Soft Tissue and Bone Tumors
Soft Tissue Tumors
Most soft tissue tumors in children are benign.1 When the mass is hard and fixed, conventional radiographs are often performed to determine whether an underlying bone lesion is present that is simulating, or is associated with, a soft tissue mass. Conventional radiographs may show secondary changes in the configuration of adjacent bones and may also reveal the presence of cortical bone erosion, periosteal reaction, and soft tissue calcifications. Occasionally, radiolucency indicates the presence of a fat-containing tumor.1
Superficial masses are often assessed by ultrasonography (US), a modality that is easily accessible, lacks ionizing radiation, and rarely requires sedation.2 In addition, color and spectral Doppler analysis can assess and characterize vascularity.
Nuclear medicine also plays a significant role in the assessment of soft tissue tumors. Many soft tissue sarcomas are detected by gallium-67 and thallium-201 scintigraphy.3,4 Technetium-99m–methylene diphosphonate (99mTc-MDP) bone scintigraphy may be performed to identify local bone involvement and distant skeletal metastases. In addition, positron emission tomography (PET) and PET computed tomography (CT) are playing an increasing role in initial tumor assessment and evaluation of tumor response and recurrence.
With continuing advances in magnetic resonance imaging (MRI), the role of CT has become limited. CT offers similar but more detailed information than that provided by conventional radiographs. Two areas in which CT has a clear advantage over MRI are in the detection of calcification, such as in myositis ossificans, and when lesions are present along the anterior abdominal wall or chest, where artifact may significantly degrade MRI quality.1 Also, CT of the lungs is usually performed to assess for metastatic disease.
Although MRI technology continues to evolve, the basic tenets of the MRI examination of a child with a soft tissue tumor have remained constant. First, images should cover the entire tumor, including its margins, and they should include any needle biopsy tracts that may have to be excised at the time of surgical resection. Care must be taken to ensure quality images and coverage of the entire lesion and important adjacent structures. The area that contains the tumor should be imaged in at least two orthogonal planes. T1- and T2-weighted images should be obtained. Fat saturation is often added to confirm the presence of fat in a lesion and/or to highlight the presence of enhancement after intravenous gadolinium administration. Short tau inversion recovery (STIR) sequences are also useful, because they inherently suppress signal from fat and do not require a homogeneous magnetic field. Gadolinium-enhanced T1-weighted images add information about tumor vascularity and are helpful in guiding percutaneous biopsy in that viable tumor can be differentiated from areas of necrosis.5 Cystic lesions may be identified by their lack of central enhancement.6 MR angiography (MRA) is useful for detecting the presence of blood flow within the tumor and for planning the optimal surgical approach when large vessels may be involved. Most soft tissue tumors have prolonged T1 and T2 signal characteristics.7 The MRI signal characteristics of most soft tissue tumors are nonspecific and usually cannot predict histology, nor can they differentiate between benign and malignant neoplasms. Biopsy is necessary for histologic diagnosis.
Benign Soft Tissue Tumors
Overview: Desmoid tumors, representing deep or aggressive fibromatosis, are rare mesenchymal neoplasms with a fibrotic texture.8,9 Although distant metastases do not occur, these tumors are locally aggressive and can lead to significant morbidity and mortality. Desmoid tumors are slightly more common in females, with an incidence of 2 to 4 cases per million per year. The peak incidence is in the third and fourth decades of life.10 When these tumors occur in younger patients, they tend to be more aggressive, with recurrence rates up to 87%.11
Etiology: Desmoid tumors are classified into superficial and deep groups. Superficial tumors are usually small and slow growing, and deep lesions may occur in the abdomen or extraabdominally. In children, extraabdominal desmoid tumors are more common than the abdominal type. Most desmoid tumors in patients with Gardner syndrome are found in the abdomen. Extraabdominal desmoid tumors, also referred to as aggressive fibromatosis, are usually solitary and arise from the fascial sheaths and aponeuroses of striated muscle. In a series of tumors studied at St. Jude Children’s Research Hospital, head and neck, trunk, and extremity involvement was seen with approximately equal frequency.12 Histologically, desmoid tumors consist of benign fibrous tissues that contain spindle cells and abundant collagen. Although they do not metastasize, desmoid tumors can infiltrate contiguous structures, including bone.
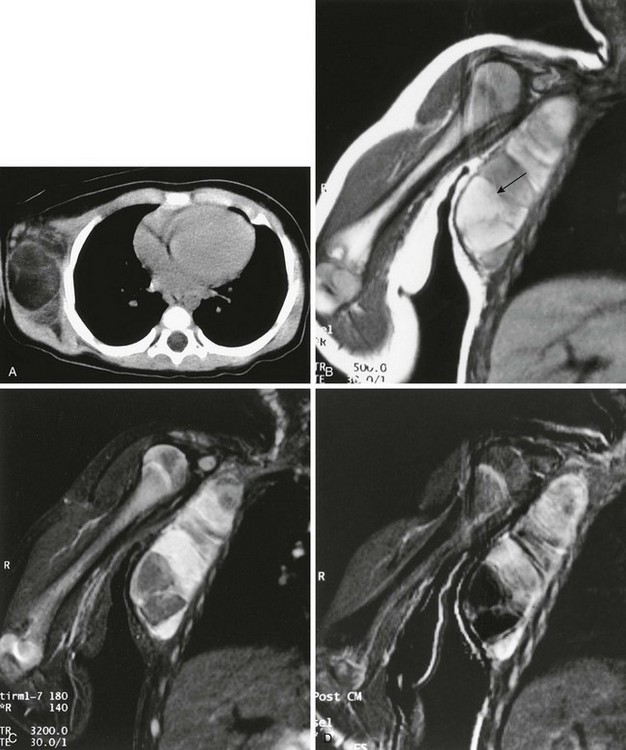
Imaging: Desmoid tumors are variably echogenic on US, and their borders may be smooth or irregular. On contrast-enhanced CT, most appear more attenuated than striated muscle (Fig. 139-1). On MRI, these tumors may be nodular with infiltrative or well-defined margins. Tumors may be homogeneous or heterogeneous with varying signal characteristics. Some lesions are low signal on T1- and T2-weighted images, but more often tumors are heterogeneous and contain areas that are hyperintense to “fat” on T2-weighted images. On T1-weighted images, these masses contain areas that are hypointense, isointense, or slightly hyperintense when compared with muscle. This variability in signal reflects differences in the relative proportions of collagen, spindle cells, and mucopolysaccharides within the lesion (e-Fig. 139-2). On T2-weighted images, low signal generally reflects collagen, and high signal reflects a greater quantity of cellular tissue.13 Tumors with high signal on T1-weighted images have been found to contain fat or myxoid material. Contrast enhancement may be homogeneous, heterogeneous, or absent and does not correlate with clinical outcome.14
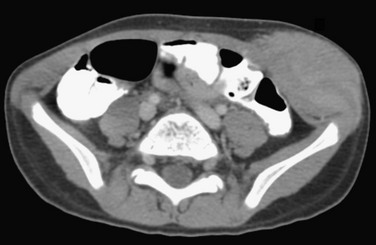
Figure 139-1 Extraabdominal desmoid tumor (Gardner fibroma) in a 5-year-old girl.
Axial contrast-enhanced computed tomographic image shows a large lenticular-shaped mass originating in the anterior abdominal wall musculature.
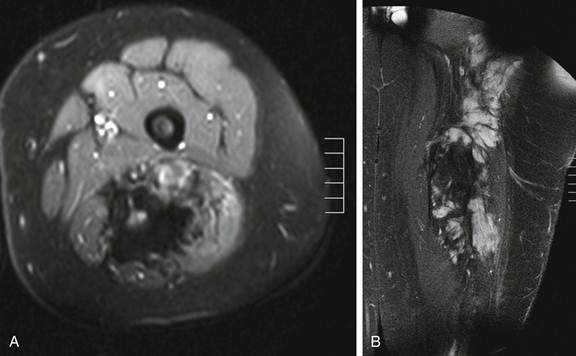
e-Figure 139-2 Multicompartmental posterior thigh desmoid tumor in a 17-year-old boy.
A, T2-weighted fat-saturated axial magnetic resonance image demonstrates hyperintense and hypointense components of the mass. Hyperintense areas likely represent cellular-dominant components of the mass, whereas hypointense areas likely represent colloid components. B, T1-weighted post-gadolinium coronal images demonstrate heterogeneous enhancement of the mass.
Treatment: Stable asymptomatic desmoid tumors are often treated conservatively and observed for changes.15 Symptomatic desmoids must be treated, and therapy often depends on anatomic location. If possible, surgical resection with wide margins is the treatment of choice.10 However, recurrence is common (19% to 77%) and is more frequent with extraabdominal desmoids (30% to 50%) than with intraabdominal desmoids (15% to 30%).8,9,16 If surgery is not possible, systemic therapy should be considered. This is often the case when desmoids are associated with familial adenomatous polyposis or Gardner syndrome. These conditions increase the likelihood of postoperative complications such as hemorrhage, short-bowel syndrome, intestinal ischemia, obstruction, or fistula formation.9,15 Nonsurgical treatment options that include radiation and systemic therapy are also available. Patients treated with chemotherapy have a lower incidence of tumor recurrence compared with those treated with radiation.13
Infantile Myofibromatosis
Infantile myofibromatosis is the most common fibrous tumor of infancy. Tumors may involve skin, muscle, bone, or viscera and may be solitary (myofibroma) or multiple (myofibromatosis). Myofibromatosis occurs in children younger than 2 years of age. The prognosis of musculoskeletal lesions is excellent, and spontaneous resolution usually occurs, although visceral involvement may portend a poorer prognosis. Lesions have a variable appearance on US and range from solid to anechoic centrally with a thick wall. On CT, myofibromas enhance to a lesser degree or similarly to muscle and often exhibit a peripheral rim of enhancement. On MRI, lesions are low signal on T1 weighting and usually high signal on T2 weighting. Some masses show decreased central signal on T2 weighting, because these masses are composed of collagen and have cellular elements. Enhancement of the fibrous and cellular components is seen with gadolinium administration (Fig. 139-3).12,17–21 Osseous lesions tend to occur in the metaphysis and have a lytic appearance (Fig. 139-4).20
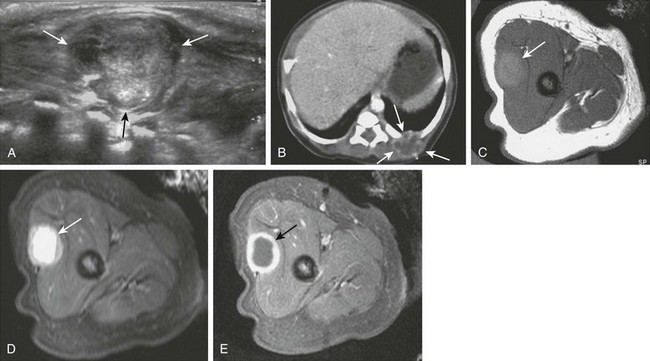
Figure 139-3 Infantile myofibromatosis in a neonate.
A, Ultrasound shows a solid, intramuscular subscapular mass (arrows). B, Axial contrast-enhanced computed tomographic image shows peripheral enhancement within the mass (arrows). C, Thigh lesion in the same patient shows hyperintense signal on T1-weighted magnetic resonance imaging. D, T2-weighted image with fat saturation demonstrates cystic changes with a thick, irregular rind of soft tissue. E, Post-gadolinium T1-weighted fat-saturated images demonstrate enhancement of the rind of soft tissue and central areas of nonenhancement.
Benign Peripheral Nerve Sheath Tumors
Overview: Benign peripheral nerve sheath tumors are divided into schwannomas, also known as neurinomas and neurilemomas, and neurofibromas. Benign neurofibromas and, less commonly, schwannomas are frequently multiple and are associated with neurofibromatosis type 1 (NF1). However, both tumors may be solitary and may occur sporadically.
Etiologies and Clinical Presentation: Schwannomas account for approximately 5% of all benign soft tissue neoplasms, and they are usually found as a solitary lesion in individuals between 20 and 50 years of age. Although they may occur virtually anywhere in the body, the head and neck, flexor surfaces of extremities (notably the ulnar and peroneal nerves), mediastinum, and retroperitoneum are the most commonly involved sites.22 If the lesion is large, pain and neurologic symptoms may be clinical manifestations; otherwise, patients are usually asymptomatic.23,24
Neurofibromas may be localized, plexiform, or diffuse, and the localized form accounts for 90% of cases and is not usually associated with NF1, unlike the diffuse and plexiform subtypes.22 Localized neurofibromas account for approximately 5% of all benign soft tissue tumors and are typically seen in younger patients between 20 and 30 years of age.22 They often come to medical attention as a painless, slowly growing mass.23
Schwannomas and localized neurofibromas are similar histologically and are composed primarily of Schwann cells, therefore they exhibit similar imaging characteristics. Microscopic examination reveals a dense central core of Schwann cells surrounded by a peripheral zone of myxoid tissue. Peripheral nerve sheath tumors have a low incidence of malignant degeneration.22
Imaging: A tumor can be suspected to have a neurogenic origin if it is located along the distribution of a peripheral nerve. Calcification in degenerating (“ancient”) schwannomas may be visible radiographically; on bone scintigrams, these tumors have been observed to take up 99mTc-MDP.25 Peripheral nerve sheath tumors may be accompanied by subtle atrophy of surrounding or distally innervated muscle. Most of these tumors are well-defined spherical or fusiform masses. On CT images, they tend to be hypoattenuated, possibly because of lipids in their Schwann cells, adipocytes, and perineural tissues.26 On MRI, the bulk of the tumor shows low intensity on T1-weighted sequences and high intensity on T2-weighted sequences. Typically, the central zone consists of collagen and neurofibroma cells and is hypointense on T2-weighted images, lending a “target” appearance to the lesion that can also be seen on contrast-enhanced T1-weighted images; it is more easily appreciated when wide window settings are used to view the images. The presence of a target sign helps to distinguish these benign tumors from their less well-organized malignant counterparts (Fig. 139-5).27–30
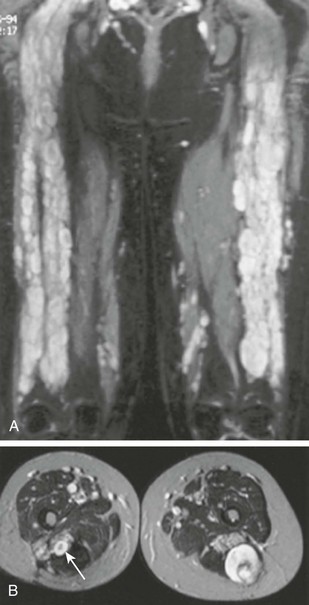
Figure 139-5 A young woman first seen at the age of 16 with a malignant peripheral nerve sheath tumor.
A, Coronal short tau inversion recovery magnetic resonance image of the thighs shows multiple plexiform neurofibromas distributed along the sciatic nerves. B, Transverse T2-weighted images show multiple, discrete, high-intensity neurofibromas. A characteristic target sign is evident (arrow). The larger lesion in the posterior left thigh, which had shown recent growth, was a low-grade malignant peripheral nerve sheath tumor.
On MRI, differentiating a schwannoma from a neurofibroma can be difficult; however, schwannomas may contain more prominent areas of hemorrhage, cystic change, and necrosis with resultant heterogeneous signal intensities. In addition, neurofibromas intimately involve and are inseparable from the normal nerve. Schwannomas are eccentric to the nerve, and this may be apparent on MRI in tumors that involve larger nerves. When smaller nerves are involved, schwannomas may obliterate the nerve of origin, and the MRI appearance is indistinguishable from that of a neurofibroma.31
Plexiform neurofibromas arise from the axis of a primary nerve and form tortuous, cordlike tumors along its axis. They are regarded as indicators of neurofibromatosis, even when they are the sole manifestation of the disease. Tumors tend to appear as lobulated, amorphous masses with ill-defined borders. Similar to solitary neurofibromas, these tumors are usually hyperintense on T2-weighted imaging and often have well-defined, central, tubular, hypointense structures, or they may form large masses that resemble a “bag of worms” on transverse images.32
Diffuse neurofibromas are particularly uncommon and are often associated with NF1 (7 of 10 patients in one series).33 These lesions occur primarily in children and young adults and most commonly involve the head and neck regions. These ill-defined, infiltrative lesions tend to be located in the skin and subcutaneous tissues. They appear as linear or reticular strands of intermediate signal on T1-weighted images and are of high signal intensity on T2-weighted images, with linear areas of enhancement after gadolinium administration.
Benign Fatty Tumors
Overview: The 2002 World Health Organization (WHO) Soft Tissue Tumor Classification includes nine types of benign fatty tumor: lipoma, lipomatosis, lipomatosis of nerve, lipoblastoma/lipoblastomatosis, angiolipoma, myolipoma of soft tissue, chondroid lipoma, spindle cell/pleomorphic lipoma, and hibernoma. Except for lipoblastoma, all of these tumors are more common in adults than in children.24
Lipoblastoma and lipoblastomatosis are benign mesenchymal tumors of immature fat that occur primarily in infants and young children, most often in boys younger than 8 years of age. The average age of patients at the time of presentation is 3.6 years.34 Lipoblastomas are usually painless superficial tumors most commonly located in the extremities and the head and neck region, but they may also involve the trunk and deeper structures, such as the mediastinum and retroperitoneum. The tumor consists of lobules of immature adipose tissue with a variable amount of myxoid stroma separated by richly vascularized septa composed of connective tissue. The discrete form, lipoblastoma, is a well-circumscribed lesion that occurs in approximately 70% of cases and involves the superficial soft tissues. Lipoblastomas eventually evolve into mature lipomas. The term lipoblastomatosis refers to the diffuse type that often infiltrates adjacent deeper tissues, such as muscle, and has a tendency to recur locally35; spontaneous resolution has also been reported.36,37
Imaging: Imaging features reflect the amount of fatty tissue present. On US images, hyperechoic fat can be clearly delineated from the myxoid component, and CT images show a similar combination of hypoattenuated fat and denser myxoid tissue. On MRI, the signal is often heterogeneous (e-Fig. 139-6), and lipomatous elements appear hyperintense to muscle on T1-weighted images and isointense to subcutaneous fat on T2-weighted images; nonfatty tissues produce lower signal intensity than fat on T1-weighted images and are more intense than fat on T2-weighted images. In addition to low-signal myxoid components on T1 imaging, the fatty mass may contain low-signal fibrous septa that show contrast enhancement.38–40
The differential diagnosis includes lipoma, which is much more prevalent in adults than in children; hibernoma, a rare tumor analogous to brown fat; and myxoid liposarcoma. Liposarcoma is rare in children younger than 5 years of age.41 Thus, a fat-containing tumor that occurs in a child younger than 2 years of age, even with nonlipomatous components, is almost invariably a lipoblastoma.
Treatment: The majority of fat-containing masses in children are benign. Although complete surgical resection is the treatment of choice for focal lesions,34 subtotal resection may therefore be considered, particularly when surgical resection may involve critical neurovascular structures, or when it may lead to significant cosmetic deformity.
Malignant Soft Tissue Tumors
Overview: The head and neck and genitourinary tract are the most frequent locations of rhabdomyosarcoma (RMS). About 20% of these tumors occur in the extremities. Most extremity RMS is of the alveolar or undifferentiated histologic types, not the embryonal or botryoid types found in the face and neck and in the genitourinary system.42 Prognosis is less favorable for patients with RMS of the extremities than for those with tumors that arise from the genitourinary system or the head and neck region.43 In the extremities, tumors tend to be deep and tend to spread along fascial planes, and RMS may cause erosion of adjacent bone.
Etiologies, Pathophysiology, and Clinical Presentation: RMS contains a mixture of rhabdomyoblasts, which are recognized by their typical cross striations, and undifferentiated cells. These tumors are thought to arise from progenitor cells for striated muscle and can occur anywhere in the body, even in areas with no striated muscle. RMS accounts for more than 60% of soft tissue sarcoma (STS) in children younger than 5 years of age but only 25% of STS in those aged 15 to 19 years.44
Imaging: Imaging is usually nonspecific but is essential for staging and surgical management. MRI has superseded CT and US in this clinical setting, because it can define the anatomic location of the tumor (unicompartmental vs. multicompartmental), indicate its relationship to important nerves and blood vessels, and reveal local involvement of bone or lymph nodes. MRI characteristics are nonspecific, and most tumors are predominantly low signal on T1-weighted images, high signal on T2-weighted images, and demonstrate central tumoral enhancement (Fig. 139-7). Only 15% to 20% of patients with RMS have clinically detectable metastases at presentation. The lungs, bone marrow, and bone are the most common sites of distant metastases. Lymph nodes may also be involved. Bone metastases resemble those that occur with neuroblastoma; they have been reported even in the absence of detectable primary tumor.
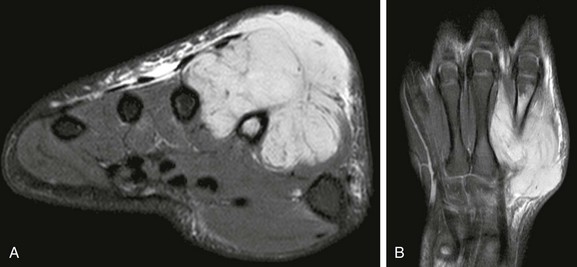
Figure 139-7 Rhabdomyosarcoma of the hand in an 11-year-old boy.
A, T2-weighted fat-saturated magnetic resonance imaging (MRI) demonstrates a multilobulated, multicompartment mass with secondary involvement of the second metacarpal bone. Note that the mass has a sharp interface with the second metacarpal cortex, an indication that the mass abuts and secondarily involves the bone rather than arising primarily from the bone. B, Post-gadolinium T1-weighted fat-saturated coronal MRI demonstrates diffuse tumoral enhancement of the mass.
