Shock, Sepsis, and Multiple Organ Dysfunction Syndrome
Shock Syndrome
Description
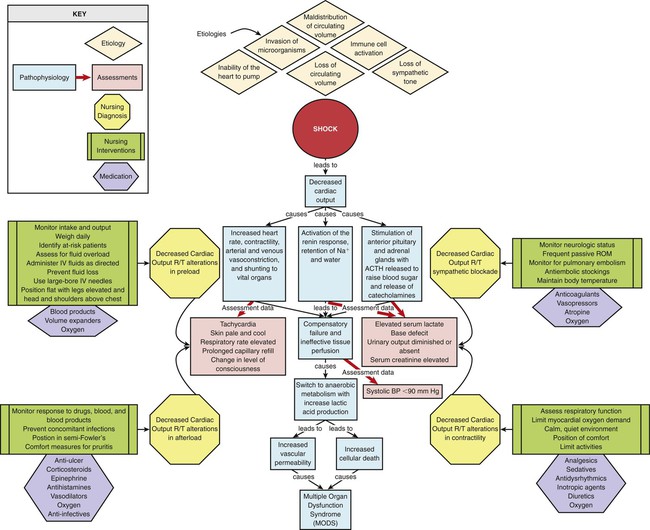
Shock is a complex pathophysiologic process that often results in MODS and death. All types of shock involve ineffective tissue perfusion and acute circulatory failure. The shock syndrome is a pathway involving a variety of pathologic processes that may be categorized as four stages: initial, compensatory, progressive, and refractory. Progression through each stage varies with the patient’s prior condition, duration of initiating event, response to therapy, and correction of underlying cause (Fig. 35-1).
Pathophysiology
During the progressive stage, the compensatory mechanisms begin failing to meet tissue metabolic needs, and the shock cycle is perpetuated. As tissue perfusion becomes ineffective, the cells switch from aerobic to anaerobic metabolism to produce energy. Anaerobic metabolism produces small amounts of energy but large amounts of lactic acid, producing lactic acidemia. Vasodilation and increased vascular permeability from endothelial and epithelial hypoxia and inflammatory mediators results in intravascular hypovolemia, tissue edema, and further decline in tissue perfusion.1,2,3 A systemic release of inflammatory mediators in response to tissue hypoxia, especially in gut tissue, produces microcirculatory impairment and derangement of cellular metabolism, facilitating progression of the shock cycle.1–4 The patient is experiencing systemic inflammatory response syndrome (SIRS), and irreversible damage begins to occur. Some cells die as a result of apoptosis, an injury-activated, preprogrammed cellular suicide. Others die as the sodium–potassium pump in the cell membrane fails, causing the cell and its organelles to swell. Cellular energy production comes to a complete halt as the mitochondria swell and rupture. At this point, the problem becomes one of oxygen use instead of oxygen delivery. Even if the cell were to receive more oxygen, it would be unable to use it because of damage to the mitochondria. The cell’s digestive organelles swell and leak destructive enzymes into the cell, accelerating cell death.4
Every system in the body is affected by this process (Box 35-1). Cardiac dysfunction develops as a result of the release of myocardial depressant cytokines.1–3 Ventricular failure eventually occurs, further perpetuating the entire process. Central nervous system (CNS) dysfunction develops as a result of cerebral hypoperfusion, leading to failure of the SNS, cardiac and respiratory depression, and thermoregulatory failure. Endothelial injury from hypoxia and inflammatory cytokines and impaired blood flow result in microvascular thrombosis. Hematologic dysfunction occurs as a result of consumption of clotting factors, release of inflammatory cytokines, and dilutional thrombocytopenia. Disseminated intravascular coagulation (DIC) eventually may develop. Pulmonary dysfunction occurs as a result of increased pulmonary capillary membrane permeability, pulmonary microemboli, and pulmonary vasoconstriction. Ventilatory failure and acute respiratory distress syndrome (ARDS) develop. Renal dysfunction develops as a result of renal vasoconstriction and renal hypoperfusion, leading to acute kidney injury (AKI). Gastrointestinal dysfunction occurs as a result of splanchnic vasoconstriction and hypoperfusion and leads to failure of the gut organs. Disruption of the intestinal epithelium releases gram-negative bacteria into the system, which further perpetuates the entire shock syndrome.5
During the refractory stage, shock becomes unresponsive to therapy and is considered irreversible. As the individual organ systems die, MODS—defined as failure of two or more body systems—occurs. Death is the final outcome. Regardless of the etiologic factors, death occurs from ineffective tissue perfusion because of the failure of the circulation to meet the oxygen needs of the cell.4
Assessment and Diagnosis
The patient with a mean arterial blood pressure (MAP) less than 60 mm Hg or with evidence of global tissue hypoperfusion is considered to be in a shock state.1,3 Because shock is a dynamic physiologic phenomenon, hypotension may occur late in the process or may normalize even when tissue perfusion is still inadequate.6–9 Clinical manifestations vary according to the underlying cause of shock, the stage of the shock, and the patient’s response to shock.
