[level-membership-for-pediatrics-category]Chapter 133
Skeletal Dysplasias and Selected Chromosomal Disorders
The abridged form of the International Skeletal Dysplasia Society skeletal dysplasia classification serves as the organization of this chapter (Box 133-1).1 The full nosology text can be found at http://isds.ch/uploads/pdf_files/Nosology2010.pdf (accessed August 12, 2012). The major genetic families are presented with a short description of the salient unifying characteristics of the diseases within each group. When known, the gene and protein involved are considered and the impact of the mechanism of action discussed. The major members of each group are then expanded on to provide a clear picture for the reader.
Radiologic Assessment
In the history of the delineation of many of the specific skeletal dysplasias, radiologic assessment plays a major role. By using an orderly approach to the radiographic analysis, the general type of the dysplasia may be elucidated. Many of the skeletal dysplasias and syndromes have distinctive radiographic features that will allow an exact diagnosis when even one of those distinctive features is identified and used as a search criterion in textbooks on skeletal dysplasia. Two such texts are Taybi and Lachman’s Radiology of Syndromes, Metabolic Disorders and Skeletal Dysplasias, which includes an excellent gamuts section, and Bone Dysplasias, An Atlas of Genetic Disorders of Skeletal Development by Spranger and colleagues, in which the images are particularly helpful.2,3 In the online version of Taybi and Lachman’s book, the gamut search may be built iteratively, with the diagnoses becoming more selective as findings are added to the search criteria. Internet searches can also be performed on the Online Mendelian Inheritance in Man database, which is accessed through the U.S. Library of Medicine portal at http://www.ncbi.nlm.nih.gov/pubmed/.
Step II: Assessment of Epiphyseal Ossification
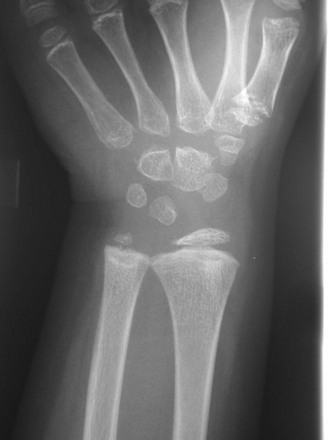
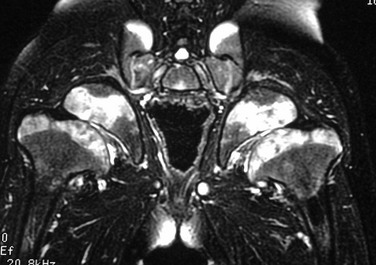
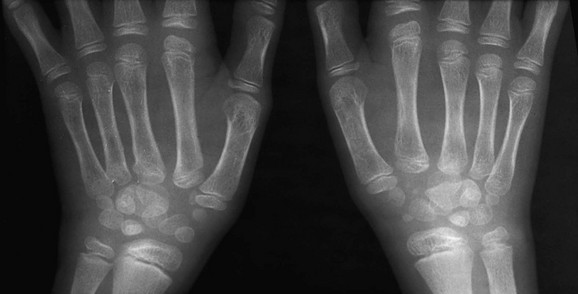
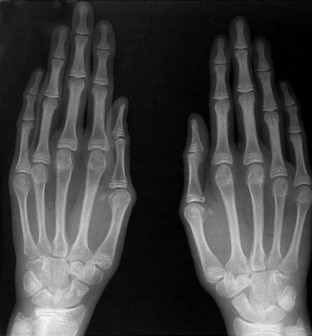
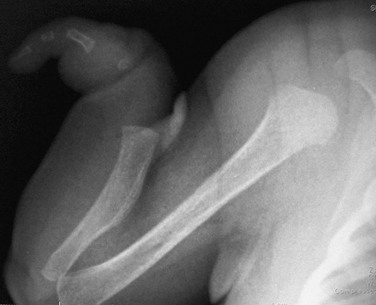
If epiphyseal ossification is delayed or if the ossified epiphyses are very small, irregular for age, or both, then an epiphyseal dysplasia of some sort is present. Carpal and tarsal bones are often affected. In diseases that can be considered pure epiphyseal dysplasias such as multiple epiphyseal dysplasia and pseudoachondroplasia, carpal and the tarsal bones are markedly crenellated and small (Fig. 133-1). Another excellent location for epiphyseal analysis is the ring apophyses of the vertebral bodies, which exhibit delayed and irregular epiphyseal ossification in epiphyseal dysplasia. Central anterior vertebral body protrusions (central tongues or beaking) noted in Morquio syndrome and pseudoachondroplasia are also disorders related to abnormalities of the ring apophyses.
Step III: Assessment of Metaphyses and Physes
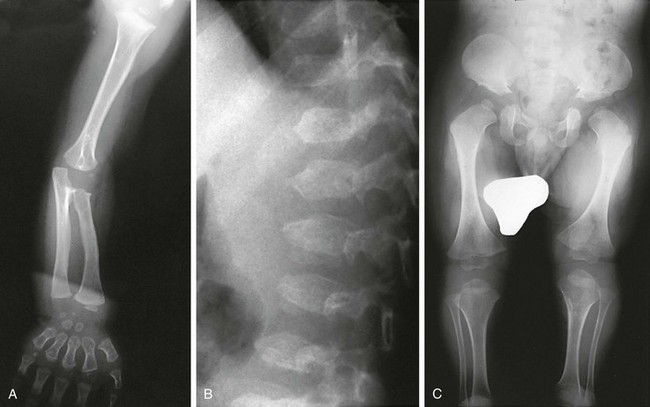
Fraying and irregularity of the physes and abnormal flaring of the metaphyses indicate disturbed endochondral ossification. Marked irregularity of the physes is characteristic of the pure metaphyseal dysplasias such as metaphyseal dysplasia, Jansen or Schmid type. When the metaphyses are merely flared and the physes are fairly normal, endochondral ossification may be slowed but the actual process of endochondral ossification progresses normally. This occurs in achondroplasia. The metaphyses are flared, whereas the physis and the zone of provisional calcification (ZPC) are sharply defined (Fig. 133-2).
Figure 133-2 A 14-year-old boy with achondroplasia.
The long bones are short and thick. Note the normal, sharp-appearing physes.
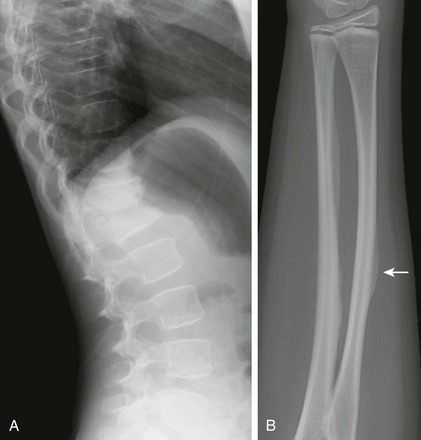
It must be kept in mind that rickets also disturbs the physis. In rickets, the physis is frayed and cupped. Except in healing rickets, the ZPC is inapparent. In metaphyseal chondrodysplasias, the ZPC is present, although it is markedly irregular (Fig. 133-3). Analysis of the sclerotic line of the ZPC is frequently an excellent differentiating feature. Other factors include prominent osteopenia in rickets with blurring of the trabeculae; clinical data are also very helpful.
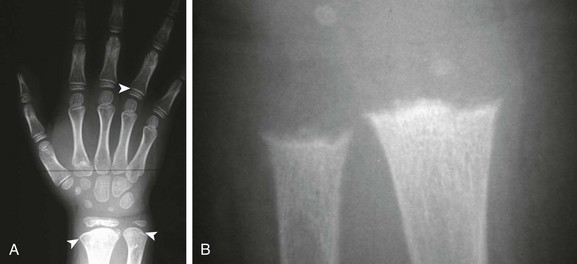
Figure 133-3 An 8-old boy with Schmid metaphyseal chondrodysplasia.
A, Note the presence of zone of provisional calcification (arrowheads). Normal laboratory values help confirm the diagnosis. B, A 15-month-old girl with rickets. The physes are frayed with mild metaphyseal cupping. The bright white line of the zone of provisional calcification is not evident.
Step V: Analysis of the Vertebral Bodies
Anisospondyly is when the vertebral body shape varies wildly (e-Fig. 133-4). Multiple ossification centers may also be present. Although rare, this is a specific finding in dyssegmental dysplasia.
Step VIII: Summation
After all radiologic findings have been established, a gamut search of some or all of these abnormalities, in conjunction with the clinical findings, may lead to the specific diagnosis. If the “group” of dysplasias has been established, then the specific diagnosis can often be made by referring to a differential diagnosis table such as that developed by Taybi and Lachman.2
Selected Skeletal Dysplasias and Syndromes
Fibroblast Growth Factor Receptor Type 3 Group
Overview: This group includes thanatophoric dwarfism and achondroplasia. The former is probably the most common lethal skeletal dysplasia and the latter the most common skeletal dysplasia. The group includes the milder variant called hypochondroplasia, and homozygous achondroplasia, which is similar to thanatophoria.4
A common genetic locus (4p16.3) is involved. Differing allelic mutations are the cause of the variable severity of expression. The protein encoded is FGFR3, which governs the velocity of endochondral growth. Although long believed that achondroplasia and thanatophoria were caused by loss of function mutations, the mutation in this group actually results in an upmodulation of FGFR3 activity, which is inversely related to the velocity of endochondral growth. FGFR3 mutations have been linked to advanced paternal age, with mutations theoretically accumulating during spermatogenesis.5
Thanatophoric Dwarfism
Radiographic Findings (Fig. 133-5):
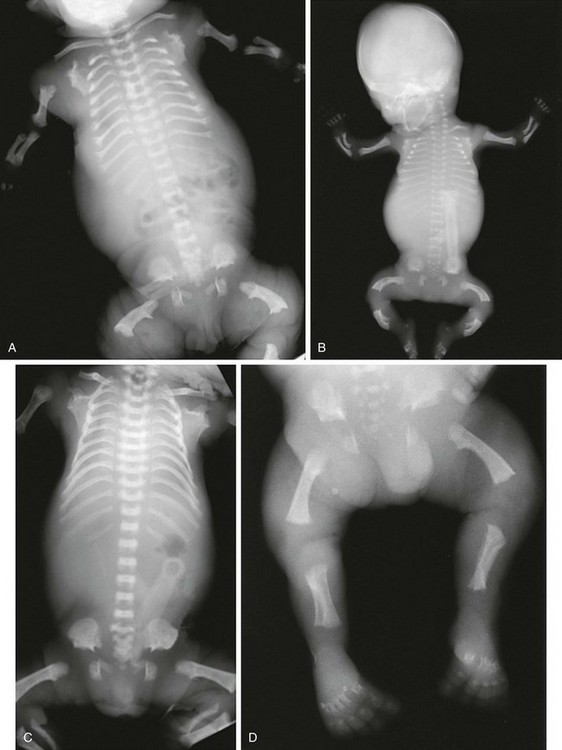
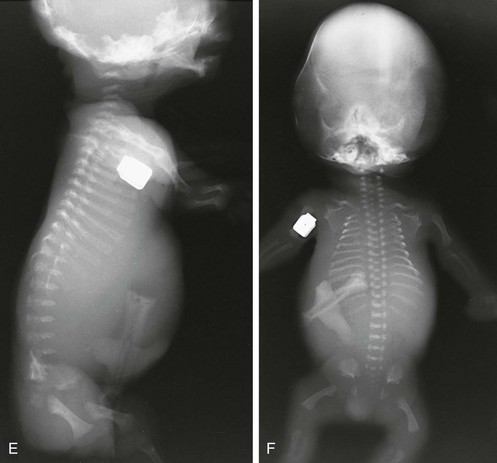
Figure 133-5 Thanatophoric dysplasia.
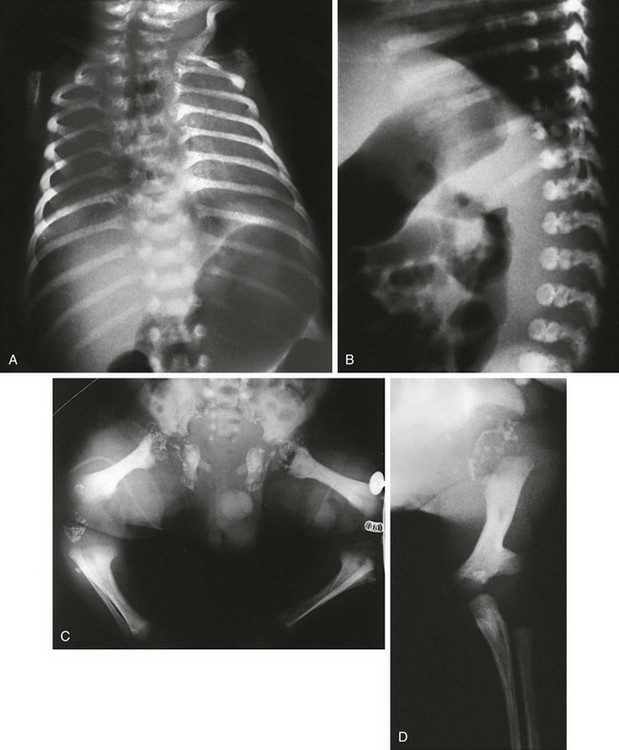
A and B, Radiographic findings in thanatophoric dysplasia type I. In an affected fetus of 30 weeks’ gestation (A), a long, narrow trunk; very short ribs; severe platyspondyly are seen. Note the H and U shapes of bodies are caused by angle of incidence of the x-ray beam; small, flared iliac wings; narrowed sacrosciatic notches; dysplastic (trident) acetabular roofs; and French telephone receiver–shaped femurs. At 22 weeks’ gestation in another affected fetus (B), a proportionally large skull, micromelia, and other findings similar to those in A are seen. C and D, Radiologic findings in thanatophoric dysplasia type II. An affected preterm fetus (C) exhibits findings similar to those in type I except for taller vertebral bodies and straighter femurs. Another affected fetus (D) also shows the same findings as in type I but with straighter femurs. E, Radiograph of affected infant with severe platyspondyly, anteriorly rounded vertebrae, straight femurs, and severely constricted skull base. F, Cloverleaf skull and almost straight femurs; otherwise, radiographic findings are similar to those in C. Note the ovoid lucency at the femoral necks in all cases.
1. Skull: proportionately large skull in relation to the body, narrow skull base, cloverleaf skull (type 1 only)
2. Thorax: long, narrow trunk; very short ribs; handlebar clavicles
3. Spine: severely flattened, small vertebral bodies with round anterior ends
4. Pelvis: small, flared iliac bones; very narrow sacrosciatic notches; flat, dysplastic acetabula
5. Extremities: generalized micromelia; ovoid lucency of femoral necks, round proximal femoral metaphyses with medial spike, curved long bones (type 1 only)
Achondroplasia
Except for the portions of the occipital bone that form the margin of the foramen magnum, all the bones of the skull are formed by membranous ossification.6 This results in an enlarged forehead and is termed frontal bossing. In contrast, the foramen magnum is narrowed and can cause cervicomedullary compression. Symptoms may include occipitocervical pain, ataxia, incontinence, apnea, paralysis, and respiratory arrest.7
Radiologic Findings (Fig. 133-6 and e-Fig. 133-7):
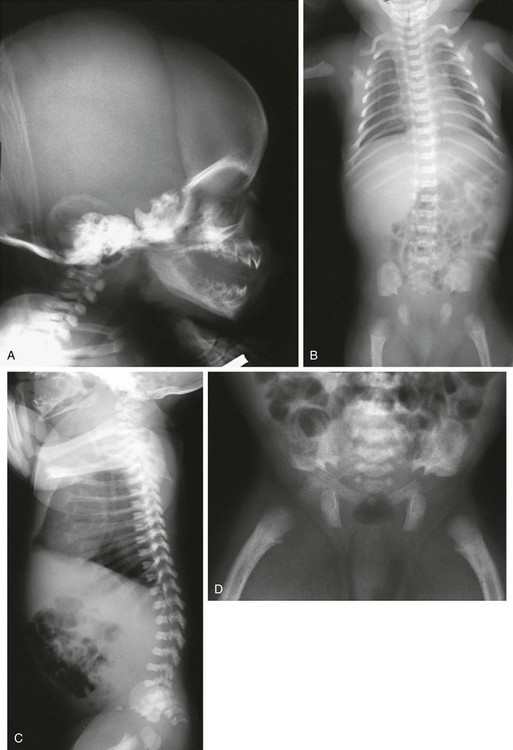
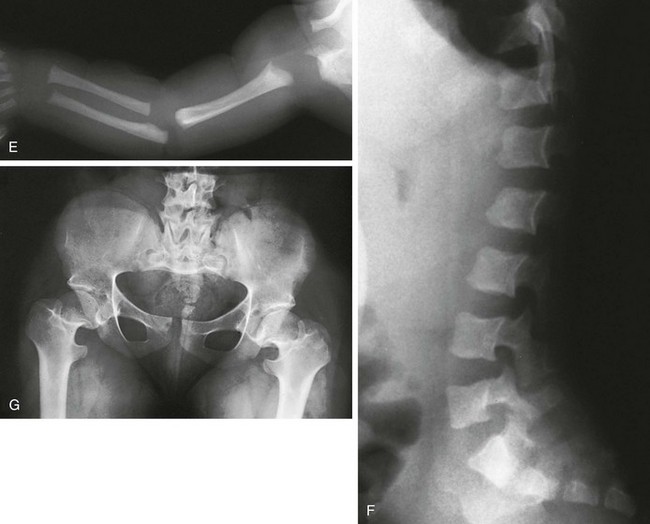
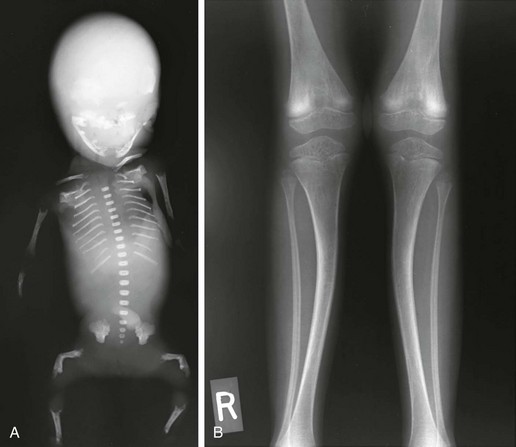
Figure 133-6 Achondroplasia.