Primitive Peripheral Neuroectodermal Tumor and Extraosseous Ewing Sarcoma
Overview: Primitive peripheral neuroectodermal tumor (PPNET) and extraosseous Ewing sarcoma (EOES) are small round cell neoplasms that belong to the Ewing sarcoma family of tumors; they can arise in both soft tissue and bone. These neoplasms are related histogenically and share a common cytogenetic characteristic, the translocation of bands 24 and 12 of the short arms of chromosomes 11 and 22, but they are often indistinguishable histologically. Also known as peripheral neuroepithelioma, PPNETs have a higher degree of neural differentiation than is seen with Ewing sarcoma; thus these two tumors can be distinguished on the basis of immunohistochemical markers. This distinction is important, because disease-free survival is poorer for patients with PPNETs than for those with EOES.46,47
Etiologies, Pathophysiology, and Clinical Presentation: Both tumors occur most commonly in truncal and paravertebral soft tissues (50% to 60% of cases) and in the extremities (25% of cases), although PPNET occurs less commonly in the extremities than does EOES, and patients with EOES are generally younger. Askin tumors are thoracic PPNETs that involve the chest wall.48 These tumors can be very large at the time of presentation and tend to be poorly circumscribed. The soft tissue mass does not calcify but may erode adjacent bone.
Imaging: On MRI, these tumors have a nonspecific imaging appearance. PPNET and EOES are typically isointense to muscle on T1-weighted images and inhomogeneously hyperintense on T2-weighted and STIR images; they show variable contrast enhancement. When these lesions outgrow their blood supply or, alternatively, when masses hemorrhage spontaneously or after minor trauma, cystic components may be seen; these lesions may superficially mimic a hematoma (Fig. 139-8). Postcontrast imaging is helpful in these circumstances to help identify solid soft tissue components and guide biopsy of viable tumor cells. Distant spread occurs to bone, lung, liver, and brain.49,50
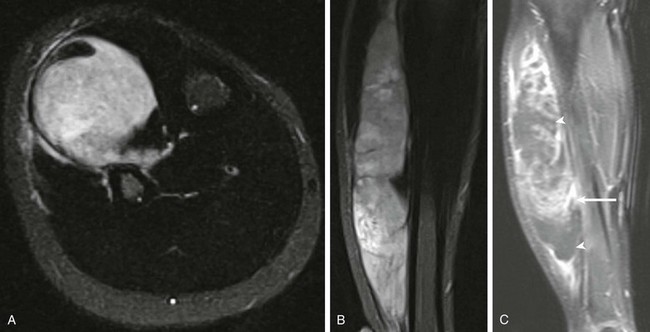
Figure 139-8 Anterior leg primitive neuroectodermal tumor in a 12-year-old boy.
Short tau inversion recovery axial (A) and coronal (B) images demonstrate an extraosseous mass with extensive peritumoral edema. C, Post-gadolinium T1-weighted fat-saturated coronal magnetic resonance image demonstrates cystic nonenhancing components of the tumor (arrowheads) and solid enhancing components (arrow).
Synovial Sarcoma
Overview, Etiologies, Pathophysiology, and Clinical Presentation: Synovial sarcomas predominantly occur in adults younger than 50 years of age, but they account for about 10% of pediatric STS. These tumors arise from undifferentiated mesenchymal cells, not from true synovial cells. A monophasic variety comprises spindle cells, and a biphasic variety consists of spindle cells and epithelial elements. Although synovial sarcomas often occur close to joints, tendons, and bursae, they are rarely intraarticular.52 About 80% of synovial sarcomas occur in the extremities, and considerably more lower than upper extremity involvement has been noted. Synovial sarcomas may spread to regional lymph nodes, and the lungs are the most common sites of distant metastasis.53
Imaging: Radiographically visible calcifications are present in 30% of cases. On MRI, synovial sarcomas are often lobulated, well-defined, deep-seated lesions, although they may be infiltrative and can encase major blood vessels. Femoral vein invasion has been described, and erosion of the cortex of adjacent bones is present in up to 20% of patients.
MRI signal characteristics are nonspecific. Synovial sarcomas are usually isointense to muscle on T1-weighted images. Foci of high T1-weighted signal may be present, and fluid-fluid levels are caused by hemorrhage; tumors generally have heterogeneous signal with areas of high intensity on T2-weighted images (Fig. 139-9). In one large series, 35% of tumors showed a triple-signal pattern on T2-weighted images; these findings are consistent with high (fluid) signal intensity, intermediate signal intensity similar to that of fat, and low signal intensity resembling that of fibrous tissue (see Fig. 139-9, B).54 Some synovial sarcomas superficially mimic ganglion cysts (e-Fig. 139-10). A multilocular appearance with fluid-fluid levels may be seen in 18% to 25% of these tumors. As a rule of thumb, a diagnosis of ganglion cyst should only be considered when a fluid-filled neck can be seen extending from the cyst to a joint or tendon sheath, and no solid components lie within the cystlike lesion. Otherwise, a diagnosis of neoplasm should be favored, with biopsy recommended.54–57
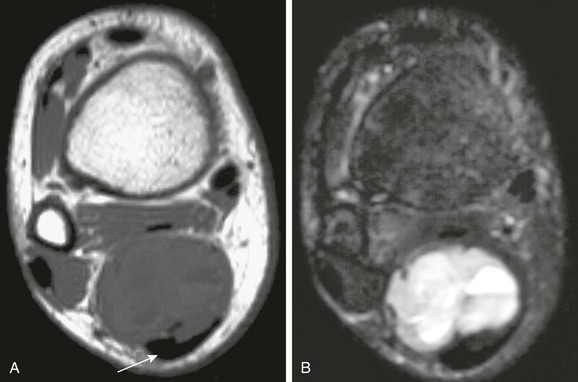
Figure 139-9 Synovial sarcoma adjacent to the Achilles tendon.
A, Transverse T1-weighted magnetic resonance imaging (MRI) shows mixed intermediate and low signal in the mass. The Achilles tendon (arrow) is shown. B, A transverse fat-saturated T2-weighted MRI at the same level shows a fluid-fluid level typical of hemorrhage.
Treatment: The treatment of choice is surgery with or without adjuvant and neoadjuvant chemotherapy. A recent multicenter study of patients under 20 years of age with a minimum of 10 years of follow-up found that wide surgical resection is the most efficacious method of treatment. Presence of tumor in the trunk and high histologic grade were negative factors for recurrence-free and metastasis-free survival.58
Malignant Peripheral Nerve Sheath Tumor
Overview: Malignant peripheral nerve sheath tumor (MPNST) is the accepted name for a spindle cell sarcoma that arises from a nerve or a neurofibroma. In contrast to other STS that is of mesenchymal cell origin, MPNSTs are of neuroectodermal origin. The MPNST designation has replaced many formerly used terms, including malignant schwannoma, neurofibrosarcoma, neurogenic sarcoma, and malignant neural neoplasm. A variant of MPNST is the so-called triton tumor (named after a salamander) that contains neural and rhabdomyosarcomatous elements.22
Etiologies, Pathophysiology, and Clinical Presentation: MPNST accounts for about 4% to 10% of STS, and they are the most common malignancy associated with NF1. Half of these tumors occur in patients with NF1; conversely, 2% to 29% of patients with NF1 develop MPNSTs—a much higher incidence than is seen in the general population. MPNST patients with NF1 are usually younger than those without associated NF1. Furthermore, in patients with NF1, MPNSTs tend to arise in preexisting benign neurofibromas; they are high-grade tumors that have a tendency to recur locally and metastasize. MPNSTs may also arise at previously irradiated sites.31
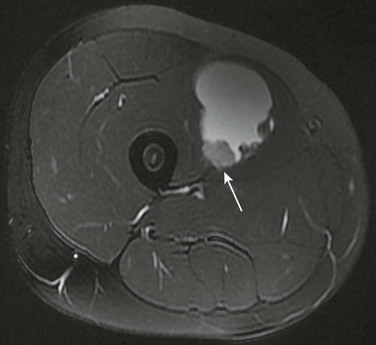
Imaging: Similar to benign neurofibromas, MPNSTs are deep soft tissue lesions that are often associated with primary nerves, especially those of the thigh and lower extremities. CT and MRI appearance is nonspecific. Tumors may be well or poorly defined, homogeneous or inhomogeneous, and they occasionally erode bone. Malignant transformation of benign neurofibromas should be considered in patients in whom the mass is painful or enlarging, or when the typical target appearance of a benign neurofibroma is absent (Fig. 139-11). Tumor uptake on gallium scintigraphy may indicate malignant transformation or progressive growth of neurofibromas; however, biopsy is usually necessary to confirm malignancy.23,24,26,28–30
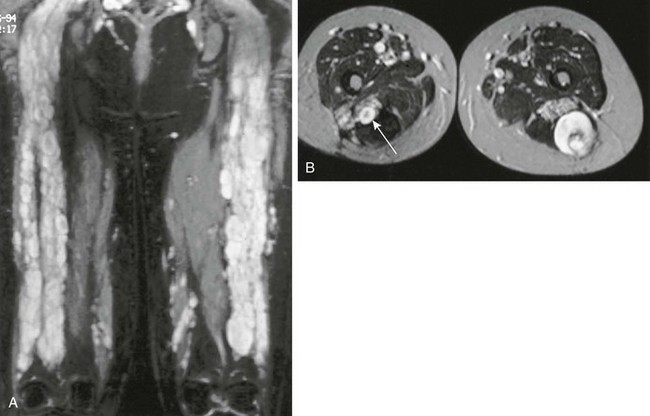
Figure 139-11 Multiple neurofibromas.
A, Coronal short tau inversion recovery magnetic resonance image of the thighs shows multiple neurofibromas distributed along the sciatic nerves of a young woman first seen at the age of 16 with a malignant peripheral nerve sheath tumor. B, Transverse T2-weighted images show multiple, discrete, high-intensity neurofibromas. A characteristic target sign is present (arrow). The larger lesion in the posterior left thigh, which had shown recent growth, was a low-grade malignant peripheral nerve sheath tumor.
Infantile Fibrosarcoma
Overview, Etiologies, Pathophysiology, and Clinical Presentation: Infantile fibrosarcoma is an uncommon tumor that contains fibroblasts and myoblasts and occurs in young children, especially in the first 3 months of life.59 This tumor is now considered a low-grade malignancy, in distinction from an adult-type fibrosarcoma that occurs in older children (10 to 15 years of age) and is more aggressive. Clinically, infantile fibrosarcomas present as enlarging, sometimes painful masses. Tumors often occur in the distal ends of the extremities and occasionally in the head, neck, and trunk. Because of their high degree of vascularization, they may be confused with hemangiomas on physical and imaging examinations.60 These tumors may rarely erode adjacent bone, and angiographic studies may reveal tumor vasculature.61,62
Imaging: Because these masses are often present during fetal life, extensive remodeling of adjacent bones may occur (e-Fig. 139-12). MRI characteristics are nonspecific. Fibrosarcomas are usually isointense to muscle on T1-weighted images and are hyperintense on T2-weighted images. They may contain hypointense foci, which correlate with fibrosis.63
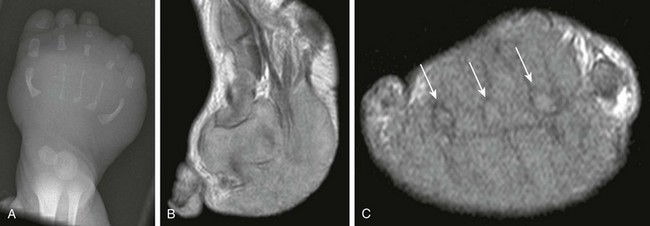
e-Figure 139-12 Infantile fibrosarcoma with rhabdoid elements in a 1-week-old boy.
A, Anteroposterior radiograph of the foot demonstrates gracile metatarsal bones and a large soft tissue density mass. T1 sagittal (B) and T1 short-axis (C) magnetic resonance image demonstrate a large multicompartment soft tissue mass enveloping metatarsal bones (arrows).
Dermatofibrosarcoma Protuberans
Overview, Etiologies, Pathophysiology, and Clinical Presentation: Dermatofibrosarcoma protuberans is an intermediate-grade malignancy that involves the dermis. This tumor is most often seen in adults; it occurs rarely in children but may be seen at birth. The tumor most commonly appears as a red-blue or pink plaque that grows slowly and may become nodular. A less common atrophic variant occurs as a depressed plaque. Lesions are typically fixed to the dermis but may extend into the underlying tissues. Lesions frequently recur locally, and metastasis occurs in 1% to 6% of patients; 75% of metastases occur in the lungs.67,68
Imaging: MRI is useful for determining the extent of disease, especially with deep tumor invasion. MRI characteristics are nonspecific. On T1-weighted images, lesions are hypointense to fat and may be isointense, hyperintense, or slightly hypointense to muscle. On T2-weighted images, lesions are isointense or hyperintense to fat. Lesions can enhance after gadolinium administration. On MRI, these tumors may be confused with subcutaneous granuloma annulare, a benign localized inflammatory dermatosis that occurs in children.69
Therapy: Treatment involves surgical resection with or without adjuvant and neoadjuvant chemoradiotherapy. One review found that only the presence of metastasis had an overall decreased patient survival rate, and those with fibrosarcomatous change or acral location had a decreased disease-free interval despite wide local resection.70
Malignant Fibrous Histiocytoma
The 2002 WHO Soft Tissue Tumor Classification has changed the designation of malignant fibrous histiocytoma (MFH). Pleomorphic MFH is no longer considered a definable or reproducible entity. As a result, many lesions that had been regarded as MFH will be classified as other entities. The term pleomorphic MFH is now synonymous with undifferentiated pleomorphic sarcoma, which is essentially a diagnosis of exclusion that accounts for approximately 5% of adult STS.71–73
Disseminated Disease of Soft Tissues
Overview, Etiologies, Pathophysiology, and Clinical Presentation: Muscle involvement by non-Hodgkin lymphoma (NHL) is usually due to metastatic spread via lymphatic and hematogeneous routes; however, it may be the result of direct extension from primary bone lymphoma. Much less commonly, muscle lymphoma occurs as a primary extranodal tumor. The disease can cross compartmental boundaries, or it may invade subcutaneous tissues. Involvement of adjacent bone and bone marrow may also be noted. Primary T-cell lymphoma of the skin is referred to as mycosis fungoides. Typical findings include focal thickening caused by dermal and epidural infiltrates and lymphadenopathy in advanced-stage disease.74
Imaging: Muscle involvement results in solitary or multiple masses detectable on CT and MRI and with the use of gallium-67 scintigraphy and fluorodeoxyglucose (FDG)-PET. On CT, muscles affected by lymphoma appear diffusely enlarged with or without obliteration of normal fat planes. The tumor may be poorly defined, and its attenuation is equal to or slightly less than that of normal muscle on contrast-enhanced and noncontrast CT images. On MRI, masses are isointense or slightly hypointense to normal muscle on T1-weighted images; they are hyperintense on T2-weighted images and markedly hyperintense on STIR images, and they enhance homogeneously with gadolinium (e-Fig. 139-13). Abnormal activity on gallium-67 and FDG-PET scans correlates well with MRI findings.75–77
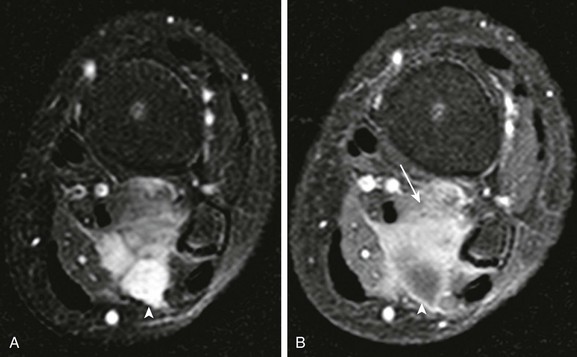
e-Figure 139-13 Extranodal non-Hodgkin lymphoma of calf muscle in a child.
A, T2-weighted fat-saturated axial magnetic resonance image shows multicompartment involvement of the soleus and popliteus muscle. B, Posterior cystic components (arrowhead) do not enhance after contrast. Solid enhancing components are noted in the anterior aspect of the mass (arrow).
Granulocytic Sarcoma
Overview, Etiologies, Pathophysiology, and Clinical Presentation: Granulocytic sarcoma is a rare solid tumor of primitive precursors of white blood cells and is also known as chloroma and extramedullary myeloblastoma. It occurs in patients with acute and chronic myelogenous leukemia and other myeloproliferative diseases. Children are more often affected than adults, and 60% of these tumors occur in children younger than 15 years of age.78
Imaging: Chloromas may be solitary or multiple, and they can involve any part of the body, including the brain and muscles. Orbits and subcutaneous tissues, however, are the most common sites (see Chapter 7). These tumors are typically isoattenuating to slightly hyperattenuating to muscle on noncontrast CT and hyperattenuating to muscle on contrast CT. On MRI, lesions are typically isointense to muscle on T1-weighted images and hyperintense to muscle on T2-weighted images. Tumors usually enhance after gadolinium administration.79–81
Metastases
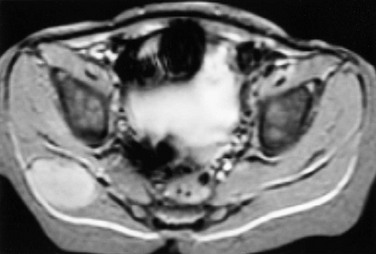
Subcutaneous tissues and muscles may be involved in metastatic lesions. Neuroblastoma, in particular, may metastasize to the skin, subcutaneous tissue, or muscle (Fig. 139-14). Despite its large volume, muscle is not a frequent site for metastatic disease. On CT, muscle metastases produce low-attenuation masses with loss of normal muscle planes. On MRI, metastases are similar in intensity to muscle on T1-weighted images and are hyperintense on T2-weighted images. Contrast-enhanced T1-weighted images show masses of high signal intensity. Focal necrosis may also occur.82,83
Bone Tumors
Radiographic Evaluation of Bone Tumors
Regardless of the site and aggressiveness of a bone lesion, the initial mode of imaging of bone tumors is radiography. Radiographs serve to confirm the presence and site of a tumor, assist in the formulation of differential diagnoses, characterize the tumor, and guide in the selection of further imaging.84,85 The decision to obtain radiographs is a clinical one, based on presentation, history, physical examination, and occasionally on laboratory tests. The differential diagnosis of a pediatric bone tumor can be narrowed down by asking a few simple questions.
How Old Is the Child?
Most bone tumors have a proclivity to occur within a certain age range. The differential diagnosis of a bone tumor in a 1-year-old infant is much different than that of a 16-year-old teenager or a 5-year-old child. Box 139-1 lists common pediatric bone tumors in accordance with the peak age at which they most commonly occur.86–88
What Is the Location of the Lesion? What Bone? What Part of the Bone?
Many bone tumors have a proclivity to affect certain bones within the skeleton or to occur at certain locations within a bone. Box 139-2 lists common pediatric bone tumors according to their preferred location in growing long bones. Lesions also vary in their centricity relative to the involved bone. Some tumors, such as a simple bone cyst, are central, whereas others are eccentric within bone (nonossifying fibroma) or juxtacortical (osteochondroma, periosteal osteosarcoma).86,87,89
Is the Lesion Unifocal or Multifocal?
Some lesions are always solitary, and others are usually multifocal. Some may be solitary or multifocal, although multifocal disease often implies a systemic disease or an underlying syndrome predisposing the patient to the development of a particular type of bone tumor. Box 139-3 lists common pediatric bone tumors that are multifocal.
Is the Lesion Aggressive or Nonaggressive in Appearance?
In general, unlike malignant lesions, benign lesions have a nonaggressive radiographic appearance. Exceptions are common, and some lesions may have both aggressive and nonaggressive features. Box 139-4 lists lesions covered in this chapter by their characteristic radiographic appearance—aggressive, nonaggressive, or indeterminate.
Radiographic features of a nonaggressive and usually benign bone tumor are well-defined margins with a narrow zone of transition, particularly with sclerosis; expansion of bone contour from slow growth; smooth, single-layered periosteal new bone; and absence of an associated soft tissue mass. Radiographic features of an aggressive and usually malignant bone tumor are poorly defined margins with a wide zone of transition, permeative or “moth-eaten” bone destruction, frank destruction of bone without remodeling, aggressive forms of periosteal new bone, interrupted periosteal new bone, and the presence of an associated soft tissue mass. Aggressive forms of periosteal reaction include layering, or “onionskin,” and “hair-on-end” periosteal new bone. Interrupted periosteal new bone may take the form of a Codman triangle, a sign of an aggressive process.90
Some benign bone tumors are adequately defined by radiography and do not require any further imaging for diagnosis or treatment. Most bone tumors, however, do require additional imaging; this may take the form of CT, MRI, scintigraphy, PET scanning, and rarely even US. The choice of imaging for a given tumor depends on the differential diagnostic considerations, possible treatment options, and whether the lesion is aggressive or nonaggressive. MRI is usually the preferred modality in the delineation of aggressive and suspected malignant lesions, and radiographs followed by MRI adequately define most bone lesions. As opposed to radiography, on MRI an aggressive lesion may have a well-defined margin, particularly with T1 weighting. Although MR is also very good at delineating nonaggressive and likely benign lesions, CT is better able to delineate ossified bone and thus occasionally may better define the characteristics and anatomy of many benign lesions. Image-guided biopsy has become a viable option to determine or confirm the diagnosis of many bone lesions. CT, US, fluoroscopy, and even MRI may be used for guidance.91–95
Benign Bone Tumors
Cartilaginous Tumors
Overview, Etiologies, Pathophysiology, and Clinical Presentation: In pathologic series, osteochondroma is the most common pediatric bone tumor. Rather than a true tumor, osteochondromas are thought to be a developmental defect of growing bone in which an injury to the perichondrium causes bone growth in an aberrant direction. It is theorized that islets of cartilage from the physis are displaced along the metaphyseal surface and then grow. Osteochondromas occur in approximately 1% of the general population. Solitary osteochondroma is slightly more common in boys than girls. Growth ceases at skeletal maturity, and lesions are usually detected in the second decade of life as they grow. Most osteochondromas are asymptomatic and are discovered incidentally. However, a host of complications can occur (Box 139-5). When one of a pair of adjacent bones is affected, the osteochondroma can cause pressure deformity of the other bone. Symptomatic presentations of osteochondroma are usually due to irritation of adjacent muscles, tendons, nerves, or rarely blood vessels.96,97 Pseudoaneurysm is a rare complication. A bursa may develop over an osteochondroma as a result of inflammation, and a pedunculated osteochondroma may fracture. Presentation with pain as a result of malignant transformation of a solitary osteochondroma in a child is extraordinarily rare.98,99
Osteochondromas develop in 6% to 12% of patients who received radiation at a young age. Latent periods vary from 3 to 16 years. Osteochondromas can occur even after low doses of radiation therapy and often occur in bones that were in the periphery of the radiation field. Multiple osteochondromas have been found in patients who received total-body irradiation as preparation for bone marrow transplantation at a young age. Sarcomatous degeneration of radiation-induced osteochondroma is very rare and of no greater incidence than with other osteochondromas.100
Patients with hereditary osteochondromatosis (multiple exostoses, diaphyseal aclasis) develop multiple osteochondromas throughout the skeleton. The disorder is autosomal dominant, and 10% of cases arise spontaneously. Patients have a mutation of the EXT1 gene family that results in an error in regulation of normal chondrocyte proliferation and maturation.101,102 In most patients, the disorder becomes manifest by 10 years of age. The multiplicity of lesions in these patients may lead to substantial deformity. Axial osteochondromas are frequently seen and may cause complications. Small lesions are common on tubular the bones of the hand. Most notable is a pseudo-Madelung deformity of the wrist because of forearm exostoses that cause ulnar shortening and angular deformity of the distal radius. Multiple metaphyseal lesions may interfere with normal modeling of the metaphyses.103
Malignant transformation of solitary osteochondromas, even those that are radiation induced, is exceedingly rare and probably occurs in fewer than 1% of patients.100,104 However, the reported incidence of transformation to chondrosarcoma in patients with hereditary osteochondromatosis is 0.5% to 25%. Wide variation reflects patient selection biases; the actual incidence is probably less than 5%. Malignant transformation does not occur until well after skeletal maturity. Clinical and imaging findings suggestive of malignant transformation include an osteochondroma that grows or begins to produce symptoms after physeal closure, a cartilaginous cap greater than 1.5 to 2 cm thick, indistinct lesion margins, new lucency within an osteochondroma, and an associated soft tissue mass. Chondrosarcomas tend to arise at the periphery of an osteochondroma and are usually of low histologic grade. If a diagnosis of chondrosarcoma is suggested based on pathology in someone in the first or second decade of life, it usually is misdiagnosed as chondroblastic conventional osteosarcoma or periosteal osteosarcoma, which are invariably chondroblastic.105,106
Dysplasia epiphysealis hemimelica (DEH), also known as Trevor disease, may be a manifestation of epiphyseal osteochondroma. Patients come to medical attention before 15 years of age, and 75% of patients are boys. Patients are seen initially with deformity, swelling, and pain. These patients form osteochondroma-like protuberances from the epiphyses. The lesions are usually confined to one side of the joint (medial more than lateral) and may occasionally involve contiguous joints in one extremity. The lower extremity (femur, tibia, talus) is usually affected, and the most commonly affected region is the ankle and hindfoot.107 Radiographs show deformity with irregular enlargement of one side of the epiphysis, and MRI is necessary to define the abnormality in younger children, because the lesions may be predominantly cartilaginous. With further ossification in older children, CT is preferred.