Compensatory mechanisms may produce normal hemodynamic values even when tissue perfusion is compromised.3,5,8,10–11 Global indicators of systemic perfusion and oxygenation include serum lactate, arterial base deficit, serum bicarbonate, and central or mixed venous oxygen saturation levels. Inadequate cellular oxygenation with anaerobic metabolism and increased metabolic lactate production increase the serum lactate level.8,12 The level and duration of this hyperlactatemia are predictive of morbidity and mortality,7,10–12 and management guided by lactate levels has been effective in improving outcomes.13–14 The base deficit derived from arterial blood gas (ABG) values also reflects global tissue acidosis and is useful to assess the severity of shock.6,9–11 Studies have demonstrated serum bicarbonate to be an equivalent alternative to arterial base deficit in predicting mortality in surgical and trauma patients.15–16 The use of mixed venous oxygen saturation (Svo2) measured by means of a pulmonary artery catheter or central venous oxygen saturation (Scvo2) measured with a central venous catheter allows assessment of the balance of oxygen delivery and oxygen consumption and the ratio of oxygen extraction.3,17–19 After years of recommended use to guide the care of patients with severe sepsis, this measure of global oxygen balance is being evaluated for use in other critically ill populations.17–21 Noninvasive indicators of regional tissue perfusion or oxygenation such as sublingual capnometry and subcutaneous or skeletal muscle tissue oxygen saturation (Sto2) measured with near-infrared spectroscopy are also being evaluated.3,8,19 The sections on different types of shock discuss clinical assessment and diagnosis of the patient in shock.
Medical Management
The major focus of the treatment of shock is the improvement and preservation of tissue perfusion. Adequate tissue perfusion depends on an adequate supply of oxygen being transported to the tissues and the cell’s ability to use it. Oxygen transport is influenced by pulmonary gas exchange, CO, and hemoglobin level. Oxygen use is influenced by the internal metabolic environment and mitochondrial function. Management of the patient in shock focuses on supporting oxygen delivery.1,3
The quantity and choice of fluid is a subject of debate and depends on the situation.3,22–27 Excessive volume expansion, more than what increases preload and stroke volume (SV), worsens organ function and may produce coagulopathy, cytokine activation, and abdominal compartment syndrome.3,24 Methods to measure preload responsiveness include respiratory or positional variation in pulse pressure, systolic pressure, and SV and are more accurate than central venous pressure (CVP).3 Fluid resuscitation with normal saline or with albumin produces similar outcomes regardless of baseline serum albumin level.26,28 Crystalloid solutions are inexpensive and effective. Advantages of colloids include faster restoration of intravascular volume and use of smaller amounts. Colloids are believed to stay in the intravascular space, unlike crystalloids, which readily leak into the extravascular space. Disadvantages include expense, allergic reactions, and difficulties in typing and cross-matching blood. Colloids also can leak out of damaged capillaries and cause a variety of additional problems, particularly in the lungs. Hypertonic or hyperoncotic fluids offer no additional benefit over isotonic crystalloids and are not recommended.3,22,27,29–30
Blood should be considered to augment oxygen transport if the patient’s hemoglobin level is critically low, although what threshold value should be used is still undetermined.3,5,31 Transfusion of stored red blood cells does not substantially increase oxygen consumption and has been associated with immunosuppression, infection, impairment of microcirculatory flow, increased pulmonary vascular resistance, coagulopathy, and increased mortality. Restrictive transfusion practice has demonstrated lower mortality.3,5,17,31–32 Transfusion-related acute lung injury (TRALI) resulting from immune and nonimmune neutrophil activation has become the leading cause of transfusion-related death and may occur with transfusion of any plasma-containing blood or blood product.32–35
Vasoconstrictor agents are used to increase afterload by increasing the systemic vascular resistance (SVR) and improving the patient’s blood pressure level. Vasodilator agents are used to decrease preload or afterload, or both, by decreasing venous return and SVR. Positive inotropic agents are used to increase contractility. Antidysrhythmic agents are used to influence heart rate. Box 35-2 provides examples of each of these agents.
