Specific Infections with Critical Care Implications
OVERWHELMING INFECTIONS OF THE CENTRAL NERVOUS SYSTEM
FULMINANT ENDOVASCULAR INFECTIONS
Staphylococcus aureus Bacteremia
Staphylococcal Toxic Shock Syndrome
Streptococcal Toxic Shock Syndrome
SERIOUS SKIN AND SKIN STRUCTURE INFECTIONS
SERIOUS GASTROINTESTINAL AND INTRA-ABDOMINAL INFECTIONS
LIFE-THREATENING INFECTIONS OF THE HEAD AND NECK
Overwhelming Infections of the Central Nervous System
Acute Bacterial Meningitis
Epidemiology, Pathogenesis, Risk Factors, and Clinical Presentation
The annual incidence of bacterial meningitis in individuals older than 16 years of age in developed countries has been steadily decreasing and was estimated at less than 2 cases per 100,000 in the United States in 2006-2007.1–3 In recent studies from the United States and the Netherlands, the predominant organisms in microbiologically confirmed cases of bacterial meningitis in adolescents and adults were Streptococcus pneumoniae (45-60%), Neisseria meningitidis (15-30%), Haemophilus influenzae (7%), and Listeria monocytogenes (5%),2–4 with no pathogen identified in approximately 10% of cases.1,2 Streptococcus agalactiae (group B β-hemolytic streptococcus) has also recently emerged as an important cause of meningitis in adults, causing over 5% of cases of microbiologically defined episodes in the United States from 1998 to 2007. In some studies, rates of culture-negative cases were as high as 25%.5 Important recent changes in microbiologic causes of bacterial meningitis reflect the impact of current vaccination strategies. These changes include the marked reduction in cases of of H. influenzae meningitis in both children and adults, and the introduction of the conjugated pediatric pneumococcal vaccine that has resulted in decreased rates of invasive childhood pneumococcal disease in children and adults.3,6 A new meningococcal vaccine, which has the potential to diminish the rates of meningococcal disease in high-risk populations, has also been approved for use in adolescents and high-risk adult populations.7 Another important trend is the increase in prevalence of nosocomial meningitis.4 The microbiology of nosocomial meningitis differs from that of community-acquired cases, including higher rates of staphylococcal infection and infection due to a variety of aerobic gram-negative organisms.
The major route of acquisition of bacterial meningitis follows colonization of the nasopharynx with subsequent hematogenous spread and invasion of cerebrospinal fluid (CSF). Less frequently, infection occurs from hematogenous dissemination from distant sites or from other localized intracranial focal infections including sinusitis, mastoiditis, or otitis, or it is secondary to trauma or neurosurgery. In addition to trauma and contiguous focal infectious processes facilitating invasion of the CSF by bacteria, there is a wide variety of immunologic deficits that result in impaired clearance of encapsulated organisms. These include organism-specific deficits such as terminal complement deficiencies predisposing to meningococcal disease, as well as more general deficits such as immunoglobulin deficiencies, splenectomy, alcoholism, cirrhosis, diabetes mellitus, and human immunodeficiency virus (HIV) infection.2,4 Patients with defects leading to impaired cell-mediated immunity including advanced age and general debility, as well as hematologic malignancies, chemotherapy, and use of tumor necrosis factor (TNF)-β inhibitors, are predisposed to Listeria infection.8
The presenting symptoms of bacterial meningitis include fever, headache, stiff neck, and altered sensorium. In the recent large series of 696 cases from the Netherlands, 95% had at least two of these four symptoms, although only 44% had all of the classic triad of headache, fever, and stiff neck.2 Other important presenting symptoms in this cohort included nausea in 74%, focal neurologic deficits in one third of cases, and Glasgow Coma Score of less than 8 in 14% of cases.2 Rash, especially a petechial or purpural rash that may be an important clue for the diagnosis of meningococcal meningitis, was seen in 26%.2 Presenting symptoms alone without CSF findings or microbiologic data cannot adequately distinguish between bacterial meningitis and viral or other aseptic meningitis. However, certain features are more suggestive of bacterial rather than viral origin including winter versus summer onset, rapid progression of disease, presentation in shock, and presence of another focal site of bacterial infection such as sinusitis, otitis, or pneumonia. In addition to viruses, other nonbacterial infections including cryptococcosis, tuberculous meningitis, rickettsial diseases, Lyme disease, and syphilis are in the differential diagnoses of acute bacterial meningitis.
Diagnostic Strategies and Early Management of Suspected Bacterial Meningitis
The early management of suspected bacterial meningitis requires careful coordination and appropriate sequencing of the procedures necessary for establishing the diagnosis (lumbar puncture and imaging studies) and the interventions necessary for optimal treatment (antibiotics and dexamethasone). Rapid initiation of therapy leads to improved outcomes but may decrease specific microbiologic yield on CSF analysis.1,5 Similarly, although lumbar puncture can usually be performed safely without any imaging studies, computed tomography (CT) scan may be required to minimize the risk of this procedure, leading to potential delays in institution of antimicrobial therapy.9 Recent reviews and published practice guidelines have tried to place these competing urgencies in perspective, using data culled from multiple recent prospective studies and randomized trials.1,5 One algorithm for early management of bacterial meningitis is shown in Figure 54.1.
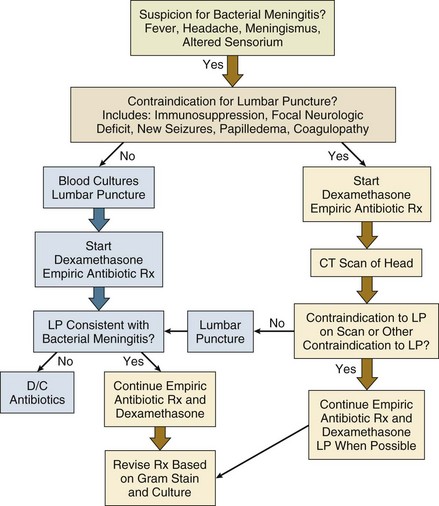
The primary tenet of these algorithms is that the initiation of treatment assumes highest priority and thus any delay in performing a lumbar puncture because of the need for imaging or because of other patient-specific contraindications should not delay the administration of antibiotic therapy. Brain herniation is a feared but rare complication of lumbar puncture when performed for diagnosis of suspected meningitis in patients with elevated intracranial pressure.10 One recent study of a cohort of 301 patients with suspected bacterial meningitis described the relative safety of lumbar puncture without CT scanning in patients without specific clinical contraindications.9 Proposed criteria for performing imaging prior to lumbar puncture include new-onset seizures, prior CNS disease, immunocompromised state, papilledema, focal neurologic deficits, or moderate to severe impairment of consciousness.5 Only approximately 45% of patients with bacterial meningitis will have criteria for neuroimaging prior to lumbar puncture, although it remains standard practice in many hospitals for all patients with suspected meningitis to undergo imaging first.1,5 The main purpose of early imaging is to find evidence of brain shift and both noncontrast CT and magnetic resonance imaging (MRI) can be used for this purpose. Considerations for the optimal imaging modality may be different when imaging tests are done to manage subsequent complications of meningitis or to better define space-occupying lesions. Other specific contraindications to lumbar puncture include coagulopathies and presence of local processeses overlying the lumbar puncture site such as stasis ulcers, burns, or cellulitis.
The diagnosis of bacterial meningitis relies heavily on analysis of CSF parameters including opening pressure, cell count, protein, glucose, and Gram stain and culture. Typically, patients with bacterial meningitis have elevated opening pressures of 200 to 500 mm H2O, including 40% with opening pressure greater than 400 mm in one recent cohort.2,5 White blood cell (WBC) counts may range from 100 to 10,000 cells/mm3, most commonly in the 1000 to 5000 range; very low CSF WBC counts are associated with a worse prognosis.4,5 Usually there is a polymorphonuclear (PMN) cell predominance of 80% or greater, although up to 10% will have a lymphocytic predominance, particularly early on. CSF-to-serum glucose ratios are less than 0.4, and CSF protein levels are nearly always increased.5 In a study comparing cohorts of patients with bacterial and viral meningitis, CSF glucose ratios of less than 0.31, total WBC counts of greater than 2000, and total PMN cell counts of greater than 1180 have been predictive of bacterial rather than viral meningitis.11 Other studies have suggested that protein values of greater than 0.5 g/L and WBC counts greater than 100 are also independently predictive of bacterial meningitis.12 Gram stains are positive in 60% to 90% of cases of bacterial meningitis, and results on Gram stain are reported to be 97% specific as to cause.5 Yield of Gram stain is higher on specimens concentrated by Cytospin centrifuge. Highest diagnostic yield from Gram staining is for S. pneumoniae meningitis; Gram stains in Listeria meningitis are positive in only one third of cases because of the lower inoculum of bacteria in the CSF. Clinicians should also be aware that preliminary “stat” Gram stains done during off-hours are more likely to be misinterpreted; thus, stains should always be reviewed by trained clinical microbiologists before modifying therapy based on a Gram stain report. In untreated patients, cultures will ultimately be positive in up to 90% of cases. Initiation of antibiotic therapy prior to lumbar puncture will not significantly alter cell count, protein, glucose, and even Gram stain results but will decrease CSF culture yield by up to 20%.5,12 Blood cultures should be performed in all patients with suspected bacterial meningitis prior to initiation of antibiotics, even if the lumbar puncture is delayed.
Additional CSF and blood tests have been used to confirm a diagnosis of bacterial meningitis or help distinguish bacterial from viral disease. Latex agglutination tests for bacterial antigens, although initially reported to have good sensitivity and specificity for diagnosis of specific bacterial meningitis pathogens, have more recently been shown to contribute little to the management of most patients with suspected meningitis.12 Polymerase chain reaction (PCR) of CSF for bacterial deoxyribonucleic acid (DNA) may be useful to confirm an etiologic diagnosis in culture-negative cases, especially those with prior antibiotic therapy, but is not routinely available in most hospitals. Elevated serum C-reactive protein (CRP) levels (>20 mg/L) and elevated serum procalcitonin (>0.5 ng/mL) are not specific for bacterial meningitis but had high predictive value in distinguishing between cohorts of children with bacterial and viral meningitis.12 The level of soluble triggering receptor on myeloid cells in CSF has also been reported to distinguish between bacterial and viral meningitis.12 Additional CSF studies may be useful for diagnosis of other specific infections such as CSF cryptococcal antigen and Venereal Disease Research Laboratory (VDRL) slide test.
Initial antibiotic therapy for suspected bacterial meningitis is most commonly initiated in the absence of culture and even Gram stain data. Initial empiric therapy must include agents active against the most likely pathogens based on the patient’s age and underlying illnesses and modified for other specific risk factors such as nosocomial acquisition (Table 54.1). Treatment regimens must take into account local rates of antimicrobial resistance, particularly rates of high-level resistance to penicillin and third-generation cephalosporins in S. pneumoniae. Standard regimens include vancomycin administered at doses targeted to achieve adequate CSF levels (serum troughs of 15 to 20 µg/mL) and a third-generation cephalosporin, either ceftriaxone or cefotaxime. In areas with increased rates of resistance to third-generation cephalosporins, rifampin may be added. When there is risk for Listeria infection based on age greater than 50 or on other specific risk factors such as alcoholism or altered immunity, high-dose ampicillin is included in the treatment regimen. For patients with nosocomial or postprocedure-related infections, cefotaxime or ceftriaxone may be changed to an agent with improved activity against nosocomial gram-negative organisms including Pseudomonas aeruginosa. Initial empiric therapy is modified on the basis of identification and susceptibility data of isolated pathogens. Standard recommended treatment durations are 7 days for meningococcal disease and H. influenzae, 10 to 14 days for S. pneumoniae, and 21 or more days for Listeria.1,5
Table 54.1
Treatment of Bacterial Meningitis in Adults*
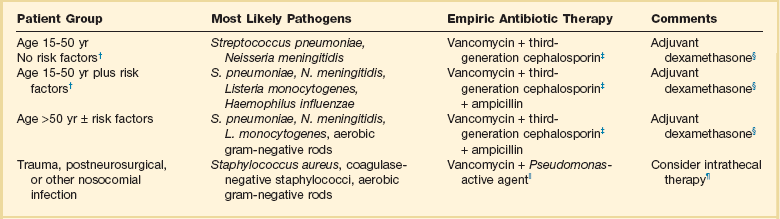
*Patients older than 15 years of age.
†Risk factors: altered immunity including human immunodeficiency virus (HIV) infection and alcoholism.
‡Third-generation cephalosporin: ceftriaxone or cefotaxime; for highly penicillin-allergic patients an alternative would be aztreonam or a fluoroquinolone.
§Evidence for benefit of adjuvant dexamethasone is limited. Consider addition of rifampin in settings with high rates of cephalosporin resistance.
||Includes cefepime or ceftazidime; alternatives include aztreonam, meropenem, and ciprofloxacin.
¶For multiresistant organisms or catheter-related infections. Most recent experience is with aminoglycosides or polymyxins.
Modified with permission from van de Beek D, de Gans J, Tunkel AR, et al: Community-acquired bacterial meningitis in adults. N Engl J Med 2006;354:44-53; and Tunkel AR, Hartman BJ, Kaplan SL, et al: Practice guidelines for the management of bacterial meningitis. Clin Infect Dis 2004;39:1267-1284.
Studies in high-income countries have demonstrated the benefits of adjuvant dexamethasone in decreasing mortality rates and neurologic sequelae in patients with bacterial meningitis, and adjuvant steroids are now part of meningitis practice guidelines in the United States and Europe.5,13,14 In one large, randomized, placebo-controlled trial of 301 adults with suspected bacterial meningitis and cloudy CSF, adjuvant dexamethasone decreased mortality rates from 15% to 7% and decreased unfavorable outcomes from 25% to 15%.13 Benefits of corticosteroids were most evident in those with pneumococcal infection and those with a moderately impaired level of consciousness; patients with meningococcal disease had generally better outcomes with or without corticosteroids. More recent trials from resource-limited settings including Malawi and Vietnam have failed to confirm the benefits of adjuvant steroids, even in those with documented pneumococcal infection, calling into question the generalizability of use of steroids in all settings.12,14 A recent meta-analysis was unable to clearly demonstrate benefits or harm with use of adjuvant corticosteroids.14 If steroids are to be used, the standard regimen based on published and animal studies is 10 mg of dexamethasone every 6 hours for 4 days and initiated prior to or concurrent with the first dose of antibiotics.1,5 Any benefit of steroids initiated after onset of antibiotic therapy is unknown. Patients with suspected or confirmed meningococcal meningitis should be placed in isolation to prevent nosocomial transmission via respiratory droplets for the first 24 hours of therapy. Meningococcal prophylaxis is indicated for household and other close contacts of meningococcal cases including first responders. Conversely, patients with Gram stains and clinical presentations consistent with nonmeningococcal disease should also be quickly identified, to eliminate unnecessary use of meningococcal prophylaxis.
Complications, ICU Monitoring, and Prognosis
Patients with bacterial meningitis often require ICU monitoring either for complications evident at the time of presentation or for observation for complications that may subsequently develop during their course. Criteria for admission to an ICU have been proposed (Table 54.2)1 and include sepsis and septic shock, respiratory compromise or pulmonary infiltrates, those with high risk of brain herniation, moderately impaired and deteriorating level of consciousness, new or evolving neurologic deficits, and presence of seizures. Some authorities feel that all patients with bacterial meningitis may benefit from intensive monitoring early on for presence of new neurologic signs or subtle seizures and for effective control of agitation.15 Patients with generally good outcomes can be defined early in their course on the basis of age, Glasgow Coma Score, APACHE II score, absence of focal neurologic abnormalities, and favorable laboratory features, but these stratification schemes will still fail to identify some patients with more complicated courses.2,16
Table 54.2
Indications for Intensive Care Unit (ICU) Monitoring and Management for Bacterial Meningitis
| Indication | ICU Management |
| Sepsis and shock | Hemodynamic support and monitoring, early goal-directed therapy |
| Pulmonary infiltrates or respiratory compromise | Airway management, monitoring of oxygenation, non-invasive or invasive ventilatory support |
| Glasgow Coma Scale score <10 | Monitoring of examination; monitor for development of increased ICP, hydrocephalus, and seizures |
| Deteriorating level of consciousness | Monitoring of examination; monitor for development of increased ICP, hydrocephalus, and seizures |
| New or evolving neurologic deficits | Monitoring of examination; monitor for development of increased ICP, hydrocephalus, and seizures |
| Evidence of increased ICP | Consider intracranial pressure monitoring; osmotic diuretics |
| Acute hydrocephalus | Repeated lumbar puncture, lumbar drain, or ventriculostomy |
| Seizures | Continuous EEG monitoring, antiepileptic agents |
| Severe agitation | Careful sedation |
| Hyperglycemia | Maintain normoglycemic state with insulin therapy |
| Hyperpyrexia | Cooling modalities for fever >40° C |
EEG, electroencephalogram; ICP, intracranial pressure.
Modified with permission from van de Beek D, de Gans J, Tunkel AR, et al: Community-acquired bacterial meningitis in adults. N Engl J Med 2006;354:44-53.