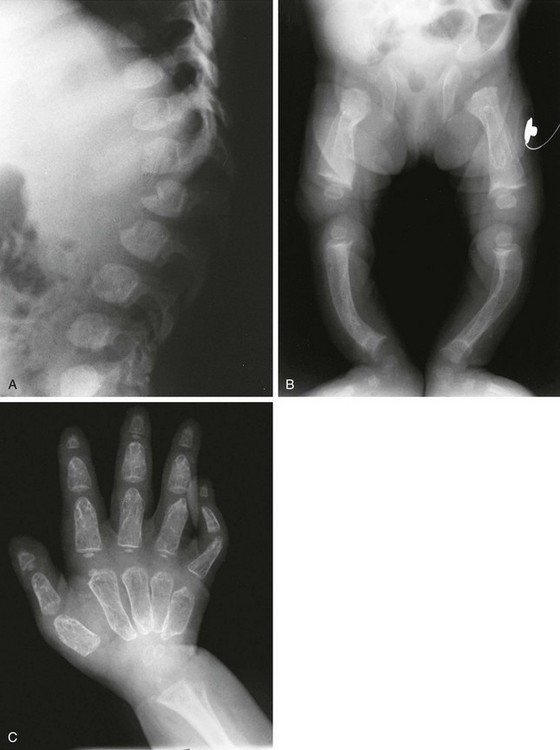
A to D, Radiographs of an affected newborn. A, Severe midface hypoplasia. B, Thorax: small thorax and short ribs. C, Thorax: short ribs with anterior scalloping and bullet-shaped vertebrae. D, Pelvis: rounded ilia (elephant ear–shaped) iliac bones, narrow sacrosciatic notches, flat acetabular roof, and proximal femoral ovoid lucency. Achondroplasia. E, Extremities: rhizomelia and mesomelia. F, Radiograph in an affected 1½-year-old with classic vertebrae with short pedicles, posterior scalloping, and somewhat short vertebral bodies. G, Radiograph in an affected woman with flat acetabular roofs, elephant ear–shaped iliac wings, and short femoral necks (compare with D). (G, From Silverman FN: Achondroplasia. Prog Pediatr Radiol. 1973;4:94-124.)
1. Skull: enlarged, with significant midface hypoplasia; hydrocephalus rarely present; small skull base with tight foramen magnum
2. Thorax: small; shortened and anteriorly splayed ribs
3. Spine: short pedicles with decreased interpediculate distance most marked in the lumbar spine moving downward; posterior vertebral body scalloping, gibbus deformity.
4. Pelvis: round iliac wings with lack of flaring (elephant ear–shaped), flattened acetabular roofs, narrow sacrosciatic notches with champagne glass shaped pelvic inlet
5. Extremities: rhizomelic micromelia
6. Hands: brachydactyly with trident hands
7. Knees: Central deep notch in growth plates (Chevron deformity)
8. Hips: proximal femoral ovoid lucency (infancy); hemispheric capital femoral epiphyses, short femoral necks
9. Legs: prominent tibial tubercle apophyseal region, fibula overgrowth
10. Arms: Cortical hyperostosis at deltoid insertion on anterolateral humerus
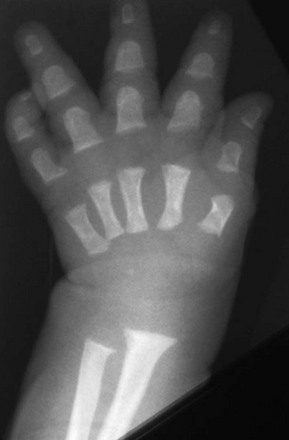
Hypochondroplasia
In hypochondroplasia radiographic and clinical findings are less severe compared with achondroplasia, and the diagnosis can be challenging. Stature is slightly shortened but is highly variable and may be normal given the range of normal stature in society. Radiographically, aside from shortening of the long bones, interpediculate narrowing is a very sensitive finding (Fig. 133-8).8
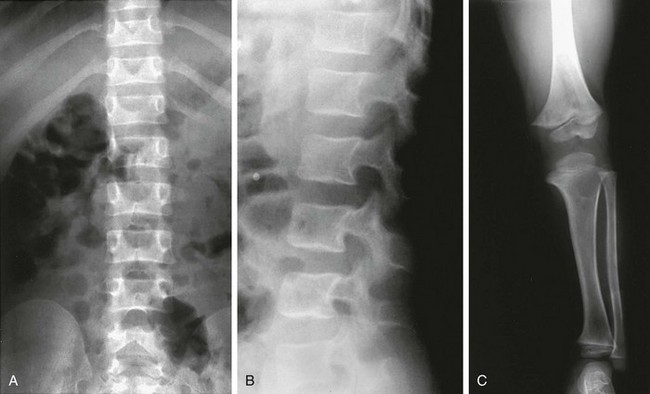
Type 2 Collagen Group
Overview: Defects in chromosome 12q13.1-13.3 result in abnormalities of type 2 collagen. Type 2 collagen is present in cartilaginous epiphyses and in the vitreous humor. Therefore, abnormalities of collagen 2 manifest with platyspondyly due to lack of normal growth at the ring epiphysis, a general delay in epiphyseal ossification, and myopia. Cleft palate completes the phenotypic picture.
Achondrogenesis type 2
Radiologic Findings (Fig. 133-9):
1. Skull: proportionately large
2. Thorax: very small and short ribs
3. Spine: almost complete lack of mineralization; cervical and sacral posterior elements also often unossified
4. Pelvis: small iliac wings with concave inferior and medial margins; absence of ischia, pubic bones, and sacral elements
5. Extremities: micromelia, mostly rhizomelia and mesomelia, with relative sparing of hands and feet; metaphyseal flaring; absence of talus and calcaneal ossification (epiphyseal equivalents)
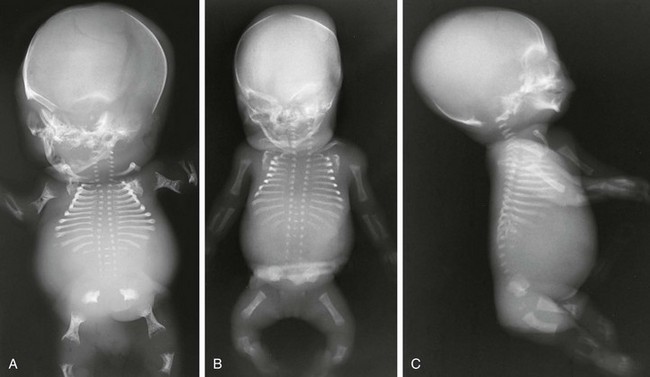
Spondyloepiphyseal Dysplasia Congenita
Radiologic Findings (Fig. 133-10):
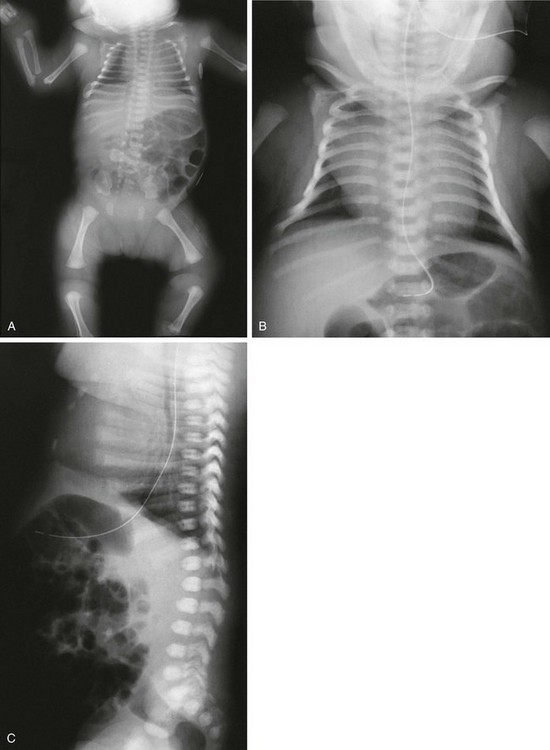
Figure 133-10 Spondyloepiphyseal dysplasia congenita.
A, Radiograph in an affected newborn with a small thorax, rounded iliac wings, vertical ischia, absence of pubic ossification, short femurs, and metaphyseal rounding of long bones. B, Radiograph in an affected newborn with bell-shaped chest, short ribs, and elongated clavicles. C, Radiograph in an affected newborn with moderately short ribs with mild anterior splaying and anteriorly rounded vertebral bodies with minimal flattening and no coronal clefts.
2. Spine: dorsally wedged or oval vertebral bodies (at birth); anteriorly rounded platyspondyly (later)
3. Pelvis: absent pubic ossification (at birth and during infancy), vertical ischia with short ilia
4. Extremities: normal tubulization with mild micromelia, significant generalized ossification delay (early) and hypoplastic-appearing or dysplastic epiphyses (later), unossified talus or calcaneus in the newborn, normal hands and feet with ossification delay (epiphyses or carpal, tarsal)
Kniest Dysplasia
The same delay in epiphyseal ossification is seen along with platyspondyly. Cloudlike dystrophic calcification is present in abnormally enlarged epiphyses as the child gets older. On magnetic resonance imaging (MRI), the areas of calcification have prolonged T2 values that are likely related to the degeneration of abnormal collagen matrix.9
Radiologic Findings (e-Fig. 133-11):
2. Spine: coronal clefts (at birth and during infancy), platyspondyly with endplate irregularity (later)
3. Extremities: dumbbell femurs; generalized ossification delay, epiphyses becoming hypoplastic or dysplastic and then later even mega-epiphyses, cloudlike irregular calcification in physeal plate regions (in late childhood and early adulthood); hands with bulbous joints (metaphyseal flaring or epiphyseal fragmentation) mimicking rheumatoid arthritis
Common Features in Type 11 Collagenopathy Group
• Similar to type 2 collagenopathy
• Myopia (except for Stickler type 3)
Abnormal Sulfation Group
The abnormal sulfation group is a molecularly defined group of disorders with a defect in the sulfate transporter gene on chromosome 5 coding for the diastrophic dysplasia sulfate transporter (DTDST) protein. This group comprises not only diastrophic dysplasia but also multiple epiphyseal dysplasia MED–multilayered patellae/brachydactyly/clubfeet, as well as achondrogenesis type IB and atelosteogenesis type II.10 These conditions are all autosomal recessive, and the severity of the phenotype is inversely related to the level of sulfation.11,12
Common Features in Abnormal Sulfation Group
Achondrogenesis Type I
Achondrogenesis type I is actually two separate disorders that appear almost identical radiographically. Achondrogenesis type IB belongs to this diastrophic dysplasia (molecular) group.11 In achondrogenesis type IA, a molecular or gene abnormality has not yet been identified. Clinically, the two types appear identical: proportionately large skull; micromelic, hydropic, pear-shaped trunk; polyhydramnios; and lethality.
Radiologic Findings (Fig. 133-12):
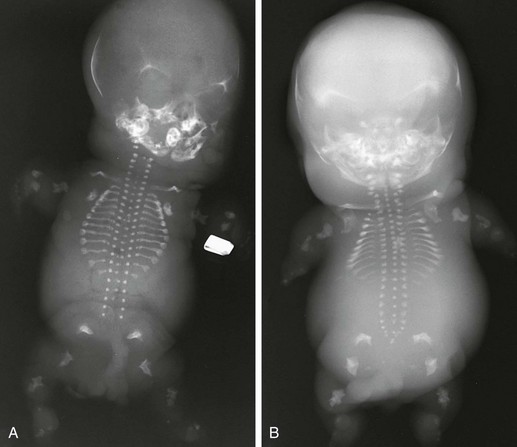
Figure 133-12 Achondrogenesis.
A, Radiograph from a stillborn infant with type IA achondrogenesis demonstrates a tiny thorax; short, anteriorly cupped ribs with beading; micromelia; wedged femurs; and poor to absent vertebral body ossification. B, Radiograph from a stillborn infant with type IB achondrogenesis. Findings are similar to those in type IA but with arched iliac wings, no rib beading, and trapezoidal femurs
1. Skull: decreased ossification
2. Thorax: tiny; very short ribs with anterior splaying
3. Spine: absent or minimal vertebral body ossification
4. Pelvis: short iliac bones with concave acetabular roofs, absent pubic (ischial) ossification
5. Extremities: severe micromelia with broadened ends of limbs, trapezoidal or wedge-shaped femurs
Diastrophic Dysplasia
Radiologic Findings (Fig. 133-13):
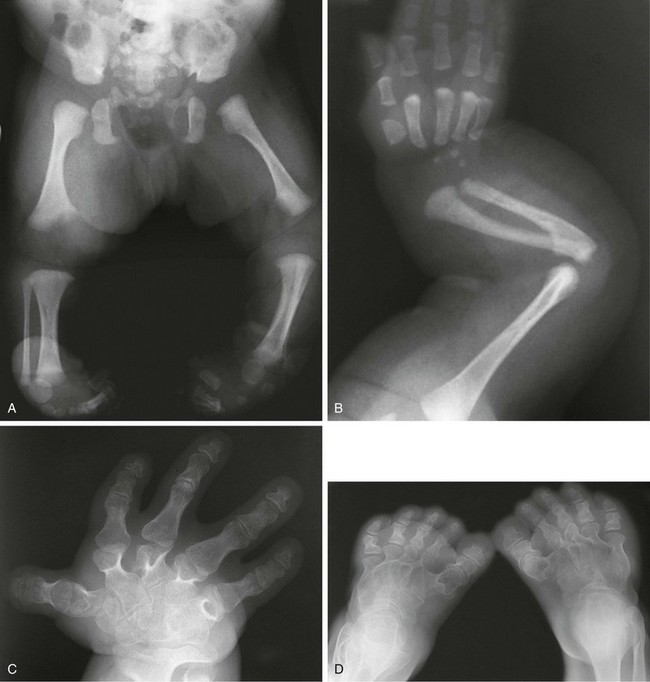
Figure 133-13 Diastrophic dysplasia.
A and B, Radiographs from an affected newborn. A, Lower extremities: rhizomelia and mesomelia and severe clubfeet deformities. B, Upper extremity: elbow dislocation and short, ovoid first metacarpal. C and D, Radiographs from an affected 21-year-old. C, Upper extremity: hitchhiker thumb, ovoid first metacarpal, brachydactyly, and irregular and extra carpal bones. D, Lower extremities: unusual clubfoot and twisted metatarsals
1. Head: ear pinna calcification, cleft or high arched palate
3. Spine: progressive scoliosis, kyphosis, upper cervical subluxation (odontoid hypoplasia), cervical kyphosis, posterior process clefting (cervical and sacral)
4. Extremities: often micromelia; short, thick tubular bones; generalized brachydactyly; short ovoid first metacarpal delta phalanx causing proximal placement of thumb (hitchhiker thumb), twisted metatarsals, accessory and irregular carpal bones; epiphyseal dysplasia with multiple joint contractures
5. Other sites: precocious costochondral and laryngeal area cartilage calcification; multiple sternal and patella centers
Multiple Epiphyseal Dysplasia: Multilayered Patellae/Brachydactyly/Clubfeet
Radiologic Findings: Extremities show the following: epiphyseal dysplasia, especially at hips (half- or quarter-moon–shaped); double-layered or multilayered patella (visible on lateral knee radiograph); mild brachydactyly; clubfeet or twisted metatarsals; mildly shortened long bones, some with mild undertubulation.
Filamin Group
The filamin group combines a wide group of dysplasias that have in common an abnormality in the number and configuration of carpal, tarsal, and vertebral bones with joint dislocations. The identification of the group is another triumph in the study of molecular genetics, as it reclassifies correctly a group of disorders described as “syndromes” within a common framework of genetically determined diseases no different from other skeletal dysplasias.13,14 The group includes oto-palato-digital (OPD) syndrome types 1 and 2, Larsen syndrome, frontometaphyseal dysplasia, Melnick-Needles osteodysplasty, and spondylo-carpal-tarsal synostosis.
OPD Syndrome
Radiographic Findings in OPD (e-Fig. 133-14):
1. Head: prominent supraorbital ridge
2. Spine: small pedicles with wide interpediculate distance
3. Extremities: accessory carpal bones, double ossification center of lunate, fusion of carpal bones especially trapezoid and scaphoid, fusion of the second metacarpal to trapezoid in adolescence, similar findings in feet; short first metatarsal and phalanges of great toe, and short and wide distal phalanx of thumb; radial head dislocation
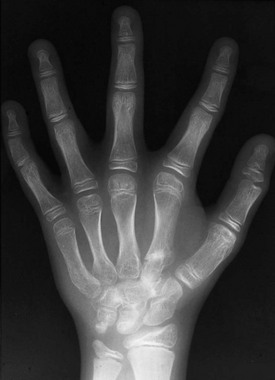
e-Figure 133-14 Oto-palato-digital syndrome.