Sodium bicarbonate is not recommended in the treatment of shock-related lactic acidosis.17,36–37 No overall benefit has been found, and the risks associated with its use are significant. They include shifting of the oxyhemoglobin dissociation curve to the left, rebound increase in lactic acid production, development of hyperosmolar state, fluid overload resulting from excessive sodium, and rapid cellular electrolyte shifts.36–37
The critically ill patient should be started on enteral nutritional support therapy within 24 to 48 hours.38 The type of nutritional supplementation initiated varies according to the cause of shock, and it should be tailored to the individual patient’s needs, as indicated by the underlying condition, laboratory data, and treatment. When enteral feeding is contraindicated parental nutrition should be considered, though a delay of seven days is recommended for better outcomes.38–41 Supplementation of enteral feeding with parenteral nutrition to increase caloric intake is a subject of debate but has not been shown to improve patient outcomes.38,41–42 A delay of one week is recommended before consideration of this strategy.38
Glucose control to a target level of 140 to 180 mg/dL is recommended for all critically ill patients.43–44 Benefits of glucose control in the critically ill include lower incidences of infection, renal failure, sepsis, and death.43–46
Nursing Management
The psychosocial needs of the patient and family dealing with shock are extremely important. These needs are based on situational, familial, and patient-centered variables. Nursing interventions for the psychosocial stress of critical illness include providing information on patient status, explaining procedures and routines, supporting the family, encouraging the expression of feelings, facilitating problem solving and shared decision making, individualizing visitation schedules, involving the family in the patient’s care, and establishing contacts with necessary resources.47 The consensus of all relevant professional organizations is that patients and families should be given the option of family presence during invasive procedures and resuscitation.47–50
Collaborative management of the patient with shock is outlined in Box 35-3.
Hypovolemic Shock
Description
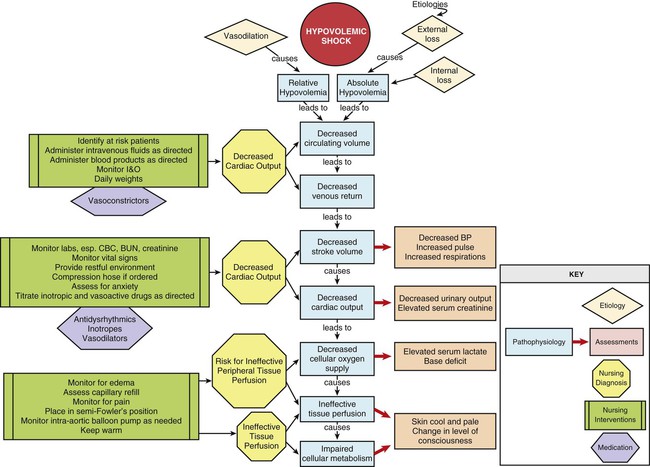
Hypovolemic shock occurs from inadequate fluid volume in the intravascular space. The lack of adequate circulating volume leads to decreased tissue perfusion and initiation of the general shock response. Hypovolemic shock is the most commonly occurring form of shock (Fig. 35-2).
Etiology
Hypovolemic shock can result from absolute or relative hypovolemia. Absolute hypovolemia occurs when there is a loss of fluid from the intravascular space. This can result from an external loss of fluid from the body or from internal shifting of fluid from the intravascular space to the extravascular space. Fluid shifts can result from a loss of intravascular integrity, increased capillary membrane permeability, or decreased colloidal osmotic pressure. Relative hypovolemia occurs when vasodilation produces an increase in vascular capacitance relative to circulating volume (Box 35-4).
