[level-membership-for-hematology-oncology-and-palliative-medicine-category]t(1:13)(p36;q14)
PAX7-FOXO1A
CREB1-EWSR1
COL1A1
IGF2
t(21;22)(q22;q12)
t(7;22)(p22;q12)
t(17;22)(q12;q12)
t(2;22)(q33;q12)
ERG-EWSR1
ETV1-EWSR1
E1AF-EWSR1
FEV-EWSR1
t(9;17)(q22;q11)
t(9;15)(q22;q21)
NR4A3-RBP56
NR4A3-TCF12
PDGFRA
t(11;16)(p11;p11)
CREB3L1-FUS
t(2;19)(p23;p13)
t(2;17)(p23;q23)
t(2;2)(p23;q13)
ALK-TPM4
ALK-CLTC
ALK-RANBP2
t(12;22)(q13;q12
DDIT3-EWSR1
SSX2-SYT
SSX4-SYT
amplification
CDK4
SAS
HMGA2
LOH, Loss of heterozygosity.
Genetics and Molecular Pathogenesis
Atypical Lipomatous Tumor/Well-Differentiated Liposarcoma
Clinical Description and Pathology
Genetics and Molecular Pathogenesis
Gastrointestinal Stromal Tumor
Clinical Description and Pathology
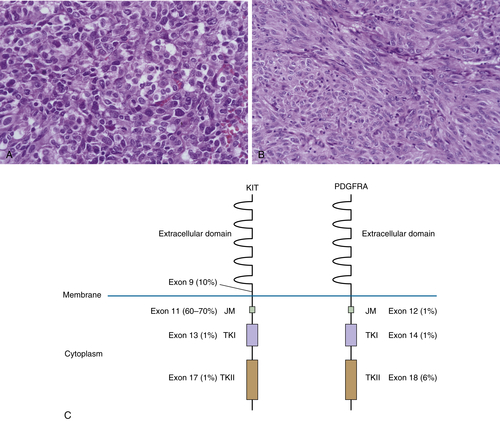
Genetics and Molecular Pathogenesis
Conclusion
1. The role of genetic testing in soft tissue sarcoma . Histopathology . 2006 ; 48 ( 1 ) : 13 – 21 .
2. Advances in sarcoma genomics and new therapeutic targets . Nat Rev Cancer . 2011 ; 11 ( 8 ) : 541 – 557 ∗ .
3. Constant p53 pathway inactivation in a large series of soft tissue sarcomas with complex genetics . Am J Pathol . 2010 ; 177 ( 4 ) : 2080 – 2090 .
4. The (epi)genetics of human synovial sarcoma . Genes Chromosomes Cancer . 2007 ; 46 ( 2 ) : 107 – 117 .
5. Synovial Sarcoma; an Analysis of 34 Tumors . Cancer . 1965 ; 18 : 613 – 627 .
6. MR imaging of synovial sarcoma . AJR Am J Roentgenol . 1991 ; 156 ( 2 ) : 337 – 340 .
7. Synovial sarcoma: a multivariate analysis of prognostic factors in 112 patients with primary localized tumors of the extremity . J Clin Oncol . 2000 ; 18 ( 10 ) : 2087 – 2094 .
8. Synovial sarcoma: identification of low and high risk groups . Cancer . 1999 ; 85 ( 12 ) : 2596 – 2607 .
9. Synovial sarcoma. A Scandinavian Sarcoma Group project . Acta Orthop Scand Suppl . 2000 ; 291 : 1 – 28 .
10. A conditional mouse model of synovial sarcoma: insights into a myogenic origin . Cancer Cell . 2007 ; 11 ( 4 ) : 375 – 388 ∗ .
11. Molecular mechanisms underlying human synovial sarcoma development . Genes Chromosomes Cancer . 2001 ; 30 ( 1 ) : 1 – 14 .
12. Histologic grade, but not SYT-SSX fusion type, is an important prognostic factor in patients with synovial sarcoma: a multicenter, retrospective analysis . J Clin Oncol . 2004 ; 22 ( 20 ) : 4040 – 4050 .
13. Impact of SYT-SSX fusion type on the clinical behavior of synovial sarcoma: a multi-institutional retrospective study of 243 patients . Cancer Res . 2002 ; 62 ( 1 ) : 135 – 140 .
14. SYT-SSX gene fusion as a determinant of morphology and prognosis in synovial sarcoma . N Engl J Med . 1998 ; 338 ( 3 ) : 153 – 160 .
15. Molecular prognostication for soft tissue sarcomas: are we ready yet? J Clin Oncol . 2004 ; 22 ( 20 ) : 4031 – 4034 .
