136 Smoke Inhalation
 Key Points
Key Points• The majority (50% to 80%) of deaths attributable to fire are caused by smoke inhalation rather than burns.
• Smoke is a combination of heated particles and gases (toxicants).
• Carbon monoxide has been implicated in more smoke inhalation deaths than any other single compound.
• In addition to inhalation of toxicants, smoke inhalation can cause upper airway burns that result in pulmonary parenchymal injury.
• The mainstay of treatment consists of ventilator support, early intubation, optimization of fluid resuscitation, pulmonary hygiene, and treatment of specific toxicants.
Epidemiology
Fires are common events in the United States. It is estimated that fire departments respond to fire alarms every 20 seconds.1 In 2007, more than 1 million fire incidents and nearly 3500 deaths were reported, with civilian fatalities occurring every 153 minutes on average.1 An estimated 50% to 80% of fire-related deaths are the result of smoke inhalation. Incident after incident, most of the victims of fires in commercial buildings, such as clubs, escape burns but suffer from smoke inhalation
Smoke inhalation injuries related to fire result from the toxic gases generated. Deaths from smoke inhalation have increased in recent years because of the abundant use of newer synthetic material in building and furnishings.2
Pathophysiology
Deaths from fires are most often caused by smoke inhalation.3,4 The injury from smoke inhalation is a result of direct thermal injury to the airway and lung parenchyma, as well as mucosal irritation, corrosive injuries, and asphyxiation from toxic gases. Toxic gases can be classified as irritant gases and asphyxiants. The danger from such toxic gases predominates in exposed fire victims. Smoke is composed of a complex mixture of suspended small particles, fumes, and gases. More than 400 toxic compounds have been demonstrated in the smoke of a typical house fire. Polyvinyl chloride, a component of many plastic goods, generates at least 75 different toxic products when burned. Of these toxic substances, carbon monoxide (CO) appears to be the most common fatal substance associated with fire victims.5,6
Cyanide is a highly toxic chemical. Hydrogen cyanide is a common by-product of the pyrolysis of wool, silk, and plastics. Cyanogenic compounds such as nitriles are used in industry as solvents and adhesives and are metabolized by the body to cyanide. Acetonitrile, which in the past was commercially available as an artificial nail glue remover, has resulted in fatality when accidentally ingested.7 With increasing concern about terrorism, it is high on the potential lists of chemical agents. Its mechanism is binding of the cytochrome aa3 site on the electron transport system of mitochondria. The result is an inability to use oxygen and subsequent cellular asphyxia.
H2S is the by-product of certain industries, such as paper factories, petroleum refineries, and dehairing of hides. It is produced naturally from decay of organic matter and from sulfur hot springs. These products have the characteristic “rotten egg” odor. Like cyanide, it is a potent inhibitor of the cytochrome oxidase system. The human nose is exquisitely sensitive to the odor of H2S, and it is easily detected at levels as low as 0.13 parts per million (ppm). Irritation of mucous membranes occurs at levels of approximately 50 to 100 ppm, and such levels are also capable of causing bronchospasm, blepharospasm, and laryngeal edema. At levels greater than 500 ppm, a phenomenon classically described as a “rapid knockdown” effect occurs, which includes immediate loss of consciousness with the potential for cardiovascular collapse and respiratory arrest. Sulfhemoglobin may result from exposure to H2S. Clinically, it may produce cyanosis. Unfortunately, it is an extremely stable compound and is eliminated only when the red blood cells are removed from the circulation. Recently, H2S was a popular means of suicide in Japan.8 Bath sulfur, a product readily available, is added to water to mimic the water of natural sulfur hot springs. However, when the bath sulfur is combined with acid, such as toilet bowl cleaner, a lethal H2S gas is liberated. Any sulfur-containing substance, including laundry detergent, is a suitable substitute for bath sulfur. Reports of suicide in the United States with this method have been described in the media.
Methemoglobin is the oxidized form of hemoglobin and can result from exposure to oxidizing agents, such as nitrites and nitrates, as well as from smoke inhalation.9 In this oxidized state, hemoglobin is no longer capable of carrying oxygen. Cyanosis is typically seen at levels approximately of 15% to 20%. Levels greater than 70% are usually lethal.
Differential Diagnosis and Medical Decision MakinG
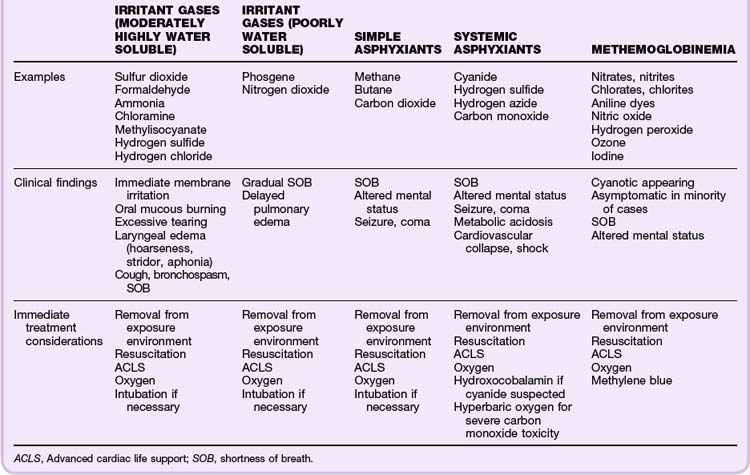
As a means of better understanding this complex problem, dividing smoke inhalation injury into toxidromes (toxic syndromes) may help identify clinically significant smoke inhalation. Aside from the potential concomitant thermal injuries associated with structural fires, these major toxidromes involve irritant gases, simple asphyxiants, systemic asphyxiants, and methemoglobinemia (Table 136.1).
