Spina bifida: A congenital spinal cord injury
KRISTIN J. KROSSCHELL, PT, MA, PCS and MARI JO PESAVENTO, PT, PCS
After reading this chapter the student or therapist will be able to:
1. Identify the various types of spina bifida.
2. Recognize the incidence and etiology of spina bifida.
3. Identify the clinical manifestations of myelomeningocele, including neurological, orthopedic, and urological sequelae.
4. Comprehend medical management in the newborn period and beyond.
5. Determine physical and occupational therapy evaluations, including manual muscle testing, range of motion, sensory testing, reflex testing, developmental and functional and mobility assessments, and perceptual and cognitive evaluations.
6. List the major physical and occupational therapy goals and appropriate therapeutic management for each of the following stages: (a) before surgical closure of sac, (b) after surgery during hospitalization, (c) preambulatory, (d) toddler through preschool age, (e) primary school age through adolescence, and (f) transition to adulthood.
7. Identify psychological adjustment to congenital spinal cord injury.
A developmental framework, the Guide to Physical Therapist Practice,1 and the International Classification of Functioning, Disability and Health (ICF) have been used to aid in understanding the sequential problems of the child with spina bifida. The developmental model, however, must always stay in line with the functional model for adult trauma because the problems of the congenitally involved child grow quickly into limitations in functional activities and participation in life of the injured adult. With concentration on the present but with an eye to the future, appropriate management goals can be achieved.
Overview of congenital spinal cord injury
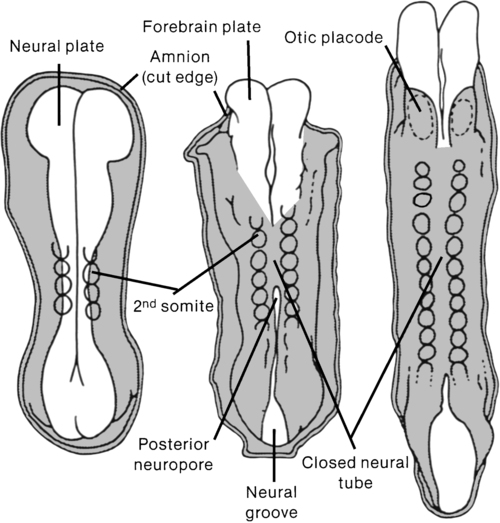
A congenital spinal cord lesion occurs in utero and is present at the time of birth. Understanding how this malformation develops requires an appreciation of normal nervous system maturation. The nervous system develops from a portion of embryonic ectoderm called the neural plate. During gestation, the neural plate develops folds that begin to close, forming the neural tube (Figure 15-1). The neural tube differentiates into the CNS, which is composed of brain and spinal cord tissue. In the normal embryo, neural tube closure begins in the cervical region and proceeds cranially and caudally. Closure is generally complete by the twenty-sixth day.

Types of spina bifida
Spina bifida involves a defect in the neural tube closure and the overlying posterior vertebral arches. The extent of the defect may result in one of two types of spina bifida: occulta or cystica. Spina bifida occulta is characterized by a failure of one or more of the vertebral arches to meet and fuse in the third month of development. The spinal cord and meninges are unharmed and remain within the vertebral canal (Figure 15-2, A). The bony defect is covered with skin that may be marked by a dimple, pigmentation, or patch of hair.2 The common site for this defect is the lumbosacral area, and it is usually associated with no disturbance of neurological or musculoskeletal functioning. Spina bifida cystica results when the neural and overlying vertebral arches fail to close appropriately. Cystic protrusion of the meninges or the spinal cord and meninges is present through the defective vertebral arches.
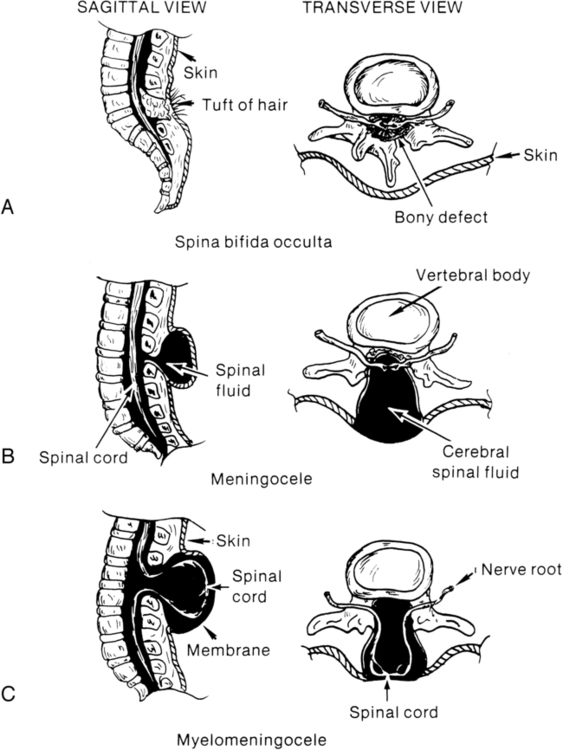