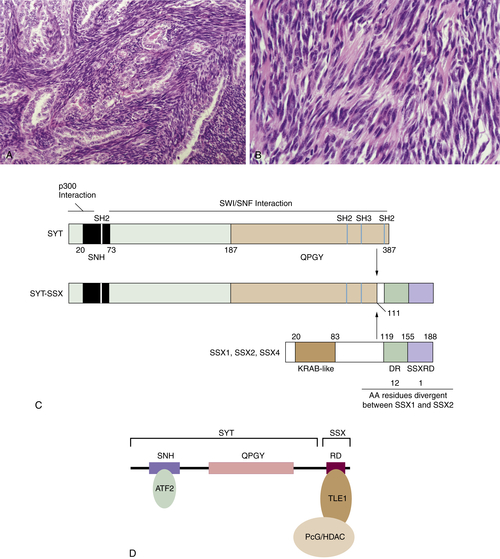
16. Conserved SNH domain of the proto-oncoprotein SYT interacts with components of the human chromatin remodelling complexes, while the QPGY repeat domain forms homo-oligomers . Oncogene . 2003 ; 22 ( 50 ) : 8156 – 8167 .
17. p300 interacts with the nuclear proto-oncoprotein SYT as part of the active control of cell adhesion . Cell . 2000 ; 102 ( 6 ) : 839 – 848 .
18. A KRAB-related domain and a novel transcription repression domain in proteins encoded by SSX genes that are disrupted in human sarcomas . Oncogene . 1998 ; 17 ( 15 ) : 2013 – 2018 .
19. Delineation of the protein domains responsible for SYT, SSX, and SYT-SSX nuclear localization . Exp Cell Res . 2000 ; 256 ( 1 ) : 192 – 202 .
20. Analysis of transforming activity of human synovial sarcoma-associated chimeric protein SYT-SSX1 bound to chromatin remodeling factor hBRM/hSNF2 alpha . Proc Natl Acad Sci U S A . 2001 ; 98 ( 7 ) : 3843 – 3848 .
21. Deconstruction of the SS18-SSX Fusion Oncoprotein Complex: Insights into Disease Etiology and Therapeutics . Cancer Cell . 2012 ; 21 ( 3 ) : 333 – 347 ∗ .
22. Immunohistochemical staining for TLE1 distinguishes synovial sarcoma from histologic mimics . Am J Clin Pathol . 2011 ; 135 ( 6 ) : 839 – 844 .
23. Prospective evaluation of TLE1 as a diagnostic immunohistochemical marker in synovial sarcoma . Am J Surg Pathol . 2009 ; 33 ( 12 ) : 1743 – 1751 .
24. TLE1 is a robust diagnostic biomarker for synovial sarcomas and correlates with t(X;18): analysis of 319 cases . Eur J Cancer . 2010 ; 46 ( 6 ) : 1170 – 1176 .
25. Histone deacetylase inhibitors reverse SS18-SSX-mediated polycomb silencing of the tumor suppressor early growth response 1 in synovial sarcoma . Cancer Res . 2008 ; 68 ( 11 ) : 4303 – 4310 .
26. Significant growth suppression of synovial sarcomas by the histone deacetylase inhibitor FK228 in vitro and in vivo . Cancer Lett . 2005 ; 224 ( 2 ) : 311 – 319 .
27. Phosphatidylinositol-3’-kinase/AKT signaling is essential in synovial sarcoma . Int J Cancer . 2011 ; 129 ( 7 ) : 1564 – 1575 .
28. Well-differentiated liposarcoma (atypical lipomatous tumors) . Semin Diagn Pathol . 2001 ; 18 ( 4 ) : 258 – 262 .
29. Dedifferentiated liposarcoma . Semin Diagn Pathol . 2001 ; 18 ( 4 ) : 263 – 266 .
30. Atypical lipomatous tumor: molecular characterization . Curr Opin Oncol . 2004 ; 16 ( 4 ) : 355 – 358 .
31. Cytogenetic and fluorescence in situ hybridization investigation of ring chromosomes characterizing a specific pathologic subgroup of adipose tissue tumors . Cancer Genet Cytogenet . 1993 ; 68 ( 2 ) : 85 – 90 .
32. MDM2 and CDK4 immunostainings are useful adjuncts in diagnosing well-differentiated and dedifferentiated liposarcoma subtypes: a comparative analysis of 559 soft tissue neoplasms with genetic data . Am J Surg Pathol . 2005 ; 29 ( 10 ) : 1340 – 1347 .
33. The MDM2-p53 interaction . Mol Cancer Res . 2003 ; 1 ( 14 ) : 1001 – 1008 .
34. Overexpression of Mdm2 in mice reveals a p53-independent role for Mdm2 in tumorigenesis . Proc Natl Acad Sci U S A . 1998 ; 95 ( 26 ) : 15608 – 15612 .
35. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2 . Science . 2004 ; 303 ( 5659 ) : 844 – 848 .
