[level-membership-for-basic-science-category]
CHAPTER 19 Single-Gene Disorders
To date, more than 10,000 single-gene traits and disorders have been identified. Most of these are individually rare, but together they affect between 1% and 2% of the general population at any one time. The management of these disorders in affected individuals and in their extended families presents the major workload challenge in clinical genetics.
Huntington Disease
Genetics
Mapping and Isolation of the HD Gene
HD was one of the first disorders to be mapped by linkage analysis using polymorphic DNA markers when, in 1983, the disorder was found to show close linkage with a probe known as G8 on the short arm of chromosome 4, greatly aided by collection of blood samples from the huge pedigree containing over 100 affected subjects living on the shores of Lake Maracaibo in Venezuela. As well as providing the first means of predictive testing for HD, this work also revealed that HD homozygotes are no more severely affected than heterozygotes. This is in contrast to many other autosomal dominant disorders (p. 109). When the gene itself was isolated in 1993, it was found to contain a highly polymorphic CAG (polyglutamine) repeat sequence located in the 5′ region. The messenger RNA (mRNA) codes for a protein of approximately 350 kDa, known as huntingtin (also IT15). Huntingtin is expressed in many different cells throughout the central nervous system, as well as other tissues, although its function remains unclear.
The Mutation in HD
Almost all individuals with HD possess an expansion of a CAG polyglutamine (triplet) repeat sequence located in the 5′ region of the HD gene, a mutational mechanism first identified in humans in contrast to almost all other types of mutation that were first reported in other species such as Drosophila and mice. A joint working party of the American College of Medical Genetics and the American Society of Human Genetics recommended that HD genes should be categorized under four headings on the basis of CAG repeat length (Table 19.1).
Table 19.1 Comparison of Genetic Aspects of Huntington Disease and Myotonic Dystrophy
| Huntington Disease | Myotonic Dystrophy | |
|---|---|---|
| Inheritance | Autosomal dominant | Autosomal dominant |
| Chromosome locus | 4p16.3 | 19q13.3 |
| Trinucleotide repeat | CAG in 5′ translated region | CTG in 3′ untranslated region |
| Repeat sizes | Normal ≤26 | Normal <37 |
| Mutable 27–35 | ||
| Reduced penetrance | Full mutation | |
| 36–39 | 50–2000+ | |
| Fully penetrant ≥40 | ||
| Protein product | Huntingtin | MD protein kinase (DMPK) |
| Early-onset form | Juvenile | Congenital |
| Usually paternally transmitted | Usually maternally transmitted |
Parent of Origin Effect in Disease Transmission
Explanations for this include the possibility that expansion is caused by slippage (p. 24) of DNA polymerase, simply reflecting the number of mitoses undergone during gametogenesis (p. 41). An alternative possibility is based on the observation that huntingtin is expressed in oocytes, so that there could be selection against oocytes with large expansions as a consequence of preferential apoptosis.
Clinical Applications and Future Prospects
Prenatal diagnosis is possible for those couples who find this acceptable, although only about 25 such tests are performed in the United Kingdom annually. Obviously there are considerable emotional and ethical issues associated with termination of pregnancy; the condition is late in onset and the couple must consider the possibility of effective therapy being available in the foreseeable future. One appealing therapeutic approach is based on the observation that large CAG repeats result in intracellular accumulation of huntingtin ‘aggregates’, which are cleaved by a protease known as caspase to form a toxic product that causes cell death (apoptosis). Caspase inhibitors have been shown to have a beneficial effect in a HD mouse model. Another therapeutic approach under consideration is fetal neuronal cell transfer into regions of the brain, such as the caudate nucleus and putamen, which become atrophic in the early stages of the disease. This approach carries ethical considerations that will be difficult for some couples.
Myotonic Dystrophy
Myotonic dystrophy (MD) is the most common form of muscular dystrophy seen in adults, with an overall incidence of approximately 1 : 8000. It shares many features in common with HD (see Table 19.1)—both show autosomal dominant inheritance with anticipation, and an early-onset form with different clinical features. However, in MD the early-onset form is transmitted almost exclusively by the mother and presents at birth, in contrast to juvenile HD, which is generally paternally transmitted with an age of onset in the teens.
Clinical Features
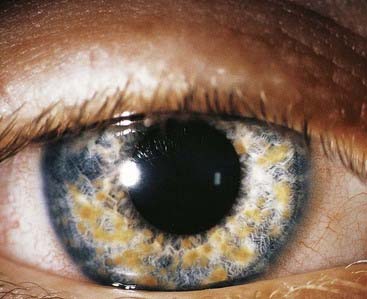
In contrast to most forms of muscular dystrophy, clinical features in MD are not limited exclusively to the neuromuscular system. Individuals with MD usually present in adult life with slowly progressive weakness and myotonia. This latter term refers to tonic muscle spasm with prolonged relaxation, which can manifest as a delay in releasing the grip on shaking hands. Other clinical features include cataracts (Figure 19.1), cardiac conduction defects, disturbed gastrointestinal peristalsis (dysphagia, constipation, diarrhea), weak sphincters, increased risk of diabetes mellitus and gallstones, somnolence, frontal balding, and testicular atrophy. The age of onset is very variable and in its mildest form usually runs a relatively benign course. However, as the age of onset becomes earlier, so the clinical symptoms increase in severity and more body systems are involved. In the ‘congenital’ form, affected babies present at birth with hypotonia, talipes, and respiratory distress that can prove life threatening (see Figure 7.19). Children who survive tend to show a lack of facial expression (‘myopathic facies’) with delayed motor development and learning difficulties (Figure 19.2).
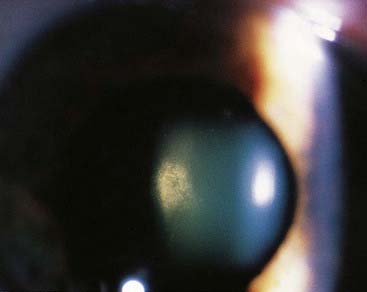
FIGURE 19.1 Refractile lens opacities in an asymptomatic person with myotonic dystrophy.
(Courtesy Mr. R. Doran and Mr. M. Geall, Department of Ophthalmology, General Infirmary, Leeds, UK.)

Genetics
It follows autosomal dominant inheritance with increasing severity in succeeding generations—anticipation (p. 120). It used to be thought that this reflected ascertainment bias, caused by the greater likelihood of detecting a mildly affected parent with a severely affected child, rather than the other way round. However, studies in the 1980s confirmed that anticipation is a real phenomenon.
Genotype–Phenotype Correlation in Myotonic Dystrophy
In unaffected persons the CTG sequence lying 3′ to the DMPK gene consists of up to 37 repeats (see Table 19.1). Affected individuals have an expansion of at least 50 copies of the CTG sequence. There is a close correlation between disease severity and the size of the expansion, which can exceed 2000 repeats. The severe congenital cases show the largest repeat copy number, with almost invariable inheritance from the mother. Thus, meiotic or germline instability is greater in the female for alleles containing large sequences. Curiously, expansion of a relatively small number of repeats appears to occur more commonly in the male, and most MD mutations are thought to have occurred originally during meiosis in the male. One possible explanation for these observations is that mature spermatozoa can carry only small expansions, whereas ova can accommodate much larger expansions.
Another puzzling feature of MD is the reported tendency for healthy individuals who are heterozygous for MD alleles in the normal size range to preferentially transmit alleles greater than 19 CTG repeats in size. This possible example of meiotic drive (p. 135) could explain the relatively high frequency of MD with constant replenishment of a reservoir of potential MD mutations.