The diagnosis of cyanide toxicity is generally based on a high index suspicion from the history. The victim will appear in a shocklike state. The classic bitter almond odor of cyanide is rarely detected. Hypoxia is a late finding because of its underlying pathophysiology. Oxygen is available, but the cells and tissues are unable to use it. Because cyanide assay is not immediately available in most institutions, indirect evaluation of the difference in the oxygen content of arterial and venous blood can provide valuable insight.4 A small arterial (PaO2)-venous (PvO2) oxygen gap would support the diagnosis. Severe lactic acidosis should prompt further investigation of cyanide toxicity. A lactic acid level greater than 10 mmol/L has been correlated with a cyanide level of approximately 40 mmol/L, a level considered significant and warranting immediate action.
Treatment
Hydroxocobalamin (Cyanokit) is a new cyanide antidote approved by the Food and Drug Administration in December 2006. It has essentially replaced the older antidote, which used sodium nitrite to generate a controlled methemoglobinemia, follow by the administration of sodium thiosulfate, a substrate required by the liver enzyme rhodanase to then detoxify the formation of cyanomethemoglobin. Use of hydroxocobalamin is indicated in any individual with cyanide toxicity or suspected cyanide toxicity. By mechanism, it binds to cyanide to form cyanocobalamin, which is vitamin B12. It appears to be safe, with just a few clinically significant adverse effects, such as allergic reactions and transient hypertension. Generalized erythema and red chromaturia because of the dark red nature of the antidote once reconstituted are commonly reported. Unfortunately, laboratory studies that depend on colorimetric analysis are affected and will give false readings. Its clinical significance is unknown. Because of its safety profile, this antidote can be administered empirically at the scene of a fire to victims and fire rescuers who may be deemed at risk for toxic exposure to cyanide.10 In at least one animal study evaluating the effect of hydroxocobalamin on mice poisoned with sodium sulfide (at a lethal dose for 90% of the population [LD90]), the antidote resulted in improved survival following its administration.11
Treatment of CO toxicity is primarily supportive with 100% supplemental oxygen. HBO therapy has been studied clinically for years, but its role in treating CO toxicity remains controversial.12,13 If considering HBO, generally accepted indications would include the following: loss of consciousness at the scene, comatose state, seizure, myocardial ischemia, and levels greater than 30% in adults and greater 15% in infants, children, or pregnant women.
Borron SJ, Baud F, Barriot P, et al. Prospective study of hydroxocobalamin for acute cyanide poisoning in smoke inhalation. Ann Emerg Med. 2007;49:794–801.
Cahalane M, Demling RH. Early respiratory abnormalities from smoke inhalation. JAMA. 1984;251:771–773.
Clark WR, Nieman GF. Smoke Inhalation. Burns. 1988;14:473–494.
Crapo RO. Smoke-inhalation injuries. JAMA. 1981;246:1694–1696.
Prien T. Toxic smoke compounds and inhalation injury—a review. Burns. 1988;14:451–460.
Wolf SJ, Lavonas EJ, Sloan EP, et al. Clinical policy: critical issues in the management of adult patients presenting to the emergency department with acute carbon monoxide poisoning. Ann Emerg Med. 2008;51:138–152.
1 Karter M. National Fire Protection Association. Fire loss in the United States in 2007. Available at www.nfpa.org
2 Bowes PC. Casualties attributed to toxic gas and smoke at fires: a survey of statistics. Med Sci Law. 1976;16:104–110.
3 Young CJ, Moss J. Smoke inhalation: diagnosis and treatment. J Clin Anesth. 1989;1:377–386.
4 Heimbach DM, Waeckerle JF. Inhalation Injuries. Ann Emerg Med. 1988;17:1316–1320.
5 Orzel RA. Toxicological aspects of fire smoke: polymer pyrolysis and combustion. Occup Med. 1993;8:414–429.
6 Zhu BL, Ishikawa T, Michiue T. Influence of inhaling carbon monoxide–containing gas in fire fatalities—an investigation of forensic autopsy cases. Chudoku Kenkyu. 2007;20:37–44.
7 Caravati EM, Litovitz TL. Pediatric cyanide intoxication and death from acetonitrile-containing cosmetic. JAMA. 1988;260:3470–3473.
8 Morii D, Miyagatani Y, Nakamae N, et al. Japanese experience of hydrogen sulfide: the suicide craze in 2008. J Occup Med Toxicol. 2010;5:28.
9 Hoffman RS, Sauter D. Methemoglobinemia resulting from smoke inhalation. Vet Hum Toxicol. 1989;31:168–170.
10 Uhl W, Nolting A, Bolor G. Safety of hydroxocobalamin in healthy volunteers in a randomized, placebo controlled study. Clin Toxicol. 2006;44:17–28.
11 Truong DH, Mihajlovic A, Gunnes P, et al. Prevention of hydrogen sulfide–induced mouse lethality and cytotoxicity by hydroxocobalamin. Toxicology. 2007;242(1-3):16–22.
12 Weaver LK, Hopkins RO, Chan KJ. Hyperbaric oxygen for acute carbon monoxide poisoning. N Engl J Med. 2002;347:1057–1067.
13 Wolf SJ, Lavonas EJ, Sloan EP, et al. Clinical policy: critical issues in management of adult patients presenting to emergency department with acute carbon monoxide poisoning. Ann Emerg Med. 2008;51:138–152.