Major complications that occur during the course of bacterial meningitis have been summarized by van de Beek and colleagues1,2,12 and include deteriorating level of consciousness, which may be caused by development of meningoencephalitis (15% to 20% of cases), seizures (15% to 23%), brain edema (6% to 10%), and hydrocephalus (3% to 8%). Mannitol, hyperventilation, repeated lumbar punctures, prophylactic lumbar drainage, prophylactic ventriculostomy, and other modalities have all been proposed for management of increasing intracranial pressure. Increased pressure is associated with worse prognosis, but no single approach for treatment has been confirmed to be effective in all cases.1,17 Patients with severely increased pressure with impending herniation may require ventriculostomy with continuous CSF pressure monitoring. All patients should be monitored for seizures including nonfocal seizures that may only manifest as worsening level of consciousness, but routine use of prophylactic antiepileptic medication is not currently recommended.1,5 Focal neurologic deficits early in the course of meningitis are most often caused by cerebrovascular complications or arteritis secondary to inflammation or less commonly to venous infarcts. The most common late neurologic complication is hearing loss, described in up to 20% of patients overall and in up to 34% of those with pneumococcal meningitis.1,2 The incidence of hearing loss may be decreased in dexamethasone-treated patients.13 Subdural empyema, brain abscess, and hemorrhage are all rare but potentially catastrophic complications of bacterial meningitis.
Repeat lumbar punctures are no longer routinely performed in patients with bacterial meningitis who are improving but are indicated in patients with worsening or suboptimal clinical response at 48 hours.5 Repeat CSF analysis is also indicated in patients with more resistant organisms such as highly cephalosporin-resistant S. pneumoniae, especially when treated with dexamethasone, because steroids may decrease antibiotic penetration into the CSF and delay sterilization.18 Repeat imaging by MRI or enhanced CT scan should be performed in patients with clinical deterioration or persistent decreased level of consciousness to assess for the complications listed earlier.
Overall mortality rates for bacterial meningitis are reported to be approximately 20% but vary by organism, and an additional 14% will have moderate or major neurologic sequelae.1 Prognosis for meningococcal meningitis is better than for pneumococcal infection. Late neuropsychiatric cognitive effects are seen in up to 10% of cases of bacterial meningitis.
Encephalitis
The other major CNS infectious syndrome besides bacterial meningitis that may present with fever, headache, altered sensorium, and meningeal signs is acute encephalitis, which is most commonly viral. Although there is significant overlap between clinical presentations of patients with bacterial meningitis and viral encephalitis, the presentation of viral encephalitis is dominated by evidence of parenchymal brain dysfunction including moderate to severely impaired sensorium, delirium, psychosis, focal neurologic findings, and seizures.19,20 The presence of meningismus is more variable, but most patients will have some evidence of meningeal enhancement on imaging and inflammatory cells in CSF.
A large number of viruses can cause encephalitis, but management strategies focus on the most common causes and those most likely to respond to specific interventions, particularly encephalitis caused by herpes simplex virus (HSV).19–22 Other important causes with worldwide distribution include other herpesviruses including varicella-zoster virus; Epstein-Barr virus; and cytomegalovirus (CMV), especially in immunocompromised patients.19,20,21 Influenza virus, mumps, measles, and enteroviruses such as enterovirus 71 are also important causes of acute viral encephalitis worldwide. Acute HIV infection can also occasionally present as a potentially treatable cause of fulminant meningoencephalitis.21 Acute presentations of progressive multifocal leukoencephaly caused by JC virus are also increasingly reported in HIV-infected and other immunocompromised patients.23 In addition to those viruses seen worldwide, there are a variety of vector-borne viruses with regional and seasonal distributions.19,21 Important pathogens include eastern, western, and Venezuelan equine encephalitides; Japanese B encephalitis; St. Louis encephalitis; La Crosse encephalitis; and Nipah encephalomyelitis. Recently, West Nile encephalitis emerged as a major pathogen in the United States. West Nile first appeared in New York City in 1999, but cases are now distributed throughout the country, demonstrating the potentially unpredictable and dramatic spread of emerging vector-borne pathogens.24,25 Rabies, although extremely rare in developed countries, also needs to be considered in the differential diagnosis of fulminant encephalitis, both for its epidemiologic implications and because of recent reports of survival for patients with this previously universally fatal infection with aggressive ICU management protocols.26 In addition to viral infections, many atypical bacteria, fungi, and protozoal organisms can present as acute meningoencephalitis.19 Particularly important to consider early in the differential diagnosis are infections that may respond to specific antimicrobial therapy including rickettsial diseases such as Rocky Mountain spotted fever (RMSF) and meningoencephalitis from spirochetal infections including neurosyphilis and Lyme disease.27,28 Important clues to these diseases include epidemiologic history and associated clinical features such as rash. Data from the California Encephalitis Project, in which cases of suspected infectious encephalitis underwent an aggressive evaluation for a variety of infectious agents, suggest that the cause of at least 50% to 60% of cases of presumed infectious encephalitis remains undefined, and in 10% a noninfectious diagnosis was ultimately established.29
Herpes simplex encephalitis is the most common sporadic cause of serious viral meningoencephalitis, causing 3% to 10% of cases of infectious encephalitis in adults and with an estimated frequency of about 1000 to 2000 cases per year in the United States.21,22,29 Ninety percent of cases are caused by HSV type 1, usually occurring in adults as reactivation disease and not primary infection; HSV type 2 commonly causes aseptic meningitis but much less commonly presents as fulminant encephalitis.22,30 Herpes encephalitis is an acute necrotizing process, typically causing hemorrhagic necrosis with particular predilection for the frontotemporal lobes and cingulated gyrus. Mortality rates are up to 70% in untreated cases, with 95% of those surviving having serious neurologic sequelae.21,22 Cases of herpes encephalitis beyond the neonatal period occur throughout life, but the highest case rates occur in younger individuals (younger than 20 years old) and in adults older than age 50 (half of cases). Presentation may include a prodromal viral syndrome in 50% of cases, followed by a generally rapid onset of headache, confusion, and altered level of consciousness. Meningismus, focal neurologic findings, and seizures are also common.20–22 In one series of patients with encephalitis, of which 37% were caused by HSV, focal CNS disease was much more likely than diffuse CNS disease to be caused by HSV.31 The approach to cases of suspected herpes encephalitis, as well as other viral or unknown encephalitides, includes early initiation of antiviral therapy and concurrent rapid clinical evaluation that includes imaging, CSF studies, and electroencephalogram (EEG) to establish a diagnosis.19,20,22 MRI is considered the imaging modality of choice. MRI is positive in more than 90% of cases, often showing changes of edema and necrosis in the medial temporal lobes, insular cortex, and cingulate gyrus as soon as 2 to 3 days into the illness.32 EEGs will be abnormal in nearly all cases, but early findings may be nonspecific and the characteristic periodic lateralizing epileptiform discharges may not be seen until later in the course.22 CSF findings include low- to moderate-grade lymphocytic pleocytosis in 85% and elevated protein in 80%; elevated red blood cell count and mildly decreased glucose are common but not universal, and cell counts may be completely normal in 8%.22,30 Likelihood of HSV encephalitis is extremely low with CSF WBC count of fewer than 5 cells/mm3 and protein levels of less than 50 mg/dL.33 The International Herpes Management Forum has issued guidelines for confirmation of the diagnosis and for treatment.22 PCR of CSF for HSV DNA has replaced brain biopsy as the diagnostic method of choice for HSV encephalitis. Based on data from two trials comparing PCR with the gold standard of brain biopsy, sensitivity of PCR was 98% and specificity was 94% for diagnosing HSV encephalitis; the negative predictive value of PCR on CSF obtained more than 72 hours into the course of illness was close to 100% for excluding HSV.22,34,35 PCR has also expanded understanding of the range of clinical presentations of HSV encephalitis including recognition of less severe cases.31 Viral culture has low sensitivity in older children and adults and is not recommended, and CSF and serum antibody studies have little role in the diagnosis.19,22 Treatment should be initiated on the basis of clinical suspicion pending PCR results using high-dose intravenous (IV) acyclovir at doses of 10 mg/kg every 8 hours and continued for 14 to 21 days, or until PCR results are available or an alternative diagnosis is established. Extrapolating from results in neonates, a positive CSF PCR for HSV at the end of treatment may be an indication to prolong therapy, especially if clinical response has been suboptimal.19
West Nile Virus Encephalitis
In the 12 years since it first appeared in the Western hemisphere, West Nile virus (WNV) has become a major infectious cause of invasive neurologic disease throughout the United States, with more than 1000 cases from 40 states reported in 2010.24,36 WNV is a mosquito-borne arbovirus affecting birds, mammals, and humans. Of human cases, 80% are asymptomatic and most of the remainder have West Nile fever, but 1% will develop neuroinvasive disease manifesting as meningitis, encephalitis, or a polio-like flaccid paralysis syndrome.24,25 Risk of encephalitis increases with age or with immunosuppression, particularly organ transplantation, and manifestations can range from mild disorientation to coma and death.24 Parkinson-like tremors are also commonly reported. The flaccid paralysis syndrome, caused by viral infection of anterior horn cells, may have abrupt onset and patients may require prolonged ventilatory support. The fatality rate is 9% for WNV neuroinvasive syndromes, and survivors may have prolonged symptoms. In some patients, flaccid paralysis has not resolved after 1 to 2 years.24 CSF findings of WNV are similar to those of other viral infections with lymphocytic pleocytosis predominating. Neuroimaging may be normal, but some scans may demonstrate abnormal signal in the basal ganglia, thalamus, and other deep brain structures or abnormal signal in anterior horn cells of the spinal cord.25,37 Diagnosis is confirmed by the finding of WNV-specific IgM antibodies in CSF and serum or by nucleic acid–based PCR of CSF, serum, or other fluids.19,24 Treatment is primarily supportive; multiple interventions including ribavirin, interferon (INF)-α, immunoglobulin, and other agents have been reported in individual cases and case series, but there are currently no good outcome data to support the use of any specific therapy.
Brain Abscess
Brain abscesses are focal infections of brain parenchyma, occurring from direct spread from local, contiguous infection (ear, sinuses, mastoid, bacterial meningitis); from hematogenous spread of infecting organisms from distant sites; or by direct inoculation from trauma or surgery.38,39 Modern imaging modalities have improved the initial management and follow-up care of patients with brain abscess, but it is less certain that this has significantly affected overall outcome. Presenting symptoms may be indistinguishable from those of other CNS infections, with fever, headache, and focal neurologic findings predominating.38–40 Seizures and decreased level of consciousness are also seen. Sepsis was noted in 18% of cases in one recent series.41 Duration of symptoms prior to presentation is typically much longer than that of bacterial meningitis or viral encephalitis. Microbiology reflects the source of the original infection and most commonly includes streptococci, staphylococci, oral anaerobes, and enteric gram-negative organisms, but multiple other bacterial, fungal, tuberculous, and atypical organisms can also be found.38–40 In areas of high HIV prevalence, opportunistic brain infections, especially toxoplasmosis, have become more prevalent than typical pyogenic abscesses, and knowledge of HIV status is crucial to initial management of suspected infectious mass lesions.42
The diagnosis of brain abscess is suggested by the clinical presentation and imaging findings by either CT or MRI showing enhancing parenchymal lesions, usually with surrounding inflammatory changes.38,39 Radiographic appearance may be indistinguishable from a malignant lesion. Management of suspected abscess includes either stereotactic aspiration or surgical drainage to both establish a microbiologic diagnosis and for primary treatment of the abscess. Smaller lesions (<1 cm) can be treated with antibiotics alone, but larger lesions will require either aspiration or open drainage.38,39 Recent studies have confirmed earlier findings that aspiration may result in lower mortality rates and less residual neurologic deficit than surgical resection.43 Duration of treatment is until resolution of the abscess on serial imaging studies. Unlike bacterial brain abscesses, toxoplasmosis lesions in patients with AIDS are usually multiple and may respond to empiric toxoplasmosis therapy without need for further invasive diagnostic procedures.42
Spinal Epidural Abscess
Spinal epidural infections most commonly arise by contiguous spread of infection into the spinal canal as a complication of vertebral infections or, less commonly, as a complication of epidural procedures such as epidural injections or epidural catheterization.44–46 Vertebral infections result from hematogenous infection of vertebral bodies or disk space or by direct introduction of organisms from surgery or trauma. In addition to affecting the vertebral bodies and intervertebral disk spaces, vertebral osteomyelitis may expand into surrounding paravertebral soft tissues and spinal canal and even rarely into the medullary spinal cord. Paravertebral and epidural infections can rapidly progress, resulting in severe neurologic symptoms from compression of nerve roots or the spinal cord, or by infarction of the spinal cord from inflammation and vasculitis of vertebral arteries.45,47 Thus an aggressive diagnostic and therapeutic approach is necessary to prevent catastrophic consequences such as irreversible paraplegia and death.
The incidence of spinal epidural infection from large retrospective reviews is reported as approximately 0.2 to 2 cases per 10,000 hospital admissions, but the frequency of this diagnosis may be increasing, perhaps because of improvements in radiographic diagnosis and increasing prevalence of risk factors.45 Incidence in adults increases with age older than 30 years; important predisposing factors include diabetes, injection drug use, alcoholism, immunosuppression, underlying spinal disorders, trauma, invasive procedures, and extraspinal sites of infection that cause significant bacteremia.45,46 Infections can occur at any level of the spinal cord, although most series report predominance of lumbar involvement. Approximately two thirds of infections are caused by Staphylococcus aureus including methicillin-resistant S. aureus (MRSA) with a variety of other gram-positive organisms (coagulase-negative staphylococci, viridans streptococci, β-hemolytic streptococci), as well as gram-negative organisms all reported in lesser numbers.44–46 Granulomatous infections including tuberculosis and brucellosis remain major causes of vertebral and epidural infection in many areas of the world.47 Approximately 10% of episodes are culture-negative.45
Presenting symptoms are extremely nonspecific and include back pain in more than 95% of cases and fever in two thirds of cases.44,45 Thirty percent of cases will present with evidence of early neurologic deficits such as muscle weakness, incontinence, or sensory deficits, and up to one third will have evidence of some paralysis on examination.44–46 Associated laboratory features include leukocytosis in two thirds of cases and elevated erythrocyte sedimentation rate (ESR) in nearly all cases. The most sensitive imaging modality is MRI with gadolinium, with reported sensitivity of greater than 95%, but most infections were also visualized on CT and unenhanced MRI.44,45 Standard therapy previously included emergent surgical drainage of the abscess and systemic antibiotic therapy. However, over the past 2 decades there has been an emerging literature on more conservative medical management employing percutaneous aspiration and antibiotics without open surgical drainage.44–46 These reports have suggested that outcomes are similar or better (i.e., less neurologic sequelae) compared to surgery.44 No trials have directly compared treatment modalities, and retrospective analyses have not identified risk factors for worse outcomes with medical therapy alone. Indications for surgery include neurologic progression on treatment or lack of clinical response, thus those managed without surgery require careful monitoring.44,45 Surgery may also be the preferred treatment for those with more severe neurologic deficits on initial presentation, though these patients have worse outcomes overall regardless of treatment modality.44,45
Fulminant Endovascular Infections
Acute Infective Endocarditis
Several large case series published since 2005 have addressed changes in the epidemiology of bacterial endocarditis over the past 2 decades.48,49 The annual incidence of endocarditis of 5 to 7 cases per 100,000 has not changed significantly over time.48 However, the International Collaboration on Endocarditis Prospective Cohort Study, a multicenter study of 1779 cases from 39 medical centers in 16 countries, has illustrated the recent changes in microbiology and clinical presentation of bacterial endocarditis.49 In this cohort, as well as other recent cohorts, S. aureus rather than viridans streptococci were the predominant pathogens and increasing numbers of cases were considered either nosocomially acquired or health care associated.49 S. aureus endocarditis is associated with a significantly higher mortality rate, higher rate of stroke and other systemic embolization, and higher rate of sustained bacteremia than endocarditis caused by other organisms.49–51 The increased antibiotic resistance in organisms causing endocarditis, especially the increasing rates of MRSA, are reflected in the recent guidelines for diagnosis, treatment, and management of complications of endocarditis from the American Heart Association.50 Other important recent epidemiologic trends are the decreased importance of congenital valvular and rheumatic disease as predispositions, the increasing age of cases, and the increased role of hemodialysis and indwelling cardiac devices such as pacemakers and implantable defibrillators as risk factors.49–52
Infectious endocarditis must be considered in the differential diagnosis of a broad range of clinical syndromes. Fever is the single most common presenting symptom, often accompanied by other constitutional symptoms of anorexia, weight loss, and fatigue. Onset may be acute or subacute with tendency toward more acute presentations with S. aureus infections. Signs and symptoms may be consequences of direct cardiac effects of infection including new or changing heart murmur, worsening congestive heart failure and valvular dysfunction, or onset of heart block from myocardial abscesses.50 The clinical presentation may also be dominated by systemic manifestations of emboli or immune complex disease including such findings as skin lesions, splenomegaly, glomerulonephritis, cerebrovascular events, and other acute vascular embolic events. Patients with acute, fulminant endocarditis, particularly S. aureus endocarditis, may present with concurrent cardiogenic and septic shock.