The milder form of spina bifida cystica, called meningocele, involves protrusion of the meninges and cerebrospinal fluid (CSF) only into the cystic sac (see Figure 15-2, B). The spinal cord remains within the vertebral canal, but it may exhibit abnormalities.3 Clinical signs vary (according to spinal cord anomalies) or may not be apparent. This is a relatively uncommon form of spina bifida cystica.
A more severe form of spina bifida cystica, called myelocele or myelocystocele, is present when the central canal of the spinal cord is dilated, producing a large, skin-covered cyst. The neural tube appears to close normally but is distended from the cystic swelling. The CSF may ceaselessly expand the neural canal. Prompt medical attention is mandatory. This form of spina bifida is also rare.4
The more common and severe form of the defect is known as myelomeningocele, in which both spinal cord and meninges are contained in the cystic sac (see Figure 15-2, C). Within the sac the spinal cord and associated neural tissue show extensive abnormalities. In incomplete closure of the neural tube (dysraphism), abnormal growth of the cord and a tortuous pathway of neural elements make normal transmission of nervous impulses abnormal. The result is a variable sensory and motor impairment at the level of the lesion and below.2 In an open myelomeningocele, nerve roots and spinal cord may be exposed, with dura and skin evident at the margin of the lesion. Exposure of the open neural tube to the amniotic fluid environment leads to neuroepithelial degeneration, with massive loss of neural tissue by the end of pregnancy.5
Although spina bifida cystica can occur at any level of the spinal cord, myelomeningoceles are most common in the thoracic and lumbosacral regions. Myelomeningocele occurs in 94% of the cases of spina bifida cystica, and two thirds of open lesions involve the thoracolumbar junction.2 The terms spina bifida, myelodysplasia, and myelomeningocele are frequently used interchangeably.
Other forms of spinal dysraphism include diastematomyelia, lipomeningocele, and sacral agenesis. Diastematomyelia is present in 30% to 40% of patients with myelomeningocele and is secondary to partial or complete clefting of the spinal cord.6 Lipomeningocele, another form of spina bifida cystica, is usually caused by a vertebral defect associated with a superficial fatty mass (lipoma or fatty tumor) that merges with the lower level of the spinal cord. No associated hydrocephalus is present, and neurological deficit is generally minimal; however, problems with urinary control and motor control of the lower extremities may be noted.7 Neurological tissue invasion may be caused by a tethered spinal cord; therefore early lipoma resection is indicated for cosmesis and to minimize neurological sequelae. Lumbosacral or sacral agenesis may occur and is caused by an absence of the caudal part of the spine and sacrum. Children with this form of dysraphism may have narrow, flattened buttocks, weak gluteal muscles, and a shortened intergluteal cleft. The normal lumbar lordosis is absent, although the lower lumbar spine may be prominent. Calf muscles may be atrophic or absent. The pelvic ring is completed with either direct opposition of the iliac bones or with interposition of the lumbar spine replacing the absent sacrum. These children may have scoliosis, motor and sensory loss, and visceral abnormalities including anal atresia, fused kidneys, and congenital heart malformations. Management is started early and is symptomatic for each system.8
Failure of fusion of the cranial end of the neural tube results in a condition known as anencephaly. In this condition some brain tissue may be evident, but forebrain development is usually absent.9 Sustained life is not possible with this neural tube defect; therefore this condition is not discussed further.
Incidence, etiology, and economic impact
Statistics about the incidence of spina bifida vary considerably in different parts of the world. Spina bifida and anencephaly, the most common forms of neural tube defects, affect about 300,000 newborns each year worldwide.10 In the United States the incidence is currently 2.48 per 10,000, down from approximately 7.23 per 10,000 births from 1974 through 1979 (before the folic acid mandate).11,12 Current worldwide folic acid fortification programs have resulted in decreased incidence of spina bifida,13,14 with annual decreases of 6600 folic acid–preventable spina bifida and anencephaly births reported since 2006.15 There was a 31% decline in spina bifida prevalence rates in the immediate postfortification period (October 1998 through December 1999).13 There was a continued decline in spina bifida prevalence rates from 1999 to 2004 of 10%.16 Studies have also demonstrated that decline varied by ethnicity and race from prefortification to optional fortification to mandatory fortification in the United States.16,17 Initially after fortification, the largest decline in prevalence was noted in Hispanic and non-Hispanic white races or ethnicities. Despite this initial decline, postfortification prevalence rates remain highest in infants born to Hispanic mothers, and less in infants born to non-Hispanic white and non-Hispanic black mothers.16 In addition to periconceptual folate supplementation, it is thought that incidence has decreased subsequent to food fortification in several countries, decreased exposure to environmental teratogens, and increased and more accurate prenatal screening for fetal anomalies.10
Spina bifida is thought to be more common in females than in males, although some studies suggest no real sex difference.3 A study of the association of race and sex with different neurological levels of myelomeningocele found the proportions of whites and females to be significantly higher in patients with thoracic-level spina bifida.4 A significant relation also has been noted between social class and spina bifida: the lower the social class, the higher the incidence.18,19
A multifactorial genetic inheritance has been proposed as the cause of spina bifida, coupled with environmental factors, of which nutrition, including folic acid intake, are key. Cytoplasmic factors, polygenic or oligogenic inheritance, chromosomal aberrations, and environmental influences (e.g., teratogens) have all been considered as possible causes.5,15 Genetic factors seem to influence the occurrence of spina bifida. The chances of having a second affected child are between 1% and 2%, whereas in the general population the percentage drops to one fifth of 1%.20,21 Although these factors are related to the incidence of spina bifida, the cause of this defect remains in question. Environmental conditions, such as hyperthermia in the first weeks of pregnancy, or dietary factors, such as eating canned meats or potatoes or drinking tea, have been implicated but not substantiated.22,23 In addition, historically, nutritional deficiencies, such as of folic acid and vitamin A, have been implicated as a cause of primary neural tube defects.24–27 Approximately 50% to 70% of neural tube defects can be prevented if a woman of childbearing age consumes sufficient folic acid daily before conception and throughout the first trimester of pregnancy. As a result of research findings in support of folic acid implementation, the U.S. Public Health Service has mandated folic acid fortification since 1998 as a public health strategy. Prenatal vitamins, especially folic acid, are recommended to discourage the condition’s development. Current fortification programs are preventing about 22,000 cases, or 9% of the estimated folic acid–preventable spina bifida and anencephaly cases.15 Genetic considerations, such as an Rh blood type, a specific gene type (HLA-B27), an X-linked gene, and variations in the many folate pathway genes have been implicated, but not conclusively.28,29 Malformations are attributed to abnormal interaction of several regulating and modifying genes in early fetal development.30 Disturbance of any of the sequential events of embryonic neurulation produces neural tube defects (NTDs), with the phenotype (i.e., spina bifida, anencephaly) varying depending on the region of the neural tube that remains exposed.5 Environmental factors combined with genetic predisposition appear to trigger the development of spina bifida, although definitive evidence is not available to support this claim.31
The incidence of spina bifida has declined since the advent of amniocentesis and the use of ultrasonography for prenatal screening. The presence of significant levels of alpha fetoprotein in the amniotic fluid has led to the detection of large numbers of affected fetuses.32 Currently, maternal serum alpha-fetoprotein levels have been effective in detecting approximately 80% of neural tube defects.33 Prenatal screening can be most effective when a combination of serum levels, amniocentesis or amniography, and ultrasonography is used.34–36 Although this screening is not yet performed routinely, it is suggested for those at risk for the defect. Knowledge of the defect allows for preparation for cesarean birth and immediate postnatal care. This includes mobilization of the interdisciplinary team that will continue to care for the child. For parents who decide to carry an involved fetus to term, adjustment to their child’s disability can begin before birth, which includes mobilizing their own support system. Education from an integrated team regarding what will follow after delivery and neurosurgical closure is imperative to aid families in decision making and to allow families to assess and understand the child’s disability and future care options.
Other advances in the field of prenatal medicine that affect spina bifida management and outcome include in utero treatment of hydrocephalus and in utero surgical repair to close the myelomeningocele. This challenging surgical procedure is practiced in only a few specialty centers and so far has been shown to offer palliation of the defect at best.37 Treatment such as this, in conjunction with prenatal diagnosis, has been shown to have a positive impact on the incidence and severity of complications associated with spina bifida.38–45 Limitations of current postnatal treatment strategies and considerations of prenatal treatment options continue to be explored. Ethics, timing of repair, and surgical procedures are all being investigated. In addition, continued assessment of outcomes from those who have undergone presurgical management requires continued exploration. The Management of Myelomeningocele Study (MOMS) was initiated in 2003 as a large randomized, clinical trial designed to compare the two approaches to the treatment of infants with spina bifida (prenatal or fetal surgery versus postnatal surgery) to determine if one approach was better than the other. The primary end point of this trial was the need for a shunt at one year, and secondary end points included neurologic function, cognitive outcome, and maternal morbidity after prenatal repair. This study had 112 patients enrolled in 2007 with a projected enrollment of 200.46–49 The trial was stopped for efficacy of prenatal surgery after enrollment of just 183 infants. Results demonstrated that prenatal surgery significantly reduced the need for shunting and improved mental and motor function at 30 months. Reduced incidence of hindbrain herniation at 12 months and successful ambulation by 30 months were also reported. While prenatal surgery was associated with improved function and reduced need for shunting, maternal and fetal risks, including preterm delivery and uterine dehiscense at delivery were reported.49a
In 1996 the lifetime cost to society per affected person with spina bifida was estimated to be $635,000.50,51 More recent estimates have not been reported; however, with an economy in flux it is likely that this value underassesses costs to society today. In addition to medical management costs per child, there are additional costs that affect both the family and society across the life span that are variable and often related to differential market forces and social welfare policies.50
In 2007, Ouyang52 reported that average medical expenditures during the first year of life for those with spina bifida during 2002 and 2003 averaged $50,000 (using MarketScan 2003 database). The majority of expenditures during infancy were from inpatient admissions secondary to surgeries being concentrated during this time period for those with spina bifida. After infancy, average medical care expenditures during 2003 ranged from $15,000 to $16,000 per year among different age groups of persons with spina bifida. Incremental expenditures associated with medical care were not stable, but decreased with increasing age, from $14,000 per year for children to $10,000 per year for adults 45 to 64 years of age.52
Clinical manifestations
The most obvious clinical manifestation of myelomeningocele is the loss of sensory and motor functions in the lower limbs. The extent of loss, while primarily dependent on the degree of the spinal cord abnormality, is secondarily dependent on a number of factors. These include the amount of traction or stretch resulting from the abnormally tethered spinal cord, the trauma to exposed neural tissue during delivery, and postnatal damage resulting from drying or infection of the neural plate.2 Specific clinical impairments that commonly lead to functional limitations for the child with spina bifida are addressed in this section.
Musculoskeletal impairment
Orthopedic deformities.
The orthopedic problems that occur with myelomeningocele may be the result of (1) the imbalance between muscle groups; (2) the effects of stress, posture, and gravity; and (3) associated congenital malformations. Decreased sensation and neurological complications also may lead to orthopedic abnormalities.53
Besides the obvious malformation of vertebrae at the site of the lesion, hemivertebrae and deformities of other vertebral bodies and their corresponding ribs also may be present.53,54 Lumbar kyphosis may be present as a result of the original deformity. In addition, as a result of the bifid vertebral bodies, the misaligned pull of the extensor muscles surrounding the deformity, as well as the unopposed flexor muscles, contributes further to the lumbar kyphosis. As the child grows, the weight of the trunk in the upright position also may be a contributing factor.54 Scoliosis may be present at birth because of vertebral abnormalities or may become evident as the child grows older. The incidence of scoliosis is lower in low lumbar or sacral level deformities.54,55 Scoliosis may also be neurogenic, secondary to weakness or asymmetrical spasticity of paraspinal muscles, tethered cord syndrome (TCS), or hydromyelia.55 Lordosis or lordoscoliosis is often found in the adolescent and is usually associated with hip flexion deformities and a large spinal defect.3,54 Many of these trunk and postural deformities exist at birth but are exacerbated by the effects of gravity as the child grows. They can compromise vital functions (cardiac and respiratory) and therefore should be closely monitored by the therapist and the family.
As has been alluded to previously, the type and extent of deformity in the lower extremities depend on the muscles that are active or inactive. In total flaccid paralysis, in utero deformities may be present at birth, resulting from passive positioning within the womb. Equinovarus (clubfoot) and “rocker-bottom” deformity are two of the most common foot abnormalities. Knee flexion and extension contractures also may be present at birth. Other common deformities are hip flexion, adduction, and internal rotation, usually leading to a subluxed or dislocated hip. Although many of these problems may be present at birth, preventing positional deformity (such as the frog-leg position), which may result from improper positioning of flaccid extremities, is of the utmost importance. Orthopedic care varies throughout the course of the child’s life. Changes in clinical orthopedic management have evolved to establish evidence-based interventions.56
Osteoporosis.
Because the paralyzed limbs of the child with spina bifida have increased amounts of unmineralized osteoid tissue, they are prone to fractures, particularly after periods of immobilization.57,58 Early mobilization and weight bearing can aid in decreasing osteoporosis.54,59 Fortunately, these fractures heal quickly with appropriate medical management.
Neurological impairment
Hydrocephalus.
Hydrocephalus develops in 80% to 90% of children with myelomeningocele.21,60 Hydrocephalus results from a blockage of the normal flow of CSF between the ventricles and spinal canal. The most obvious effect of the buildup of CSF is abnormal increase in head size, which may be present at birth because of the great compliance of the cranial sutures in the fetus, or it may develop postnatally.61 Other signs of hydrocephalus include bulging fontanels and irritability. Internally, a concomitant dilation of the lateral ventricles and thinning of the cerebral white matter are usually present. Without reduction of the buildup of CSF, increased brain damage and death may result.
Chiari malformation.
Patients with myelomeningocele have a 99% chance of having an associated Chiari II malformation.6 Cardinal features of the Chiari II malformation include myelomeningocele in the thoracolumbar spine, venting of the intracranial CSF through the central canal, hypoplasia of the posterior fossa, herniation of the hindbrain into the cervical spinal canal, and compressive damage to cranial nerves. This malformation is a congenital anomaly of the hindbrain that involves herniation of the medulla and at times the pons, fourth ventricle, and inferior aspect of the cerebellum into the upper cervical canal. The herniation usually occurs between C1 and C4 but may extend down to T1.6,62,63 In those with Chiari II malformations and spina bifida there is a significant reduction in cerebellar volume, and within the cerebellum the anterior lobe is enlarged and the posterior lobe is reduced.64 Not all Chiari II malformations are symptomatic. As a result of a symptomatic Chiari malformation, problems with respiratory and bulbar function may be evident in the child with spina bifida.2 Paralysis of the vocal cords occurs in a small percentage of patients and is associated with respiratory stridor. Apneic episodes also may be evident, although their direct cause remains in question. Children with spina bifida also may exhibit difficulty in swallowing and have an abnormal gag reflex.2 Problems with aspiration, weakness and cry, and upper-extremity weakness also may be present in children with a symptomatic Chiari II malformation.65,66 Thus, depending on the orthopedic deformities present and the neurological involvement, severe respiratory involvement is possible in the affected child. These symptoms may be caused by significant compression of the hindbrain structures or dysplasia of posterior fossa contents, which can also occur in patients with Chiari II malformation.6,67 This complex hindbrain malformation is a common cause of death in children with myelomeningocele despite surgical intervention and aggressive medical management.68
Association pathways.
Diffusion tensor tractography studies of association pathways in children with spina bifida have revealed characteristics of abnormal development, impairment in myelination, and abnormalities in intrinsic axonal characteristics and extraaxonal or extracellular space. These changes in diffusion metrics observed in children with spina bifida are suggestive of abnormal white matter development and persistent degeneration with increased age.69
Hydromyelia.
Twenty percent to 80% of patients with myelomeningocele have hydromyelia.6,70,71 Hydromyelia signifies dilation of the center canal of the spinal cord as hydrocephalus signifies dilation of the ventricles of the brain. The area of hydromyelia may be focal, multiple, or diffuse, extending throughout the spinal cord. The hydromyelia may be a consequence of untreated or inadequately treated hydrocephalus with resultant transmission of CSF through the obex into the central canal, with distention a result of increased hydrostatic pressure from above.6 The increased collection of fluid may cause pressure necrosis of the spinal cord, leading to muscle weakness and scoliosis. Common symptoms of hydromyelia include rapidly progressive scoliosis, upper-extremity weakness, spasticity, and ascending motor loss in the lower extremities.6,72 Aggressive treatment of hydromyelia at the onset of clinical signs of increasing scoliosis is mandatory and may lead to improvement in or stabilization of the curve in 80% of cases. Surgical interventions may include revision of a CSF shunt, posterior cervical decompression, or a central canal to pleural cavity shunt with a flushing device.6,67
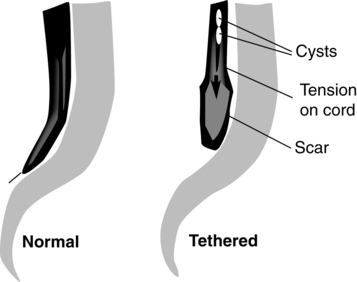
Tethered cord.
Tethered spinal cord is defined as a pathological fixation of the spinal cord in an abnormal caudal location (Figure 15-3). This fixation produces mechanical stretch, distortion, and ischemia with daily activities, growth, and development.73 Ischemic injury from traction of the conus directly correlates with degree of oxidative metabolism and degree of neurologic compromise. In addition to ischemic injury, traction of the conus by the filum may also mechanically alter the neuronal membranes, resulting in altered electrical activity.74–78 The presence of tethered cord syndrome (TCS) should be suspected in any patient with abnormal neurulation (including patients with myelomeningocele, lipomeningocele, dermal sinus, diastematomyelia, myelocystocele, tight filum terminale, and lumbosacral agenesis). Presenting symptoms may include decreased strength (often asymmetrical), development of lower-extremity spasticity, back pain at the site of sac closure, early development of or increasing degree of scoliosis (especially in the low lumbar or sacral level),79,80 or change in urological function.68,81–83 Approximately 10% to 30% of children will develop TCS after repair of a myelomeningocele. Because essentially all children with repaired myelomeningocele will have a tethered spinal cord, as demonstrated on magnetic resonance imaging (MRI), the diagnosis of TCS is made based on clinical criteria. The six common clinical presentations of TCS are increased weakness (55%), worsening gait (54%), scoliosis (51%), pain (32%), orthopedic deformity (11%), and urological dysfunction (6%).84 This clinical spectrum may be primarily associated with these dysraphic lesions or may be caused by spinal surgical procedures.73 The cord may be tethered by scar tissue or by an inclusion epidermoid or lipoma at the repair site.6 The primary goal of surgery is to detach the spinal cord where it is adherent to the thecal sac, relieving the stretch on the terminal portion of the cord. Surgery to untether the spinal cord (tethered cord release [TCR]) is performed to prevent further loss of muscle function, decrease the spasticity, help control the scoliosis,80,85 or relieve back pain.86,87

