23 Skull-Base Tumors
Chemodectomas, Nonchromaffin Paragangliomas, Chordomas, and Chondrosarcomas*
Paragangliomas
Paragangliomas are benign neoplasms of neural crest origin that arise from small (0.1-0.5 mm) collections of neuroepithelial cells called paraganglia or glomus bodies.1 The paraganglion system is important during fetal development to supply catecholamines to the adrenal medulla as it matures and becomes functional.2 Most paraganglia subsequently regress, except along the autonomic nervous system and in certain organs. Paragangliomas are classified as chromaffin and nonchromaffin, depending on whether they produce catecholamines and thereby react with chromic acid.3 Adrenal paragangliomas are usually referred to as pheochromocytomas and are chromaffin-reaction positive, whereas tumors of the extra-adrenal paraganglionic tissue are not. Extra-adrenal paragangliomas of the head and neck are typically nonchromaffin and arise in the cervical region and temporal bone. Terms considered synonymous with paraganglioma include chemodectoma and glomus body tumor. Paragangliomas are named by their site of origin (e.g., carotid paraganglioma, jugular paraganglioma, tympanic paraganglioma, etc.), although the terms glomus-jugulare, glomus-tympanicum, glomus-vagale, and carotid-body tumors are still commonplace.4
Epidemiology and Genetics
Approximately 90% of all paragangliomas arise in the adrenal gland and are called pheochromocytomas. Most (85%) extra-adrenal paragangliomas arise in the abdomen. Approximately 3% occur in the head and neck.2
Paragangliomas of the head and neck are rare, accounting for 0.012% of all tumors5 and 1 out of every 30,000 head and neck tumors.6 Carotid-body tumors tend to be equally distributed between men and women, whereas temporal bone and vagale tumors occur more often in women.2,7–9 Some investigators suggest that higher altitudes and hypoxia may contribute to the development of carotid-body tumors.10,11 Paragangliomas generally occur between ages 50 and 60,7 but may occur earlier in patients with a family history. Between 1% and 3% have endocrine activity that causes symptoms similar to pheochromocytomas or carcinoid tumors.12 Although paragangliomas are normally benign, a small subset (10% or fewer) are malignant and metastasize.8,13 They are multifocal in 10% to 20% of cases, although multifocality can be as high as 33% to 55% in familial cases.9,14 Familial paragangliomas account for approximately 10% of all cases, and are much more likely to be multicentric and bilateral than sporadic tumors.3
Familial paragangliomas are inherited in an autosomal-dominant pattern, but display genomic imprinting with paternal inheritance, because the phenotype is expressed only when transmitted by the father.2,3,15–17 The responsible gene is PGL, which codes for SDH, a mitochondrial enzyme complex that plays an important role in oxidative phosphorylation and intracellular oxygen sensing and signaling.18 Within these complexes are specific subunits coded for using three distinct genes: SDHB, SDHC, and SDHD. Genetic analysis has identified three different genetic types of paraganglioma: paraganglioma 1 (PGL1) 11q23, paraganglioma 2 (PGL2) 11q13, and paraganglioma 3 (PGL3).19–22 PGL1 and PGL2 display the genetic imprinting pattern of inheritance, whereas PGL3 does not and is transmitted from either parent.19,22 Paragangliomas occur in syndromes containing multiple tumors, including multiple endocrine neoplasia type II,23 von Hippel-Lindau disease,24 neurogenic disorders such as von Recklinghausen neurofibromatosis type I,25 and Carney syndrome.26
Anatomy
The carotid body is composed of multiple chemoreceptors located at the bifurcation of the common carotid artery, which are responsible for detecting changes in the partial pressures of oxygen and carbon dioxide in arterial blood. It is the most common location for head and neck paragangliomas, comprising between 60% and 70% of cases.3 The temporal bone is the next most common location. Tumors arising along the superior jugular bulb are referred to as glomus-jugulare tumors, whereas those from the tympanic (Jacobson nerve) nerve of cranial nerve IX or the auricular (Arnold nerve) nerve of cranial nerve X are called glomus-tympanicum tumors. The former runs along the tympanic canaliculus from the inferior petrous portion of the temporal bone between the jugular fossa and the carotid canal to the floor of the tympanic cavity. The Arnold nerve (X) runs from the jugular fossa laterally into the mastoid process. Glomus-vagale tumors may arise along the vagus nerve in any of three vagal ganglia, although usually from the most inferior (nodose) ganglia. Other possible sites include the ciliary ganglion, nasal cavity, larynx, trachea, periaortic region, and fallopian tubes.27
Pathologic Conditions
Paraganglia are composed of two types of cells.2 Type I, also called chief or granular cells, are part of the amino precursor and uptake decarboxylase system; they have catecholamine-containing granules and can be identified immunohistochemically by staining with neuron-specific enolase, chromogranin A, and synaptophysin. Type II, known as supporting or sustentacular cells, stain positive for S-1000 and glial fibrillary acidic protein.2
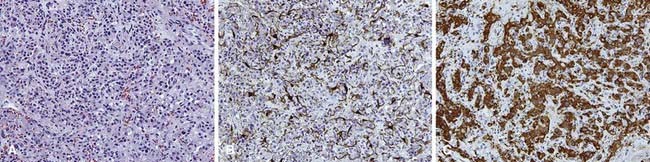
Nonchromaffin paragangliomas are notoriously hypervascular with well-defined edges and a pseudocapsule. Areas of necrosis or hemorrhage are not typically seen except in cases of malignancy.2 Histologically, they appear benign with predominantly type I cells, but also type II cells and capillaries. They have nests of round, polygonal, or spindle-shaped epithelioid cells surrounded by an elaborate vasculature.28 Nuclear atypia is variable and does not correlate with clinical behavior. Figure 23-1 shows the typical histologic appearance of a paraganglioma. Head and neck paragangliomas are almost always negative for chromaffin reaction, which is not sensitive enough to identify the small amounts of catecholamines they contain. Alternatively, immunohistochemical staining for neuron-specific enolase, chromogranin A, synaptophysin, and serotonin can be used to identify type I cells and S-100 and glial fibrillary acidic protein staining can identify type II cells.2,28
Malignancy is typically diagnosed clinically, as there are no established pathologic criteria for diagnosis. Attempts have been made to use mitoses, nuclear polymorphism, or capsular invasion to reliably predict metastatic potential.29 Others have shown that most carotid-body tumors exhibit capsular invasion,30 whereas some metastatic carotid-body tumors do not contain mitoses.31
Clinical Presentation
Carotid-body tumors generally present as a slow-growing, palpable neck mass at the bifurcation of the common carotid artery that has been present for several years.32 Other symptoms or signs may include neck discomfort, true-vocal-cord immobility that may result in hoarseness (from cranial nerve X involvement), tongue weakness and atrophy (from hypoglossal nerve XII involvement), or Horner syndrome (from involvement of the sympathetic chain).14 A very large mass may displace the soft tissue of the airway, resulting in airway symptoms like stridor extension into the parapharyngeal space that may rarely lead to dysphagia. Carotid sinus syndrome has also been reported, as in bilateral neck involvement.
Glomus-vagale tumors are less common than carotid-body tumors and appear more cephalad in the neck. They present as an upper lateral neck mass, often visible only intraorally, displacing the oropharynx anteromedially. Involvement of cranial nerves X and XII or Horner syndrome, as described in carotid-body tumors and accessory nerve weakness, are present in approximately 50% of cases.7
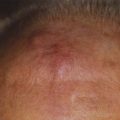
Glomus-tympanicum tumors usually arise in the tympanic cavity or floor of the middle ear, and are typically small and well circumscribed. They may appear as a blue or reddish lesion behind the tympanic membrane. The most common symptoms are progressive conductive hearing loss and pulsatile tinnitus.33 Other symptoms include aural pressure or fullness, vertigo, and headache.
Routes of Spread
Most paragangliomas are benign, but may be locally destructive, invading into bone and displacing or invading adjacent soft tissue. Carotid-body paragangliomas may affect adjacent cranial nerves X and XII and the sympathetic chain. Temporal-bone paragangliomas may cause destruction of the temporal bone and posterior cranial fossa. They may extend medially to the internal auditory canal and involve cranial nerves VII and VIII. Laterally, they may progress to or through the tympanic membrane and appear as a middle-ear mass or a mass in the external auditory canal. It has been reported that 5% of temporal bone and 10% to 15% of carotid-body and vagale paragangliomas are malignant, determined clinically rather than histologically.34,35 Malignant paragangliomas can spread to lymph nodes or distant sites. Caution must be taken to avoid confusing multifocality with metastases or extensive growth, cranial nerve involvement, or recurrence as a sign of malignancy.
Diagnostic Studies
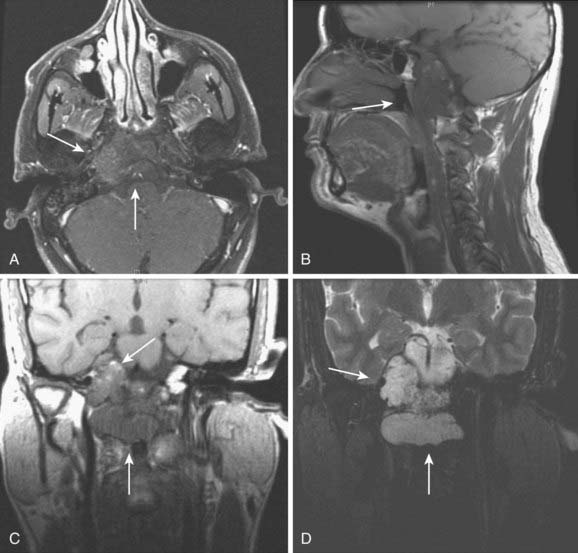
Paragangliomas are often diagnosed presumptively by their typical radiographic appearance rather than by fine-needle aspiration (FNA) or open biopsy, which are associated with the risk of bleeding. Radiographic evaluation also details the extent of the lesion and its relationship to adjacent critical structures. Because paragangliomas are hypervascular, they typically appear as strongly enhanced and well circumscribed on magnetic resonance imaging (MRI) or contrast-enhanced computed tomography (CT). If there is skull-base bone involvement, a high-resolution temporal bone CT will supplement the MRI in delineating tumor extent at the eroded jugular fossa (Fig. 23-2A and Fig. 23-2B) and through the numerous vascular channels and bony foramina.
