Sickle cell anemia: Anesthetic implications
Although the term sickle cell crisis is commonly used, in reality, a variety of crises are subsumed under this term, including vaso-occlusive, splenic sequestration, aplastic, and hemolytic crises. Patients with SCA have increased perioperative morbidity and mortality rates, as compared with the general population, likely due to vaso-occlusion from sickled erythrocytes, resulting in acute tissue injury and chronic organ damage (Table 206-1). People with SCA also undergo more operations and at an earlier age; the most commonly performed surgical procedures are cholecystectomy (due to an increased rate of formation of pigmented gallstones and cholelithiasis in this population), splenectomy (because of splenic sequestration and splenic pooling), and hip arthroplasty (related to the 50% rate of osteonecrosis in the femoral head among individuals with SCA who are 35 years of age and older). In addition, postoperative hospital length of stay is typically longer in this population.
Table 206-1
Effects of Vaso-occlusive Insults from Sickled Erythrocytes on Organ Systems in Patients with Sickle Cell Disease
| System | Effect | Cause | Finding(s) |
| Cardiac | Cardiomegaly Hyperdynamic circulation | Long-term increased cardiac output and gradual occlusion of pulmonary vascular bed | Full pulses Murmurs |
| Pulmonary | ↓ Total lung capacity ↓ Vital capacity ↑ Risk of contracting pneumococcal pneumonia |
In patients with SCD, 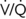 mismatch and pulmonary sickling add to their likelihood of developing pulmonary infarction and infection perioperatively. When compared with the general population, these patients have a 10-fold higher risk of serious perioperative pulmonary complications. mismatch and pulmonary sickling add to their likelihood of developing pulmonary infarction and infection perioperatively. When compared with the general population, these patients have a 10-fold higher risk of serious perioperative pulmonary complications. |
Resting SaO2 70-90 mm Hg |
| Renal | Papillary necrosis and nephrotic syndrome | The relatively hypoxic, hypertonic, and acidotic environment in the medulla leads to sickling of RBCs with consequent vaso-occlusion, resulting in decreased renal medullary blood flow. Hematuria, which often occurs in patients with sickle cell nephropathy, increases venous pressure, further worsening the ischemia of the renal medulla and predisposing the patient to further RBC sickling. |
Large dilute urine volume |
| Hepatic | Cirrhosis, hepatitis, hepatic sequestration, hepatosplenomegaly, and cholelithiasis Hemosiderosis Hemochromatosis | Both the vascular complications from the sickling process itself and the fact that patients with SCD have often received multiple transfusions increase their risk for developing viral hepatitis, iron overload, and (combined with the effects of chronic hemolysis) pigmented gallstones. | A combination of the following findings, depending on the hepatic cause: fever, pain, jaundice, elevated AST/ALT, cutaneous leukocytoclastic vasculitis, essential mixed cryoglobulinemia with purpura, arthralgias, glomerulonephritis, and peripheral neuropathy, positive PCR assay for viral RNA, elevated serum ferritin, Kupffer cell hyperplasia with erythrophagocytosis, sinusoidal distention with aggregates of sickled erythrocytes, and fine fibrosis in the space of Disse |
 , ventilation-perfusion.
, ventilation-perfusion.