The effectiveness of a TCR may be demonstrated by an increase in muscle function, relief of back pain, and stabilization or reversal of scoliosis.80,85,87 It has been reported that scoliosis response to untethering and progression of scoliosis after untethering vary with location of tethering80,87 as well as Risser grade88 and Cobb angle.89 Those with Risser grade 3 to 5 and Cobb angle less than 40 degrees are less likely to experience curve progression after untethering. Those with Risser grades 0 to 2 and Cobb angle greater than 40 degrees are at higher risk of recurrence.74,89 Spasticity, however, is not always alleviated in all patients.90 Selective posterior rhizotomy has been advocated for patients whose persistent or progressive spastic status after tethered cord repair continues to interfere with their mobility and functional independence.68,70
Bowel and bladder dysfunction.
Because of the usual involvement of the sacral plexus, the child with spina bifida commonly deals with some form of bowel and bladder dysfunction. Besides various forms of incontinence, incomplete emptying of the bladder remains a constant concern because infection of the urinary tract and possible kidney damage may result.91 Regulation of bowel evacuation must be established so that neither constipation nor diarrhea occurs. Negative social aspects of incontinence can be minimized by instituting intervention that emphasizes patient and family education and a regular, consistently timed, reflex-triggered bowel evacuation.92
Cognitive impairment and learning issues.
The last major clinical manifestation resulting from the neurological involvement of myelomeningocele is impaired intellectual function. Although children with spina bifida without hydrocephalus may have normal intellectual potential, children with hydrocephalus, particularly those who have shunt infections, are likely to have below-average intelligence.93–95 These children often demonstrate learning disabilities and poor academic achievement.96 Even those with a normal IQ show moderate to severe visual-motor perceptual deficits.97 The inability to coordinate eye and hand movements affects learning and may interfere with activities of daily living (ADLs), such as buttoning a shirt or opening a lunchbox.98 Difficulties with spatial relations, body image, and development of hand dominance may also be evident.2,98 Children with myelomeningocele demonstrate poorer hand function than age-matched peers. This decreased hand function appears to be caused by cerebellar and cervical cord abnormalities rather than hydrocephalus or a cortical pathological condition (see Chapter 21).99
Prenatal studies have shown that the CNS as a whole is abnormally developed in fetuses with myelomeningocele.100–103 The impairment of intellectual and perceptual abilities has been linked to damage to the white matter caused by ventricular enlargement.2 This damage to association tracts, particularly in the frontal, occipital, and parietal areas, could account for the often severe perceptual-cognitive deficits noted in the child with spina bifida.69,104 Lesser involvement of the temporal areas may account for the preservation of speech, whereas the semantics of speech, which depends on association areas, is impaired. The “cocktail party speech” of children with spina bifida can be deceptive because they generally use well-constructed sentences and precocious vocabulary. A closer look, however, reveals a repetitive, inappropriate, and often meaningless use of language not associated with higher intellectual functioning. Research on learning difficulties in children with spina bifida and hydrocephalus suggests that many of these children experience difficulties. Tasks and skills affected include memory, reasoning, math, handwriting, organization, problem solving, attention, sensory integration, auditory processing, visual perception, and sequencing.101–103
Integumentary impairment
Latex allergy and sensitivity have been noted with increasing frequency in children with myelomeningocele, with frequent reports of intraoperative anaphylaxis.105–109 These children have also been reported to have a higher than expected prevalence of atopic disease.110 A 1991 Food and Drug Administration Medical Bulletin estimated that 18% to 40% of patients with spina bifida demonstrate latex sensitivity,105,111 with others reporting an incidence of 20% to 67%.112,113 Within latex is 2% to 3% of a residual-free protein material that is thought to be the antigenic agent.107 Frequent exposure to this material results in the development of the immunoglobulin E antibody. Children with spina bifida are more likely to develop the immunoglobulin E sensitivity because of repeated parental or mucosal exposure to the latex antigen.114 Because of the risk of an anaphylactic reaction, exposure to any latex-containing products such as rubber gloves, therapy balls, pacifiers, spandex, dental dams, elastic or rubber bands, balloons, adhesive bandages, or exercise bands should be avoided. Latex-free gloves, therapy balls, treatment mats, and exercise bands are now widely available and should be considered for standard use in all clinics treating children with spina bifida. Spina bifida, even in the absence of multiple surgical interventions, may be an independent risk factor for latex sensitivity. Foods reported to be highly associated with latex allergy include avocado, banana, chestnut, and kiwi.115 Latex-free precautions from birth are more effective in preventing latex sensitization than are similar precautions instituted later in life.115–117 Latex sensitization decreased from 26.7% to 4.5% in children treated in a latex-free environment from birth.117
The presence of paralysis and lack of sensation on the skin places the child with spina bifida at major risk for pressure sores and decreased skin integrity. Various types of skin breakdown have occurred in 85% to 95% of all children with spina bifida by the time they reach young adulthood.118 Common areas at risk for pressure sores include the lower back, kyphotic or scoliotic prominences, heels, feet, toes, and perineum. A pressure sore may result from excessive skin pressure that can cause reduced capillary flow, tissue anoxia, and eventual skin necrosis. Excessive pressure may manifest itself early as reactive hyperemia, a blister, and later as an open sore or overt necrosis. Chronic, untreated sores may lead to osteomyelitis and eventual sepsis.110 Pressure sores often result in loss of time from school and work and can lead to financial hardship from medical treatment and hospitalizations. These negative consequences can largely be prevented with attention to education and instruction of the child and family. The goal of such education is to foster an understanding of the causes of skin breakdown and the necessary meticulous attention to skin care that must be carried out on a regular basis.
Growth and nutrition
Nutritional intake and weight gain and loss have been found to be problematic in children with myelomeningocele. Early on, infants with spina bifida may have feeding issues as a result of an impaired gag reflex, swallowing difficulties, and a high incidence of aspiration.2,66 Altered oral-motor function has been attributed to the Chiari II malformation.119 These impairments may lead to nutritional issues and delayed growth and weight gain. Speech, physical, and occupational therapists as a team are often needed to address these issues.
Conversely, obesity can be a significant issue for children with spina bifida. This problem is complex and multifactorial.120 Mobility limitations and decreased energy expenditure result in lower physical activity levels. In addition, decreased lower limb mass diminishes the ability to burn calories, which leads to weight gain. Decreased caloric intake as well as a lifelong engagement in rewarding and physically challenging physical activities are both necessary to enhance weight control and control obesity.
Children with myelomeningocele are short in stature. Growth in these children may be influenced by growth-retarding factors as a result of a neurological deficit such as tethered cord.121 Endocrine disorders and growth hormone deficiency have also been found to contribute to short stature in this population.122 As a result of complex CNS anomalies (midline defects, hydrocephalus, Arnold-Chiari malformation), these children are at risk for hypothalamopituitary dysfunction leading to growth hormone deficiency.123,124 Treatment with recombinant human growth hormone has proven successful in fostering growth acceleration in these children.123,125,126
Medical management
Neurosurgical management
Since the early 1960s the presence of a myelomeningocele has been treated as a life-threatening situation, and sac closure most often takes place within the first 24 to 48 hours of life.2,127 Recent advances in treatment have led to investigational treatment in utero to repair the defect before birth.38 The aim of either surgery is to replace the nervous tissue into the vertebral canal, cover the spinal defect, and achieve a watertight sac closure.128 This early management has decreased the possibility of infection and further injury to the exposed neural cord.24,128,129
Progressive hydrocephalus may be evident at birth in a small percentage of children born with myelomeningocele. A greater majority, however, have hydrocephalus 5 to 10 days after the back lesion has been closed.128,130–132 With the advent of computed tomography (CT), early diagnosis of hydrocephalus can be made in the newborn without the need for clinical examination.
Although clinical signs are not always definitive, hydrocephalus may be suspected if (1) the fontanels become full, bulging, or tense; (2) the head circumference increases rapidly; (3) a separation of the coronal and sagittal sutures is palpable; (4) the infant’s eyes appear to look downward only, with the cornea prominent over the iris (“sunsetting sign”); and (5) the infant becomes irritable or lethargic and has a high-pitched cry, persistent vomiting, difficult feeding, or seizures (Table 15-1).21,61,133
TABLE 15-1 
SIGNS AND SYMPTOMS OF SHUNT MALFUNCTION
| Infants | Bulging fontanelSwelling along the shunt tract Prominent veins on scalp Downward eye deviation (“sunsetting”) Vomiting or change in appetite Irritability or drowsiness Seizures High-pitched cry |
| Toddler | HeadacheVomiting or change in appetite Lethargy or irritability Swelling along the shunt tract Seizures Onset of or increased strabismus |
| Older child | All the above, plus:Deterioration in school performance Neck pain or pain over myelomeningocele site Personality change Decrease in sensory or motor functions Incontinence that begins or worsens Onset of or increased spasticity |
If the results of CT confirm hydrocephalus, a ventricular shunt is indicated. This procedure involves diverting the excess CSF from the ventricles to some site for absorption. In general, two types of procedures—the ventriculoatrial (VA) and ventriculoperitoneal (VP) shunt—are currently used, the latter being the most common (Figure 15-4). The shunt apparatus is constructed from Silastic tubing and consists of three parts: a proximal catheter, a distal catheter, and a one-way valve. As CSF is pumped from the ventricles toward its final destination, backflow is prevented by the valve system. In this manner intracranial pressure is controlled, CSF is regulated, and hydrocephalus is prevented from causing damage to brain structures. An alternate means of controlling hydrocephalus may be the use of endoscopic third ventriculostomy (EVT). EVT is a procedure that, in selected patients with obstructive hydrocephalus, allows egress of CSF from the ventricles to the subarachnoid space. This can decompress the ventricles and allow normal intracranial pressures and brain growth. This procedure is typically reserved for last resort.134
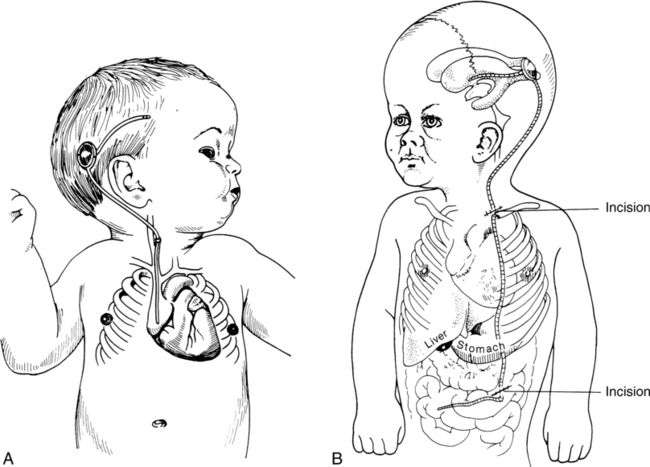

Unfortunately for children with spina bifida, their problems do not end after the back is surgically closed and a shunt is in place. Management strategies in the care of shunted hydrocephalus vary.135 Shunt complications occur frequently and require an average of two revisions before age 10 years.60 The most common causes of complications are shunt obstruction and infection.2,136 Revising the blocked end of the shunt can clear obstructions. Infections may be handled by external ventricular drainage and courses of antibiotic therapy followed by insertion of a new shunting system.2 The problem of separation of shunt components has been largely overcome by the use of a one-piece shunting system. The single-piece shunt decreases the complications of shunting procedures.
Prophylactic antibiotic therapy 6 to 12 hours before surgery and 1 to 2 days postoperatively is effective in controlling infection for both sac repair and shunt insertion.71 This brief course of antibiotics has not led to resistant organisms. The main cause of death in children with myelomeningocele remains increased intracranial pressure and infections of the CNS. With the use of antibiotics, shunting, and early sac closure, the survival rate has increased from 20% to 85%.61,94,137
Urological management
Initial newborn workup should include a urological assessment. The urology team aims to preserve renal function and promote efficient bladder management. An early start to therapy helps to preserve renal function for children with spina bifida.138 Initially, a renal and bladder ultrasound is performed to assess those structures.100 Urodynamic testing can be performed to determine any blockage in the lower urinary tract. Functioning of the bladder outlet and sphincters, as well as ureteric reflux, also can be evaluated. These tests, plus clinical observations of voiding patterns, help the urologist classify the infant’s bladder function. If the bladder has neither sensory nor motor supply, a constant flow of urine is present. In this case infection is rare because the bladder does not store urine and the sphincters are always open.139
If no sensation but some involuntary muscle control of the sphincter exists, the bladder will fill, but emptying will not occur properly. Overflow or stress incontinence results in dribbling urine until the pressure is relieved. Because of constant residual urine, infection is a potential problem and kidney damage may result.139 When some voluntary muscle control but no sensation is present, the bladder will fill and empty automatically. The child can eventually be taught to empty the bladder at regular intervals to avoid unnecessary accidents.
A program of clean intermittent catheterization (CIC) done every 3 to 4 hours prevents infection and maintains the urological system.140–143 Parents are taught this method and can then begin to take on this aspect of their child’s care. At the age of 4 or 5 years, children with spina bifida can be taught CIC; thus they become independent in bladder care at a young age. Achieving this form of independence adds to the normal psychological development of these children. Some children may require urinary diversion through the abdominal wall (ileal conduit) or through the appendix (Mitrofanoff principle appendicovesicotomy)144–146 or other, less common methods, such as intravesical transurethral bladder stimulation, to handle their urinary condition.140,147 Although CIC is not possible for all children with spina bifida, it remains the method of choice for bladder management.
Bowel management and training programs should be started early. Medications, enemas, and attention to fiber content in the diet are all of value in establishing a bowel management program. The Malone antegrade continence enema (ACE) procedure is an important adjunct in the case of adults and children with problems of fecal elimination in whom standard medical therapies have failed.148,149
Orthopedic management
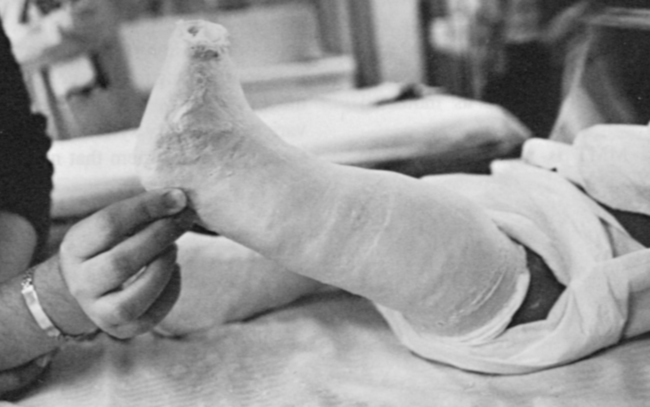
Orthopedic management of the newborn with a myelomeningocele will generally concentrate on the feet and hips. Soft tissue releases of the feet may take place during surgery for sac closure. Casting the feet (Figure 15-5) and performing early aggressive taping are also effective in the management of clubfoot deformities.150,151 Short-leg posterior splints (ankle-foot orthoses [AFOs]) may be used to maintain range and prevent foot deformities.