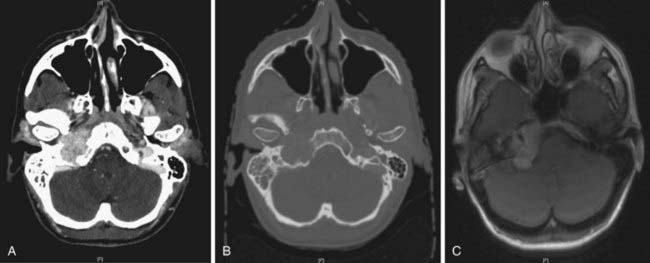
MRI provides important information about tumors and adjacent soft tissues and vascular channels. Flow voids within tumors7 produce the characteristic “salt and pepper” appearance commonly seen on a T2-weighted MRI sequence, representing areas of hemorrhage, slow-flowing blood, and tumor cells.36 MRI is superior to CT in determining skull-base involvement, intracranial extension, dural sinuses, and encasement of the internal carotid artery and internal jugular vein,7,37,38 although CT better determines middle- and inner-ear involvement and bony erosion of the jugular fossa or temporal bone.7 Figure 23-2C shows an MRI of a glomus jugulare.
Ultrasound has been used in the diagnosis of carotid-body or vagale tumors, but is of limited value for temporal-bone tumors and provides much less information than MRI. The classic appearance is that of a hypoechoic and heterogenous hypervascular lesion. For carotid-body tumors, splaying of the internal and external carotid artery may be seen.39 A Doppler-flow study showing upward movement of intratumoral blood flow at the carotid bifurcation is diagnostic of a carotid-body tumor, whereas downward flow of a neck mass indicates a glomus vagale.40
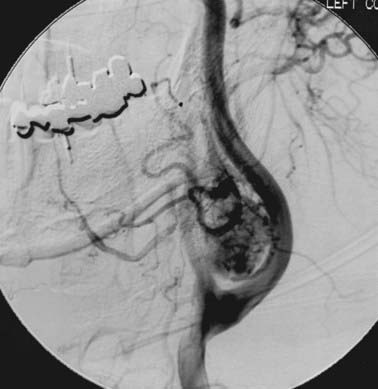
Angiography will confirm the hypervascularity of these tumors and also is an option to detect multifocality. Figure 23-3 shows a typical angiogram with intense tumoral vascularity and splaying of the internal and external carotid arteries.
A family history of paragangliomas requires a more extensive radiographic workup because of the higher likelihood of multifocal disease. Because paragangliomas share similar histologic features with other neuroendocrine tumors, including somatostatin receptors, octreotide scintigraphy may be another way to detect multifocality (or metastases).41,42 Radiolabeled metaiodobenzylguanidine, an analog of norepinephrine, is taken up and stored in the intracellular vesicles of neuroendocrine tumors and can be useful for diagnosing tumors of neuroendocrine origin, such as neuroblastoma,43 pheochromocytoma,44 and other paragangliomas.45
Staging
There is no universally accepted staging system for carotid-body and glomus-vagale tumors, but several have been published for temporal-bone paragangliomas. Some authors have used the staging system proposed by McCabe and Fletcher.46 The Glasscock-Jackson system is also available.47 The classification by Fisch48 is often used for surgical outcomes.
Standard Therapeutic Approaches
Radiation and surgery can be used as definitive treatment for paragangliomas. When the entire tumor cannot be removed, postoperative radiation is usually recommended. The decision of which modality to use is based on a number of factors such as patient age and overall health, and on the potential short-term and long-term risks of treatment complications. In general, surgery is preferred when complete resection is anticipated with a minimal surgical risk and in younger patients. This is especially so for early-stage, temporal-bone tumors, including middle-ear tumors and small-volume neck tumors that have a low attendant risk of permanent postoperative complications. Larger and locally invasive tumors may be treated with postoperative irradiation, or may be treated with primary radiotherapy because of potential cranial nerve or vascular injury associated with resection. When treated with irradiation, paragangliomas usually remain stable or may regress slowly, resulting in a persistent radiographic or palpable mass.49 Hence local control is defined as the lack of tumor progression. Symptoms caused by the tumor often improve with radiation.
Surgery
The role of preoperative embolization is debated.17 Advocates argue that surgical blood loss is decreased and operative time shortened. Critics cite the potential morbidity of angiography and the possible distortion of tissue planes during dissection. In addition, there is a small risk of embolization of an unintended vessel, and numerous reviews fail to show a perioperative benefit in reducing blood loss.50
It is advisable to obtain proximal and distal (caudal and cephalad) control of the internal carotid artery, external carotid artery, and common carotid artery prior to extensive dissection of the tumor itself. Resection of the external carotid artery at times may be required to facilitate this.51 Similarly, caudal identification of the cranial nerves X, XI, and XII, as well as the sympathetic chain facilitates their safe dissection from the tumor during subsequent periadventitial (sometimes referred in the literature as subadventitial) dissection of the paraganglioma. Nerves macroscopically involved by tumor are best resected to achieve the 90% to 95% complete resection often reported.
Chemotherapy
The role of chemotherapy for paragangliomas remains undefined, and is usually reserved for rare metastatic disease and unresectable postirradiation recurrences. Solitary case reports have demonstrated activity for several agents, including carboplatin, cisplatin, cyclophosphamide, gemcitabine, and etoposide.52–55 Some series have reported combination therapy with cyclophosphamide, vincristine, doxorubicin, and dacarbazine (DTIC),56 as well as with cyclophosphamide, cisplatin, doxorubicin, and DTIC with mixed results.57
Somatostatin analogs58 and radiopharmaceuticals based on somatostatin analogs such as octreotide and lanreotide have been evaluated because of the expression of somatostatin receptors on paragangliomas. Several radiopharmaceuticals have been applied, including 111In-pentetreotide/111In-DOTA octreotide, 90Y-DOTA-octreotide and 177Lu-DOTA-octreotate, and 111In and 90Y DOTAlanreotide.59–61
A molecularly targeted approach using imatinib mesylate, a selective inhibitor of the ABL, platelet-derived growth factor receptor (PDGFR), and stem-cell-ligand receptor (c-kit) tyrosine kinases, in 15 adult patients with disseminated endocrine tumors did not prove to be effective.62 Based on preclinical data, other strategies (e.g., targeting RAF) are being explored, but clinical evaluation remains to be performed.63
Simulation and Treatment Planning
Intensity-modulated radiation therapy (IMRT) can provide a highly conformal dose distribution that decreases the dose to surrounding normal tissues (Fig. 23-4). These techniques can be especially important for glomus-jugulare and tympanicum tumors to spare the adjacent cochlea. Although the dose normally required for these tumors is moderate, minimizing the integral dose is an important goal to reduce these long-term risks. Acute toxicities can also be reduced by limiting the dose to oral cavity structures and salivary glands. Diagnostic information that can help in defining the gross tumor volume (GTV)64 should be reviewed, including CT, MRI, and angiograms. The GTV encompasses the visible gross tumor on diagnostic imaging scans. The clinical target volume (CTV) only requires a small margin of approximately 0.5 cm to account for microscopic extension. Regional lymph-node spread is rare, so the regional nodes are not electively included in the target volume unless malignancy is suspected. The planning target volume (PTV), taken into account for set-up uncertainty and tumor motion, typically adds another 0.5 cm. If there is evidence of lymph-node spread (malignancy), the regional nodes are included in the CTV. The PTV may be extended slightly superiorly and inferiorly if the tumor extent is indistinct on the planning scan. Sensitive surrounding critical normal structures should be contoured to avoid exceeding normal-tissue tolerances, including the globes, brainstem, salivary glands, and bilateral cochleas.
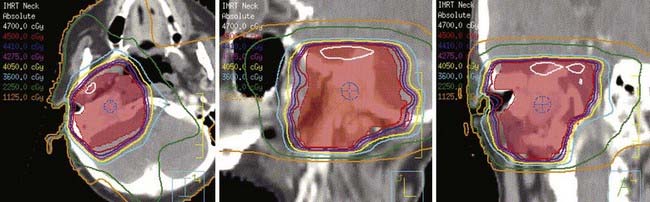
FIGURE 23-4 • Intensity-modulated radiation therapy plan showing conformality of isodose curves around the planning target volume (red).
Moderate doses of 45 to 50 Gy at 1.8 to 2 Gy per fraction adequately control benign paragangliomas without exceeding the tolerances of adjacent neurologic and optic structures such as the spinal cord, brainstem, and optic chiasm. Local control appears to be reduced with doses lower than 40 Gy.65
Stereotactic Radiosurgery
The use of stereotactic radiosurgery (SRS) to treat temporal-bone tumors is increasing, because it can deliver a large dose of radiation in few fractions and minimize normal-tissue doses.66–71 Treatment planning should again use contrast-enhanced CT, MRI, and angiography to help delineate tumor extent. A variety of doses ranging from 15 to 27 Gy in a single fraction have been published in the literature. It appears that 15 to 18 Gy in a single fraction adequately controls the disease and minimizes the risk of complications.66–71
Outcomes
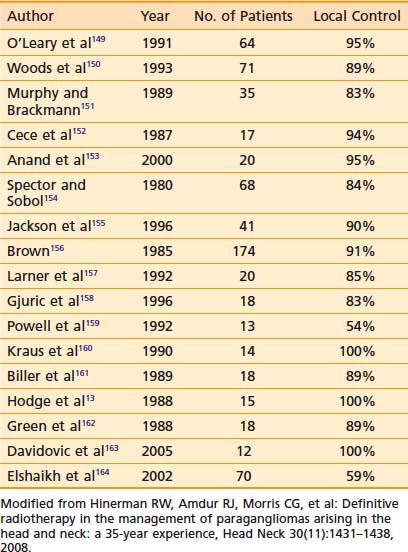
Results with any treatment approach are excellent. Table 23-1 and Table 23-2 show that the local control rates of selected patients treated with surgical resection and radiation are comparable, typically greater than 85% to 95%.72 Local control in radiation series is generally defined as lack of tumor progression after treatment. Although there is less data for carotid-body tumors, the available data suggests that control rates equal those for temporal-bone tumors. Valdagni and Amichetti reported on 7 patients with 13 carotid-body tumors treated with radiation and found 100% local control.73 Verniers and colleagues reported a series including 17 carotid-body tumors, none of which recurred after radiotherapy, with a mean follow-up of 10 years.74 In a series reported by Hinerman and colleagues, 18 patients with 25 carotid-body tumors or glomus-vagale tumors were treated with radiotherapy. The 15-year local control rate was 92%.75
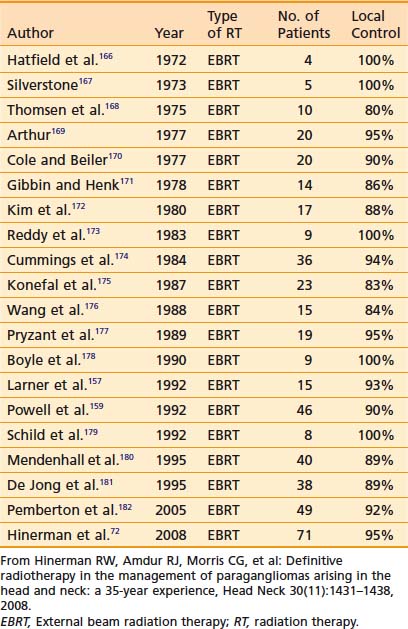
Because SRS is relatively new, less data on treatment outcomes is available and follow-up is shorter. Early results show excellent local control (Table 23-3). However, because this disease can have late failures, longer follow-up is needed to show results comparable to surgery or fractionated radiotherapy.