Assessment and Diagnosis
The clinical manifestations of hypovolemic shock depend on the severity of fluid loss and the patient’s ability to compensate for it. Clinical classes developed by the American College of Surgeons to describe the levels of severity of hypovolemic shock in the trauma setting have been widely accepted, but a recent test of the validity of these classes suggests that modifications are necessary.51 A simpler approach of classifying hypovolemic shock as mild, moderate, or severe is also commonly used. Class I, or mild shock, indicates a fluid volume loss up to 15% or an actual volume loss up to 750 mL. Compensatory mechanisms maintain CO, and the patient appears free of symptoms other than possibly slight anxiety.1,5,51
Class II hypovolemia occurs with a fluid volume loss of 15% to 30% or an actual volume loss of 750 to 1500 mL. As volume loss worsens, the patient moves from mild to moderate hypovolemic shock. Falling CO activates more intense compensatory responses. Anxiety increases.1 The heart rate may increase to more than 100 beats/minute in response to increased SNS stimulation unless blocked by pre-existing beta-blocker therapy. The pulse pressure narrows as the diastolic blood pressure increases because of vasoconstriction. Postural hypotension develops.1 The respiratory rate increases as blood loss worsens and ABG specimens drawn during this phase may reveal respiratory alkalosis, as evidenced by a low partial pressure of carbon dioxide (Paco2). Urine output starts to decline to 20 to 30 mL/hour as renal perfusion decreases. The urine sodium level decreases, whereas urinary osmolality and specific gravity increase as the kidneys start to conserve sodium and water. The patient’s skin becomes pale and cool with delayed capillary refill because of peripheral vasoconstriction. Jugular veins appear flat as a result of decreased venous return.1,5
Hypovolemic shock that is class III occurs with a fluid volume loss of 30% to 40% or an actual volume loss of 1500 to 2000 mL. This level of severity may produce the progressive stage of shock as compensatory mechanisms become overwhelmed and ineffective tissue perfusion develops. Blood pressure decreases, but often after tissue hypoperfusion is already significant.6,9 The heart rate may increase to more than 120 beats/minute, and dysrhythmias may develop as myocardial ischemia ensues. During this phase serum lactate levels increase and ABG values reveal metabolic acidosis evidenced by a low bicarbonate (HCO3−) and elevated base deficit. Decreased renal perfusion results in the development of oliguria. Blood urea nitrogen (BUN) and serum creatinine levels start to rise as the kidneys begin to fail. The patient’s skin becomes ashen, cold, and clammy, with marked delayed capillary refill. The patient may appear confused as cerebral perfusion decreases.1,5,51
Class IV hypovolemic shock is severe shock and usually refractory in nature. It occurs with a fluid volume loss of greater than 40% or an actual volume loss of more than 2000 mL. As the compensatory mechanisms of the body become insufficient, tachycardia and hemodynamic instability worsen and hypotension ensues. Severe lactic acidosis is present. Peripheral pulses and capillary refill become absent because of marked peripheral vasoconstriction. The skin may appear cyanotic, mottled, and extremely diaphoretic. Organ failure occurs. Urine output ceases. The patient may be confused and agitated, eventually becoming unresponsive. Various clinical manifestations associated with failure of the different body systems will develop.1,5
Assessment of the hemodynamic parameters of a patient in hypovolemic shock varies by stage but commonly reveals a decreased CO and cardiac index (CI). Loss of circulating volume leads to a decrease in venous return to the heart, which results in a decrease in the preload of the right and left ventricles. This is evidenced by a decline in the CVP or right atrial pressure (RAP) and pulmonary artery occlusion pressure (PAOP). Vasoconstriction of the arterial system results in an increase in the afterload of the heart, as evidenced by an increase in the SVR. This vasoconstriction may produce inaccurate systolic and diastolic blood pressure values when measured by arterial catheter or noninvasive oscillometry. MAP is more accurate in this low-flow state.18
Medical Management
Aggressive fluid resuscitation in trauma and surgical patients is the subject of great debate. The benefit of limited or hypotensive (systolic blood pressure 60 to 80 mm Hg or MAP 40 to 60 mm Hg) volume resuscitation in patients with uncontrolled hemorrhage is postulated to lessen bleeding and improve survival22,52–54 and has been demonstrated in the preliminary results of a randomized controlled trial.53 The type and amount of solutions used for fluid resuscitation and the rate of administration influence immune function, inflammatory mediator release, coagulation, and the incidence of cardiac, pulmonary, renal, and gastrointestinal complications.3,22,24–25,27 Consensus on the optimal resuscitative strategy for hypovolemic shock is lacking and is likely situation specific.18,22,24,52,54
Nursing Management
Management of the patient in hypovolemic shock requires continuous evaluation of intravascular volume, tissue perfusion, and response to therapy. The patient in hypovolemic shock may have any number of nursing diagnoses, depending on the progression of the process (Box 35-5). Nursing interventions include minimizing fluid loss, administering volume replacement, assessing response to therapy, providing comfort and emotional support, and preventing and maintaining surveillance for complications.