The orthopedist also will evaluate the stability of the hips. In children with lower-level lesions, attempts to prevent dislocation are made by using a hip abductor brace (Figure 15-6, A) or a total-body splint (Figure 15-6, B) for a few months after birth. With higher-level lesions, dislocated hips are no longer treated because they do not appear to have an effect on later rehabilitation efforts.133,152–154
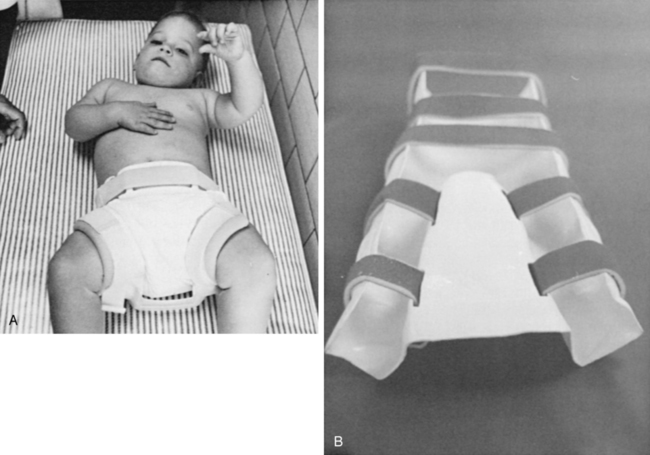
 A, Hip abductor brace. B, Total body splint.
A, Hip abductor brace. B, Total body splint.Hip dislocation.
Hip dislocations may occur at any level of neurologic deficit.155 The goal of treatment for those with hip dislocation should be maximum function, not radiographic realignment. The most important factor in determining ability to walk is the level of neural involvement and not the status of the hip.153,156–159 A level pelvis and good hip range of motion (ROM) are more important than hip relocation. In those with lower lumbar lesions and asymmetry caused by contracture, treatment will be directed at releasing the contracture and no attempts will be made to reduce the hip. Hip dislocations in those with sacral level lesions should be considered as lever-arm dysfunction, and surgical hip relocation is indicated.56,155,157,158 Immobilization after hip dislocation may lead to a frozen immobile joint from an open reduction procedure, redislocation from a lack of significant dynamic forces available for joint stability around the hip joint, and an increased fracture risk. Recently a questionnaire, the Spina Bifida Hips Questionnaire (SBHQ), to evaluate the ADLs that are important to children with spina bifida and dislocated hips and their families has been developed and has demonstrated construct validity as well as reliability.160
Knee valgus stress.
Many children with spina bifida who walk have excessive trunk and pelvic movement, knee flexion contractures, and rotational malalignment that may lead to excessive knee valgus stress. The most common deformities leading to this problem are rotational malalignment of the femur and femoral anteversion in association with excessive anterior tibial torsion. These deformities should be addressed via surgical correction as excessive knee valgus stress can lead to knee pain and arthritis in adult life.56,159,161,162 In addition, the PT may need to reassess the child’s gait pattern and use of assistive aids and bracing to minimize stress and maintain long-term joint viability for those with spina bifida over the life span.
Scoliosis.
The prevalence of scoliosis in spina bifida is estimated to be as high as 50%. Increasing scoliosis can lead to loss of trunk stability when curves are greater than 40 degrees and when associated pelvic obliquity becomes 25 degrees or more. Surgical intervention, often recommended to prevent further progression, may improve or further impair sitting balance, ambulation, and performance of ADLs.163 Various authors have reported that although surgery can improve curves by up to 50%, surgical morbidity must be considered and complications may be as high as 40% to 50%. Functional benefits are largely unsubstantiated owing to poorly constructed studies.164–166 Wai166 suggests that spinal deformity may not affect overall physical function or self-perception. After surgical correction it may take up to 18 months to appreciate functional improvement, and walking may be difficult for those who were just exercise ambulators before correction. Although surgical repair of scoliosis does improve quality of life in patients with cerebral palsy and muscular dystrophy, this has not been demonstrated in those with spina bifida.167 Interventions such as chair modifications to shift the trunk to improve balance in the coronal plane and reduce pelvic obliquity and truncal asymmetry should be considered as a first option, before surgical correction.163,167
Back pain.
Back pain needs to be efficaciously evaluated in those with spina bifida who report back pain. Knowing when the patient experiences pain, what increases pain, what positions exacerbate pain, and what region of the body is affected can help lead to appropriate referral, testing, and management. Knowing if your patient has a shunt, spinal rods, and/or a Chiari malformation will also be important to your assessment and management. Pain in the neck, shoulders, and upper back with associated weakness and/or abnormal sensory findings should be evaluated by the treating neurosurgeon to rule out shunt malfunction. Spinal rods that have broken or that are breaking through the skin may also be a source of pain in this area. Pain not caused by rods, a shunt, Chiari issues, or a syrinx may have a mechanical cause and could be a result of poor posture, tension, or weight gain. A patient who reports low back pain may have a symptomatic tethered cord if the patient is also reporting changes in gait, increased tripping or falling, bladder changes, and/or pain shooting down the legs. Manual muscle testing (MMT) and urodynamic testing (refer to Chapter 29) are appropriate at this point and should be compared with baseline testing findings. Mechanical low back pain may be a result of abnormal gait mechanics, asymmetrical strength, and use of older orthotics that no longer fit. Assessment of seating and support systems, including cushions, and gait mechanics and use of orthotics and ambulatory aids are mandatory to increase stability and redistribute balance over stressed joints and to maximize reduction of the patient’s pain and discomfort. Strengthening, particularly of the gluteal muscles, for those who are ambulatory may also be indicated. In addition, programs aimed at weight reduction may be necessary to alleviate stress and pain to preserve long-term viability of tissues. In addition, for women the chest may cause tension on the upper back, and breast reduction has been advocated for some to relieve this tension.168–170
Foot deformity.
The goal of treatment of the foot in spina bifida should be a flexible and supple foot. An insensate flail foot often becomes rigid over time, and foot management can become complicated by pressure sores. Up to 95% of patients will use an orthosis, and a supple flail foot will be easier to manage over time. Surgeries that are extraarticular with avoidance of arthrodesis, as well as simple tenotomies versus tendon releases and lengthenings, may best manage outcomes for bracing and ambulation.56 Equinovarus deformities may be managed with early and intensive taping in the newborn period, known as the French method,171,172 stretching and casting, and surgical intervention. The Ponsetti method, advocated by some, also has been reported to have positive outcomes; however, the significant investment in time and commitment by the family for frequent cast changes may affect the ability to carry out other ADLs without disruption.155 In those with lipomas, foot deformity that may be acquired over time is best managed in a similar manner. Maintaining a supple and plantigrade foot with adequate muscle balance with use of soft tissue correction through tendon lengthening, tendon transfer, and plantar fascial release is recommended until 8 years of age. After that time, deformities may become more rigid and may necessitate more bony procedures.173
Osteoporosis, osteopenia, and fracture.
Osteoporosis (thinning of the bone) and osteopenia (low bone mineral density [BMD]) in the legs and spine have been described in children and teens with spina bifida. These conditions increase the risk of fracture, increase the time for healing after fracture, and may lead to back pain. A study by Valtonen and colleagues in 2006 documented the occurrence of osteoporosis in adults with spina bifida. This condition often is not recognized.174 Medical factors such as physical inactivity, decreased vitamin D, diminished exposure to sunlight, urinary diversion, renal insufficiency, hypercalciuria, medication for epilepsy, and oral cortisone treatment for more than 3 months increase the risk of osteoporosis.59,175,176 It can be assumed that patients with meningomyelocele are at potential risk to develop osteoporosis at a younger age because of impaired walking ability and subsequent low physical loading of the lower limbs. Older age and higher levels have been associated with increased numbers of fractures in spina bifida.174 The optimal strategies for prevention and treatment of osteoporosis in this population have not been established. Further research is required to see if the methods used to prevent and treat osteoporosis in individuals without spina bifida also work for teens and adults who have spina bifida. Considering the effects of prolonged immobilization on independence in daily activities and quality of life, there should be no disagreement that all efforts are necessary to prevent these fractures. Furthermore, osteoporotic fracture may lead to a vicious cycle of immobilization, decreased bone density, and repeated fractures.174 Annual incidence of fracture is 0.029% in adolescents and 0.018% in adults.177 Studies have shown promising results of regular functional electric stimulation–assisted training, but this is often nearly impossible to carry out in daily life.178 The effects of standing programs on bone density are unclear.179,180 The prevention of fractures should be among the major goals in the rehabilitation of people with meningomyelocele. The assessment of BMD is worthwhile in patients with risk factors for osteoporosis, because low BMD is a known risk factor for fractures.175
Postoperative management.
Care should be taken to avoid postoperative complications such as skin breakdown and postimmobilization fractures in the postoperative period. To decrease the risk of nonunion and allow for early mobilization and weight bearing, one should consider rigid internal fixation versus Kirschner wire fixation. After surgery, immobilization in a custom-molded body splint rather than a hip spica cast is preferred. Postoperative physical therapy should begin as soon as wounds are stable and healing is occurring. Therapy should focus on ROM (active and passive) and early weight bearing. Crawling should be strictly forbidden for a minimum of 3 to 4 weeks postimmobilization to reduce the risk of fracture.159
Evaluations
Manual muscle testing
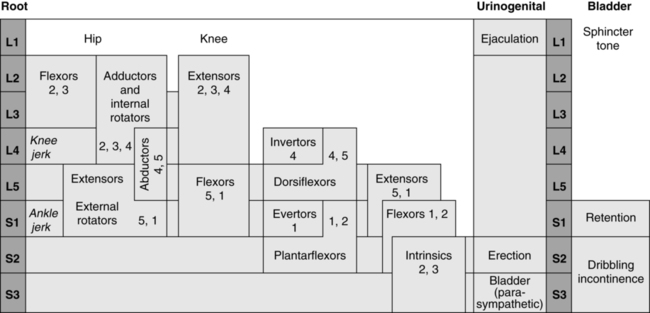
Muscle strength is generally graded for groups of muscles and can be graded by using either a numerical (1 to 5) or an alphabetical designation (Figure 15-7) or simply by noting presence or absence of muscular contraction by a plus or a minus on the muscle test form. The last method may be sufficient initially, but as the child matures a more definitive muscle grade should be determined.


By use of a MMT form that lists the spinal segmental level for each muscle group, an approximate level of lesion can be determined from the test results (see Figure 15-7). Because the spinal cord is often damaged asymmetrically, MMT does not always accurately reflect the level of the lesion. If reflex activity is also noted on the form, the presence of distal sparing of the spinal cord can be determined. Muscle testing of the newborn gives the clinician an appreciation of muscle function and possible potential for later ambulation as well as an awareness of possible deforming forces. For example, if hip extensors or abductors are not functioning, then the action of hip flexors and adductors must be countered to prevent future deformities.
Muscle testing of the toddler or young child may require some of the techniques previously described. In addition, developmental positions can be used to assess muscle strength in an uncooperative youngster. For example, strength of hip extensors and abductors can be assessed as a child attempts to creep up steps or onto a low mat table. With addition of resistance to movements, fairly accurate muscle grades can be determined. To elicit hip flexor action in sitting, if an interesting toy or object is placed on the child’s ankle or between the toes, the child will often lift the leg spontaneously to reach for it. Ingenuity and creativity are prerequisites for muscle testing in the young child. Reliability of MMT in children with spina bifida younger than 5 years is difficult but has been demonstrated in a clinic setting where all therapists were trained in specific MMT technique to ascertain consistency in testing.181 By the age of 4 or 5 years, muscle grades can generally be determined by traditional testing techniques, although the reliability of the test results will increase with the age of the child.182 In most clinics MMT is used to assess strength and changes in strength over time. Reliability of MMT may be called into question when trying to assess meaningful detectable changes in power against gravity. If that is the case, one can use hand-held dynamometry (HHD) to test muscles with a grade of 3 or greater. Excellent intertester reliability of HHD for children with spina bifida has been demonstrated.183
Muscle testing is indicated before and after any surgical procedure and at periodic intervals of 6 months to 1 year to detect any change in muscle function. Timely detection of any loss in strength is critical, as the child may encounter increased weakness resulting from tethering of the spinal cord or shunt malfunction as he or she grows. The level of innervation should not decrease throughout the life of the child with spina bifida. In the growing child or adolescent, an increasing weakness resulting from shunt malfunction, tethering of the spinal cord, or hydromyelia frequently can be substantiated by a muscle test of the lower extremities. The MMT is also valuable in determining the motor level so that potential future functional level can be determined (Figure 15-8).
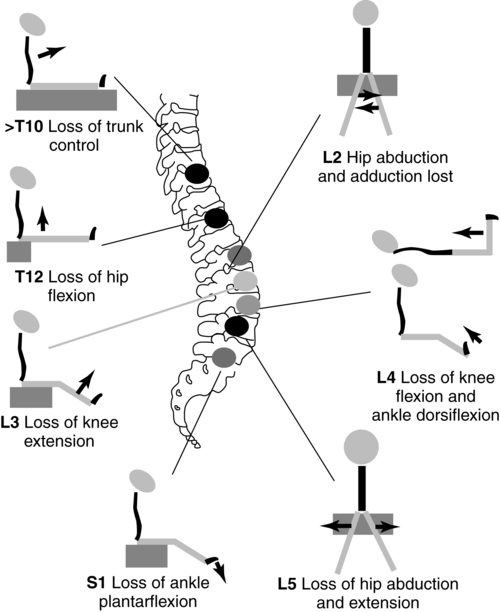

Sensory testing
Because of dermatome innervation the pin is usually drawn from the anal area across the buttocks, down the posterior thigh and leg, then to the anterior surface of the leg and thigh, and finally across the abdominal muscles. Reactions to be noted are a facial grimace or cry, which indicates that the painful sensation has reached a cortical level. Care must be taken to see that each sensory dermatome has been evaluated. Results can be recorded by shading in the dermatomes where sensation is present (Figure 15-9).
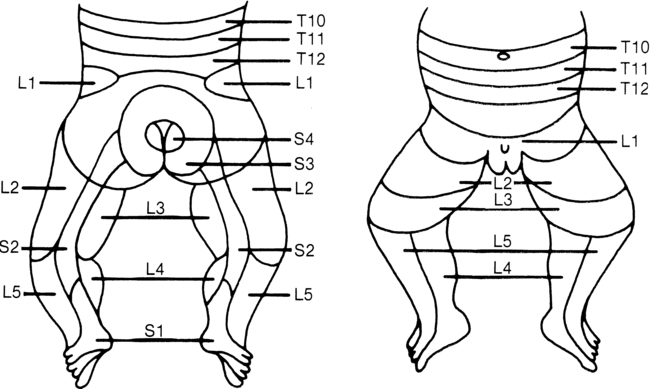
 Lower-limb dermatomes. (From Brocklehurst G: Spina bifida for the clinician. Clin Dev Med 57:53, 1976.)
Lower-limb dermatomes. (From Brocklehurst G: Spina bifida for the clinician. Clin Dev Med 57:53, 1976.)The therapist may be called on to evaluate the newborn before surgical closure of the spinal meningocele. Although sensory and motor levels can be determined as previously described, the infant’s general condition should be considered in interpreting test findings. Any medication taken by the mother during labor and delivery may influence the neonate’s performance and thus should be noted. In addition, the physiological disorganization normally seen in all infants during the first few days after birth may also affect testing.184 At best, this presurgical evaluation establishes a tentative baseline, but significant changes in the infant’s neurological status in the first few weeks of life should not be surprising to the clinician.
In the young child from 2 to 7 years of age, light touch sensation and position sense can be tested in addition to pain sensation. Again, to elicit an appropriate response and reliable test results, the ingenuity of the therapist will be required. Using games such as “Tell me when the puppet touches you” may be more effective for the young child than traditional testing methods. Sensory dermatome mapping using the chart in Figure 15-9, or a similar form such as the WeeSTeP once the child with spina bifida gets older, can aid in establishing sensory level as well as insensate areas that may be at high risk of injury.185
After testing, a survey of the sensory dermatome chart should indicate whether sensation is normal, absent, or impaired. MMT and sensory testing (dermatomes) can assist in determining spinal level of function (Figure 15-10).