36. Gene expression profiling of liposarcoma identifies distinct biological types/subtypes and potential therapeutic targets in well-differentiated and dedifferentiated liposarcoma . Cancer Res . 2007 ; 67 ( 14 ) : 6626 – 6636 ∗ .
37. A subgroup of malignant fibrous histiocytomas is associated with genetic changes similar to those of well-differentiated liposarcomas . Cancer Genet Cytogenet . 2002 ; 139 ( 1 ) : 24 – 29 .
38. ASK1 (MAP3K5) as a potential therapeutic target in malignant fibrous histiocytomas with 12q14-q15 and 6q23 amplifications . Genes Chromosomes Cancer . 2004 ; 40 ( 1 ) : 32 – 37 .
39. JUN oncogene amplification and overexpression block adipocytic differentiation in highly aggressive sarcomas . Cancer Cell . 2007 ; 11 ( 4 ) : 361 – 374 ∗ .
40. c-Jun amplification and overexpression are oncogenic in liposarcoma but not always sufficient to inhibit the adipocytic differentiation programme . J Pathol . 2009 ; 218 ( 3 ) : 292 – 300 .
41. Molecular abnormalities of mdm2 and p53 genes in adult soft tissue sarcomas . Cancer Res . 1994 ; 54 ( 3 ) : 794 – 799 .
42. Molecular abnormalities of the p53 pathway in dedifferentiated liposarcoma . J Pathol . 1997 ; 181 ( 1 ) : 8 – 13 .
43. Distinct mdm2/p53 expression patterns in liposarcoma subgroups: implications for different pathogenetic mechanisms . J Pathol . 1997 ; 181 ( 1 ) : 14 – 24 .
44. MDM2 amplification and loss of heterozygosity at Rb and p53 genes: no simultaneous alterations in the oncogenesis of liposarcomas . J Cancer Res Clin Oncol . 1998 ; 124 ( 10 ) : 532 – 540 .
45. Gastrointestinal stromal tumours: an update . Histopathology . 2006 ; 48 ( 1 ) : 83 – 96 .
46. Gastrointestinal stromal tumors: pathology and prognosis at different sites . Semin Diagn Pathol . 2006 ; 23 ( 2 ) : 70 – 83 .
47. A novel monoclonal antibody against DOG1 is a sensitive and specific marker for gastrointestinal stromal tumors . Am J Surg Pathol . 2008 ; 32 ( 2 ) : 210 – 218 .
48. Monoclonal antibody DOG1.1 shows higher sensitivity than KIT in the diagnosis of gastrointestinal stromal tumors, including unusual subtypes . Am J Surg Pathol . 2009 ; 33 ( 3 ) : 437 – 446 .
49. Gastrointestinal pacemaker cell tumor (GIPACT): gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal . Am J Pathol . 1998 ; 152 ( 5 ) : 1259 – 1269 .
50. Familial gastrointestinal stromal tumours with germline mutation of the KIT gene . Nat Genet . 1998 ; 19 ( 4 ) : 323 – 324 .
51. The biology of stem cell factor and its receptor C-kit . Int J Biochem Cell Biol . 1999 ; 31 ( 10 ) : 1037 – 1051 .
52. W/kit gene required for interstitial cells of Cajal and for intestinal pacemaker activity . Nature . 1995 ; 373 ( 6512 ) : 347 – 349 .
53. Signal transduction via the stem cell factor receptor/c-Kit . Cell Mol Life Sci . 2004 ; 61 ( 19-20 ) : 2535 – 2548 .
54. PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib . J Clin Oncol . 2005 ; 23 ( 23 ) : 5357 – 5364 .
55. Gastrointestinal stromal tumors in a mouse model by targeted mutation of the Kit receptor tyrosine kinase . Proc Natl Acad Sci U S A . 2003 ; 100 ( 11 ) : 6706 – 6711 .
56. A knock-in mouse model of gastrointestinal stromal tumor harboring kit K641E . Cancer Res . 2005 ; 65 ( 15 ) : 6631 – 6639 .
57. The triad of paragangliomas, gastric stromal tumours and pulmonary chondromas (Carney triad), and the dyad of paragangliomas and gastric stromal sarcomas (Carney-Stratakis syndrome): molecular genetics and clinical implications . J Intern Med . 2009 ; 266 ( 1 ) : 43 – 52 .
58. The triad of gastric leiomyosarcoma, functioning extra-adrenal paraganglioma and pulmonary chondroma . N Engl J Med . 1977 ; 296 ( 26 ) : 1517 – 1518 .
59. Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD . Eur J Hum Genet . 2008 ; 16 ( 1 ) : 79 – 88 .
60. Familial gastrointestinal stromal tumors and germ-line mutations . N Engl J Med . 2007 ; 357 ( 10 ) : 1054 – 1056 ∗ .
61. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations . Proc Natl Acad Sci U S A . 2011 ; 108 ( 1 ) : 314 – 318 .
62. SDHA loss-of-function mutations in KIT-PDGFRA wild-type gastrointestinal stromal tumors identified by massively parallel sequencing . J Natl Cancer Inst . 2011 ; 103 ( 12 ) : 983 – 987 .
63. Succinate dehydrogenase-deficient GISTs: a clinicopathologic, immunohistochemical, and molecular genetic study of 66 gastric GISTs with predilection to young age . Am J Surg Pathol . 2011 ; 35 ( 11 ) : 1712 – 1721 .
64. “Pediatric-type” gastrointestinal stromal tumors in adults: distinctive histology predicts genotype and clinical behavior . Am J Surg Pathol . 2011 ; 35 ( 4 ) : 495 – 504 .
65. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor . J Clin Oncol . 2003 ; 21 ( 23 ) : 4342 – 4349 .
66. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor . J Clin Oncol . 2008 ; 26 ( 33 ) : 5352 – 5359 .
67. Immunohistochemistry for SDHB divides gastrointestinal stromal tumors (GISTs) into 2 distinct types . Am J Surg Pathol . 2010 ; 34 ( 5 ) : 636 – 644 .
68. “Pediatric-type” gastrointestinal stromal tumors are SDHB negative (“type 2”) GISTs . Am J Surg Pathol . 2011 ; 35 ( 8 ) : 1245 – 1247 author reply 1247-1248 .
69. Succinate Dehydrogenase Subunit B (SDHB) Is Expressed in Neurofibromatosis 1-Associated Gastrointestinal Stromal Tumors (Gists): Implications for the SDHB Expression Based Classification of Gists . J Cancer . 2011 ; 2 : 90 – 93 .
70. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation . Clin Cancer Res . 2005 ; 11 ( 11 ) : 4182 – 4190 .
71. Heterogeneity of kinase inhibitor resistance mechanisms in GIST . J Pathol . 2008 ; 216 ( 1 ) : 64 – 74 .
72. Imatinib upregulates compensatory integrin signaling in a mouse model of gastrointestinal stromal tumor and is more effective when combined with dasatinib . Mol Cancer Res . 2010 ; 8 ( 9 ) : 1271 – 1283 .
73. Mechanisms of oncogenic KIT signal transduction in primary gastrointestinal stromal tumors (GISTs) . Oncogene . 2004 ; 23 ( 22 ) : 3999 – 4006 .
74. KIT oncogenic signaling mechanisms in imatinib-resistant gastrointestinal stromal tumor: PI3-kinase/AKT is a crucial survival pathway . Oncogene . 2007 ; 26 ( 54 ) : 7560 – 7568 ∗ .
75. Novel V600E BRAF mutations in imatinib-naive and imatinib-resistant gastrointestinal stromal tumors . Genes Chromosomes Cancer . 2008 ; 47 ( 10 ) : 853 – 859 .
76. Validated prediction of clinical outcome in sarcomas and multiple types of cancer on the basis of a gene expression signature related to genome complexity . Nat Med . 2010 ; 16 ( 7 ) : 781 – 787 ∗ .
77. New insights in sarcoma oncogenesis: a comprehensive analysis of a large series of 160 soft tissue sarcomas with complex genomics . J Pathol . 2011 ; 223 ( 1 ) : 64 – 71 .
78. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy . Nat Genet . 2010 ; 42 ( 8 ) : 715 – 721 ∗ .
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]t(1:13)(p36;q14)
PAX7-FOXO1A
CREB1-EWSR1
COL1A1
IGF2
t(21;22)(q22;q12)
t(7;22)(p22;q12)
t(17;22)(q12;q12)
t(2;22)(q33;q12)
ERG-EWSR1
ETV1-EWSR1
E1AF-EWSR1
FEV-EWSR1
t(9;17)(q22;q11)
t(9;15)(q22;q21)
NR4A3-RBP56
NR4A3-TCF12
PDGFRA
t(11;16)(p11;p11)
CREB3L1-FUS
t(2;19)(p23;p13)
t(2;17)(p23;q23)
t(2;2)(p23;q13)
ALK-TPM4
ALK-CLTC
ALK-RANBP2
t(12;22)(q13;q12
DDIT3-EWSR1
SSX2-SYT
SSX4-SYT
amplification
CDK4
SAS
HMGA2
LOH, Loss of heterozygosity.
Genetics and Molecular Pathogenesis
Atypical Lipomatous Tumor/Well-Differentiated Liposarcoma
Clinical Description and Pathology
Genetics and Molecular Pathogenesis
Gastrointestinal Stromal Tumor
Clinical Description and Pathology
Genetics and Molecular Pathogenesis
[/not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