Clinical criteria have been developed and prospectively evaluated for the diagnosis of infective endocarditis.53,54 The most current version of these, the Modified Duke Criteria for Diagnosis of Infective Endocarditis (Box 54.1), relies primarily on blood culture data and echocardiographic findings as the two major clinical criteria for diagnosis; additional minor clinical criteria (fever, underlying predisposition, embolic phenomena, immunologic phenomena) can also be used as supporting evidence for determining confirmed or suspected cases.54 Patients with endocarditis have sustained bacteremia; in the absence of prior antibiotic therapy the yield of at least one positive blood culture after three sets are drawn is greater than 90%, and typically multiple cultures are positive. Thus two to three separate sets of blood culture should always be obtained in suspected endocarditis prior to initiation of therapy. Culture-negative cases may be attributed to prior antimicrobial therapy or to infection caused by fastidious or difficult-to-cultivate organisms.50 Follow-up blood cultures are critical to document sterilization of blood and should be repeated frequently until negative. Echocardiography should be performed on all patients with suspected endocarditis.50,52 When possible, transesophageal echocardiography (TEE) is the preferred modality in adults. Sensitivity of TEE is reported to be as high as 95% for native valve infection compared with 60% to 75% for transthoracic echocardiography (TTE) and is significantly better than TTE for diagnosis of prosthetic valve infection and for detection of complications such as perivalvular abscess that may affect management.50 In children and uncomplicated adult cases, in which TTE is diagnostic, TEE may not be necessary. If initial studies are negative, TEE should be repeated in 7 to 10 days if clinical suspicion remains high.50
Embolic complications may occur at any time during the course of infective endocarditis with overall incidence of 20% to 50%; risk decreases but still remains significant after initiation of antimicrobial therapy, especially with larger vegetations, and remains high for up to 2 to 3 weeks.50,55,56 Risks for embolization include specific organisms, especially S. aureus, Candida, HACEK* organisms, and Abiotrophia; increased vegetation size (>10 mm) and mobility as determined by echocardiography; mitral versus aortic valve involvement; and anterior versus posterior location on the mitral valve.50 More than half of clinically manifesting emboli involve the CNS, most commonly in the middle cerebral artery distribution.50,55 In addition to emboli, other risks for a fatal outcome include congestive heart failure, abnormal mental status, comorbid conditions, higher APACHE score, and S. aureus infection.49–51 Congestive heart failure appears to have the highest association with death.49–51 Medical therapy (versus surgical therapy) was also associated with higher 6-month mortality rates in recent studies.51,52
The decision to perform emergent or urgent valvular surgery on patients with active endocarditis remains complex. Indications for surgery can be broadly classified into those related to management of heart failure, management of uncontrolled infection, and management of embolization, and decisions are also impacted by patient status and available surgical expertise. Traditional indications for surgery include refractory congestive heart failure; persistent infection despite optimal antimicrobial therapy; fungal or other difficult-to-treat organisms; one or more emboli during the first weeks of antimicrobial therapy; and valvular complications of dehiscence, perforation, fistula, and large perivalvular abscesses (Table 54.3).50 Vegetation size and location are also emerging as possible independent indications for surgery.50 The risk of infecting a new prosthetic valve during surgery for active endocarditis is only 2% to 3%; thus active infection is not a contraindication to surgery that is indicated for complications associated with high mortality rate.50 Reviews of recent studies have confirmed the decreased mortality rates of cohorts of patients who were managed with early surgical intervention, especially those with left-sided S. aureus endocarditis, though there are no randomized controlled trials completed that address this.50,52 The optimal surgical procedure and the role of more conservative procedures such as vegetation resection remain incompletely defined, though resection of vegetations is frequently done in isolated, refractory right-sided endocarditis. The majority of patients who survive an episode of left-sided endocarditis who are not operated on initially will ultimately require valve replacement within the next 15 years.50
Table 54.3
Possible Indications for Emergent or Urgent Surgical Intervention for Acute Infective Endocarditis
| Indication | Details/Comments |
| Clinical Indications | |
| Refractory congestive heart failure | Most common single indication for surgery |
| Severe valvular decompensation | Usually acute mitral or aortic insufficiency but also valve obstruction. Less commonly, severe tricuspid regurgitation |
| Emboli and recurrent emboli | Especially with 1 or more events occurring during first 1 to 2 weeks of antimicrobial therapy |
| Persistent infection/sepsis | Persistent positive blood cultures after 7 days of optimal antibiotic treatment and no other source |
| Difficult-to-eradicate organisms | Fungal endocarditis, highly resistant bacteria; staphylococcal or gram-negative prosthetic valve endocarditis (controversial) |
| Extension of infection into myocardium | Evidence of new heart block, echocardiographic findings as listed below |
| Echocardiographic Indications | |
| Evidence of valve decompensation | Dehiscence, rupture, perforation, fistula, perivalvular abscess |
| Vegetation characteristics | >10 mm in size, especially on anterior mitral valve and emboli; increase in size despite treatment; >15 mm in size on aortic, mitral, or prosthetic valve; >20 mm in size on tricuspid valve |
| Other Possible Indications | |
| Complicated prosthetic valve infection | As defined by TEE findings; high failure rate for medical therapy alone |
| ?Any left-sided Staphylococcus aureus infection | Early surgical management may be associated with lower mortality |
TEE, transesophageal echocardiography.
Modified from Baddour LM, Wilson WR, Bayer AS, et al: Infective endocarditis: Diagnosis, antimicrobial therapy, and management of complications, a statement for healthcare professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, and the Councils on Clinical Cardiology, Stroke, and Cardiovascular Surgery and Anesthesia, American Heart Association. Circulation 2005;111:e394-e433; and Thuny F, Grisoli D, Collart F, et al: Management of infective endocarditis: Challenges and perspectives. Lancet 2012;379 (9819): 965-975.
Anticoagulation in patients with active endocarditis can present difficult management dilemmas. Anticoagulation has been considered to be relatively contraindicated in active endocarditis because of the risk of hemorrhagic CNS events.50,57 However, data from a recent case cohort study suggest that risk of warfarin therapy in acute left-sided native valve endocarditis has been overestimated.57 In patients already on anticoagulation for mechanical valves or other specific indications, warfarin should be switched to heparin therapy during the initial phase of therapy, especially in cases in which surgical intervention is being considered. Anticoagulation should be discontinued if possible after acute CNS embolic events. Neurologic events, most commonly embolic events but also mycotic aneurysms, are the second leading cause of death in acute endocarditis after heart failure. Decisions on timing of surgery and management of anticoagulation perioperatively and postoperatively in patients with CNS events are particularly challenging.
Optimal antibiotic therapy for patients with infective endocarditis is outlined in the American Heart Association guidelines, though there are more recent specific recommendations for treatment of MRSA valvular infections.50,58 Individual treatment regimens including choice of antibiotic, need for combination therapy, and duration of therapy are based on the organism, susceptibility data, location of infected valve, and whether the infection is complicated or uncomplicated. Left-sided S. aureus endocarditis is generally treated for 6 weeks with a parenteral β-lactam or vancomycin. Addition of gentamicin for native valve disease is no longer recommended due to risk of nephrotoxicity.59 MRSA infections require treatment with vancomycin or daptomycin. Uncomplicated right-sided endocarditis in injection drug users may have a more benign course, and there is evidence for use of shorter courses of therapy and even for use of oral therapy in selected patients. Treatment of viridans streptococcal endocarditis is 4 weeks of a β-lactam or vancomycin, and gentamicin is added for disease caused by relatively penicillin-resistant strains. Selected patients with uncomplicated viridans streptococcal infections have been treated with 2 weeks of combination therapy. β-Lactam therapy for susceptible isolates is always preferred to vancomycin in nonallergic patients. Enterococcal endocarditis is usually treated for 4 to 6 weeks with synergistic combinations of a penicillin or vancomycin and an aminoglycoside, but regimens need to be modified on the basis of strain resistance patterns in view of increasing rates of penicillin, vancomycin, and high-level aminoglycoside resistance. Prosthetic valve infections require longer treatment courses and use of combination therapy, and failure rates of medical therapy for prosthetic valve disease are high.50
Device-Related Endovascular Infections
Endovascular infections other than valvular infections have become increasingly common as causes of bacteremia and sepsis. This is related to the increased use of indwelling devices including vascular catheters for chemotherapy and other long-term parenteral therapies, use of accesses for hemodialysis, and the expanding indications for implantable cardiac devices for patients with congestive heart failure and arrhythmias. Infection is the most common serious complication of peripherally inserted central venous catheters (PICCs), tunneled catheters, and totally implanted intravascular devices.60 Patients presenting with sepsis and focal findings related to the catheter such as tunnel infection, port abscess, or cellulitis; those with associated venous thrombosis; and those with concurrent endocarditis or osteomyelitis require removal of the catheter.60 Management is more difficult in patients with sepsis or bacteremia without local signs of infection when removal of the catheter is not a trivial procedure. Diagnosis of the catheter as the source of a bacteremia can sometimes be made with use of paired central and peripheral blood cultures. Positive central and negative peripheral cultures or central cultures that are positive significantly earlier than peripheral cultures implicate the catheter as the source.60 The majority of infected permanent catheters require removal; selected uncomplicated catheter infections in nonseptic patients whose bloodstream has sterilized on antibiotics can be treated with catheter retention and antibiotic therapy, especially infections caused by coagulase-negative staphylococci and other less virulent organisms.60 Algorithms for the management of suspected and confirmed line infections in patients with long-term access devices are shown in Figure 54.2A. Antibiotic lock therapy is a strategy in which antibiotic solutions are used to fill the catheter lumen to maintain high antibiotic concentrations in the catheter and catheter hub and yield higher rates of catheter salvage than parenteral antibiotics alone.60 Hemodialysis catheter infections pose particular management challenges because of the high prevalence of infection in these patients as well as the need to preserve dialysis access. An algorithm for management of tunneled hemodialysis catheters is shown in Figure 54.2B. Hemodialysis has emerged as a major risk factor for endocarditis in recent studies. Catheter salvage strategies and guidewire exchange strategies are employed more frequently in hemodialysis catheter–associated infections than in other populations.60,61
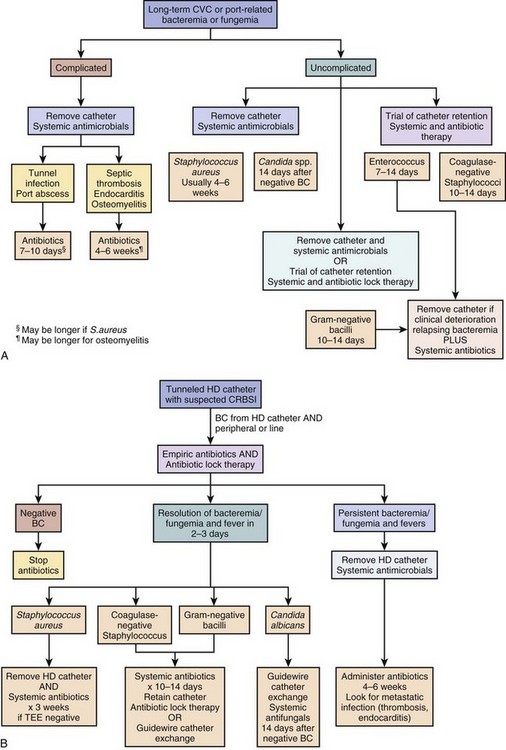
Indications for placement of implantable cardioverter-defibrillator devices (ICDs) in patients with cardiac disease have recently been expanded, resulting in increased number of ICDs and increased prevalence of pacemaker and ICD infections.62,63 Rates of pacemaker and ICD infections have increased out of proportion to increased rates of ultilization of these devices, and in population-based studies infection rates range from 1 to 3 infections per 1000 device years.62 Infections include site and generator pocket infections and infections of the leads. Lead infections, which account for probably half of all infections, may be complicated by sepsis and septic shock, suppurative thrombophlebitis, and endocarditis and almost always require explantation of the device.62 Diagnosis is typically made by blood cultures and TEE, which may show vegetations on the leads or associated endocarditis. The most common organisms are coagulase-negative staphylococci and S. aureus in 80% of cases. Most infected leads can now be removed through percutaneous extraction procedures rather than open surgical explantation, but the expertise and technology for this are not available at all hospitals. Devices should not be replaced until bacteremia has completely resolved.
Primary Bacteremias
In 10% to 30% of patients presenting from the community with bacteremia and sepsis, the primary source of the bacteremia remains unknown, even after careful clinical and radiographic evaluation.64,65 This includes patients with infections manifesting with a primary bacteremia, as well as those with probable secondary bacteremia from an occult focus. Bacteremia of unknown cause is associated with higher mortality rate.64 Primary bacteremias are more common with certain microorganisms and with specific host predispositions. These syndromes, if unrecognized, have particularly high morbidity and mortality rates. Patients with impairment in immunoglobulin production or function and impaired or absent splenic function are predisposed to infections with encapsulated organisms such as S. pneumoniae, N. meningitidis, H. influenzae, and group B streptococcus.66 Patients with advanced liver disease and cirrhosis are predisposed to these, as well as a variety of other spontaneous bacteremias.67 Patients with cell-mediated immune defects may present with other unusual bacteremias such as Listeria and Salmonella sepsis. Severe neutropenia greatly increases the risk of spontaneous bacteremia and sepsis from endogenous gastrointestinal flora, and the management of fever in neutropenic patients includes empiric initiation of broad-spectrum antibacterial therapy.68 Several important specific bacteremia syndromes are described as follows.
Meningococcemia and Meningococcal Sepsis
Invasive meningococcal disease can present as either bacterial meningitis or as the syndrome of meningococcemia with meningococcal sepsis without meningitis.7 The proportion of primary meningococcemia cases and severity of disease manifestations depend in part on the serotypes present in the community but are also heavily influenced by host genetic makeup. The annual incidence of meningococcal disease in the United States is approximately 1 case per 100,000, with mortality rate of 10% to 14% and significant residual morbidity in up to 20%.7 Besides young children, adolescents and young adults ages 11 to 19 are at increased risk for meningococcal disease, but risk persists throughout life and is increased in those with certain immune deficiencies such as terminal complement deficiency and cirrhosis.7 Patients with meningococcemia present with fever, headache, malaise, vomiting, and myalgias.69,70 Most patients will also present with rapid development of a characteristic nonblanching petechial or purpuric rash, but the rash can be maculopapular or absent. Early symptoms that may be important clues to the diagnosis are leg pains and cold hands and feet.69 If untreated, disease may progress rapidly within 24 hours to fulminant purpura and hemorrhagic shock. The keys to improved outcome are early recognition and aggressive therapy. Early institution of antibiotic therapy with a penicillin or third-generation cephalosporin can significantly improve outcomes. PCR of blood is useful for diagnosis, especially in patients who received antibiotics prior to hospitalization.7,69 A tetravalent conjugated polysaccharide vaccine is available in the United States and is recommended for routine vaccination of older children and adolescents and other high-risk populations.7
Primary Pneumococcal Bacteremia
Pneumococcal bacteremia is most commonly a consequence of pneumococcal pneumonia, but 5% to 10% of episodes occur without an identified underlying focus.71 Severe episodes may present with fulminant sepsis and purpura fulminans clinically indistinguishable from meningococcal sepsis. Important risk factors for pneumococcal sepsis include asplenia and hyposplenism, HIV infection, sickle cell disease, alcoholism, malignancy, and other immunocompromised states, although this syndrome can occur in otherwise healthy adults and children without predisposing risks.72 S. pneumoniae causes 50% to 90% of cases of the fulminant sepsis syndrome that occurs in postsplenectomy or functionally asplenic patients.67 Other causes of the postsplenectomy sepsis syndrome include N. meningitidis; H. influenzae; and, less commonly, Capnocytophaga canimorsus, Escherichia coli, Salmonella, and the vector-borne parasitic infections malaria and babesiosis. Immunization with pneumococcal, meningococcal, and H. influenzae vaccines is recommended for those at risk for postsplenectomy sepsis.66
Staphylococcus aureus Bacteremia
S. aureus bacteremia can occur from localized staphylococcal disease such as skin and soft tissue infection or pneumonia, endovascular infection, and endocarditis. It can also be of occult origin. Regardless of the initial source of bacteremia, there is significant risk for development of late complications including endocarditis, bone and joint disease, infections of prosthetic devices, or other metastatic foci in up to one third of cases.58,73 Features associated with complicated S. aureus bacteremia include prolonged bacteremia, prolonged fever, and embolic lesions. Longer-course therapy (at least 4 weeks) is recommended for patients with complicated disease, whereas 2 weeks of treatment may be adequate for selected patients with uncomplicated bacteremia when endocarditis has been excluded.74 TEE is recommended to exclude endocarditis in patients with otherwise clinically uncomplicated S. aureus bacteremia when short-course therapy is being considered.73,74
Toxin-Mediated Infections
Staphylococcal Toxic Shock Syndrome
S. aureus produces multiple virulence factors that contribute to its success as a human pathogen. These virulence factors potentiate local adherence, tissue invasion, and avoidance of host defenses, all features that are important in the pathogenesis of localized skin and soft tissue infection, pneumonia, bacteremia, and metastatic infections. Staphylococci also produce a variety of exotoxins that are released into the systemic circulation to act at distant sites.75 Some of these exotoxins are directly pathogenic to specific cells, such as exfoliatoxin B, which causes staphylococcal scalded skin syndrome. Other exotoxins function as potent superantigens, antigens that bypass the intermediate T-cell antigen processing steps by binding directly to Vβ domains on T-cell receptors. This causes direct activation of multiple T-cell classes resulting in unopposed release of large amounts of cytokines including interleukin (IL) 2, IL-4, IL-6, IFN-γ, TNF-α, and IL-1β.75 The resulting “cytokine storm” can lead to septic shock, multiorgan failure, and death.
