NASCIS III 48-hour protocol should be applied. If therapy cannot be administered within 8 hours of injury, or there is a penetrating SCI, then methylprednisolone should not be administered for neuroprotection. The decision to administer methylprednisolone must account for patient medical comorbid conditions. For example, the risks of administration may outweigh the benefits in a patient with diabetes mellitus and a complete thoracic SCI injury. Blood glucose levels must be carefully monitored during the course of methylprednisolone therapy, and hyperglycemia aggressively managed with an insulin infusion.
Evidence for Early Surgical Decompression
One of the parameters to consider when studying the effect of surgical decompression after SCI is the timing of this decompression. There is no definition of this timing although most authors and spinal surgeons would agree that early surgery is that which is performed within 24 hours of initial injury. A great deal of the preclinical animal literature focuses on timing of surgical decompression at much earlier times following injury, in the range of 8 to 24 hours. These animal studies consistently report improved neurological outcomes with early decompression. There have been a number of recent systematic literature reviews that address previous preclinical and clinical research.44,50,51 Perhaps the main force behind these reviews is not only to get a firm grasp on what has been accomplished to date but to form a clear rationale for proceeding with clinical trials in the future. Preliminary results from the Surgical Treatment of Acute Spinal Cord Injuries Trial (STASCIS) indicate that decompression within 24 hours of injury may actually improve outcome in patients with isolated SCI.52 Based on both animal studies and recent clinical investigations guidelines have been formed that recommend surgical decompression within 24 hours for cervical SCI. Very early decompression, within 12 hours of injury, should be strongly considered for patients suffering incomplete cervical SCI or those who are deteriorating neurologically.
Fehlings M.G., Baptiste D.C. Current status of clinical trials for acute spinal cord injury. Injury. 2005;36(Suppl 2):B113-122.
Fehlings M.G., Perrin R.G. The timing of surgical intervention in the treatment of spinal cord injury: a systematic review of recent clinical evidence. Spine. 2006;31:S28-35. discussion S36
Hawryluk G.W., Rowland J., Kwon B.K., Fehlings M.G. Protection and repair of the injured spinal cord: a review of completed, ongoing, and planned clinical trials for acute spinal cord injury. Neurosurg Focus. 2008;25:E14.
Miyanji F., Furlan J.C., Aarabi B., et al. Acute cervical traumatic spinal cord injury: MR imaging findings correlated with neurologic outcome—prospective study with 100 consecutive patients. Radiology. 2007;243:820-827.
Tator C.H., Koyanagi I. Vascular mechanisms in the pathophysiology of human spinal cord injury. J Neurosurg. 1997;86:483-492.
Please go to expertconsult.com to view the complete list of references.
1. Sekhon L.H., Fehlings M.G. Epidemiology, demographics, and pathophysiology of acute spinal cord injury. Spine. 2001;26:S2-12.
2. Ackery A., Tator C., Krassioukov A. A global perspective on spinal cord injury epidemiology. J Neurotrauma. 2004;21:1355-1370.
3. Kraus J.F., Franti C.E., Riggins R.S., et al. Incidence of traumatic spinal cord lesions. J Chron Dis. 1975;28:471-492.
4. Priebe M.M., Chiodo A.E., Scelza W.M., et al. Spinal cord injury medicine. 6. Economic and societal issues in spinal cord injury. Arch Phys Med Rehabil. 2007;88:S84-88.
5. Tator C.H., Fehlings M.G. Review of the secondary injury theory of acute spinal cord trauma with emphasis on vascular mechanisms. J Neurosurg. 1991;75:15-26.
6. Tator C.H., Koyanagi I. Vascular mechanisms in the pathophysiology of human spinal cord injury. J Neurosurg. 1997;86:483-492.
7. Kwon B.K., Tetzlaff W., Grauer J.N., et al. Pathophysiology and pharmacologic treatment of acute spinal cord injury. Spine. 2004;4:451-464.
8. Kakulas B.A. Neuropathology: the foundation for new treatments in spinal cord injury. Spinal Cord. 2004;42:549-563.
9. Schanne F.A., Kane A.B., Young E.E., Farber J.L. Calcium dependence of toxic cell death: a final common pathway. Science. 1979;206:700-702.