 Box 35-5 Nursing Diagnoses
Box 35-5 Nursing Diagnoses
Hypovolemic Shock
• Deficient Fluid Volume related to active blood loss, p. 1132
• Deficient Fluid Volume related to interstitial fluid shift, p. 1132
• Decreased Cardiac Output related to alterations in preload, p. 1128
• Imbalanced Nutrition: Less Than Body Requirements related to increased metabolic demands or lack of exogenous nutrients, p. 1143
• Anxiety related to threat to biologic, psychologic, and/or social integrity, p. 1125
• Compromised Family Coping related to a critically ill family member, p. 1127
Measures to minimize fluid loss include limiting blood sampling, observing lines for accidental disconnection, and applying direct pressure to bleeding sites. Measures to facilitate the administration of volume replacement include insertion of large-bore peripheral intravenous catheters, rapid administration of prescribed fluids, and positioning the patient with the legs elevated, trunk flat, and head and shoulders above the chest. Monitoring the patient for clinical manifestations of fluid overload or complications related to fluid and blood product administration is essential for preventing further problems. It is also essential to monitor the patient for SIRS, which may occur for up to several days after resuscitation.55
Cardiogenic Shock
Description
Cardiogenic shock is the result of failure of the heart to effectively pump blood forward. It can occur with dysfunction of the right or the left ventricle, or both. The lack of adequate pumping function leads to decreased tissue perfusion and circulatory failure (Fig. 35-3). It occurs in approximately 5% to 8% of the patients with an ST-segment myocardial infarction (MI), and it is the leading cause of death of patients hospitalized with MI.56–58 The mortality rate for cardiogenic shock has decreased with the advent of early revascularization therapy and is currently about 47% to 65%.56–60
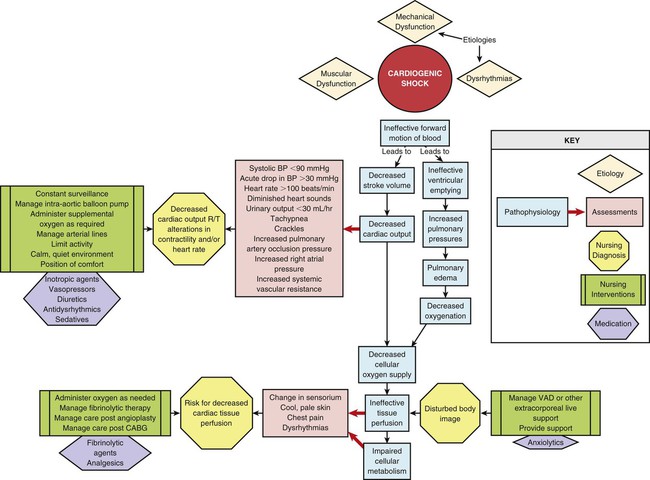
Etiology
Cardiogenic shock can result from problems affecting the muscular function or the mechanical function of the heart or the cardiac rhythm.57–59,61 The most common cause is acute MI resulting in the loss of 40% or more of the functional myocardium. It can occur with ST-elevation or non–ST-elevation MI.57–59 The damage to the myocardium may occur after one massive MI (usually of the anterior wall), or it may be cumulative as a result of several smaller MIs or a small MI in a patient with pre-existing ventricular dysfunction.56–57 Cardiomyopathy may cause cardiogenic shock as left ventricular function becomes unable to maintain adequate CO. Examples of problems affecting the mechanical function of the heart to fill and eject adequately include severe valvular disease; acute papillary muscle, chordal, or septal rupture; cardiac tamponade; and massive pulmonary embolus (Box 35-6).57–59,61
Pathophysiology
Cardiogenic shock results from the impaired ability of the ventricle to pump blood forward, which leads to a decrease in SV and an increase in the blood left in the ventricle at the end of systole. The decrease in SV results in a decrease in CO, which leads to decreased cellular oxygen supply and ineffective tissue perfusion. Typically, myocardial performance spirals downward as compensatory vasoconstriction increases myocardial afterload and low blood pressure worsens myocardial ischemia. As left ventricular contractility declines and ventricular compliance decreases, an increase in end-systolic volume results in blood backing up into the pulmonary system and the subsequent development of pulmonary edema. Pulmonary edema causes impaired gas exchange and decreased oxygenation of the arterial blood, which further impair tissue perfusion. In a substantial number of patients, the pathophysiology may follow a different course due to activation of inflammatory cytokines. A SIRS response results with systemic vasodilation and, possibly, normalization of the CO.57–58,62 Whether this process contributes to the genesis or the outcome of cardiogenic shock is uncertain, but it is thought to be activated by acute MI and to facilitate development of sepsis.57,62 Death due to cardiogenic shock results from cardiopulmonary collapse or multiple organ failure.59