The radiograph at right shows typical findings of fusion of the accessory ossification centers of the second metacarpal with the adjacent deformed carpal bones. Widening and abnormal tubulation of the metacarpals are seen. The distal phalanx of the thumb is short with a coned-shaped epiphysis. Clinodactyly is present.
Larsen Syndrome
In Larsen syndrome, multiple joint dislocations are present. In keeping with the common theme of filamin abnormalities, supernumerary carpal bones are common along with other digital changes. A doubled calcaneal ossification center is a helpful clue to accurate diagnosis. Scoliosis is common. This is a filamin type B abnormality. A similar filamin type B abnormality causes spondylo-carpal-tarsal synostosis syndrome, whose name describes the pattern of skeletal involvement.15
Radiographic Features (Fig. 133-15 and e-Fig. 133-16):
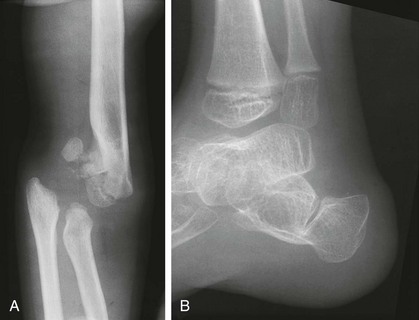
Figure 133-15 A 5-year-old girl with Larsen syndrome.
A, Anteroposterior elbow radiograph shows chronic dislocation with epiphyseal deformities. B, Lateral radiograph of an ankle in a 5-year-old boy shows a bifid calcaneus and deformity of the talus and distal tibial epiphysis.
1. Spine: cervical spine kyphosis
2. Extremities: multiple joint dislocations, double or triple calcaneal ossification center, accessory carpal bones, broad irregular metacarpals
TRPV4 Group
TRPV4 (transient receptor potential cation channel, subfamily 5, member 4) is a calcium permeable nonselective cation channel that appears to play an important role in chondrogenesis. This channelopathy is also the cause of several other nonskeletal syndromes such as Charcot Marie-Tooth disease, scapula-peroneal spinal muscular atrophy, and congenital distal spinal muscular atrophy.16,17
Metatropic Dysplasia
Radiologic Findings (Fig. 133-17):
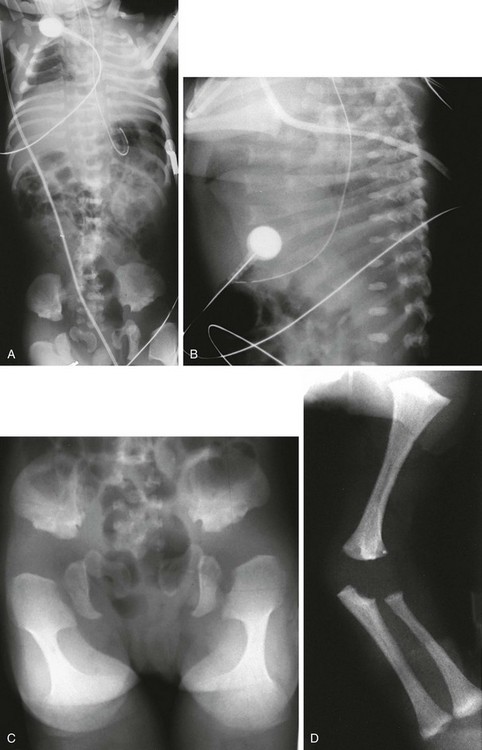
Figure 133-17 A newborn with metatropic dysplasia.
A, Thorax: long trunk and small chest. B, Spine: dense wafer vertebral bodies and short ribs with anterior splaying. C, Pelvis: short iliac wings, narrow sciatic notches, irregular acetabular roofs, and rounded, enlarged proximal femoral metaphyses (halberd shaped) with markedly flared distal metaphyses (trumpet-shaped) D, Upper extremities: flared proximal humeral and distal radial and ulnar metaphyses; shortened long bones.
2. Spine: dense wafer vertebral bodies (newborns), reconstituted platyspondyly (children and adults), scoliosis (adults)
3. Pelvis and hips: short, squared iliac wings; flat, irregular acetabular roof; narrow sacrosciatic notches; widening of proximal femoral metaphyses with a pronounced medial aspect about lesser trochanter (halberd (hunting ax)–shaped proximal femurs)
4. Extremities: flared metaphyses (trumpet shaped), with epiphyseal dysplasia and shortening (dumbbell shape).
Spondylometaphyseal Dysplasia Koslowski Type
Characteristic radiographic findings (e-Fig. 133-18) include severe platyspondyly with overfaced pedicles. The extremities show sclerosis, flaring, and irregularity at the metaphyses. Carpal bone ossification is delayed and may not be apparent until age 5 to 6 years.
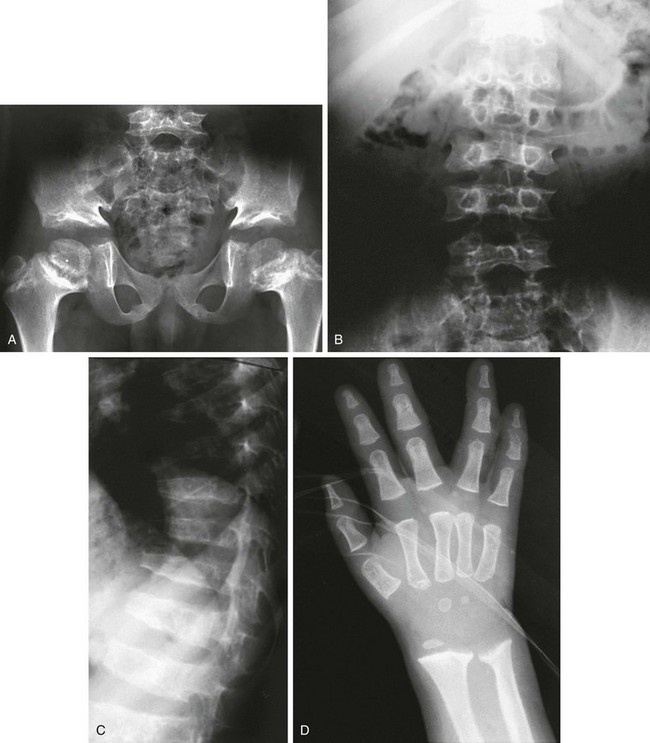
e-Figure 133-18 Kozlowski-type spondylometaphyseal dysplasia.
A to C, Radiographs from an affected 8-year-old. A, Pelvis: narrow sacrosciatic notches, ossification defect (acetabular roofs), and widened epiphyseal plates. B and C, Spine: overfaced vertebral bodies (“open staircase,” or closely set pedicles) and flattened, irregular vertebral bodies (platyspondyly). D, An affected 2-year-old with metaphyseal widening and irregularity at the wrist and short tubular bones of the hands, with almost no carpal bone ossification. Note the similarities in proximal femurs to metatropic dysplasia (see Fig. 133-17).
Short Rib–Polydactyly Group
Some members of this group are now known to be ciliopathies.18 Several types of SRP and types of ATD are caused by mutations in genes encoding for normal dynein heavy chains or other aspects of the ciliogenesis.19,20 It is interesting to note that situs abnormalities are a feature of these dysplasias attesting to the important role cilial transport plays in body situs. In patients with primary cilial dyskinesia (immotile cilia syndrome), approximately 50% have situs inversus (Kartagener syndrome).
Short Rib–Polydactyly Dysplasia
Radiologic Findings (Fig. 133-19):
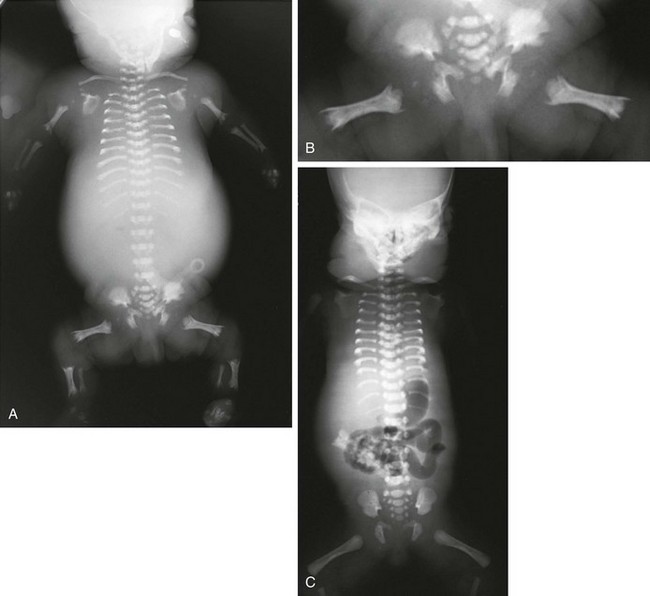
Figure 133-19 Short rib–polydactyly dysplasia.
A, Radiograph from a stillborn infant with type I/III form, demonstrating very short ribs and, handlebar clavicles. B, Magnified, coned view of pelvis shows hypoplastic pelvis with notched acetabula, and metaphyseal-spiked femurs. C, Radiograph from a stillborn infant with short rib–polydactyly dysplasia type II. Findings are similar to those in type I/III but with round-ended femurs and hypoplastic acetabula.
1. Thorax: small; very short horizontal ribs
2. Spine: relatively normally shaped
3. Pelvis: small, dysplastic ilia
4. Extremities: micromelia; medial and lateral spurs at metaphyses; ovoid or tiny, normal-shaped tibias; severe brachydactyly with hypoplastic middle and distal phalanges; polydactyly commonly present
Asphyxiating Thoracic Dysplasia (Jeune Syndrome)
ATD is a genetically heterogeneous disorder with a mixed prognosis. Many affected patients die in the perinatal period from respiratory complications related to a small chest. Survivors may die from renal complications (progressive nephropathy) later in life. Other internal organs may also be involved. Sometimes, postaxial polydactyly is present. Definite radiographic (but not clinical) similarities to chondroectodermal dysplasia are evident. An allelic relationship has been considered but remains unproven.21 Some cases are so alike radiologically that they are best termed ATD/Ellis–van Creveld syndrome complex.
Radiologic Findings (Fig. 133-20):
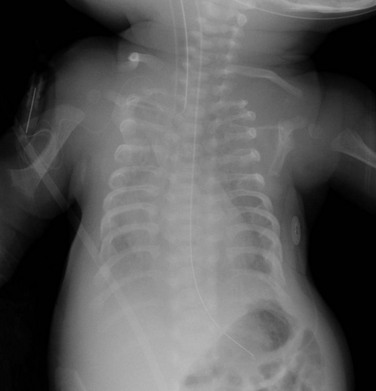
Figure 133-20 Newborn with asphyxiating thoracic dysplasia.
Note the short ribs (although not as short as ribs in short rib–polydactyly (see Fig. 133-19) and metaphyseal flaring and irregularity in proximal humeri.
1. Thorax: long and barrel-shaped, handlebar clavicles, short horizontal ribs with bulbous anterior ends
3. Pelvis: small; short, flared iliac wings; trident acetabular roof; narrowed sacrosciatic notches
4. Extremities: generalized shortening, precocious proximal femoral epiphyseal ossification, cone-shaped epiphyses in hands, metaphyseal flaring with irregularity(more pronounced in child)
Chondroectodermal Dysplasia (Ellis–van Creveld Syndrome)
Radiologic Findings (e-Fig. 133-21):
1. Thorax: small; moderately short ribs
2. Pelvis: small; short, flared iliac wings; trident acetabula; narrowed sacrosciatic notches
4. Extremities: generalized shortening with more mesomelia and acromelia; premature ossification of capital femoral epiphyses; delayed ossification of proximal tibial epiphyses; humeral and femoral bowing; exostosis of proximal/medial portion of tibia
5. Hands: characteristic postaxial polydactyly, capitate/hamate (and other carpal) fusions, extra carpal bone, cone-shaped epiphyses
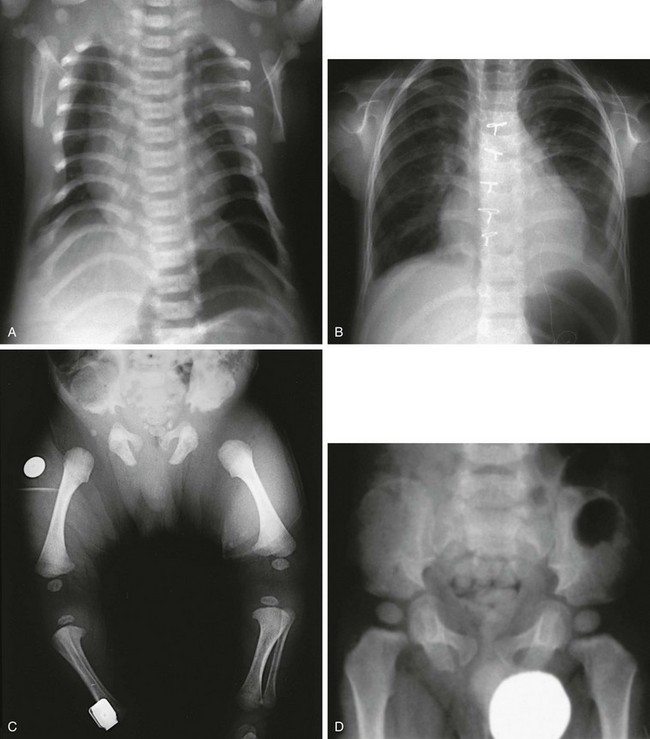
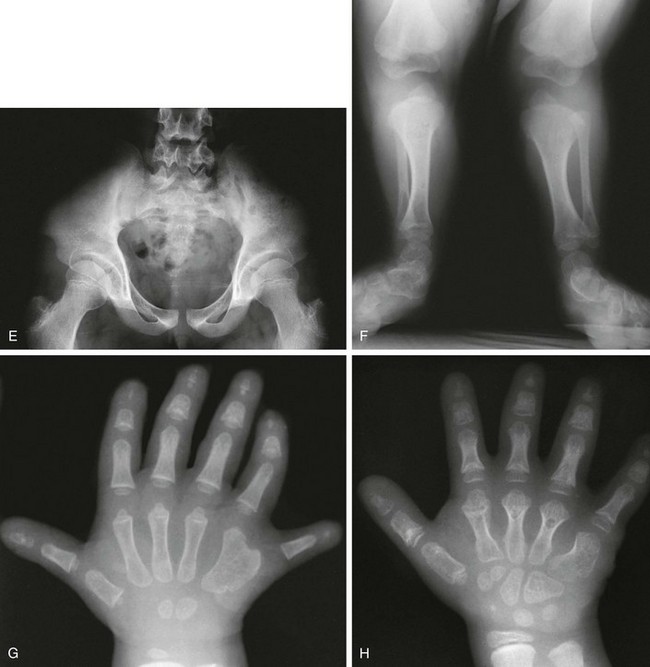
e-Figure 133-21 Chondroectodermal dysplasia (Ellis–van Creveld syndrome).
A, Radiograph from an affected 3-month-old infant with a narrow thorax, short ribs, anterior rib flaring, and a normal spine. B, Radiograph from an affected 5-year-old with similar chest configuration; cardiomegaly is present, with sternal sutures from a primum defect repair. C, Radiograph from an affected 3-month-old with short, flared iliac wings; trident acetabula; and mesomelia and rhizomelia. D and E, Radiographs from an affected child at 2½ (D) and 12 years of age (E), showing near normalization of pelvis. Chondroectodermal dysplasia (Ellis–van Creveld syndrome). F, Radiograph from an affected 2½-year-old with remarkable mesomelia. G, Radiograph from an affected 2-year-old with characteristic hands: polydactyly, middle and distal phalangeal hypoplasia, cone-shaped epiphyses of middle phalanges, and beginning carpal coalition. H, Radiograph from the same child as in G at 6 years of age. After polydactyly repair, a residual wide fifth metacarpal is seen; other findings are similar except that no carpal coalition is present.
Multiple Epiphyseal Dysplasia and Pseudoachondroplasia Group
Multiple Epiphyseal Dysplasia
Radiologic Findings (Fig. 133-22 and e-Fig. 133-23):
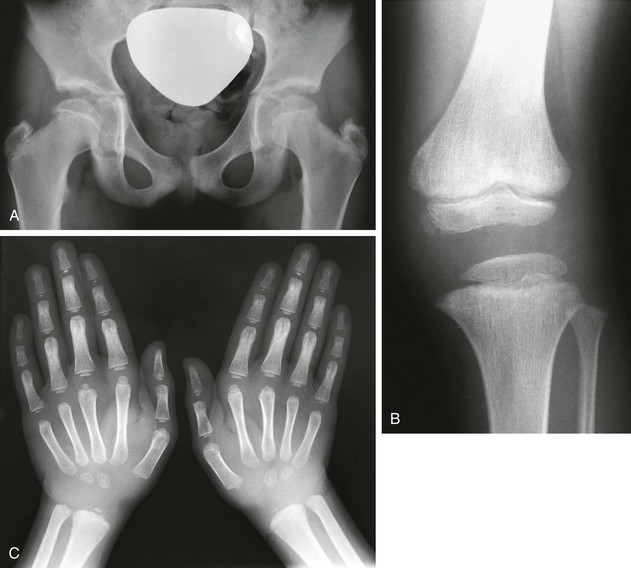
Figure 133-22 Fairbanks-type multiple epiphyseal dysplasia.