Subungual exostosis is a broad-based irregular osteochondroma of the tuft of the finger under the nail bed. The lesion is most common in males in the second decade of life and most commonly affects the great toe. Unlike conventional osteochondroma, there is no medullary continuity of the exostosis with the underlying bone.108,109
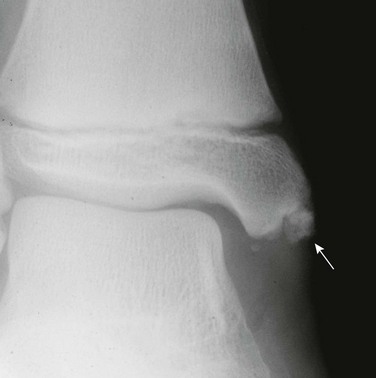
Imaging: On radiography, CT, and MRI, a hallmark of osteochondroma is continuity of the cortex and medullary space from the underlying bone into the lesion. On radiography, osteochondromas are most often found on the long-bone metaphyses, and 35% occur at the knee. Lesions in younger patients tend to be closer to the growth plates. Osteochondromas may also form on the pelvis, ribs, and scapulae; spinal lesions are rare. Underlying metaphyses are broadened as a result of disturbance of normal modeling. The shape of osteochondromas varies from sessile, plaquelike lesions (Fig. 139-15) to pedunculated lesions (Fig. 139-16) with a long stalk. The stalk of a pedunculated lesion is directed away from the adjacent joint. En face, osteochondromas may be mistaken for sclerotic intramedullary lesions.
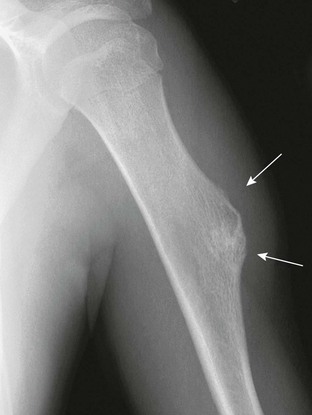
Figure 139-15 Sessile osteochondroma (arrows) of the proximal humeral diaphysis in a 16-year-old girl.
CT nicely demonstrates the morphology of the lesions and usually confirms the diagnosis; however, MRI much better demonstrates the cartilaginous cap characteristic of the lesion (Fig. 139-17). On T2-weighted images and with other cartilage-sensitive sequences, the cartilaginous cap is seen as a well-defined, thin, high-signal crescent that caps the osteochondroma. The role of MRI is not to define the presence or absence of malignancy but to identify pathologic fractures or overlying soft tissue impingement. Pedunculated osteochondromas tend to fracture and cause soft tissue impingement, whereas sessile osteochondromas tend to only cause soft tissue impingement.110
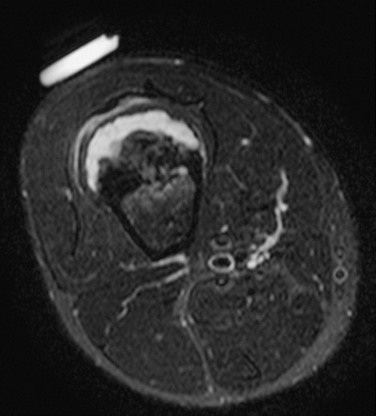
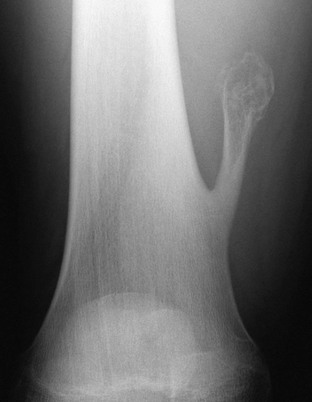
Figure 139-17 Sessile osteochondroma in a 14-year-old boy.
T2-weighted magnetic resonance image with fat saturation shows a broad, high-signal cartilaginous cap that covers the osteochondroma. Note the developing, overlying pseudobursa. The anterior cortex under the cartilaginous cap is thickened. An external vitamin E marker was placed anteriorly over the palpable abnormality for localization.
In DEH, radiographs show deformity with irregular enlargement of one side of the epiphysis (Fig. 139-18). MRI is necessary to define the abnormality in younger children, because the lesions may be predominantly cartilaginous. With further ossification in older children, CT is preferred.
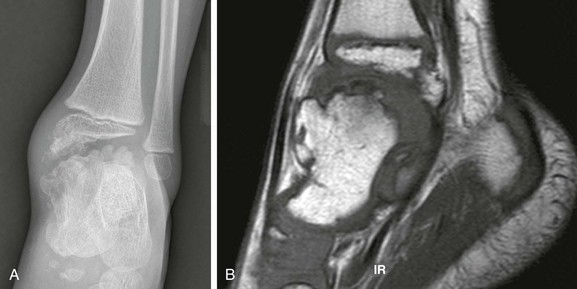
Figure 139-18 Trevor disease (dysplasia epiphysealis hemimelica) in a 3-year-old boy.
A, Radiography demonstrates “kissing” epiphyseal osteochondromas at the level of the tibiotalar joint. B, T1-weighted sagittal magnetic resonance image shows talar marrow continuity with the epiphyseal osteochondromas.
Treatment: Osteochondromas are treated nonoperatively, unless soft tissue impingement is significant, or the lesion causes biomechanical alignment disorders. Treatment of epiphyseal osteochondromas are problematic, but nonarticular components may be resected. The articular components of epiphyseal osteochondromas usually are smooth, and the affected joint typically adapts over time.111
Overview, Etiologies, Pathophysiology, and Clinical Presentation: Enchondromas form owing to a failure of normal endochondral ossification adjacent to a physis. Tumors are composed of cartilage cells derived from the neighboring physis. Enchondromas are most frequently located in the small tubular bones of the hands and feet and in the metaphyses and metadiaphyses of the long bones. Enchondromas represent 80% of primary hand tumors in children and can form in any bone that forms in cartilage. Rib and vertebral lesions are uncommon. Enchondromas become more common with age, with a peak age for diagnosis in the third decade.
Enchondroma protuberans is an enchondroma variant that can resemble either a periosteal chondroma or a sessile osteochondroma (e-Fig. 139-19).112 It has been described as an exophytic, exaggerated, eccentric from of enchondroma. This tumor arises in the medulla and expands eccentrically through the cortex so that the tumor eventually protrudes beyond it.113 Rather than a cartilaginous cap, the tumor is covered by a thin layer of cortex and periosteum. This enchondroma variant most commonly occurs in the proximal humerus and in the hand.114
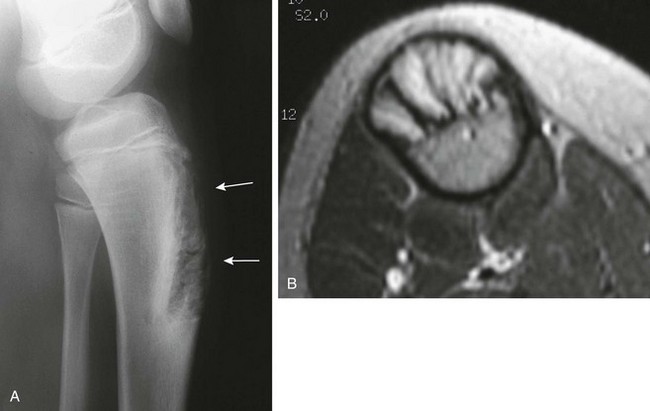
e-Figure 139-19 Enchondroma protuberans in an 11-year-old boy.
A, Radiograph shows an ill-defined lucent lesion in the anterior cortex of the proximal tibia (arrows) abutting the physeal equivalent region of the tibial tuberosity. B, Axial T2-weighted magnetic resonance image shows a well-defined, high-signal lesion in the anterior tibial cortex and intramedullary space.
Metachondromatosis is a very rare disorder that is a combination of enchondromatosis and osteochondromatosis.115
Imaging: On radiography, as with other cartilaginous tumors, enchondromas exhibit a lobulated growth pattern that results in asymmetric expansion of the medullary cavity and endosteal scalloping (e-Fig. 139-20). The lesions may have characteristic channel-lytic lucencies that are perpendicular to the physis. Lesions are oval, well-circumscribed, and lucent with thin eggshell-like margins (Fig. 139-21). Focal, punctate calcifications may be evident on radiographs but are better appreciated with CT. The cartilaginous “ring and arc” pattern may be seen on CT. Margins of the lesion are sclerotic. The lesion may scallop the endosteum, erode cortex, and expand or distort the bone. Periosteal reaction is absent. The tumor is isointense with muscle on T1-weighted MRI and exhibits a heterogeneous, predominantly high T2-weighted signal. Signal intensity of the lesion parallels cartilage on all sequences. Enhancement with gadolinium varies; some lesions enhance peripherally, whereas others enhance more homogeneously. Adjacent bone marrow edema and enhancement are typically absent, but bone scans typically show increased activity.116
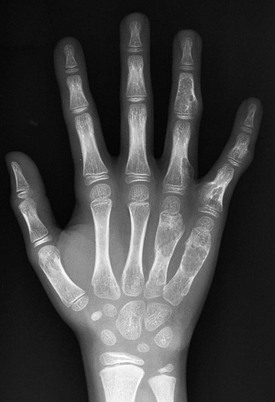
Figure 139-21 Enchondromatosis in a 13-year-old girl.
Multiple, well-defined, lucent, expansile lesions are present in the metacarpals and phalanges of the ring and little fingers. A pathologic fracture is seen through an enchondroma in the distal fourth metacarpal.
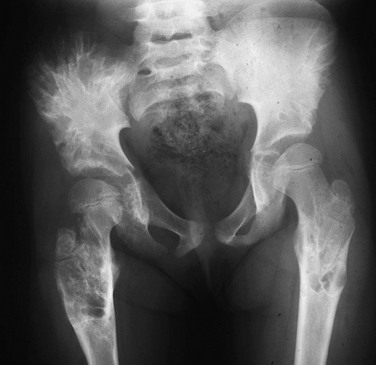
e-Figure 139-20 Enchondromatosis in an 8-year-old girl.
Multiple lesions are present within the proximal femurs and the iliac bones. The iliac lesions on the right are confluent and produce a radial pattern of tubular lytic channels. The proximal femoral diaphyses are mildly expanded with rim sclerosis, and some speckled cartilaginous calcification is present in some of the lesions.
Periosteal (Juxtacortical) Chondroma:
Overview, Etiologies, Pathophysiology, and Clinical Presentation: This rare tumor is a surface variant of an enchondroma that arises from the periosteal surface of the cortex of the large and small tubular bones. One theory holds that the tumor is posttraumatic in origin, located under the periosteum and external to the cortex. Some tumors, such as within the femoral neck, are not covered with periosteum and are better labeled “juxtacortical.” Periosteal chondroma most commonly occurs in the proximal humerus metaphysis, phalanges of the hands and feet, femur, and proximal tibia. Patients are usually 10 to 30 years of age, and peak incidence is in the second decade of life; most come to medical attention with mild pain and swelling.117 The lesion is more frequent in boys, and periosteal chondromas may be seen in patients with Ollier disease.
Imaging: Radiographically, although periosteal chondroma may bear superficial similarity to a sessile osteochondroma, it is associated with sclerosis and external cortical scalloping, forming a periosteal shelf (e-Fig. 139-22). Focal calcifications of matrix within the lesion may be seen. Cross-sectional imaging delineates the underlying cortex and clearly distinguishes the lesion from a sessile osteochondroma. CT may show chondroid calcification (Fig. 139-23). MRI shows chondroid composition of the lesion, and peripheral enhancement is usually seen with gadolinium.118 The tumor is usually 1 to 3 cm in size. A shell of reactive bone may be seen around the lesion, adjacent cortex is eroded or saucerized, and reactive bone sclerosis and buttressing is seen.
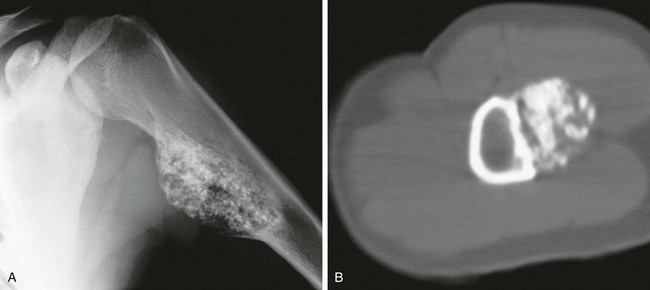
Figure 139-23 Periosteal chondroma in a 19-year-old girl.
A, Anteroposterior view of the humerus shows a broad-based tumor with a lobular mineralized matrix. B, Computed tomography shows the superficial nature of the lesion, which is separated from the medulla by cortical bone.
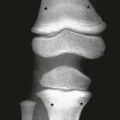
Overview, Etiologies, Pathophysiology, and Clinical Presentation: Chondroblastoma, an uncommon tumor composed of primitive cartilage cells, usually occurs in the second decade of life. Roughly half occur before the physes close. The most specific feature of chondroblastoma is its location in the epiphysis of a long bone, most often the proximal humerus, distal femur, or proximal tibia. Chondroblastoma may also occur in epiphyseal equivalent regions such as apophyses, the patella, and carpal and tarsal bones. Larger lesions often may extend into an adjacent metaphysis.119 Up to 15% of chondroblastomas have a component of aneurysmal bone cyst (ABC). Larger lesions may extend into an adjacent metaphysis, particularly in skeletally mature patients. The tumor evokes a striking inflammatory response, which may help to distinguish it from other lesions.120,121
Imaging: Radiographically, a chondroblastoma is seen as an eccentric, lucent, well-defined smooth or lobulated lesion with sclerotic borders within an epiphysis or epiphyseal equivalent (Fig. 139-24). The lesion may expand the bone, but the cortex is usually intact. Periosteal reaction distant from the lesion is another common feature suggestive of an accompanying inflammatory process.122 Periosteal reaction on an adjacent metaphysis is seen in 30% to 50% of cases. Approximately one third of chondroblastomas have a calcified chondroid matrix. This is better demonstrated by CT, which may also show cortical destruction (see Fig. 139-24). On MRI, chondroblastoma typically parallels cartilage signal intensity on all sequences. Signal intensity varies with the degree of calcification in the lesion. The rim of the tumor may have a lower intensity, and some foci give no signal because of calcification. Adjacent inflammatory changes consist of bone marrow and soft tissue edema and joint effusion and are usually prominent (see Fig. 139-24).123,124
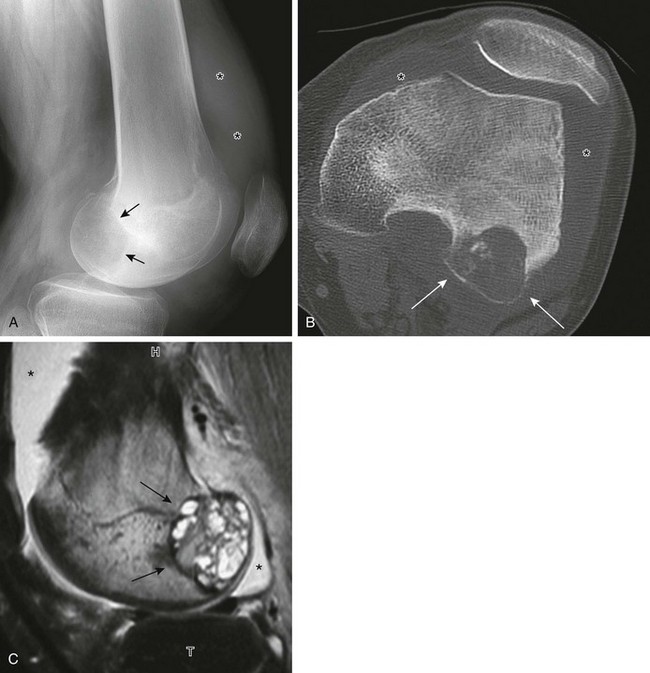
Figure 139-24 Chondroblastoma in a 17-year-old boy.
A, Radiograph shows a lucent lesion within the posterior aspect of the lateral femoral condyle (arrows). An effusion distends the suprapatellar pouch (asterisks). B, Computed tomography image shows a well-defined lesion (arrows) with some calcified cartilaginous matrix and effusion (asterisks). C, Sagittal T2-weighted magnetic resonance image with fat saturation shows the lesion to be of mixed high-signal intensity with multiple cysts consistent with secondary aneurysmal bone cyst formation (arrows). Note internal regions of low signal intensity consistent with chondroid matrix. Bone marrow edema is seen in the femur (compare with the tibia [T]). Knee-joint effusion (asterisk) and adjacent soft tissue edema are noted.
Chondroblastomas superficially will mimic an epiphyseal Brodie abscess. MRI is useful in this circumstance. Both chondroblastomas and epiphyseal Brodie abscesses may demonstrate exuberant adjacent marrow and soft tissue edema, effusions, and reactive bone proliferation. However, on postcontrast imaging, chondroblastomas will demonstrate central enhancement, whereas Brodie abscesses will not.125
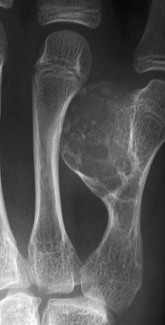
Overview, Etiologies, Pathophysiology, and Clinical Presentation: Chondromyxoid fibroma (CMF) is a benign rare tumor that predominantly affects males in the second or third decade of life. Patients come to medical attention with pain. The lesion is a rubbery mix of fibrous, myxoid, and chondroid tissue. CMF most frequently arises within ilium, long bones at the knee, and tubular bones of the foot, although the proximal tibia is the most common site. The tumor is metaphyseal and often extends into metadiaphysis, but very rarely does it extend past a physis.126,127
Imaging: On radiography, CMF has a characteristic but nonspecific appearance of a solitary, eccentric, lucent, well-defined lesion with sclerotic margins, and septations may be evident within the lesion. In the short bones of the hands and feet, the lesion appears more central. The underlying cortex may be expanded, thinned, and occasionally absent. The lesion may appear bubbly, similar to an ABC.128 Most lesions are elongated and oriented parallel to the long axis of the involved bone (Fig. 139-25). Matrix calcification and periosteal new bone formation usually do not occur. On MRI, CMF produces variable and often heterogeneous signal intensity depending on the composition of the lesion. In general, the lesion is of low signal on T1- and intermediate to high signal on T2-weighted imaging.126
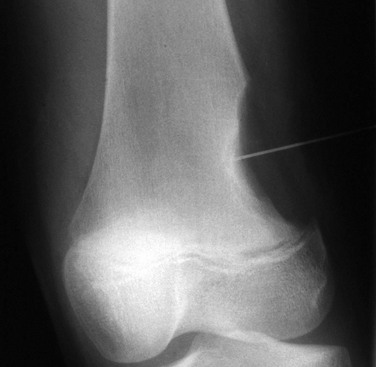
Cysts
Overview, Etiologies, Pathophysiology, and Clinical Presentation: A simple bone cyst is also referred to as a solitary cyst or unicameral bone cyst (UBC), although the latter term is a misnomer, because these cysts may be septated. One theory holds that bone cysts arise because of a defect in endochondral bone formation or altered hemodynamics with venous occlusion that elevates intraosseous pressure and leads to cyst formation. Bone cysts have a membrane of loose vascular connective tissue and contain osteoclast-like giant cells and accumulations of fibrinoid material. The cyst space is usually filled with yellow, sometimes bloody fluid.