The most well-studied staphylococcal superantigens are the pyrogenic antigens TSST-1 and TSST-2 and enterotoxins B and C. TSST-1 and TSST-2 are the primary exotoxins causing the staphylococcal toxic shock syndrome.75 First described in 1979 as a unique syndrome primarily associated with menstruating women using super absorbent brands of tampons, staphylococcal toxic shock syndrome is now known to occur potentially from infection or colonization at any site with an exotoxin-producing strain.76,77 Disease is associated with conditions leading to exotoxin production in a host who lacks preexisting antibodies to TSST-1. The overall incidence of this syndrome, as well as the proportion of cases associated with menstruating women, has decreased since 1980.76 Toxic shock syndrome is characterized by the acute onset of fever, hypotension, myalgia, scarlatiniform or erythroderma-like rash, nausea, vomiting, diarrhea, and development of multiple-organ failure including renal failure, elevated liver enzymes, and disseminated intravascular coagulopathy.76,77 Typically the rash will evolve and cause late desquamation of hands and feet. Specific criteria for the diagnosis of staphylococcal toxic shock syndrome have been described (Box 54.2).76 Treatment is primarily supportive, but antistaphylococcal agents are administered to treat the underlying staphylococcal colonization or infection; some evidence suggests that immunoglobulin may be beneficial by binding exotoxin and attenuating the cytokine response. Staphylococcal strains producing the staphylococcal enterotoxins B and C have been implicated in some nonmenstrual-associated cases.75
Streptococcal Toxic Shock Syndrome
Group A β-hemolytic streptococci are also capable of producing a toxic shock–like syndrome analogous to staphylococcal toxic shock syndrome.78,79 Most often this is seen in the setting of severe bacteremic streptococcal soft tissue infection or with streptococcal necrotizing fasciitis. In one large survey of invasive streptococcal infections in Ontario, 13% were complicated by toxic shock syndrome, and the mortality rate of these infections was 81%.79 The pathogenesis of this syndrome is attributed to streptococcal pyrotoxins functioning as superantigens but may also involve binding of streptococcal M protein-fibrinogen complexes to PMN cells causing PMN activation and endothelial damage. Clinical criteria for the diagnosis of streptococcal toxic shock syndrome have been defined (see Box 54.2).78 In addition to use of antibiotics that decrease toxin production in vitro, such as clindamycin and linezolid, IV immunoglobulin (IVIG) has been used for treatment though clinical evidence for benefit of IVIG is limited.80 Other streptococcal species have also been reported to produce a toxic shock–like illness including viridans streptococci; groups B, C, and G β-hemolytic streptococci; and Streptococcus suis.
Clostridial Toxic Shock Syndrome
A rare, generally fatal syndrome of toxic shock has been reported following medical abortion with oral mifepristone and vaginal misoprostol.81 This syndrome has also been seen complicating endometritis after live births and is due to Clostridium sordellii. Clinical findings include tachycardia, hypotension, edema, hemoconcentration, and profound leukocytosis, and generally the absence of fever. Optimal management of this syndrome remains poorly defined.
Tetanus
Tetanus is a syndrome of increased muscle rigidity and convulsive spasms caused by a toxin produced by the environmental spore-forming anaerobic bacterium Clostridium tetani.82 Since the introduction of routine immunization, tetanus has become increasingly rare, with the number of U.S. cases decreasing from 560 cases in 1947 to only 233 cases from 2001 to 2008 with case fatality rate of 13%.82 Clinical disease is caused by contamination of wounds, most commonly traumatic wounds, with bacterial spores. Spores then germinate and produce toxins including tetanospasmin, a highly potent neurotoxin that inhibits neurotransmitter release, resulting in blockage of inhibitor impulses and unopposed muscle contractions. Cases predominate in the summer or wet season, and disease is now most commonly seen in older adults because of either missed primary immunization or waning effects of childhood immunization.82 The most common syndrome is that of generalized tetanus with descending symptoms of trismus (lockjaw), difficulty swallowing, muscle rigidity, and spasms. Symptoms may persist for several weeks, and complete recovery may take months. Complications include laryngospasm, fractures, hypertension, nosocomial infections, and death. Treatment is primarily supportive, although metronidazole or penicillin may be given to treat potentially infected or colonized wounds. Patients require admission to an ICU for control of rigidity and spasms with benzodiazepines or neuromuscular blocking agents and for ventilator support. Human tetanus immunoglobulin may be administered, but this only binds free toxin, so there may be little benefit by the time of clinical presentation. Routine childhood and adult immunization with tetanus toxoid remains the primary strategy for preventing this rare but often fatal condition. Guidelines for use of immunoglobulin and vaccination in management of potentially infected wounds have been published.82
Botulism
Clostridium botulinum is an anaerobic spore-forming bacterium that produces botulinum toxin, a family of closely related but immunologically distinct polypeptide toxins that are among the most potent neurologically acting poisons known.83 Other, less common clostridial species can also produce botulinum toxin. Botulinum toxin binds irreversibly to synaptic complexes, affecting ganglionic synapses, presympathetic synapses, and neuromuscular junctions. Clinical syndromes include those caused by ingestion of preformed toxin such as food botulism and those resulting from acquisition of the organism and production of toxin in vivo such as infant botulism in very young children and wound botulism from contaminated wounds, most commonly seen in injection drug users. Patients present initially with cranial nerve dysfunction and evolve to descending paralysis without sensory or cognitive effects. Diagnosis is made on clinical grounds; testing of food products or human samples in the United States is performed by the Centers for Disease Prevention and Control (CDC). Treatment is primarily supportive, but equine type-specific antitoxin is available through the CDC.83 Antitoxin is only effective in binding free toxin if given in the first 72 hours. Because of its potency, environmental stability, and potential for aerosolization, botulinum toxin is considered a potential agent of bioterrorism.83
Diphtheria
Diphtheria is caused by toxigenic strains of the bacterial species Corynebacterium diphtheriae. This disease has become extremely rare in the United States secondary to universal vaccination with diphtheria toxoid vaccine, with an average of one confirmed case per year.84 Patients with respiratory diphtheria present with sore throat; low-grade fever; occasional neck swelling; and the characteristic grayish adherent membrane covering the tonsils, throat, or nose.84 Complications include myocarditis, polyneuritis, and airway obstruction, with mortality rates of 5% to 10%. Outbreaks of diphtheria continue to occur in many parts of the world.
Serious Skin and Skin Structure Infections
Serious skin and soft tissue infections are characterized by rapidly progressive inflammation and necrosis of skin, subcutaneous fat, and fascia. Occasionally, muscle is also involved. Several terms have been used to describe these infections: necrotizing fasciitis; synergistic necrotizing cellulitis; progressive bacterial synergistic gangrene; anaerobic cellulitis; and, when muscle is involved with clostridial infection, clostridial myonecrosis (gas gangrene).85 Differentiating among these entities is difficult and somewhat artificial. A variety of features of some of the most important necrotizing skin and soft tissue infections are shown in Table 54.4. Microbiologically, these infections may be caused by a single pathogen such as group A β-hemolytic streptococcus; however, they are more frequently polymicrobial in nature. Risk factors for these infections include diabetes, old age, peripheral vascular disease, malignancy, alcoholism, renal failure, and immunosuppressive therapy. Infection may follow traumatic injuries that become contaminated by soil. The primary traumatic event may be relatively minor. Initial signs and symptoms may be similar to a severe cellulitis. Early findings include fever, tachycardia, moderate to severe pain, and swelling and induration of the skin.86 Late findings include severe pain or even anesthesia, skin necrosis, hemorrhagic bullae, crepitus, drainage, and signs of systemic inflammatory response syndrome or severe sepsis including multiorgan failure.86 Laboratory tests usually demonstrate leukocytosis and metabolic acidosis. Plain radiographs may reveal gas in the soft tissues. CT or MRI is useful in delineating extent of disease or the presence of soft tissue gas. Diagnostic evaluation should also include blood cultures, Gram staining of tissue exudates, and aerobic and anaerobic cultures obtained at surgery or from a needle aspiration. Incision and exploration or biopsy can even be done at the bedside to obtain material for Gram stain, culture, and histologic evaluation.
Table 54.4
Differentiating Features of Necrotizing Skin and Soft Tissue Infections
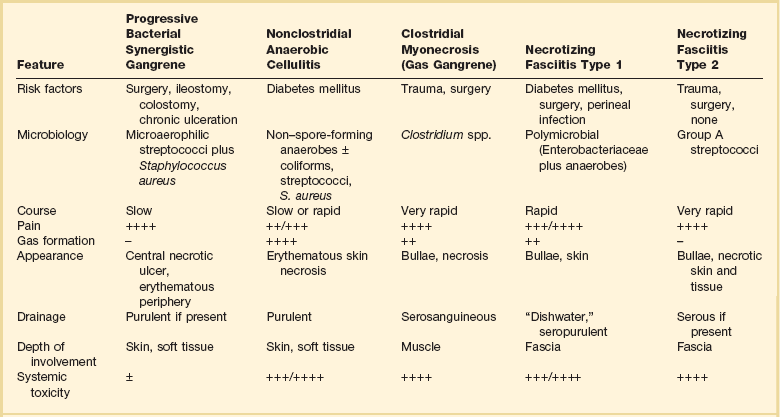
−, absent; ±, occasionally present; +, minimal; ++, mild; +++, moderate; ++++, marked or severe.
Once a necrotizing soft tissue infection has been identified, prompt therapy is important. Early aggressive surgical débridement is essential.87 Because most cases of necrotizing skin infection are polymicrobial, empiric antibiotic coverage should be sufficiently broad spectrum to cover gram-positive cocci, gram-negative bacilli, and anaerobes. A reasonable empirical regimen might consist of a β-lactam/β-lactamase inhibitor or a carbapenem combined with vancomycin or another agent effective against MRSA. Antibiotic therapy can subsequently be adjusted on the basis of results of cultures and sensitivities.88 In cases of group A β-hemolytic streptococcal infection, penicillin plus clindamycin is the therapy of choice. Clindamycin is effective in turning off toxin production. Linezolid also has similar activity.89 IVIG may be a useful adjuvant therapy for streptococcal toxic shock.80,90
Necrotizing Fasciitis
Necrotizing fasciitis is an uncommon, severe infection that causes necrosis of the subcutaneous tissue and fascia with sparing of the underlying muscle. Two predominant types, based on microbiology, are described. In type I, at least one anaerobic species is isolated along with one or more facultative anaerobes and members of the Enterobacteriaceae.85,91 This form of necrotizing fasciitis most commonly affects the extremities but may involve the abdominal wall, postoperative wounds, perianal area, and groin. It occurs following trauma or a variety of surgical procedures, perirectal abscess, decubitus ulcer, or perforation of the intestines. Patients at increased risk include those with diabetes mellitus, alcoholism, and injection drug use. The involved area is initially erythematous and painful, but over several days skin changes include color changes, formation of bullae, and cutaneous gangrene. The involved area becomes anesthetic secondary to thrombosis of small blood vessels and destruction of superficial nerves. Anesthesia may develop before the appearance of skin necrosis and is an important clue to the presence of necrotizing fasciitis rather than simple cellulitis.86 Subcutaneous gas is often present, and systemic toxicity is common.91 When the lesion is probed at the bedside or in the operating room, there is no resistance along tissue planes. In type II (also known as hemolytic streptococcal gangrene), group A streptococci are generally isolated alone or in combination with S. aureus.92 These strains usually produce pyrogenic exotoxin A. Periodically, group C or group G streptococci are causative organisms. Infection usually develops at a site of trauma but may occur in the absence of an obvious portal of entry.91 The involved area is extremely painful, erythematous, and edematous. Infection spreads widely in deep fascial planes with relative sparing of the overlying skin and therefore may not be recognized. This form of necrotizing fasciitis is present in approximately 50% of cases of streptococcal toxic shock syndrome.92 Over several days, the skin becomes dusky and bullae develop. Bullae then rupture and evolve into an area covered by necrotic eschar, often resembling a third-degree burn. Streptococci can usually be cultured from fluid of the early bullae and frequently from blood. Complications include metastatic abscess formation, and the mortality rate from this infection is high.
Fournier’s gangrene is a form of necrotizing fasciitis that involves the male perineum, usually the scrotum, but can involve the penis, the perineum, or the abdominal wall. It is caused by anaerobic streptococci along with other bacteria such as E. coli; S. aureus; β-hemolytic streptococci; Proteus species; a variety of anaerobes; and, on occasion, Pseudomonas species.93 The first symptoms are commonly scrotal swelling and pain followed by progressive necrosis of scrotal skin and subcutaneous tissues.93 The patient may appear toxic. Initially the patient may be erroneously diagnosed with an acute abdomen unless the genitalia are examined. Gangrene of the perineum and sometimes the penis may develop. Urgent and aggressive surgery is necessary along with broad-spectrum antibiotics.
Clostridial Myonecrosis (Gas Gangrene)
Gas gangrene is a life-threatening infection of skeletal muscle caused by Clostridium species. It should be suspected when the Gram stain of drainage reveals gram-positive bacilli in a patient who is critically ill. Penicillin G is the drug of choice with or without clindamycin. Aggressive surgical exploration and débridement is the mainstay of therapy. The role of hyperbaric oxygen therapy in the treatment of clostridial myonecrosis or in the treatment of necrotizing fasciitis remains controversial. If used, it must be used as an adjunct to surgery.94
Vibrio Infections
Various Vibrio species including Vibrio vulnificus have caused mild to severe cellulitis in patients who sustained lacerations or puncture wounds when in contact with saltwater in the southeastern United States and the Gulf of Mexico. Septicemia with secondary necrotizing soft tissue infection may occur after ingestion of raw or undercooked shellfish in immunocompromised hosts, especially those with cirrhosis.95 An increased incidence of Vibrio infections has been described as a result of flooding after natural disasters. Doxycycline, third-generation cephalosporins, and fluoroquinolones are effective therapies, along with aggressive surgical débridement of necrotic tissue.
Community-Acquired MRSA
During the past decade, community-associated MRSA (CA-MRSA) infections among persons without health care–associated risk factors have emerged in several geographic areas and have become endemic in some areas.96 Many of these strains carry the staphylococcal cassette chromosome mec type IV element and the gene encoding Panton-Valentine leukocidin, a toxin that promotes tissue destruction.97,98 Although most CA-MRSA infections are mild skin and soft tissue infections, severe, life-threatening cases of necrotizing fasciitis, myonecrosis, necrotizing pneumonia, and sepsis have occurred.97,98 For patients with invasive infections caused by CA-MRSA, vancomycin and linezolid are appropriate therapeutic options.99 CA-MRSA strains are often susceptible to clindamycin, doxycycline, and trimethoprim/sulfamethoxazole.
Serious Gastrointestinal and Intra-Abdominal Infections
Bacteremia Associated with Diarrheal Illness
Of the enteric pathogens, Salmonella species are most likely to cause bacteremia and serious infection. Enteric fever is usually caused by Salmonella enterica serotype typhi and rarely by Salmonella paratyphi, Salmonella choleraesuis, Yersinia enterocolitica, or Campylobacter fetus.100 Features of classic typhoidal fever caused by S. enterica include sustained fever, bacteremia, headache, and abdominal pain. Physical findings include “rose spots,” which are 2- to 4-mm discrete, irregular, blanching pink macules that are often seen on the anterior chest; hepatosplenomegaly; and relative bradycardia. Multiorgan system dysfunction can occur as a consequence of metastatic infection or immune complex deposition.101 Intestinal bleeding or perforation may occur as a result of hyperplasia of the lymphoid tissue in the terminal ileum. Diarrhea is seen in less than 50% of cases and only early in the illness. Constipation is a frequent later complaint. The following specimens should be sent for culture: stool, blood, or bone marrow. Serologic findings may provide supportive evidence or may be useful in epidemiologic evaluation.
Therapeutic options include third-generation cephalosporins, fluoroquinolones, and trimethoprim/sulfamethoxazole. Antimicrobial resistance has been reported. Treatment for uncomplicated cases is 12 to 14 days; 30 days of therapy may be necessary for metastatic foci. Metastatic foci are relatively common in the setting of bacteremia. Infection may involve gallbladder, spleen, bone, joints, and the meninges. There is a propensity to infect preexisting intravascular lesions such as atherosclerotic plaques and aneurysms. Sickle cell anemia and the presence of an orthopedic prosthesis are risk factors for osteomyelitis.101 Meningitis tends to occur in young children.101
Peritonitis
Peritonitis is a localized or general inflammation of the peritoneal cavity that is generally caused by bacteria or fungi but may be caused by a variety of noninfectious agents such as gastric contents, talc, or bile salts. Infective peritonitis has been classified as primary, secondary, or tertiary. Peritonitis is primary when it is not related directly to other intra-abdominal processes.102 This classification includes spontaneous bacterial peritonitis (SBP), which occurs in patients with underlying ascites from cirrhosis or nephrotic syndrome, and tuberculous peritonitis. Secondary peritonitis most often arises from an enteric source or pelvic focus and includes peritonitis following an acute perforation of the gastrointestinal tract, intestinal necrosis, postoperative peritonitis that may be secondary to an anastomotic leak, and posttraumatic peritonitis following blunt or penetrating abdominal trauma. Intestinal ischemia and frank necrotic bowel may be caused by a variety of processes including malignancies, vascular insufficiency, volvulus, or intussusception.102 Rupture of an abscess in the pancreas, liver, or spleen or, rarely, rupture of a distended gallbladder can also cause peritonitis. Localized lower abdominal peritonitis can also result from gynecologic infections such as salpingitis and endometritis. Tertiary peritonitis is described as occurring when clinical and systemic signs of peritonitis persist or recur after treatment for secondary peritonitis. A distinct form of device-associated peritonitis is seen in patients undergoing peritoneal dialysis.