Clinical Applications and Future Prospects
Presymptomatic genetic testing and prenatal diagnosis can be offered to those families for whom it is appropriate and acceptable. This is particularly relevant for couples who have had a child with the severe congenital form, for whom the risk of recurrence is relatively high. As in HD, presymptomatic testing should not be undertaken without an offer of long-term support and medical care, and a discussion about possible difficulty in obtaining life and health insurance (p. 366).
Hereditary Motor and Sensory Neuropathy
Clinical Features
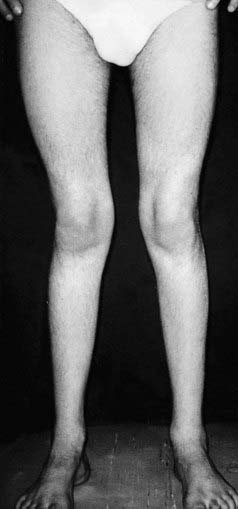
In autosomal dominant HMSN-I—the most common form—there is onset of slowly progressive distal muscle weakness and wasting in the lower limbs between the ages of 10 and 30 years, followed later by the upper limbs in many patients, and associated ataxia and tremor. The appearance of the lower limbs has been likened to that of an ‘inverted champagne bottle’ (Figure 19.3). With age, locomotion becomes more difficult and the feet tend to show exaggeration of their normal arch, known as ‘pes cavus’. Despite these quite striking changes, many patients retain reasonable muscle strength and are not too seriously disabled. Other faculties such as vision, hearing, and intellect are not impaired. Palpable thickening of peripheral nerves can sometimes be detected.
Genetics
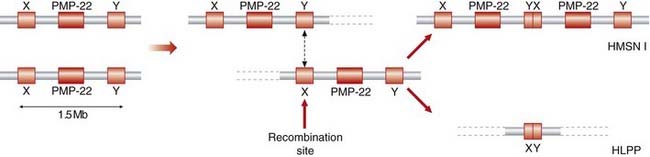
HMSN can show autosomal dominant, autosomal recessive or X-linked inheritance, although autosomal dominant forms are by far the most common. More than 70% of cases of HMSN-I are due to a DNA duplication of 1.5 Mb chromosome 17p that encompasses the peripheral myelin protein-22 (PMP22) gene. The glycoprotein product is present in the myelin membranes of peripheral nerves, where it helps to arrest Schwann cell division. HMSN-I in humans is therefore thought to be the result of a PMP22 dosage effect, though point mutations can be found some patients. The duplication is generated by misalignment and subsequent recombination between homologous sequences that flank the PMP22 gene (Figure 19.4); this event usually occurs in male gametogenesis (rather than in the female, which is the case in Duchenne muscular dystrophy [p. 307]). The reciprocal deletion product of this misaligned recombination event, giving rise to haploinsufficiency, causes a relatively mild disorder known as hereditary neuropathy with liability to pressure palsies. Minor nerve trauma, such as pressure from prolonged sitting on a long-haul flight, causes focal numbness and weakness. The same misalignment recombination mechanism occurs in Hb Lepore and anti-Lepore (see Figure 10.3; p. 157), congenital adrenal hyperplasia (p. 174), and deletion 22q11 syndrome (p. 282), to name but a few.
Other Forms of Hereditary Motor and Sensory Neuropathy
A more rare form of HMSN shows X-linked dominant inheritance, with males having typical HMSN-I features and females being more mildly affected—sometimes with HMSN-II characteristics. This is due to mutations in the gene that encodes a gap junction protein called GJB1 (previously Connexin 32).
Neurofibromatosis
There are two main types of neurofibromatosis, NF1 and NF2. Both conditions, especially NF2, could be included under familial cancer syndromes (see Chapter 14) but are covered in more detail here. NF1 has a birth incidence of approximately 1 : 3000; NF2 approximately 1 : 35,000 and a prevalence of around 1 : 200,000.
Clinical Features
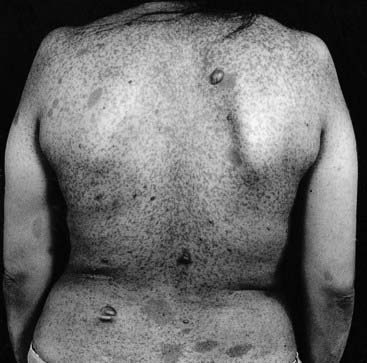
The most notable features of NF1 are small pigmented skin lesions, known as café-au-lait (CAL) spots, and small soft fleshy growths known as neurofibromata (Figure 19.5). CAL spots first appear in early childhood and continue to increase in both size and number until puberty. A minimum of six CAL spots at least 5 mm in diameter is required to support the diagnosis in childhood, and axillary and/or inguinal freckling should be present. Neurofibromata are benign tumors that arise most commonly in the skin, usually appearing in adolescence or adult life, and increasing in number with age.
Other clinical findings include relative macrocephaly (large head) and Lisch nodules. These are small harmless raised pigmented hamartomata of the iris (Figure 19.6). The most common complication, occurring in a third of childhood cases, is mild developmental delay characterized by a non-verbal learning disorder. For many, significant improvement is seen through the school years. Most individuals with NF1 enjoy a normal life and are not unduly inconvenienced by their condition. However, a small number of patients develop one or more major complications, such as epilepsy, a central nervous system tumor, or scoliosis.
Genetics
NF1 shows autosomal dominant inheritance with virtually 100% penetrance by the age of 5 years. The effects are very variable and affected members of the same family can show striking differences in disease severity. The features in affected MZ twins are usually very similar, so the variability in family members with the same mutation may be due to modifying genes at other loci. Approximately 50% of cases of NF1 are due to new mutations, with the estimated mutation rate being approximately 1 per 10,000 gametes. This is around 100 times greater than the average mutation rate per generation per locus in humans.
There are a few reports of more than one affected child born to unaffected parents—the result of gonadal mosaicism (p. 121), usually paternal in origin. Somatic mosaicism in NF1 can manifest with features limited to a particular part of the body. This is referred to as segmental NF.
The Neurofibromatosis Type 1 Gene and its Product
The NF1 gene, neurofibromin, was successfully mapped to chromosome 17, adjacent to the centromere, in 1987. Its isolation was aided by the identification of two patients who both had a balanced translocation with a breakpoint at 17q11.2. A cosmid clone was identified containing both translocation breakpoints and a search for transcripts from this region yielded four genes, one of which was shown to be neurofibromin. It is large, spanning over 350 kilobases (kb) of genomic DNA and comprising at least 59 exons. The other three genes identified in this region were found to lie within a single intron of the neurofibromin gene, where they are transcribed in the opposite direction from the complementary strand (p. 14).
The neurofibromin protein encoded by this gene shows structural homology to the guanosine triphosphatase (GTPase)-activating protein (GAP), which is important in signal transduction (p. 214) by downregulating RAS activity. The place of neurofibromin in the RAS-MAPK pathway is shown in Figure 16.12, highlighting the link with Noonan syndrome (p. 254). Loss of heterozygosity (p. 215) for chromosome 17 markers has been observed in several malignant tumors in patients with NF1, as well as in a small number of benign neurofibromata. These observations indicate that the neurofibromin gene functions as a tumor suppressor (p. 214). It has been shown to contain a GAP-related domain (GRD), which interacts with the RAS proto-oncogene product. A mRNA editing site exists in the neurofibromin gene and edited transcript causes GRD protein truncation, which inactivates the tumor suppressor function. A higher range of editing is seen in more malignant tumors.