Cysts are more common in boys than girls by threefold, and 75% are seen in patients younger than 25 years of age; 25% are found incidentally. Bone cysts typically occur centrally in the metaphyses of the long bones and most commonly involve the proximal humerus (50%) and proximal femur (20%). Cysts have an “active phase,” during which they increase in size and remain in close proximity to the physis. “Latent phase” cysts are found farther from the physis and usually do not continue to grow. Cysts may appear to “migrate” into the diaphysis, but actually it is the growth plate that migrates away from the cyst. In older patients, pelvic and calcaneal bone cysts become more common.129
Solitary UBCs are occasionally found in the calcanei of pediatric patients.130 Often, these cysts are painless and are first detected by radiography of acute injuries to the feet. Calcaneal bone cysts are nearly always located near the base of the neck of the calcaneus. The thin, overlying lateral cortical wall of the calcaneus forms a well-defined bony border that allows differentiation from the “physiologic” pseudocystic radiolucent areas observed in the same region of normal bones.
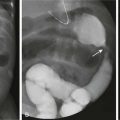
Imaging: On radiography, bone cysts have a central, medullary location within the metaphysis. Most cysts are less than 3 cm in diameter but may be much larger in long axis. The cyst wall is well defined and sclerotic; the overlying cortex is thinned, and the lesion may be mildly expansile.131 With fracture, a fragment of bone may be seen dependently within the cyst. This “fallen fragment” sign is considered pathognomonic for a simple bone cyst (Fig. 139-26). CT delineates the cyst and confirms a fallen fragment, but the study is rarely necessary. In atypical cases, MRI is performed and confirms the cystic nature of the lesion. The fluid contents are low signal on T1- and high signal on T2-weighted imaging. With contrast, the cyst lining enhances, but the contents do not (Fig. 139-27).132
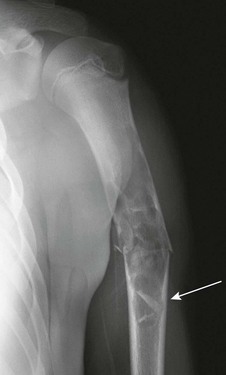
Figure 139-26 Simple bone cyst with pathologic fracture in a 12-year-old boy.
The cyst has thinned and scalloped the overlying cortex. A fallen fragment is noted (arrow).
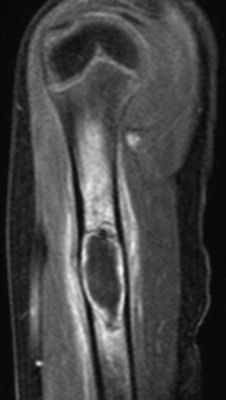
Figure 139-27 Simple bone cyst in an 8-year-old boy.
Sagittal T1-weighted magnetic resonance image with fat saturation post gadolinium shows a lesion that contains fluid with an enhancing rim. Enhancement is also seen in the adjacent marrow and soft tissues. The degree of enhancement is increased in this patient owing to a healing pathologic fracture through the lesion.
Treatment: Fractured cysts tend to heal spontaneously; however, larger cysts with or without fracture are usually treated with curettage and bone grafting. The prognosis is excellent, although 35% to 50% of bone cysts recur, in some cases multiple times. Treated cysts often have a complex appearance with mixed sclerosis and lucency, septations, and mild expansion and deformity of the involved bone. Premature growth plate closure may occur as a complication of treatment or pathologic fracture but not as a result of the cyst itself. Cyst aspiration with corticosteroid injection or sclerotherapy has also been used for treatment.
Overview, Etiologies, Pathophysiology, and Clinical Presentation: ABC is a pseudolesion that occurs as a result of intraosseous or subperiosteal hemorrhage, or it occurs as a transitional lesion secondary to an underlying primary bone tumor. Histologically an ABC is composed of anastomosing channels that contain blood and are variably lined with fibrous walls that contain red blood cells, hemosiderin granules, foreign body giant cells, and spicules of reactive bone. The etiology of an ABC is poorly understood and may be primary or, more commonly, secondary and/or reactive. A wide variety of lesions may act as the nidus for ABC development, and underlying lesions are pathologically identified in one third of cases. A large ABC may obscure the underlying lesion, or it may represent only a small component of a larger tumor. In lesions without an underlying tumor, the role of antecedent trauma acting as a nidus has been proposed. ABC is slightly more common in girls. Whether primary or secondary, the lesion is most common in the first three decades of life, and it is rare in patients younger than 5 years. Patients usually come to medical attention with nonspecific pain and swelling, and 10% come in with pathologic fracture.133 ABC is most common in the metaphyses of long bones, the craniofacial bones, and the spine; spinal lesions occur in the posterior elements. Long-bone lesions can be subclassified as either intramedullary or juxtacortical (cortical or subperiosteal). A subperiosteal ABC is rare and mimics other subperiosteal tumors and pathologies.134–136
An unusual solid variant of ABC has radiographic features similar to those of the typical ABC.137,138 The solid variant lacks cavernous, blood-containing spaces and is characterized histologically by the solid elements—proliferating fibrous tissue, benign giant cells, and newly formed osteoid matrix—found in typical ABCs. A third of these tumors are not “aneurysmal.” The solid variant of ABCs is histologically indistinguishable from extragnathic giant cell (reparative) granuloma. It is most common in the second and third decade. The lesion favors the axial skeleton over appendicular locations, and the most common locations are in the craniofacial bones, small tubular bones of the hands and feet, and the femur.
Imaging: On radiography, ABC appears as a lucent, expansile, “blowout” or “soap-bubble” lesion with thin, smooth, bony walls (e-Figs. 139-28 and 139-29). As a general rule, if the lesion diameter is greater than the widest part of the affected normal bone, an ABC should be considered (see e-Fig. 28). If the diameter is less than the widest part of the affected normal bone, a UBC should be considered. Lesions are multiloculated, and the cortex is usually intact but may be markedly thinned to the point of being invisible, and periosteal new bone may be present. Both CT and MRI demonstrate fluid-fluid levels (see e-Fig. 139-28, B), which are characteristic of the lesion.139,140 This finding is due to sedimentation of degraded blood products, especially methemoglobin, which has a much shorter T1 relaxation time than that of hemoglobin. Fluid-fluid levels may be single or multiple and may be seen as varying horizontal levels within separate loculations (see e-Figs. 139-28 and 139-29). If the loculations are very small, fluid-fluid levels may be less apparent. The signal characteristics of the cyst contents are variable and are probably dependent on the relative age and concentration of the blood components. Abundant hemosiderin may produce foci of low signal, which may be diffuse throughout the lesion. Cyst contents do not enhance, but the septations do.
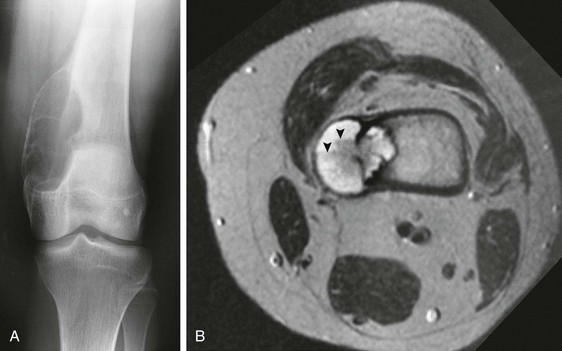
Figure 139-29 Aneurysmal bone cyst in a 13-year-old.
A, Radiograph of the knee shows an eccentric, expansile, lucent lesion with a thin, bony shell that involves the medial aspect of the distal femur. B, On a transverse T2-weighted magnetic resonance image, the lesion is seen to penetrate the cortex of the femur and extend into the adjacent soft tissues. Several fluid-fluid levels are demonstrated (arrowheads).
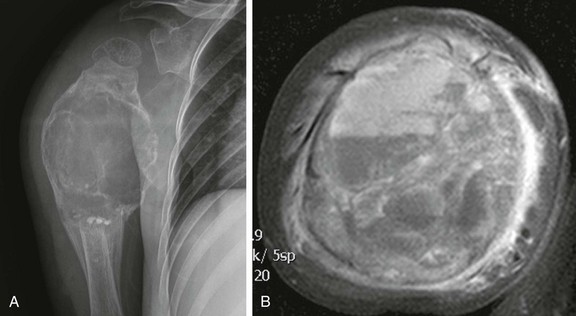
e-Figure 139-28 Recurrent aneurysmal bone cyst (ABC) in a 4-year-old boy.
A, Radiograph of the humerus shows a “blowout” lesion, whose width is wider than the widest part of normal bone. Note the bone graft material in the dependent components of the ABC. B, T2-weighted fat-saturated axial magnetic resonance image demonstrates multiple fluid-fluid levels.
It may not be possible to differentiate primary and secondary ABCs. Secondary ABCs can occur with numerous benign and malignant bone lesions, including fibrous dysplasia, chondroblastoma, GCT, nonossifying fibroma, simple bone cyst, and osteosarcoma, particularly the telangiectatic variant.141,142 Any solid component suggests an underlying tumor. Lesions composed of a greater percentage composition of fluid-fluid levels are more likely benign in origin. Differentiation of ABC from telangiectatic osteosarcoma is particularly difficult and at times cannot be achieved by imaging. Greater bone destruction may be evident with telangiectatic osteosarcoma. It should be noted that ABC may also develop within a conventional osteosarcoma
Overview, Etiologies, Pathophysiology, and Clinical Presentation: GCT, also known as osteoclastoma, is an uncommon neoplasm that rarely occurs before skeletal maturity. Approximately 5% of cases are reported before skeletal maturity,143 most in the second decade, and rarely in the first decade of life.144 The lesion is most common in the long bones, particularly the distal femur and proximal tibia; it is less common in the short bones of the hands and feet, and in children, it rarely occurs elsewhere.145 In skeletally mature individuals, the lesion is uniformly within the epiphysis with variable extension into the adjacent metaphysis. Epiphyseal lesions abut the articular surface. In skeletally immature patients, the lesion is almost uniformly metaphyseal and usually abuts the physis. Epiphyseal involvement is very rare before physeal closure, and multifocal GCT is very rare in children. Patients with GCT come to medical attention with pain and tenderness, swelling, and limited range of motion of the adjacent joint.146
Imaging: On radiographs, GCT appears as a geographic, lytic lesion (Fig. 139-30). Margins vary from sclerotic to ill defined. Frequently, a relatively sharp but nonsclerotic margin is apparent. Periosteal new bone, expansion of bone, and pathologic fracture are common. The metaphyseal end of the lesion tends to be less well defined, although CT delineates the lesion and its margins. No calcified or ossified matrix is seen.
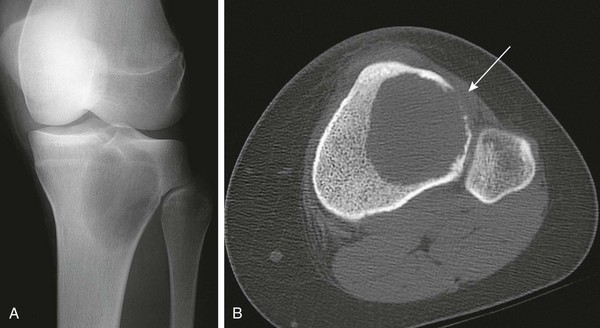
Figure 139-30 Giant cell tumor in a 14-year-old girl.
A, Radiograph shows a relatively well-defined lucent lesion in the proximal tibial metaphysis and epiphysis. The lesion does not have sclerotic margins and is near the articular surface but does not abut it. B, Axial computed tomography shows a large lucent lesion of the tibia. The lesion margins are well-defined but not sclerotic. The anterolateral cortex is destroyed (arrow).
MRI findings vary. In one large series, 56% of tumors were solid or solid with cystic change, and 44% were cystic.143 Solid areas tend to have intermediate T1 and T2 signal. Hemosiderin from intratumoral hemorrhage may produce foci of low signal. Occasionally, a GCT may have a more aggressive appearance, with cortical penetration and soft tissue extension. Approximately 15% of GCTs have an associated component of ABC and appear more expansile.147 The prognosis of GCT is excellent, although up to 25% of tumors recur locally. Malignant GCT and metastasizing GCT have rarely been reported in children. The pulmonary “implants” from GCT are usually of self-limiting growth potential, but recurrence and disease progression is possible.148
Fibrous Tumors
Fibrous Cortical Defect and Nonossifying Fibroma (Fibroxanthoma):
Overview, Etiologies, Pathophysiology, and Clinical Presentation: Fibrous cortical defect (FCD) and nonossifying fibroma (NOF) are extremely common tumors that occur in the metaphyses of the long bones of children. FCDs are essentially a normal variant and are found in up to 40% of children during development. FCD and NOF are histologically identical and are composed of highly cellular stroma with spindle-shaped fibroblasts, osteoclast-like multinucleated giant cells, and foam or xanthoma cells. Arbitrarily, lesions smaller than 2 cm are considered an FCD, and those larger than 2 cm are called NOFs. Lesions probably represent a developmental defect in the periosteum of cortical bone and are seen in the latter part of the first decade until shortly after skeletal maturation. The average time from diagnosis to spontaneous regression is 29 to 52 months; lesions become inactive after skeletal maturity, although rarely, an NOF may persist into adulthood.150,151
FCD and NOF are usually detected incidentally. The lesions are asymptomatic, with the exception of very large lesions, which may cause dull pain. Uncommonly, an NOF may be large enough to cause a pathologic fracture or lead to a stress fracture. FCD and NOF are most common in the metaphyses of the long bones of the lower extremity, especially at the knee. Lesions are more commonly posterior. FCDs and cortical desmoids of the posteromedial distal femoral metaphysis are histologically similar.152
Multiple NOFs may be seen with neurofibromatosis. In Jaffe-Campanacci syndrome, disseminated NOFs are associated with cystic lesions of the jaw, café-au-lait skin lesions, mental retardation, ocular anomalies, hypogonadism, and cardiovascular anomalies in the absence of other signs of neurofibromatosis. There is debate that Jaffe-Campanacci syndrome is a forme fruste of neurofibromatosis.153–156
Imaging: On radiography, FCDs are seen as small, well-defined, ovoid, cortically based lesions. NOFs appear to be similar but are larger (Fig. 139-31), more lobular, and multilocular with a characteristic “soap bubble” appearance. Usually, the lesions extend inward; however, the outer cortex may be thinned and bulging. In thinner bones, such as the fibula, the lesion may occupy the entire width of the bone. FCDs and NOFs originate in metaphysis near the growth plate and migrate into metadiaphysis and diaphysis with maturation, and their radiographic appearance is sufficiently specific that neither additional imaging nor biopsy is indicated.
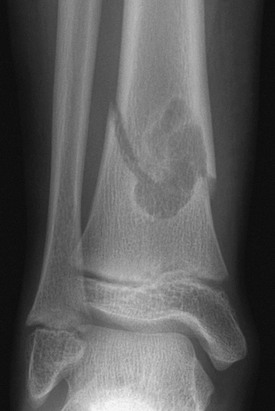
Figure 139-31 Pathologic fracture through a nonossifying fibroma in a 10-year-old boy.
The lesion has well-defined, minimally sclerotic margins.
The radiographic features of FCD and NOF are also manifest on CT. On MRI, lesions are well defined, lobular, and cortically based. Low signal is seen on T1, and T2 signal and enhancement with gadolinium varies with the stage of lesion development. Active, early lesions are high signal on T2-weighted imaging, and they enhance. Involuting lesions are low signal on T2 and do not enhance, and no peritumoral edema is seen.157 Active, early lesions may also show uptake on bone scans and PET scans.158
Overview, Etiologies, Pathophysiology, and Clinical Presentation: Although it is not a true neoplasm, fibrous dysplasia that involves a long bone may mimic a bone tumor or cyst, especially when it causes localized expansion of the bone and is monostotic.159 Pathologically, fibro-osseous tissue replaces the normal medullary space. Fibrous dysplasia can be monostotic or polyostotic and monomelic or polymelic, although 70% to 80% of cases are monostotic. Fibrous dysplasia is more common in girls than in boys, and most patients with focal disease are adolescents or young adults, but patients are occasionally seen in the first decade of life. Any bone may be affected. The most common presentation is monostotic disease that affects craniofacial bones, especially the skull base; a long bone, most commonly the femur; or a rib. Lesions may involve metaphysis and diaphysis but spare the epiphysis before physeal fusion. Patients with solitary lesions come to medical attention with pain, edema, deformity, or pathologic or fatigue fracture. Patients with polyostotic disease are seen with similar signs and symptoms, but often at a younger age (i.e., first decade). Polyostotic disease predominates on one side, is often syndrome related, and may be suspected based on other clinical findings.160,161
In 2% to 3% of patients, fibrous dysplasia is associated with endocrine disorders, mostly of hypothalamic dysfunction. In McCune-Albright syndrome, female patients are seen with precocious puberty, cutaneous café-au-lait spots, and unilateral polyostotic fibrous dysplasia. Mazabraud syndrome, characterized by polyostotic fibrous dysplasia and intramuscular myxoma, is rare in children.162
Imaging: Fibrous dysplasia in the long bones causes expansion of the medullary cavity, endosteal scalloping, coarse trabeculation, and sclerotic margins that form a “rind.” Bowing of the affected bone may occur. In the femur, the resulting deformity is called a shepherd’s crook (Fig. 139-32). Lesions may be central or eccentric. Because the radiographic opacity of a fibrous dysplasia lesion depends on the relative amount of dysplastic bone and fibrous material within the lesion, the appearance varies, from having the look of ground glass to appearing radiolucent. The ground-glass appearance is due to matrix that contains a fine meshlike pattern of delicate bone spicules (Fig. 139-33; see also Fig. 132). It is relatively specific for fibrous dysplasia but may be simulated by other lesions that replace the medullary trabeculae. Lucent lesions usually have a sclerotic margin, and small cartilaginous foci within the lesion may develop chondroid calcification (osteocartilaginous fibrous dysplasia). In addition, a single lesion or involved bone may demonstrate varying appearances within different areas. Active, early lesions tend to be radiolucent, whereas older lesions may be more sclerotic. No periosteal reaction is present unless there is a fracture. Similar findings are visible with CT.
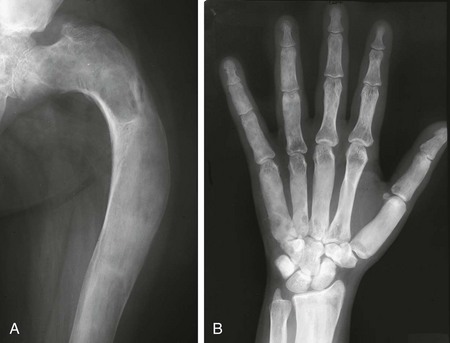
Figure 139-32 Polyostotic fibrous dysplasia in a 22-year-old woman.
A, The femur is expanded and bowed with a “shepherd’s crook” deformity. The femoral trabeculae are replaced by “ground glass” matrix. B, Diffuse sclerosis is seen in the hand and wrist with mild expansion and indistinct transition from cortex to medullary space.
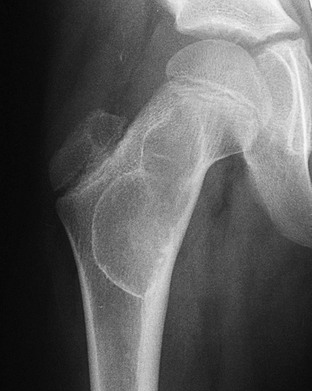
Figure 139-33 Focal fibrous dysplasia in an 11-year-old boy.
The lesion has well-defined, sclerotic margins and a ground-glass matrix.
On MRI, lesions are similar in signal intensity to muscle on T1-weighted imaging. On T2, lesion signal intensity varies depending on the composition of the lesion; pure fibrous tissue is hypointense on T2. However, lesions of fibrous dysplasia are often hyperintense163 because of the inhomogeneous nature of the lesion, which consists of spindle cells, trabeculae of immature woven bone with osteoid seams, and small cysts. Fluid-fluid levels have been reported,164 but soft tissue extension is rare. Central or, less frequently, peripheral enhancement may occur with gadolinium, and lesion uptake on bone scans is variable and may even be normal. Fibrous dysplasia lesions of the face or skull may be quite debilitating and clinically challenging.
Treatment: Treatment is supportive for this benign lesion. Imaging findings are diagnostic, and biopsy is rarely necessary. Surgical treatment is predicated on the presence or absence of pathologic fracture or impending biomechanical failure. Orthopedic implants are used to reinforce and stabilize affected bones.
Overview, Etiologies, Pathophysiology, and Clinical Presentation: Osteofibrous dysplasia (OFD), also known as extragnathic ossifying fibroma or intracortical fibrous dysplasia, is a proliferation of fibro-osseous tissue. OFD is usually sporadic, although a kindred with six affected members by autosomal-dominant inheritance has been reported.165 Most cases occur during the first decade of life, and some tumors have been found in newborns.166,167 OFD is a rare lesion that is usually confined to the diaphysis and metadiaphysis of the tibia but can also involve the fibula, sometimes synchronously in the same patient. Rarely, lesions may be multiple and bilateral. OFD is usually painless and is characterized by deformity that can progress until physeal fusion occurs. Patients may come to medical attention with anterior bowing, and fracture or pseudoarthrosis may complicate the disease’s course.
OFD is histologically similar to fibrous dysplasia in that it contains well-differentiated fibroblasts, collagen, and bony trabeculae. The main differentiating feature is the presence of active osteoblasts in OFD. Differentiated adamantinoma and adamantinoma are closely related to OFD, because they share similar histochemical properties.168 Especially in the second decade, radiologic and even pathologic distinction between OFD, differentiated adamantinoma, and adamantinoma is difficult, although OFD tends to occur at a younger age than adamantinoma. This differentiation is discussed later in this chapter.
Imaging: By radiography, OFD is an eccentric, lucent, solitary or multiloculated lesion that involves the anterior cortex of the tibia (Fig. 139-34). The lesion may be rounded in appearance or ovoid and may be longitudinally oriented with the tibial shaft; it may be purely intraosseous, or it may have a minor extraosseous component. Involvement of the tibia is more commonly proximal, but distal lesions occur and may be complicated by pseudarthrosis. The cortex is expanded, and larger lesions will expand posteriorly and may replace the medullary cavity. Lesions appear lucent or similar to ground glass and are associated with cortical thickening and anterior bowing of the bone. Cross-sectional imaging is very helpful in determining the lesion’s intracortical location, an important feature in distinguishing OFD from fibrous dysplasia, which is intramedullary.