Assessment and Diagnosis
A variety of clinical manifestations occur in the patient in cardiogenic shock, depending on etiologic factors, the patient’s underlying medical status, and the severity of the shock state. Some clinical manifestations are caused by failure of the heart as a pump, whereas many are related to the overall shock response (Box 35-7).
Initially, clinical manifestations reflect the decline in CO. These signs and symptoms include systolic blood pressure less than 90 mm Hg or an acute drop in systolic or mean blood pressure of 30 mm Hg or more; decreased sensorium; cool, pale, moist skin; and urine output of less than 30 mL/hour.57,59,63 The patient also may complain of chest pain. Tachycardia develops to compensate for the decrease in CO. A weak, thready pulse develops, and diminished S1 and S2 heart sounds may occur as a result of the decreased contractility. The respiratory rate increases to improve oxygenation. ABG values at this point indicate respiratory alkalosis, as evidenced by a decrease in Paco2. Urinalysis findings demonstrate a decrease in urine sodium level and an increase in urine osmolality and specific gravity as the kidneys start to conserve sodium and water. Serum B-type natriuretic peptide (BNP) levels will likely be elevated.
As the left ventricle fails, auscultation of the lungs may disclose crackles and rhonchi, indicating the development of pulmonary edema. Hypoxemia occurs, as evidenced by a fall in Pao2 and Sao2 as measured by ABG values. Heart sounds may reveal an S3 and S4. Jugular venous distention is evident with right-sided failure. The patient also may experience dysrhythmias in response to tissue hypoxia, the underlying problem, and drug therapy.58–59
Assessment of the hemodynamic parameters of a patient in cardiogenic shock reveals a decreased CO with a CI less than 2.2 L/min/m2 in the presence of an elevated PAOP of more than 15 mm Hg.57,59,63 A proportional pulse pressure (systolic BP/pulse pressure) less than 25% is indicative of left ventricular failure and a CI less than 2.2 and may be useful when direct measurement of CI is unavailable.64 Increased filling pressures are necessary to rule out hypovolemia as the cause of circulatory failure. The increase in PAOP reflects an increase in the left ventricular end-diastolic pressure (LVEDP) and left ventricular end-diastolic volume (LVEDV) resulting from decreased SV. With right ventricular failure, the RAP also increases. Compensatory vasoconstriction typically results in an increase in the afterload of the heart, as evidenced by an increase in the SVR, unless SIRS produces vasodilation and a normal or decreased SVR. Echocardiography confirms the diagnosis of cardiogenic shock, provides noninvasive estimates of PAOP and ejection fraction, and often clarifies etiologic factors.57–59
Medical Management
Treatment of the patient in cardiogenic shock requires an aggressive approach. The major goals of therapy are to treat the underlying cause, enhance the effectiveness of the pump, and improve tissue perfusion. This approach includes identifying and treating the etiologic factors of heart failure and administering pharmacologic agents or using mechanical devices to enhance CO. Inotropic agents are used to increase contractility and maintain adequate blood pressure and tissue perfusion. A vasopressor may be necessary to maintain blood pressure when hypotension is severe.57–59 As both of these therapies increase myocardial oxygen demand, the lowest possible doses should be used.57,59 Diuretics may be used for preload reduction. After blood pressure has been stabilized, vasodilating agents are used for preload and afterload reduction. Antidysrhythmic agents should be used to suppress or control dysrhythmias that can affect CO. Intubation and mechanical ventilation are usually necessary to support oxygenation.