Other genes, including TP53 (p. 218) on the short arm of chromosome 17, are also involved in tumor development and progression in NF1. Conversely, it is also known that the neurofibromin gene is implicated in the development of sporadic tumors not associated with NF, including carcinoma of the colon, neuroblastoma, and malignant melanoma. These observations confirm that the neurofibromin gene plays an important role in cell growth and differentiation.
Genotype–Phenotype Correlation
Many different mutations have been identified in the neurofibromin gene, which include deletions, insertions, duplications, and point substitutions (p. 23). Most lead to severe truncation of the protein or complete absence of gene expression. To date, there is little evidence for a clear genotype-phenotype relationship with the exception of one specific mutation, a 3-bp inframe deletion in exon 17, which has recurred in different cases and families, and affected individuals do not appear to develop cutaneous neurofibromata. Generally, NF1 shows quite striking intrafamilial variation, suggesting the possibility of modifier genes. Patients with large deletions that include the entire neurofibromin gene tend to be more severely affected, with significant intellectual impairment, a somewhat marfanoid habitus, and a larger than average number of cutaneous neurofibromata.
Clinical Applications and Future Prospects
At present there is no cure for NF1. Drug therapy aimed at upregulating neurofibromin GAP activity or downregulating RAS activity could prove beneficial in the absence of effective gene therapy. However, it is difficult to envisage how this could be applied to diverse target tissues, including the central nervous system. Nevertheless, there is great interest in the whole RAS-MAPK pathway (see Figure 16.12, p. 256), especially its role in tumor formation, and whether drugs can effectively modify its activity.
Marfan Syndrome
The original patient described by the French pediatrician Bernard Marfan, in 1896, probably had the similar but rarer condition now known as Beal syndrome, or congenital contractual arachnodactyly (p. 301). In clinical practice physicians often consider the diagnosis of Marfan syndrome (MFS) for any patient who is tall with subjective features of long limbs and fingers. However, it is essential to be objective in clinical assessment because a number of conditions have ‘marfanoid’ features, and many tall, thin people are entirely normal. Detailed diagnostic criteria, referred to as the Gent criteria, are in general use by geneticists (Table 19.2).
Table 19.2 Revised (Gent) Criteria for Making a Diagnosis of Marfan Syndrome
| System | Major Criteria | Minor Criteria |
|---|---|---|
| Skeletal | Four of these should be present: | |
| Pectus carinatum | Pectus excavatum | |
| Pectus excavatum requiring surgery | Joint hypermobility | |
| Reduced upper to lower segment body ratio or span : height ratio >1.05 | High arched palate with dental crowding | |
| Hypermobility of wrist and thumbs Medial displacement of medial malleolus |
Facial features, including down-slanting palpebral fissures causing pes planus | |
| Radiological protrusio acetabulae | ||
| Ocular | Ectopia lentis | Flat cornea Increased axial length of the globe Hypoplastic iris |
| Cardiovascular | Dilatation of the ascending aorta | Mitral valve prolapse |
| Dissection of the ascending aorta | Dilatation or dissection of descending thoracic or abdominal aorta under 50 years | |
| Pulmonary | None | Spontaneous pneumothorax Apical blebs |
| Skin/connective tissue | None | |
| Dura | Lumbosacral dural ectasia | None |
| Family history/genetics | First-degree relative who meets criteria | None |
| Presence of FBN1 mutation, or high-risk haplotype in MFS family | None |
Clinical Features
MFS is a disorder of fibrous connective tissue, specifically a defect in type 1 fibrillin, a glycoprotein encoded by the FBN1 gene. In the classic presentation affected individuals are tall compared with unaffected family members, have joint laxity, a span : height ratio greater than 1.05, a reduced upper to lower segment body ratio, pectus deformity, and scoliosis (Figure 19.7). The connective tissue defect gives rise to ectopia lentis (lens subluxation) in a proportion of (but not all) families and, very importantly, dilatation of the ascending aorta, which can lead to dissection. The latter complication is obviously life threatening, and for this reason alone care must be taken over the diagnosis. Aortic dilatation may be progressive but the rate of change can be reduced by β-adrenergic blockade (if tolerated) and there is great hope for angiotensin-II receptor antagonists (similar properties to angiotensin-converting enzyme inhibitors), whose trials are under way. Surgical replacement should be undertaken if the diameter reaches 50 to 55 mm. Pregnancy is a risk factor for a woman with MFS who already has some dilatation of the aorta, and monitoring is very important.


A diagnosis of MFS requires careful clinical assessment, body measurements looking for evidence of disproportion, echocardiography, ophthalmic evaluation, and, in some doubtful cases, lumbar magnetic resonance imaging to look for evidence of dural ectasia (see Table 19.2). The metacarpophalangeal index, a radiological measurement of the ratio of these hand bone lengths, does not feature in the revised criteria. Where the family history is non-contributory, a positive diagnosis is made when the patient has a minimum of two major criteria plus involvement of a third organ system; for a person with a close relative who is definitely affected, it is sufficient to have one major criterion plus involvement of a second organ system.
Genetics
MFS follows autosomal dominant inheritance and the majority of cases are linked to the large FBN1 gene on 15q21, with 65 exons spanning 200 kb and containing five distinct domains. The largest of these, occupying about 75% of the gene, comprises about 46 epidermal growth factor repeats (see p. 190). Finding the causative mutations in affected patients was initially very difficult, but hundreds have now been reported. Most are missense and have a dominant-negative effect, resulting in less than 35% of the expected amount of fibrillin in the extracellular matrix. Mutations have also occasionally been found in related phenotypes such as neonatal MFS, familial ectopia lentis, Shrintzen-Goldberg syndrome, and the MASS phenotype (mitral valve prolapse, myopia, borderline aortic enlargement, non-specific skin and skeletal findings).
Loeys-Dietz Syndrome
Familial aortic aneurysm is not confined to MFS and a new, distinct, condition has been delineated. This also follows autosomal dominant inheritance and aneurysms can be aggressive and occur before major aortic dilatation. Additional findings include cleft palate or bifid uvula, craniosynostosis, mental retardation, and generalized arterial tortuosity with aneurysms occurring elsewhere in the circulation. Some individuals have features overlapping with MFS but they do not fulfill the accepted Gent diagnostic criteria. The condition is now known as Loeys-Dietz syndrome, and the gene was identified through a candidate approach. Transforming growth factor (TGF) signaling (p. 85) had been shown to be important in vascular and craniofacial development in mouse models; this led Loeys and colleagues to sequence the TGF-β receptor 2 (TGFBR2) gene in a series of families. Heterozygous mutations were found in most of these, and in the others missense mutations were found in the related gene, TGFBR1.
Cystic Fibrosis
Clinical Features
In 85% of people with CF, pancreatic function is impaired, with reduced enzyme secretion from blockage of the pancreatic ducts by inspissated secretions. This leads to malabsorption with an increase in the fat content of the stools but is satisfactorily treated with oral supplements of pancreatic enzymes.