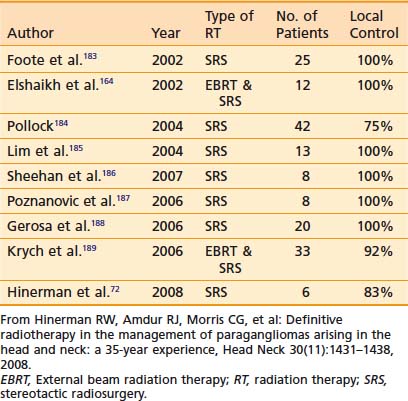
Treatment Toxicity
Table 23-4 shows surgical complications during the preceding two decades. Most surgical morbidity is related to cranial-nerve deficits, which can be either temporary or permanent. Other complications are related to wound infections or vascular damage. With appropriate selection and modern surgical and anesthesiologic methods, permanent cranial-nerve deficits are approximately 10% to 15%, and vascular injury is rare. Isolated X or XII deficits are well tolerated, and compensatory rehabilitation is available with good results when there are combined deficits.72,76
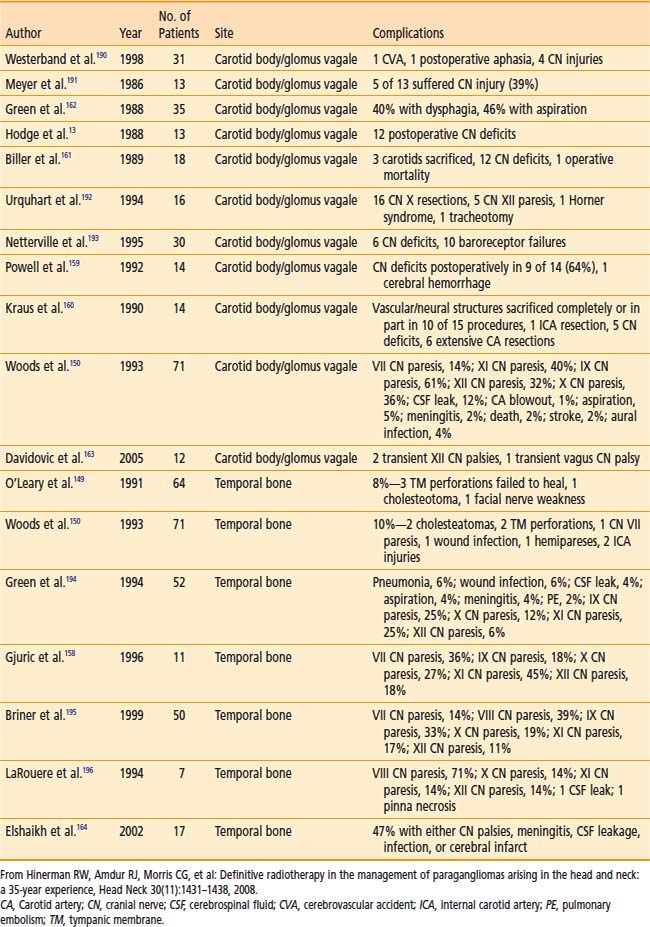
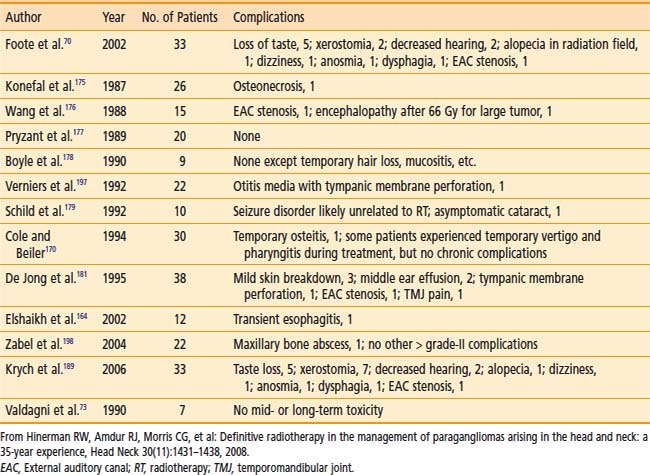
Table 23-5 shows the reported treatment-related morbidity with radiotherapy. Cranial-nerve deficits and bone necrosis are occasionally seen after radiotherapy. Late radiation-induced malignancy, though possible, has not been reported. It is hoped that complication rates will continue to improve as imaging and radiation delivery technologies improve.
Chordoma and Chondrosarcoma
In a study that extracted data from the Survival, Epidemiology, and End Results database, McMaster and colleagues found that 32% of chordomas arise in the skull, 33% in the spine, 29% in the sacrum, and only 6% elsewhere.77 Noel and colleagues examined 47 cases of chordomas in the skull or cervical spine. Of these, 23 tumors (49%) arose in the clivus, 15 (32%) in the sphenoclival region, 4 (9%) in the petroclival region, 3 (6%) in the cervical spine, and 2 (4%) in other areas.78 They observed an incidence of 0.08 patients per 100,000. The incidence of chordoma has been stable during the preceding three decades.77 The median age of diagnosis for chordomas is in the sixth to seventh decade.79 There is a slight male predominance, and the disease rarely occurs in blacks.79
Histologically, chordomas appear as benign cells, but their clinical behavior belies that microscopic appearance. They were first described microscopically as physaliferous cells because of their large vacuoles.79 The cells are uniform in appearance with small, oval nuclei containing dense chromatin (Fig. 23-5A). There are three subtypes of chordomas: (1) classic, (2) dedifferentiated, or (3) chondroid. The classic and chondroid variants are the most commonly seen subtypes in skull-base tumors. Chondroid chordoma has been thought to carry a better prognosis than chondrosarcoma, but they are often difficult to distinguish. Data also suggest that the outcomes for the chondroid and classic variants are similar.80,81 In comparison, dedifferentiated chordomas are more aggressive, with more mitotic activity and cellular atypia, similar to a round-cell tumor or spindle-cell sarcoma. Dedifferentiation occurs in fewer than 4% of cases at presentation82 and most commonly occur in the sacrococcygeal region.79
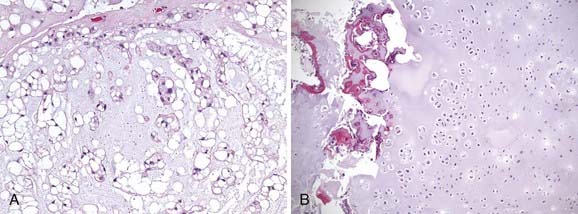
Genetic changes linked to the development of chordomas have been mapped to chromosomes 1p, 3p, and 7q. Scheil and colleagues determined that mismatched repair genes on 1p and 3p as well as oncogenes found at 7q may be involved in the development of chordomas.83 Kelley and colleagues performed linkage analysis on 22 members of a family, 10 of whom had a chordoma, and mapped a locus for familial chordoma at 7q33. In addition, they found an autosomal-dominant pattern of inheritance.84
Chondrosarcomas are neoplasms of cartilage that typically arise in the long bones or the pelvis, with fewer than 10% occurring in the head and neck region.85 They compose only 0.15% of primary intracranial tumors.86 In the base of the skull, they are thought to arise from cartilage at the synchondroses.86 Histologically, three different types have been described: classical (62%), myxoid (8%), and mesenchymal (30%).87
Classical chondrosarcoma has three grades based on mitoses, cellularity, and the amount of chondroid matrix, which determine the clinical course.88 Grade I typically has low cellularity and a high amount of hyaline cartilage matrix (Fig. 23-5B), whereas grade III has more mitoses, more cellularity, and more mucomyxoid matrix.80,88 Clinically, grade III chondrosarcomas are more aggressive and can have a metastasis rate of up to 70%.89 Most tumors of the skull base are classic low-grade chondrosarcomas.80
Chondrosarcoma may be associated with the genetic Maffucci syndrome,90 characterized by multiple benign enchondromas and hemangiomas. Genetic abnormalities have been identified in peripheral chondrosarcomas, mainly with mutations in the exostosis genes.88
Both skull-base chordomas and chondrosarcomas are clinically characterized by aggressive local growth and bone destruction. Overall, low-grade chondrosarcomas have a good prognosis and behave indolently with little risk of metastases. Chordomas, on the other hand, display a wide range of clinical behaviors ranging from indolent to aggressive, following a continuum from relatively slow growth pattern to fatality within 5 years. They can rarely metastasize, typically several years after initial diagnosis, to lung and lymph nodes, for example,91 and may also recur locally in bone and along prior routes of surgical access (even to skin).92 Chordomas are hence particularly challenging as their histopathology is not adequately clinically predictive and a better understanding of their underlying biology is needed.
Clinical Presentation
The presenting symptoms of chordoma and chondrosarcoma are similar and depend on the site of origin. The most common symptoms of skull-base tumors are headaches and diplopia,93,94 with cranial nerve VI involvement most common followed by cranial nerves III and IV,95 with possible ptosis as well as limited extraocular mobility. The time between the onset of symptoms and diagnosis ranges from weeks to years, with the average being 3.5 years.79 Neuralgia may also result, most commonly of the trigeminal nerve.79
The primary pattern of growth is local bony extension from the bone of origin, although chordomas, which typically arise at the midline, may rarely invade rather than simply displace soft tissue or brain parenchyma.96 Most chondrosarcomas originate in the petroclival area, involving both the clivus and petrous bone. This pattern indicates that chondrosarcomas tend to originate laterally and grow centrally, whereas chordomas, on the other hand, appear to originate near the midline.80
Staging
There is no accepted staging system for chordomas or chondrosarcomas, but the American Joint Committee on Cancer staging system for soft-tissue sarcomas may be used, although skull-base lesions are generally T1-N0, so pathologic grade plays an important role in attempting to predict prognosis. Al-Mefty and Borba proposed a classification scheme for chordomas that uses anatomic extent and the complexity of surgical approach required for resection.97
Diagnostic Studies
MRI and CT are the most important imaging studies used to evaluate the central skull base. However, chordomas and chondrosarcomas of the skull base are virtually indistinguishable by radiologic imaging. On CT, they appear as an enhancing, well-circumscribed, soft-tissue lesion in the clivus with surrounding bone destruction. Chordomas are similar in density to brain parenchyma. On MRI, both tumors appear hypointense on T1 and hyperintense on T2 signal, and they frequently enhance with gadolinium (Fig. 23-6 and Fig. 23-7). MRI is superior to CT in determining soft-tissue relationships of the tumor, but often CT is complementary in delineating extension toward the petrous bone.