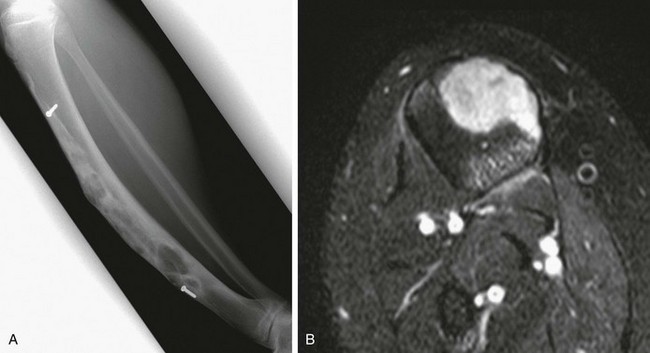
Figure 139-34 Osteofibrous dysplasia in a 15-year-old boy.
Biopsies obtained several years prior had led to a diagnosis of adamantinoma, for which surgical excision with grafting had been performed (screws are related to the prior graft). Symptoms recurred. A, Lateral radiograph shows anterior tibial bowing with mixed sclerosis and cystic lucencies centered anteriorly within the tibia. B, Axial T2-weighted magnetic resonance image with fat saturation shows homogeneous, high-signal soft tissue within the lesion. Percutaneous and excisional biopsies both showed osteofibrous dysplasia.
Overview, Etiologies, Pathophysiology, and Clinical Presentation: Long-bone adamantinomas are rare tumors in children, and patients are often seen initially with pain and anterior tibial bowing.169 As opposed to OFD, adamantinoma is potentially progressive and malignant acting. Up to 15% of affected patients die of metastatic disease. On the basis of clinical, radiologic, histologic, and histochemical characteristics, long-bone adamantinomas (unrelated to the jaw lesion of the same name) have recently been divided into two groups: classic and differentiated (OFD-like). Both types involve the tibia, the fibula, or both bones. The classic form occurs almost exclusively in adults and is found in either the cortex or the medulla, but it can expand through the cortex and periosteum. The differentiated form is seen in children and young adults up to 20 years of age and has been reported in newborn infants. The tumor is limited to the bony cortex and has a radiographic appearance identical to that of OFD. However, adamantinoma are OFD pathologically distinct: epithelial and mesenchymal cells, which express immunoreactive cytokeratin and vimentin, are present in adamantinomas. Epithelial elements are absent in classic OFD and are minimal in differentiated adamantinoma.170
Clinical, radiologic, and pathologic distinction of adamantinoma from OFD is challenging. The concept of a differentiated form implies a continuum between OFD and adamantinoma and suggests that OFD may be a regressive phase of adamantinoma. Some well-documented cases in the literature support this contention. Czerniak and colleagues171,172 suggested that patients with the differentiated type have a more favorable prognosis than those with classic adamantinoma—no differentiated tumors have been known to metastasize—and that long-bone adamantinoma could be included among the few neoplasms capable of spontaneous regression.
Imaging: Radiographic findings of long-bone adamantinoma are analogous to those of OFD. Variable features that might raise suspicion for adamantinoma are periosteal reaction, moth-eaten destruction, and soft tissue extension. MRI of adamantinoma is nonspecific. The T1- and T2-weighted signal characteristics are similar to those of other tumors. Soft tissue extension may also be evident.168,173,174
Treatment: Treatment of adamantinoma is usually en bloc resection. Treatment for OFD and differentiated adamantinoma is much more conservative. If surgery is to be performed for OFD or differentiated adamantinoma, it should be performed after the child reaches puberty, and only if the disease is extensive and has caused deformity.168
Langerhans Cell Histiocytosis:
Overview, Etiologies, Pathophysiology, and Clinical Presentation: The cause of Langerhans cell histiocytosis (LCH) is unknown. An infectious etiology was postulated in the past, but recent evidence of clonal proliferation of Langerhans cells suggests a neoplastic etiology. The unifying pathologic feature of LCH is in an inappropriate proliferation of Langerhans cells. Like the cells of monocyte-macrophage lineage, the Langerhans cell originates from CD34+ stem cells of the bone marrow. Histologically, the tumors are composed of Langerhans histiocytes that contain characteristic cleaved nuclei, and on electron microscopy, racquet-shaped Birbeck granules are seen in the cytoplasm adjacent to the cell membrane; lesions also contain ordinary histocytes and eosinophils. Immunohistochemical staining for S100 protein and CD1a antigen are used in making the diagnosis.175,176
Focal LCH is a relatively common pediatric bone tumor and represents a continuum of disease that ranges from a single, indolent, self-limiting osseous lesion to a fulminant disseminated disorder that involves multiple organs systems. Approximately 80% of patients with LCH have osseous involvement. In the past, patients with LCH pathology were divided into three diagnoses: eosinophilic granuloma referred to localized skeletal disease, usually a single lesion, and 70% of patients fall into this category; children with Hand-Schüller-Christian disease had the triad of geographic bone lesions, proptosis, and diabetes insipidus—a triad rarely seen in an individual patient; and Letterer-Siwe disease referred to the fulminant, disseminated, often fatal multisystem form. Fewer than 10% of patients fall into this category. It is now recognized that LCH is a spectrum of disease rather than these three distinct entities.177,178
LCH is more common in whites, is twice as common in boys, and is seen from the neonatal period into adulthood. Most patients are younger than 15 years at presentation, with a peak incidence from 1 to 5 years of age. Focal lesions tend to be found in children who are slightly older, with an average age of 10 to 12 years. Multifocal and systemic disease is most common in infants and younger children. Fulminant life-threatening LCH is usually seen in the first two decades of life and is rare beyond 3 years of age. Disseminated disease may cause lymphadenopathy, hepatosplenomegaly, skin lesions (purpuric rash), diabetes insipidus, exophthalmos, thrombocytopenia, and anemia. Pulmonary involvement of LCH in children is seen almost always in the setting of disseminated disease, in which osseous lesions vary from relatively few to a diffuse, confluent involvement.179,180
The skull is the most frequent site of LCH, followed by the femur, mandible, pelvis, ribs, and spine; in addition, 70% of lesions occur in flat bones, and 30% occur in tubular bones (long bones, clavicle, hands, and feet). Lesions are usually located in the medulla. Primary lesions of the cortex are rare; however, the cortex is often secondarily affected by expansion of an intramedullary lesion. Long-bone lesions occur in the diaphysis or metaphysis, with rare involvement of the epiphysis. Rarely, lesions may cross an open growth plate. Multiple bone lesions occur in approximately 25% of patients.181,182
Imaging: In the extremities, most LCH lesions are purely osteolytic with well-defined, minimally sclerotic borders. Many lesions are mildly expansile. Endosteal scalloping may be prominent and may progress to cortical disruption. Some lesions are permeative, and periosteal new bone formation may occur, giving them an aggressive appearance. Periosteal new bone may be in single or multiple layers. Although it is often said that LCH can have any radiographic appearance, lesions often have both aggressive and nonaggressive features. When such an indeterminate lesion is identified, LCH is high on the list of differential diagnoses.
Radiographically, LCH of the calvaria typically appears as a lytic bone lesion with a well-defined “punched out” appearance. Skull lesions appear geographic with beveled borders as a result of differential destruction of the inner and outer tables (Fig. 139-35; see also Chapter 21). A so-called button sequestrum may be seen, and lesions of the maxilla and mandible produce “floating teeth” (e-Fig. 139-36). Vertebral lesions produce compression deformities, most commonly in the thoracic spine, followed by the lumbar and cervical regions. LCH often produces vertebra plana with marked loss of height of a single vertebral body (Fig. 139-37). With vertebral involvement, soft tissue extension into the spinal canal may be apparent and is best assessed by MRI. The height of affected vertebral segments tends to reconstitute after therapy. Involvement of the posterior element may occur but is less common than vertebral body involvement.183–185
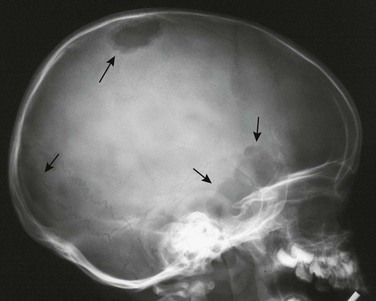
Figure 139-35 Langerhans cell histiocytosis in a 2-year-old girl.
Lateral skull radiograph shows three lytic lesions in the skull. The lesion at the vertex has beveled edges.
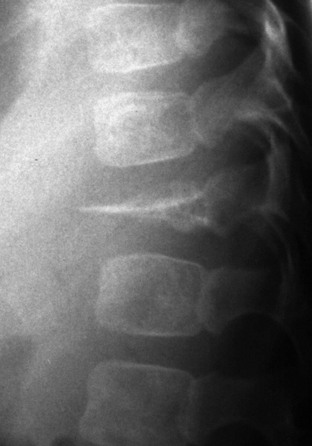
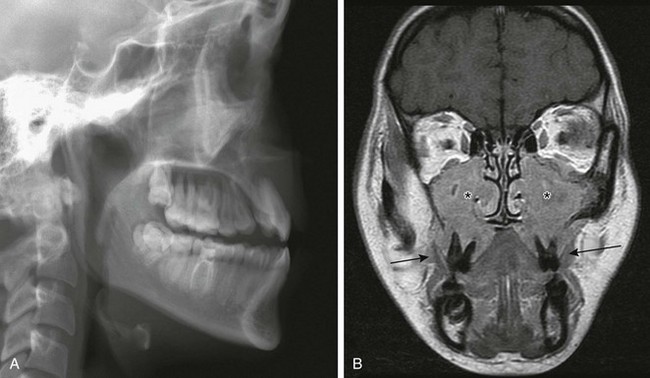
e-Figure 139-36 Langerhans cell histiocytosis in a 16-year-old girl.
Lateral radiograph (A) and coronal T1-weighted magnetic resonance image with gadolinium show “floating” maxillary teeth (arrows in B). Enhancing soft tissue (asterisks in B) fills the maxillary sinuses and replaces the maxilla.
The imaging appearance of LCH depends on the stage and activity of the lesions. The natural history is for spontaneous involution of some lesions to occur. Even at the time of diagnosis, some lesions may be present that are already in a state of involution or quiescence. Involuting lesions will be less well defined and will show sclerosis. Both CT and MRI can be used for further delineation of osseous lesions. On CT, active lesions usually have a well-defined, nonsclerotic margin. A cortical break and soft tissue mass may be seen. MRI is preferred to CT, owing to the lack of ionizing radiation and superior tissue contrast. On MRI, active lesions are composed of soft tissue, which is low signal on T1- and high signal on T2-wieghted imaging, and it enhances relatively homogeneously (see e-Fig. 139-36). About half of the lesions will be hyperintense to muscle on T1-weighted images, and extensive bone marrow and soft tissue edema produces high signal on T2-weighted images and enhances (Fig. 139-38). This prominent inflammatory reaction produces an aggressive appearance suggestive of a malignant process. LCH lesions that disrupt the cortex may also produce a sizable soft tissue mass (seen in 30% of cases on MRI) and may simulate malignancy. Older, involuted, or involuting lesions appear as low signal on T1- and T2-weighted images. Also, a multiplicity of lesions is very suggestive of LCH, as opposed to many other entities that are usually unifocal. If LCH is a consideration, a radiographic skeletal survey may be beneficial in identifying other lesions. Often, the presenting lesion is not at the best site for biopsy, and other unsuspected sites may prove more suitable for tissue diagnosis. Because multiple skeletal lesions may coexist, skeletal imaging is necessary at the time of diagnosis and for purposes of follow-up. Some controversy exists about the relative accuracy of radiographic skeletal surveys and radionuclide bone scintigraphy, because up to 35% of lesions may be missed by bone scans.
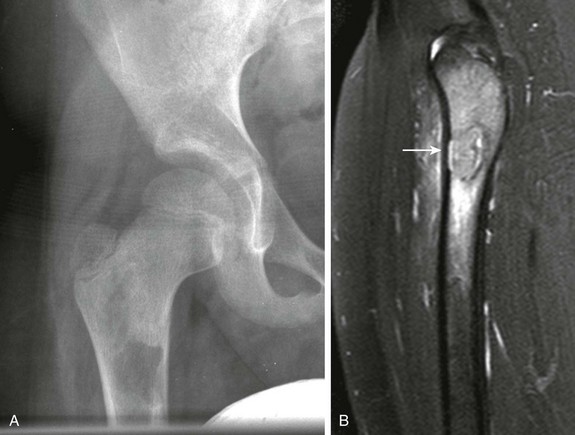
Figure 139-38 Biopsy-proven Langerhans cell histiocytosis in an 8-year-old boy.
A, Radiograph demonstrates a lytic lesion in the right subtrochanteric femur. B, Short tau inversion recovery sagittal magnetic resonance image of the right femur demonstrates the primary mass (arrow) with extensive adjacent bone and soft tissue edema.
Whole-body MRI may become an alternative method for identifying and following multifocal disease. PET scans have also shown utility in identifying sites of active disease and for following response to therapy.186–188
Treatment: LCH localized to the skeleton carries a favorable prognosis. Recurrences are not uncommon. Solitary lesions are watched and usually involute spontaneously. More aggressive therapy may be occasionally warranted, depending on symptoms and lesion characteristics. Curettage and ablative techniques have been used, and children with multiple lesions and/or associated systemic disease are treated more aggressively with steroids and chemotherapy.189 The disease is thus treated in a manner similar to that for a malignant process. The morbidity in children with systemic disease varies depending on the organs involved and the histologic pattern of disease. Children in whom organ function is unaffected and who respond well to initial therapy show a better long-term prognosis. Mortality of disseminated disease is approximately 10%.
Osseous Tumors
Overview, Etiologies, Pathophysiology, and Clinical Presentation: Osteoid osteoma is a common, benign tumor of bone that has a characteristic presentation and radiologic appearance. These tumors occur predominantly in boys, usually those in the second decade of life; however, it is not uncommon in both sexes from the first decade until the mid fourth decade, with a strong prevalence in whites. Pathologically, osteoid osteoma consists of a nidus that is usually surrounded by dense sclerotic bone. The nidus contains interlacing trabeculae at various stages of ossification within a stroma of loose, vascular connective tissue. Three types of osteoid osteoma are recognized: cortical (most common), cancellous or medullary, and subperiosteal (least common). The latter two types produce less sclerotic bone than cortical lesions, making radiologic diagnosis difficult. Osteoid osteomas can also be subdivided into extraarticular and intraarticular types. The incidence of medullary and subperiosteal types is higher at juxtaarticular and intraarticular locations.
Osteoid osteomas may develop in any bone. The single most common location of osteoid osteoma is the femur, and specifically, the femoral neck. The tibia is the next most frequent site. Osteoid osteomas occur less frequently in the upper extremities than in the lower extremities but frequently affect the tubular bones of the hands and feet. Osteoid osteomas most commonly affect the metaphysis or metadiaphysis, less commonly the diaphysis, and rarely the epiphysis. They are less common in flat bones and the spine, where they tend to affect the vertebral posterior arches. In such cases, patients usually are seen initially with painful scoliosis.190 The classic presentation of osteoid osteoma is well-localized pain that is especially severe at night and relieved by aspirin or another nonsteroidal antiinflammatory drugs (NSAIDs); aspirin relieves the pain in 75% of patients. Lesions near a joint may mimic arthritis.
Imaging: Radiographically, the nidus may be purely radiolucent or may contain a dense center. The lucent nidus ranges from a few millimeters to 15 mm in diameter. With cortical osteoid osteomas the nidus is encased by a broad zone of dense bone (Fig. 139-39).191 Intraarticular osteoid osteomas tend to have little reactive bone or periosteal new bone formation. This type most frequently affects the hip, where it causes osteopenia and joint effusion.192 Regardless of location, the nidus of an osteoma may be difficult to see on radiography.
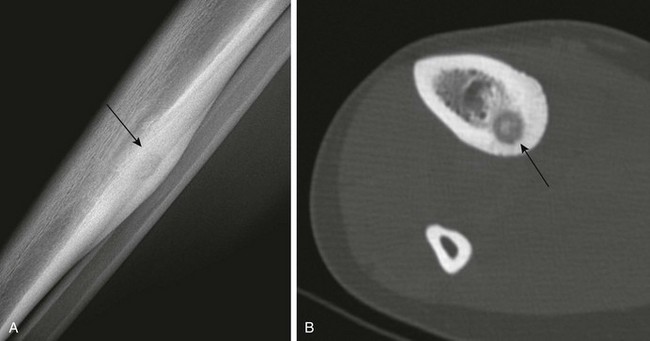
Figure 139-39 Osteoid osteoma of the tibia in a 15-year-old girl.
A, Radiograph shows cortical thickening posteriorly. The lucent nidus is faintly seen (arrow). B, Computed tomography better shows the nidus (arrow). A small radiopaque sequestrum is present within the nidus.
CT is valuable in showing the lesion, confirming the diagnosis, and determining the anatomic location of the lesion before percutaneous therapy or surgical excision. The CT appearance of a lucent nidus with central sequestrum opacity and surrounding sclerosis is usually diagnostic of osteoid osteoma, particularly if the clinical presentation is classic (see Fig. 139-39). Intramedullary and subperiosteal lesions show less sclerosis. Subperiosteal osteoid osteomas lie on top of the cortex and may erode it,193 and they may or may not have ossification within the nidus. Without the ossification, findings are less specific. CT shows the nidus of an osteoid osteoma better than MRI. On MRI, low intensity may be seen on both T1 and T2 images, depending on the relative amount of ossification and fibrovascular tissue. Although there is no signal from the sclerotic bone, the inherent contrast resolution of MRI provides precise definition of potentially extensive reactive changes in the bone marrow and adjacent soft tissues. Osteoid osteoma may produce an intense inflammatory response within adjacent bone, joint, and soft tissues. If the nidus is not identified, a more aggressive process can be suggested.194 Both the nidus and the adjacent inflamed tissues enhance with gadolinium.195 Intraarticular osteoid osteomas produce joint effusions and synovial proliferation that also appear of high intensity on T2-weighted images and enhance after gadolinium.
Treatment: The traditional treatment for osteoid osteoma has been surgical excision, however, localization of the lesion and confirmation of excision may be challenging in the operating room. Recently, percutaneous methods of treatment have been developed. Other percutaneous methods include excision with a large-bore needle or drill and cryoablation,196 but percutaneous radio frequency ablation is now the preferred method of treatment of osteoid osteoma.197 Complex lesions or lesions located where percutaneous methods are contraindicated may still require surgical excision. Surgical or percutaneous methods are successful in approximately 90% of patients on the initial attempt.
Overview, Etiologies, Pathophysiology, and Clinical Presentation: Osteoblastomas are closely related to osteoid osteomas and have in the past been considered a larger version of that tumor (giant osteoid osteoma). The two lesions are nearly identical histologically.198 Osteoblastoma consists of numerous osteoblast-lined trabeculae that contain osteoid.199 However, these trabeculae are less organized than those in osteoid osteoma. Size is an important consideration in distinguishing between the two types of tumors: those less than 1.5 cm in diameter are considered to be osteoid osteomas, whereas tumors larger than 1.5 cm are usually osteoblastomas.200 The two types of tumors can also be distinguished by differences in clinical presentation, anatomic location, and imaging features. Osteoblastoma most commonly occurs in patients in the second and third decades of life,201 and it is more common in boys. Osteoblastomas are not associated with a typical pattern of pain, and if painful, they do not typically respond to NSAIDs. Many osteoblastomas are found in the posterior elements of the vertebrae, where they can cause scoliosis and neurologic deficits. Nearly half of these tumors occur in the appendicular skeleton, primarily in the proximal femoral diaphysis and metaphysis. Tibial lesions are next most common. Long-bone lesions may be cortical or medullary in location. Another characteristic location for osteoblastoma is the neck of the talus, usually at its dorsal margin.
Imaging: Osteoblastoma appears as three distinct radiologic variants: 1) an osteoid osteoma–like appearance, but larger; 2) an ABC-like appearance, most commonly in the spine; and 3) an aggressive appearance that mimics a malignancy, which is uncommon.202 Osteoblastomas vary from 1 to 10 cm in size. Most are lytic with a well-defined sclerotic margin, but the tumor usually lacks the wide, dense rim of sclerosis typical of osteoid osteoma. Lesions in the spine are often expansile (Fig. 139-40). Adjacent sclerosis with spinal lesions may be minimal or absent. In the long bones, osteoblastomas appear radiologically as round or oval lucent tumors, eccentric within the medulla or cortex. Some sclerosis is seen adjacent to the lesion, and mineralization of the matrix is frequently present. Periosteal reaction is common and is solid or layered. Talar lesions may expand into the soft tissues, and osteoporosis of the talus and other bones of the foot may be an associated finding. The calcified or ossified matrix of an osteoblastoma and the tumor’s thin, bony outer shell are especially well visualized on CT images. Edema in the soft tissues or marrow may give high signal on T2-weighted MRI, but signal characteristics are not specific; osteoid within the lesion may cause areas of decreased signal, and bone scans may help in determining the location of a lesion. In addition, osteoblastomas are relatively vascular, hence angiography may reveal a dense capillary blush.
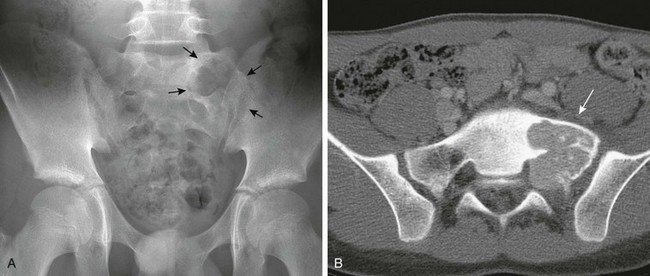
Malignant Bone Tumors
Approximately 50% of all bone tumors in the pediatric age group are malignant; nearly two thirds of these are osteosarcomas. Ewing sarcomas of bone comprise most of the remainder. Other types of osseous malignancies, such as chondrosarcoma, are extremely rare in children, although non-Hodgkin lymphoma occasionally occurs as a primary bone neoplasm. Osteosarcoma and Ewing sarcoma differ sufficiently, both in their clinical and imaging presentation, such that they can usually be distinguished from one another. Their characteristic features, discussed in this chapter, are summarized in Table 139-1.