Genetics
Mapping and Isolation of the Cystic Fibrosis Gene
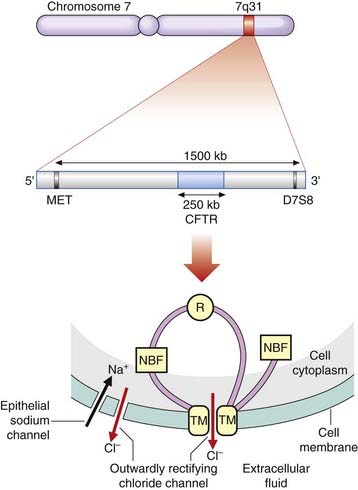
The mapping and isolation of the CF was a celebrated milestone in the history of human molecular genetics and it is easy to forget how very difficult and time consuming such research was just 25 years ago. The CF locus was mapped to chromosome 7q31 in 1985 by the demonstration of linkage to the gene for a polymorphic enzyme known as paraoxonase. Shortly afterward, two polymorphic DNA marker loci, known as MET and D7S8, were shown to be closely linked flanking markers. The region between these markers was scrutinized for the presence of HTF or CpG islands, which are known to be present close to the 5′ end of many genes (p. 75). This led to the identification of several new DNA markers that were shown to be very tightly linked to the CF locus with recombination frequencies of less than 1%. These loci were found to be in linkage disequilibrium (p. 138) with the CF locus, and one CF mutation was found to be associated with one particular haplotype in 84% of cases, consistent with the concept of a single original mutation being responsible for a large proportion of all CF genes. The identification of loci tightly linked to the CF locus narrowed its location down to a region of approximately 500 kb. The CF gene was eventually cloned by two groups of scientists in North America in 1989 by a combination of chromosome jumping, physical mapping, isolation of exon sequences and mutation analysis. It was named the CF transmembrane conductance regulator (CFTR) gene, spans a genomic region of approximately 250 kb, and contains 27 exons.
The Cystic Fibrosis Transmembrane Conductance Regulator Protein
The structure of CFTR is consistent with a protein product containing 1480 amino acids with a molecular weight of 168 kDa. It is thought to consist of two transmembrane (TM) domains that anchor it to the cell membrane, two nucleotide binding folds (NBFs) that bind ATP, and a regulatory (R) domain, which is phosphorylated by protein kinase-A (Figure 19.8).
Mutations in the Cystic Fibrosis Transmembrane Conductance Regulator Gene
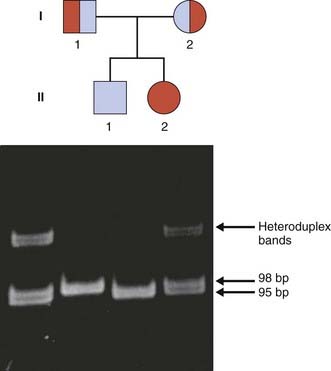
The first mutation to be identified in CFTR was a deletion of three adjacent base pairs at the 508th codon which results in the loss of a phenylalanine residue. This mutation is now known as Phe508del (previously deltaF508) and accounts for approximately 70% of all mutations in CFTR, the highest incidence of 88% being in Denmark (Table 19.3). The mutation can be demonstrated very simply by PCR using primers that flank the 508th codon (Figure 19.9).
Table 19.3 Contribution of Phe508del Mutation to All CF Mutations
| Country | % |
|---|---|
| Denmark | 88 |
| Netherlands | 79 |
| UK | 78 |
| Ireland | 75 |
| France | 75 |
| USA | 66 |
| Germany | 65 |
| Poland | 55 |
| Italy | 50 |
| Turkey | 30 |
Data from European Working Group on CF Genetics (EWGCFG) gradient of distribution in Europe of the major CF mutation and of its associated haplotype. Hum Genet 1990; 85:436–441, and worldwide survey of the Phe508del mutation—report from the Cystic Fibrosis Genetic Analysis Consortium. Am J Hum Genet 1990; 47:354–359
More than 1500 other mutations in the CFTR gene have been identified. These include missense, frameshift, splice-site, nonsense, and deletion mutations (p. 23). Most of these are extremely uncommon, although a few can account for a small but significant proportion of mutations in a particular population. For example, the G542X and G551D mutations account for 12% and 3%, respectively, of all CF mutations in the Ashkenazi Jewish and North American Caucasian populations. Commercial multiplex PCR-based kits have been developed that detect approximately 90% of all carriers. Using these it is possible to reduce the carrier risk for a healthy individual from a population risk of 1 in 25 to less than 1 in 200.
Genotype-Phenotype Correlation
Mutations in CFTR can influence the function of the protein product by:
The relationship between genotype and phenotype is complex. Homozygotes for Phe508del almost always have severe classical CF, as do compound heterozygotes with Phe508del and G551D or G542X. The outcome for other compound heterozygote combinations can be much more difficult to predict. The complexity of the interaction between CFTR alleles is illustrated by the IVS8-6 poly T variant. This contains a polythymidine tract in intron 8 that influences the splicing efficiency of exon 9, resulting in reduced synthesis of normal CFTR protein. Three variants consisting of 5T, 7T, and 9T have been identified. The 9T variant is associated with normal activity but the 5T allele leads to a reduction in the number of transcripts containing exon 9. The 5T variant has a population frequency of approximately 5%, but is more often found in patients with CBAVD (40–50%) or disseminated bronchiectasis (30%). Curiously, it has been shown that the number of thymidine residues influences the effect of another mutation, R117H. When R117H is in cis with 5T (i.e., in the same allele) it causes the PS (pancreatic sufficiency) form of CF when another CF mutation is present on the other allele. However, in compound heterozygotes (e.g., Phe508del /R117H) where R117H is in cis with 7T, it can result in a milder but variable phenotype, ranging from CBAVD to PS CF. The milder phenotype is likely to result from the expression of higher levels of full-length R117H protein with some residual activity. The increasing number of CFTR mutations and variability of the associated phenotypes has led some authors to propose a spectrum of ‘CFTR disease’, recognizing that a label of CF may be inappropriate for patients with milder symptoms.
Clinical Applications and Future Prospects
Before the mapping of the CF locus and the subsequent isolation of CFTR, it was not possible to offer either carrier detection or reliable prenatal diagnosis. Now parents of an affected child can almost always be offered prenatal diagnosis by direct mutation analysis. Similarly, knowledge of one or both of the mutations in an affected child now permits the offer of carrier detection to close family relatives. In many parts of the world, it is now standard practice to offer cascade screening to all families in which a mutation has been identified. Population screening for carriers of CF (p. 318) and neonatal screening for CF homozygotes (p. 320) have been widely implemented.
Inherited Cardiac Arrhythmias and Cardiomyopathies
Inherited Arrhythmias
Clinical Features
In LQTS, also known as Romano-Ward syndrome, the ECG findings are dominated by, as the name suggests, a QT interval outside the normal limits, remaining long when the heart rate increases. They are classified according to the gene involved (Table 19.4). The inheritance is overwhelmingly autosomal dominant but a rare recessive form exists, combined with sensorineural deafness, which is known as Jervell and Lange-Nielsen syndrome. The ECG changes may be evident from a young age and a cardiac event occurs by age 10 years in about 50%, and by age 20 years in 90%. First cardiac events tend to be later in LQT2 and LQT3. Predictive genetic testing, where possible, is helpful to identify those at risk in affected families, and decisions about prophylactic β-blockade can be made. β-Blockers are particularly useful in LQT1 but less so in LQT2 and LQT3; indeed, it is possible that β-blockers may be harmful in LQT3.