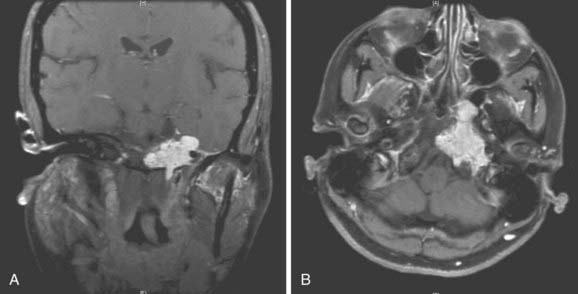
Obtaining tissue is usually required to clarify the diagnosis via either an FNA biopsy when accessible,98 or an open biopsy, such as at the beginning of a planned resection. Because chondrosarcoma is associated with a better prognosis than chordoma,99 it is helpful to the patient to distinguish chordoma, particularly chondroid chordoma, from low-grade chondrosarcoma.80,100 Morphologically, the two histologies appear very similar and immunohistochemically there are some shared staining characteristics. Both chordomas and chondrosarcomas can stain positively for vimentin and S-100.101 Chordomas stain positively for epithelial membrane antigen and cytokeratin, whereas chondrosarcomas do not.102,103 In addition, chordomas are more likely than chondrosarcomas to stain positively for galectin-3, (a β-galactosidase binding protein,104) and adhesion molecules E-cadherin, β-catenin, and γ-catenin, as well as neural-cell adhesion molecule.105
Standard Therapeutic Approaches
As with other rare histologies such as paragangliomas, there have been no randomized studies to define treatment for chordomas and chondrosarcomas. Standard therapy involves modern skull-base surgical resection, with a planned gross total resection when there is a high likelihood of success without undue morbidity. It is estimated that approximately 50% of patients with chordoma can achieve a gross total resection,106 but up to 50% require multiple procedures.93,99,107
Because of the tumor location and the proximity of normal structures, conventional radiotherapy cannot achieve tumoricidal doses without an associated high risk of complications. Therefore, this type of tumor is particularly well-suited to IMRT, SRS, or proton-beam therapy. The characteristic Bragg Peak of a proton therapy dose distribution offers the distinct advantage of allowing a concentrated dose to the target volume as it keeps doses to normal tissue within tolerance levels, greatly increasing the therapeutic ratio (see Chapter 69). Figure 23-8 shows a comparison of isodose plans for a patient using IMRT and protons.
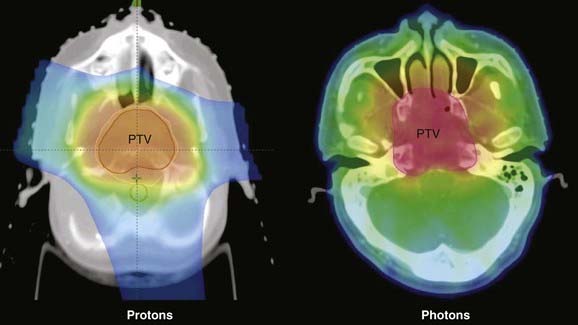
Chemotherapy for Chordomas
Razis et al.108 and Harwick and Miller109 reported symptomatic improvement in isolated patients treated with 2 mg of vincristine sulfate intravenously weekly. Cyclophosphamide,110 actinomycin D,111 decarbazine,112 methotrexate,113 and platinum compounds114 given as single agents have been ineffective both in terms of response and symptomatic pain relief, whereas combination therapy has been effective in isolated cases only. In a phase-II study, a topoisomerase I inhibitor, 9-nitro-camptothecin (9-NC), was used to treat 15 patients with chordomas. Although only one (7%) objective response rate was observed, 9-NC appeared to delay progression of disease, with a median 3-month progression-free survival rate of 47%, and a 6-month progression-free survival rate of 33%.115
Another strategy used razoxane, a radiosensitizer that blocks dividing cells in G2/M phase of the cell cycle, and has shown favorable results in soft-tissue sarcoma. In a prospective trial, some benefit was seen in five patients with sacral and skull-base chordomas. Objective tumor regressions were noted in three of four patients with measurable disease, and all patients remained locally controlled and survived at least 5 years.116
More recent studies have explored the use of molecularly targeted agents for chordoma. Imatinib mesylate is an inhibitor of some tyrosine kinases, mainly BCR-ABL, KIT, platelet-derived growth factor receptor A−(PDGFRA), and platelet-derived growth factor receptor B−(PDGFRB).117–120 Dysregulation of other signaling pathways found in chordoma121 have resulted in the off-label use of antiangiogenic agents and other targeted therapies such as cetuximab, gefitinib, and erlotinib.
Chemotherapy for Chondrosarcomas
Conventional chemotherapy regimens for the vast majority of grade-II and grade-III chondrosarcomas generally result in low therapeutic response rates.122 Reports of the use of chemotherapy for base-of-skull chondrosarcomas are anecdotal and limited to case reports of agents such as doxorubicin and temozolomide.123–125 The dedifferentiated chondrosarcoma has a natural history analogous to osteogenic sarcoma and generally responds to agents that are appropriate for the latter.126,127 Mechanisms of chemotherapy resistance could be related to expression of P-glycoprotein128 similar to what has been previously shown in osteosarcoma.129
Insight into the molecular and biologic characteristics of chondrosarcoma may lead to the development of novel strategies. Deletion of the methylthioadenosine phosphorylase gene, important for the production of a ubiquitous enzyme essential for methionine synthesis, has been seen in 50% of chondrosarcoma samples and provides the rationale for the testing of pemetrexed, a multitargeted antifolate.130 Investigation of potential molecular targets, such as expression for PDGFRA and PDGFRB, supports the testing of receptor tyrosine kinases either as single agents or in combination with chemotherapy.131 Preclinical studies of the use of histone deacetylase inhibitors in chondrosarcoma cell lines also suggest that clinical evaluation of these agents with chemotherapy in this disease may be of interest.132
Radiation Technique
Radiotherapy for skull-base chordomas and chondrosarcomas requires high doses that exceed normal tissue tolerance for adjacent critical structures, such as the brainstem, spinal cord, and optic structures. Traditionally, the radiation technique for these tumors involved conventional lateral and anterior-posterior fields, which limited the amount of radiation that could be given without causing a risk of complications. More complex 3-dimensional conformal beam arrangements and IMRT provide superior dose conformality around the target volume, reducing the dose to normal structures. Because these tumors are often invasive and adjacent to critical structures, however, it is not always possible to completely exclude the organs at risk. SRS, either with specialized equipment or a linear accelerator, has also been used.106,133,134
The doses that have been used for both chordoma and chondrosarcoma are in the range of 55 to 80 Gy. Rich and colleagues found a local control rate of 28% with doses less than 60 Gy and advocated 65 to 70 Gy for treatment of chordomas, but with a risk of late complications.94 The failure rate was 47% for doses less than 40 Gy, 18% for doses between 40 to 60 Gy, and 10% for doses higher than 60 Gy. In a study by Pearlman and Friedman, a decreased rate of local failures was seen with increasing doses up to 60 Gy.135 Hug and colleagues prescribed a mean dose of 71.9 cobalt gray equivalent (CGE) within a range of 66.6 to 79.2 CGE for chordomas, and 69.3 CGE within a range of 64.8 to 72 CGE for chondrosarcomas using protons or a combination of protons and photons.136 The authors did not note any dose response effect on local control with these doses, all of which exceeded 64 CGE. Noel and colleagues137 found that a minimal dose of 56 CGE with combined photon and proton therapy was associated with worse local control.
Results
The outcomes for skull-base chordomas are difficult to assess because, as is typical with rare tumors, series often span many years, report few patients, include surgery, often combine initial and recurrent disease, and have selection bias that determined the use of postoperative radiation that includes tumor mass, a factor that correlates with the likelihood of overall control with irradiation.136,138 Other prognostic factors that have been reported are recurrence, age,78 and gender,138,139 with males having better local control than females.
Surgery
Gay and colleagues reported on 46 patients with chordoma and 14 patients with chondrosarcoma of the skull base who were treated with resection at the University of Pittsburgh from 1984 to 1993.93 Of these, 30 patients (50%) had previous surgery. Gross total resection was achieved in 47%, near-total resection in 20%, subtotal resection (≥90% of the tumor removed) in 23%, and partial resection (≤90% of the tumor removed) in 10%. Postoperative radiation was given in 20% of patients. Outcome was better for chondrosarcoma than chordoma. With a median follow-up of 3.9 years (in a range of 1-11 years), the 5-year, recurrence-free survival was 65% for chordoma and 90% for chondrosarcoma (p = 0.09). Al-Mefty and Borba published a study on a series of 25 patients with skull-base chordomas treated between 1990 and 1996.97 They performed 33 surgical procedures in 23 patients. Of these, 10 patients (43%) received a gross total resection, 11 (48%) had a subtotal resection (>90% of the tumor removed), and 2 (8%) had a partial resection (<90% of the tumor removed); 17 patients received adjuvant radiotherapy with proton and photon-beam radiation with a mean dose of 68.8 CGE. The median follow-up was 25.4 months. Of the 21 patients followed for more than 3 months, 15 were alive without disease, 1 died of intercurrent disease, 3 were alive with recurrence, and 2 died with disease. Maira and colleagues reported on 12 patients with clival chordomas treated surgically, 2 of whom received postoperative radiotherapy.140 The median follow-up was 40 months (in a range of 14-86 months). Eight patients (67%) had gross total resection and were disease-free upon last follow-up, and 4 patients (33%) had partial resection; 2 of them developed a local recurrence.
Radiotherapy
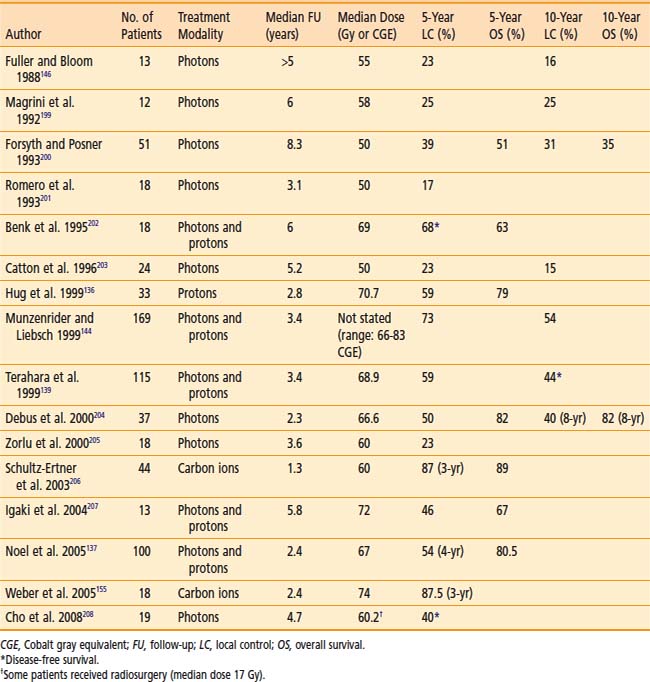
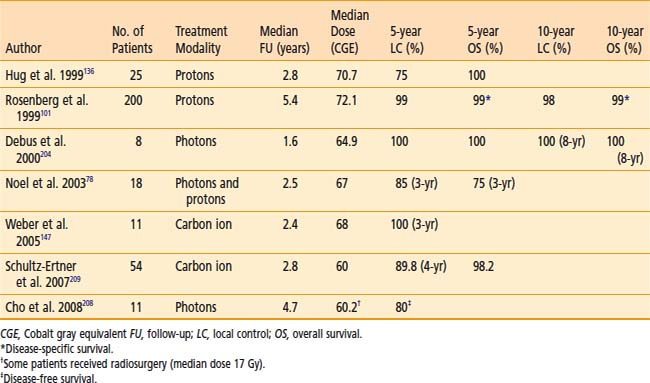
Table 23-6 and Table 23-7 show the published results of both photon and particle-beam radiation for skull-base chordomas and chondrosarcomas. Most of these series include patients treated with irradiation alone or after subtotal resection. Two important generalizations can be made from the data: (1) The outcome of chondrosarcomas is superior to that of chordomas, and (2) particle-beam radiation, of which proton therapy has the most published experience, seems to offer superior local control, probably because of improved dose-distribution characteristics that allow for higher and more effective doses to be delivered safely. The follow-up for many of these studies is limited, however, and longer-term data is needed to have a more complete understanding of the benefits of proton therapy compared with photon irradiation.