Intra-aortic balloon pump (IABP) support should be instituted quickly if drug therapy does not immediately reverse the shock state.57–58 The IABP is a temporary mechanical device used to decrease myocardial workload by improving myocardial supply and decreasing myocardial demand. It achieves this goal by improving coronary artery perfusion and reducing left ventricular afterload. Chapter 16 provides more information about IAPB therapy.
As soon as the cause of pump failure has been identified, measures should be taken to correct the problem if possible. In the setting of acute MI, early and complete revascularization by percutaneous coronary intervention or coronary artery bypass surgery provides significant survival benefit.57–60 Fibrinolytic agents may be used in select patients. Procedural or surgical intervention may be necessary to remedy mechanical etiology. When conventional therapies fail, a ventricular assist device (VAD) or extracorporeal life support with a membrane oxygenator (ECMO) may be used to support the patient in acute cardiogenic shock.57–59 These mechanical circulatory assist devices provide an external means to sustain effective organ perfusion, allowing time for the patient’s ventricle to heal or for cardiac transplantation to take place.
Nursing Management
The patient in cardiogenic shock may have any number of nursing diagnoses, depending on the progression of the process (Box 35-8). Nursing interventions include limiting myocardial oxygen demand, enhancing myocardial oxygen supply, maintaining adequate tissue perfusion, providing comfort and emotional support, and preventing and maintaining surveillance for complications. Measures to limit myocardial oxygen demand include administering analgesics, sedatives, and agents to control afterload and dysrhythmias; positioning the patient for comfort; limiting activities; providing a calm and quiet environment and offering support to reduce anxiety; and teaching the patient about the condition. Measures to enhance myocardial oxygen supply include administering supplemental oxygen, monitoring the patient’s respiratory status, administering prescribed medications, and managing device therapy.
 Box 35-8 Nursing Diagnoses
Box 35-8 Nursing Diagnoses
Cardiogenic Shock
• Decreased Cardiac Output related to alterations in contractility, p. 1128
• Decreased Cardiac Output related to alterations in heart rate, p. 1128
• Imbalanced Nutrition: Less Than Body Requirements related to increased metabolic demands or lack of exogenous nutrients, p. 1143
• Disturbed Body Image related to functional dependence on life-sustaining technology, p. 1136
• Compromised Family Coping related to a critically ill family member, p. 1127
Patients who require mechanical device therapy (IABP, LVAD, or ECMO) therapy need to be observed frequently for complications. Complications of cardiac mechanical assist devices may include infection, bleeding, thrombocytopenia, hemolysis, embolus, stroke, device malfunction, circulatory compromise of a cannulated extremity, SIRS, and sepsis.63
Anaphylactic Shock
Description
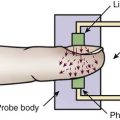
Anaphylactic shock, a type of distributive shock, is the result of an immediate hypersensitivity reaction. It is a life-threatening event that requires prompt intervention. The severe and systemic response leads to decreased tissue perfusion and initiation of the general shock response (Fig. 35-4).