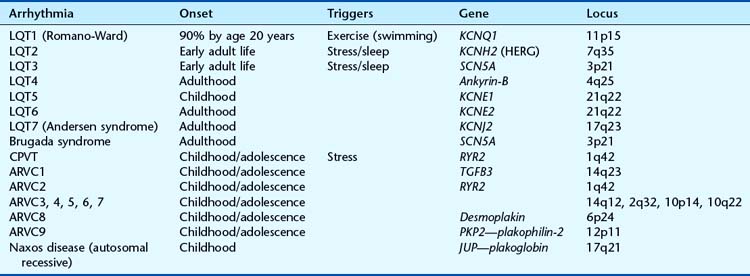
Brugada syndrome also follows autosomal dominant inheritance and was first described in 1992. The cardiac event is characterized by a proneness to idiopathic ventricular tachycardia (VT), and there may be abnormal ST-wave elevation in the right chest leads with incomplete right bundle branch block. In at-risk family members with a normal ECG, the characteristic abnormalities can usually be unmasked by the administration of potent sodium channel blockers such as flecainide. The condition is relatively common in Southeast Asia; there is a male predominance of 8 : 1, and the average age of arrhythmic events is 40 years. The definitive treatment is an implantable defibrillator and exercise is not a particular risk factor. Mutations in the SCN5A gene are found in about 20% of Brugada syndrome patients, as well as some cases of LQT3 (see Table 19.4). In some families both arrhythmias occur.
ARVC, which follows mainly dominant inheritance, is characterized by localized or diffuse atrophy and fatty infiltration of the right ventricular myocardium. It can lead to VT and sudden cardiac death in young people, especially athletes with apparently normal hearts. The ECG shows right precordial T-wave inversion and prolongation of the QRS complex. ARVC appears to demonstrate substantial genetic heterogeneity (see Table 19.4) with five genes identified, one of which, encoding plakoglobin, is implicated in the rare recessive form found on the island of Naxos. The RYR2 gene, for ARVC2, is also mutated in catecholaminergic polymorphic ventricular tachycardia (CPVT), also known as Coumel’s VT. Individuals with CPVT present with syncopal events, sometimes in childhood or adolescence, and reproducible stress-induced ventricular tachycardia, without a prolonged QT interval; the heart is structurally normal.
Genetics
These are genetically heterogeneous conditions. Nearly all follow autosomal dominant inheritance; the genes and their loci are summarized in Table 19.4. In some cases, however, there is evidence for biallelic inheritance (p. 119)—i.e., patients require mutations at two different loci to have clinical symptoms and ECG changes. This poses very significant difficulties in relation to genetic testing strategies, interpretation of mutation tests, and the usefulness of predictive genetic testing based on the findings at one locus. The same problem may also apply to some cases of cardiomyopathy.
Inherited Cardiomyopathies
Dilated cardiomyopathy is characterized by cardiac dilatation and reduced systolic function. Causes include myocarditis, coronary artery disease, systemic and metabolic diseases, and toxins. When these are excluded the prevalence of idiopathic dilated cardiomyopathy is 35 to 40 per 100,000 and familial cases account for about 25%. As with the inherited cardiac arrhythmias, they are genetically heterogeneous but nearly always follow autosomal dominant inheritance. They are also very variable, and within the same family affected members may show symptoms in childhood at one end of the spectrum, whereas in other individuals the onset of cardiac symptoms may not occur until late in adult life. At least 10 different loci have been mapped in different family studies. One cause is the result of mutations in the LMNA gene (which encodes lamin A/C), noted for its pleiotropic effects (p. 112), of which dilated cardiomyopathy is one and may occasionally be isolated.
Genetic testing is now available within clinical services, but the vast genetic heterogeneity means that the pick-up rate for mutations is low. After a diagnosis has been made in an index case, a detailed family history is indicated and investigation by ECG and echocardiogram should be offered. Screening may need to continue well into adult life. Among the causes of cardiomyopathy that can be detected relatively easily by a biochemical test is X-linked fabry disease, for which enzyme replacement is available (see Table 23.1; p. 350).
Spinal Muscular Atrophy
Clinical Features
Genetics
Mapping and Isolating the Spinal Muscular Atrophy Gene
All three childhood forms of SMA were mapped to chromosome 5q in 1990 using linkage. Detailed mapping narrowed the locus to a 1000 kb consisting of a 500-kb inverted duplication (Figure 19.10). This region is noted for its high rate of instability, with several DNA duplications and a relatively large number of pseudogenes (p. 17).
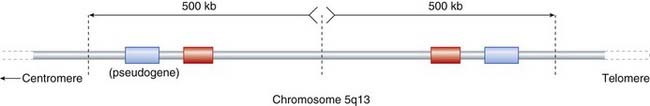
Within the candidate region two distinct genes were isolated that show a high incidence of deletion in patients with SMA—SMN and NAIP—each present as two almost identical copies. The SMN genes are now referred to as SMN1 and SMN2 (the pseudogene of SMN1 that shares ~99% homology). SMN1 shows homozygous deletion of exons 7–8 in 95% to 98% of all patients with childhood-onset SMA. Point mutations in SMN1 have been identified in 1% to 2% of patients with childhood SMA who do not show the exons 7–8 deletion on one allele. The number of copies of SMN2, arranged in tandem in cis configuration on each chromosome, varies between zero and five. It produces a similar transcript to SMN1 but this is not sufficient to fully compensate. Nevertheless, the presence of copies of SMN2 modifies the phenotype, causing milder forms of SMA.
Duchenne Muscular Dystrophy
Clinical Features
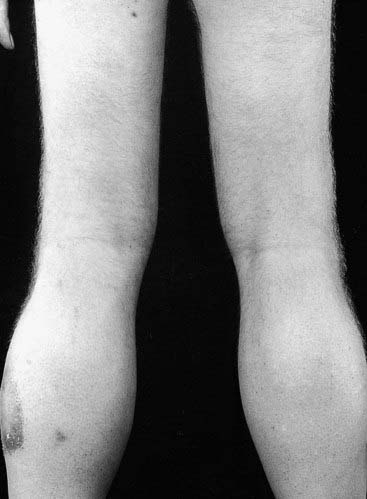
On examination, boys with DMD show an apparent increase in the size of the calf muscles, which is actually due to replacement of muscle fibers by fat and connective tissue—referred to as pseudohypertrophy (Figure 19.11). DMD is sometimes known as pseudohypertrophic muscular dystrophy. In addition, approximately one-third of boys with DMD show mild-moderate intellectual impairment, with the mean IQ being 83.
Genetics
Both DMD and BMD show X-linked recessive inheritance. Males with DMD rarely, if ever, reproduce. Therefore, as genetic fitness equals zero, the mutation rate equals the incidence in affected males divided by 3 (p. 133), which approximates to 1 : 10,000—one of the highest known mutation rates in humans.
Isolation of the Gene for DMD
The isolation of the gene for DMD—the dystrophin gene—represented a major scientific achievement at the time, because of a successfully applied positional cloning strategy. The initial clue to the site of the DMD locus was provided by reports of several females affected with DMD who had a balanced X-autosome translocation with a common X-chromosome breakpoint at Xp21. In these women, those cells in which the derivative X chromosome is randomly inactivated are at a major disadvantage because of inactivation of the autosomal segment (Figure 7.16; p. 117). Consequently, cells in which the normal X chromosome has been randomly inactivated are more likely to survive. The net result is that the derivative X autosome is active in most cell lines, and if the breakpoint has damaged an important gene, in this case dystrophin, the woman will be affected by the disease in question.