Toxicity
Surgery
The risk of surgical morbidity has improved significantly with advances in radiology, image-guided surgery, and surgical approaches themselves, but remains significant and correlates generally with the extent of the tumor and surgical procedure. In the 1995 series by Gay and colleagues,93 a 30% rate of postoperative leakage was observed, which increased the risk of permanent complications. Prior radiotherapy increased the risk of postoperative death. In the 1997 series by Al-Mefty and Borba,97 1 of the 25 patients died of a cardiovascular accident postoperatively after the internal carotid artery was sacrificed, 2 patients had permanent neurologic dysfunction, and 3 patients had radiation necrosis. Pamir and Ozduman reported a 28.2% rate of surgical complications and a 2.6% surgical mortality.79 Complications included hydrocephalus (4 patients), transient and permanent cranial nerve palsy (8 patients), CSF leak (2 patients), hemiparesis (1 patient), vertebral artery rupture (1 patient), and craniocervical instability (3 patients). Stuer, reporting on 13 patients, all of whom also received postoperative irradiation, noted 7 patients with transient cranial-nerve deficits and an overall rate of 63% for transient morbidity.141
Radiotherapy
Permanent neurologic complications are a recognized risk of radiation therapy. Santoni and colleagues found a 2- and 5-year temporal lobe complication rate of 8% and 13%, respectively, in 96 patients treated with photon and proton radiation for chordoma or chondrosarcoma.142 Debus and colleagues examined brainstem toxicity in 367 patients treated for chordoma or chondrosarcoma of the skull base with photon and proton radiation.143 They found 17 patients (5%) with brainstem toxicity and a 5- and 10-year toxicity rate of 6% and 12%, respectively. In another report from the same institution, Munzenrider and Liebsch found a 2- and 5-year rate of temporal lobe injury of 8% and 13%, respectively, in 96 patients with chordomas, 8 of whom had moderate to severe complications and 3 of whom required temporal lobe resection.144
Optic pathway toxicity has also been reported. Habrand showed a 10% rate of optic complications for doses of 55 CGE to the optic nerve or chiasm and 20% when the optic nerve received 65 CGE.145 Munzenrider and Liebsch reported on 12 out of 274 patients (4.4%) who developed optic neuropathy after receiving 63.4 to 79.4 CGE.144 The median dose of the optic structures in patients with neuropathy was 62.1 CGE. Finally, Noel and colleagues found 8 patients out of 100 (8%) with optic complications: 1 with optic chiasm necrosis at 48 CGE and 7 with decreased visual acuity.137
Another risk of radiation is hormonal dysfunction from damage to the pituitary/hypothalamic axis. Fuller and Bloom found 2 out of 13 (15%) cases of hypopituitarism observed at 2 and 5 years after treatment with photon radiation to a median dose of 55 Gy.146 Similarly, Weber and colleagues found 4 of 29 patients with hypopituitarism (14%) after carbon-ion radiation therapy.147 Munzenrider and Liebsch found a 40% risk of pituitary dysfunction after proton radiation.144 Noel and colleagues137 found a 16% rate of pituitary insufficiency in 100 patients after photon and proton radiation: 7 patients had partial dysfunction and 9 had complete dysfunction. A dose of up to 60 CGE correlated with the risk of this complication and the risk appeared to decrease above those levels. Pai and colleagues examined the dose-response relationship of radiation to the hypothalamic/pituitary axis and the risk of developing hormonal insufficiency after photon and proton radiation for skull-base tumors.148 They found 5- and 10-year rates of 72% and 84% for hyperprolactinemia, 30% and 63% for hypothyroidism, 29% and 36% for hypogonadism, and 19% and 28% for hypoadrenalism, respectively. They also found that a minimum dose of 50 CGE to the pituitary, a maximum dose of 70 CGE to the pituitary, and a minimum dose of 50 CGE to the hypothalamus correlated with a higher rate of dysfunction.
1 Lawson W. The neuroendocrine nature of the glomus cells: An experimental, ultrastructural, and histochemical tissue culture study. Laryngoscope. 1980;90:120-144.
2 Wasserman PG, Savargaonkar P. Paragangliomas: Classification, pathology, and differential diagnosis. Otolaryngol Clin North Am. 2001;34:845-862. vi
3 Sobol SM, Dailey JC. Familial multiple cervical paragangliomas: report of a kindred and review of the literature. Otolaryngol Head Neck Surg. 1990;102:382-390.
4 Myssiorek D. Head and neck paragangliomas: an overview. Otolaryngol Clin North Am. 2001;34:829-836. v
5 Lack EE, Cubilla AL, Woodruff JM, et al. Paragangliomas of the head and neck region: A clinical study of 69 patients. Cancer. 1977;39:397-409.
6 Mariman EC, van Beersum SE, Cremers CW, et al. Fine mapping of a putatively imprinted gene for familial non-chromaffin paragangliomas to chromosome 11q13.1: Evidence for genetic heterogeneity. Hum Genet. 1995;95:56-62.
7 Lustrin ES, Palestro C, Vaheesan K. Radiographic evaluation and assessment of paragangliomas. Otolaryngol Clin North Am. 2001;34:881-906. vi
8 Parry DM, Li FP, Strong LC, et al. Carotid body tumors in humans: genetics and epidemiology. J Natl Cancer Inst. 1982;68:573-578.
9 Van der Mey AG, Frijns JH, Cornelisse CJ, et al. Does intervention improve the natural course of glomus tumors? A series of 108 patients seen in a 32-year period. Ann Otol Rhinol Laryngol. 1992;101:635-642.
10 Saldana MJ, Salem LE, Travezan R. High altitude hypoxia and chemodectomas. Hum Pathol. 1973;4:251-263.
11 Rodriguez-Cuevas S, Lopez-Garza J, Labastida-Almendaro S. Carotid body tumors in inhabitants of altitudes higher than 2000 meters above sea level. Head Neck. 1998;20:374-378.
12 Farrior JBIII, Hyams VJ, Benke RH, et al. Carcinoid apudoma arising in a glomus jugulare tumor: review of endocrine activity in glomus jugulare tumors. Laryngoscope. 1980;90:110-119.
13 Hodge KM, Byers RM, Peters LJ. Paragangliomas of the head and neck. Arch Otolaryngol Head Neck Surg. 1988;114:872-877.
14 Van der Mey AG, Jansen JC, van Baalen JM. Management of carotid body tumors. Otolaryngol Clin North Am. 2001;34:907-924. vi
15 Tran LP, Velanovich V, Kaufmann CR. Familial multiple glomus tumors: report of a pedigree and literature review. Ann Plast Surg. 1994;32:89-91.
16 Van Baars F, Cremers C, van den BP, et al. Genetic aspects of nonchromaffin paraganglioma. Hum Genet. 1982;60:305-309.
17 Gujrathi CS, Donald PJ. Current trends in the diagnosis and management of head and neck paragangliomas. Curr Opin Otolaryngol Head Neck Surg. 2005;13:339-342.
18 Pawlu C, Bausch B, Neumann HP. Mutations of the SDHB and SDHD genes. Fam Cancer. 2005;4:49-54.
19 Niemann S, Steinberger D, Muller U. PGL3, a third, not maternally imprinted locus in autosomal dominant paraganglioma. Neurogenetics. 1999;2:167-170.
20 Heutink P, van der Mey AG, Sandkuijl LA, et al. A gene subject to genomic imprinting and responsible for hereditary paragangliomas maps to chromosome 11q23-qter. Hum Mol Genet. 1992;1:7-10.
21 Mariman EC, van Beersum SE, Cremers CW, et al. Analysis of a second family with hereditary non-chromaffin paragangliomas locates the underlying gene at the proximal region of chromosome 11q. Hum Genet. 1993;91:357-361.
22 Niemann S, Muller U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet. 2000;26:268-270.
23 Maier W, Marangos N, Laszig R. Paraganglioma as a systemic syndrome: pitfalls and strategies. J Laryngol Otol. 1999;113:978-982.
24 Schimke RN, Collins DL, Rothberg PG. Functioning carotid paraganglioma in the von Hippel-Lindau syndrome. Am J Med Genet. 1998;80:533-534.
25 DeAngelis LM, Kelleher MB, Post KD, et al. Multiple paragangliomas in neurofibromatosis: A new neuroendocrine neoplasia. Neurology. 1987;37:129-133.
26 Carney JA. Gastric stromal sarcoma, pulmonary chondroma, and extra-adrenal paraganglioma (Carney Triad): natural history, adrenocortical component, and possible familial occurrence. Mayo Clin Proc. 1999;74:543-552.
27 Trombetta M, Silverman J, Colonias A, et al. Paraganglioma: a potentially challenging tumor. Oncology. 2008;22:341-352.
28 Enzinger FAA, Weiss SW. Paraganglioma. In: Gay S, editor. Soft Tissue Tumors. 3rd ed. Mosby Year-Book; 1995:965-990.
29 Harrington SW, Clagett OT, Dockerty MB. Tumors of the carotid body: clinical and pathologic considerations of twenty tumors affecting nineteen patients (one bilateral). Ann Surg. 1941;114:820-833.
30 Bestler JM, Toomey JM. Malignant carotid body tumor. Report of a case. Arch Otolaryngol. 1969;89:752-755.
31 Zbaren P, Lehmann W. Carotid body paraganglioma with metastases. Laryngoscope. 1985;95:450-454.
32 Krupski WC, Effeney DJ, Ehrenfeld WK, et al. Cervical chemodectoma. Technical considerations and management options. Am J Surg. 1982;144:215-220.