Table 139-1
Differentiating Features of Osteosarcoma and Ewing Sarcoma
| Feature | Osteosarcoma | Ewing Sarcoma |
| Age range | 15 to 25 years | 0 to 25 years |
| Incidence | More common | Less common |
| Rare in nonwhites | ||
| Location | Metaphyses of long bones | Metadiaphysis of long bones |
| Axial, flat-bone involvement more common than osteosarcoma | ||
| Matrix | Usually “cloudlike” mineralization | Not mineralized or sclerotic |
| Periosteal reaction | “Sunburst” | “Onionskin” |
| Codman triangles | ||
| Metastases | Lung (80%) | Lung, rarely bone, marrow, lymph nodes |
| Bone (20%) | ||
| Other | Occasional multifocal disease | 11 : 22 translocation |
| Association with retinoblastoma | Radiation sensitive |
Osteosarcoma
Overview, Etiologies, Pathophysiology, and Clinical Presentation: Osteosarcoma is the most common malignant primary bone tumor in the pediatric population. The peak incidence of osteosarcoma is in patients 15 to 25 years of age, but the youngest patient described to date with osteosarcoma was a 19-month-old girl.203 The tumors are slightly more common in males, and the long bones are affected in approximately 70% of cases; more than half of osteosarcomas are distributed about the bones of the knee, and the face, mandible, cranium, and axial skeleton are among the less commonly affected sites (Fig. 139-41). Most osteosarcomas are single, primary neoplasms that arise from the medullary cavity of the metaphyses of the long bones. The diaphyses alone are less commonly involved, which occurs in 2% to 11% of cases; an epiphyseal origin of osteosarcoma is extremely rare. Osteosarcomas can involve multiple skeletal sites synchronously, a condition known as osteosarcomatosis.204,205 Extremely rare in children, extraskeletal osteosarcomas are found in various organs or in the soft tissues of the extremities of adults.206,207
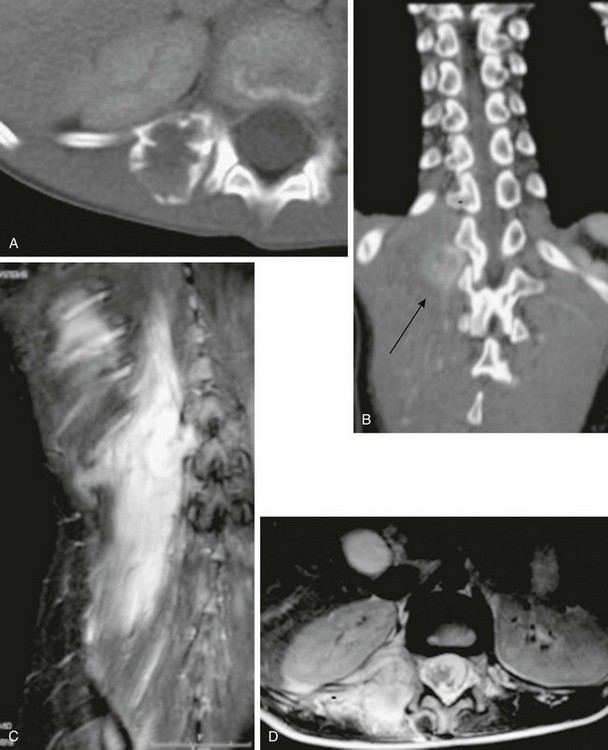
Figure 139-41 Osteosarcoma of the spine in an 8-year-old girl.
A, Axial contrast-enhanced computed tomography through T12 after 10 weeks of chemotherapy shows a peripherally sclerotic expansile mass arising from the right costovertebral junction. Note disruption of the cortex posteriorly. B, Coronal reconstruction shows the relationship of the mass (arrow) to the apex of the scoliosis. C, Coronal short tau inversion recovery magnetic resonance imaging (MRI) demonstrates massive edema in the paraspinal muscles in response to the osteosarcoma. D, Axial contrast-enhanced T1-weighted MRI with fat saturation demonstrates intense tumor enhancement and moderate enhancement of the surrounding paraspinous musculature.
Intramedullary Osteosarcoma
Overview, Etiologies, Pathophysiology, and Clinical Presentation: Approximately 75% of osteosarcomas are the high-grade variant, and 75% of cases occur in patients between the ages of 15 and 25 years. The histologic hallmark of osteosarcoma is the presence of an osteoid matrix produced by sarcoma cells. In most cases, there is extensive immature bone formation. However, other tissues may predominate in the tumor matrix. The three main types of osteosarcoma, based on matrix type, are osteoblastic, chondroblastic, and fibroblastic. These tumors have differing mineral content.207
Imaging: Radiography remains the primary method of diagnosis for conventional osteosarcoma. Other imaging methods are used mainly for staging purposes and to assist with surgical planning. Osteosarcoma typically manifests as a large, mixed sclerotic–lytic mass with a cloudlike matrix that involves the long-bone metaphyses. The tumors cause cortical erosion and destruction rather than expansion. Resultant periosteal new bone formation, often of the spiculated “sunburst” variety (Fig. 139-42), and periosteal elevation are often observed, frequently with Codman triangles (Fig. 139-43). However, conventional osteosarcomas are occasionally purely lytic and exhibit no periosteal reaction. Osteosarcomas with chondroblastic elements tend to be more lytic on radiography (see Fig. 139-43), and osteoblastic dominant osteosarcomas will demonstrate increased osteoid matrix (see Fig. 139-42).
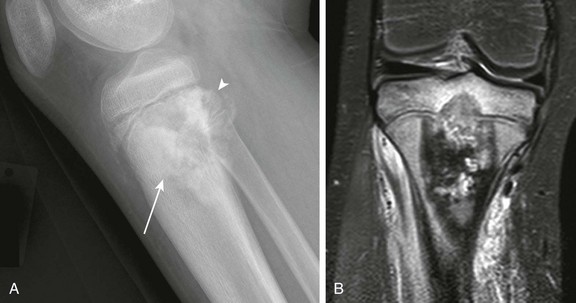
Figure 139-42 Proximal tibial osteosarcoma in an 11-year-old boy.
A, Radiograph demonstrates osteoblastic osteosarcoma with osteoid matrix (arrow) and “sunburst” periostitis (arrowhead). B, Corresponding short tau inversion recovery coronal magnetic resonance image shows low signal intensity throughout the majority of the mass, indicative of osteoid matrix. Note the presence also of transphyseal transgression through the central physis.
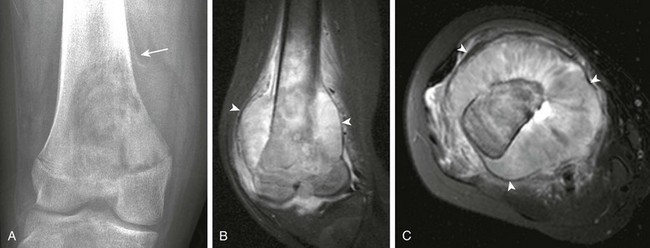
Figure 139-43 Osteosarcoma in a 13-year-old girl.
A, Radiograph demonstrates permeative lytic destruction in the distal femur metaphysis with aggressive periosteal reaction with a Codman triangle (arrow). Short tau inversion recovery coronal (B) and axial (C) magnetic resonance image demonstrates distal femoral metaphyseal mass with transphyseal transgression into the epiphysis with a large extraosseous tumor component with intermediate signal intensity, consistent with osteoid matrix. Note the displaced periosteal envelope (arrowheads).
CT is superior to radiographs for delineating osteoid matrix. Like radiographs, CT often underestimates the true extent of bone involvement (Fig. 139-44).
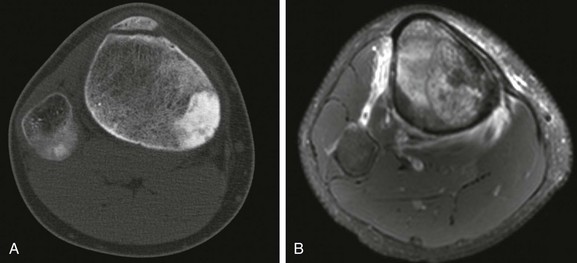
Figure 139-44 Tibial osteosarcoma in a 14-year-old boy.
A, Axial computed tomography demonstrates intraosseous osteoblastic matrix replacing a portion of the medullary cavity. B, Short tau inversion recovery axial magnetic resonance image through the same level demonstrates near complete intramedullary involvement.
MRI is used extensively to evaluate osteosarcoma. The longitudinal extent of marrow involvement, an important determinant of surgical therapy, is accurately shown as well-defined hypointense signal within hyperintense fatty marrow on T1-weighted images obtained in either coronal or sagittal planes. It is important to obtain a longitudinal T1-weighted image of the entire bone to measure intramedullary tumor length, to assess possible epiphyseal involvement (seen in as many as 80% of metaphyseal tumors), and to detect skip metastases that occur in a small percentage of cases. On T2-weighted images, the tumor-containing marrow can be either hyperintense or, if there is sufficient bone formation, hypointense to normal fat (see Fig. 139-42). The soft tissue component is usually of heterogeneous, mainly high-intensity signals that contrast greatly with surrounding structures. STIR images are very sensitive to the water content of tumors, thus they markedly increase the conspicuity of most osteosarcomas; the intramedullary length of tumor extension may be overestimated on STIR sequences.208 Contrast-enhanced T1-weighted images, preferably obtained with fat saturation, provide similar contrast and better signal-to-noise ratios than T2-weighted images. Furthermore, contrast-enhanced T1-weighted images are especially useful in determining the relationship between the tumor and the major blood vessels, in detecting joint involvement in the presence of effusion,209 and in estimating the amount of necrosis within the tumor. Osteosarcoma is frequently accompanied by edema of the adjacent soft tissues that is hyperintense to muscle on T2-weighted STIR images and contrast-enhanced T1-weighted images. This edema can be localized to the tumor periphery, or it can involve whole muscle groups, as in the case of larger tumors. The latter finding has been associated with a poor prognosis.210
Overview, Etiologies, Pathophysiology, and Clinical Presentation: Telangiectatic osteosarcoma comprises about 2% of all osteosarcomas. Like conventional osteosarcomas, telangiectatic osteosarcoma tends to occur in the long bones adjacent to the knee and is seen more frequently in boys than in girls. These tumors contain little osteoid and do not form bone but rather are composed of single or multiple cavities that contain blood or necrotic tumor with septa of anaplastic cells.211,212 Telangiectatic osteosarcomas by definition are composed of approximately 90% cystic components prior to treatment.213
Imaging: Radiographically, telangiectactic osteosarcomas tend to be lytic, and they tend to expand, rather than destroy, the cortex; they may also be associated with a soft tissue mass. Except for the presence of malignant cells that are often at the periphery of the cavity, the pathologic appearance of telangiectatic osteosarcoma mimics that of an ABC. Indeed, on MRI, the appearance of telangiectatic osteosarcoma and ABC may be identical; both may have single or multiple fluid-fluid levels produced by blood products of differing ages that are best demonstrated on T2-weighted images.214 Telangiectatic osteosarcomas are characterized by enhancing soft tissue in the periphery and septations of the tumor, features absent in ABCs.215 Biopsy is necessary to differentiate telangiectatic osteosarcoma from an ABC. The prognosis of patients with telangiectatic osteosarcoma is similar to that of patients with conventional osteosarcoma.
Overview, Etiologies, Pathophysiology, and Clinical Presentation: Surface osteosarcomas are classified by histologic grade as low-, intermediate-, and high-grade lesions.216 Parosteal osteosarcoma is more common in girls than in boys, tends to occur after skeletal maturity has been achieved, and is of low histologic grade. Periosteal osteosarcoma probably arises from the deep layers of periosteum or the outer cortex and is classified as an intermediate-grade osteosarcoma; most periosteal osteosarcomas are chondroblastic,217 and they may be mistaken for a chondrosarcoma by pathologists unfamiliar with pediatric orthopedic oncology. High-grade surface osteosarcomas comprise dedifferentiated parosteal osteosarcoma and high-grade surface osteosarcoma. The origin of high-grade surface osteosarcoma is controversial; histologically, it is difficult to differentiate from the much more common, conventional, intramedullary high-grade osteosarcomas.
Imaging: Parosteal osteosarcomas are very osteoblastic and are slow growing. They demonstrate mature periosteal ossification and most commonly occur in the posterior aspect of the distal femur (Fig. 139-45). Superficially, parosteal osteosarcoma may mimic a sessile osteochondroma. MRI is used to define the degree of intramedullary extension, if present, and adjacent neurovascular and muscle involvement.
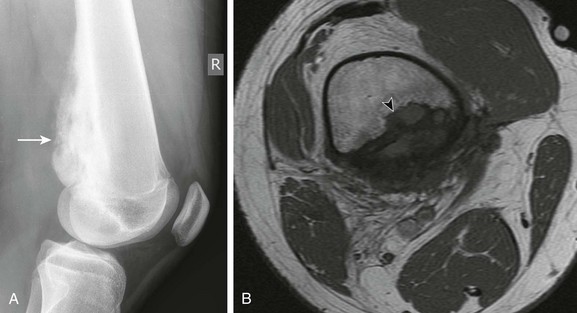
Figure 139-45 Parosteal osteosarcoma in a 14-year-old boy.
A, Lateral radiograph demonstrates a surface-based osteoblastic mass in the distal femur (arrow). B, T1-weighted axial magnetic resonance image through the distal femur demonstrates the surface-based mass with intramedullary extension (arrowhead).
Periosteal osteosarcomas also arise from the surface and may have adjacent intramedullary marrow edema, but true invasion is rare.217 Because these lesions are invariably chondroblastic, they often have little to no matrix on radiography and are hyperintense on fluid-sensitive sequences (Fig. 139-46). Superficially, these lesions may mimic a juxtacortical chondroma.
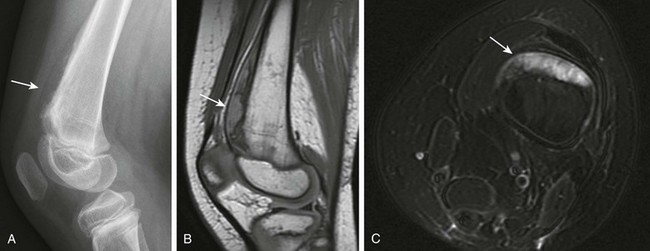
Figure 139-46 Periosteal osteosarcoma in an 11-year-old boy.
A, Lateral radiograph demonstrates aggressive, perpendicularly oriented periosteal reaction with little to no matrix (arrow). B, Sagittal proton-density magnetic resonance imaging (MRI) demonstrates an intermediate signal lesion. C, On T2-weighted fat-saturated axial MRI, it is hyperintense and surface based, indicative of its chondroblastic composition (arrow).
Treatment: Osteosarcomas are not radiosensitive. Thus, treatment consists of preoperative chemotherapy; extirpation of resectable lesions, usually by limb-sparing surgery; and postoperative chemotherapy. MRI serves as a surgical road map to define the presence or absence of neurovascular involvement, physeal and epiphyseal extension, and joint involvement. If the physis is spared, physeal-sparing surgery can be attempted with an intercalary graft. When the epiphysis is involved, morbidity is higher, and a joint prosthesis is necessary.
Prognosis is influenced by response to initial chemotherapy, which is evaluated postoperatively by histologic estimation of necrosis within the treated tumor. Little change in tumor size is expected, even in tumors that respond well to chemotherapy. Imaging methods of assessing the effects of therapy include thallium-201 scintigraphy and dynamic contrast-enhanced MRI. Although PET and PET-CT appear to be useful in both staging and monitoring response, experience is currently limited in its use for osteosarcoma and other bone tumors. Long-term follow-up of osteosarcoma and other malignant bone tumors in children is essential and extends over many months.218,219
Overview, Etiologies, Pathophysiology, and Clinical Presentation: In 1921, James Ewing described a radium-sensitive bone tumor, which he called an endothelioma, that consisted of sheets of small polyhedral cells of probable endothelial origin.220 Although the origin of this undifferentiated tumor has been debated since its initial description, what we now know as Ewing sarcoma has proved to be a distinct entity with characteristic histologic, radiologic, and cytogenetic features. The Ewing family of tumors includes Ewing sarcoma of bone, extraosseous Ewing sarcoma (EOES), and primitive neuroectodermal tumor (PNET), also known as peripheral neuroepithelioma. PNETs exhibit neural differentiation and may arise from either bone or soft tissues. The Ewing family of tumors shares a distinctive cytogenetic feature: reciprocal translocation of chromosome bands q24 and q12 of chromosomes 11 and 22. This same translocation is also found in Askin tumor of the thorax.221–223
In younger patients, Ewing sarcoma of bone occurs less frequently than osteosarcoma, with 2.9 cases per million diagnosed in patients younger than 20 years of age. Most cases are detected in patients between 10 and 25 years of age (median 15 years). Like osteosarcoma, Ewing sarcoma occurs slightly more often in boys and is much more common in whites. Fever, leukocytosis, and elevation of the erythrocyte sedimentation rate may accompany these neoplasms. More than 50% of Ewing sarcomas involve a single long bone; Ewing sarcoma involving the bones of the hands and feet, often initially diagnosed as an infection, is rare. Within the long bones, the metaphysis and diaphysis are the usual locations. The flat bones, especially the ribs and pelvis, are also commonly involved. Most Ewing sarcomas appear to arise from the medullary cavity. Multifocal osseous involvement at the time of diagnosis is rare.223–225
Imaging: The typical radiographic appearance of Ewing sarcoma in the long bones is that of a permeative lesion with a lamellar “onionskin” periosteal reaction (see Fig. 139-47). However, nearly 40% of these tumors display diffuse sclerosis, sometimes with a mixed lytic–sclerotic pattern. The sclerosis correlates histologically with the presence of dead bone.226 Because Ewing sarcomas do not ossify, soft tissue extension is often poorly detected by radiographs but is almost always apparent on CT or MRI (see Fig. 139-48). Indeed, the soft tissue masses tend to be disproportionately large in comparison to the amount of bone destruction, and they are especially extensive in Ewing sarcoma of the pelvis (Fig. 139-49). Cortical permeation and destruction may be visible on CT or MRI. However, these tumors can permeate haversian canals and can grow into the soft tissues without causing large areas of cortical loss. Extensive marrow involvement is particularly well shown as nonspecific low signal, isointense to muscle on T1-weighted images. It has been observed that most Ewing sarcomas found in the medullary cavity display intermediate signal that is isointense to that of fat on T2-weighted images. Rarely, Ewing sarcoma can arise from the surface of the bone rather than from the medullary cavity. In this periosteal or subperiosteal location, Ewing sarcoma resembles other surface malignancies, such as periosteal osteosarcoma with periosteal elevation and Codman triangles.227 The affected cortex is typically excavated or saucerized,228 and cross-sectional imaging is required to exclude medullary involvement. Patients with this form of Ewing sarcoma are thought to have a relatively favorable prognosis.
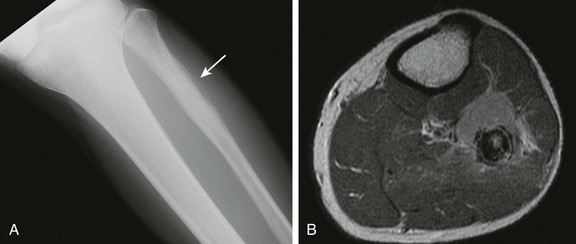
Figure 139-47 Ewing sarcoma of the fibula in a 17-year-old.
A, Anteroposterior radiograph of the leg demonstrates aggressive “onionskin” periostitis of the proximal fibula (arrow). B, T1-weighted axial postcontrast magnetic resonance image demonstrates a large extraosseous mass that arises from the fibula with cortical destruction.
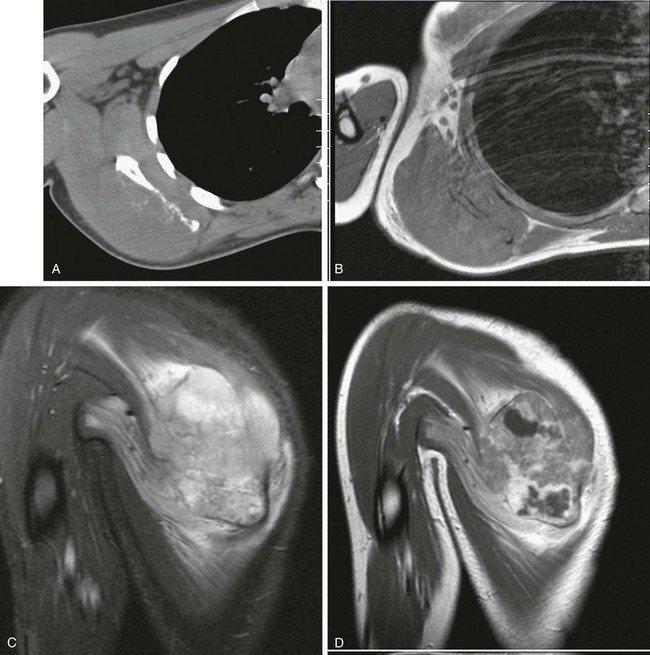
Figure 139-48 Ewing sarcoma of the right scapula in a 16-year-old girl.
A, Axial computed tomographic image shows a permeative pattern to the scapula with subtle dystrophic calcifications within the soft tissue mass. B, T1 axial magnetic resonance imaging (MRI) shows marrow replacement and cortical destruction by the mass. C, Short tau inversion recovery sagittal MRI demonstrates a large heterogeneous mass with cortical destruction and intramuscular invasion. D, T1 postcontrast MRI demonstrates heterogeneous central tumoral enhancement with areas of nonenhancement consistent with tumoral necrosis.
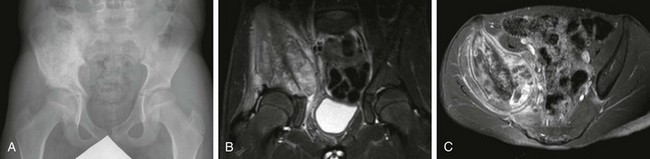
Figure 139-49 Ewing sarcoma of the right iliac bone in an 11-year-old girl.
A, Frontal radiograph of the pelvis demonstrates a mixed lytic and blastic lesion of the right iliac bone. B, Short tau inversion recovery coronal magnetic resonance imaging (MRI) demonstrates a right iliac mass with extraosseous extension. C, Postcontrast T1-weighted axial MRI demonstrates heterogeneous enhancement of the mass, as it better delineates both extraperitoneal extension and posterior extension into the pelvic abductor muscles.