Range-of-motion evaluation
In the child with spina bifida, contractures may be evident at multiple joints at birth because of unopposed musculature (Figure 15-11). Hip adduction should not be tested beyond the neutral position to avoid dislocation of hips, which are often unstable. Range should be done slowly and without excessive force to avoid fractures so often experienced in paralytic lower extremities. ROM should be checked with the same frequency as MMT. Active ROM of the upper extremities can be assessed by observation and handling the infant. A formal ROM evaluation for the upper extremities is not usually indicated. A baseline ROM and tone assessment of the upper extremities should be completed.

Reflex testing
As the primitive reflexes (initially needed for survival and to experience movement) become integrated, they are replaced by more mature and functional reactions. The righting and equilibrium reactions help the child attain the erect position and counteract changes in the center of gravity. Because these reactions depend on an intact CNS as well as a certain level of postural control, they may be delayed, incomplete, or absent in the child with spina bifida. For example, a child with a low thoracic spinal cord lesion may show an incomplete equilibrium reaction in sitting. This may be caused by the lack of a stable postural base or by lack of initiation of the reaction centrally. Both the neurological and muscular components of these reactions must be considered. Reflex testing for the child with spina bifida may not be as intensive as that for a child with cerebral palsy. It may, however, provide a check on the progress of normal development and as such reflect the integrity of the CNS (see Chapters 3 and 16).
Developmental and functional evaluations
The way in which a task is accomplished is as important to evaluate as the accomplishment itself. For example, in rolling, is head righting sufficient to keep the head off the supporting surface? From the hands-knees position, can reciprocal crawling be initiated without the lower extremities being held in wide abduction? Can the child pull to stand easily by using trunk rotation? Assessing the quality of the child’s abilities will assist the clinician in determining where therapeutic measures should begin and what the goals of such intervention will be. Standardized assessments may provide the families with guidelines and a record of motor skills over time (see Chapter 3).
There are no standardized functional or motor assessments specific to those with spina bifida. Some assessments look at development relative to standardized norms and may guide the family and therapist in determining treatment goals and challenges. This information may be invaluable in determining bracing needs and other equipment needs as well as timing of various interventions. If a standardized assessment were desired to use with the infant with spina bifida, the Alberta Infant Motor Scale (AIMS) might be appropriate. The AIMS186 is designed to measure motor development from birth to 18 months of age. It is a 58-item observational test of infants in supine, prone, sitting, and standing positions. Each item includes detailed descriptions of the weight-bearing surface, the infant’s posture, and antigravity movements expected of the infant in that position. The AIMS requires minimal handling of the infant and can be completed in 20 to 30 minutes. The test was normed on a cross-sectional sample of 2200 infants in Alberta, Canada. Interrater and test-retest reliability are high (0.95 to 0.99), as is concurrent validity with the Peabody Developmental Motor Scales (PDMS) (0.99) and the Bayley Scales of Infant Development (0.97). Predictive validity of the AIMS appears to be fair.187 For the child with spina bifida the AIMS could be used to assess current motor development and track progress in motor development over time.
The Milani-Comparetti Motor Development Screening Test for Infants and Young Children may also be useful in assessing the functional level of the child with spina bifida. This screening examination is designed to evaluate motor development from birth to 2 years of age (Figure 15-12).188 It requires no special equipment and can be administered in 4 to 8 minutes. The test evaluates both spontaneous behavior and evoked responses. Spontaneous behavior includes postural control of the head and body in various positions as well as a sequence of active movement patterns. Primitive reflexes, righting, and equilibrium reactions constitute the evoked responses. The Milani-Comparetti test was normed on a sample of 312 children from Omaha, Nebraska. Interrater reliability percent of agreement was 89% to 95%. Test-retest reliability percent agreement was 82% to 100%. Predictive validity of the test has not been well established.188 The Milani-Comparetti test should assist the clinician in evaluating each child’s underlying postural mechanisms and his or her ability to attain the erect position. The test manual provides information on special examination procedures and scoring.