33 Alford BR, Guilford FR. A comprehensive study of tumors of the glomus jugulare. Laryngoscope. 1962;72:765-805.
34 Gallivan MV, Chun B, Rowden G, et al. Laryngeal paraganglioma. Case report with ultrastructural analysis and literature review. Am J Surg Pathol. 1979;3:85-92.
35 Kahn LB. Vagal body tumor (nonchromaffin paraganglioma, chemodectoma, and carotid body-like tumor) with cervical node metastasis and familial association: ultrastructural study and review. Cancer. 1976;38:2367-2377.
36 Olsen WL, Dillon WP, Kelly WM, et al. MR imaging of paragangliomas. AJR Am J Roentgenol. 1987;148:201-204.
37 Holliday RA, Reede DL. MRI of mastoid and middle ear disease. Radiol Clin North Am. 1989;27:283-299.
38 Swartz JD, Korsvik H. High-resolution computed tomography of paragangliomas of the head and neck. J Comput Tomogr. 1984;8:197-202.
39 Shulak JM, O’Donovan PB, Paushter DM, et al. Color flow Doppler of carotid body paraganglioma. J Ultrasound Med. 1989;8:519-521.
40 Stoeckli SJ, Schuknecht B, Alkadhi H, et al. Evaluation of paragangliomas presenting as a cervical mass on color-coded Doppler sonography. Laryngoscope. 2002;112:143-146.
41 Kau R, Arnold W. Somatostatin receptor scintigraphy and therapy of neuroendocrine (APUD) tumors of the head and neck. Acta Otolaryngol. 1996;116:345-349.
42 Bustillo A, Telischi F, Weed D, et al. Octreotide scintigraphy in the head and neck. Laryngoscope. 2004;114:434-440.
43 Geatti O, Shapiro B, Sisson JC, et al. Iodine-131 metaiodobenzylguanidine scintigraphy for the location of neuroblastoma: preliminary experience in ten cases. J Nucl Med. 1985;26:736-742.
44 Swensen SJ, Brown ML, Sheps SG, et al. Use of 131I-MIBG scintigraphy in the evaluation of suspected pheochromocytoma. Mayo Clin Proc. 1985;60:299-304.
45 Kopf D, Bockisch A, Steinert H, et al. Octreotide scintigraphy and catecholamine response to an octreotide challenge in malignant phaeochromocytoma. Clin Endocrinol (Oxf). 1997;46:39-44.
46 McCabe BF, Fletcher M. Selection of therapy of glomus jugulare tumors. Arch Otolaryngol. 1969;89:182-185.
47 Jackson CG, Glasscock MEIII, Harris PF. Glomus tumors. Diagnosis, classification, and management of large lesions. Arch Otolaryngol. 1982;108:401-410.
48 Fisch U. Infratemporal fossa approach for glomus tumors of the temporal bone. Ann Otol Rhinol Laryngol. 1982;91:474-479.
49 Mukherji SK, Kasper ME, Tart RP, et al. Irradiated paragangliomas of the head and neck: CT and MR appearance. AJNR Am J Neuroradiol. 1994;15:357-363.
50 Litle VR, Reilly LM, Ramos TK. Preoperative embolization of carotid body tumors: When is it appropriate? Ann Vasc Surg. 1996;10:464-468.
51 Kotelis D, Rizos T, Geisbusch P, et al. Late outcome after surgical management of carotid body tumors from a 20-year single-center experience. Langenbecks Arch Surg. 2009;394:339-344.
52 Cairnduff F, Smith IE. Carboplatin chemotherapy for malignant paraganglioma. Lancet. 1986;2:982.
53 Mertens WC, Grignon DJ, Romano W. Malignant paraganglioma with skeletal metastases and spinal cord compression: response and palliation with chemotherapy. Clin Oncol (R Coll Radiol). 1993;5:126-128.
54 Kimura S, Iwai M, Fukuda T, et al. Combination chemotherapy for malignant paraganglioma. Intern Med. 1997;36:35-39.
55 Pipas JM, Krywicki RF. Treatment of progressive metastatic glomus jugulare tumor (paraganglioma) with gemcitabine. Neuro Oncol. 2000;2:190-191.
56 Patel SR, Winchester DJ, Benjamin RS. A 15-year experience with chemotherapy of patients with paraganglioma. Cancer. 1995;76:1476-1480.
57 Massey V, Wallner K. Treatment of metastatic chemodectoma. Cancer. 1992;69:790-792.
58 Duet M, Guichard JP, Rizzo N, et al. Are somatostatin analogs therapeutic alternatives in the management of head and neck paragangliomas? Laryngoscope. 2005;115:1381-1384.
59 Kaltsas GA, Papadogias D, Makras P, et al. Treatment of advanced neuroendocrine tumours with radiolabelled somatostatin analogues. Endocr Relat Cancer. 2005;12:683-699.
60 Chrisoulidou A, Kaltsas G, Ilias I, et al. The diagnosis and management of malignant phaeochromocytoma and paraganglioma. Endocr Relat Cancer. 2007;14:569-585.
61 Forrer F, Riedweg I, Maecke HR, et al. Radiolabeled DOTATOC in patients with advanced paraganglioma and pheochromocytoma. Q J Nucl Med Mol Imaging. 2008;52:334-340.
62 Gross DJ, Munter G, Bitan M, et al. The role of imatinib mesylate (Glivec) for treatment of patients with malignant endocrine tumors positive for c-kit or PDGF-R. Endocr Relat Cancer. 2006;13:535-540.
63 Kappes A, Vaccaro A, Kunnimalaiyaan M, et al. ZM336372, a Raf-1 activator, inhibits growth of pheochromocytoma cells. J Surg Res. 2006;133:42-45.
64 Giraud P, Elles S, Helfre S, et al. Conformal radiotherapy for lung cancer: different delineation of the gross tumor volume (GTV) by radiologists and radiation oncologists. Radiother Oncol. 2002;62:27-36.
65 Kim JA, Elkon D, Lim ML, et al. Optimum dose of radiotherapy for chemodectomas of the middle ear. Int J Radiat Oncol Biol Phys. 1980;6:815-819.
66 Liscak R, Vladyka V, Simonova G, et al. Leksell gamma knife radiosurgery of the tumor glomus jugulare and tympanicum. Stereotact Funct Neurosurg. 1998;70(Suppl 1):152-160.
67 Liscak R, Vladyka V, Wowra B, et al. Gamma knife radiosurgery of the glomus jugulare tumour—early multicentre experience. Acta Neurochir (Wien). 1999;141:1141-1146.
68 Eustacchio S, Leber K, Trummer M, et al. Gamma knife radiosurgery for glomus jugulare tumours. Acta Neurochir (Wien). 1999;141:811-818.
69 Jordan JA, Roland PS, McManus C, et al. Stereotactic radiosurgery for glomus jugulare tumors. Laryngoscope. 2000;110:35-38.
70 Foote RL, Pollock BE, Gorman DA, et al. Glomus jugulare tumor: tumor control and complications after stereotactic radiosurgery. Head Neck. 2002;24:332-338.
71 Feigenberg SJ, Mendenhall WM, Hinerman RW, et al. Radiosurgery for paraganglioma of the temporal bone. Head Neck. 2002;24:384-389.
72 Hinerman RW, Amdur RJ, Morris CG, et al. Definitive radiotherapy in the management of paragangliomas arising in the head and neck: a 35-year experience. Head Neck. 2008;30:1431-1438.
73 Valdagni R, Amichetti M. Radiation therapy of carotid body tumors. Am J Clin Oncol. 1990;13:45-48.
74 Verniers DA, Keus RB, Schouwenburg PF, et al. Radiation therapy, an important mode of treatment for head and neck chemodectomas. Eur J Cancer. 1992;28A:1028-1033.
75 Hinerman RW, Mendenhall WM, Amdur RJ, et al. Definitive radiotherapy in the management of chemodectomas arising in the temporal bone, carotid body, and glomus vagale. Head Neck. 2001;23:363-371.
76 Koskas F, Vignes S, Khalil I, et al. Carotid chemodectomas: long-term results of subadventitial resection with deliberate external carotid resection. Ann Vasc Surg. 2009;23:288-289.
77 McMaster ML, Goldstein AM, Bromley CM, et al. Chordoma: incidence and survival patterns in the United States, 1973–1995. Cancer Causes Control. 2001;12:1-11.
78 Noel G, Habrand JL, Jauffret E, et al. Radiation therapy for chordoma and chondrosarcoma of the skull base and the cervical spine. Prognostic factors and patterns of failure. Strahlenther Onkol. 2003;179:241-248.
79 Pamir MN, Ozduman K. Tumor-biology and current treatment of skull-base chordomas. Adv Tech Stand Neurosurg. 2008;33:35-129.
80 Almefty K, Pravdenkova S, Colli BO, et al. Chordoma and chondrosarcoma: Similar, but quite different, skull base tumors. Cancer. 2007;110:2457-2467.
81 Mitchell A, Scheithauer BW, Unni KK, et al. Chordoma and chondroid neoplasms of the spheno-occiput. An immunohistochemical study of 41 cases with prognostic and nosologic implications. Cancer. 1993;72:2943-2949.
82 Tomlinson FH, Scheithauer BW, Forsythe PA, et al. Sarcomatous transformation in cranial chordoma. Neurosurgery. 1992;31:13-18.
83 Scheil S, Bruderlein S, Liehr T, et al. Genome-wide analysis of sixteen chordomas by comparative genomic hybridization and cytogenetics of the first human chordoma cell line, U-CH1. Genes Chromosomes Cancer. 2001;32:203-211.
84 Kelley MJ, Korczak JF, Sheridan E, et al. Familial chordoma, a tumor of notochordal remnants, is linked to chromosome 7q33. Am J Hum Genet. 2001;69:454-460.
85 Gadwal SR, Fanburg-Smith JC, Gannon FH, et al. Primary chondrosarcoma of the head and neck in pediatric patients: a clinicopathologic study of 14 cases with a review of the literature. Cancer. 2000;88:2181-2188.
86 Chandler JP, Yashar P, Laskin WB, et al. Intracranial chondrosarcoma: a case report and review of the literature. J Neurooncol. 2004;68:33-39.
87 Korten AG, ter Berg HJ, Spincemaille GH, et al. Intracranial chondrosarcoma: Review of the literature and report of 15 cases. J Neurol Neurosurg Psychiatry. 1998;65:88-92.
88 Gelderblom H, Hogendoorn PC, Dijkstra SD, et al. The clinical approach towards chondrosarcoma. Oncologist. 2008;13:320-329.
89 Evans HL, Ayala AG, Romsdahl MM. Prognostic factors in chondrosarcoma of bone: A clinicopathologic analysis with emphasis on histologic grading. Cancer. 1977;40:818-831.
90 Tachibana E, Saito K, Takahashi M, et al. Surgical treatment of a massive chondrosarcoma in the skull base associated with Maffucci’s syndrome: a case report. Surg Neurol. 2000;54:165-169.