Treatment: With appropriate multimodal therapy, the long-term survival rate of patients with nonmetastatic medullary Ewing sarcoma approaches that of patients with osteosarcoma (about 65%). In some series, tumor volume has been shown to influence prognosis. Survival of patients with pelvic tumors may be somewhat shortened and is much reduced in patients with metastatic disease. As many as 25% of patients with Ewing sarcoma have detectable metastases at the time of diagnosis; most of these are found in the lung, therefore chest CT is necessary for disease staging. Local and regional lymph node involvement may rarely occur, and metastases in bone or bone marrow are less frequent, although skip metastases have been reported.229
Percentage change in size of the soft tissue mass appears to be an important prognostic indicator.230 T2-weighted MRI shows an increase in intensity of the treated medullary component because of serous atrophy, with increased interstitial fluid of the yellow marrow and replacement of cytoplasmic lipid with serous material. High T2-weighted signal intensity caused by radiation-induced inflammatory reactions may also be observed in those patients treated with radiation therapy.231–234
The surgical approach to resection of Ewing sarcoma is similar to osteosarcoma.
 What the Clinician Needs to Know
What the Clinician Needs to KnowLaor, T. MR imaging of soft tissue tumors and tumor-like lesions. Pediatr Radiol. 2004;34(1):24–37.
Levine, SM, Lambiase, RE, Petchprapa, CN. Cortical lesions of the tibia: characteristic appearances at conventional radiography. Radiographics. 2003;23:157–177.
Murphey, MD, Robbin, MR, McRae, GA, et al. The many faces of osteosarcoma. Radiographics. 1997;17:1205–1231.
Navarro, OM, Laffan, EE, Ngan, B-Y. Pediatric soft-tissue tumors and pseudo-tumors: MR imaging features with pathologic correlation: part 1. Imaging approach, pseudotumors, vascular lesions, and adipocytic tumors. Radiographics. 2009;29(3):887–906.
O’Donnell, P, Saifuddin, A. The prevalence and diagnostic significance of fluid-fluid levels in focal lesions of bone. Skeletal Radiol. 2004;33:330–336.
References
1. Brisse, H, Orbach, D, Klijanienko, J, et al. Imaging and diagnostic strategy of soft tissue tumors in children. European Radiology. 2006;16(5):1147–1164.
2. Finlay, K, Probyn, L. Ultrasonography of lumps and bumps. Can Assoc Radiol J. 2002;53(1):39–49.
3. Bitran, JD, Bekerman, C, Golomb, HM, et al. Scintigraphic evaluation of sarcomata in children and adults by Ga67 citrate. Cancer. 1978;42(4):1760–1765.
4. Howman-Giles, R, Uren, RF, Shaw, PJ. Thallium-201 scintigraphy in pediatric soft-tissue tumors. J Nucl Med. 1995;36(8):1372–1376.
5. Navarro, OM, Laffan, EE, Ngan, B-Y. Pediatric soft-tissue tumors and pseudo-tumors: MR imaging features with pathologic correlation: part 1. Imaging approach, pseudotumors, vascular lesions, and adipocytic tumors. Radiographics. 2009;29(3):887–906.
6. van Rijswijk, CSP, Geirnaerdt, MJA, Hogendoorn, PCW, et al. Soft-tissue tumors: Value of static and dynamic gadopentetate dimeglumine-enhanced MR imaging in prediction of malignancy. Radiology. 2004;233(2):493–502.
7. Laor, T. MR imaging of soft tissue tumors and tumor-like lesions. Pediatr Radiol. 2004;34(1):24–37.
8. Dinauer, PA, Brixey, CJ, Moncur, JT, et al. Pathologic and MR imaging features of benign fibrous soft-tissue tumors in adults. Radiographics. 2007;27(1):173–187.
9. Shinagare, AB, Ramaiya, NH, Jagannathan, JP, et al. A to Z of desmoid tumors. AJR Am J Roentgenol. 2011;197(6):W1008–W1014.
10. Shields, CJ, Winter, DC, Kirwan, WO, et al. Desmoid tumours. Eur J Surg Oncol. 2001;27(8):701–706.
11. Romero, JA, Kim, EE, Kim, CG, et al. 11 Different biologic features of desmoid tumors in adult and juvenile patients: MR demonstration. J Comput Assist Tomogr. 2006;19(5):782–787.
12. Rao, BN, Horowitz, ME, Parham, DM, et al. Challenges in the treatment of childhood fibromatosis. Arch Surg. 1987;122(11):1296–1298.
13. McCarville, MB, Hoffer, FA, Adelman, CS, et al. MRI and biologic behavior of desmoid tumors in children. AJR. 2007;189:633–640.
14. Casillas, J, Sais, GJ, Greve, JL, et al. Imaging of intra- and extraabdominal desmoid tumors. Radiographics. 1991;11(6):959–968.
15. Sturt, NJH, Clark, SK. Current ideas in desmoid tumours. Fam Cancer. 2006;5(3):275–285. [discussion 287-288].
16. Murphey, MD, Ruble, CM, Tyszko, SM, et al. From the archives of the AFIP: musculoskeletal fibromatoses: radiologic-pathologic correlation. Radiographics. 2009;29(7):2143–2173.
17. Koujok, K, Ruiz, RE, Hernandez, RJ. Myofibromatosis: imaging characteristics. Pediatr Radiol. 2004;35(4):374–380.
18. Kransdorf, MJ, Jelinek, JS, Moser, RP, et al. Magnetic resonance appearance of fibromatosis. A report of 14 cases and review of the literature. Skeletal Radiol. 1990;19(7):495–499.
19. Patrick, LE, O’Shea, P, Simoneaux, SF, et al. Fibromatoses of childhood: the spectrum of radiographic findings. AJR Am J Roentgenol. 1996;166(1):163–169.
20. Robbin, MR, Murphey, MD, Temple, HT, et al. Imaging of musculoskeletal fibromatosis. Radiographics. 2001;21(3):585–600.
21. Soper, J, De Silva, M. Infantile myofibromatosis: a radiological review. Pediatr Radiol. 1993;23(3):189–194. [Springer].
22. Woertler, K. Tumors and tumor-like lesions of peripheral nerves. Semin Musculoskelet Radiol. 2010;14(5):547–558.
23. Murphey, MD, Smith, WS, Smith, SE, et al. From the archives of the AFIP. Imaging of musculoskeletal neurogenic tumors: radiologic-pathologic correlation. Radiographics. 1999;19(5):1253–1280.
24. Vilanova, JC, Woertler, K, Narváez, JA, et al. Soft-tissue tumors update: MR imaging features according to the WHO classification. European Radiology. 2006;17(1):125–138.
25. Mandell, GA, Herrick, WC, Harcke, HT, et al. Neurofibromas: location by scanning with Tc-99m DTPA. Work in progress. Radiology. 1985;157(3):803–806.
26. Kumar, AJ, Kuhajda, FP, Martinez, CR, et al. Computed tomography of extracranial nerve sheath tumors with pathological correlation. J Comput Assist Tomogr. 1983;7(5):857–865.
27. Huang, G-S, Huang, C-W, Lee, H-S, et al. On the AJR viewbox. Diffuse neurofibroma of the arm: MR characteristics. AJR Am J Roentgenol. 2005;184(5):1711–1712.
28. Levine, E, Huntrakoon, M, Wetzel, LH. Malignant nerve-sheath neoplasms in neurofibromatosis: distinction from benign tumors by using imaging techniques. AJR Am J Roentgenol. 1987;149(5):1059–1064.
29. Lin, J, Martel, W. Cross-sectional imaging of peripheral nerve sheath tumors: characteristic signs on CT, MR imaging, and sonography. AJR Am J Roentgenol. 2001;176(1):75–82.
30. Stull, MA, Moser, RP, Kransdorf, MJ, et al. Magnetic resonance appearance of peripheral nerve sheath tumors. Skeletal Radiol. 1991;20(1):9–14.
31. Suh, JS, Abenoza, P, Galloway, HR, et al. Peripheral (extracranial) nerve tumors: correlation of MR imaging and histologic findings. Radiology. 1992;183(2):341–346.
32. Ros, PR, Eshaghi, N. Plexiform neurofibroma of the pelvis: CT and MRI findings. Magnetic Resonance Imaging. 1991;9(3):463–465.
33. Hassell, DS, Bancroft, LW, Kransdorf, MJ, et al. Imaging appearance of diffuse neurofibroma. AJR Am J Roentgenol. 2008;190(3):582–588.
34. Collins, MH, Chatten, J. Lipoblastoma/lipoblastomatosis: a clinicopathologic study of 25 tumors. Am J Surg Pathol. 1997;21(10):1131–1137.
35. Moholkar, S, Sebire, NJ, Roebuck, DJ. Radiological-pathological correlation in lipoblastoma and lipoblastomatosis. Pediatr Radiol. 2006;36(8):851–856.
36. Chung, EB, Enzinger, FM. Benign lipoblastomatosis. An analysis of 35 cases. Cancer. 1973;32(2):482–492.
37. Cowling, MG, Holmes, SJ, Adam, EJ. Benign chest wall lipoblastoma of infancy producing underlying bone enlargement. Pediatr Radiol. 1995;25(1):54–55.
38. Fisher, MF, Fletcher, BD, Dahms, BB, et al. Abdominal lipoblastomatosis: radiographic, echographic, and computed tomographic findings. Radiology. 1981;138(3):593–596.
39. Katz, DS, Merchant, N, Beaulieu, CF, et al. Clinical image. Lipoblastoma of the thigh: MR appearance. J Comput Assist Tomogr. 1996;20(6):1002–1003.
40. Murphey, MD, Carroll, JF, Flemming, DJ, et al. From the archives of the AFIP: Benign musculoskeletal lipomatous lesions. Radiographics. 2004;24(5):1433–1466.
41. Shmookler, BM, Enzinger, FM. Liposarcoma occurring in children. An analysis of 17 cases and review of the literature. Cancer. 1983;52(3):567–574.
42. Arndt, CAS, Crist, WM. Common musculoskeletal tumors of childhood and adolescence. N Engl J Med. 1999;341:342–352.
43. Gehan, EA, Glover, FN, Maurer, HM. Prognostic factors in children with rhabdomyosarcoma. Natl Cancer Inst Monogr. 1981;56:83–92.
44. Maurer, HM. The intergroup rhabdomyosarcoma study: update, November 1978. Natl Cancer Inst Monogr. 1981;56:61–68.
45. Shapeero, LG, Couanet, D, Vanel, D, et al. Bone metastases as the presenting manifestation of rhabdomyosarcoma in childhood. Skeletal Radiol. 1993;22:433–438.
46. Dehner, LP. Primitive neuroectodermal tumor and Ewing’s sarcoma. Am J Surg Pathol. 1993;17:1–13.
47. Ibarburen, C, Haberman, JJ, Zerhouni, EA. Peripheral primitive neuroectodermal tumors: CT and MRI evaluation. Eur J Radiol. 1996;21:225–232.
48. Askin, FB, Rosai, J, Sibley, RK, et al. Malignant small cell tumor of the thoracopulmonary region in childhood: a distinctive clinicopathologic entity of uncertain histogenesis. Cancer. 1979;43:2438–2451.
49. Kushner, BH, Hajdu, SI, Gulati, SC, et al. Extracranial primitive neuroectodermal tumors: the Memorial Sloan-Kettering Cancer Center experience. Cancer. 1991;67:1825–1829.
50. Schmidt, D, Herrmann, C, Jürgens, H, et al. Malignant peripheral neuroectodermal tumor and its necessary distinction from Ewing’s sarcoma. Cancer. 1991;68:2251–2259.
51. Orr, WS, Denbo, JW, Billups, CA, et al. Analysis of prognostic factors in extraosseous Ewing sarcoma family of tumors: Review of St. Jude Children’s Research Hospital experience. Ann Surg Oncol. 2012;19:3816–3822.
52. Kan, JH, Hernanz-Schulman, M, Damon, BM, et al. MRI features of three paediatric intra-articular synovial lesions: a comparative study. Clin Radiol. 2008;63(7):805–812.
53. Cadman, NL, Soule, EH, Kelly, PJ. Synovial sarcoma: an analysis of 134 tumors. Cancer. 1965;18:613–627.
54. Jones, BC, Sundaram, S, Kransdorf, MJ. Synovial sarcoma: MR imaging findings in 34 patients. AJR Am J Roentgenol. 1993;161:827–830.
55. Blacksin, MF, Siegel, JR, Benevenia, J, et al. Synovial sarcoma: frequency of non-aggressive MR characteristics. J Comput Assist Tomogr. 1997;21:785–789.
56. Mahajan, H, Lorigan, JG, Shirkhoda, A. Synovial sarcoma: MR imaging. Magn Rason Imaging N Am. 1989;7:211–216.
57. Morton, MJ, Berquist, TH, McLeod, RA, et al. MR imaging of synovial sarcoma. AJR Am J Roentgenol. 1991;156:337–340.
58. Speth, BM, Krieg, AH, Kaelin, A, et al. Synovial Sarcoma in patients under 20 years of age: a multicenter study with a minimum follow-up of 10 years. J Child Orthop. 2011;5:335–342.
59. Dahlin, DC. Case report 189: infantile fibrosarcoma (congenital fibrosarcoma-like fibromatosis). Skeletal Radiol. 1982;8:77–78.
60. Boon, LM, Fishman, SJ, Lund, DP, et al. Congenital fibrosarcoma masquerading as congenital hemangioma: report of two cases. J Pediatr Surg. 1995;30:1378–1381.
61. Exelby, PR, Knapper, WH, Huvos, AG, et al. Soft-tissue fibrosarcoma in children. J Pediatr Surg. 1973;8:415–420.
62. Lilleng, PK, Monge, OR, Walløe, A, et al. Fibrosarcoma in children. Acta Oncol. 1997;36:438–440.
63. Lee, MJ, Cairns, RA, Munk, PL, et al. Musculoskeletal radiology: congenital-infantile fibrosarcoma: magnetic resonance imaging findings. Can Assoc Radiol J. 1996;47:121–125.
64. Ninane, J, Gosseye, S, Panteon, E, et al. Congenital fibrosarcoma: preoperative chemotherapy and conservative surgery. Cancer. 1986;58:1400–1406.
65. Soule, EH, Pritchard, DJ. Fibrosarcoma in infants and children: a review of 110 cases. Cancer. 1977;40:1711–1721.
66. McCarville, MB, Kaste, SC, Pappo, AS. Soft tissue malignancies in infancy. AJR. 1999;173:973–977.
67. Mendenhall, WM, Zlotecki, RA, Scarborough, MT. Dermatofibrosarcoma protuberans. Cancer. 2004;101:2503–2508.
68. Weinstein, JM, Drolet, BA, Esterly, NB, et al. Congenital dermatofibrosarcoma protuberans. Arch Dermatol. 2003;139:207–211.
69. Chung, S, Frush, DP, Prose, NS, et al. Subcutaneous granuloma annulare: MR imaging features in six children and literature review. Radiology. 1999;210:845–849.
70. Clarke, LE. Fibrous and fibrohistiocytic neoplasms: an update. Dermatol Clin. 2012;30:643–656.
71. Munk, PL, Sallomi, DF, Janzen, DL, et al. Malignant fibrous histiocytoma of soft tissue imaging with emphasis on MRI. J Comput Assist Tomogr. 1998;22:819–826.
72. Raney, RB, Jr., Allen, A, O’Neill, J, et al. Malignant fibrous histiocytoma of soft tissue in childhood. Cancer. 1986;57:2198–2201.
73. Ros, PR, Viamonte, M, Jr., Rywlin, AM. Malignant fibrous histiocytoma: mesenchymal tumor of ubiquitous origin. AJR Am J Roentgenol. 1984;142:753–759.
74. Lee, VS, Martinez, S, Coleman, RE. Primary muscle lymphoma: clinical and imaging findings. Radiology. 1997;203:237–244.
75. Eustace, S, Winalski, CS, McGowen, A, et al. Skeletal muscle lymphoma: observations at MR imaging. Skeletal Radiol. 1996;23:425–430.
76. Malloy, PC, Fishman, EK, Magid, D. Lymphoma of bone, muscle, and skin: CT findings. AJR Am J Roentgenol. 1992;159:805–809.
77. Metzler, JP, Fleckenstein, JL, Vuitch, F, et al. Skeletal muscle lymphoma: MRI evaluation. Magn Reson Imaging. 1992;10:491–494.
78. Willemze, R, Jaffe, ES, Burg, G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768–3785.
79. Gomez, N, Ocon, E, Friera, A, et al. Magnetic resonance imaging features of chloroma of the shoulder. Skeletal Radiol. 1997;26:70–72.
80. Guermazi, A, Feger, C, Rousselot, P, et al. Granulocytic sarcoma (chloroma) imaging findings in adults and children. AJR Am J Roentgenol. 2002;178:319–325.
81. Pui, MH, Fletcher, BD, Langston, JW. Granulocytic sarcoma in childhood leukemia: imaging features. Radiology. 1992;190:698–702.
82. Schultz, SR, Bree, RL, Schwab, RE, et al. CT detection of skeletal muscle metastases. J Comput Assist Tomogr. 1986;10:81–83.
83. Williams, JB, Youngberg, RA, Bui-Mansfield, LT, et al. MR imaging of skeletal muscle metastases. AJR Am J Roentgenol. 1997;168:555–557.
84. Dorfman, HD, Czerniak, B. Bone tumors. St. Louis: Mosby; 1998.
85. Mirra, JM, Picci, P, Gold, RH. Bone tumors: clinical, radiologic, and pathologic correlations. Philadelphia: Lea & Febiger; 1989.
86. Levine, SM, Lambiase, RE, Petchprapa, CN. Cortical lesions of the tibia: characteristic appearances at conventional radiography. Radiographics. 2003;23:157–177.
87. Lodwick, GS. A probabilistic approach to the diagnosis of bone tumors. Radiol Clin North Am. 1965;3:487–497.
88. Senac, MO, Jr., Isaacs, H, Gwinn, JL. Primary lesions of bone in the 1st decade of life: retrospective survey of biopsy results. Radiology. 1986;160:491–495.
89. Seeger, LL, Yao, L, Eckardt, JJ. Surface lesions of bone. Radiology. 1998;206:17–33.
90. Hayes, CW, Conway, WF, Sundaram, M. Misleading aggressive MR imaging appearance of some benign musculoskeletal lesions. Radiographics. 1992;12:1119–1134.
91. Aoki, J, Watanabe, H, Shinozaki, T, et al. FDG PET of primary benign and malignant bone tumors: standardized uptake value in 52 lesions. Radiology. 2001;219:774–777.
92. Daldrup-Link, HE, Franzius, C, Link, TM, et al. Whole-body MR imaging for detection of bone metastases in children with skeletal scintigraphy and FDG PET. AJR Am J Roentgenol. 2001;177:229–236.
93. Franzius, C, Schober, O. Assessment of therapy response by FDG PET in pediatric patients. Q J Nucl Med. 2003;47:41–45.
94. Gyorke, TA, Zajic, TB, Lange, AB, et al. Impact of FDG PET for staging of Ewing sarcomas and primitive neuroectodermal tumours. Nucl Med Commun. 2006;27:17–24.
95. Hawkins, DS, Rajendran, JG, Conrad, EU, III., et al. Evaluation of chemotherapy response in pediatric bone sarcomas by [F-18]-fluorodeoxy-d-glucose positron emission tomography. Cancer. 2002;94:3277–3284.
96. Karasick, D, Schweitzer, ME, Eschelman, DJ. Symptomatic osteochondromas: imaging features. AJR Am J Roentgenol. 1997;168:1507–1512.
97. Vasseur, M-A, Fabre, O. Vascular complications of osteochondromas. J Vasc Surg. 2000;31:532–538.
98. Brien, EW, Mirra, JM, Kerr, R. Benign and malignant cartilage tumors of bone and joint: their anatomic and theoretical basis with an emphasis on radiology, pathology and clinical biology. I. The intramedullary cartilage tumors. Skeletal Radiol. 1997;26:325–353.
99. Brien, EW, Mirra, JM, Luck, JV, Jr. Benign and malignant cartilage tumors of bone and joint: their anatomic and theoretical basis with an emphasis on radiology, pathology and clinical biology. II. Juxtacortical cartilage tumors. Skeletal Radiol. 1999;28:1–20.
100. Jaffe, N, Ried, HL, Cohen, M, et al. Radiation induced osteochondroma in long-term survivors of childhood cancer. Int J Radiat Oncol Biol Phys. 1983;9:665–670.
101. Pierz, KA, Womer, RB, Dormans, JP. Pediatric bone tumors: osteosarcoma, Ewing’s sarcoma, and chondrosarcoma associated with multiple hereditary osteochondromatosis. J Pediatr Orthop. 2001;21:412–418.
102. Vanhoenacker, FM, Van Hul, W, Wuyts, W, et al. Hereditary multiple exostoses: from genetics to clinical syndrome and complications. Eur J Radiol. 2001;40:208–217.
103. Stieber, JR, Dormans, JP. Manifestations of hereditary multiple exostoses. J Am Acad Orthop Surg. 2005;13:110–120.
104. Libshitz, HI, Cohen, MA. Radiation-induced osteochondromas. Radiology. 1982;142:643–647.
105. Mahboubi, S, Dormans, JP, D’Angio, G. Malignant degeneration of radiation-induced osteochondroma. Skeletal Radiol. 1997;26:195–198.
106. Marcovici, PA, Berdon, WE, Liebling, MS. Osteochondromas and growth retardation secondary to externally or internally administered radiation in childhood. Pediatr Radiol. 2007;37:301–304.
107. Azouz, EM, Slomic, AM, Marton, D, et al. The variable manifestations of dysplasia epiphysealis hemimelica. Pediatr Radiol. 1985;15:44–49.
108. Richardson, RR. Variants of exostosis of the bone in children. Semin Roentgenol. 2005;40:380–390.
109. Vázquez-Flores, H, Domínguez-Cherit, J, Vega-Memije, ME, et al. Subungual osteochondroma: clinical and radiologic features and treatment. Dermatol Surg. 2004;30:1031–1034.
110. Murphey, MD, Choi, JJ, Kransdorf, MJ, et al. Imaging of osteochondroma: Variants and complications with radiologic-pathologic correlation. Radiographics. 2000;20:1407–1434.
111. Saglik, Y, Altay, M, Unal, VS, et al. Manifestations and management of osteochondromas: a retrospective analysis of 382 patients. Acta Orthop Belg. 2006;72:748–755.