The Ages and Stages Questionnaire (ASQ) is a screening assessment that assesses developmental and social-emotional delays during crucial early ages of life. This test is available in English and Spanish and can be completed in 10 to 15 minutes.189 It was developed and validated on 15,138 children in all 50 states and several U.S. territories. The test-retest reliability (0.92), interrater reliability (0.93), validity (0.82 to 0.88), sensitivity (0.86), and specificity (0.85) have been well documented.189 This test provides parents and providers with a checklist to easily assess change over time.
The PDMS-2 is another standardized assessment that may prove helpful in evaluating a child with congenital spinal cord injury.190 The PDMS-2 was developed using item response theory (IRT) and consists of six gross and fine motor subtests from birth through 6 years of age. The test takes 45 to 60 minutes to complete or 20 to 30 minutes per subtest. The two scales allow a comparison of the child’s motor performance with a normative sample of children at various age levels. A stratified sample of 2003 children from 46 states in the United States was used to develop PDMS-2 test norms. Test-retest and interrater reliability are high. Content, construct, and concurrent validity have been well established. Although the child with activity limitations would not be expected to succeed on many of the gross motor items at the later age levels, the scale still serves as a reminder of expected gross motor performance at each age. The fine motor scale offers a chance to assess fine motor performance of children with congenital spinal cord injury. This area has been frequently overlooked in children with myelomeningocele. Fine motor development, however, may be affected because of congenital abnormalities in brain development associated with myelomeningocele or related to tethering of the spinal cord that can result in fine motor paresis. In addition, the PDMS-2 offers guidelines for administering the test to children with various activity limitations.190
The Bruininks-Oseretsky Test of Motor Proficiency, second edition (BOT-2) can be used to evaluate the higher functioning ambulatory child with lower lumbar or sacral level spina bifida.191 Fine manual control, manual coordination, body coordination, and strength and agility subtests can be used to assist in evaluating areas of fine motor control, balance, and coordination difficulties. This test has been standardized on a sample of 1520 subjects from age 4 through 21 years.191
The Movement Assessment Battery for Children, second edition (Movement ABC-2), can be used to identify children who are significantly behind their peers in motor development, assist in planning an intervention program in either a school or a clinical setting, and measure change as a result of intervention or can serve as a measurement instrument in research involving motor development. This tool may be useful to assess children with lower lumbar and sacral level myelomeningocele, as well as children with lipomeningocele. The Movement ABC identifies and evaluates the movement problems that can determine a child’s participation and social adjustment at home or school. The Movement ABC Checklist provides classroom assessment of movement difficulties, screening for “at risk” children (ages 5 to 12 years), and systematic monitoring of treatment programs. It provides a comprehensive assessment for those identified as “at risk” (3 to 16 years, 11 months), yielding both normative and qualitative measures of movement competence, manual dexterity, ball skills, and static and dynamic balance.192
Finally, the Pediatric Evaluation of Disability Inventory (PEDI) is a comprehensive assessment of function in children aged 6 months to 7 years.193 The PEDI measures both capability and performance of functional activities in three areas: self-care, mobility, and social function. Capability is a measure of the functional skills for which the child has demonstrated mastery. Functional performance is measured by the level of caregiver assistance needed to accomplish a task. A modifications scale provides a measure of environmental modifications and equipment needed in daily functioning. The PEDI has been standardized on a normative sample of 412 children from New England. Some data from clinical samples (N = 102) are also available. Interrater reliability of the PEDI is high as demonstrated by high intraclass correlation coefficients (ICCs = 0.96 to 0.99). Concurrent validity of the PEDI with the WeeFIM (child’s version of the Functional Independence Measure) was also high (r = .80 to 0.97).193 The PEDI can be administered in approximately 45 minutes by clinicians or educators familiar with the child or by structured interview of the parent. The PEDI should provide a descriptive measure of the functional level of the child with myelomeningocele as well as a method for tracking change over time. The PEDI has had a rich tradition in helping to document functional development, and new methods proposed for the next generation of the PEDI include using item banks and computer adaptive testing. The computer adaptive testing feature and the revised and expanded content of the new PEDI will enable therapists to more efficiently assess children’s functioning to a broader age group of children.194,195
Another assessment of motor performance that may be commonly used with the school-age child with spina bifida is the School Function Assessment (SFA). The SFA is standardized and was conceptually developed to reflect the functional abilities and needs of a student in elementary school. The three areas assessed include the student’s participation in school activities, task supports required by the student for participation, and the student’s activity performance.196,197 It was designed to facilitate collaborative program planning for students with a variety of disabling conditions. The instrument is a judgment-based (questionnaire) assessment that is completed by one or more school professionals who know the student well and have observed his or her typical performance on the school-related tasks and activities being assessed. Items have been written in measurable, behavioral terms that can be used directly in the student’s Individualized Educational Plan (IEP).196
Gait analysis
Formal computerized gait analysis was initially used to evaluate children with cerebral palsy. Increasingly it is being used to evaluate children with meningomyelocele once they have established a gait pattern to determine factors leading to changes in gait, including changes in alignment, muscle length, muscle torque, and symmetry. The gait analysis may aid in decision making regarding orthotic and orthopedic interventions. Whether it is useful to do formal gait analyses in all children with spina bifida remains to be determined.198 Gait analyses have also been useful in establishing a database of trends in kinetics and kinematics for various levels of spina bifida.
Perceptual and cognitive evaluations
For the newborn from 3 to 30 days old, the Brazelton Neonatal Behavioral Assessment scale may be adapted to assess the infant’s organization in terms of physiological response to stress, state control, motoric control, and social interaction.184 Ideally the infant should be medically stable and free from CNS-depressant drugs before evaluation. Generally this evaluation will occur after the back lesion has been closed and a shunt has been positioned to relieve the hydrocephalic condition.
Repeated administration of the Brazelton Neonatal Behavioral Assessment scale in the first month of life may help monitor the infant’s progress in organization and reflect the curve of recovery. Although the manual for this behavioral assessment is complete, proper administration, scoring, and interpretation require direct training with someone already proficient in using the scale.199 Excellent training videos for the Brazelton Neonatal Behavioral Assessment scale are available through the Brazelton Institute for purchase or through the local university’s learning resource centers.200
A full developmental evaluation appropriate for the infant and toddler with spina bifida is the Bayley Scales of Infant and Toddler Development, Third Edition (BSID-III).201 The Bayley Scales, consisting of a mental and motor scale and a behavioral rating scale, can be used to test children from age 1 month to 42 months. The test provides information on gross motor, fine motor, language, social-emotional, adaptive, and cognitive development.
The BSID-III provides the clinician with a broader view of the child’s total development. The gross motor information from this developmental assessment will not be specific enough for a therapist evaluating a child with spina bifida. The additional information on fine motor, language, personal-social, and cognitive development, however, is sufficient and will be important in planning a comprehensive intervention program.201
Various tests are available as screening tools to test visual-motor integration and perception. The Beery-Buktenica Developmental Test of Visual-Motor Integration, 6th Edition (Beery VMI) is an early screening tool to aid in diagnosis of learning problems in children. It assesses integration of visual perception and motor control of children from age 2 years through 18 years. The test takes 10 to 15 minutes to complete and requires the child to be able to copy designs. The Beery VMI is norm referenced and was standardized on a large sample of children chosen from throughout the United States. There is also an adult version that can be used with individuals 19 to 100 years of age that facilitates identification of neurological and related problems in the adult.202
Children with spina bifida often exhibit upper-extremity weakness in addition to probable sensory dysfunction. As a result, fine motor skills in children with spina bifida are often impeded by slowness and inadequate adjustment of manipulative forces, and a non–motor-perceptual test is often desired.203–205 The Motor-Free Visual Perception Test, Third Edition (MVPT-3)206 and the Test of Visual Perceptual Skills, Non-Motor, Third Edition (TVPS-3)207 can be used to determine the child’s visual perceptual processing skills on the basis of a non–motor assessment of these skills. Both tests evaluate visual discrimination, visual memory, spatial relations, figure-ground, and visual closure. The TVPS-3 also evaluates form constancy and sequential memory. The MVPT-3 can be used with individuals from 4 to 70 years of age, and the TVPS-3 can be used with children from 4 to 18 years of age. The TVPS-3 has two levels; the lower level tests children from ages 4 to 12 years, and the upper level tests children from ages 12 years to 17 years, 11 months. Both tests are easy and quick to administer (less than 15 minutes) and, based on the examiner’s experience and training, interpretations can be made with prescription for remediation. The MVPT-3 was standardized on a nationally representative sample. The test-retest reliability of the MVPT-3 was 0.81.206 Performance on the motor-free test has been shown to be independent of the degree of motor involvement when compared with other tests of visual perception.206 The TVPS-3 was standardized on a nationally stratified sample of 2000 children across the United States.
Treatment planning and rehabilitation related to significant stages of development
Newborn to toddler (preambulatory phase)
Stage 1: before closure of myelomeningocele—early newborn period
Physical therapy management of the infant in stage 1 is limited by his or her medical condition (Table 15-2). Therapists are called on a regular basis in large tertiary care centers to carry out preoperative MMT to help to ascertain functional motor level. Physicians (neurosurgeons and orthopedic surgeons) on the spina bifida care team rely on this assessment to guide their discussion with the families regarding care and prognosis. When carrying out the preoperative MMT, great care must be taken to avoid contaminating an open sac, which is usually covered with a Telfa nonadherent dressing or a wet sterile dressing that must be kept moist with a saline solution.
TABLE 15-2 
SUMMARY OF TREATMENT PLANNING AND REHABILITATION RELATED TO SIGNIFICANT STAGES OF DEVELOPMENT
| STAGE OF RECOVERY | MAJOR PHYSICAL THERAPY GOALS | PHYSICAL THERAPY MANAGEMENT |
| NEWBORN TO TODDLER (PREAMBULATORY PHASE) | ||
| Stage 1: before surgical closure of myelomeningocele—newborn | Determine functional motor level | Preoperative manual muscle testing |
| Stage 2: after surgery, during hospitalization—newborn to infant | Confirm functional motor levelPrevent contracture and deformity Encourage normal sensorimotor development |
Postoperative manual muscle testingROM exercises taught to hospital personnel and family Positioning in prone and side lying Provide toys of various colors, textures, and shapes Graded auditory and visual stimuli: music boxes, squeaky toys, brightly colored objects Therapeutic handling to encourage good head and trunk control |
| Stage 3: condition stabilized—infant to toddler | Confirm functional motor levelEncourage normal development sequence | Manual muscle testing once or twice per yearWork in sitting on head righting and equilibrium reactions Eye-hand coordination activities Early weight bearing on lower extremities Encourage prone progression Weight shifting in standing frame Comprehensive home program |
| TODDLER THROUGH ADOLESCENT (AMBULATORY PHASE) | ||
| Stage 4: toddler through preschool | Confirm functional motor levelBegin ambulation Continue development in cognitive and psychosocial areas Collaborate on goals with other team members |
Manual muscle testing once or twice per yearChoose appropriate orthotic device Gait training Development and strengthening of righting and equilibrium reactions Consider referral to EI program Public preschool program Continue home program Open communication with other team members |
| Stage 5: primary school through adolescence | Confirm functional motor levelReevaluate ambulation potential Maintain present level of functioning Prevent skin breakdown as child becomes more sedentary Promote independence in self-care skills Remediate any perceptual-motor problems Provide appropriate adaptive devices Promote self-esteem and social-sexual adjustment |
Postoperative manual muscle testingReplace orthotic device as necessary Wheelchair prescriptions as necessary Teach locomotion activities Maintain strength in trunk and extremities Teach skin care Work with team members to teach dressing, feeding, hygiene, and bowel and bladder care Provide program and activities for sensorimotor integration Check for fit and proper use of adaptive devices Collaborate with other team members in counseling efforts |
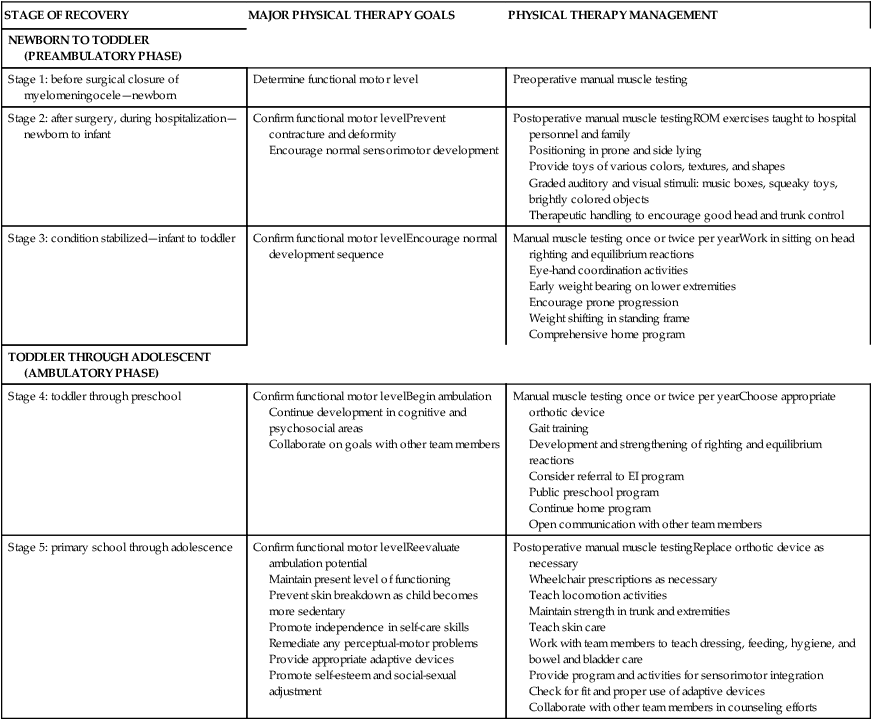
Stage 2: after surgery, during hospitalization, and transition to home—newborn through early infancy
Therapeutic intervention after surgical back closure during stage 2 is often limited by the infant’s neurological and orthopedic status. A major goal during this stage is to prevent contractures and maintain ROM while giving stimulation to provide as normal an environment as possible. Traditional ROM exercises can be taught to nursing staff and family. They also can be carried out while the child is being held at the adult’s shoulder or prone over the adult’s lap. These positions allow closeness between the caregiver and infant, thus encouraging maximal relaxation and interaction between them. ROM movements and positioning in prone or side lying may be initiated to prevent or decrease contractures in the lower extremities. If clubfeet are present, soft tissue stretching may be indicated. Stretching begins distally on the soft tissue of the forefoot and proceeds proximally toward the calcaneus. This is done to take advantage of the pliability of soft tissue structures and to minimize fixed deformity later. In addition, taping may be used to maintain optimal ROM and alignment between periods of stretching.150 In treating the newborn after surgery, great care must be taken to avoid contaminating the surgical dressing, which is usually covered with Xeroform Petrolatum Gauze (3% bismuth tribromophenate in a special petrolatum blend on fine mesh gauze). This dressing is nonadherent and clings and conforms to all body contours. The Xeroform dressing is covered with Telfa. This postoperative dressing remains on for 2 weeks.
Stage 3: condition stabilized—infant to toddler (preambulatory)
Following a normal developmental sequence, the child with spina bifida will usually begin some form of prone progression as trunk and upper-extremity stability improve. This is a significant phase of development because it allows for the development of a sensorimotor base as the child expands environmental horizons. During this phase of high mobility, insensate skin must be checked for injury frequently and often must be protected by heavier clothing. This may help prevent any major skin breakdown, which could significantly delay the rehabilitation process. For some children with high-level lesions in whom prone mobility is not safe or practical for long distances, a Star Car (Tash) (Figure 15-13), the Ready Racer (Tumble Forms), or the PlasmaCar may be used. These provide the child with a means of exploring the environment safely but independently.

 Caster cart used for independent mobility.
Caster cart used for independent mobility.Early weight bearing is also of utmost importance, both physiologically and psychologically. The upright position has beneficial effects on circulation and renal and bladder functioning as well as on the promotion of bone growth and density.59,176,208,209 Psychologically, weight bearing in an upright posture allows a normal view of the world and contributes to more normal perceptual, cognitive, and emotional growth. One way to achieve this weight bearing is in the kneeling position. This is developmentally appropriate because children 8 to 10 months old frequently use kneeling as a transition from all fours to standing.
Because young infants are frequently held in the standing position and bounced on their parents’ laps, this form of weight bearing on the lower extremities is appropriate from birth onward. Failure to promote weight bearing in this manner may deprive the child with spina bifida of the normal experience of standing at a very early age. When standing these children, however, care must be taken to see that the lower extremities are in good alignment and that undue pressure is not exerted on them (Figure 15-14). In this way the risk of fractures is minimized and a normal weight-bearing experience is provided.
 Assisted standing with normal postural alignment.
Assisted standing with normal postural alignment.Also in this phase of preambulation, transitions from one position to another should be assessed and facilitated. Teaching the child strategies for transitions will enhance his or her optimal functional independence. Compensations may be taught to substitute for weakened musculature. In addition, adaptive equipment and mobility devices may be recommended to enhance acquisition of age-appropriate milestones. Providing appropriate facilitation of mobility at a level similar to that of a child’s peers is important for psychosocial growth and development (Figure 15-15).
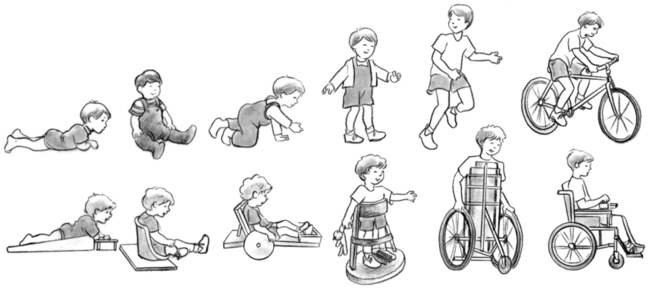