Bacterial peritonitis is typically caused by flora of the large intestine including aerobes, with E. coli being the most frequent, and anaerobes, of which Bacteroides fragilis is the predominant isolate.103 Common symptoms include localized or generalized abdominal pain, nausea, and vomiting. Treatment with corticosteroids may mask typical signs and symptoms, delaying the diagnosis. Signs may include abdominal rigidity, distention, fever, and an overall toxic appearance. A rigid abdomen may be seen in the early stages of an acute peritonitis, although it may be absent in a peritonitis that progresses more slowly, such as that caused by tuberculosis, or when sterile bile, pancreatic fluid, or urine leaks into the peritoneal cavity. As intraperitoneal fluid accumulates, abdominal distention and an ileus occur. The WBC count is usually elevated. Bacteremia may occur, and the sepsis syndrome may develop as peritonitis evolves. Aspiration of peritoneal fluid is an essential part of the evaluation for peritonitis. Laparoscopy or laparotomy may be necessary. Studies performed on peritoneal fluid should include a cell count with differential blood cell count, amylase, Gram stain and aerobic and anaerobic culture, acid-fast smear and culture, and fungal smear and culture.
The primary cause of secondary peritonitis should be sought and eliminated if possible. Liver function tests and a serum amylase level may define a source in the liver, gallbladder, or pancreas. A plain film of the abdomen or a chest radiograph may reveal free air under the diaphragm in the case of a ruptured viscus. A CT scan of the abdomen may reveal an underlying intra-abdominal abscess or other focal process. Antibiotic therapy to cover gram-negative bacillary organisms and anaerobes should be initiated. Options for empiric therapy include combinations such as a third- or fourth-generation cephalosporin or a fluoroquinolone with metronidazole or monotherapy with a β-lactam/β-lactamase inhibitor combination, a carbapenem, or a glycylcycline such as tigecycline.104–107 The optimal duration of therapy is not well defined, although nonbacteremic patients are generally treated for 7 to 10 days.103 Regimens as short as 2 days of treatment may be adequate in uncomplicated situations with adequate surgical source control and in those with penetrating trauma. Bacteremic patients are generally treated for a total of 14 days using a combination of IV followed by oral therapy.
SBP is caused by the translocation of enteric organisms to regional lymph nodes, which produce bacteremia and ultimately seeding of ascitic fluid.108 Periodically a urinary tract infection may be the source of bacteremia. SBP occurs most commonly in adults with cirrhosis, nephrotic syndrome, or systemic lupus erythematosus.108,109 Coliforms are the most common pathogens in adults, accounting for 70% of infections, with E. coli being the most common isolate followed by Klebsiella species. Gram-positive cocci may be seen in up to 20% of cases and anaerobes in less than 5%. Generally SBP is monomicrobial in contrast to the polymicrobial nature of most other forms of peritonitis. Findings of SBP may be subtle, so a high index of suspicion is necessary. Ascites is almost always present.108 Fever and abdominal pain are seen in the majority of patients. New onset or worsening of hepatic encephalopathy may be seen. Some patients may have abdominal tenderness or rebound tenderness, but these findings are less frequent than in other forms of peritonitis. The most useful diagnostic test is a paracentesis.108–110 SBP is defined by an ascitic segmented neutrophil count of at least 250 cells/mm3 with or without a positive fluid culture and no obvious intra-abdominal source of infection.109,110 The pH of ascitic fluid is low in SBP, whereas that of sterile ascitic fluid is the same as in serum.110 Despite the low sensitivity (≈33%), Gram staining of ascitic fluid should be performed. Patients with other infections of the abdomen, such as tuberculous peritonitis, secondary bacterial peritonitis caused by perforation, or peritonitis caused by noninfectious processes including pancreatitis or malignancy, also may show elevated neutrophil counts. Presence of a single organism generally supports the diagnosis of SBP, although there is a variant of SBP caused by traumatic entry into the bowel during paracentesis that may be polymicrobial, and conversely, gram-negative bacillary organisms may occur as sole pathogens in secondary peritonitis. If mixed gram-positive and gram-negative bacteria are seen, an intestinal perforation is the more likely source.110 Peritoneal fluid should be cultured aerobically and anaerobically. Blood cultures should also be performed and are positive in up to 75% of patients with SBP.109 Other than for symptom relief, a repeat paracentesis is necessary only when the patient fails to have a clinical response or when an unusual organism is isolated. Treatment is usually continued for a total of 7 to 10 days, although some experts have suggested that a 5- to 7-day course of IV therapy may be adequate.109,111,112 Most studies show a high mortality rate of 30% to 40% for this syndrome, and after an initial episode of SBP, the probability of recurrence within 1 year is 70%.108,111
Intra-abdominal Abscess
Considering the presence of a potential intra-abdominal abscess in febrile patients without any obvious cause of fever is important, especially if there is a predisposing condition such as diverticulitis, inflammatory bowel disease, or a history of recent abdominal surgery or abdominal trauma. Intra-abdominal abscess formation may complicate either primary or secondary peritonitis. The processes that typically predispose to intra-abdominal abscess formation are the same as those that cause secondary peritonitis and include perforation, complicated acute cholecystitis, suppurative cholangitis, acute appendicitis, diverticulitis, intestinal malignancy, surgical procedures, blunt or penetrating trauma, or intestinal ischemia from a mesenteric vascular occlusion, an intestinal obstruction, or a volvulus. Most commonly, abscesses are postoperative complications of trauma or of gastrointestinal or biliary surgery.113 Abscesses can be found anywhere within the abdomen including the retroperitoneal space. Generally their location is in proximity with the original site of contamination, but they may develop at distant sites.114 One example of distal infection is subphrenic abscess, which may be a consequence of a perforated appendix. Symptoms of intra-abdominal abscess may include fever, chills, anorexia, weight loss, and abdominal pain. Unexplained fever may be the only sign of an occult intra-abdominal abscess. An abdominal CT scan is the imaging modality of choice for diagnosis of abdominal abscess. Drainage is essential to establish the diagnosis, obtain microbiology to target antimicrobial therapy, and achieve therapeutic success. This may be accomplished by the insertion of percutaneous catheters, or by laparoscopic or operative drainage.113,114 Large abscesses cannot be eradicated by antibiotic therapy alone. Criteria for considering percutaneous drainage include the presence of a well-defined fluid collection and a safe percutaneous route of access.115 Drains should remain in place until drainage volume is minimal, usually less than 10 mL in a 24-hour period.
A repeat CT scan should be performed to demonstrate complete collapse of the abscess cavity.113 Indications for operative surgical drainage rather than percutaneous drainage include the following situations: (1) Percutaneous drainage cannot be performed safely; (2) percutaneous drainage fails; (3) there are multiple interloop abscesses; (4) there is a coagulopathy; or (5) there is infected pancreatic necrosis. Antibiotic therapy is indicated to treat inflammation in the surrounding tissue and to prevent metastatic infection and sepsis from bacteremia. Antibiotic coverage should be directed at abdominal flora including aerobic and anaerobic organisms even if anaerobes are not isolated.103,112,113 Mortality rate with undrained pancreatic, hepatic, or retroperitoneal abscesses is reported to be 45% to 100%.114 An approach to the management of intra-abdominal abscesses is outlined in Figure 54.3.
Biliary Tract Infections
Infections of the biliary tract, including the gallbladder and the common bile duct, are usually associated with obstruction to the flow of bile. The biliary tract of healthy individuals is sterile. In acute cholecystitis, infection of the gallbladder is most commonly caused by microorganisms that are generally part of the normal intestinal flora.116 Most biliary infections are polymicrobial, and anaerobes are more frequently isolated in the elderly and in those with common bile duct manipulation or prior biliary procedures. The source of bacteria is presumed to be the duodenum.116 Many antibiotics achieve good levels in the bile in the absence of obstruction; however, when obstruction of the hepatic ducts or the common bile duct is present, the levels of such antibiotics are often subtherapeutic. Antibiotic therapy is adjunctive to decompression/drainage, which can be accomplished surgically, percutaneously, or endoscopically via endoscopic retrograde cholangiopancreatography (ERCP). Antibiotics prevent the development of bacteremia, progression of infection, and development of liver abscesses. Cultures of blood and bile should be obtained. Empiric antibiotic therapy should generally be directed at gram-negative bacteria. In patients who are critically ill, the elderly, and those with prior common bile duct and complex biliary procedures, antianaerobic and antienterococcal therapy should also be instituted.
Acute acalculous cholecystitis is an acute inflammation of the gallbladder that occurs in the absence of gallstones. Individuals at risk include debilitated, hospitalized patients such as those who have had major surgical procedures, prolonged intensive care stays, hyperalimentation, or predisposing conditions that result in bile stasis, cholecystoparesis, or gallbladder ischemia.116 The symptoms and findings are similar to those of calculous cholecystitis and include fever, right upper quadrant abdominal pain, nausea, vomiting, leukocytosis, and elevated liver function tests with an obstructive pattern. Diagnostic imaging modalities include ultrasonography, CT scanning, and hepatoiminodiacetic acid (HIDA) scanning. If an open or laparoscopic cholecystectomy cannot be accomplished because of the patient’s underlying condition, then urgent percutaneous cholecystostomy should be performed. In addition, broad-spectrum antibiotics should be instituted.
Acute cholangitis is a serious infection with high morbidity and mortality rates. The most common cause of acute cholangitis is obstruction and subsequent infection associated with stones in the common bile duct, which have usually migrated from the gallbladder. Other causes of biliary tract infection include malignant obstruction of the bile duct secondary to pancreatic cancer, cholangiocarcinoma, cancer of the papilla of Vater, or portahepatic metastases. Periodically biliary strictures, pancreatitis, and infection with the intestinal nematode Ascaris lumbricoides can cause obstruction. Bacteria enter the bile duct from the gastrointestinal tract through the bloodstream or lymphatics.117 The most common presenting symptoms are fever, abdominal pain, and jaundice, also known as Charcot’s triad. Septic shock may occur if treatment is delayed. Blood cultures should be obtained, and ultrasound or CT scanning should be performed. Broad-spectrum antibiotics should be initiated, and biliary decompression accomplished either endoscopically with an endoscopic sphincterotomy, via a percutaneous transhepatic biliary drainage procedure, or by surgical decompression. Endoscopic or percutaneous procedures may be the initial treatment of choice in patients who are seriously ill and at high risk for complications. Elective surgery, which generally includes cholecystectomy and bile duct exploration, can be deferred to a later date when the patient has stabilized.
Pancreatic Infections
Although acute pancreatitis is usually a sterile inflammatory process, infectious complications may occur. In addition, a variety of viral infections (rubella; coxsackievirus type B; mumps; Epstein-Barr virus; CMV; and hepatitis A, B, and C); Mycoplasma pneumoniae; and parasites such as A. lumbricoides have been implicated as causes of pancreatitis.118 Some patients may develop sterile or infected pancreatic or peripancreatic necrosis, infected pancreatic pseudocysts, or abscess formation as complications of acute pancreatitis.118 Sterile necrosis of either the parenchyma or the duct system usually occurs very early in the clinical course of severe pancreatitis. Risk for infection increases as necrosis becomes more extensive. Of patients with acute pancreatitis, only approximately 5% develop pancreatic infections; however, the highest mortality rate from pancreatitis occurs in these patients.118 The incidence of infection increases over the first few weeks and peaks during the third and fourth weeks. Patients with extensive necrosis, those who are very ill, and those with early infection have the highest mortality rates.118 CT scan with high-dose contrast agent is the most useful modality for predicting who is at risk for the development of infection. Between 40% and 70% of those with more than 30% necrosis seen on CT scan will ultimately become infected.118 If gas bubbles are seen in the region of the pancreas, it should be presumed that infection is present. Clinically, it is extremely difficult to determine whether patients with necrotizing pancreatitis have superimposed infection because sterile pancreatic necrosis may cause leukocytosis and fever even in the absence of infection, and approximately 50% of infected patients may not show early clinical signs of infection. CT-guided fine-needle aspiration of necrotic pancreatic tissue for Gram stain and aerobic and anaerobic culture is a useful procedure to determine the presence of infection.118 The microbiologic picture resembles that of the intestinal flora. E. coli is most frequently isolated, followed by Klebsiella pneumoniae, Enterococcus species, Pseudomonas species, and S. aureus.102 Fifteen percent of organisms isolated are intestinal anaerobes. Treatment of pancreatic infection usually involves débridement of infected tissue or drainage of infected pseudocysts or abscesses combined with antimicrobial therapy based on cultures of the infected site. Surgical management often requires multiple staged procedures. Antibiotics that penetrate into pancreatic tissue include the carbapenems, especially imipenem, fluoroquinolones, piperacillin, advanced generation cephalosporins, and metronidazole.103
Clostridium difficile Colitis
Manifestations of Clostridium difficile infection may range from asymptomatic carriage to a fulminant, relapsing, or life-threatening colitis.119,120 Antibiotics or chemotherapeutic agents have been associated with alteration of normal bowel flora and growth of C. difficile.119,120 Diarrhea may develop while a patient is receiving antibiotics or several weeks after completion of a course of antibiotics. Only strains that produce toxins are capable of causing diarrhea or colitis.119,120 Common symptoms include fever, which may be either low grade or high, crampy abdominal pain, and diarrhea, which is watery, profuse, and foul smelling. Approximately 50% of patients will have leukocytes in smears of the stool.119,120 Leukocytosis is common. The diagnosis is established by assaying the stool for C. difficile toxin. Complications of severe disease include electrolyte derangements, dehydration, toxic megacolon, and colonic perforation. Some patients may have little or no diarrhea but present with toxic megacolon, colonic perforation, peritonitis, or even septic shock without other localizing symptoms. An epidemic strain of C. difficile, NAP1/027, has enhanced toxin production and causes outbreaks of illness characterized by increased severity, poorer response to antibiotic therapy, and frequent relapse.121 Oral metronidazole remains first-line treatment for mild to moderate disease; empiric therapy with oral metronidazole should be started while testing for C. difficile toxin is being performed. If patients do not have a favorable response within 3 to 5 days, they should be switched to oral vancomycin. Oral vancomycin should be administered to those with severe infection or unresponsiveness to or intolerance of metronidazole. For patients who are unable to tolerate oral medication, IV metronidazole, vancomycin retention enemas, or vancomycin instillation through a colonic catheter should be administered. Surgery is indicated for complications such as severe toxic megacolon and colonic perforation. A variety of surgical procedures including diverting ileostomy, cecostomy, colostomy, and subtotal colectomy have been performed to manage toxic megacolon. Subtotal colectomy is considered the procedure of choice for the management of fulminant toxic megacolon.
Life-Threatening Infections of the Head and Neck
Ludwig’s Angina, Lateral Pharyngeal Space Infections, and Peritonsillar Abscess
Infections arising from the flora of the mouth and posterior pharynx can involve the fascial planes of the neck and have the potential to progress rapidly and cause serious life-threatening illness.122,123 In general, these infections are polymicrobial and the microbiology reflects the normal flora of the mouth. These infections spread rapidly through contiguous fascial spaces and it is essential that they be diagnosed and treated expeditiously to prevent serious sequelae such as airway obstruction, hematogenous infection, and mediastinitis. Potentially life-threatening infections of the head and neck may involve three cervical spaces. Fascial planes both separate and connect these areas. The spaces and infectious manifestations are as follows, and representative images are shown in Figure 54.4:
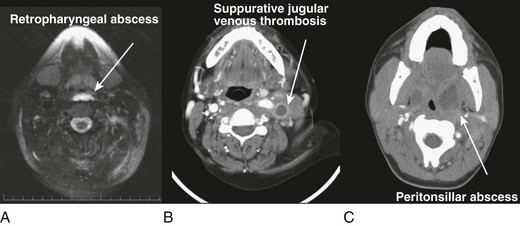
1. The submandibular space may be affected by infection involving the flora of the mouth and tongue. A bilateral cellulitis of the soft tissue, known as Ludwig’s angina, is the most important type of infection in the submandibular space. Manifestations of this infection include enlargement of the tongue and submandibular swelling. An odontogenic focus is the origin of infection of 70% to 90% of cases.123,124
2. The lateral pharyngeal space consists of an anterior and a posterior compartment divided by the styloid process. The anterior compartment is composed of musculature, and the posterior compartment has nerves and blood vessels. Infection in the anterior compartment causes soft tissue swelling that results in unilateral trismus caused by irritation of the internal pterygoid muscle, induration and swelling along the angle of the jaw, bulging of the palatine tonsil into the posterior pharynx, and systemic toxicity. Patients may present with unilateral neck or jaw pain along with ear pain and dysphagia. Pain may worsen when the head is turned because of compression of infected tissue. Dental infection, upper respiratory infection, pharyngitis, and otitis media with mastoiditis may all cause lateral pharyngeal space infection.125 The carotid sheath is within the posterior compartment and contains the internal carotid artery, the internal jugular vein, the vagus nerve, cranial nerves IX through XII, and lymph nodes. When infection occurs in this space, patients most commonly present with signs of sepsis without localizing signs at the neck. Generally signs and symptoms are related to complications from involvement of the neurovascular structures. The most common complication is suppurative jugular venous thrombosis. Bacteremia and septic emboli may be seen. The carotid artery may also rupture. The carotid sheath is dense and not easily penetrated, so generally arterial erosion is usually a complication of infections of longer duration (1 to 2 weeks). Intermittent bleeding from the mouth or nose may precede rupture.124 Cranial nerve palsies or Horner’s syndrome may occur.