10. Lipton S.A., Rosenberg P.A. Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med. 1994;330:613-622.
11. Xiong Y., Rabchevsky A.G., Hall E.D. Role of peroxynitrite in secondary oxidative damage after spinal cord injury. J Neurochem. 2007;100:639-649.
12. Donnelly D.J., Popovich P.G. Inflammation and its role in neuroprotection, axonal regeneration and functional recovery after spinal cord injury. Exp Neurol. 2008;209:378-388.
13. Bao F., Liu D. Peroxynitrite generated in the rat spinal cord induces apoptotic cell death and activates caspase-3. Neuroscience. 2003;116:59-70.
14. Noble L.J., Wrathall J.R. Distribution and time course of protein extravasation in the rat spinal cord after contusive injury. Brain Res. 1989;482:57-66.
15. Schnell L., Fearn S., Schwab M.E., et al. Cytokine-induced acute inflammation in the brain and spinal cord. J Neuropathol Exp Neurol. 1999;58:245-254.
16. Pineau I., Lacroix S. Proinflammatory cytokine synthesis in the injured mouse spinal cord: multiphasic expression pattern and identification of the cell types involved. J Comp Neurol. 2007;500:267-285.
17. Fleming J.C., Norenberg M.D., Ramsay D.A., et al. The cellular inflammatory response in human spinal cords after injury. Brain. 2006;129:3249-3269.
18. Popovich P.G., Wei P., Stokes B.T. Cellular inflammatory response after spinal cord injury in Sprague-Dawley and Lewis rats. J Comp Neurol. 1997;377:443-464.
19. Yan P., Li Q., Kim G.M., et al. Cellular localization of tumor necrosis factor-alpha following acute spinal cord injury in adult rats. J Neurotrauma. 2001;18:563-568.
20. Bethea J.R., Nagashima H., Acosta M.C., et al. Systemically administered interleukin-10 reduces tumor necrosis factor-alpha production and significantly improves functional recovery following traumatic spinal cord injury in rats. J Neurotrauma. 1999;16:851-863.
21. Kim G.M., Xu J., Song S.K., et al. Tumor necrosis factor receptor deletion reduces nuclear factor-kappaB activation, cellular inhibitor of apoptosis protein 2 expression, and functional recovery after traumatic spinal cord injury. J Neurosci. 2001;21:6617-6625.
22. Keane R.W., Kraydieh S., Lotocki G., et al. Apoptotic and anti-apoptotic mechanisms following spinal cord injury. J Neuropathol Exp Neurol. 2001;60:422-429.
23. Crowe M.J., Bresnahan J.C., Shuman S.L., et al. Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat Med. 1997;3:73-76.
24. Yong C., Arnold P.M., Zoubine M.N., et al. Apoptosis in cellular compartments of rat spinal cord after severe contusion injury. J Neurotrauma. 1998;15:459-472.
25. Casha S., Yu W.R., Fehlings M.G. FAS deficiency reduces apoptosis, spares axons and improves function after spinal cord injury. Exp Neurol. 2005;196:390-400.
26. Chu G.K., Yu W., Fehlings M.G. The p75 neurotrophin receptor is essential for neuronal cell survival and improvement of functional recovery after spinal cord injury. Neuroscience. 2007;148:668-682.
27. Quencer R.M., Bunge R.P. The injured spinal cord: imaging, histopathologic clinical correlates, and basic science approaches to enhancing neural function after spinal cord injury. Spine. 1996;21:2064-2066.
28. Totoiu M.O., Nistor G.I., Lane T.E., Keirstead H.S. Remyelination, axonal sparing, and locomotor recovery following transplantation of glial-committed progenitor cells into the MHV model of multiple sclerosis. Exp Neurol. 2004;187:254-265.
29. Norenberg M.D., Smith J., Marcillo A. The pathology of human spinal cord injury: defining the problems. J Neurotrauma. 2004;21:429-440.
30. Hawryluk G.W., Rowland J., Kwon B.K., Fehlings M.G. Protection and repair of the injured spinal cord: a review of completed, ongoing, and planned clinical trials for acute spinal cord injury. Neurosurg Focus. 2008;25:E14.