91 Chambers PW, Schwinn CP. Chordoma. A clinicopathologic study of metastasis. Am J Clin Pathol. 1979;72:765-776.
92 Fischbein NJ, Kaplan MJ, Holliday RA, et al. Recurrence of clival chordoma along the surgical pathway. AJNR Am J Neuroradiol. 2000;21:578-583.
93 Gay E, Sekhar LN, Rubinstein E, et al. Chordomas and chondrosarcomas of the cranial base: Results and follow-up of 60 patients. Neurosurgery. 1995;36:887-897.
94 Rich TA, Schiller A, Suit HD, et al. Clinical and pathologic review of 48 cases of chordoma. Cancer. 1985;56:182-187.
95 Aydin P, Ozek M, Kansu T. Intermittent diplopia in chordoma. Ann Ophthalmol. 1994;26:20-22.
96 Oikawa S, Kyoshima K, Goto T, et al. Histological study on local invasiveness of clival chordoma. Case report of autopsy. Acta Neurochir (Wien). 2001;143:1065-1069.
97 Al-Mefty O, Borba LB. Skull base chordomas: a management challenge. J Neurosurg. 1997;86:182-189.
98 Kay PA, Nascimento AG, Unni KK, et al. Chordoma: cytomorphologic findings in 14 cases diagnosed by fine needle aspiration. Acta Cytol. 2003;47:202-208.
99 Colli B, al-Mefty O. Chordomas of the craniocervical junction: follow-up review and prognostic factors. J Neurosurg. 2001;95:933-943.
100 Mendenhall WM, Mendenhall CM, Lewis SB, et al. Skull base chordoma. Head Neck. 2005;27:159-165.
101 Rosenberg AE, Nielsen GP, Keel SB, et al. Chondrosarcoma of the base of the skull. A clinicopathologic study of 200 cases with emphasis on its distinction from chordoma. Am J Surg Pathol. 1999;23:1370-1378.
102 Heffelfinger MJ, Dahlin DC, MacCarty CS, et al. Chordomas and cartilaginous tumors at the skull base. Cancer. 1973;32:410-420.
103 Ishida T, Dorfman HD. Chondroid chordoma versus low-grade chondrosarcoma of the base of the skull: can immunohistochemistry resolve the controversy? J Neuro-Oncol. 1994;18:199-206.
104 Juliao SF, Rand N, Schwartz HS. Galectin-3: a biologic, marker and diagnostic aid for chordoma. Clin Orthop Related Res. 2002;397:70-75.
105 Naka T, Oda Y, Iwamoto Y, et al. Immunohistochemical analysis of E-cadherin, alpha-catenin, beta-catenin, gamma-catenin, and neural cell adhesion molecule (NCAM) in chordoma. J Clin Pathol. 2001;54:945-950.
106 Pamir MN, Kilic T, Ture U, et al. Multimodality management of 26 skull-base chordomas with 4-year mean follow-up: experience at a single institution. Acta Neurochir (Wien). 2004;146:343-354.
107 Al-Mefty O, Borba LB. Skull base chordomas: a management challenge. J Neurosurg. 1997;86:182-189.
108 Razis DV, Tsatsaronis A, Kyriazides I, et al. Chordoma of the cervical spine treated with vincristine sulfate. J Med. 1974;5:274-277.
109 Harwick RD, Miller AS. Craniocervical chordomas. Am J Surg. 1979;138:512-516.
110 Rissanen PM, Holsti LR. Sacrococcygeal chordomas and their treatment. Radiol Clin Biol. 1967;36:153-164.
111 Pearlman AW, Singh RK, Hoppenstein R, et al. Chordoma: combined therapy with radiation and surgery: case report and new operative approach. Bull Hosp Joint Dis. 1972;33:47-57.
112 Spratt JS, Martin AE, McKeown J. Sacral chordoma: a case study and review. J Surg Oncol. 1981;18:101-103.
113 Cummings BJ, Esses S, Harwood AR. The treatment of chordomas. Cancer Treat Rev. 1982;9:299-311.
114 Fuller DB, Bloom JG. Radiotherapy for chordoma. Int J Radiat Oncol Biol Phys. 1988;15:331-339.
115 Chugh R, Dunn R, Zalupski MM, et al. Phase II study of 9-nitro-camptothecin in patients with advanced chordoma or soft tissue sarcoma. J Clin Oncol. 2005;23:3597-3604.
116 Rhomberg W, Bohler FK, Novak H, et al. A small prospective study of chordomas treated with radiotherapy and razoxane. Strahlenther Onkol. 2003;179:249-253.
117 Casali PG, Messina A, Stacchiotti S, et al. Imatinib mesylate in chordoma. Cancer. 2004;101:2086-2097.
118 Tamborini E, Miselli F, Negri T, et al. Molecular and biochemical analyses of platelet-derived growth factor receptor (PDGFR) B, PDGFRA, and KIT receptors in chordomas. Clin Cancer Res. 2006;12:6920-6928.
119 Orzan F, Terreni MR, Longoni M, et al. Expression study of the target receptor tyrosine kinase of imatinib mesylate in skull base chordomas. Oncol Rep. 2007;18:249-252.
120 Burger H, den Bakker MA, Kros JM, et al. Activating mutations in c-KIT and PDGFRalpha are exclusively found in gastrointestinal stromal tumors and not in other tumors overexpressing these imatinib mesylate target genes. Cancer Biol Ther. 2005;4:1270-1274.
121 Weinberger PM, Yu Z, Kowalski D, et al. Differential expression of epidermal growth factor receptor, c-Met, and HER2/neu in chordoma compared with 17 other malignancies. Arch Otolaryngol Head Neck Surg. 2005;131:707-711.
122 Rizzo M, Ghert MA, Harrelson JM, et al. Chondrosarcoma of bone: analysis of 108 cases and evaluation for predictors of outcome. Clin Orthop Relat Res. 2001:224-233.
123 Yoshimoto T, Sawamura Y, Ikeda J, et al. Successful chemoradiation therapy for high-grade skull base chondrosarcoma in a child. Childs Nerv Syst. 1995;11:250-253.
124 La Rocca RV, Morgan KW, Paris K, et al. Recurrent chondrosarcoma of the cranial base: A durable response to ifosfamide-doxorubicin chemotherapy. J Neurooncol. 1999;41:281-283.
125 Aksoy S, Abali H, Kilickap S, et al. Successful treatment of a chemoresistant tumor with temozolomide in an adult patient: report of a recurrent intracranial mesenchymal chondrosarcoma. J Neurooncol. 2005;71:333-334.
126 Dickey ID, Rose PS, Fuchs B, et al. Dedifferentiated chondrosarcoma: the role of chemotherapy with updated outcomes. J Bone Joint Surg Am. 2004;86-A:2412-2418.
127 Mitchell AD, Ayoub K, Mangham DC, et al. Experience in the treatment of dedifferentiated chondrosarcoma. J Bone Joint Surg Br. 2000;82:55-61.
128 Terek RM, Schwartz GK, Devaney K, et al. Chemotherapy and P-glycoprotein expression in chondrosarcoma. J Orthop Res. 1998;16:585-590.
129 Baldini N, Scotlandi K, Barbanti-Brodano G, et al. Expression of P-glycoprotein in high-grade osteosarcomas in relation to clinical outcome. N Engl J Med. 1995;333:1380-1385.
130 Chow WA, Bedell V, Gaytan P, et al. Methylthioadenosine phosphorylase gene deletions are frequently detected by fluorescence in situ hybridization in conventional chondrosarcomas. Cancer Genet Cytogenet. 2006;166:95-100.
131 Lagonigro MS, Tamborini E, Negri T, et al. PDGFRalpha, PDGFRbeta and KIT expression/activation in conventional chondrosarcoma. J Pathol. 2006;208:615-623.
132 Yamamoto S, Tanaka K, Sakimura R, et al. Suberoylanilide hydroxamic acid (SAHA) induces apoptosis or autophagy-associated cell death in chondrosarcoma cell lines. Anticancer Res. 2008;28:1585-1591.
133 Feigl GC, Bundschuh O, Gharabaghi A, et al. Evaluation of a new concept for the management of skull base chordomas and chondrosarcomas. J Neurosurg. 2005;102(Suppl):165-170.
134 Krishnan S, Foote RL, Brown PD, et al. Radiosurgery for cranial base chordomas and chondrosarcomas. Neurosurgery. 2005;56:777-784.
135 Pearlman AW, Friedman M. Radical radiation therapy of chordoma. Am J Roentgenol Radium Ther Nucl Med. 1970;108:332-341.
136 Hug EB, Loredo LN, Slater JD, et al. Proton radiation therapy for chordomas and chondrosarcomas of the skull base. J Neurosurg. 1999;91:432-439.
137 Noel G, Feuvret L, Calugaru V, et al. Chordomas of the base of the skull and upper cervical spine. One hundred patients irradiated by a 3D conformal technique combining photon and proton beams. Acta Oncol. 2005;44:700-708.
138 O’Connell JX, Renard LG, Liebsch NJ, et al. Base of skull chordoma. A correlative study of histologic and clinical features of 62 cases. Cancer. 1994;74:2261-2267.
139 Terahara A, Niemierko A, Goitein M, et al. Analysis of the relationship between tumor dose inhomogeneity and local control in patients with skull base chordoma. Int J Radiat Oncol Biol Phys. 1999;45:351-358.
140 Maira G, Pallini R, Anile C, et al. Surgical treatment of clival chordomas: the transsphenoidal approach revisited. J Neurosurg. 1996;85:784-792.
141 Stuer C, Schramm J, Schaller C. Skull base chordomas: management and results. Neurol Med Chir (Tokyo). 2006;46:118-124.
142 Santoni R, Liebsch N, Finkelstein DM, et al. Temporal lobe (TL) damage following surgery and high-dose photon and proton irradiation in 96 patients affected by chordomas and chondrosarcomas of the base of the skull. Int J Radiat Oncol Biol Phys. 1998;41:59-68.
143 Debus J, Hug EB, Liebsch NJ, et al. Brainstem tolerance to conformal radiotherapy of skull base tumors. Int J Radiat Oncol Biol Phys. 1997;39:967-975.
144 Munzenrider JE, Liebsch NJ. Proton therapy for tumors of the skull base. Strahlenther Onkol. 1999;175(Suppl 2)):57-63.
145 Habrand IL, Austin-Seymour M, Birnbaum S, et al. Neurovisual outcome following proton radiation therapy. Int J Radiat Oncol Biol Phys. 1989;16:1601-1606.
146 Fuller DB, Bloom JG. Radiotherapy for chordoma. Int J Radiat Oncol Biol Phys. 1988;15:331-339.
147 Weber DC, Rutz HP, Pedroni ES, et al. Results of spot-scanning proton radiation therapy for chordoma and chondrosarcoma of the skull base: the Paul Scherrer Institut experience. Int J Radiat Oncol Biol Phys. 2005;63:401-409.