3. The retropharyngeal space contains an area that extends from the base of the skull to the diaphragm and hence is a portal for neck infections to extend into the chest. Infections of the retropharyngeal space or prevertebral space may spread as a result of extension through this area. Retropharyngeal space abscesses are relatively uncommon and are most often seen in young children. These abscesses usually result from odontogenic infection, penetrating trauma, or peritonsillar abscess. Peritonsillar abscess, also known as “quinsy,” is an unusual complication of acute tonsillitis seen predominantly in adolescents and young adults. Patients usually present with fever, pharyngitis, odynophagia, dysphagia, trismus, drooling, and a muffled voice that has been described as “hot potato” in quality. On examination there is usually swelling of the anterior tonsillar pillars and soft palate. The most common symptoms of retropharyngeal space infection in adults are fever, dysphagia, pharyngeal pain, dyspnea, noisy breathing, and stiff neck. A lateral radiograph of the neck may reveal prevertebral soft tissue swelling. Any deep neck infection has the potential to spread to the mediastinum via the retropharyngeal space.
Other potential complications of infection of the oral cavity are aspiration pneumonia and lung abscess. Because most of these infections originate from an odontogenic focus, the microbiology reflects polymicrobial oral flora and generally includes Bacteroides species, aerobic streptococci, microaerophilic streptococci, peptostreptococci, fusobacteria, Veillonella species, and Actinomyces species. On occasion, enteric gram-negative bacilli, P. aeruginosa, or S. aureus may play a role.125
Deep neck infections are medical and surgical emergencies. Complications include hematogenous dissemination with sepsis syndrome, airway obstruction, necrotizing pneumonia or empyema, osteomyelitis of the mandible or maxilla, mediastinitis, or intracranial extension and cavernous sinus thrombosis. A contrast CT scan or MRI of the neck is important to help define the anatomy including the vascular structures and to indicate potential need for drainage. Consultation should be obtained from an otolaryngologist. When patients have dyspnea, stridor, or an inability to handle secretions, an artificial airway should be established. Airway obstruction is most likely to occur in infections of the submandibular space. Surgically obtained specimens should be cultured aerobically and anaerobically. For patients with peritonsillar abscess, high-dose IV penicillin is the therapy of choice. Other treatment options include clindamycin or ampicillin-sulbactam. Patients with peritonsillar abscess should undergo incision and drainage to prevent spontaneous rupture, aspiration pneumonia, airway obstruction, or dissection of infection into the lateral retropharyngeal space. Surgical drainage is especially important for infections involving the retropharyngeal and lateral pharyngeal space. Approximately half of cases of Ludwig’s angina in the submandibular space can be cured without surgical intervention.124
Lemierre Syndrome
Lemierre syndrome (postanginal sepsis) is a fulminant infectious syndrome caused by acute oropharyngeal infection that is complicated by secondary septic thrombophlebitis of the internal jugular vein. It is usually seen in healthy adolescents or young adults. Fusobacterium species are most commonly implicated. Complications include septicemia, pneumonia, empyema, meningitis, brain abscess, and vocal cord paralysis. This infection may complicate a routine case of infectious mononucleosis.126 Therapy usually consists of surgical drainage of the focus and broad-spectrum IV antibiotic therapy.
Epiglottitis
Acute infectious epiglottitis is an inflammatory process of the epiglottis, supraglottis, and surrounding soft tissues.127 Since the near disappearance of invasive H. influenzae infections in children following universal immunization, acute epiglottitis has become primarily a disease of adults, with an annual incidence of 1 case per 100,000 and peak incidence from ages 40 to 50. Patients present with severe pharyngitis; pain on swallowing; fever; and, less commonly, shortness of breath, hoarseness, and muffled voice. Findings on examination include marked anterior neck tenderness, lymphadenopathy, drooling, and respiratory distress.127,128 The standard of diagnosis for suspected epiglottitis in adults is visualization of the epiglottis with indirect laryngoscopy.127 Radiography has low sensitivity and specificity. Treatment includes maintenance of an airway; depending on severity and rapidity of onset of symptoms, this may include emergent tracheostomy, elective intubation, or only close observation in an ICU for the mildest cases.127 Antibiotics active against the most commonly implicated pathogens, H. influenzae and β-hemolytic streptococci, are administered, and corticosteroids are generally recommended.127,128 Disease may be more aggressive in HIV-infected or other immunocompromised individuals.
Mediastinitis
Acute mediastinitis is an infection of mediastinal structures that can develop from direct extension of pharyngeal and neck infections (descending necrotizing mediastinitis), from esophageal trauma or rupture, or as a complication of cardiothoracic surgical procedures.129 Descending necrotizing mediastinitis and mediastinitis from esophageal procedures have both become uncommon, and most cases are seen as complications of cardiothoracic surgery procedures or from trauma.129,130 Symptoms of mediastinitis may initially be mild, but as disease progresses, patients will develop chest pain; dysphagia; and respiratory distress, as well as fever, tachycardia, crepitus, and localized swelling. Sepsis is common. Patients with postcardiothoracic mediastinitis will generally have evidence of local or deep sternal wound infection. Leukocytosis is typical. Plain films may show mediastinal widening; mediastinal air-fluid levels; and subcutaneous, mediastinal, or pericardial air. CT is more sensitive than plain radiographs for diagnosis; contrast esophagography with water-soluble contrast is the optimal study for esophageal perforation.129
Treatment of infection related to descending neck infection or esophageal perforation requires broad-spectrum antibiotics directed at oropharyngeal flora including streptococci, oral anaerobes, and gram-negative bacilli in addition to surgical intervention.129 Mediastinitis following cardiothoracic surgery is most commonly caused by staphylococci, but a large variety of other gram-positive and gram-negative organisms have been implicated. Treatment requires surgical drainage and débridement along with antibiotic therapy.130
Serious Vector-Borne Infections
Rocky Mountain Spotted Fever
Tick-borne rickettsial diseases cause severe illness and death in otherwise healthy individuals. Although the various rickettsial diseases may have distinct epidemiologic and etiologic differences, they are clinically similar.131 RMSF is caused by Rickettsia rickettsii, which is a gram-negative, obligate, intracellular bacterium. Infection is transmitted to humans by a variety of ticks but most frequently by the dog tick, Dermacentor variabilis. The incidence is greatest in the southeastern and south central United States. The vast majority of cases occur from April to September. Reported risks for infection include living in wooded areas and exposure to dogs. R. rickettsii infects endothelial cells, resulting in vasculitis, which leads to the characteristic rash and involvement of the lungs, brain, and other organs.131–134 Following an incubation period of 3 to 12 days, patients frequently present with fever, rash, and evidence or history of a tick bite, although 30% to 40% of patients do not recall a tick bite.131 Other symptoms include chills; myalgias; nausea; vomiting; abdominal pain that may be severe enough to mimic an acute abdomen; diarrhea; headache; photophobia; mental status changes; conjunctival injection; and, periodically, cough or arrhythmias secondary to myocarditis. The rash is classically an erythematous macular rash that appears initially on the ankles/soles and wrists/palms and spreads centripetally to the arms, legs, trunk, neck, and face.132–134 The lesions evolve to become petechial. The rash typically appears after 2 to 6 days of illness. Although rash is the hallmark of this illness, up to 20% of patients are “spotless” or have an atypical rash at presentation.135 The most characteristic laboratory abnormality is thrombocytopenia. The WBC count is usually normal but more than two thirds of cases have increased band forms. The creatine phosphokinase level may be elevated, as may transaminases and bilirubin.131 The chest radiograph may reveal infiltrates consistent with pneumonitis or acute respiratory distress syndrome (ARDS). RMSF is frequently a severe illness. Serious complications, in addition to ARDS, include renal failure, disseminated intravascular coagulopathy (DIC), hemophagocytic syndrome, meningoencephalitis, and gangrene.131
Rickettsial infections may be difficult to diagnose. The best way to establish the diagnosis of RMSF in patients with a rash is by obtaining a skin biopsy for immunohistochemical or direct immunofluorescent staining. This test has a sensitivity of 70% and specificity of 100%.132 PCR can also be performed on tissue specimens.131 However, both immunohistochemical staining and RMFS PCR may not be readily available in many institutions. Serologic testing by indirect immunofluorescence or enzyme-linked immunosorbent assay (ELISA) may also be performed. Empiric treatment with doxycycline should be instituted for suspected RMSF before laboratory confirmation of a diagnosis. Distinguishing RMSF from meningococcemia is especially important. Gastrointestinal symptoms, a pulse-temperature disparity, periorbital edema, edema of the extremities, conjunctival injection, hepatosplenomegaly, and elevated serum transaminases are more likely in RMSF. If there is any doubt as to which infection is present, treatment for both should be instituted empirically.
Ehrlichioses and Anaplasmosis
Illnesses that cause fever and rash are listed in Box 54.3. Other serious rickettsial diseases include ehrlichioses and anaplasmosis, caused by Ehrlichia chaffeensis, E. ewingii, and Anaplasma phagocytophilium, respectively.131 They have a similar presentation, but rash is much less commonly seen than in RMSF. These infections are transmitted by ticks and are distributed across the United States and Europe. All are intracellular pathogens that infect leukocytes. Common laboratory abnormalities include leukopenia, relative lymphopenia, presence of atypical lymphocytes, eosinopenia, and thrombocytopenia.136 Anemia and renal involvement are rare. PCR of serum is now the rapid diagnostic test of choice. Blood smear microscopy might reveal the presence of morulae in infected leukocytes, which is highly suggestive of anaplasmosis or, less commonly, ehrlichiosis.131 Serologic results can also be diagnostic. Complications include ARDS, bleeding, rhabdomyolysis, and myocarditis.136
Malaria
Malaria is a protozoan infection transmitted by female anopheline mosquitoes. The severity of malaria infection depends on a variety of factors including host immunity and age and the species of malaria. Of the four main human pathogens, Plasmodium falciparum causes the most serious infection. Chloroquine-resistant P. falciparum has spread through many parts of the world. The incubation period of P. falciparum is approximately 12 days. It parasitizes all ages of red blood cells and causes the highest degree of parasitemia of any of the species.137 P. falciparum has worldwide distribution and causes a severe illness frequently termed blackwater fever. Common symptoms and signs include fever, chills, headache, myalgia, arthralgia, and hepatosplenomegaly. Other symptoms include jaundice, vomiting, diarrhea, and nonproductive cough.137 Severe malaria is a multisystem illness with a mortality rate of up to 25% in the nonimmune, untreated patient.137 Defining criteria for severe malaria include (1) severe normocytic anemia, (2) renal failure, (3) pulmonary edema, (4) hypoglycemia, (5) shock, (6) DIC, (7) metabolic acidosis, and (8) cerebral involvement with coma or generalized seizures. Other signs or symptoms frequently present in severe disease include altered mental status, prostration, jaundice, and high-grade fever.137,138 Additional laboratory features include thrombocytopenia, elevated transaminases, hyperbilirubinemia, evidence of coagulopathy, elevated blood urea nitrogen (BUN) and creatinine levels, and macroscopic hemoglobinuria. Levels of parasitemia are often high. Patients may develop pulmonary edema, or pulmonary edema may occur after successful treatment of parasitemia. ARDS or secondary infection with bacterial pneumonia may also occur.
The differential diagnosis is broad and includes bacterial sepsis, meningitis, rickettsial infections, pneumonia, viral hemorrhagic fever, leptospirosis, severe influenza, meningococcemia, typhoid fever, and viral hepatitis. The diagnostic test of choice is the thick/thin peripheral Giemsa-stained blood smear, which confirms the diagnosis. Rapid diagnostic tests are becoming increasingly useful. However, patients from an endemic area may occasionally present with another serious illness that may be erroneously attributed to malaria because of incidental parasitemia. Treatment for severe malaria regardless of species consists of IV quinine (not available in the United States) or IV quinidine combined with IV doxycycline or IV clindamycin.139,140 When using quinidine, continuous cardiac monitoring should be performed. IV artesunate is available as an investigational new drug through the CDC for those with severe malaria who are unable to obtain IV quinidine or who are intolerant to it, have a contraindication, or have failed therapy. IV therapy should be continued until the parasite density is less than 1% and oral therapy can be tolerated. Adjunctive therapy for severe malaria may include exchange transfusion, although this is controversial.139 In addition, broad-spectrum antibiotics for bacterial sepsis and pneumonia should be given if there is concern for secondary bacterial infection.
Dengue
Hemorrhagic fever is caused by a variety of viruses, and the hallmark is bleeding. Generally dengue has a geographic endemicity, being seen predominantly in Africa, South America, and Asia. Dengue is transmitted by mosquitoes and has no other reservoir except humans. It is now endemic in at least 112 countries worldwide including many parts of the Caribbean, Mexico, Puerto Rico, and Central America and has recently reemerged in the United States in the Florida Keys and along the Mexican border.141–144 In many areas the mosquito vector is Aedes aegypti. This mosquito species has adapted to man-made conditions, and therefore urban transmission is frequent. The virus has four serotypes, each with a number of genotypes.143 Dengue is generally divided into four clinical syndromes: a mild influenza-like illness; classic dengue (characterized by fever, retro-orbital headache, severe bone pain and myalgia, maculopapular rash, and nausea and vomiting); dengue hemorrhagic fever (DHF); and dengue shock syndrome (DSS).141 DHF or DSS may manifest after a few days of typical dengue symptoms, and classically symptoms start as the temperature normalizes.144 Those with DHF have bleeding, petechiae, ascites, pleural effusion, and sometimes encephalopathy. Laboratory features include hemoconcentration, leukopenia, elevated transaminases, and thrombocytopenia.144 The differential diagnosis includes many of the infectious entities also in the differential diagnosis of malaria. Noninfectious illnesses in the differential diagnosis include hemolytic uremic syndrome and thrombotic thrombocytopenic purpura. Diagnosis is established serologically, and treatment is supportive.143
Severe Viral Infections
Hantavirus Pulmonary Syndrome
Acute infections caused by species of hantavirus are transmitted to humans from rodents and are characterized by nephritis and hemorrhage or by a syndrome of acute noncardiogenic pulmonary edema.145 Four hantaviruses are associated with hantavirus pulmonary syndrome (HPS). This syndrome was first recognized in the southwestern United States. Rodents, especially deer mice, are the host. Transmission to humans occurs by inhalation of aerosols of rodent urine or feces. Initial symptoms of HPS resemble those of influenza and consist of fever, myalgia, headache, and gastrointestinal symptoms. Two to 15 days later, acute noncardiogenic pulmonary edema and shock develop.146–148 Laboratory findings at this stage include leukocytosis, hemoconcentration, and thrombocytopenia. Chest radiographic findings include increased vascular markings consistent with pulmonary edema, bilateral infiltrates, and pleural effusions.149 Treatment is supportive and consists of ventilator support and treatment of shock. This syndrome has a high mortality rate of 50% to 70%, but those who survive improve rapidly after 5 to 7 days and often have complete recovery within 2 to 3 weeks.
Influenza
Influenza results from infection with influenza A or B virus. Infection occurs in yearly epidemics, typically during the winter in temperate climates, with occasional worldwide epidemics referred to as pandemics, which occur when there is antigenic shift (a major antigenic change resulting in a new subtype of influenza A).150 These viruses are spread from person to person primarily through coughing and sneezing.151 Onset of symptoms is abrupt and occurs after an incubation period of a day or two.152 Symptoms include fever, chills, headache, myalgia, sore throat, and malaise.153 Respiratory symptoms, especially a dry cough, are usually present. As systemic signs and symptoms decrease, respiratory complaints become more prominent. Of these, cough is the most frequent and may persist 1 to 2 weeks after fever resolves. Leukocytosis is common early in the illness, and mild leukopenia may be observed later. Most cases are not associated with any significant complications, but when complications do occur, pulmonary complications are the most frequent. Two types of pulmonary complications are recognized: primary influenza viral pneumonia and secondary bacterial pneumonia.154 Primary influenza viral pneumonia occurs mainly in individuals with cardiovascular disease or in pregnant women. Rapid progression of fever, cough, dyspnea, and hypoxemia usually occurs. Chest radiographs reveal bilateral findings consistent with pulmonary edema. Patients may develop ARDS. Culture of the sputum fails to reveal significant bacteria, whereas molecular diagnostic tests and viral cultures will demonstrate influenza virus. Severe influenza viral pneumonia requires intensive monitoring and support. Mortality rate is high.