A, Radiograph from an affected 10-year-old with small ossified proximal femoral epiphyses (ossification defect). B and C, Radiographs from an affected 6-year-old. B, Similar epiphyseal ossification defects in the knee. C, Small epiphyses of the short tubular bones of the hands and carpal ossification delay (epiphyseal equivalents), but no brachydactyly.
1. Spine: in young adults, disk herniations into vertebral end plates (Schmorl nodes)
2. Extremities: small, irregular, flattened ossification centers (epiphyses); small, irregular carpal (and tarsal) centers
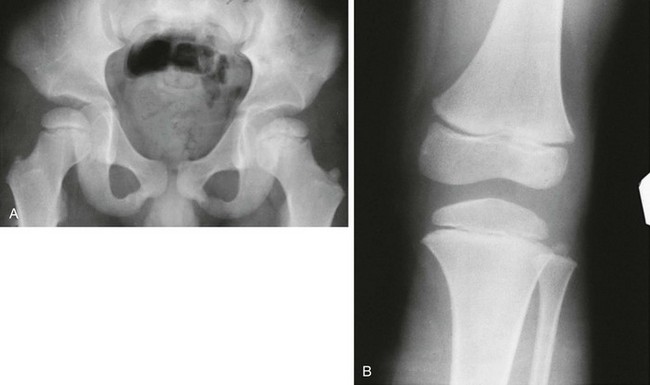
Pseudoachondroplasia
Radiologic Findings (Fig. 133-24):
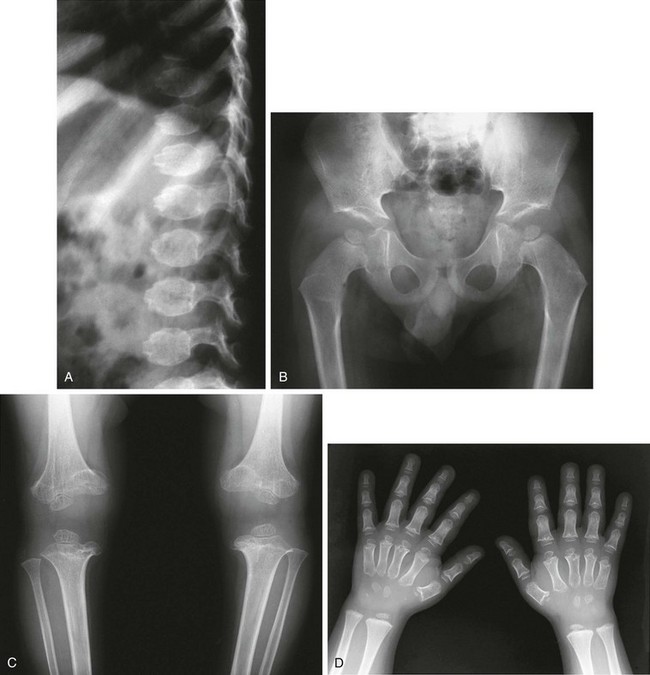
Figure 133-24 Pseudoachondroplasia.
Radiologic findings at ages 3 years (A and C) and 4 years (B and D). A, Central anterior tonguing and superior and inferior rounding of vertebral bodies. B, Acetabular roof hypoplasia and mini-epiphyses. C, Small knee epiphyses and metaphyseal widening with ossification defects. D, Proximal metacarpal rounding, small epiphyseal centers, metaphyseal widening and irregularity, and carpal ossification delay.
2. Thorax: mild anterior rib widening
3. Spine: superiorly and inferiorly rounded vertebral bodies, anterior central tongue (unossified ring epiphyses), normalization of vertebrae (later)
4. Pelvis: rounded iliac wings; hypoplastic, poorly formed acetabular roofs
5. Extremities: mini-epiphyses in the hips, moderate to severe generalized epiphyseal “dysplasia” (small, irregular, poorly ossified), mushroomlike metaphyseal widening and irregularity in the knees, proximally rounded metacarpals with mini-epiphyses in hands, irregular carpal and tarsal bones
Metaphyseal Chondrodysplasia Group
Jansen-Type Metaphyseal Chondrodysplasia
This is the severest form of MCD. The presentation is in the neonatal period or during late infancy, with marked short stature and a waddling gait. This is a distinct autosomal-dominant disorder with an abnormality in a parathyroid receptor gene (PTHR), leading to hypercalcemia and its complications.22 However, the radiographic findings in the skeleton are not those of typical hyperparathyroidism or hypoparathyroidism.23
Radiologic Findings (Fig. 133-25):
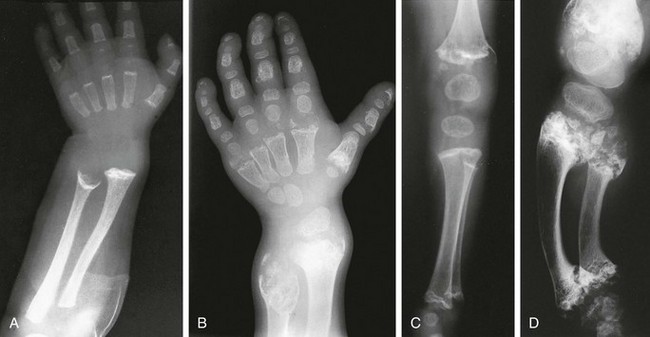
Figure 133-25 Radiographic findings in Jansen-type metaphyseal chondrodysplasia.
A, At 1 year, severe metaphyseal cupping and splaying are present at the wrists and also in the hand bones. B, At 7 years, increasing metaphyseal change is seen at the wrists with enlarged epiphysis; enlarged epiphyses with wide epiphyseal plates are also present in the hands. C, At 1 year, severe metaphyseal irregularities at the knees and ankles (femur, tibia, and fibula) and enlarged, rounded epiphyses are present. D, At 7 years, severely fragmented, sclerotic metaphyses, wide epiphyseal plates, and enlarged epiphyses are present.
Schmid-Type Metaphyseal Chondrodysplasia
Radiologic Findings (Fig. 133-26):
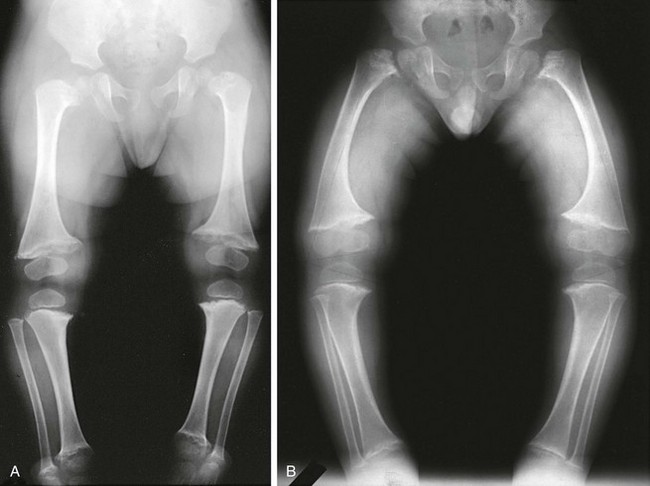
Figure 133-26 Schmid-type metaphyseal chondrodysplasia.
A, Radiograph from an affected 19-month-old with severe coxa vara and moderate metaphyseal changes (cupping irregularity, widening) at the knees (and, not pictured here, ankles). B, Radiograph from an affected 3-year-old with coxa vara, genu varum, and moderate metaphyseal changes at hips and knees (widening, irregularity).
McKusick-Type Metaphyseal Chondrodysplasia
Cartilage-hair hypoplasia, as this entity is also known, is an autosomal recessive disorder. The genetic defect is at the 9p region (RMRP gene), with a high frequency among the Amish and Finnish populations.24–26 The presentation is of variable short-limbed dwarfism in early childhood. Significant clinical features indicate the diagnosis and are important for medical management: sparse, thin, light-colored hair; Hirschsprung disease; immunological problems; and increased incidence of malignancy.
Radiologic Findings (e-Fig. 133-27):
1. Thorax: anterior rib widening/flaring
2. Spine: slightly small square vertebral bodies
3. Extremities: flaring, cupping, and fragmentation of metaphyses (especially knees), hips usually spared; brachydactyly with cone shaped epiphysis
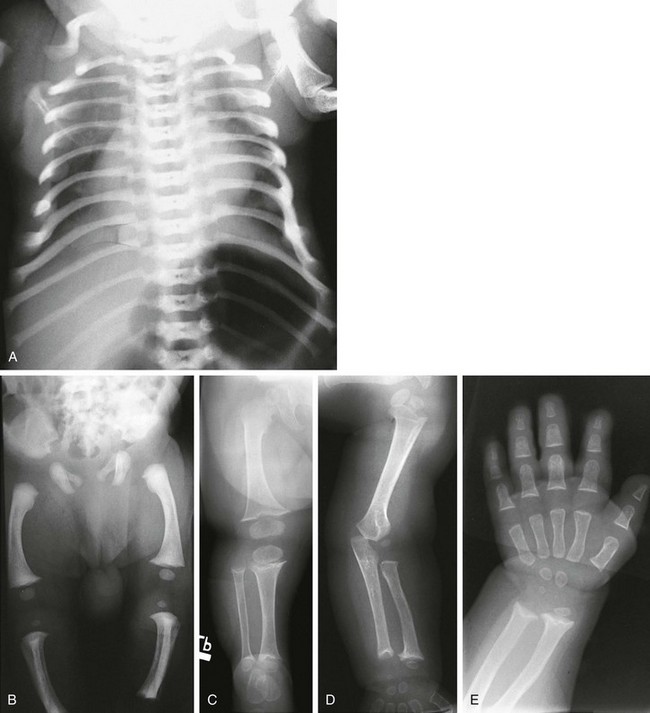
e-Figure 133-27 McKusick-type metaphyseal chondrodysplasia (cartilage-hair hypoplasia).
A and B, Radiographs from an affected newborn. A, Thorax: anterior rib end widening and cupping. B, Lower extremities: mild femoral bowing with minimal distal femoral flare. C and D, Radiographic findings in late infancy. C, Lower extremity: metaphyseal flaring and irregularity at the knees and ankles (but the hips are spared). D, Upper extremity: similar metaphyseal changes at the wrists (the shoulders are spared). E, At 6 months of age, metaphyseal widening and cupping of the short tubular bones of the hand, with similar metaphyseal changes in the wrist, are seen.
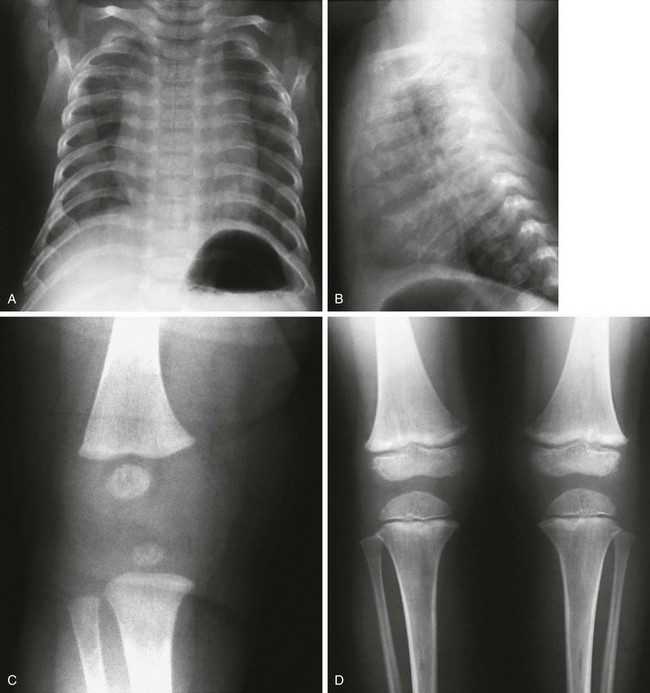
Shwachman-Diamond Dysplasia
Radiologic Findings (e-Fig. 133-28):
1. Thorax: anterior rib irregularity or splaying
2. Extremities: metaphyseal irregularity and sclerosis, especially at knees and hips
3. Gastrointestinal: malabsorption pattern on small bowel examination; lipomatosis of pancreas
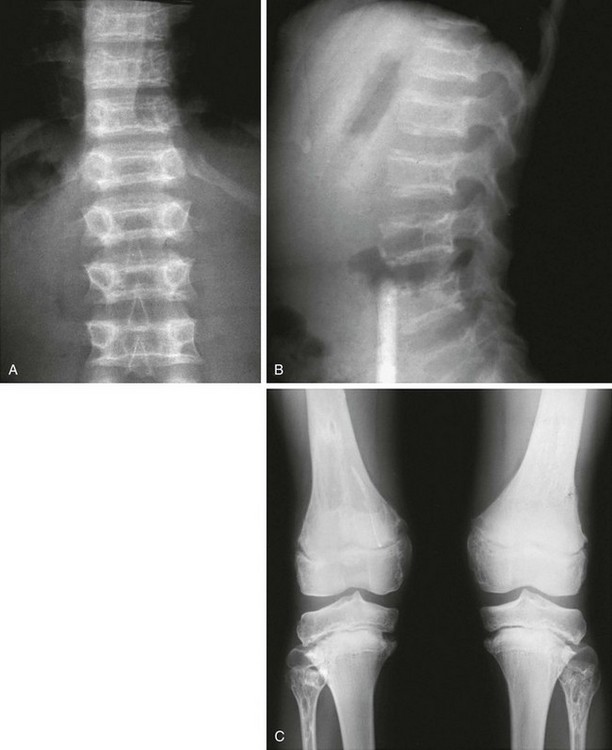
Spondylometaphyseal Dysplasia Group
Radiologic Findings (e-Fig. 133-29):
1. Spine: severe platyspondyly with endplate irregularity
2. Extremities: enchondromatosis in long bones, but rarely involving hands and feet
Acromelic/Acromesomelic Dysplasia Group
Trichorhinophalangeal Syndrome Types I and II
Both of these disorders have been located on the long arm of chromosome 8. The gene implicated in TRPS type I (also known as Giedion syndrome) is TRPS1. TRPS type II (also known as Langer-Giedion syndrome) is slightly more complicated. TRPS type II is the result of a contiguous gene abnormality resulting from the loss of not only TRPS1 but also EXT1, a major cause of multiple hereditary exostoses located distal to TRPS1. TRPS type I is autosomal dominant, whereas most cases of TRPS type II are sporadic. The clinical manifestations of both disorders include mild short stature; sparse, slow-growing hair; pear-shaped nose (“hose nose”); and short, crooked fingers. The contiguous gene abnormality explains the added features in TRPS type II, which include multiple exostoses and mental retardation.27
Radiologic Findings (Fig. 133-30):
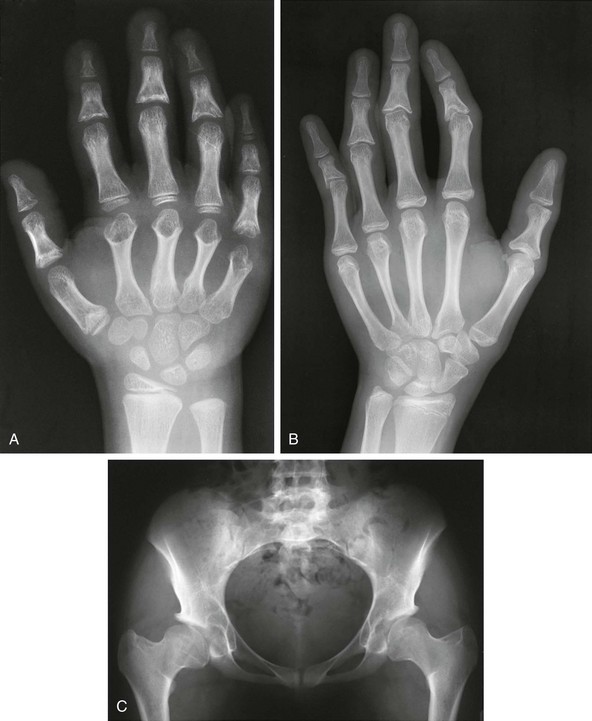
Figure 133-30 Radiographic findings in trichorhinophalangeal syndrome type I.
A, In an affected 8-year-old, cone-shaped epiphyses involving the first metacarpal, the proximal fifth phalanx, and all middle phalanges and metacarpals in the second through fifth digits (early fusion) are present. B, The radiograph from an affected 18-year-old shows similar changes. C, In an affected young adult, coxa vara and small capital femoral epiphyses are present.
Acromesomelic Dysplasia of Maroteaux
Radiologic Findings (e-Fig. 133-31):
1. Spine: oval vertebral bodies (early), anterior beaking and posterior wedging (later), gibbus, kyphoscoliosis, or all of these ultimately
2. Extremities: shortening of all tubular bones, especially radius or ulna and tibia or fibula; brachydactyly of hands and feet with cone-shaped epiphyses but with relatively large great toes
e-Figure 133-31 Radiographs from a 2½-year-old with acromesomelic dysplasia of Maroteaux.
A, Upper extremities: primarily mesomelia and acromelia (short radius and ulna, short tubular bones of the hand). B, Spine: almost ovoid vertebral bodies with anterior central spike. C, Lower extremities: mild rhizomelia but primarily mesomelia.