When the child attempts to pull to a standing position or would be expected to do so normally (at 10 to 12 months of age), the use of a standing device is indicated. Generally a standing frame is the first orthosis chosen. This is a relatively inexpensive tubular frame to which adjustable parts are attached (Figure 15-16). Because it is not custom made, it can be fitted fairly quickly, although adjustments may be necessary to accommodate spinal deformities. This standing device offers support of the trunk, hips, and knees and leaves the hands free for other activities. Time spent in the standing frame should be increased gradually. This allows the child to adjust to the upright position in terms of muscle strength, endurance, blood pressure, and pressure on skin surfaces.
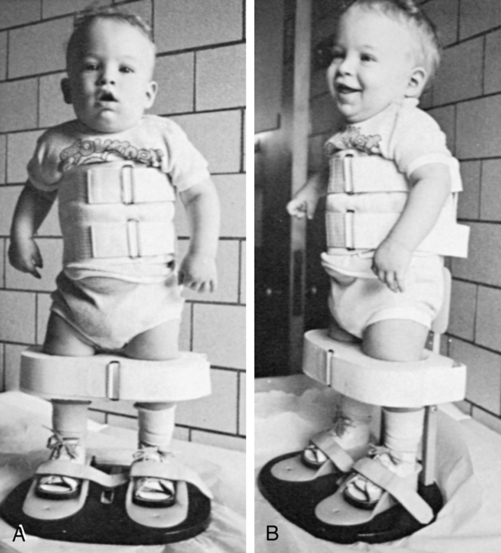
 Standing frame. A, Anterior view. B, Lateral view.
Standing frame. A, Anterior view. B, Lateral view.Toddler through adolescent (ambulatory phase)
Stage 4: toddler through preschool
Thus far, the child has learned to shift weight in the standing frame. By rotating the trunk toward the weighted side, the non–weight-bearing side can be shifted forward (Figure 15-17). By reversing the weight shift, the opposite side can be moved forward and a type of “pivoting forward” progression can be accomplished. To maintain balance while shifting, the child may initially use a two-wheeled walker. The therapist may help initiate weight shift and trunk rotation by alternately pulling the arms forward.210 Once the child has gained this form of mobility, the type of permanent bracing chosen will depend on the level of the lesion and a variety of other factors.
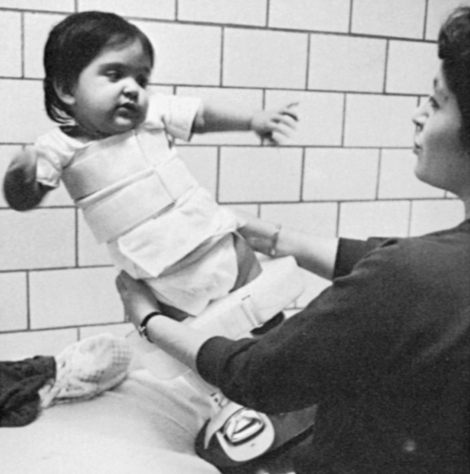
 Weight shift and forward rotation in standing frame.
Weight shift and forward rotation in standing frame.The overall goal for ambulatory training is to promote efficient, independent mobility with the least amount of bracing while maintaining optimal joint integrity. Ambulatory potential as well as choice of bracing depends on many factors, including neurosegmental level of the lesion, motor power at the neurosegmental level, extent and degree of orthopedic deformity, balance, age, height, weight, sex, motivation, spasticity, design and effectiveness of the orthosis, effectiveness of PT intervention, environmental factors, upper-extremity strength and control, and cognitive level.163,211 The best prognosis for ambulation is most often seen in the child who is not shunted and has good cognition, good quadriceps power, no deformity, a stable neurological condition, and hands-free sitting balance. Factors that may limit potential for ambulation include hydrocephalus, high-level lesions, kyphosis or kyphoscoliosis, and unstable neurology.
Developmental progression preceding ambulation in spina bifida shows a high degree of variability.212 A study by Bartonek in 2010212 confirmed that those with greater muscle power of the lower limb muscles ambulated earlier and more frequently. Those engaged in physical therapy programs aimed at achieving the specific goal of walking are also likely to make earlier strides toward ambulation, although this has not been formally studied. Those who have been enrolled in early treadmill body-weight support training (BWST) programs as infants have demonstrated early stepping responsiveness, and it has been suggested that BWST training intervention could promote muscle strengthening and take advantage of neural plasticity to promote development of the neuromuscular patterns necessary to support the onset of gait. There is also potential to improve bone density, cardiovascular function, and the integrity of lower spinal sensorimotor function. Infants with spina bifida demonstrate developmental delay as early as 3 months of age, and BWST intervention during the first postnatal year has the potential to reduce this delay and promote earlier onset of gait in the population with spina bifida.213 Efficacy of enhanced sensory inputs during treadmill stepping in children with spina bifida has also been examined. Increasing friction by using Dycem matting and enhancing visual flow by using a checkerboard pattern on the treadmill belt both appear to be more effective than the standard black treadmill belt in eliciting stepping.214 Teulier and colleagues215 studied stepping responses in infants with spina bifida. They reported a decrease in number of steps per minute compared with typically developing peers (14.4 versus 40.8 steps per minute) and a decrease in frequency of alternating steps. In contrast to these interlimb coordination differences, they reported that within-limb step parameters in infants with spina bifida were quite similar to those in children who were typically developing.
For thoracic and high-level lumbar lesions, a parapodium is often chosen. The parapodium was developed by the Ontario Crippled Children’s Centre in 1970 and is similar to the standing frame except that hinges at the hips and knees allow for sitting and standing.210 It also can be adjusted for growth and can accommodate orthopedic deformities. As with the standing frame, proper alignment of the parapodium is critical. The therapist, in conjunction with the orthotist, should check for correct standing alignment. The prevention of additional orthopedic deformities, development of good muscular control, and normal body image depend on a well fitting orthosis.
Another type of orthosis for the child with a thoracic or high lumbar lesion is the Orthotic Research and Locomotion Assessment Unit (ORLAU) swivel walker. It consists of modular design similar to that of the standing frame, with a chest strap and knee blocks attached to swiveling footplates.216 Rather than the whole base moving forward, as when weight is shifted in the parapodium, in the swivel walker each footplate is spring-loaded and is able to swivel forward independently. This allows for independent balance on one foot and therefore crutchless ambulation. The ORLAU swivel walker is manufactured in the United Kingdom, and assembly kits may be ordered; however, availability is limited (Appendix 15-A).217 Another parapodium in use is the Rochester parapodium. Separate hip and knee joints allow a variety of free movement for sitting and bending down. The lower portion remains rigid and supportive when hips are flexed. Therefore a child can bend and pick up objects from the floor, and the unlockable hip and knee joints will relock automatically on extension.218
There is a perception that although children with myelomeningocele use orthoses effectively, very few continue ambulation into adulthood. Two studies of patients with thoracic-level myelomeningocele219 demonstrated that the ORLAU can be used effectively into the adult years. Both studies noted a 58% to 59.4% compliance rate. Patients in the studies who started using the ORLAU after 11 years of age continued use for 3 to 24 years. Mazur and colleagues219 noted that those with myelomeningocele who did not ambulate had a fivefold increase in pressure sores and twice the number of fractures compared who those who did ambulate. Vigorous walking programs should be considered to assist long-term health.
Both the parapodium and swivel walker have had some problems with instability, ease of application, and cosmesis. New designs attempt to correct these problems.220 Nevertheless, existing limitations in the parapodium and swivel walker, particularly energy cost of walking, slow rate of locomotion, and cosmesis, have limited their use, primarily to the younger child. These devices, however, remain an effective means of preventing musculoskeletal deformities caused by long-term sitting, wheelchair positioning, and general immobility. They also enhance social-emotional development gained from the upright position.216,221 Another option for the child with a higher-level lesion and good sitting balance is the reciprocating gait orthosis (RGO). This brace consists of bilateral long-leg braces with a pelvic band and thoracic extension, if necessary. The hip joints are connected by a cable system that can work in two ways: If the child has active hip flexors, he or she can activate the cable system by shifting weight and flexing the non–weight-bearing extremity. This brings the weight-bearing extremity into relative extension in preparation for the next step. Without hip flexors, the child extends his or her trunk over one extremity, thus positioning it in relative extension. By virtue of the cable system, the non–weight-bearing extremity moves into flexion, thus initiating a step. Several types of the RGO are in use, including the dual-cable LSU222 and the horizontal-cable type.223
Most recently the Isocentric Reciprocating Gait Orthosis (I-RGO) (Center for Orthotics Design, Campbell, California) has been used for children with high-level spina bifida. It has a more cosmetic and efficient design compared with the dual-cable LSU or horizontal-cable–type RGO. This cableless brace has two to three times less friction and therefore is more energy efficient. The brace stabilizes the hip, knee, and ankle joints and balances the person, enabling him or her to stand hands free without the use of crutches or a walker (Figure 15-18).223 Leg advancement for walking occurs through use of hip flexor or lower abdominal muscle contraction or through use of active or passive trunk extension. In a study of 15 patients with lesions from T10-L3, use of the RGO produced favorable results. It was used effectively by 13 of the 15 patients. Initial use of the RGO was initiated at 5 years, and eight of the 15 discontinued use at 10 years of age. During the period of use, four became community ambulators, nine were household ambulators, and two remained nonfunctional (standing only). Average daily use ranged from 6 hours for those ambulating in the community to 30 minutes for those who were nonfunctional ambulators. Six of the 15 had no quadriceps power yet were able to functionally use the RGO for ambulation. Strong motivation and realistic goals are important to successful use.224
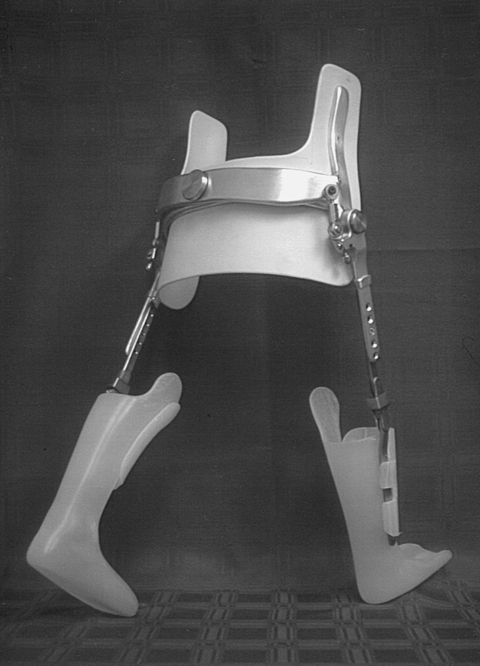
 Reciprocating gait orthosis.
Reciprocating gait orthosis.The type of orthosis chosen (long-leg, with or without pelvic band, or short-leg) depends on the level of the myelomeningocele and the muscle power within that level (Table 15-3). Because lesions are frequently incomplete, muscle strength must be accurately assessed before bracing is prescribed. Independent sitting balance with hands free also is a prerequisite for use of long- or short-leg braces. Even children with L3 to L4 lesions who demonstrate incomplete knee extension may be able to use a short-leg brace with an anterior shell rather than requiring long-leg bracing.225 This crouch-control AFO (CCAFO) will prevent a crouching gait pattern by improving knee extension during gait (Figure 15-19).226 Another alternative to a standard solid ankle AFO may be the carbon fiber spring AFO. This brace provides dynamic assist, supports the patient through the entire stance phase, and increases the energy return during the third rocker phase of push-off, simulating the natural push-off action.227 For children demonstrating excessive knee valgus caused by hip adduction, use of a Ferrari KAFO (FKAFO) may be considered.228 The PT must work in conjunction with the orthopedist and orthotist to have each child fitted with the minimal amount of bracing that allows for joint stability and a good gait pattern (see Chapter 34).
TABLE 15-3 
COMMON GAIT PATTERNS AND LEVELS OF ASSISTANCE REQUIRED IN MYELOMENINGOCELE
| LEVEL OF LESION | MUSCLE PERFORMANCE | RECOMMENDED LEVEL OF ASSISTANCE AND BRACING | AMBULATORY PROGRESSION |
| T8-L1 and above | Flaccid LEs with fair to poor trunk | Parapodium: ORLAU, Toronto, RochesterAssistive devices often unnecessary with ORLAU but may improve function with Toronto or Rochester braces. | |
| L1-L2 | Flaccid LEs with hip flexors present | Parapodium with progression to RGORGO, ambulating with hips locked. | Begin ambulating with a walker, progress to forearm crutches.Four-point or swing-through gait. |
| L3-L4 | Fair quadriceps with weak or absent hamstrings | HKAFOs may be used with severe lordosis because of weak or absent gluteal musculature and decreased trunk control or to control rotation and abduction and adduction.If quadriceps are less than fair strength, KAFOs may be needed. As the patient progresses he or she may be cut down from KAFOs to AFOs; AFOs may be used with or without twister cables. |
Begin ambulating with a walker, progress to forearm crutches.Four-point gait. Begin ambulating with a walker and progress to forearm crutches. In some rare cases the patient may progress to no assistive device at all depending on the gait pattern. With increased use of trunk reversal the patient should be returned to forearm crutches to allow for a pattern that is more cosmetic and energy efficient. Four-point gait pattern. |
| L5 | Good hip flexors and quadriceps; fair anterior tibialis; weak gluteus medius and maximus, toe extensors and gastrocsoleus | AFOs with or without twisters depending on gluteal strength.AFO is used to prevent a crouch gait pattern from weak gastrocsoleus. | Forearm crutches or no assistive devices.Four-point gait. |
| S1 | Good hip flexors, quadriceps, gluteus medius, and toe extensors; weak gluteus maximus and gastrocsoleus | AFO | Generally no assistive device is used unless decreased balance reactions or excessive lateral trunk flexion is present. |
| S2-S3 | Good hip flexors, quadriceps, gluteus medius and maximus, and gastrocsoleus | Often no bracing needed. | Often no assistive devices needed. |
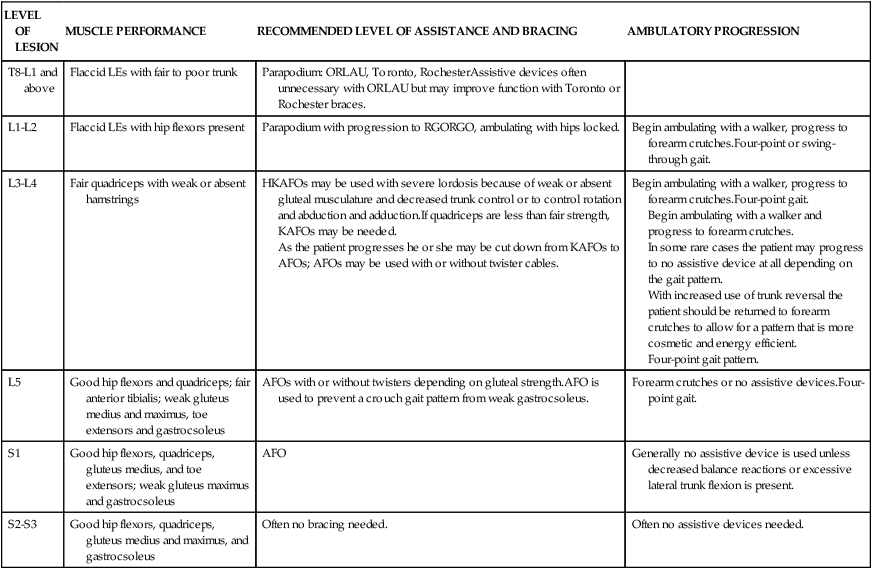