Secondary bacterial pneumonia is more common than primary viral pneumonia. It occurs most often in the elderly or those with preexisting pulmonary disease. Following a classic influenza syndrome and a period of improvement of a few days, there is recrudescence of fever and cough accompanied by sputum production and consolidation on chest radiograph. Gram stain and culture of sputum most often demonstrate S. pneumoniae, H. influenzae, or S. aureus. Other rare complications of influenza include Reye syndrome, which is an often fatal CNS and hepatic complication, myositis, transverse myelitis, myocarditis, and pericarditis.155 Influenza virus is readily isolated from nasal or throat specimens, sputum, or tracheal secretions in the first 2 or 3 days of illness. The neuraminidase inhibitors, inhaled zanamivir and oral oseltamivir, are active against influenza A and B viruses and are effective in treating acute influenza if started early in the illness.150 Secondary bacterial pneumonia should be treated with antibiotics. A pandemic with H1N1 occurred in 2009 and public health readiness continues for the next pandemic.156 Respiratory syncytial virus is emerging as an important cause of serious illness in the elderly and high-risk adults with clinical manifestations, length of hospital stay, use of ICU, and mortality rates similar to influenza.157
Potential Agents of Bioterrorism
Anthrax
Bacillus anthracis, the causative agent of anthrax, is an aerobic, gram-positive, sporulating bacillus. When human infection occurs, spores germinate in blood and tissue. Human infection can be of three types: (1) cutaneous, (2) inhalational, and (3) gastrointestinal.158 Cutaneous is the most common and characteristically appears as a painless papule that evolves to a vesicular stage and then to a depressed black eschar surrounded by a ring of vesicles. Untreated, it carries a mortality rate of approximately 20%.158
The inhalational form is the form most likely to be encountered in the critical care setting. After an incubation period of generally 1 to 7 days but potentially up to 60 days, patients present with fever, malaise, dry cough, and an influenza-like illness. Progression to severe respiratory distress and septic shock occurs. The hallmark of this infection is a hemorrhagic mediastinitis.158,159 Mortality rate has been as high as 85%. In virtually all cases, chest radiographs are abnormal and show either a widened mediastinum or pleural effusions. CT scan is particularly sensitive in detecting mediastinal changes.160 Blood cultures are positive in 70% of cases. Clinical suspicion should be raised by the sudden appearance of multiple cases of severe influenza-like illness with a fulminant course and high mortality rates. The diagnosis is generally established by culturing the organism from blood, CSF, pleural fluid, or vesicular fluid; by PCR; or by biopsy. Therapy is initially empiric and consists of the combination of ciprofloxacin or doxycycline plus clindamycin and rifampin.158,159
Smallpox
Smallpox is caused by the variola virus. This serious infection is highly contagious and fatal in about 30% of cases. Two major clinical forms exist, with the most common being variola major. This is a severe form with extensive rash and high fever. After an incubation period of 12 to 14 days, patients develop high fever, malaise, headache, myalgias, and vomiting. The rash appears initially as small intraoral spots and within 24 hours develops on the face, then spreads to the legs, feet, arms, and hands.161 The rash appears as papules, which are filled with a thick, opaque fluid and have a depressed center. They evolve into pustules, which are raised, round, and firm to the touch. The differential diagnosis includes varicella. Treatment is supportive. The antiviral agent cidofovir may have activity.162
Plague
The causative agent of plague is Yersinia pestis, an aerobic gram-negative bacillus. The three clinical forms are (1) bubonic, (2) pneumonic, and (3) septicemic.163,164 Pneumonic plague is transmitted person to person through inhalation of contaminated aerosols and is highly contagious. After an incubation period of 2 to 3 days, patients develop fever, chills, headache, hemoptysis, dyspnea, stridor, cyanosis, respiratory failure, circulatory collapse, and bleeding.163,164 Diagnosis is based on clinical suspicion and confirmed by culture. Treatment is with streptomycin and doxycycline.163,164
Tularemia
Tularemia is caused by Franciscella tularensis, which is a gram-negative coccobacillus. Types of infection include ulceroglandular, typhoidal, and pneumonic. If there were to be an intentional release of this agent, infection would likely occur via the aerosol route.165,166 Symptoms include cough, substernal pain, abdominal pain, prostration, fever, chills, and headache. Diagnosis is established by culture onto special media or by serologic testing. Treatment is with streptomycin.
Viral Hemorrhagic Fevers
Viral hemorrhagic fevers are caused by several different families of viruses. Symptoms generally include fever, myalgia, hemorrhage, shock, coma, seizures, and possibly renal failure. The diagnosis is established by viral isolation or serologically. Treatment involves supportive care. The antiviral agent ribavirin may have a role in treatment.167
References
1. van de Beek, D, de Gans, J, Tunkel, AR, et al. Community-acquired bacterial meningitis in adults. N Engl J Med. 2006; 354:44–53.
2. van de Beek, D, de Gans, J, Spanjaard, L, et al. Clinical features and prognostic factors in adults with bacterial meningitis. N Engl J Med. 2004; 352:1849–1859.
3. Thigpen, MC, Whitney, CG, Messonnier, NE, et al. Bacterial meningitis in the United States 1998-2007. N Engl J Med. 2011; 364(21):2016–2025.
4. Durand, ML, Calderwood, SB, Weber, DJ, et al. Acute bacterial meningitis in adults: A review of 493 episodes. N Engl J Med. 1993; 328:21–28.
5. Tunkel, AR, Hartman, BJ, Kaplan, SL, et al. Practice guidelines for the management of bacterial meningitis. Clin Infect Dis. 2004; 39:1267–1284.
6. Lexau, CA, Lynefield, R, Danila, R, et al. Changing epidemiology of invasive pneumococcal disease among older adults in the era of pediatric pneumococcal conjugate vaccine. JAMA. 2005; 294:2043–2051.
7. Bilukha, OO, Rosenstein, N, National Center for Infectious Diseases, Centers for Disease Control and Prevention (CDC). Prevention and control of meningococcal disease. Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2005; 54(RR-7):1–21.
8. Mylonakis, E, Hohmann, EL, Calderwood, SB. Central nervous system infection with Listeria monocytogenes: 33 years’ experience at a general hospital and review of 776 episodes from the literature. Medicine (Baltimore). 1998; 77:313–336.
9. Hasbun, R, Abrahams, J, Jekel, J, et al. Computed tomography of the head before lumbar puncture in adults with suspected meningitis. N Engl J Med. 2001; 345:1727–1733.
10. Korein, J, Cravisto, H, Leicach, M. Reevaluation of lumbar puncture: A study of 129 patients with papilledema or intracranial hypertension. Neurology. 1959; 9:290–297.
11. Spanos, A, Harrell, FE, Jr., Durack, DT. Differential diagnosis of acute meningitis: An analysis of the predictive value of initial observations. JAMA. 1989; 262:2700–2707.
12. Brouwer, MC, Tunkel, AR, van de Beek, D. Epidemiology, diagnosis and antimicrobial treatment of acute bacterial meningitis. Clin Microbiol Rev. 2010; 23(3):467–492.
13. de Gans, J, van de Beek, D, European Dexamethasone in Adulthood Bacterial Meningitis Study Investigators. Dexamethasone in adults with bacterial meningitis. N Engl J Med. 2002; 347:1549–1556.
14. van de Beek, D, Farrar, JJ, de Gans, J, et al. Adjunctive corticosteroids in bacterial meningitis: A meta-analysis of individual patient data. Lancet Neurol. 2010; 9(3):254–263.
15. Wijdicks, EFM. The Clinical Practice of Critical Care Neurology, 2nd ed. New York: Oxford University Press; 2003.
16. Flores-Cordero, JM, Amaya-Villar, R, Rincon-Ferrari, MD, et al. Acute community-acquired bacterial meningitis in adults admitted to the intensive care unit: Clinical manifestations, management and prognostic factors. Intensive Care Med. 2003; 29:1967–1973.
17. Lindvall, P, Ahlm, C, Ericsson, M, et al. Reducing intracranial pressure may increase survival among patients with bacterial meningitis. Clin Infect Dis. 2004; 38:384–390.
18. Kaplan, SL, Mason, EO, Jr. Management of infections due to antibiotic-resistant Streptococcus pneumoniae. Clin Microbiol Rev. 1998; 11:628–644.
19. Tunkel, AR, Glaser, CA, Bloch, KC, et al. The management of encephalitis: Clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2008; 47(3):303–327.
20. Steiner, I, Budka, H, Chaudhuri, A, et al. Viral encephalitis: A review of diagnostic methods and guidelines for management. Euro J Neurol. 2005; 12:331–343.
21. Whitley, RJ, Gnann, JW. Viral encephalitis: Familiar infections and emerging pathogens. Lancet. 2002; 359:507–513.
22. Tyler, KL. Herpes simplex virus infections of the central nervous system: Encephalitis and meningitis, including Mollaret’s. Herpes. 2004; 11(Suppl 2):57A–64A.
23. Lima, MA, Koralnik, IJ. New features of progressive multifocal leukoencephalopathy in the era of highly active antiretroviral therapy and natalizumab. J Neurovirol. 2005; 11(Suppl 3):52–57.
24. Hayes, EB, Sejvar, JJ, Zaki, SR, et al. Virology, pathology, and clinical manifestations of West Nile virus disease. Emerg Infect Dis. 2005; 11:1174–1179.
25. Sejvar, JJ, Haddad, MB, Tierney, BC, et al. Neurologic manifestations and outcome of West Nile virus infection. JAMA. 2003; 290:511–515.
26. Willoughby, RE, Jr., Tieves, KS, Hoffman, GM, et al. Survival after treatment of rabies with induction of coma. N Engl J Med. 2005; 352:2508–2514.
27. Gunther, G, Haglund, M. Tickborne encephalopathies: Epidemiology, diagnosis, treatment and prevention. CNS Drugs. 2005; 19:1009–1032.
28. Marra, CM. Neurosyphilis. Curr Neurol Neurosci Rep. 2004; 4:435–440.
29. Glaser, CA, Gilliam, S, Schnurr, D, et al. In search of encephalitis etiologies: Diagnostic challenges in the California Encephalitis Project, 1998-2000. Clin Infect Dis. 2003; 36:731–742.
30. Aurelius, E, Johansson, B, Skoldenberg, B, et al. Encephalitis in immunocompetent patients due to herpes simplex virus type 1 or 2 as determined by two specific polymerase chain reaction and antibody assays of cerebrospinal fluid. J Med Virol. 1993; 39:179–186.
31. Domingues, RB, Tsanaclis, AM, Pannuti, CS, et al. Evaluation of the range of clinical presentations of herpes simplex encephalitis by using polymerase chain reaction assay of cerebrospinal fluid samples. Clin Infect Dis. 1997; 25:86–91.
32. McCabe, K, Tyler, KL, Tanabe, J. Diffusion-weighted MRI abnormalities as a clue to the diagnosis of herpes simplex encephalitis. Neurology. 2003; 61:1015–1016.
33. Hanson, KE, Alexander, BD, Woods, C, et al. Validation of laboratory screening criteria for herpes simplex virus testing of cerebrospinal fluid. J Clin Microbiol. 2007; 45(3):721–724.
34. Aurelius, E, Johansson, B, Skoldenberg, B, et al. Rapid diagnosis of herpes simplex encephalitis by nested polymerase chain reaction assay of cerebrospinal fluid. Lancet. 1991; 337:189–192.
35. Lakeman, FD, Whitley, RJ. Diagnosis of herpes simplex encephalitis: Application of polymerase chain reaction to cerebrospinal fluid from brain-biopsied patients and correlation with disease. J Infect Dis. 1995; 171:857–863.
36. Centers for Disease Control and Prevention (CDC). West Nile virus disease and other arboviral diseases—United States, 2010. MMWR Morb Mortal Wkly Rep. 2011; 60(30):1009–1013.
37. Li, J, Loeb, JA, Shy, ME, et al. Asymmetric flaccid paralysis: A neuromuscular presentation of West Nile virus infection. Ann Neurol. 2003; 53:703–710.
38. Calfee, DP, Wispelwey, B. Brain abscess. Semin Neurol. 2000; 20:353–360.
39. Mathisen, GE, Johnson, JP. Brain abscess. Clin Infect Dis. 1997; 25:763–781.
40. Tonon, E, Scotton, PG, Gallucci, M, et al. Brain abscess: Clinical aspects of 100 patients. Int J Infect Dis. 2006; 10:103–109.
41. Lu, CH, Chang, WN, Lin, YC, et al. Bacterial brain abscess: Microbiological features, epidemiological trends and therapeutic outcomes. Q J Med. 2002; 95:501–509.
42. Mamidi, A, DeSimone, JA, Pomerantz, RJ. Central nervous system infections in individuals with HIV-1 infection. J Neurovirol. 2002; 8:158–167.
43. Ratnaike, TE, Das, S, Gregson, BA, Mendelow, AD. A review of brain abscess surgical treatment—78 years: Aspiration versus excision. World Neurosurg. 2011; 76(5):431–436.
44. Siddiq, F, Chowfin, A, Tight, R, et al. Medical vs. surgical management of spinal epidural abscess. Arch Intern Med. 2004; 164:2409–2412.
45. Reishaus, E, Waldbaur, H, Seeling, W. Spinal epidural abscess: A meta-analysis of 915 cases. Neurosurg Rev. 2000; 232:175–202.
46. Maslen, D, Jones, SR, Crislip, MA. Spinal epidural abscess: Optimizing patient care. Arch Intern Med. 1993; 153:1713–1721.
47. Tsiodras, V, Falagas, ME. Clinical assessment and medical treatment of spine infections. Clin Orthop Res. 2006; 444:38–50.
48. Tleyjeh, IM, Steckelberg, JM, Murad, HS, et al. Temporal trends in infective endocarditis: A population-based study in Olmsted County, Minnesota. JAMA. 2005; 293:3022–3028.
49. Fowler, VG, Miro, JM, Hoen, B, et al. Staphylococcus aureus endocarditis: A consequence of medical progress. JAMA. 2005; 293:3012–3021.
50. Baddour, LM, Wilson, WR, Bayer, AS, et al. Infective endocarditis: Diagnosis, antimicrobial therapy, and management of complications, a statement for healthcare professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, and the Councils on Clinical Cardiology, Stroke, and Cardiovascular Surgery and Anesthesia, American Heart Association. Circulation. 2005; 111:e394–e433.
51. Hasbun, R, Vikram, HR, Barakat, LA, et al. Complicated left-sided native valve endocarditis in adults: Risk classification for mortality. JAMA. 2003; 289:1933–1940.
52. Thuny, F, Grisoli, D, Collart, F, et al. Management of infective endocarditis: Challenges and perspectives. Lancet. 2012; 379(9819):965–975.
53. Durack, DT, Lukes, AS, Bright, DK. New criteria for diagnosis of infective endocarditis: Utilization of specific echocardiographic findings. Am J Med. 1994; 96:200–209.
54. Li, JS, Sexton, DJ, Mick, N, et al. Proposed modifications to the Duke criteria for the diagnosis of infective endocarditis. Clin Infect Dis. 2000; 30:633–638.
55. Heiro, M, Nikoskelainen, J, Engblom, E, et al. Neurologic manifestations of infective endocarditis: A 17-year experience in a teaching hospital in Finland. Arch Intern Med. 2000; 160:2781–2787.
56. Vilacosta, I, Graupner, C, San Roman, JA, et al. Risk of embolization after institution of antibiotic therapy for infective endocarditis. J Am Coll Cardiol. 2002; 39:1489–1495.
57. Snygg-Martin, U, Rasmussen, RV, Hassager, C, et al. Warfarin therapy and the incidence of cerebrovascular complications of left-sided native valve endocarditis. Eur J Clin Microbiol and Infect Dis. 2011; 30(2):151–157.
58. Liu, C, Bayer, A, Cosgrove, SE, et al. Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin Infect Dis. 2011; 52(3):e18–e55.
59. Cosgrove, SE, Vigliani, GA, Campion, M, et al. Initial low dose gentamicin for Staphylococcus aureus bacteremia and endocarditis is nephrotoxic. Clin Infect Dis. 2009; 48(6):713–721.
60. Mermel, LA, Allon, M, Bouzon, E, et al. Clinical practice guidelines for the diagnosis and management of intravascular catheter-related infection: 2009 update by the Infectious Diseases Society of America. Clin Infect Dis. 2009; 49(1):1–45.
61. Saxena, AK, Panhotra, BR. Haemodialysis catheter-related bloodstream infections: Current treatment options and strategies for prevention. Swiss Med Wkly. 2005; 135:127–138.
62. Baddour, LM, Epstein, AE, Erickson, CC, et al. Update on cardiovascular implantable electronic device infections and their management: A scientific statement from the American Heart Association. Circulation. 2010; 121(3):458–477.
63. Greenspon, AJ, Patel, JD, Lau, E, et al. 16-year trends in the infection burden for pacemakers and implantable cardioverter-defibrillators in the United States 1993-2008. J Am Coll Cardiol. 2011; 58(10):1001–1006.
64. Weinstein, MP, Towns, ML, Quartey, SM, et al. The clinical significance of positive blood cultures in the 1990s: A prospective comprehensive evaluation of the microbiology, epidemiology and outcome of bacteremia and fungemia in adults. Clin Infect Dis. 1997; 24:584–602.
65. Leiboici, L, Konisberger, H, Pitlik, SD, et al. Bacteremia and fungemia of unknown origin in adults. Clin Infect Dis. 1992; 14:436–443.
66. Melles, DC, de Marie, S. Prevention of infections in hyposplenic and asplenic patients: An update. Neth J Med. 2004; 62:45–52.
67. Johnson, DH, Cunha, B. Infections in cirrhosis. Infect Dis Clin North Am. 2001; 15:363–371.
68. Bow, EJ. Management of the febrile neutropenic cancer patient: Lessons from 40 years of study. Clin Microbiol Infect. 2005; 11(Suppl 5):24–29.
69. Thompson, MJ, Ninis, N, Perera, R, et al. Clinical recognition of meningococcal disease in children and adolescents. Lancet. 2006; 367:397–403.