Mesomelic Dysplasia Group
Dyschondrosteosis
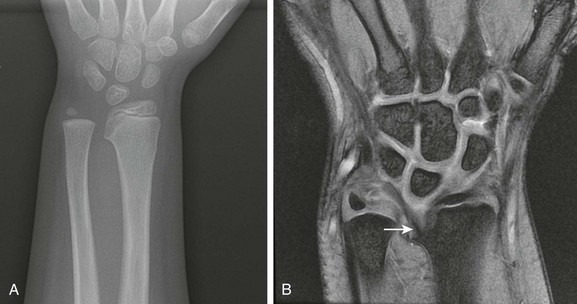
This skeletal dysplasia, also known as Leri-Weill syndrome, is an autosomal-dominant condition. It consists of a pseudoautosomal homeobox gene (SHOX gene) found on the short arm of the X chromosome. Dyschondrosteosis manifests with mild to moderate short stature, usually with both forearm and calf shortening. Madelung deformity is the major marker for this disease. Interestingly, Madelung deformity is also common in Turner syndrome because of the lack of two copies of SHOX since only one X chromosome is present (see discussion of Turner syndrome).
MRI is important here from a therapeutic point of view. A thickened ligament has been described by Vickers and Nielsen, which appears to tether the medial radial physis.28 It can be found on the volar side of the joint and may be an abnormally thickened volar radiolunotriquetral ligament. Operative lysis of this structure if performed early can ameliorate the Madelung-type deformity. Radiographically, the ligament should be suspected when a triangular lucency is seen at the medial aspect of the distal radial metaphysis. On MRI, the ligament is clearly visible as a thick hypointense band of tissue originating at the medial radial physis.29
Chondrodysplasia Punctata Group
Rhizomelic Chondrodysplasia Punctata
Bent-Bone Dysplasia Group
Campomelic Dysplasia
Radiologic Findings (Fig. 133-34):
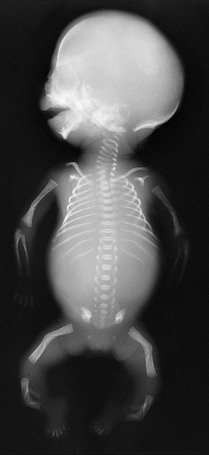
Figure 133-34 A fetus of 21 weeks’ gestation with campomelic dysplasia.
Radiographic findings include a large skull with a small face; hypoplastic or absent scapular bodies; 11 ribs; poorly ossified thoracic pedicles; tall, narrow iliac wings; and short extremities with proportionately long, bent femurs.
1. Skull: enlarged, narrow with a small face
2. Thorax: mildly short ribs, numbering 11; severe hypoplasia of the bodies of the scapulae
3. Spine: nonossification of thoracic pedicles, cervical kyphosis, hypoplasia of cervical vertebral bodies
4. Pelvis: narrow, tall iliac wings
5. Extremities: proportionately long, bowed femurs with shortened bowed tibias; shortened upper extremity long bones
Disorders of Increased Bone Density Without Modification of Bone Shape
Osteopetrosis
Radiologic Findings (e-Fig. 133-35 and Fig. 133-36):
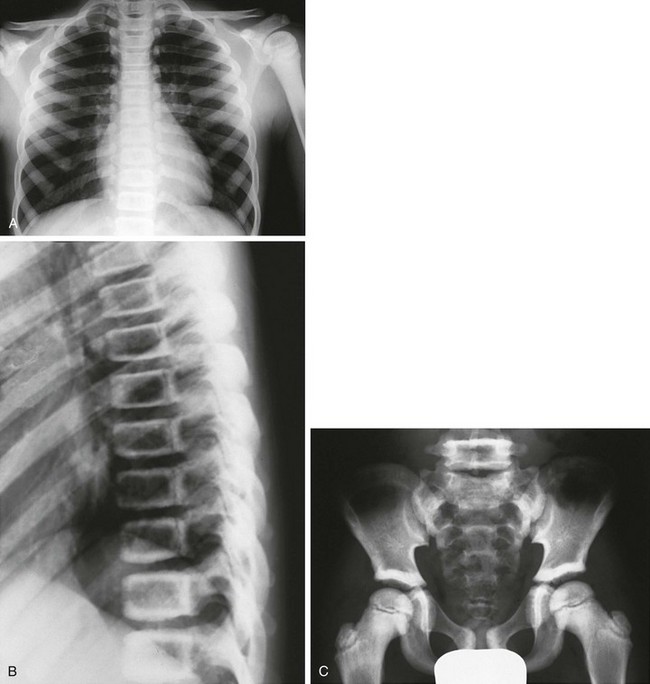
Figure 133-36 An 8-year-old with delayed-type osteopetrosis (autosomal dominant).
A, Radiographs reveal dense thoracic bones without medullary encroachment of left humerus. B, “Sandwich,” almost “picture frame” vertebral bodies (dense outer borders). C, Increased bone density outlining ileum, including supraacetabular regions, and pubic symphysis region, as well as dense proximal femoral epiphyses and femoral necks with sparing of lower medullary space.
1. Generalized increased bone density
2. Skull: thick and dense, especially at the base
3. Thorax: splayed anterior ribs
4. Spine: “sandwich” vertebral bodies, “picture frame” vertebral bodies
5. Extremities: splayed metaphyses, bone-within-bone configuration, dense metaphyseal bands
6. Central nervous system: carbonic anhydrase II deficiency has diffuse dense cerebral calcifications
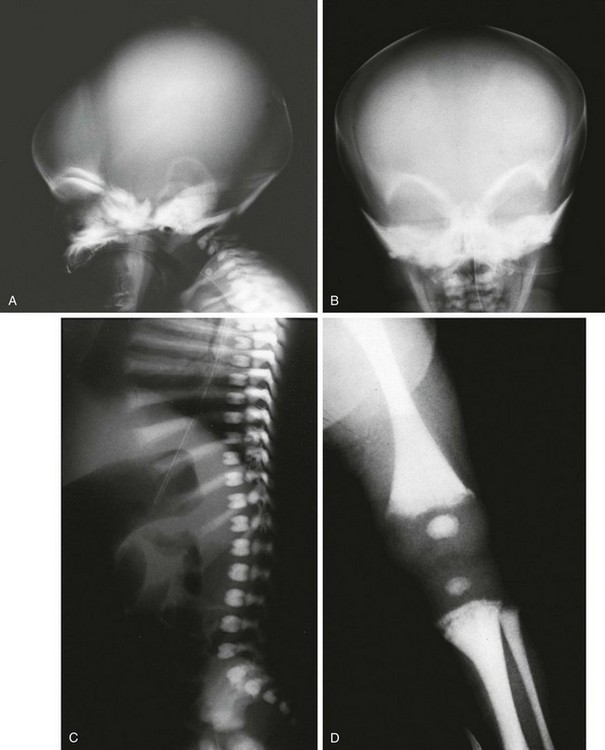
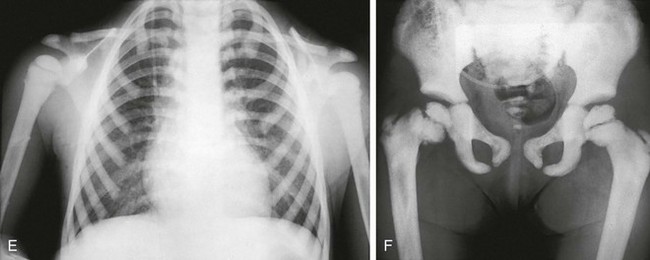
e-Figure 133-35 Precocious-type osteopetrosis (autosomal recessive).
A to C, Radiographs from a newborn. A and B, Dense skull and face, especially skull base. C, Dense spine and ribs. D, At 3 months, very dense long bones, medullary obliteration, and frayed metaphyses (rickets) are present. Precocious-type osteopetrosis (autosomal recessive). E and F, Radiographic findings at 4 years. E, Dense thorax (ribs, clavicles, scapulas) and upper extremity long bones (note: less medullary obliteration). F, Dense pelvis and lower extremity long bones (including capital femoral epiphyses), and bilateral pathologic femoral neck fractures.
Pyknodysostosis
Pyknodysostosis is an autosomal-recessive disorder that often manifests in infancy. Clinical findings include short-limbed dwarfism, micrognathia, fractures, and short fingertips. The impressionist painter Toulouse-Lautrec likely had this condition.30
Radiologic Findings (Fig. 133-37):
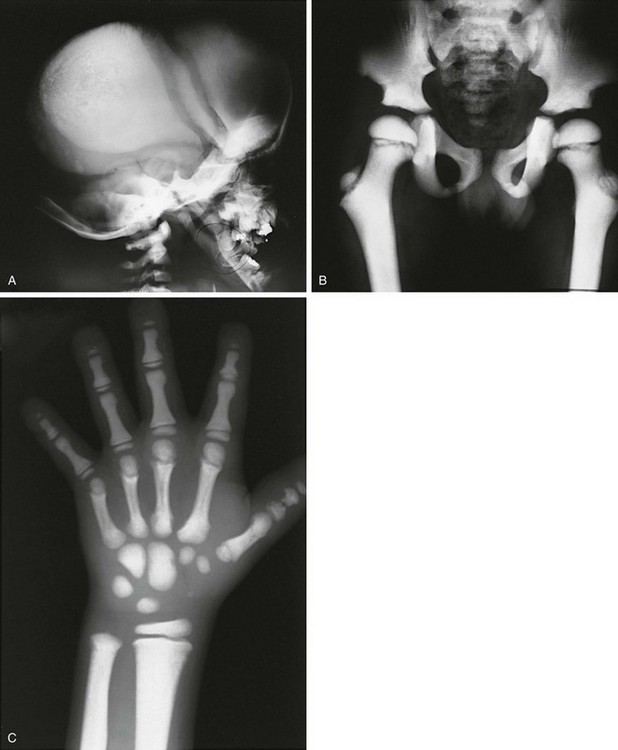
Figure 133-37 An 8-year-old with pyknodysostosis.
A, Radiographis reveal dense skull convexity and base, widely separated sutures with open fontanel, and absence of mandibular angle. B, Hip and pelvis: generalized increased bone density with long, overmodeled (resorbed) femoral necks. C, Hands: dense bones, overmodeled metacarpals and phalanges, phalangeal tuft resorption.
Osteopoikilosis
This is an autosomal-dominant condition caused by mutations in LEMD3.31 The gene function and its relationship to this disease are not clear. It is often asymptomatic and identified on routine radiographs. These lesions often show increased uptake on bone scans. When skin lesions of dermatofibrosis are also present, the combination is called Buschke-Ollendorff syndrome.
Osteopathia Striata
Radiologic Findings (Fig. 133-39):
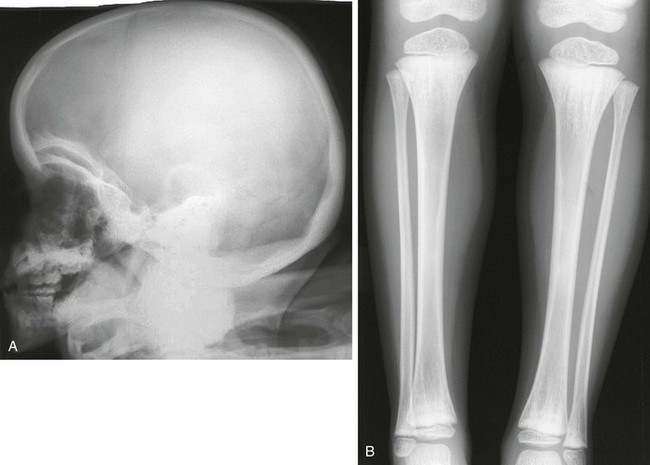
Figure 133-39 Radiographic findings in osteopathia striata with cranial sclerosis.
A, Lateral skull film revealing dense thickening of the calvaria with increased basilar and orbital sclerosis. B, An anteroposterior radiograph of the lower extremity reveals linear striations of the metadiaphyseal regions of both ends of the tibias and fibulas.
Melorheostosis
Increased Bone Density Group with Metaphyseal and Diaphyseal Involvement
Craniodiaphyseal Dysplasia
Radiologic Findings (Fig. 133-41):
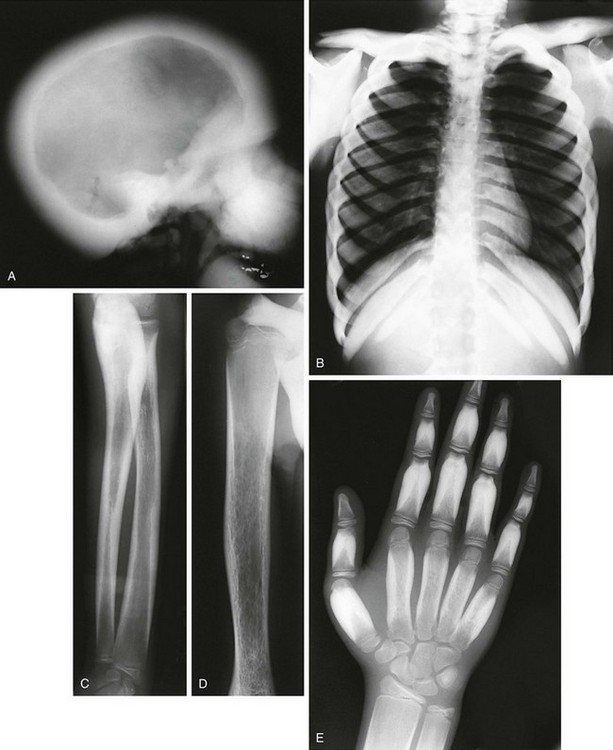
Figure 133-41 Radiographic findings in a 13-year-old with craniodiaphyseal dysplasia.
A, Extremely dense bone filling in the facial region and thickening the diploic space. B, Diffusely dense, thickened ribs and clavicles. C and D, Diffuse cortical long bone diaphyseal thickening and diaphyseal undermodeling. E, “Flame” sclerosis (cortical thickening) of the tubular bones of the hand.
1. Skull: marked thickening and sclerosis of calvaria and facial bones, obliteration of foramina and sinuses
2. Thorax: diffusely widened, sclerotic ribs and clavicles
3. Extremities: straightened, undermodeled long bone with diaphyseal widening with metaphyseal sparing; “flame” sclerosis (cortical thickening) of the short-tubular bones (hands)
Craniometaphyseal Dysplasia
Pyle Dysplasia
Radiologic Findings (Fig. 133-43):
Figure 133-43 Radiographic findings in pyle dysplasia.
A and B, In an affected 17-year-old, markedly broad, undertubulated distal femurs (A) and markedly broad, undertubulated proximal and distal tibias with mild medial bowing proximally (B). C, In a different patient, distal flaring of metacarpals and proximal flaring of phalanges are seen. The distal radial and ulnar metaphyses are broadened.
1. Skull: mild skull and facial involvement, minimal base-of-skull sclerosis, prominent supraorbital ridging
2. Thorax: mildly thickened clavicles and ribs, mild platyspondyly
3. Pelvis: thickened ischium and pubis
4. Extremities: marked undertubulation of long bones, especially distal femurs (Erlenmeyer flask deformity); distal flaring of metacarpals and proximal flaring of phalanges
Osteogenesis Imperfecta and Decreased Bone Density
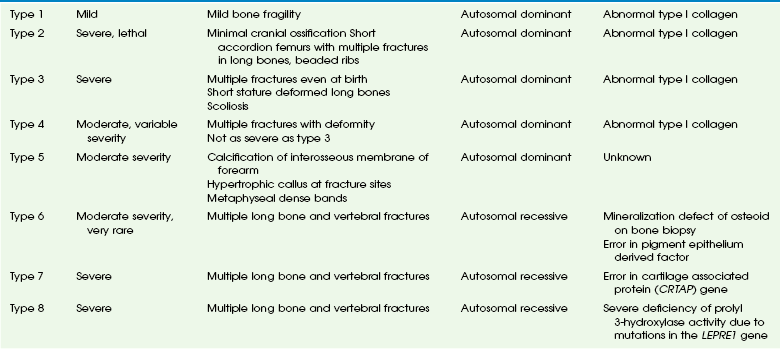
The initial classification of OI was divided into the congenita and tarda forms. With the recognition that all forms are genetically determined, the congenita/tarda system was discontinued. Since 1979, OI is classified according to the Sillence classification. Originally including only four types, this system has now grown to eight types (Table 133-1). The Sillence classification describes a spectrum of disease rather than a strict system based on objective scientific identities such as molecular genetics. In fact, we know now that multiple allelic mutations affect collagen I and cause OI, although other OI types are unrelated to collagen I abnormalities.
Radiologic Findings, Severe Type (Fig. 133-44):
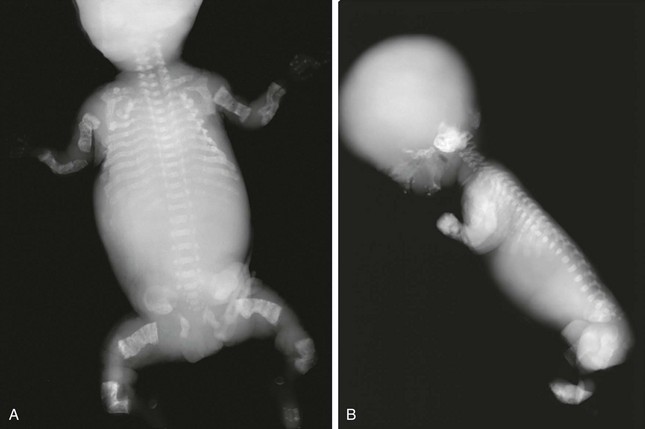
Figure 133-44 Radiographic findings in a stillborn full-term fetus with osteogenesis imperfecta type II.