148 Pai HH, Thornton A, Katznelson L, et al. Hypothalamic/pituitary function following high-dose conformal radiotherapy to the base of skull: Demonstration of a dose-effect relationship using dose-volume histogram analysis. Int J Radiat Oncol Biol Phys. 2001;49:1079-1092.
149 O’Leary MJ, Shelton C, Giddings NA, et al. Glomus tympanicum tumors: a clinical perspective. Laryngoscope. 1991;101:1038-1043.
150 Woods CI, Strasnick B, Jackson CG. Surgery for glomus tumors: the Otology Group experience. Laryngoscope. 1993;103:65-70.
151 Murphy TP, Brackmann DE. Effects of preoperative embolization on glomus jugulare tumors. Laryngoscope. 1989;99:1244-1247.
152 Cece JA, Lawson W, Biller HF, et al. Complications in the management of large glomus jugulare tumors. Laryngoscope. 1987;97:152-157.
153 Anand VK, Leonetti JP, Al-Mefty O. Neurovascular considerations in surgery of glomus tumors with intracranial extensions. Laryngoscope. 1993;103:722-728.
154 Spector GJ, Sobol S. Surgery for glomus tumors at the skull base. Otolaryngol Head Neck Surg. 1980;88:524-530.
155 Jackson CG, Haynes DS, Walker PA, et al. Hearing conservation in surgery for glomus jugulare tumors. Am J Otolaryngol. 1996;17:425-437.
156 Brown JS. Glomus jugulare tumors revisited: a ten-year statistical follow-up of 231 cases. Laryngoscope. 1985;95:284-288.
157 Larner JM, Hahn SS, Spaulding CA, et al. Glomus jugulare tumors. Long-term control by radiation therapy. Cancer. 1992;69:1813-1817.
158 Gjuric M, Seidinger L, Wigand ME. Long-term results of surgery for temporal bone paraganglioma. Skull Base Surg. 1996;6:147-152.
159 Powell S, Peters N, Harmer C. Chemodectoma of the head and neck: results of treatment in 84 patients. Int J Radiat Oncol Biol Phys. 1992;22:919-924.
160 Kraus DH, Sterman BM, Hakaim AG, et al. Carotid body tumors. Arch Otolaryngol Head Neck Surg. 1990;116:1384-1387.
161 Biller HF, Lawson W, Som P, et al. Glomus vagale tumors. Ann Otol Rhinol Laryngol. 1989;98:21-26.
162 Green JD, Olsen KD, DeSanto LW, et al. Neoplasms of the vagus nerve. Laryngoscope. 1988;98:648-654.
163 Davidovic LB, Djukic VB, Vasic DM, et al. Diagnosis and treatment of carotid body paraganglioma: 21 years of experience at a clinical center of Serbia. World J Surg Oncol. 2005;3:10.
164 Elshaikh MA, Mahmoud-Ahmed AS, Kinney SE, et al. Recurrent head and neck chemodectomas: A comparison of surgical and radiotherapeutic results. Int J Radiat Oncol Biol Phys. 2002;52:953-956.
165 Mendenhall WM, Morris CG, Amdur RJ, et al. Radiotherapy alone or after subtotal resection for benign skull base meningiomas. Cancer. 2003;98:1473-1482.
166 Hatfield PM, James AE, Schulz MD. Chemodectomas of the glomus jugulare. Cancer. 1972;30:1164-1168.
167 Silverstone SM. Radiation therapy of glomus jugulare tumors. Arch Otolaryngol. 1973;97:43-48.
168 Thomsen K, Elbrond O, Andersen AP. Glomus jugulare tumours. (A series of 21 cases). J Laryngol Otol. 1975;89:1113-1121.
169 Arthur K. Radiotherapy in chemodectoma of the glomus jugulare. Clin Radiol. 1977;28:415-417.
170 Cole JM, Beiler D. Long-term results of treatment for glomus jugulare and glomus vagale tumors with radiotherapy. Laryngoscope. 1994;104:1461-1465.
171 Gibbin KP, Henk JM. Glomus jugulare tumours in South Wales—a twenty-year review. Clin Radiol. 1978;29:607-609.
172 Kim JA, Elkon D, Lim ML, et al. Optimum dose of radiotherapy for chemodectomas of the middle ear. Int J Radiat Oncol Biol Phys. 1980;6:815-819.
173 Reddy EK, Mansfield CM, Hartman GV. Chemodectoma of glomus jugulare. Cancer. 1983;52:337-340.
174 Cummings BJ, Beale FA, Garrett PG, et al. The treatment of glomus tumors in the temporal bone by megavoltage radiation. Cancer. 1984;53:2635-2640.
175 Konefal JB, Pilepich MV, Spector GJ, et al. Radiation therapy in the treatment of chemodectomas. Laryngoscope. 1987;97:1331-1335.
176 Wang ML, Hussey DH, Doornbos JF, et al. Chemodectoma of the temporal bone: a comparison of surgical and radiotherapeutic results. Int J Radiat Oncol Biol Phys. 1988;14:643-648.
177 Pryzant RM, Chou JL, Easley JD. Twenty year experience with radiation therapy for temporal bone chemodectomas. Int J Radiat Oncol Biol Phys. 1989;17:1303-1307.
178 Boyle JO, Shimm DS, Coulthard SW. Radiation therapy for paragangliomas of the temporal bone. Laryngoscope. 1990;100:896-901.
179 Schild SE, Foote RL, Buskirk SJ, et al. Results of radiotherapy for chemodectomas. Mayo Clin Proc. 1992;67:537-540.
180 Mendenhall WM, Parsons JT, Stringer SP, et al. Radiotherapy in the management of temporal bone chemodectoma. Skull Base Surg. 1995;5:83-91.
181 De Jong AL, Coker NJ, Jenkins HA, et al. Radiation therapy in the management of paragangliomas of the temporal bone. Am J Otolaryngol. 1995;16:283-289.
182 Pemberton LS, Swindell R, Sykes AJ. Radical radiotherapy alone for glomus jugulare and tympanicum tumours. Oncol Rep. 2005;14:1631-1633.
183 Foote RL, Pollock BE, Gorman DA, et al. Glomus jugulare tumor: tumor control and complications following stereotactic radiosurgery. Head Neck. 2002;24:332-338.
184 Pollock BE. Stereotactic radiosurgery in patients with glomus jugulare tumors. Neurosurg Focus. 2004;17:E10.
185 Lim M, Gibbs IC, Adler JR, et al. Efficacy and safety of stereotactic radiosurgery for glomus jugulare tumors. Neurosurg Focus. 2004;17:E11.
186 Sheehan J, Yen CP, Arkha Y, et al. Gamma knife surgery for trigeminal schwannoma. J Neurosurg. 2007;106:839-845.
187 Poznanovic SA, Cass SP, Kavanagh BD. Short-term tumor control and acute toxicity after stereotactic radiosurgery for glomus jugulare tumors. Otolaryngol Head Neck Surg. 2006;134:437-442.
188 Gerosa M, Visca A, Rizzo P, et al. Glomus jugulare tumors: the option of gamma knife radiosurgery. Neurosurgery. 2006;59:561-569.
189 Krych AJ, Foote RL, Brown PD, et al. Long-term results of irradiation for paraganglioma. Int J Radiat Oncol Biol Phys. 2006;65:1063-1066.
190 Westerband A, Hunter GC, Cintora I, et al. Current trends in the detection and management of carotid body tumors. J Vasc Surg. 1998;28:84-93.
191 Meyer FB, Sundt TM, Pearson BW. Carotid body tumors: a subject review and suggested surgical approach. J Neurosurg. 1986;64:377-385.
192 Urquhart AC, Johnson JT, Myers EN, et al. Glomus vagale: paraganglioma of the vagus nerve. Laryngoscope. 1994;104:440-445.
193 Netterville JL, Reilly KM, Robertson D, et al. Carotid body tumors: a review of 30 patients with 46 tumors. Laryngoscope. 1995;105:115-126.
194 Green JD, Brackmann DE, Nguyen CD, et al. Surgical management of previously untreated glomus jugulare tumors. Laryngoscope. 1994;104:917-921.
195 Briner HR, Linder TE, Pauw B, et al. Long-term results of surgery for temporal bone paragangliomas. Laryngoscope. 1999;109:577-583.
196 LaRouere MJ, Zappia JJ, Wilner HI, et al. Selective embolization of glomus jugulare tumors. Skull Base Surg. 1994;41:21-25.
197 Verniers DA, Keus RB, Schouwenburg PF, et al. Radiation therapy, an important mode of treatment for head and neck chemodectomas. Eur J Cancer. 1992;28A:1028-1033.
198 Zabel A, Milker-Zabel S, Huber P, et al. Fractionated stereotactic conformal radiotherapy in the management of large chemodectomas of the skull base. Int J Radiat Oncol Biol Phys. 2004;58:1445-1450.
199 Magrini SM, Papi MG, Marletta F, et al. Chordoma-natural history, treatment and prognosis. The Florence Radiotherapy Department experience (1956–1990) and a critical review of the literature. Acta Oncol. 1992;31:847-851.
200 Forsyth PA, Posner JB. Headaches in patients with brain tumors: a study of 111 patients. Neurology. 1993;43:1678-1683.
201 Romero J, Cardenes H, la Torre A, et al. Chordoma: results of radiation therapy in eighteen patients. Radiother Oncol. 1993;29:27-32.
202 Benk V, Liebsch NJ, Munzenrider JE, et al. Base of skull and cervical spine chordomas in children treated by high-dose irradiation. Int J Radiat Oncol Biol Phys. 1995;31:577-581.
203 Catton C, O’Sullivan B, Bell R, et al. Chordoma: long-term follow-up after radical photon irradiation. Radiother Oncol. 1996;41:67-70.
204 Debus J, Schulz-Ertner D, Schad L, et al. Stereotactic fractionated radiotherapy for chordomas and chondrosarcomas of the skull base. Int J Radiat Oncol Biol Phys. 2000;47:591-596.
205 Zorlu F, Gurkaynak M, Yildiz F, et al. Conventional external radiotherapy in the management of clivus chordomas with overt residual disease. Neurol Sci. 2000;21:203-207.
206 Schulz-Ertner D, Nikoghosyan A, Thilmann C, et al. Carbon ion radiotherapy for chordomas and low-grade chondrosarcomas of the skull base. Results in 67 patients. Strahlenther Onkol. 2003;179:598-605.
207 Igaki H, Tokuuye K, Okumura T, et al. Clinical results of proton beam therapy for skull base chordoma. Int J Radiat Oncol Biol Phys. 2004;60:1120-1126.
208 Cho YH, Kim JH, Khang SK, et al. Chordomas and chondrosarcomas of the skull base: comparative analysis of clinical results in 30 patients. Neurosurg Rev. 2008;31:35-43.
209 Schulz-Ertner D, Nikoghosyan A, Hof H, et al. Carbon ion radiotherapy of skull base chondrosarcomas. Int J Radiat Oncol Biol Phys. 2007;67:171-177.