Mutations in the Dystrophin Gene
Deletions that cause DMD usually disturb the translational reading frame (p. 20), but those seen in males with BMD usually do not alter the reading frame (i.e., they are ‘in-frame’). This means that the amino-acid sequence of the protein product downstream of the deletion is normal, explaining the relatively mild features in BMD. Mutations in the remaining one-third of boys with DMD include stop codons, frameshift mutations, altered splicing signals and promoter mutations. Most lead to premature translational termination, resulting in the production of little, if any, protein product. In contrast to deletions, point mutations in the dystrophin gene often arise in paternal meiosis, most probably because of a copy error in DNA replication. Full sequencing of the dystrophin gene is now available as a service, which has transformed molecular diagnosis of DMD and carrier detection.
The Gene Product Dystrophin
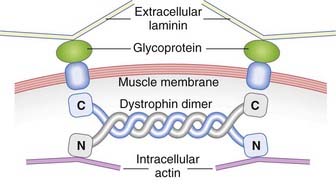
Dystrophin binds to a glycoprotein complex in the muscle membrane through its C-terminal domain (Figure 19.12). This glycoprotein complex consists of several subunits, abnormalities of which cause other rare genetic muscle disorders, including several different types of autosomal recessive limb girdle muscular dystrophy, as well as congenital muscular dystrophy.
Carrier Detection
Before DNA analysis, carrier detection was based on pedigree information combined with serum creatine kinase (CK) assay (p. 314). CK levels are grossly increased in boys with DMD, and marginally raised in approximately two-thirds of all carriers (see Figure 20.1; p. 314). CK levels are only occasionally useful today, as DNA testing has become increasingly sophisticated. Linkage studies may still be useful in circumstances where no DNA is available from an affected male, but perhaps available from normal males in the same family; each situation has to be assessed individually. Care has to be taken when using linkage for carrier detection because of the high recombination rate of 12% across the DMD gene.
Prospects for Treatment
Gene therapy offers the only realistic hope of a cure in the short to medium term. Several approaches have been tried experimentally in transgenic and naturally occurring mutant mice with dystrophin-negative muscular dystrophy. These include direct injection of recombinant DNA, myoblast implantation, and transfection with retroviral or adenoviral vectors carrying a dystrophin minigene containing only those sequences that code for the important functional domains. One approach is antisense technology to block an exon splicing enhancer sequence and generate a protein with an in-frame deletion that encodes a protein with some residual function (i.e., a BMD rather than a DMD phenotype). That mice with dystrophin-negative muscular dystrophy can show spontaneous muscle repair indicates that there could be a way of switching on an alternative compensatory protein such as utrophin. This is expressed in the fetus rather than dystrophin, with which it shares a large degree of homology. Genetically engineered mice deficient for both dystrophin and utrophin develop a typical DMD dystrophy. If the utrophin gene could be reactivated as in the dystrophin-negative mouse, this may have a therapeutic benefit.
Hemophilia
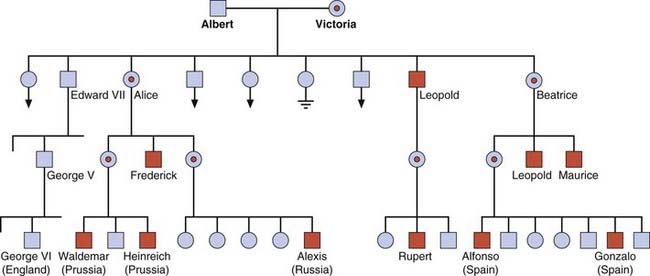
There are two forms of hemophilia: A and B. Hemophilia A is the most common severe inherited coagulation disorder, with an incidence of 1 : 5000 males. It is caused by a deficiency of factor VIII, which, together with factor IX, plays a critical role in the intrinsic pathway activation of prothrombin to thrombin. Thrombin then converts fibrinogen to fibrin, which forms the structural framework of clotted blood. The existence of hemophilia was recognized in the Talmud, and the tendency for males to be affected much more often than females was acknowledged by the Jewish authorities 2000 years ago when they excused from circumcising the sons of the sisters of a mother who had an affected son. Queen Victoria was a carrier and, as well as having an affected son—Leopold Duke of Albany—she transmitted the disorder through two of her daughters to most of the royal families of Europe (Figure 19.13).
Clinical Features
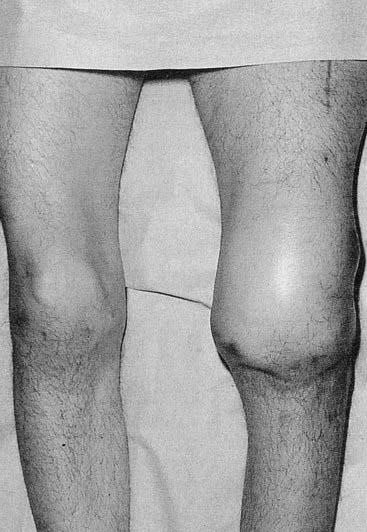
These are similar in both forms of hemophilia and vary from mild bleeding following major trauma or surgery to spontaneous hemorrhage into muscles and joints. The degree of severity shows a close correlation with the reduction in factor VIII or IX activity. Levels below 1% are usually associated with a severe hemorrhagic tendency from birth. Hemorrhage into joints causes severe pain and swelling which, if recurrent, causes a progressive arthropathy with severe disability (Figure 19.14). Affected family members generally show the same degree of severity.
Genetics
Hemophilia A
The factor VIII comprises 26 exons and spans 186 kb with a 9-kb mRNA transcript. Deletions account for 5% of all cases and usually cause complete absence of factor VIII expression. In addition, hundreds of frameshift, nonsense, and missense mutations have been described, besides insertions and a ‘flip’ inversion, which represented a new form of mutation when first identified in hemophilia A in 1993. Inversions account for 50% of all severe cases with <1% factor VIII activity. They are caused by recombination between a small gene called A located within intron 22 of the factor VIII gene and other copies of the A gene, which are located upstream near the telomere (Figure 19.15). The inversion disrupts the factor VIII gene, resulting in very low factor VIII activity. The genetic test is straightforward but detection of the numerous other mutations may require direct sequencing.
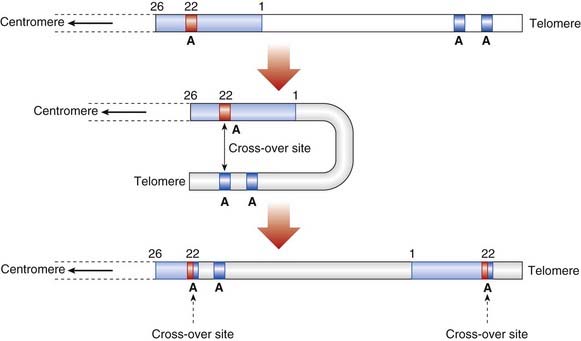
As in DMD, point mutations usually originate in male germ cells whereas deletions arise mainly in the female. The ‘flip’ inversions show a greater than 10-fold higher mutation rate in male compared with female germ cells, probably because Xq does not pair with a homologous chromosome in male meiosis—so that there is much greater opportunity for intrachromosomal recombination to occur via looping of the distal long arm (see Figure 19.15).
Biros I, Forrest S. Spinal muscular atrophy: untangling the knot? J Med Genet. 1999;36:1-8.
Bolton-Maggs PHB, Pasi KJ. Haemophilias A and B. Lancet. 2003;361:1801-1809.
Brown T, Schwind EL. Update and review: cystic fibrosis. J Genet Counseling. 1999;8:137-162.
A useful review of recent genetic developments in cystic fibrosis.
Collinge J. Human prion diseases and bovine spongiform encephalopathy (BSE). Hum Mol Genet. 1997;6:1699-1705.
A clear account of human prion diseases and their known causes with particular reference to BSE.