A and B, Findings include generalized osteoporosis, absence of skull ossification, beaded ribs, and crumpled long bones.
1. Skull: very poor to no ossification
2. Thorax: small, narrow chest; beaded ribs from healing fractures
3. Spine: severe deossification, collapsed vertebral bodies
4. Extremities: generalized osteoporosis with or without fractures; shortened, widened long bones with thin cortices; accordion-shaped femurs
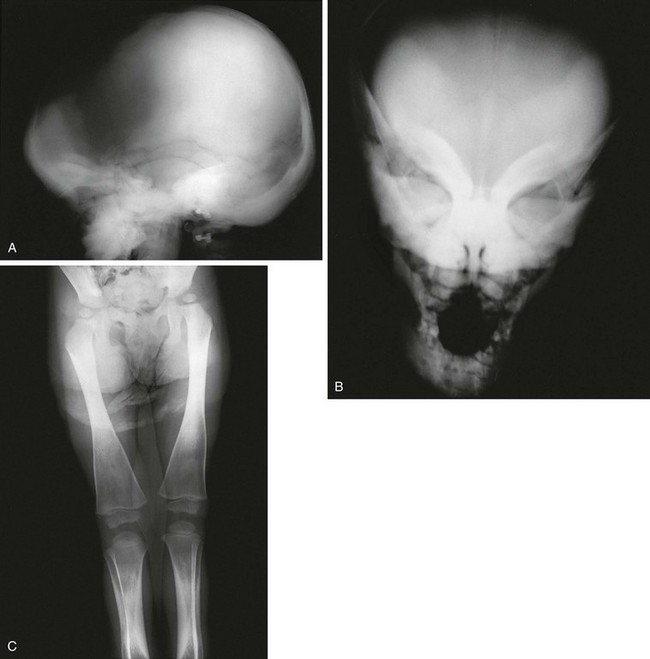
Radiologic Findings, Mild Types (e-Fig. 133-45):
1. Skull: abnormal number of wormian bones (>8 to 10), variable decrease in ossification
2. Spine: wedged or collapsed vertebrae
3. Extremities: at least some osteoporosis, variable number of fractures (especially pathologic fractures)
e-Figure 133-45 Osteogenesis imperfecta type 5 in a 12-year-old girl.
A, Lateral view of the spine showing multiple vertebral body compression fractures. B, The anteroposterior view of the forearm shows hyperostosis of the interosseous membrane (arrow) and increased density at the zones of provisional calcification at the metaphyses of the distal radius and ulna.
Abnormal Mineralization Group
Hypophosphatasia
Radiologic Findings (e-Fig. 133-46):
e-Figure 133-46 Hypophosphatasia.
A, The radiograph from a fetus with perinatal lethal type demonstrates island-like skull ossification (parietal); thin, wavy ribs; platyspondyly and missing cervical vertebrae ossification; no pedicles; and bent femurs. B, The radiograph from a 7-year-old with adult-type hypophosphatasia demonstrates osteopenia, bent tibias, and punched-out metaphyseal lesions.
Perinatal lethal/infantile form:
1. Skull: decreased ossification with single island-like centers for frontal occipital and parietal bones
2. Thorax: poorly ossified ribs; thin, wavy, fractured ribs; clavicles not affected
3. Spine: sporadic unossified vertebral bodies, dense and osteopenic vertebrae, sporadic platyspondyly, butterfly-shaped vertebral bodies, sporadic missing pedicles
4. Extremities: generalized decreased ossification, chromosome-shaped femurs, metaphyseal cupping and irregularity, central lucent defect, “campomelic femurs,” sporadic “missing” short tubular bones of hands and feet
Adult form:
Lysosomal Storage Diseases
Hurler or Hurler Syndrome (Mucopolysaccharidosis Type IH & II)
Radiologic Findings (Dysostosis Multiplex) (Fig. 133-47):
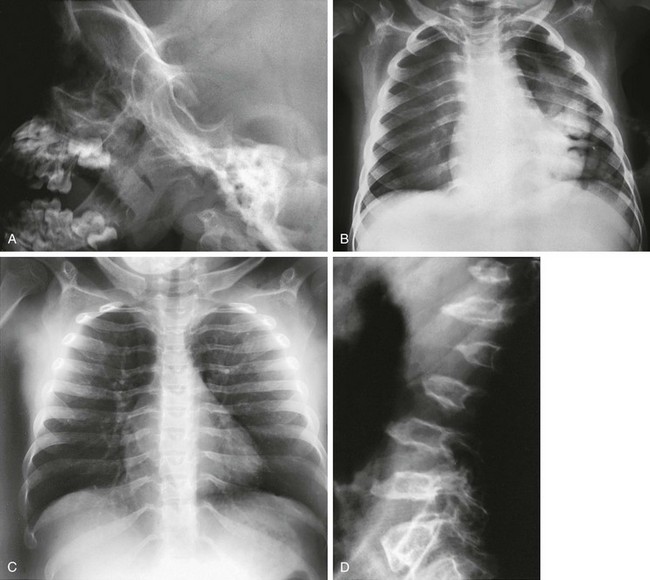
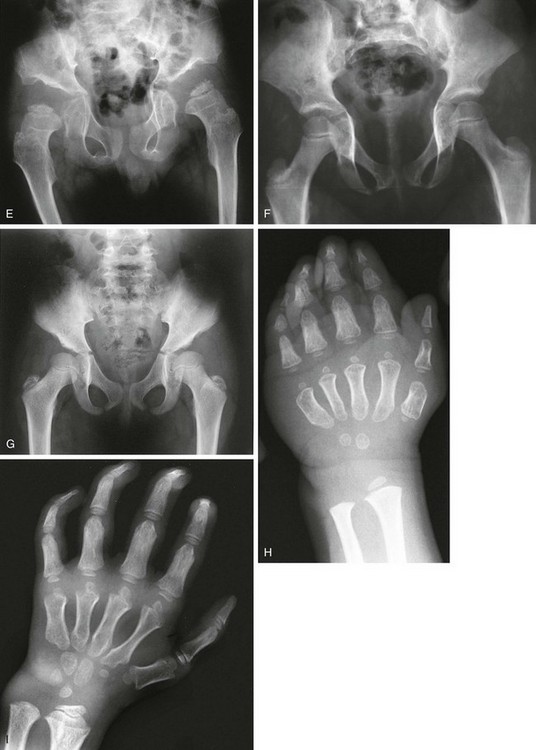
Figure 133-47 Hurler syndrome (mucopolysaccharidosis type IH).
A, Skull of an affected 3-year-old with abnormal, excavated J-shaped sella turcica. B and C, Thoraces of an affected 3-year-old (B) and an affected 8-year-old (C) with thick clavicles and paddle-shaped ribs (thin posteriorly and thick anteriorly). D, Spine of an affected 8-year-old with superiorly notched (inferiorly beaked) vertebral bodies. Radiographic findings in Hurler syndrome (mucopolysaccharidosis type IH). E to G, Pelvis of an affected 8-year-old (E), another affected 8-year-old (F), and an affected 12-year-old (G) with small iliac wings with inferior tapering, and a slanted, irregular acetabular roof (E and G). H and I, Hands of an affected 6-year-old (H) and an affected 10-year-old (I) with proximal metacarpal pointing and epiphyseal ossification delay.
1. Skull: enlarged neurocranium, abnormal J-shaped sella
2. Thorax: short, thick clavicles; paddle (oar)–shaped ribs; hypoplastic glenoid
3. Spine: gibbus, superior notched (inferior beaked) thoracolumbar vertebral bodies, upper cervical subluxation
4. Pelvis: flared, small iliac wings with inferior tapering; steep acetabular roofs
5. Extremities: diaphyseal widening of long bones; hands characteristically exhibit brachydactyly, proximal metacarpal “pointing,” diaphyseal widening of metacarpals and proximal or middle phalanges, small irregular carpal bones
Morquio Syndrome (Mucopolysaccharidosis Types IVA and IVB)
Radiologic Findings (Differentiating Features from Other Mucopolysaccharidoses) (Fig. 133-48):
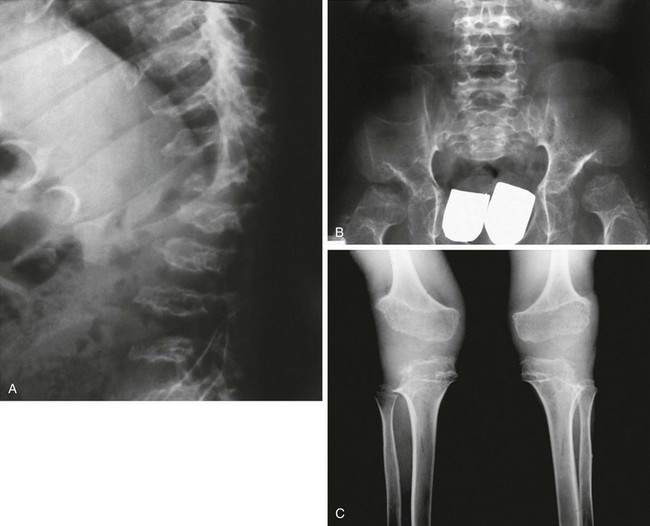
Figure 133-48 Morquio syndrome (mucopolysaccharidosis types IVA and IVB).
A, Radiograph from an affected 7-year-old with platyspondyly with central beaking (tongue). B, Radiograph from an affected 18-year-old with severe capital femoral epiphyseal and acetabular dysplasia but no inferior iliac tapering. C, Radiograph from an affected 15-year-old with lateral distal femoral and proximal tibial epiphyseal ossification defects with genu varum.
2. Thorax: widened, not oar shaped, ribs
3. Spine: central vertebral beaking or tongue; odontoid hypoplasia and cervical instability
4. Pelvis: no tapering of ileum, steep acetabular roofs
5. Extremities: proximal metacarpal rounding, genu valgus; epiphyseal dysplasia especially at carpal bones and femoral heads
Mucolipidosis Type II (I-Cell Disease)
Radiologic Findings (e-Fig. 133-49):
1. Extremities: severe osteopenia with metaphyseal cupping and fraying, poorly defined cortices, “periosteal cloaking” or reaction seen diffusely with areas of cortical bone destruction; diaphyseal expansion
2. Pelvis: wide iliac flare with hypoplastic lower iliac segment, steep acetabular roofs
3. Spine: biconvex end plates with anterior concavity of the vertebral bodies
Osteolysis Group
Hadju-Cheney Syndrome
Distinctive transverse (bandlike) acro-osteolysis is present; skull features, including persistence of the skull sutures, a J-shaped sella, and wormian bones, characterize HCS. More important, however, is the progressive osteoporosis that occurs and can lead to vertebral body fractures. Many patients with confirmed HCS have an elongated and gracile twisting fibula and polycystic kidneys. Serpentine fibula polycystic kidney syndrome had been considered a separate genetic syndrome, but recently, the same genetic defect in the NOTCH2 signaling pathway has been found to cause both syndromes, unifying the picture.32
NOTCH2 is interesting for its effects on the skeleton and tumorogenesis. The defect in HCS causes an upmodulation in NOTCH signaling, which inhibits endochondral growth and osteoblastic differentiation resulting in osteopenia33. Errors in the NOTCH signaling pathway have been associated with T-cell leukemia and lymphoma (NOTCH1).34 Dysregulated NOTCH signaling is known to occur in multiple myeloma. Recently, enhanced NOTCH2 signaling has been associated with osteosarcoma and is associated with greater tumor invasiveness.35
Overgrowth Syndromes with Skeletal Involvement
Marfan Syndrome and Congenital Contractural Arachnodactyly
These two congenital syndromes are caused by errors in fibrilin. Fibrilin is a glycoprotein that functions as a structural scaffold for elastic microfibrils. It can be found in abundance in the connective tissues of the walls of large vessels, lungs, bones, and eyes. The marfanoid body habitus is a constant feature along with long spidery fingers (hence the term arachnodactyly) and tall stature. The loss of normal fibrillin may allow liberation of transforming growth factor-β from the connective tissues, thereby allowing greater expressivity as tall stature.36,37
Proteus Syndrome
Radiologic Findings (e-Fig. 133-52):
1. Skull: macrocrania with cortical hyperostosis, dolichocephaly, and facial asymmetry
2. Thorax: scoliosis, kyphosis, large and asymmetric vertebral bodies
3. Extremities: asymmetric bone and soft tissue overgrowth
4. Other: mixed vascular malformations, hydrocephalus, emphysematous lung disease (12%)
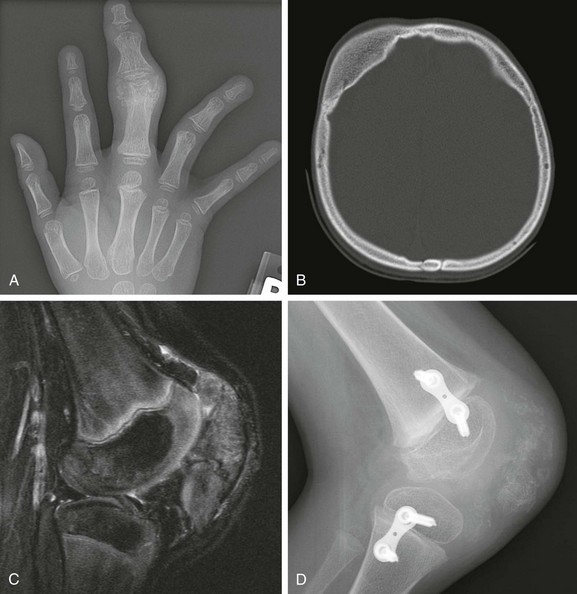
e-Figure 133-52 A 5-year-old male with proteus syndrome.
A, Anteroposterior view of the hand with osseous and soft tissue overgrowth of the third finger. B, An axial computed tomography scan shows osseous overgrowth of the right frontal bone. C, The lateral view of the knee shows osseous irregular overgrowth of the patella. D, A sagittal proton density fat-saturated magnetic resonance imaging scan shows that the cartilaginous patella is enlarged and very irregular.
Other Disorders
Radiologic Findings (Fig. 133-53):
1. Skull: large, brachycephalic; wormian bones; wide sutures; persistently open anterior fontanel
Figure 133-53 A 15-year-old with cleidocranial dysplasia.
A, Skull: large open anterior fontanel and multiple wormian bones. B, Thorax: asymmetric hypoplastic or absent clavicles and downward-sloping ribs. C, Pelvis: tall, narrow ilia and hypoplastic pubic bones. D, Spine: posteriorly wedged but otherwise normal vertebral bodies.
2. Thorax: absence or hypoplasia of clavicles, mildly shortened ribs with downward slope, 11 ribs
3. Spine: significant posterior wedging of thoracic vertebrae
4. Pelvis: high narrow iliac wings, absence or hypoplasia of pubic bones
5. Extremities: numerous pseudoepiphyses of metacarpals and tapered distal phalanges in the hands
Currarino Triad
Currarino triad (hereditary sacral agenesis syndrome) consists of imperforate or stenotic anus, osseous sacral defect, and a presacral mass.38 The presacral mass may be a teratoma (two thirds of cases), a lipoma, a dermoid cyst, an enteric cyst, or an anterior meningocele. The first sacral segment is not affected. The remainder of the sacrum is deformed into a sickle shape because of partial agenesis. Life-threatening meningitis and sepsis may occur, particularly with presacral anterior meningoceles. Moreover, approximately 50% of presacral tumors communicate with the spinal canal in Currarino triad, making surgical repair difficult without neurologic complications. The syndrome is another homeobox type mutation, this one at 7q36 affecting the HLXB9 homeobox gene.
Radiologic Findings (e-Fig. 133-54):
1. Congenital anal stenosis or low imperforate anus
2. Sickle or scimitar hemisacrum
3. Presacral mass (teratoma, lipoma, dermoid cyst, enteric cyst, or anterior meningocele)39
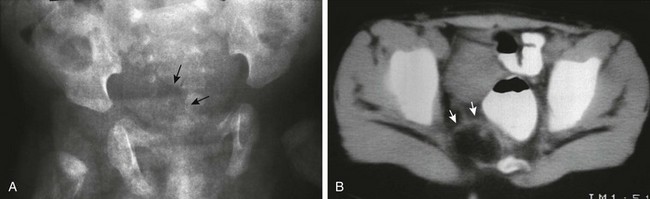
e-Figure 133-54 Currarino triad.
A, Anteroposterior radiograph of the pelvis. A crescentic defect is present in the right inferior sacrum (arrows), producing a so-called scimitar sacrum. B, A computed tomography scan of the pelvis at the level of the sacral defect in a patient with anal stenosis demonstrates a small fat-containing mass (arrows) at the level of the sacral defect. This was surgically removed and proved to be a teratoma.
Brachydactyly Group
Rubinstein-Taybi syndrome is characterized by short stature, distinctive facial features, mental retardation, and broad, short thumbs and great toes. It is caused by sporadic mutations, the majority affecting the CREB-binding protein, which plays an important role in embryonic development. In slightly more than half of the patients, a cytogenetic abnormality can be identified. Radiographically, a delta-phalanx (longitudinal epiphyseal bracket) is seen at the first proximal phalanx (Fig. 133-55). The first distal phalanx is short and broad. At times, it may have a central lucency indicating an attempt at duplication. Other findings include congenital heart defects, agenesis of the corpus callosum, and vertebral and sternal anomalies. Patients have an increased risk of tumors, mainly meningioma, leukemia, and lymphoma.