112. Caballes, RL. Enchondroma protuberans masquerading as osteochondroma. Hum Pathol. 1982;13:734–739.
113. Keating, RB, Wright, PW, Staple, TW. Enchondroma protuberans of the rib. Skeletal Radiol. 1985;13:55–58.
114. Crim, JR, Mirra, JM. Enchondroma protuberans: report of a case and its distinction from chondrosarcoma and osteochondroma adjacent to an enchondroma. Skeletal Radiol. 1990;19:431–434.
115. Hunter, AG, Kozlowski, K, Hochberger, O. Metachondromatosis. Can Assoc Radiol J. 1995;46:202–208.
116. Woertler, K, Lindner, N, Gosheger, G, et al. Osteochondroma: MR imaging of tumor-related complications. Eur Radiol. 2000;10:832–840.
117. Lewis, MM, Kenan, S, Yabut, SM, et al. Periosteal chondroma: a report of ten cases and review of the literature. Clin Orthop. 1990;256:185–192.
118. Woertler, K, Blasius, S, Brinkschmidt, C, et al. Periosteal chondroma: MR characteristics. J Comput Assist Tomogr. 2001;25:425–430.
119. Bloem, JL, Mulder, JD. Chondroblastoma: a clinical and radiological study of 104 cases. Skeletal Radiol. 1985;14:1–9.
120. Bloem, JL, Mulder, JD. Chondroblastoma: a clinical and radiological study of 104 cases. Skeletal Radiol. 1985;14:1–9.
121. Ramappa, AJ, Lee, FYI, Tang, P, et al. Chondroblastoma of bone. J Bone Joint Surg Am. 2000;82:1140–1145.
122. Brower, AC, Moser, RP, Kransdorf, MJ. The frequency and diagnostic significance of periostitis in chondroblastoma. AJR Am J Roentgenol. 1990;154:309–314.
123. Oxtoby, JW, Davies, AM. MRI characteristics of chondroblastoma. Clin Radiol. 1996;51:22–26.
124. Weatherall, PT, Maale, GE, Mendelsohn, DB, et al. Chondroblastoma: classic and confusing appearance at MR imaging. Radiology. 1994;190:467–474.
125. Yamamura, S, Sato, K, Sugiura, H, et al. Inflammatory reaction in chondroblastoma. Skeletal Radiol. 1996;25:371–376.
126. Marin, C, Gallego, C, Manjón, P, et al. Juxtacortical chondromyxoid fibroma: imaging findings in three cases and a review of the literature. Skeletal Radiol. 1997;26:642–649.
127. Wu, CT, Inwards, CY, O’Laughlin, S, et al. Chondromyxoid fibroma of bone: a clinicopathologic review of 278 cases. Hum Pathol. 1998;29:438–446.
128. Wilson, AJ, Kyriakos, M, Ackerman, LV. Chondromyxoid fibroma: radiographic appearance in 38 cases and in a review of the literature. Radiology. 1991;179:513–518.
129. Baker, DM. Benign unicameral bone cyst: a study of forty-five cases with long-term follow up. Clin Orthop Relat Res. 1970;71:140–151.
130. Van Linthoudt, D, Lagier, R. Calcaneal cysts: a radiological and anatomico-pathological study. Acta Orthop Scand. 1978;49:310–316.
131. Jaffe, HL, Lichtenstein, L. Solitary unicameral bone cyst: with emphasis on the roentgen picture, the pathologic appearance and the pathogenesis. Arch Surg. 1942;44:1004–1025.
132. Margau, R, Babyn, P, Cole, W, et al. MR imaging of simple bone cysts in children: not so simple. Pediatr Radiol. 2000;30:551–557.
133. Kransdorf, MJ, Sweet, DE. Aneurysmal bone cyst: concept, controversy, clinical presentation, and imaging. AJR Am J Roentgenol. 1995;164:573–580.
134. Campanacci, M, Capanna, R, Picci, P. Unicameral and aneurysmal bone cysts. Clin Orthop Relat Res. 1986;204:25–26.
135. Mankin, HJ, Hornicek, FJ, Ortiz-Cruz, E, et al. Aneurysmal bone cyst: a review of 150 patients. J Clin Oncol. 2005;23:6756–6762.
136. Woertler, K, Brinkschmidt, C. Imaging features of subperiosteal aneurysmal bone cyst. Acta Radiol. 2002;43:336–339.
137. Bertoni, F, Bacchini, P, Capanna, R, et al. Solid variant of aneurysmal bone cyst. Cancer. 1993;71:729–734.
138. Ilaslan, H, Sundaram, M, Unni, KK. Solid variant of aneurysmal bone cysts in long tubular bones: giant cell reparative granuloma. AJR Am J Roentgenol. 2003;180:1681–1687.
139. Hudson, TM. Fluid levels in aneurysmal bone cysts: a CT feature. AJR Am J Roentgenol. 1984;142:1001–1004.
140. Munk, PL, Helms, CA, Holt, RG, et al. MR imaging of aneurysmal bone cysts. AJR Am J Roentgenol. 1989;153:99–101.
141. O’Donnell, P, Saifuddin, A. The prevalence and diagnostic significance of fluid-fluid levels in focal lesions of bone. Skeletal Radiol. 2004;33:330–336.
142. Tsai, JC, Dalinka, MK, Fallon, MD, et al. Fluid-fluid level: a nonspecific finding in tumors of bone and soft tissue. Radiology. 1990;175:779–782.
143. Kransdorf, MJ, Sweet, DE, Buetow, PC, et al. Giant cell tumor in skeletally immature patients. Radiology. 1992;184:233–237.
144. Stratil, PG, Stacy, GS. Multifocal metachronous giant cell tumor in a 15-year-old boy. Pediatr Radiol. 2005;35:444–448.
145. Krajca-Radcliffe, JB, Thomas, JR, Nicholas, RW. Giant-cell tumor of bone: a rare entity in the hands of children. J Pediatr Orthop. 1994;14:776–780.
146. Schütte, HE, Taconis, WK. Giant cell tumor in children and adolescents. Skeletal Radiol. 1993;22:173–176.
147. Schajowicz, F, Granato, DB, McDonald, DJ, et al. Clinical and radiological features of atypical giant cell tumours of bone. Br J Radiol. 1991;64:877–889.
148. Murphey, MD, Nomikos, GC, Flemming, DJ, et al. Imaging of giant cell tumor and giant cell reparative granuloma of bone: radiologic-pathologic correlation. Radiographics. 2001;21:1283–1309.
149. Thomas, DM, Skubitz, KM. Giant cell tumour of bone. Curr Opin Oncol. 2009;21:338–344.
150. Betsy, M, Kupersmith, LM, Springfield, DS. Metaphyseal fibrous defects. J Am Acad Orthop Surg. 2004;12:89–95.
151. Moser, RP, Jr., Sweet, DE, Haseman, DB, et al. Multiple skeletal fibroxanthomas: radiologic-pathologic correlation of 72 cases. Skeletal Radiol. 1987;16:353–359.
152. Parekh, SG, Donthineni-Rao, R, Ricchetti, E, et al. Fibrous dysplasia. J Am Acad Orthop Surg. 2004;12:305–313.
153. Caffey, J. On fibrous defects in cortical walls of growing tubular bones: their radiologic appearance, structure, prevalence, natural course, and diagnostic significance. Adv Pediatr. 1955;7:13–51.
154. Campanacci, M, Laus, M. Osteofibrous dysplasia of the tibia and fibula. J Bone Joint Surg Am. 1981;63:367–375.
155. Colby, RS, Saul, RA. Is Jaffe-Campanacci syndrome just a manifestation of neurofibromatosis type 1? Am J Med Genet A. 2003;123:60–63.
156. Mirra, JM, Gold, RH, Rand, F. Disseminated nonossifying fibromas in association with cafe-au-lait spots (Jaffe-Campanacci syndrome). Clin Orthop Relat Res. 1982:192–205.
157. Jee, WH, Choe, BY, Kang, HS, et al. Nonossifying fibroma: characteristics at MR imaging with pathologic correlation. Radiology. 1998;209:197–202.
158. Goodin, GS, Shulkin, BS, Kaufman, RA, et al. PET/CT characterization of fibroosseous defects in children: 18F-FDG uptake can mimic metastatic disease. AJR Am J Roentgenol. 2006;187:1124–1128.
159. Bloem, JL, van der Heul, RO, Schuttevaer, HM, et al. Fibrous dysplasia vs adamantinoma of the tibia: differentiation based on discriminant analysis of clinical and plain film findings. AJR Am J Roentgenol. 1991;156:1017–1023.
160. DiCaprio, MR, Enneking, WF. Fibrous dysplasia. Pathophysiology, evaluation, and treatment. J Bone Joint Surg Am. 2005;87:1848–1864.
161. Fitzpatrick, KA, Taljanovic, MS, Speer, DP, et al. Imaging findings of fibrous dysplasia with histopathologic and intraoperative correlation. AJR Am J Roentgenol. 2004;182:1389–1398.
162. Cabral, CEL, Guedes, P, Fonseca, T, et al. Polyostotic fibrous dysplasia associated with intramuscular myxomas: Mazabraud’s syndrome. Skeletal Radiol. 1998;27:278–282.
163. Utz, JA, Kransdorf, MJ, Jelinek, JS, et al. MR appearance of fibrous dysplasia. J Comput Assist Tomogr. 1989;13:845–851.
164. Fisher, AJ, Totty, WG, Kyriakos, M. MR appearance of cystic fibrous dysplasia. J Comput Assist Tomogr. 1994;18:315–318.
165. Karol, LA, Brown, DS, Wise, CA, et al. Familial osteofibrous dysplasia: a case series. J Bone Joint Surg Am. 2005;87:2297–2307.
166. Hindman, BW, Bell, S, Russo, T, et al. Neonatal osteofibrous dysplasia: report of two cases. Pediatr Radiol. 1996;26:303–306.
167. Komiya, S, Inoue, A. Aggressive bone tumorous lesion in infancy: osteofibrous dysplasia of the tibia and fibula. J Pediatr Orthop. 1993;13:577–581.
168. Kahn, LB. Adamantinoma, osteofibrous dysplasia and differentiated adamantinoma. Skeletal Radiol. 2003;32:245–258.
169. Hazelbag, HM, Taminiau, AH, Fleuren, GJ, et al. Adamantinoma of the long bones. A clinicopathological study of thirty-two patients with emphasis on histological subtype, precursor lesion, and biological behavior. J Bone Joint Surg Am. 1994;76:1482–1499.
170. Ishida, T, Iijima, T, Kikuchi, F, et al. A clinicopathological and immunohistochemical study of osteofibrous dysplasia, differentiated adamantinoma, and adamantinoma of long bones. Skeletal Radiol. 1992;21:493–502.
171. Czerniak, B, Rojas-Corona, RR, Dorfman, HD. Morphologic diversity of long bone adamantinoma. The concept of differentiated (regressing) adamantinoma and its relationship to osteofibrous dysplasia. Cancer. 1989;64:2319–2334.
172. Van Rijn, R, Bras, J, Schaap, G, et al. Adamantinoma in childhood: report of six cases and review of the literature. Pediatr Radiol. 2006;36:1068–1074.
173. Van der Woude, HJ, Hazelbag, HM, Bloem, JL, et al. MRI of adamantinoma of long bones in correlation with histopathology. AJR Am J Roentgenol. 2004;183:1737–1744.
174. Yao, L, Eckardt, JJ, Seeger, LL. Fibrous dysplasia associated with cortical bony destruction: CT and MR findings. J Comput Assist Tomogr. 1994;18:91–94.
175. Egeler, RM, D’Angio, GJ. Langerhans cell histiocytosis. J Pediatr. 1995;127:1–11.
176. Kransdorf, MJ, Smith, SE. Lesions of unknown histiogenesis: Langerhans cell histiocytosis and Ewing sarcoma. Semin Musculoskel Radiol. 2000;4:113–125.
177. Azouz, EM, Saigal, G, Rodriguez, MM, et al. Langerhans’ cell histiocytosis: pathology, imaging and treatment of skeletal involvement. Pediatr Radiol. 2005;35:103–115.
178. Kilpatrick, SE, Wenger, DE, Gilchrist, GS, et al. Langerhans’ cell histiocytosis (histiocytosis X) of bone: a clinicopathologic analysis of 263 pediatric and adult cases. Cancer. 1995;76:2471–2484.
179. Kilborn, TN, Teh, J, Goodman, TR. Paediatric manifestations of Langerhans cell histiocytosis: a review of the clinical and radiological findings. Clin Radiol. 2003;58:269–278.
180. Schmidt, S, Eich, G, Hanquinet, S, et al. Extra-osseous involvement of Langerhans’ cell histiocytosis in children. Pediatr Radiol. 2004;34:313–321.
181. Meyer, JS, Harty, MP, Mahboubi, S, et al. Langerhans cell histiocytosis: presentation and evolution of radiologic findings with clinical correlation. Radiographics. 1995;15:1135–1146.
182. Stull, MA, Kransdorf, MJ, Devaney, KO. Langerhans cell histiocytosis of bone. Radiographics. 1992;12:801–823.
183. David, R, Oria, RA, Kumar, R, et al. Radiologic features of eosinophilic granuloma of bone. AJR Am J Roentgenol. 1989;153:1021–1026.
184. Hindman, BW, Thomas, RD, Young, LW, et al. Langerhans cell histiocytosis: unusual skeletal manifestations observed in thirty-four cases. Skeletal Radiol. 1998;27:177–181.
185. Van Nieuwenhuyse, JP, Clapuyt, P, Malghem, J, et al. Radiographic skeletal survey and radionuclide bone scan in Langerhans cell histiocytosis of bone. Pediatr Radiol. 1996;26:734–738.
186. Beltran, J, Aparisi, F, Bonmati, LM, et al. Eosinophilic granuloma: MRI manifestations. Skeletal Radiol. 1993;22:157–161.
187. Goo, HW, Yang, DH, Ra, YS, et al. Whole-body MRI of Langerhans cell histiocytosis: comparison with radiography and bone scintigraphy. Pediatr Radiol. 2006;36:1019–1031.
188. Kaste, SC, Rodriguez-Galindo, C, McCarville, ME, et al. PET-CT in Langerhans cell histiocytosis. Pediatr Radiol. 2007;37:615–622.
189. Windebank, K, Nanduri, V. Langerhans cell histiocytosis. Arch Dis Child. 2009;94:904–908.
190. Azouz, EM, Kozlowski, K, Marton, D, et al. Osteoid osteoma and osteoblastoma of the spine in children: report of 22 cases with brief literature review. Pediatr Radiol. 1986;16:25–31.
191. Kransdorf, MJ, Stull, MA, Gilkey, FW, et al. Osteoid osteoma. Radiographics. 1991;11:671–696.
192. Allen, SD, Saifuddin, A. Imaging of intra-articular osteoid osteoma. Clin Radiol. 2003;58:845–852.
193. Shankman, S, Desai, P, Beltran, J. Subperiosteal osteoid osteoma: radiographic and pathologic manifestations. Skeletal Radiol. 1997;26:457–462.
194. Assoun, J, Richardi, G, Railhac, J-J, et al. Osteoid osteoma: MR imaging versus CT. Radiology. 1994;191:217–223.
195. Yamamura, S, Sato, K, Sugiura, H, et al. Magnetic resonance imaging of inflammatory reaction in osteoid osteoma. Arch Orthop Trauma Surg. 1994;114:8–13.
196. Torriani, M, Rosenthal, DI. Percutaneous radiofrequency treatment of osteoid osteoma. Pediatr Radiol. 2002;32:615–618.
197. Ghanem, I. The management of osteoid osteoma: updates and controversies. Curr Opin Pediatr. 2006;18:36–41.
198. Jambhekar, NA, Desai, S, Khapake, D. Osteoblastoma: a study of 12 cases. Indian J Pathol Microbiol. 2006;49:487–490.
199. Lucas, DR, Unni, KK, McLeod, RA, et al. Osteoblastoma: clinicopathologic study of 306 cases. Hum Pathol. 1994;25:117–134.
200. Greenspan, A. Benign bone-forming lesions: osteoma, osteoid osteoma, and osteoblastoma: clinical, imaging, pathologic, and differential considerations. Skeletal Radiol. 1993;22:485–500.
201. Kroon, HM, Schurmans, J. Osteoblastoma: clinical and radiologic findings in 98 new cases. Radiology. 1990;175:783–790.
202. McLeod, RA, Dahlin, DC, Beabout, JW. The spectrum of osteoblastoma. AJR Am J Roentgenol. 1976;126:321–325.
203. Graham, NJ, Cairns, RA, Anderson, RA. Osteosarcoma in a 19-month-old-girl. Can Assoc Radiol J. 1996;47:33–35.
204. Hopper, KD, Moser, RP, Haseman, DB, et al. Osteosarcomatosis. Radiology. 1990;175:233–239.
205. Olson, PN, Prewitt, L, Griffiths, HJ, et al. Case report 703: multifocal osteosarcoma. Skeletal Radiol. 1991;20:624–627.
206. Doud, TM, Moser, RP, Jr., Giudici, MAI, et al. Case report 704: extraskeletal osteosarcoma of the thigh with several suspected skeletal metastases and extensive metastases to the chest. Skeletal Radiol. 1991;20:628–632.
207. Goorin, AM, Abelson, HT, Frei, E, III. Osteosarcoma: fifteen years later. N Engl J Med. 1985;313:1637–1643.
208. Onikul, E, Fletcher, BD, Parham, DM, et al. Accuracy of MR imaging for estimating intraosseous extent of osteosarcoma. AJR Am J Roentgenol. 1996;167:1211–1215.
209. Schima, W, Amann, G, Stiglbauer, R, et al. Preoperative staging of osteosarcoma: efficacy of MR imaging in detecting joint involvement. AJR Am J Roentgenol. 1994;163:1171–1175.
210. Hanna, SL, Fletcher, BD, Parham, DM, et al. Muscle edema in musculoskeletal tumors: MR imaging characteristics and clinical significance. J Magn Reson Imaging. 1991;1:441–449.
211. Gomes, H, Menanteau, B, Gaillard, D, et al. Telangiectatic osteosarcoma. Pediatr Radiol. 1986;16:140–143.
212. Weiss, A, Khoury, JD, Hoffer, FA, et al. Telangiectatic osteosarcoma: the St. Jude Children’s Research Hospital’s experience. Cancer. 2007;109:1627–1637.
213. Huvos, AG, Rosen, G, Bretsky, SS, et al. Telangiectatic osteogenic sarcoma: A clinico-pathologic study of 124 patients. Cancer. 1982;49:1679–1689.
214. Kaufman, RA, Towbin, RB. Telangiectatic osteosarcoma simulating the appearance of an aneurysmal bone cyst. Pediatr Radiol. 1981;11:102–104.
215. Murphey, MD, Robbin, MR, McRae, GA, et al. The many faces of osteosarcoma. Radiographics. 1997;17:1205–1231.
216. Klein, MJ, Siegal, GP. Osteosarcoma: anatomic and histologic variants. Am J Clin Pathol. 2006;125:555–581.
217. Murphey, MD, Jelinek, JS, Temple, HT, et al. Imaging of periosteal osteosarcoma: radiologic-pathologic comparison. Radiology. 2004;233:129–138.
218. Bloem, JL, Taminiau, AHM, Eulderink, F, et al. Radiologic staging of primary bone sarcoma: MR imaging, scintigraphy, angiography, and CT correlated with pathologic examination. Radiology. 1988;169:805–810.
219. Brown, ML. The role of radionuclides in the patient with osteogenic sarcoma. Semin Roentgenol. 1989;24:185–192.
220. Ewing, J. Diffuse endothelioma of bone. Proc N Y Pathol Soc. 1921;21:17. [reprinted in Clin Orthop 1984;185:2-5].
221. Hameed, M. Small round cell tumors of bone. Arch Pathol Lab Med. 2007;13:192–204.
222. Jaffe, R, Santamaria, M, Yunis, EJ, et al. The neuroectodermal tumor of bone. Am J Surg Pathol. 1984;8:885–898.
223. Kretschmar, CS. Ewing’s sarcoma and the “peanut” tumors. N Engl J Med. 1994;331:325–327.
224. Coombs, RJ, Zeiss, J, McCann, K, et al. Case report 360: multifocal Ewing tumor of the skeletal system. Skeletal Radiol. 1986;15:254–257.
225. Coombs, RJ, Zeiss, J, Paley, KJ, et al. Case report 802: Ewing’s tumor of the proximal phalanx of third finger with radiographic progression documented over a 6-year-period. Skeletal Radiol. 1993;22:460–463.
226. Shirley, SK, Gilula, LA, Siegal, GP, et al. Roentgenographic-pathologic correlation of diffuse sclerosis in Ewing sarcoma of bone. Skeletal Radiol. 1984;12:69–78.
227. Shapeero, LG, Vanel, D, Sundaram, M, et al. Periosteal Ewing sarcoma. Radiology. 1994;191:825–831.
228. Mueller, DL, Grant, RM, Riding, MD, et al. Cortical saucerization: an unusual imaging finding of Ewing sarcoma. AJR Am J Roentgenol. 1994;163:401–403.
229. Davies, AM, Makwana, NK, Grimer, RJ, et al. Skip metastases in Ewing’s sarcoma: a report of three cases. Skeletal Radiol. 1997;26:379–384.
230. Reinus, WR, Gilula, LA, Donaldson, S, et al. Prognostic features of Ewing sarcoma on plain radiograph and computed tomography scan after initial treatment: Pediatric Oncology Group Study (8346). Cancer. 1993;72:2503–2510.
231. Daugaard, S, Sunda, LM, Kamby, C, et al. Ewing’s sarcoma: a retrospective study of prognostic factors and treatment results. Acta Oncol. 1987;26:281–287.
232. Jürgens, H, Exner, U, Gadner, H, et al. Multidisciplinary treatment of primary Ewing’s sarcoma of bone: a 6-year experience of a European cooperative trial. Cancer. 1988;61:23–32.
233. Lemmi, MA, Fletcher, BD, Marina, NM, et al. Use of MR imaging to assess results of chemotherapy for Ewing sarcoma. AJR Am J Roentgenol. 1990;155:343–346.
234. Taber, DS, Libshitz, HI, Cohen, MA. Treated Ewing sarcoma: radiographic appearance in response, recurrence, and new primaries. AJR Am J Roentgenol. 1983;140:753–758.