De Paepe A, Devereux RB, Hennekam RCM, et al. Revised diagnostic criteria for the Marfan syndrome. Am J Med Genet. 1996;62:417-426.
Essential reading for those required to make a diagnosis of Marfan syndrome.
Emery AEH. Duchenne muscular dystrophy, 2nd edn. Oxford, UK: Oxford University Press; 1993.
Harper PS. Huntington’s disease, 2nd ed. London: WB Saunders; 1996.
A comprehensive review of the clinical and genetic aspects of Huntington disease.
Harper PS. Myotonic Dystrophy, 3rd ed. London: WB Saunders; 2001.
A comprehensive review of the clinical and genetic aspects of myotonic dystrophy.
Huson SM, Hughes RAC, editors. The neurofibromatoses. London: Chapman & Hall, 1994.
Karpati G, Pari G, Molnar MJ. Molecular therapy for genetic muscle diseases—status 1999. Clin Genet. 1999;55:1-8.
Kay MA, Manno CS, Ragni MV, et al. Evidence for gene transfer and expression of factor IX in haemophilia B patients treated with an AV vector. Nature Genet. 2000;24:257-261.
Report of provisional encouraging results of gene therapy in patients with hemophilia B.
Lakich D, Kazazian HH, Antonarakis SE, Gitschier J. Inversions disrupting the factor VIII gene are a common cause of severe haemophilia A. Nature Genet. 1993;5:236-241.
The first report showing how the common ‘flip’ inversion is generated.
Elements
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
CHAPTER 19 Single-Gene Disorders
To date, more than 10,000 single-gene traits and disorders have been identified. Most of these are individually rare, but together they affect between 1% and 2% of the general population at any one time. The management of these disorders in affected individuals and in their extended families presents the major workload challenge in clinical genetics.
Huntington Disease
Genetics
Mapping and Isolation of the HD Gene
HD was one of the first disorders to be mapped by linkage analysis using polymorphic DNA markers when, in 1983, the disorder was found to show close linkage with a probe known as G8 on the short arm of chromosome 4, greatly aided by collection of blood samples from the huge pedigree containing over 100 affected subjects living on the shores of Lake Maracaibo in Venezuela. As well as providing the first means of predictive testing for HD, this work also revealed that HD homozygotes are no more severely affected than heterozygotes. This is in contrast to many other autosomal dominant disorders (p. 109). When the gene itself was isolated in 1993, it was found to contain a highly polymorphic CAG (polyglutamine) repeat sequence located in the 5′ region. The messenger RNA (mRNA) codes for a protein of approximately 350 kDa, known as huntingtin (also IT15). Huntingtin is expressed in many different cells throughout the central nervous system, as well as other tissues, although its function remains unclear.
The Mutation in HD
Almost all individuals with HD possess an expansion of a CAG polyglutamine (triplet) repeat sequence located in the 5′ region of the HD gene, a mutational mechanism first identified in humans in contrast to almost all other types of mutation that were first reported in other species such as Drosophila and mice. A joint working party of the American College of Medical Genetics and the American Society of Human Genetics recommended that HD genes should be categorized under four headings on the basis of CAG repeat length (Table 19.1).
Table 19.1 Comparison of Genetic Aspects of Huntington Disease and Myotonic Dystrophy
| Huntington Disease | Myotonic Dystrophy | |
|---|---|---|
| Inheritance | Autosomal dominant | Autosomal dominant |
| Chromosome locus | 4p16.3 | 19q13.3 |
| Trinucleotide repeat | CAG in 5′ translated region | CTG in 3′ untranslated region |
| Repeat sizes | Normal ≤26 | Normal <37 |
| Mutable 27–35 | ||
| Reduced penetrance | Full mutation | |
| 36–39 | 50–2000+ | |
| Fully penetrant ≥40 | ||
| Protein product | Huntingtin | MD protein kinase (DMPK) |
| Early-onset form | Juvenile | Congenital |
| Usually paternally transmitted | Usually maternally transmitted |
Parent of Origin Effect in Disease Transmission
Explanations for this include the possibility that expansion is caused by slippage (p. 24) of DNA polymerase, simply reflecting the number of mitoses undergone during gametogenesis (p. 41). An alternative possibility is based on the observation that huntingtin is expressed in oocytes, so that there could be selection against oocytes with large expansions as a consequence of preferential apoptosis.
Clinical Applications and Future Prospects
Prenatal diagnosis is possible for those couples who find this acceptable, although only about 25 such tests are performed in the United Kingdom annually. Obviously there are considerable emotional and ethical issues associated with termination of pregnancy; the condition is late in onset and the couple must consider the possibility of effective therapy being available in the foreseeable future. One appealing therapeutic approach is based on the observation that large CAG repeats result in intracellular accumulation of huntingtin ‘aggregates’, which are cleaved by a protease known as caspase to form a toxic product that causes cell death (apoptosis). Caspase inhibitors have been shown to have a beneficial effect in a HD mouse model. Another therapeutic approach under consideration is fetal neuronal cell transfer into regions of the brain, such as the caudate nucleus and putamen, which become atrophic in the early stages of the disease. This approach carries ethical considerations that will be difficult for some couples.
Myotonic Dystrophy
Myotonic dystrophy (MD) is the most common form of muscular dystrophy seen in adults, with an overall incidence of approximately 1 : 8000. It shares many features in common with HD (see Table 19.1)—both show autosomal dominant inheritance with anticipation, and an early-onset form with different clinical features. However, in MD the early-onset form is transmitted almost exclusively by the mother and presents at birth, in contrast to juvenile HD, which is generally paternally transmitted with an age of onset in the teens.
Clinical Features
In contrast to most forms of muscular dystrophy, clinical features in MD are not limited exclusively to the neuromuscular system. Individuals with MD usually present in adult life with slowly progressive weakness and myotonia. This latter term refers to tonic muscle spasm with prolonged relaxation, which can manifest as a delay in releasing the grip on shaking hands. Other clinical features include cataracts (Figure 19.1), cardiac conduction defects, disturbed gastrointestinal peristalsis (dysphagia, constipation, diarrhea), weak sphincters, increased risk of diabetes mellitus and gallstones, somnolence, frontal balding, and testicular atrophy. The age of onset is very variable and in its mildest form usually runs a relatively benign course. However, as the age of onset becomes earlier, so the clinical symptoms increase in severity and more body systems are involved. In the ‘congenital’ form, affected babies present at birth with hypotonia, talipes, and respiratory distress that can prove life threatening (see Figure 7.19). Children who survive tend to show a lack of facial expression (‘myopathic facies’) with delayed motor development and learning difficulties (Figure 19.2).
FIGURE 19.1 Refractile lens opacities in an asymptomatic person with myotonic dystrophy.
(Courtesy Mr. R. Doran and Mr. M. Geall, Department of Ophthalmology, General Infirmary, Leeds, UK.)
Genetics
It follows autosomal dominant inheritance with increasing severity in succeeding generations—anticipation (p. 120). It used to be thought that this reflected ascertainment bias, caused by the greater likelihood of detecting a mildly affected parent with a severely affected child, rather than the other way round. However, studies in the 1980s confirmed that anticipation is a real phenomenon.