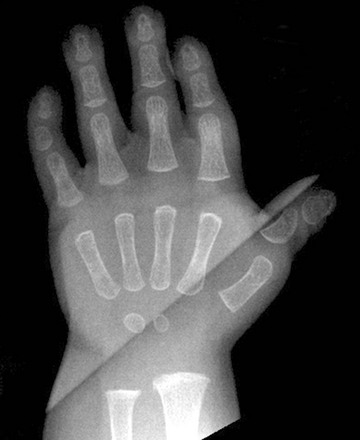
Poland Syndrome
Radiologic Findings (Fig. 133-56):
Figure 133-56 Poland syndrome.
A, A hand radiograph shows generalized hypoplasia of the right hand, with soft tissue syndactyly affecting the second through fourth digits. B, The anteroposterior chest radiograph on a different patient shows relative lucency of the left hemithorax with loss of the pectoral shadow. C, An axial T1-weighted magnetic resonance imaging scan shows absence of the left anterior chest wall musculature (arrowheads indicate normal musculature on the right).
Limb Hypoplasia—Reduction Defects Group
The brachydactyly classification system most commonly used was described by Bell in 1951 and refined by Temtamy and McKusick in 1978.40–42 Since these descriptions, the genetic loci associated with many of the types of isolated brachydactyly have been elucidated. Some of the patterns of shortening of the bones of the hand will be easily recognized by the practicing radiologist. Brachdactyly type A3 in which the fifth middle phalanx is short is especially common. This can be differentiated from (1) Kirner deformity, with radial bowing of the distal phalanx, and (2) camptodacytly, with a flexion contracture of the interphalangeal joints.
Brachmann-De Lange (Cornelia De Lange) Syndrome
Miscellaneous and Chromosomal Disorders
VACTERL Association
V: vertebral (fusion and segmentation anomalies)
A: anorectal (imperforate anus)
C: cardiac (septal defects, tetralogy of Fallot, transposition of the great vessels)
T: tracheoesophageal (esophageal atresia)
Hydrocephalus associated with the VACTERL association is known to have a high rate of recurrence in subsequent pregnancies. This is referred to as VACTERL-H association, with hydrocephalus added to the acronym. VACTERL-H is frequently an X-linked disorder, particularly when aqueductal stenosis is present and the prognosis is poor. (Fig. 133-58)
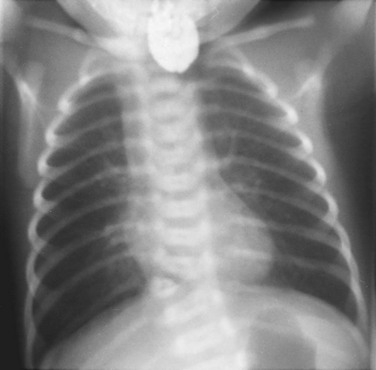
Figure 133-58 VACTERL association (vertebral, anorectal, cardiac, tracheoesophageal, renal, limb).
The chest radiograph of a newborn with tracheoesophageal fistula and esophageal atresia. Contrast medium is in the proximal esophageal pouch. Air in the stomach indicates the presence of a distal tracheoesophageal fistula. A hemivertebra is noted. The patient also had a pelvic kidney.
Trisomy 21 (Down Syndrome)
Both occipitoatlantal and atlantoaxial instability is found in Down syndrome. Anteroposterior occipitoatlantal instability is defined as more than 2 mm of motion on extension of the occipitoatlantal joints. If present, a neck MRI is recommended to evaluate for signal changes in the cord. An atlantoaxial distance of 4.5 mm or less is considered normal. With a distance from 4.5 to 10 mm and a normal neurologic exam, avoidance of high-risk sports (diving, football) is recommended. If more than 4.5 mm and with a neurological deficit, activities are restricted and MRI recommended to evaluate for cord changes.43 However, the poor reproducibility of findings and both intraobserver and interobserver variability may make it difficult to base surgical and clinical treatment protocols for upper cervical spine instability on measurements alone.
Clinical and Imaging Findings:
• Spine: occipitoatlantal and atlantoaxial instability
• Thorax: hypersegmented manubrium, 11 rib pairs, small bell shaped chest with short ribs
• Pelvis: flared iliac wings, flat acetabular roofs
• Gastrointestinal tract: duodenal atresia, Hirschsprung disease, malrotation, tracheoesophageal fistula, imperforate anus
• Cardiac: endocardial cushion defect most common but other types also occur
• Other: increased risk for leukemia (acute lymphoblastic leukemia)
Turner Syndrome
Turner syndrome is most commonly caused by a 45,XO chromosomal pattern. In 15% of cases, one full X chromosome is present as well as an X isochromosome that contains only the long arms of chromosome X. Although originally it was thought that in the normal 46,XX female, complete inactivation of the second X chromosome occurs, some genes on the short arm of the inactivated X chromosome remain activated and are necessary for proper development, which explains why a patient with an X isochromosome and the classic XO are phenotypically similar.44 The locus involved on the short arm of the X chromosome is in the pseudoautosomal region at Xp22 at a gene termed the short stature homeobox gene (SHOX). SHOX was originally determined to be associated with some patients with idiopathic short stature syndrome who have a significantly short stature (>2 SDS), a persistently low growth rate for age and no identifiable cause of a specific metabolic growth retarding condition. Subsequently, SHOX has been found to be active in Turner syndrome.45 Later, homozygous loss was found to be the cause of Langer mesomelic dysplasia and heterozygous loss the cause of dyschondrosteosis (Leri-Weill syndrome).46,47 In each of these last three conditions, the common thread of short stature, a short fourth metacarpal, and a varying degree of Madelung deformity is present.
Alman, BA. Skeletal dysplasias and the growth plate. Clin Genet. 2008;73(1):24–30.
Ikegawa, S. Genetic analysis of skeletal dysplasia: recent advances and perspectives in the post-genome-sequence era. J Hum Genet. 2006;51(7):581–586.
McAlister, WH, Herman, TE. Osteochondrodysplasias, dysostoses, chromosomal aberrations, mucopolysaccharidoses, and mucolipidoses. In Resnick D, ed.: Bone and joint disorders, 4th ed, Philadelphia, PA: Elsevier, 2002.
Superti-Furga, A, Bonafe, L, Rimoin, DL. Molecular-pathogenetic classification of genetic disorders of the skeleton. Am J Med Genet. 2001;106(4):282–293.
Rimoin, DL, Cohn, D, Krakow, D, et al. The skeletal dysplasias: clinical-molecular correlations. Ann N Y Acad Sci. 2007;1117:302–309.
References
1. Warman, ML, Cormier-Daire, V, Hall, C, et al. Nosology and classification of genetic skeletal disorders: 2010 revision. Am J Med Genet A. 2011;155A(5):943–968.
2. Lachman, RS. Taybi and Lachman’s radiology of syndromes, metabolic disorders and skeletal dysplasias, 5th ed. Philadelphia, PA: Mosby; 2007.
3. Spranger, JW, Brill, PW, Poznanski, A. Bone dysplasias: an atlas of genetic disorders of skeletal development, 2nd ed. New York: Oxford University Press; 2002.
4. Bonaventure, J, Rousseau, F, Legeai-Mallet, L, et al. Common mutations in the fibroblast growth factor receptor 3 (FGFR 3) gene account for achondroplasia, hypochondroplasia, and thanatophoric dwarfism. Am J Med Genet. 1996;63(1):148–154.
5. Dakouane Giudicelli, M, Serazin, V, et al. Increased achondroplasia mutation frequency with advanced age and evidence for G1138A mosaicism in human testis biopsies. Fertil Steril. 2008;89(6):1651–1656.
6. Shapiro, R, Robinson, F. Embryogenesis of the human occipital bone. AJR Am J Roentgenol. 1976;126(5):1063–1068.
7. Gordon, N. The neurological complications of achondroplasia. Brain Dev. 2000;22(1):3–7.
8. Matsui, Y, Yasui, N, Kimura, T, et al. Genotype phenotype correlation in achondroplasia and hypochondroplasia. J Bone Joint Surg Br. 1998;80(6):1052–1056.
9. Dwek, JR. Kniest dysplasia: MR correlation of histologic and radiographic peculiarities. Pediatr Radiol. 2005;35(2):191–193.
10. Hastbacka, J, Superti-Furga, A, Wilcox, WR, et al. Atelosteogenesis type II is caused by mutations in the diastrophic dysplasia sulfate-transporter gene (DTDST): evidence for a phenotypic series involving three chondrodysplasias. Am J Hum Genet. 1996;58(2):255–262.
11. Superti-Furga, A, Rossi, A, Steinmann, B, et al. A chondrodysplasia family produced by mutations in the diastrophic dysplasia sulfate transporter gene: genotype/phenotype correlations. Am J Med Genet. 1996;63(1):144–147.
12. Karniski, LP. Mutations in the diastrophic dysplasia sulfate transporter (DTDST) gene: correlation between sulfate transport activity and chondrodysplasia phenotype. Hum Mol Genet. 2001;10(14):1485–1490.
13. Robertson, SP. Filamin A: phenotypic diversity. Curr Opin Genet Dev. 2005;15(3):301–307.
14. Robertson, SP. Molecular pathology of filamin A: diverse phenotypes, many functions. Clin Dysmorphol. 2004;13(3):123–131.
15. Krakow, D, Robertson, SP, King, LM, et al. Mutations in the gene encoding filamin B disrupt vertebral segmentation, joint formation and skeletogenesis. Nat Genet. 2004;36(4):405–410.
16. Dai, J, Kim, OH, Cho, TJ, et al. Novel and recurrent TRPV4 mutations and their association with distinct phenotypes within the TRPV4 dysplasia family. J Med Genet. 2010;47(10):704–709.
17. Dai, J, Cho, TJ, Unger, S, et al. TRPV4-pathy, a novel channelopathy affecting diverse systems. J Hum Genet. 2010;55(7):400–402.
18. Hall, T, Bush, A, Fell, J, et al. Ciliopathy spectrum expanded? Jeune syndrome associated with foregut dysmotility and malrotation. Pediatr Pulmonol. 2009;44(2):198–201.
19. Dagoneau, N, Goulet, M, Genevieve, D, et al. DYNC2H1 mutations cause asphyxiating thoracic dystrophy and short rib-polydactyly syndrome, type III. Am J Hum Genet. 2009;84(5):706–711.
20. Mill, P, Lockhart, PJ, Fitzpatrick, E, et al. Human and mouse mutations in WDR35 cause short-rib polydactyly syndromes due to abnormal ciliogenesis. Am J Hum Genet. 2011;88(4):508–515.
21. Krakow, D, Salazar, D, Wilcox, WR, et al. Exclusion of the Ellis-van Creveld region on chromosome 4p16 in some families with asphyxiating thoracic dystrophy and short-rib polydactyly syndromes. Eur J Hum Genet. 2000;8(8):645–648.
22. Kruse, K, Schutz, C. Calcium metabolism in the Jansen type of metaphyseal dysplasia. Eur J Pediatr. 1993;152(11):912–915.
23. Bastepe, M, Raas-Rothschild, A, Silver, J, et al. A form of Jansen’s metaphyseal chondrodysplasia with limited metabolic and skeletal abnormalities is caused by a novel activating parathyroid hormone (PTH)/PTH-related peptide receptor mutation. J Clin Endocrinol Metab. 2004;89(7):3595–3600.
24. Hermanns, P, Tran, A, Munivez, E, et al. RMRP mutations in cartilage-hair hypoplasia. Am J Med Genet A. 2006;140(19):2121–2130.
25. Hermanns, P, Bertuch, AA, Bertin, TK, et al. Consequences of mutations in the non-coding RMRP RNA in cartilage-hair hypoplasia. Hum Mol Genet. 2005;14(23):3723–3740.
26. Sulisalo, T, Francomano, CA, Sistonen, P, et al. High-resolution genetic mapping of the cartilage-hair hypoplasia (CHH) gene in Amish and Finnish families. Genomics. 1994;20(3):347–353.
27. Bowen, P, Biederman, B, Hoo, JJ. The critical segment for the Langer-Giedion syndrome: 8q24.11-q24.12. Ann Genet. 1985;28(4):224–227.
28. Vickers, D, Nielsen, G. Madelung deformity: surgical prophylaxis (physiolysis) during the late growth period by resection of the dyschondrosteosis lesion. J Hand Surg Br. 1992;17(4):401–407.
29. Cook, PA, Yu, JS, Wiand, W, et al. Madelung deformity in skeletally immature patients: morphologic assessment using radiography, CT, and MRI. J Comput Assist Tomogr. 1996;20(4):505–511.
30. Bartsocas, CS. Pycnodysostosis: Toulouse-Lautrec’s and Aesop’s disease? Hormones (Athens). 2002;1(4):260–262.
31. Ben-Asher, E, Zelzer, E, Lancet, D. LEMD3: the gene responsible for bone density disorders (osteopoikilosis). Isr Med Assoc J. 2005;7(4):273–274.
32. Isidor, B, Le Merrer, M, Exner, GU, et al. Serpentine fibula-polycystic kidney syndrome caused by truncating mutations in NOTCH2. Hum Mutat. 2011;32(11):1239–1242.
33. Zanotti, S, Smerdel-Ramoya, A, Stadmeyer, L, et al. Notch inhibits osteoblast differentiation and causes osteopenia. Endocrinology. 2008;149(8):3890–3899.
34. Zanotti, S, Canalis, E. Notch and the skeleton. Mol Cell Biol. 2010;30(4):886–896.
35. Zhang, P, Yang, Y, Zweidler-McKay, PA, et al. Critical role of notch signaling in osteosarcoma invasion and metastasis. Clin Cancer Res. 2008;14(10):2962–2969.
36. Ramirez, F. Fibrillln mutations in Marfan syndrome and related phenotypes. Curr Opin Genet Dev. 1996;6(3):309–315.
37. Ramirez, F, Pereira, L, Zhang, H, et al. The fibrillin-Marfan syndrome connection. Bioessays. 1993;15(9):589–594.
38. Currarino, G, Coln, D, Votteler, T. Triad of anorectal, sacral, and presacral anomalies. AJR Am J Roentgenol. 1981;137(2):395–398.
39. Kirks, DR, Merten, DF, Filston, HC, et al. The Currarino triad: complex of anorectal malformation, sacral bony abnormality, and presacral mass. Pediatr Radiol. 1984;14(4):220–225.
40. Bell, J, On brachydactyly and Symphalangism. Treasury of human inheritance. Pensore, LS, eds. Treasury of human inheritance, London, U.K., Cambridge University Press, 1951;vol 5:1–31.
41. Temtamy, SA, McKusick, VA, Bergsma, D Genetics of hand malformations, New York, A. R. Liss, 1978;vol 14.
42. Temtamy, SA, Aglan, MS. Brachydactyly. Orphanet J Rare Dis. 2008;3:15.
43. Pizzutillo, PD, Herman, MJ. Cervical spine issues in Down syndrome. J Pediatr Orthop. 2005;25(2):253–259.
44. Brown, CJ, Willard, HF. Localization of a gene that escapes inactivation to the X chromosome proximal short arm: implications for X inactivation. Am J Hum Genet. 1990;46(2):273–279.
45. Rao, E, Weiss, B, Fukami, M, et al. Pseudoautosomal deletions encompassing a novel homeobox gene cause growth failure in idiopathic short stature and Turner syndrome. Nat Genet. 1997;16(1):54–63.
46. Belin, V, Cusin, V, Viot, G, et al. SHOX mutations in dyschondrosteosis (Leri-Weill syndrome). Nat Genet. 1998;19(1):67–69.
47. Shears, DJ, Vassal, HJ, Goodman, FR, et al. Mutation and deletion of the pseudoautosomal gene SHOX cause Leri-Weill dyschondrosteosis. Nat Genet. 1998;19(1):70–73.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]Chapter 133
Skeletal Dysplasias and Selected Chromosomal Disorders
The abridged form of the International Skeletal Dysplasia Society skeletal dysplasia classification serves as the organization of this chapter (Box 133-1).1 The full nosology text can be found at http://isds.ch/uploads/pdf_files/Nosology2010.pdf (accessed August 12, 2012). The major genetic families are presented with a short description of the salient unifying characteristics of the diseases within each group. When known, the gene and protein involved are considered and the impact of the mechanism of action discussed. The major members of each group are then expanded on to provide a clear picture for the reader.
Radiologic Assessment
In the history of the delineation of many of the specific skeletal dysplasias, radiologic assessment plays a major role. By using an orderly approach to the radiographic analysis, the general type of the dysplasia may be elucidated. Many of the skeletal dysplasias and syndromes have distinctive radiographic features that will allow an exact diagnosis when even one of those distinctive features is identified and used as a search criterion in textbooks on skeletal dysplasia. Two such texts are Taybi and Lachman’s Radiology of Syndromes, Metabolic Disorders and Skeletal Dysplasias, which includes an excellent gamuts section, and Bone Dysplasias, An Atlas of Genetic Disorders of Skeletal Development by Spranger and colleagues, in which the images are particularly helpful.2,3 In the online version of Taybi and Lachman’s book, the gamut search may be built iteratively, with the diagnoses becoming more selective as findings are added to the search criteria. Internet searches can also be performed on the Online Mendelian Inheritance in Man database, which is accessed through the U.S. Library of Medicine portal at http://www.ncbi.nlm.nih.gov/pubmed/.