 Crouch control ankle-foot orthosis.
Crouch control ankle-foot orthosis.Children with lower-level lesions (L5-S1) who use below-knee bracing often develop the ability to or choose to ambulate without assistive devices. However, recent studies have shown that crutch use may decrease excessive pelvic motion, which results in reducing abnormal joint forces.162,229 Use of crutches may prevent abnormal joint forces, maintain joint integrity, and decrease the risk of additional orthopedic complications.
Literature has suggested that crutch-assisted ambulation may result in long-term pathology. In patients with higher lumbar lesions (L3-L4) who use Lofstrand crutches, the dynamics and kinematics of upper-extremity function were explored during swing-through and four-point reciprocal modes of gait. Although there were better joint kinematics in the shoulder and other upper-extremity joints during swing-through gait, kinetics were more problematic with increased force and torque in shoulder and wrist joints in those using a swing-through gait. Whereas the swing-through gait allows a potentially faster mode of ambulation, long-term use of this pattern may lead to increased upper-extremity pathology. Careful monitoring of all joints, including upper-extremity joints, during gait reassessment should be considered in order to deter and manage these potential issues that may compromise overall joint integrity and function.230
The excessive femoral torsion present in all newborns at birth does not decrease with growth and development in the child with spina bifida because of abnormal gait and activity.159 Children ambulating with AFOs often show excessive rotation at the knee because of the lack of functioning lateral hamstrings. Rather than going to a higher level of bracing, a twister cable can be added, which often decreases the rotary component during gait.159 Twister cables can be heavy-duty torsion or more flexible elastic webbing, depending on function. Typically, the young child who is just beginning to pull to stand and remains reliant on floor mobility as the primary means of mobility should have elastic twisters prescribed to allow for ease of creeping and transitions. The older and more active child will require heavy-duty torsion cables. Rotational stresses may eventually lead to onset of late degenerative changes around the knee. A tibial derotation osteotomy may be indicated to prevent these changes from occurring.159,231,232
For children with low lumbar or sacral lesions who have at least fair strength in their dorsiflexors and plantar flexors, often a University of California Biomechanics Laboratory (UCBL) or polypropylene shoe insert to control foot position is the only bracing needed. These inserts fit snuggly inside the shoe and help control calcaneal and forefoot instabilities. A supramalleolar orthosis (SMO) will also fit easily inside the shoe but will provide additional medial and lateral support and stability that an insert would not provide. Even though a child may be able to ambulate without an assistive device or bracing, consideration must be given to the stresses that occur at the joints that over time may lead to orthopedic deformity. The greatest risk of joint instability often occurs at the knee. Barefoot walking versus use of an AFO has shown increased instability, joint stress, and pain at the hip and knee as well as increased energy expenditure.233–235 Even though children may be able to ambulate without the use of crutches, comparison of gait kinetics and kinematics of walking with crutches has shown a significant decrease of valgus forces at the knee and better overall alignment of the lower extremities.229 Treatment aimed at strengthening the gluteus medius and maximus to aid in increasing pelvic stability and reducing kinematic compensations can also be important in the management of patients with lesions at this level to enhance their efficiency during ambulation.236,237
Gait training, begun as the child first starts to stand, can now continue in a more formalized manner. By using the appropriate orthosis and assistive devices (walker, crutches, or cane), each child must be helped to achieve the most efficient and effective gait pattern possible (see Table 15-3). As a part of gait training, the child should be taken out of the bracing and “challenged” so that righting and equilibrium reactions can be developed to their maximum. For example, having a child maintain balance while sitting on a ball or other movable surface (tilt board, trampoline) requires the participation of all available musculature, especially abdominal and trunk extensor muscles (Figure 15-20). Strengthening available musculature is a primary objective in this phase of treatment. Slow gains in muscle strength are often the result of continued emphasis on strengthening during physical therapy. In addition to trunk muscles, the gluteus medius, gluteus maximus, and quadriceps are often targeted. Prone activities such as picking up toys while over a Swiss ball or moving around while prone on a scooter board require use of these muscles while providing an enjoyable exercise for the child. Participation in hippotherapy and aquatherapy programs at this age can also be beneficial. These treatment approaches can be used to improve muscle strength in general and the gait pattern when bracing is reapplied. Regardless of the strengthening activities chosen, the pediatric therapist has the special task of using creativity to involve the child in therapeutic play activities. The ideas for creative activities are limitless but essential for combining therapy with age-appropriate cognitive abilities.


Studies examining the effects of strength and endurance training in those with spina bifida238–240 have demonstrated enhanced functional outcomes. These studies included individuals in wheelchairs as well as ambulators. Interventions aimed at strengthening those with spina bifida, including electrical stimulation,241,242 behavioral treatment,243,244 and motor skills training243 have been reported. The evidence supporting strengthening, using exercise, electrical stimulation, and motor skills training was recently examined by Dagenais and colleagues in a systemic review.245 This review synthesized six studies supporting interventions in strengthening.240–246 The level of evidence for the studies was determined using the American Academy of Cerebral Palsy and Developmental Medicine Levels of evidence.247 Levels of evaluated studies were graded and noted to be II (smaller randomized controlled trials with wider confidence intervals, n <100), IV (case series or cohort study without concurrent control, case-control study) or V (case study). The total number of subjects in all of the studies was 26, with 50% of those subjects in the Andrade and colleagues246 study. All six studies reported improvements in strength; however, only three noted statistically significant results. Although the evidence suggests the possibility of being able to increase muscle strength using these modalities, in this population one must interpret the results with caution owing to lack of rigor, small subject populations, and variability across studies.
Studies using dual-emission x-ray absorptiometry (DEXA) have shown that those with spina bifida have significantly decreased lean body mass as compared with controls.248 Children with spina bifida likely have lower metabolic rates secondary to decreased muscle mass and decreased ability to burn calories. Those with higher-level lesions often have greater issues with obesity owing to decreased lower-extremity muscle activity, decreased physical activity, and decreased overall muscle mass. Adult wheelchair users have been shown to require 1500 fewer calories per day, and overall nutritional intake should be lower for those with spina bifida than their typically developing peers.249–251 Interventions to address physical inactivity and obesity may include increased physical activity, regular exercise, behavioral modifications, nutrition education programs, and attention to weight control. Exercise programs targeting upper-extremity resistance combined with aerobic activities such as swimming and arm-cycling using arm-ergometry can be helpful in addressing obesity in those with spina bifida. Adolescents with spina bifida who used an upper-extremity cycling program that was integrated with video gaming three times per week for 16 weeks improved oxygen uptake and maximum work capability.248,252,253
Bladder training usually consists of transferring the job of CIC from the parents to the child. Children as young as 3 years, but certainly by the age of 5 years, can learn CIC in a short period.254 Children may first practice on dolls with male and female genitalia. Next, using mirrors to understand their own genital anatomy, they are able to accomplish the technique on themselves. CIC in conjunction with pharmacotherapy is useful in achieving continence in children with spina bifida.140 Another method of bladder training recently being used in the United States is intravesical transurethral bladder stimulation. This technique has allowed children with neurogenic bladder to rehabilitate their bladder function so that they can detect bladder fullness and generate effective detrusor contractions, leading to improved continence.100,255
Bowel training can be achieved through proper diet, regular evacuation times, and appropriate use of stool softeners and suppositories.100 Constipation (and resulting bypass diarrhea) can be prevented by proper habit training and use of fiber supplements. Stool softeners (not laxatives and enemas) and suppositories should be used to keep the stools soft and help stimulate evacuation. Finally, toilet training, which amounts to scheduled toileting in time with the stool stimulants, usually achieves bowel continence. Surgical procedures, such as the Malone ACE procedure,91 may be necessary when other interventions have failed. The Malone ACE procedure, performed in conjunction with a Mitrofanoff procedure to gain urinary continence, can help these patients attain a better quality of life. Consistency at each step along the way is the key to successful bowel training. A therapist may be called on to assist the parents and child in achieving independence in this ADL.
Mobility and bladder and bowel dysfunction in toddlers and school-age children represent ongoing stressors for parents of children with spina bifida. It has been noted by many that spina bifida represents a considerable challenge to all family members, particularly mothers. Family climate, parents’ partner relationships, and social support networks play a considerable role in balancing stress and psychological adjustment for parents. Awareness of available systems of support for the patient and family as well as resources to which parents and their children can be referred for psychological and social support as needed is important for all health care practitioners.256,257
Stage 5: primary school through adolescence
Emotionally, the 6- to 10-year-old is in a period of relative calm. Children are interested in schoolwork and are eager to produce. This is a period during which they are developing relationships outside of the family and beginning to assume an identity and autonomy. Problem-solving and decision-making skills are at the crux of this time period. However, it is also challenging to promote independence and minimize self-reliant behaviors. Social passivity may ensue as “learned helplessness” behaviors emerge. Therefore professionals, including both PTs and OTs, should begin targeting independent function early, and before adolescence.258 Relevant family education regarding this issue should be inherent in all care plans, and independent behaviors should be promoted from very early on. Engagement in family decision making and opportunities for active problem solving have been linked to increased positive self-esteem and ego development.259 During this time period, children with spina bifida are at risk for developmental delays in social functioning.258
During this period, they are building the skills of the future, preparing them for adult work. This is a prime time to introduce and teach new skills while fostering increased autonomy and independence. Autonomy is difficult for the youngster with spina bifida. Motor skills impeding progress toward autonomy vary for each child and are dependent on the level of spina bifida as well as any cognitive impairment. However, it has been observed that the motor skills hardest to attain are those that involve motor planning and that the process skills hardest to attain are related to adaptation of performance and initiation of new steps. Thus guidance to learn not only how to do things but how to get things done is important.260 Interventions targeting independence have been embedded into several camp programs throughout the country geared specifically toward those with spina bifida. O’Mahar and colleagues261 report on one such camp program focused on campers 7 to 14 years of age in northern Illinois. Camp Ability emphasized individualized collaborative (i.e., parent and camper) goal setting, group sessions with psychoeducation and cognitive tools, and goal monitoring by the camp counselor. Campers reported significant gains in individual goals, management of spina bifida responsibilities, and independence. Medium effect sizes in goal attainment and progress from the start to the end of the camp session were noted.261 It appears that these camps may have significant benefit in addressing management of one’s disability as well as independence in self-care skills. An added benefit is the social interaction and physical activity that the camp participants engaged in while working toward their goals.
Usually during this stage, if it has not occurred previously, the evaluation of future ambulation potential occurs. The child whose larger size and limited abilities make ambulation more difficult each day frequently requests this evaluation. Strength does not increase in the same proportion as body weight.127 Ambulation, although possible for the young child, may be impossible for that same person as a young adult.
Although no guidelines include every patient, generally children with thoracic-level lesions are rarely ambulators by the late teens.152,262,263 Those with upper lumbar lesions may be household ambulators with long-leg bracing but will require wheelchairs for quick and efficient mobility as adults. With low lumbar lesions, most adults can become community ambulators. Patients with sacral-level lesions are usually able to ambulate freely within the community. Many require minimal bracing and ambulate without assistive devices.152,159 It must be remembered that ambulatory status is not determined by level of the lesion alone. The muscle power available; degree of orthopedic deformity; age, height, and weight of the patient; and, of course, motivation are also determining factors.82,154,159,264
Generally, in late childhood or early adolescence, orthopedic deformities that have been gradually developing require surgical intervention. Progressive scoliosis or kyphosis may require internal fixation when conservative methods fail.265 Sectioning of tight or contracted muscles at the hip and knee is often required.152 The iliopsoas, adductors, and hamstrings are frequently the offending muscles. These surgeries, followed by strengthening exercises and gait training, often add to the ambulatory life of the child with spina bifida. For example, in a child who displays an extreme lordotic posture, hip flexion contractures may be present and surgical lengthening of the tight muscles may be required to allow improved biomechanical alignment for standing and balance. Strengthening of the hip extensors and abdominals also helps prevent future muscle imbalances that may lead to contractures and tightness. A postoperative therapeutic program might include periods of prone lying to prevent future contractures and strengthening of hip extensors and abdominals that were previously overstretched by the lordotic position.
Of primary importance during this stage is preparing the child for independence in ADLs, which may be broken down into self-care, locomotion-related, and social interaction activities. In conjunction with the nurse, PT, and OT, self-care skills of dressing, eating, and food preparation; general hygiene; and bowel and bladder care can be addressed. Because the adolescent is so concerned with achieving independence, he or she is more likely to comply with a regimen of strengthening exercises if shown how they relate to functional independence. A creative therapist may, for example, incorporate trunk stability and upper-extremity strengthening work in activities such as making popcorn or getting ready for a dance. In addition, fostering social and recreational independence through adaptive sports and fitness programs and leisure activities should not be overlooked. Participation in adaptive sports can aid immensely in improving strength, endurance, and self-esteem. Community adaptive recreational programs may include T-ball, martial arts, swimming, tennis, basketball, skiing, bowling, and many other common sports and leisure activities (Figures 15-21 through 15-23).






Achievement of independence in ADLs for the child and adult with spina bifida does not depend solely on the level of paralysis. Also important are psychosocial and environmental factors. Mean ages for the achievement of various ADL activities have been developed and may assist the therapist in establishing realistic therapeutic goals in this area.266
Also as previously discussed, children with spina bifida have a general perceptual deficit that can manifest itself in a variety of ways. First, the child may have difficulty recognizing objects and the relations that they have to one another. He or she may therefore perceive the world in a distorted manner and have reactions that are unstable and unpredictable. These perceptual difficulties will most likely affect academic learning, and the child may associate failure with the learning process. Difficulties in attaining independence in ADLs are also linked to perceptual problems. Finally, emotional disturbances may be attributed in part to the perceptual difficulties of the child with spina bifida.98
Remedial programs, such as the Frostig Program for the Development of Visual Perception, have been effective in improving the visual perception of children with spina bifida.98 Programs of this type are most effective when remediation begins early—preferably at or before the time the child enters school. Developmental optometry examination and remediation programs focusing on vision training may also be of benefit.
Children with spina bifida may also have difficulties with tasks requiring sensorimotor integration. Children requiring programs for sensorimotor integration should be referred to a therapist certified in this area. If one is not available, many appropriate activities for sensorimotor integration may be adapted from Ayres267 or Montgomery and Richter.268