31. Yong V.W., Wells J., Giuliani F., et al. The promise of minocycline in neurology. Lancet Neurol. 2004;3:744-751.
32. Wells J.E., Hurlbert R.J., Fehlings M.G., Yong V.W. Neuroprotection by minocycline facilitates significant recovery from spinal cord injury in mice. Brain. 2003;126:1628-1637.
33. Miller R.G., Mitchell J.D., Lyon M., Moore D.H. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev. 2007:CD001447.
34. Levi A.D., Green B.A., Wang M.Y., et al. Clinical application of modest hypothermia after spinal cord injury. J Neurotrauma. 2009;26:407-415.
35. David S., Aguayo A.J. Axonal elongation into peripheral nervous system “bridges” after central nervous system injury in adult rats. Science. 1981;214:931-933.
36. Caroni P., Schwab M.E. Two membrane protein fractions from rat central myelin with inhibitory properties for neurite growth and fibroblast spreading. J Cell Biol. 1988;106:1281-1288.
37. Fouad K., Dietz V., Schwab M.E. Improving axonal growth and functional recovery after experimental spinal cord injury by neutralizing myelin associated inhibitors. Brain Res Brain Res Rev. 2001;36:204-212.
38. Dergham P., Ellezam B., Essagian C., et al. Rho signaling pathway targeted to promote spinal cord repair. J Neurosci. 2002;22:6570-6577.
39. Sykova E., Homola A., Mazanec R., et al. Autologous bone marrow transplantation in patients with subacute and chronic spinal cord injury. Cell Transplant. 2006;15:675-687.
40. Keirstead H.S., Nistor G., Bernal G., et al. Human embryonic stem cell-derived oligodendrocyte progenitor cell transplants remyelinate and restore locomotion after spinal cord injury. J Neurosci. 2005;25:4694-4705.
41. Lebkowski J.S., Gold J., Xu C., et al. Human embryonic stem cells: culture, differentiation, and genetic modification for regenerative medicine applications. Cancer J. 2001;7(Suppl 2):S83-93.
42. American Spinal Injury Association (ASIA) standard forms are available without cost at. http://www.asia-spinalinjury.org/publications/2006_Classif_worksheet.pdf.
43. Miyanji F., Furlan J.C., Aarabi B., et al. Acute cervical traumatic spinal cord injury: MR imaging findings correlated with neurologic outcome—prospective study with 100 consecutive patients. Radiology. 2007;243:820-827.
44. Fehlings M.G., Perrin R.G. The timing of surgical intervention in the treatment of spinal cord injury: a systematic review of recent clinical evidence. Spine. 2006;31:S28-35.
45. Fehlings M.G., Baptiste D.C. Current status of clinical trials for acute spinal cord injury. Injury. 2005;36(Suppl 2):B113-122.
46. Bracken M.B., Shepard M.J., Hellenbrand K.G., et al. Methylprednisolone and neurological function 1 year after spinal cord injury. Results of the National Acute Spinal Cord Injury Study. J Neurosurg. 1985;63:704-713.
47. Bracken M.B., Shepard M.J., Collins W.F., et al. A randomized, controlled trial of methylprednisolone or naloxone in the treatment of acute spinal-cord injury. Results of the Second National Acute Spinal Cord Injury Study. N Engl J Med. 1990;322:1405-1411.
48. Bracken M.B., Shepard M.J., Holford T.R., et al. Administration of methylprednisolone for 24 or 48 hours or tirilazad mesylate for 48 hours in the treatment of acute spinal cord injury. Results of the Third National Acute Spinal Cord Injury Randomized Controlled Trial. National Acute Spinal Cord Injury Study. JAMA. 1997;277:1597-1604.
49. Bracken M.B. Methylprednisolone and acute spinal cord injury: an update of the randomized evidence. Spine. 2001;26:S47-54.
50. Fehlings M.G., Tator C.H. An evidence-based review of decompressive surgery in acute spinal cord injury: rationale, indications, and timing based on experimental and clinical studies. J Neurosurg. 1999;91:1-11.
51. Gunnarsson T., Fehlings M.G. Acute neurosurgical management of traumatic brain injury and spinal cord injury. Curr Opin Neurol. 2003;16:717-723.
52. Fehlings M.G., Arvin B. The timing of surgery in patients with central spinal cord injury. J Neurosurg Spine. 2009;10:1-2.