Step II: Assessment of Epiphyseal Ossification
If epiphyseal ossification is delayed or if the ossified epiphyses are very small, irregular for age, or both, then an epiphyseal dysplasia of some sort is present. Carpal and tarsal bones are often affected. In diseases that can be considered pure epiphyseal dysplasias such as multiple epiphyseal dysplasia and pseudoachondroplasia, carpal and the tarsal bones are markedly crenellated and small (Fig. 133-1). Another excellent location for epiphyseal analysis is the ring apophyses of the vertebral bodies, which exhibit delayed and irregular epiphyseal ossification in epiphyseal dysplasia. Central anterior vertebral body protrusions (central tongues or beaking) noted in Morquio syndrome and pseudoachondroplasia are also disorders related to abnormalities of the ring apophyses.
Step III: Assessment of Metaphyses and Physes
Fraying and irregularity of the physes and abnormal flaring of the metaphyses indicate disturbed endochondral ossification. Marked irregularity of the physes is characteristic of the pure metaphyseal dysplasias such as metaphyseal dysplasia, Jansen or Schmid type. When the metaphyses are merely flared and the physes are fairly normal, endochondral ossification may be slowed but the actual process of endochondral ossification progresses normally. This occurs in achondroplasia. The metaphyses are flared, whereas the physis and the zone of provisional calcification (ZPC) are sharply defined (Fig. 133-2).
Figure 133-2 A 14-year-old boy with achondroplasia.
The long bones are short and thick. Note the normal, sharp-appearing physes.
It must be kept in mind that rickets also disturbs the physis. In rickets, the physis is frayed and cupped. Except in healing rickets, the ZPC is inapparent. In metaphyseal chondrodysplasias, the ZPC is present, although it is markedly irregular (Fig. 133-3). Analysis of the sclerotic line of the ZPC is frequently an excellent differentiating feature. Other factors include prominent osteopenia in rickets with blurring of the trabeculae; clinical data are also very helpful.
Figure 133-3 An 8-old boy with Schmid metaphyseal chondrodysplasia.
A, Note the presence of zone of provisional calcification (arrowheads). Normal laboratory values help confirm the diagnosis. B, A 15-month-old girl with rickets. The physes are frayed with mild metaphyseal cupping. The bright white line of the zone of provisional calcification is not evident.
Step V: Analysis of the Vertebral Bodies
Anisospondyly is when the vertebral body shape varies wildly (e-Fig. 133-4). Multiple ossification centers may also be present. Although rare, this is a specific finding in dyssegmental dysplasia.
Step VIII: Summation
After all radiologic findings have been established, a gamut search of some or all of these abnormalities, in conjunction with the clinical findings, may lead to the specific diagnosis. If the “group” of dysplasias has been established, then the specific diagnosis can often be made by referring to a differential diagnosis table such as that developed by Taybi and Lachman.2
Selected Skeletal Dysplasias and Syndromes
Fibroblast Growth Factor Receptor Type 3 Group
Overview: This group includes thanatophoric dwarfism and achondroplasia. The former is probably the most common lethal skeletal dysplasia and the latter the most common skeletal dysplasia. The group includes the milder variant called hypochondroplasia, and homozygous achondroplasia, which is similar to thanatophoria.4
A common genetic locus (4p16.3) is involved. Differing allelic mutations are the cause of the variable severity of expression. The protein encoded is FGFR3, which governs the velocity of endochondral growth. Although long believed that achondroplasia and thanatophoria were caused by loss of function mutations, the mutation in this group actually results in an upmodulation of FGFR3 activity, which is inversely related to the velocity of endochondral growth. FGFR3 mutations have been linked to advanced paternal age, with mutations theoretically accumulating during spermatogenesis.5
Thanatophoric Dwarfism
Radiographic Findings (Fig. 133-5):
Figure 133-5 Thanatophoric dysplasia.
A and B, Radiographic findings in thanatophoric dysplasia type I. In an affected fetus of 30 weeks’ gestation (A), a long, narrow trunk; very short ribs; severe platyspondyly are seen. Note the H and U shapes of bodies are caused by angle of incidence of the x-ray beam; small, flared iliac wings; narrowed sacrosciatic notches; dysplastic (trident) acetabular roofs; and French telephone receiver–shaped femurs. At 22 weeks’ gestation in another affected fetus (B), a proportionally large skull, micromelia, and other findings similar to those in A are seen. C and D, Radiologic findings in thanatophoric dysplasia type II. An affected preterm fetus (C) exhibits findings similar to those in type I except for taller vertebral bodies and straighter femurs. Another affected fetus (D) also shows the same findings as in type I but with straighter femurs. E, Radiograph of affected infant with severe platyspondyly, anteriorly rounded vertebrae, straight femurs, and severely constricted skull base. F, Cloverleaf skull and almost straight femurs; otherwise, radiographic findings are similar to those in C. Note the ovoid lucency at the femoral necks in all cases.
1. Skull: proportionately large skull in relation to the body, narrow skull base, cloverleaf skull (type 1 only)
2. Thorax: long, narrow trunk; very short ribs; handlebar clavicles
3. Spine: severely flattened, small vertebral bodies with round anterior ends
4. Pelvis: small, flared iliac bones; very narrow sacrosciatic notches; flat, dysplastic acetabula
5. Extremities: generalized micromelia; ovoid lucency of femoral necks, round proximal femoral metaphyses with medial spike, curved long bones (type 1 only)
Achondroplasia
Except for the portions of the occipital bone that form the margin of the foramen magnum, all the bones of the skull are formed by membranous ossification.6 This results in an enlarged forehead and is termed frontal bossing. In contrast, the foramen magnum is narrowed and can cause cervicomedullary compression. Symptoms may include occipitocervical pain, ataxia, incontinence, apnea, paralysis, and respiratory arrest.7
Radiologic Findings (Fig. 133-6 and e-Fig. 133-7):
Figure 133-6 Achondroplasia.
A to D, Radiographs of an affected newborn. A, Severe midface hypoplasia. B, Thorax: small thorax and short ribs. C, Thorax: short ribs with anterior scalloping and bullet-shaped vertebrae. D, Pelvis: rounded ilia (elephant ear–shaped) iliac bones, narrow sacrosciatic notches, flat acetabular roof, and proximal femoral ovoid lucency. Achondroplasia. E, Extremities: rhizomelia and mesomelia. F, Radiograph in an affected 1½-year-old with classic vertebrae with short pedicles, posterior scalloping, and somewhat short vertebral bodies. G, Radiograph in an affected woman with flat acetabular roofs, elephant ear–shaped iliac wings, and short femoral necks (compare with D). (G, From Silverman FN: Achondroplasia. Prog Pediatr Radiol. 1973;4:94-124.)
1. Skull: enlarged, with significant midface hypoplasia; hydrocephalus rarely present; small skull base with tight foramen magnum
2. Thorax: small; shortened and anteriorly splayed ribs
3. Spine: short pedicles with decreased interpediculate distance most marked in the lumbar spine moving downward; posterior vertebral body scalloping, gibbus deformity.
4. Pelvis: round iliac wings with lack of flaring (elephant ear–shaped), flattened acetabular roofs, narrow sacrosciatic notches with champagne glass shaped pelvic inlet
5. Extremities: rhizomelic micromelia
6. Hands: brachydactyly with trident hands
7. Knees: Central deep notch in growth plates (Chevron deformity)
8. Hips: proximal femoral ovoid lucency (infancy); hemispheric capital femoral epiphyses, short femoral necks
9. Legs: prominent tibial tubercle apophyseal region, fibula overgrowth
10. Arms: Cortical hyperostosis at deltoid insertion on anterolateral humerus
Hypochondroplasia
In hypochondroplasia radiographic and clinical findings are less severe compared with achondroplasia, and the diagnosis can be challenging. Stature is slightly shortened but is highly variable and may be normal given the range of normal stature in society. Radiographically, aside from shortening of the long bones, interpediculate narrowing is a very sensitive finding (Fig. 133-8).8
Type 2 Collagen Group
Overview: Defects in chromosome 12q13.1-13.3 result in abnormalities of type 2 collagen. Type 2 collagen is present in cartilaginous epiphyses and in the vitreous humor. Therefore, abnormalities of collagen 2 manifest with platyspondyly due to lack of normal growth at the ring epiphysis, a general delay in epiphyseal ossification, and myopia. Cleft palate completes the phenotypic picture.
Achondrogenesis type 2
Radiologic Findings (Fig. 133-9):
1. Skull: proportionately large
2. Thorax: very small and short ribs
3. Spine: almost complete lack of mineralization; cervical and sacral posterior elements also often unossified
4. Pelvis: small iliac wings with concave inferior and medial margins; absence of ischia, pubic bones, and sacral elements
5. Extremities: micromelia, mostly rhizomelia and mesomelia, with relative sparing of hands and feet; metaphyseal flaring; absence of talus and calcaneal ossification (epiphyseal equivalents)
Spondyloepiphyseal Dysplasia Congenita
Radiologic Findings (Fig. 133-10):
Figure 133-10 Spondyloepiphyseal dysplasia congenita.
A, Radiograph in an affected newborn with a small thorax, rounded iliac wings, vertical ischia, absence of pubic ossification, short femurs, and metaphyseal rounding of long bones. B, Radiograph in an affected newborn with bell-shaped chest, short ribs, and elongated clavicles. C, Radiograph in an affected newborn with moderately short ribs with mild anterior splaying and anteriorly rounded vertebral bodies with minimal flattening and no coronal clefts.
2. Spine: dorsally wedged or oval vertebral bodies (at birth); anteriorly rounded platyspondyly (later)
3. Pelvis: absent pubic ossification (at birth and during infancy), vertical ischia with short ilia
4. Extremities: normal tubulization with mild micromelia, significant generalized ossification delay (early) and hypoplastic-appearing or dysplastic epiphyses (later), unossified talus or calcaneus in the newborn, normal hands and feet with ossification delay (epiphyses or carpal, tarsal)
Kniest Dysplasia
The same delay in epiphyseal ossification is seen along with platyspondyly. Cloudlike dystrophic calcification is present in abnormally enlarged epiphyses as the child gets older. On magnetic resonance imaging (MRI), the areas of calcification have prolonged T2 values that are likely related to the degeneration of abnormal collagen matrix.9
Radiologic Findings (e-Fig. 133-11):
2. Spine: coronal clefts (at birth and during infancy), platyspondyly with endplate irregularity (later)
3. Extremities: dumbbell femurs; generalized ossification delay, epiphyses becoming hypoplastic or dysplastic and then later even mega-epiphyses, cloudlike irregular calcification in physeal plate regions (in late childhood and early adulthood); hands with bulbous joints (metaphyseal flaring or epiphyseal fragmentation) mimicking rheumatoid arthritis
Common Features in Type 11 Collagenopathy Group
• Similar to type 2 collagenopathy
• Myopia (except for Stickler type 3)
Abnormal Sulfation Group
The abnormal sulfation group is a molecularly defined group of disorders with a defect in the sulfate transporter gene on chromosome 5 coding for the diastrophic dysplasia sulfate transporter (DTDST) protein. This group comprises not only diastrophic dysplasia but also multiple epiphyseal dysplasia MED–multilayered patellae/brachydactyly/clubfeet, as well as achondrogenesis type IB and atelosteogenesis type II.10 These conditions are all autosomal recessive, and the severity of the phenotype is inversely related to the level of sulfation.11,12
Common Features in Abnormal Sulfation Group
Achondrogenesis Type I
Achondrogenesis type I is actually two separate disorders that appear almost identical radiographically. Achondrogenesis type IB belongs to this diastrophic dysplasia (molecular) group.11 In achondrogenesis type IA, a molecular or gene abnormality has not yet been identified. Clinically, the two types appear identical: proportionately large skull; micromelic, hydropic, pear-shaped trunk; polyhydramnios; and lethality.
Radiologic Findings (Fig. 133-12):
Figure 133-12 Achondrogenesis.
A, Radiograph from a stillborn infant with type IA achondrogenesis demonstrates a tiny thorax; short, anteriorly cupped ribs with beading; micromelia; wedged femurs; and poor to absent vertebral body ossification. B, Radiograph from a stillborn infant with type IB achondrogenesis. Findings are similar to those in type IA but with arched iliac wings, no rib beading, and trapezoidal femurs
1. Skull: decreased ossification
2. Thorax: tiny; very short ribs with anterior splaying
3. Spine: absent or minimal vertebral body ossification
4. Pelvis: short iliac bones with concave acetabular roofs, absent pubic (ischial) ossification
5. Extremities: severe micromelia with broadened ends of limbs, trapezoidal or wedge-shaped femurs
Diastrophic Dysplasia
Radiologic Findings (Fig. 133-13):
Figure 133-13 Diastrophic dysplasia.
A and B, Radiographs from an affected newborn. A, Lower extremities: rhizomelia and mesomelia and severe clubfeet deformities. B, Upper extremity: elbow dislocation and short, ovoid first metacarpal. C and D, Radiographs from an affected 21-year-old. C, Upper extremity: hitchhiker thumb, ovoid first metacarpal, brachydactyly, and irregular and extra carpal bones. D, Lower extremities: unusual clubfoot and twisted metatarsals
1. Head: ear pinna calcification, cleft or high arched palate
3. Spine: progressive scoliosis, kyphosis, upper cervical subluxation (odontoid hypoplasia), cervical kyphosis, posterior process clefting (cervical and sacral)
4. Extremities: often micromelia; short, thick tubular bones; generalized brachydactyly; short ovoid first metacarpal delta phalanx causing proximal placement of thumb (hitchhiker thumb), twisted metatarsals, accessory and irregular carpal bones; epiphyseal dysplasia with multiple joint contractures
5. Other sites: precocious costochondral and laryngeal area cartilage calcification; multiple sternal and patella centers
Multiple Epiphyseal Dysplasia: Multilayered Patellae/Brachydactyly/Clubfeet
Radiologic Findings: Extremities show the following: epiphyseal dysplasia, especially at hips (half- or quarter-moon–shaped); double-layered or multilayered patella (visible on lateral knee radiograph); mild brachydactyly; clubfeet or twisted metatarsals; mildly shortened long bones, some with mild undertubulation.
Filamin Group
The filamin group combines a wide group of dysplasias that have in common an abnormality in the number and configuration of carpal, tarsal, and vertebral bones with joint dislocations. The identification of the group is another triumph in the study of molecular genetics, as it reclassifies correctly a group of disorders described as “syndromes” within a common framework of genetically determined diseases no different from other skeletal dysplasias.13,14 The group includes oto-palato-digital (OPD) syndrome types 1 and 2, Larsen syndrome, frontometaphyseal dysplasia, Melnick-Needles osteodysplasty, and spondylo-carpal-tarsal synostosis.
OPD Syndrome
Radiographic Findings in OPD (e-Fig. 133-14):
1. Head: prominent supraorbital ridge
2. Spine: small pedicles with wide interpediculate distance
3. Extremities: accessory carpal bones, double ossification center of lunate, fusion of carpal bones especially trapezoid and scaphoid, fusion of the second metacarpal to trapezoid in adolescence, similar findings in feet; short first metatarsal and phalanges of great toe, and short and wide distal phalanx of thumb; radial head dislocation
e-Figure 133-14 Oto-palato-digital syndrome.
The radiograph at right shows typical findings of fusion of the accessory ossification centers of the second metacarpal with the adjacent deformed carpal bones. Widening and abnormal tubulation of the metacarpals are seen. The distal phalanx of the thumb is short with a coned-shaped epiphysis. Clinodactyly is present.
Larsen Syndrome
In Larsen syndrome, multiple joint dislocations are present. In keeping with the common theme of filamin abnormalities, supernumerary carpal bones are common along with other digital changes. A doubled calcaneal ossification center is a helpful clue to accurate diagnosis. Scoliosis is common. This is a filamin type B abnormality. A similar filamin type B abnormality causes spondylo-carpal-tarsal synostosis syndrome, whose name describes the pattern of skeletal involvement.15
Radiographic Features (Fig. 133-15 and e-Fig. 133-16):
Figure 133-15 A 5-year-old girl with Larsen syndrome.
A, Anteroposterior elbow radiograph shows chronic dislocation with epiphyseal deformities. B, Lateral radiograph of an ankle in a 5-year-old boy shows a bifid calcaneus and deformity of the talus and distal tibial epiphysis.
1. Spine: cervical spine kyphosis
2. Extremities: multiple joint dislocations, double or triple calcaneal ossification center, accessory carpal bones, broad irregular metacarpals
TRPV4 Group
TRPV4 (transient receptor potential cation channel, subfamily 5, member 4) is a calcium permeable nonselective cation channel that appears to play an important role in chondrogenesis. This channelopathy is also the cause of several other nonskeletal syndromes such as Charcot Marie-Tooth disease, scapula-peroneal spinal muscular atrophy, and congenital distal spinal muscular atrophy.16,17
Metatropic Dysplasia
Radiologic Findings (Fig. 133-17):
Figure 133-17 A newborn with metatropic dysplasia.
A, Thorax: long trunk and small chest. B, Spine: dense wafer vertebral bodies and short ribs with anterior splaying. C, Pelvis: short iliac wings, narrow sciatic notches, irregular acetabular roofs, and rounded, enlarged proximal femoral metaphyses (halberd shaped) with markedly flared distal metaphyses (trumpet-shaped) D, Upper extremities: flared proximal humeral and distal radial and ulnar metaphyses; shortened long bones.
[/not-level-membership-for-pediatrics-category]