70. Rosenstein, N, Perkins, B, Stephens, D, et al. Meningococcal disease. N Engl J Med. 2001; 344:1378–1388.
71. Trampuz, A, Widmer, AF, Fluckiger, U, et al. Changes in the epidemiology of pneumococcal bacteremia in a Swiss university hospital during a 15-year period, 1986-2000. Mayo Clin Proc. 2004; 79:604–612.
72. Taylor, SN, Sanders, CY. Unusual manifestations of invasive pneumococcal infection. Am J Med. 1999; 107(Suppl 1A):12–27.
73. Fowler, VG, Jr., Olsen, MK, Corey, GR, et al. Clinical identifiers of complicated Staphylococcus aureus bacteremia. Arch Intern Med. 2003; 163:2066–2072.
74. Mitchell, DH, Howden, BP. Diagnosis and management of Staphylococcus aureus bacteriemia. Intern Med J. 2005; 35(Suppl 2):S17–S24.
75. McCormick, JK, Yarwood, JM, Schlievert, PM. Toxic shock syndrome and bacterial superantigens: An update. Annu Rev Microbiol. 2001; 55:77–104.
76. Haijeh, RA, Reingold, A, Weil, A, et al. Toxic shock syndrome in the United States: Surveillance update, 1979-1996. Emerg Infect Dis. 1999; 5:807–810.
77. Shands, KN, Schmid, GP, Dan, BB, et al. Toxic-shock syndrome in menstruating women: Association with tampon use and Staphylococcus aureus and clinical features in 52 cases. N Engl J Med. 1980; 303:1436–1442.
78. Stevens, DI. Streptococcal toxic-shock syndrome: Spectrum of disease, pathogenesis, and new concepts in treatment. Emerg Infect Dis. 1995; 1:69–78.
79. Davies, HD, McGeer, A, Schwartz, B, et al. Invasive group A streptococcal infections in Ontario, Canada. Ontario Group A Streptococcal Study Group. N Engl J Med. 1996; 335:547–554.
80. Kaul, R, McGeer, A, Norrby-Teglund, A, et al. Intravenous immunoglobulin therapy for streptococcal toxic shock syndrome—a comparative observational study. Clin Infect Dis. 1999; 28:800–807.
81. Fischer, M, Bhatnagar, J, Guarner, J, et al. Fatal toxic shock syndrome associated with Clostridium sordellii after medical abortion. N Engl J Med. 2005; 353(22):2352–2360.
82. Centers for Disease Conrol and Prevention (CDC). Tetanus surveillance—United States, 2001-2008. MMWR Morb Mortal Wkly Rep. 2011; 60(12):365–369.
83. Villar, RG, Elliott, SP, Davenport, KM. Botulism: The many faces of botulinum toxin and its potential for bioterrorism. Infect Dis Clin North Am. 2006; 20:313–327.
84. Broder, KR, Cortese, MM, Iskander, JK, et al. Preventing tetanus, diphtheria, and pertussis among adolescents: Use of tetanus toxoid, reduced diphtheria toxoid and acellular pertussis vaccines recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2006; 55(RR-3):1–34.
85. Ustin, JS, Malangoni, MA. Necrotizing soft tissue infections. Crit Care Med. 2011; 39(9):2156–2162.
86. Hasham, S, Matteucci, P, Stanley, PRW, et al. Necrotising fasciitis. BMJ. 2005; 330:830–833.
87. Liu, YM, Chi, CY, Ho, MW, et al. Microbiology and factors affecting mortality in necrotizing fasciitis. J Microbiol Immunol Infect. 2005; 38(6):430–435.
88. Young, MH, Engleberg, NC, Mulla, ZD, et al. Therapies for necrotizing fasciitis. Expert Opin Biol Ther. 2006; 6:155–165.
89. Plosker, GL, Figgitt, DP. Linezolid: A pharmacoeconomic review of its use in serious gram-positive infections. Pharmacoeconomics. 2005; 23:945–964.
90. Darabi, K, Abdel-Wahab, O, Dzik, WH. Current usage of intravenous immune globulin and the rationale behind it: The Massachusetts General Hospital data and a review of the literature. Transfusion. 2006; 46:741–753.
91. Vinh, DC, Embil, JM. Rapidly progressive soft tissue infections. Lancet Infect Dis. 2005; 5:501–513.
92. Bisno, AL, Stevens, DL. Streptococcal infections of skin and soft tissues. N Engl J Med. 1996; 334:240–244.
93. Olsofka, JN, Carrillo, EH, Spain, DA, et al. The continuing challenge of Fournier’s gangrene in the 1990s. Am Surg. 1999; 65(12):1156–1159.
94. Jallali, N, Withey, S, Butler, PE. Hyperbaric oxygen as adjuvant therapy in the management of necrotizing fasciitis. Am J Surg. 2005; 189:462–466.
95. Chiang, SR, Chuang, YC. Vibrio vulnificus infection: Clinical manifestations, pathogenesis, and antimicrobial therapy. J Microbiol Immunol Infect. 2003; 36(2):81–88.
96. Drews, TD, Temte, JL, Fox, BC. Community-associated methicillin-resistant Staphylococcus aureus: Review of an emerging public health concern. WMJ. 2006; 105:52–57.
97. Kluytmans-Vandenbergh, MF, Kluytmans, JA. Community-acquired methicillin-resistant Staphylococcus aureus: Current perspectives. Clin Microbiol Infect. 2006; 1:9–15.
98. Kollef, MH, Micek, ST. Methicillin-resistant Staphylococcus aureus: A new community-acquired pathogen? Curr Opin Infect Dis. 2006; 19:161–168.
99. Maltezou, HC, Giamarellou, H. Community-acquired methicillin-resistant Staphylococcus aureus infections. Int J Antimicrob Agents. 2006; 27:87–96.
100. Thielman, NM, Crump, JA, Guerrant, RL. Enteric fever and other causes of abdominal symptoms with fever. In: Mandell GL, Bennett JE, Dolin R, eds. Principles and Practice of Infectious Diseases. 7th ed. Philadelphia: Elsevier; 2010:1399–1412.
101. Sanchez-Vargas, FM, Abu-El-Haija, MA, Gomez-Duarte, OG. Salmonella infections: An update on epidemiology, management, and prevention. Travel Med Infect Dis. 2011; 9(6):263–277.
102. Marshall, JC, Innes, M. Intensive care unit management of intra-abdominal infection. Crit Care Med. 2003; 31:2228–2237.
103. Solomkin, JS, Mazuski, JE, Bradley, JS, et al. Diagnosis and management of complicated intra-abdominal infection in adults and children: Guidelines by the Surgical Infection Society and the Infectious Diseases Society of America. Clin Infect Dis. 2010; 50(2):133–164.
104. Tellado, JM, Wilson, SE. Empiric treatment of nosocomial intra-abdominal infections: A focus on the carbapenems. Surg Infect. 2005; 6:329–343.
105. Blot, S, De Waele, JJ. Critical issues in the clinical management of complicated intra-abdominal infections. Drugs. 2005; 65:1611–1620.
106. Wong, PF, Gilliam, AD, Kuman, S, et al. Antibiotic regimens for secondary peritonitis of gastrointestinal origin in adults. Cochrane Database Syst Rev. 18(2), 2005.
107. Minton, J, Stanley, P. Intra-abdominal infections. Clin Med. 2004; 4:519–523.
108. Wiest, R, Krag, A, Gerbes, A. Spontaneous bacterial peritonitis: Recent guidelines and beyond. Gut. 2012; 61(2):297–310.
109. Bernardi, M. Spontaneous bacterial peritonitis: From pathophysiology to prevention. Intern Emerg Med. 2010; 5(Suppl 1):S37–S44.
110. Wilcox, CM, Dismukes, WE. Spontaneous bacterial peritonitis: A review of pathogenesis, diagnosis and treatment. Medicine (Baltimore). 1987; 66:447–456.
111. Kamani, L, Mumtaz, K, Ahmed, US, et al. Outcomes in culture positive and culture negative ascitic fluid infection in patients with viral cirrhosis: Cohort study. BMC Gastroenterol. 2008; 8:59.
112. Cheadle, WG, Spain, DA. The continuing challenge of intraabdominal infection. Am J Surg. 2003; 186:15S–22S.
113. Levison, ME, Bush, LM. Peritonitis and intraperitoneal abscesses. In: Mandell GL, Bennett JE, Dolin R, eds. Principles and Practice of Infectious Diseases. 7th ed. Philadelphia: Elsevier; 2010:1011–1034.
114. Stafford, RE, Weigelt, JA. Surgical infections in the critically ill. Curr Opin Crit Care. 2002; 8:449–452.
115. Solomkin, JS, Mazuski, J. Intra-abdominal sepsis: Newer interventional and antimicrobial therapies. Infect Dis Clin North Am. 2009; 23(3):593–608.
116. Sifri, CD, Madoff, LC. Infections of the liver and biliary system. In: Mandell GL, Bennett JE, Dolin R, eds. Principles and Practice of Infectious Diseases. 7th ed. Philadelphia: Elsevier; 2010:1035–1044.
117. Hanau, LH, Steigbigel, NH. Acute (ascending) cholangitis. Infect Dis Clin North Am. 2000; 14:521–546.
118. Sakorafas, GH, Sampanis, D, Lappas, C, et al. Necrotizing acute pancreatitis current status—emerging new strategies in surgical management. Infect Disord Drug Targets. 2012; 12(2):138–143.
119. Bobo, LD, Dubberke, ER, Kollef, M. Clostridium difficile in the ICU: The struggle continues. Chest. 2011; 140(6):1643–1653.
120. Thielman, NM, Wilson, KH. Antibiotic-associated colitis. In: Mandell GL, Bennett JE, Dolin R, eds. Principles and Practice of Infectious Diseases. 7th ed. Philadelphia: Elsevier; 2010:1375–1388.
121. Warny, M, Pepin, J, Fang, A, et al. Toxin production by an emerging strain of Clostridium difficile associated with outbreaks of severe disease in North America and Europe. Lancet. 2005; 366:1079–1084.
122. Reynolds, SC, Chow, AW. Life-threatening infections of the peripharyngeal and deep fascial spaces of the head and neck. Infect Dis Clin North Am. 2007; 21(2):557–576.
123. Rana, RS, Moonis, G. Head and neck infection and inflammation. Radiol Clin North Am. 2011; 49(1):165–182.
124. Chow, AW. Infections of the oral cavity, neck, and head. In: Mandell GL, Bennett JE, Dolin R, eds. Principles and Practice of Infectious Diseases. 7th ed. Philadelphia: Elsevier; 2010:855–871.
125. Huang, TT, Liu, TC, Chen, PR, et al. Deep neck infection: Analysis of 185 cases. Head Neck. 2004; 26:854–860.
126. Karkos, PD, Asrani, S, Karkos, CD, et al. Lemierre’s syndrome: A systematic review. Laryngoscope. 2009; 119(8):1552–1559.
127. Al-Qudah, M, Shetty, S, Alomari, M, et al. Acute adult supraglottitis: Current management and treatment. South Med J. 2010; 103(8):800–804.
128. Syed, I, Odutoye, T, Lee, MS, et al. Management of acute epiglottis in adults. Br J Hosp Med (Lond). 2011; 72(5):M74–M76.
129. van Schooneveld, TC, Rupp, ME. Mediastinitis. In: Mandell GL, Bennett JE, Dolin R, eds. Principles and Practice of Infectious Diseases. 7th ed. Philadelphia: Elsevier; 2010:1173–1182.
130. Robiscek, F. Postoperative sterno-mediastinitis. Am Surg. 2000; 66:184–192.
131. McGinley-Smith, DE, Tsao, SS. Dermatoses from ticks. J Am Acad Dermatol. 2003; 49(3):363–392.
132. Dantas-Torres, F. Rocky Mountain spotted fever. Lancet Infect Dis. 2007; 7(11):724–732.
133. Walker, DH, Paddock, CD, Dumler, JS. Emerging and re-emerging tick-transmitted rickettseial and ehrlichial infections. Med Clin North Am. 2008; 92(6):1345–1361.
134. Minniear, TD, Buckingham, SC. Managing Rocky Mountain spotted fever. Expert Rev Anti Infect Ther. 2009; 7(9):1131–1137.
135. Sexton, DJ, Corey, GR. Rocky Mountain “spotless” and “almost spotless” fever: A wolf in sheep’s clothing. Clin Infect Dis. 1992; 15:439–448.
136. Olano, JP, Walker, DH. Human ehrlichioses. Med Clin North Am. 2002; 86:375–392.
137. Trampuz, A, Jereb, M, Muzlovic, I, et al. Clinical review: Severe malaria. Crit Care. 2003; 7:315–323.
138. Bledsoe, GH. Malaria primer for clinicians in the United States. South Med J. 2005; 98:1197–1204.
139. Pasvol, G. The treatment of complicated and severe malaria. Br Med Bull. 2006; 75-76:29–47.
140. Sarkar, PK, Ahluwalia, G, Vijayan, VK, et al. Critical care aspects of malaria. J Intensive Care Med. 2010; 25(2):93–103.
141. Ross, TM. Dengue virus. Clin Lab Med. 2010; 30(1):149–160.
142. Malavige, GN, Fernando, S, Fernando, DJ, et al. Dengue viral infections. Postgrad Med J. 2004; 80:588–601.
143. Castleberry, JS, Mahon, Cr. Dengue fever in the Western Hemisphere. Clin Lab Sci. 2003; 16:34–38.
144. Isturiz, RE, Gubler, DJ, Brea del Castillo, J. Dengue and dengue hemorrhagic fever in Latin America and the Caribbean. Infect Dis Clin North Am. 2000; 14:121–140.
145. Stollenwerk, N, Harper, RW, Sandrock, CE. Bench-to-bedside review: Rare and common viral infections in the intensive care unit—linking pathophysiology to clinical presentation. Crit Care. 2008; 12(4):219.
146. Vinh, DC, Embil, JM. Hantavirus pulmonary syndrome: A concise clinical review. South Med J. 2009; 102(6):620–625.
147. Simpson, SQ, Spikes, L, Patel, S, et al. Hantavirus pulmonary syndrome. Infect Dis Clin North Am. 2010; 24(1):159–173.
148. Peters, CJ, Khan, AS. Hantavirus pulmonary syndrome: The new American hemorrhagic fever. Clin Infect Dis. 2002; 34:1224–1231.
149. Bui-Mansfield, LT, Cressler, DK. Imaging of hemorrhagic fever with renal syndrome: A potential bioterrorism agent of military significance. Milit Med. 2011; 176(11):1327–1334.
150. Fiore, AE, Fry, A, Shay, D, et al. Antiviral agents for treatment of chemoprophylaxis of influenza. MMWR. 2011; 60(RR-01):1–24.
151. Wright, PF, Neumann, G, Rawaoka, Y. Orthomyxoviruses. In: Knipe DM, Howley PM, eds. Fields Virology. 5th ed. Philadelphia: Lippincott; 2007:1691–1740.
152. Cox, NJ, Subbarao, K. Influenza. Lancet. 1999; 354:1277–1282.
153. Barry, MA. A 20-year-old woman with flu-like symptoms: Review of influenza diagnosis and treatment. JAMA. 2010; 304(6):671–678.
154. Rothberg, MB, Haessler, SD. Complications of seasonal and pandemic influenza. Crit Care Med. 2010; 38(4 Suppl):e91–e97.
155. Thompson, WW, Shay, DK, Weintraub, E, et al. Influenza-associated hospitalizations in the United States. JAMA. 2004; 292:1333–1340.
156. Ferguson, NM, Cummings, DAT, Cauchemez, S, et al. Strategies for containing an emerging influenza pandemic in Southeast Asia. Nature. 2005; 437:209–214.
157. Falsey, AR, Hennessey, PA, Formica, MA, et al. Respiratory syncytial virus infection in elderly and high-risk adults. N Engl J Med. 2005; 352:1749–1759.
158. Bartlett, JG, Inglesby, TV, Borio, L. Management of anthrax. Clin Infect Dis. 2002; 35:851–858.
159. Swartz, M. Recognition and management of anthrax—an update. N Engl J Med. 2001; 345:1621–1626.
160. IDSA website. Clinical pathway: Inhalational anthrax. www.idsociety.org, 2002. [Available at].
161. Damon, I. Orthopoxviruses: Vaccinia (smallpox vaccine), variola (smallpox), monkeypox, and cowpox. In: Mandell GL, Bennett JE, Dolin R, eds. Principles and Practice of Infectious Diseases. 7th ed. Philadelphia: Elsevier; 2010:1923–1932.
162. Bray, M, Martinez, M, Smee, DF, et al. Cidofovir protects mice against lethal aerosol or intranasal cowpox virus challenge. J Infect Dis. 2000; 181:10–19.
163. Koirala, J. Plague: Disease, management, and recognition of act of terrorism. Infect Dis Clin North Am. 2006; 20(2):273–287.
164. Inglesby, TV, Dennis, DT, Henderson, DA, et al. Plague as a biological weapon: Medical and public health management. JAMA. 2000; 283:2281–2290.
165. Casadevall, A. The future of biological warfare. Microb Biotechnol. 2012; 5(5):584–587.
166. Dennis, DT, Inglesby, TV, Henderson, DA, et al. Tularemia as a biological weapon: Medical and public health management. JAMA. 2001; 285:2763–2773.
167. Boria, L, Inglesby, T, Peters, CJ, et al. Hemorrhagic viruses as biological weapons. JAMA. 2002; 287:2391–2405.