Genotype–Phenotype Correlation in Myotonic Dystrophy
In unaffected persons the CTG sequence lying 3′ to the DMPK gene consists of up to 37 repeats (see Table 19.1). Affected individuals have an expansion of at least 50 copies of the CTG sequence. There is a close correlation between disease severity and the size of the expansion, which can exceed 2000 repeats. The severe congenital cases show the largest repeat copy number, with almost invariable inheritance from the mother. Thus, meiotic or germline instability is greater in the female for alleles containing large sequences. Curiously, expansion of a relatively small number of repeats appears to occur more commonly in the male, and most MD mutations are thought to have occurred originally during meiosis in the male. One possible explanation for these observations is that mature spermatozoa can carry only small expansions, whereas ova can accommodate much larger expansions.
Another puzzling feature of MD is the reported tendency for healthy individuals who are heterozygous for MD alleles in the normal size range to preferentially transmit alleles greater than 19 CTG repeats in size. This possible example of meiotic drive (p. 135) could explain the relatively high frequency of MD with constant replenishment of a reservoir of potential MD mutations.
Clinical Applications and Future Prospects
Presymptomatic genetic testing and prenatal diagnosis can be offered to those families for whom it is appropriate and acceptable. This is particularly relevant for couples who have had a child with the severe congenital form, for whom the risk of recurrence is relatively high. As in HD, presymptomatic testing should not be undertaken without an offer of long-term support and medical care, and a discussion about possible difficulty in obtaining life and health insurance (p. 366).
Hereditary Motor and Sensory Neuropathy
Clinical Features
In autosomal dominant HMSN-I—the most common form—there is onset of slowly progressive distal muscle weakness and wasting in the lower limbs between the ages of 10 and 30 years, followed later by the upper limbs in many patients, and associated ataxia and tremor. The appearance of the lower limbs has been likened to that of an ‘inverted champagne bottle’ (Figure 19.3). With age, locomotion becomes more difficult and the feet tend to show exaggeration of their normal arch, known as ‘pes cavus’. Despite these quite striking changes, many patients retain reasonable muscle strength and are not too seriously disabled. Other faculties such as vision, hearing, and intellect are not impaired. Palpable thickening of peripheral nerves can sometimes be detected.
Genetics
HMSN can show autosomal dominant, autosomal recessive or X-linked inheritance, although autosomal dominant forms are by far the most common. More than 70% of cases of HMSN-I are due to a DNA duplication of 1.5 Mb chromosome 17p that encompasses the peripheral myelin protein-22 (PMP22) gene. The glycoprotein product is present in the myelin membranes of peripheral nerves, where it helps to arrest Schwann cell division. HMSN-I in humans is therefore thought to be the result of a PMP22 dosage effect, though point mutations can be found some patients. The duplication is generated by misalignment and subsequent recombination between homologous sequences that flank the PMP22 gene (Figure 19.4); this event usually occurs in male gametogenesis (rather than in the female, which is the case in Duchenne muscular dystrophy [p. 307]). The reciprocal deletion product of this misaligned recombination event, giving rise to haploinsufficiency, causes a relatively mild disorder known as hereditary neuropathy with liability to pressure palsies. Minor nerve trauma, such as pressure from prolonged sitting on a long-haul flight, causes focal numbness and weakness. The same misalignment recombination mechanism occurs in Hb Lepore and anti-Lepore (see Figure 10.3; p. 157), congenital adrenal hyperplasia (p. 174), and deletion 22q11 syndrome (p. 282), to name but a few.
Other Forms of Hereditary Motor and Sensory Neuropathy
A more rare form of HMSN shows X-linked dominant inheritance, with males having typical HMSN-I features and females being more mildly affected—sometimes with HMSN-II characteristics. This is due to mutations in the gene that encodes a gap junction protein called GJB1 (previously Connexin 32).
Neurofibromatosis
There are two main types of neurofibromatosis, NF1 and NF2. Both conditions, especially NF2, could be included under familial cancer syndromes (see Chapter 14) but are covered in more detail here. NF1 has a birth incidence of approximately 1 : 3000; NF2 approximately 1 : 35,000 and a prevalence of around 1 : 200,000.
Clinical Features
The most notable features of NF1 are small pigmented skin lesions, known as café-au-lait (CAL) spots, and small soft fleshy growths known as neurofibromata (Figure 19.5). CAL spots first appear in early childhood and continue to increase in both size and number until puberty. A minimum of six CAL spots at least 5 mm in diameter is required to support the diagnosis in childhood, and axillary and/or inguinal freckling should be present. Neurofibromata are benign tumors that arise most commonly in the skin, usually appearing in adolescence or adult life, and increasing in number with age.
Other clinical findings include relative macrocephaly (large head) and Lisch nodules. These are small harmless raised pigmented hamartomata of the iris (Figure 19.6). The most common complication, occurring in a third of childhood cases, is mild developmental delay characterized by a non-verbal learning disorder. For many, significant improvement is seen through the school years. Most individuals with NF1 enjoy a normal life and are not unduly inconvenienced by their condition. However, a small number of patients develop one or more major complications, such as epilepsy, a central nervous system tumor, or scoliosis.
Genetics
NF1 shows autosomal dominant inheritance with virtually 100% penetrance by the age of 5 years. The effects are very variable and affected members of the same family can show striking differences in disease severity. The features in affected MZ twins are usually very similar, so the variability in family members with the same mutation may be due to modifying genes at other loci. Approximately 50% of cases of NF1 are due to new mutations, with the estimated mutation rate being approximately 1 per 10,000 gametes. This is around 100 times greater than the average mutation rate per generation per locus in humans.
There are a few reports of more than one affected child born to unaffected parents—the result of gonadal mosaicism (p. 121), usually paternal in origin. Somatic mosaicism in NF1 can manifest with features limited to a particular part of the body. This is referred to as segmental NF.
The Neurofibromatosis Type 1 Gene and its Product
The NF1 gene, neurofibromin, was successfully mapped to chromosome 17, adjacent to the centromere, in 1987. Its isolation was aided by the identification of two patients who both had a balanced translocation with a breakpoint at 17q11.2. A cosmid clone was identified containing both translocation breakpoints and a search for transcripts from this region yielded four genes, one of which was shown to be neurofibromin. It is large, spanning over 350 kilobases (kb) of genomic DNA and comprising at least 59 exons. The other three genes identified in this region were found to lie within a single intron of the neurofibromin gene, where they are transcribed in the opposite direction from the complementary strand (p. 14).
The neurofibromin protein encoded by this gene shows structural homology to the guanosine triphosphatase (GTPase)-activating protein (GAP), which is important in signal transduction (p. 214) by downregulating RAS activity. The place of neurofibromin in the RAS-MAPK pathway is shown in Figure 16.12, highlighting the link with Noonan syndrome (p. 254). Loss of heterozygosity (p. 215) for chromosome 17 markers has been observed in several malignant tumors in patients with NF1, as well as in a small number of benign neurofibromata. These observations indicate that the neurofibromin gene functions as a tumor suppressor (p. 214). It has been shown to contain a GAP-related domain (GRD), which interacts with the RAS proto-oncogene product. A mRNA editing site exists in the neurofibromin gene and edited transcript causes GRD protein truncation, which inactivates the tumor suppressor function. A higher range of editing is seen in more malignant tumors.
Other genes, including TP53 (p. 218) on the short arm of chromosome 17, are also involved in tumor development and progression in NF1. Conversely, it is also known that the neurofibromin gene is implicated in the development of sporadic tumors not associated with NF, including carcinoma of the colon, neuroblastoma, and malignant melanoma. These observations confirm that the neurofibromin gene plays an important role in cell growth and differentiation.
Genotype–Phenotype Correlation
Many different mutations have been identified in the neurofibromin gene, which include deletions, insertions, duplications, and point substitutions (p. 23
[/not-level-membership-for-basic-science-category]