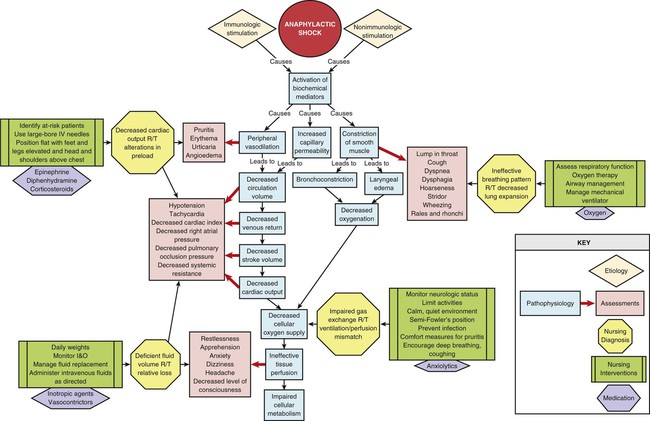
Etiology
Anaphylaxis is a serious allergic reaction caused by an immunologic antibody–antigen response or nonimmunologic activation of mast cells and basophils.65–69 A number of triggers have been identified that, when introduced by injection or ingestion or through the skin or respiratory tract, can cause a reaction. This list includes foods, food additives, diagnostic agents, biologic agents, environmental agents, medications, and venoms (Box 35-9).67–69 Anaphylaxis can also be triggered by physical factors and can even be idiopathic in nature with no known trigger.67–68 In the hospital environment, latex was once an extremely problematic antigen for patients and health care providers, but efforts to limit and prevent exposure have been highly successful.68
Immunologic anaphylactic reactions can be IgE-mediated or non–IgE-mediated responses. IgE is an antibody that is formed as part of the immune response. The first time an antigen enters the body, an antibody IgE, specific for the antigen, is formed. The antigen-specific IgE antibody is then stored by attachment to mast cells and basophils. This initial contact with the antigen is known as a primary immune response. The next time the antigen enters the body, the preformed IgE antibody reacts with it, and a secondary immune response occurs. This reaction triggers the release of biochemical mediators from the mast cells and basophils and initiates the cascade of events that precipitates anaphylactic shock.66,68–69 Some immunologic anaphylactic reactions are non–IgE-mediated. These can be IgG mediated, occur as a result of direct activation of the mast cells, or be mediated by activation of the complement system.68
Nonimmunologic direct activation of the mast cells and basophils may occur with exercise or exposure to cold, heat, sunlight, ethanol, or medications. In some cases, multiple mechanisms contribute to the anaphylactic response.68 The term anaphylactoid reaction was formerly used to denote all non–IgE-mediated reactions, but it is no longer recommended as there is no clinical distinction between the various mechanisms.65,67–69
Pathophysiology
Both immunologic and nonimmunologic activation of the mast cells and basophils result in the release of biochemical mediators. These mediators include histamine, tryptase, chymase, carboxypeptidase A3, platelet-activating factor (PAF), heparin, leukotrienes, prostaglandins, and cytokines such as IL-6, IL-33, and TNF-alpha, among others.68–69 The activation of the biochemical mediators causes vasodilation; increased capillary permeability; laryngeal edema; bronchoconstriction; excessive mucus secretion; coronary vasoconstriction; inflammation; cutaneous reactions; and constriction of the smooth muscle in the intestinal wall, bladder, and uterus. Coronary vasoconstriction causes severe myocardial depression. Cutaneous reactions cause stimulation of nerve endings, followed by itching and pain.68
Peripheral vasodilation results in relative hypovolemia and decreased venous return. Increased capillary membrane permeability results in the loss of intravascular volume into the interstitial space, as much as 35% within 10 minutes, worsening the hypovolemic state.67 Decreased venous return results in decreased end-diastolic volume and SV. The decline in SV leads to decreased CO and ineffective tissue perfusion. Death may result from airway obstruction or cardiovascular collapse, or both.65–66,68,70
Assessment and Diagnosis
Anaphylactic shock is a severe systemic reaction that can affect multiple organ systems. A variety of clinical manifestations occur in the patient in anaphylactic shock, depending on the extent of multisystem involvement. The symptoms usually start to appear within minutes of exposure to the antigen, but they may not occur for hours (Box 35-10).65–66,68 Symptoms may also reappear after a 1- to 72-hour window of resolution in what is termed a biphasic reaction.68,71–72 These late-phase reactions may be similar to the initial anaphylactic response, milder, or more severe.72 In protracted anaphylaxis, symptoms may last up to 32 hours.67
The cutaneous effects may appear first and include pruritus, generalized erythema, urticaria, and angioedema. Commonly seen on the face and in the oral cavity and lower pharynx, angioedema develops as a result of fluid leaking into the interstitial space. The patient may appear restless, uneasy, apprehensive, and anxious and may complain of being warm. Respiratory effects include the development of laryngeal edema, bronchoconstriction, and mucous plugs. Clinical manifestations of laryngeal edema include inspiratory stridor, hoarseness, a sensation of fullness or a lump in the throat, and dysphagia. Bronchoconstriction causes dyspnea, wheezing, and chest tightness.68,70 Gastrointestinal and genitourinary manifestations, which may develop as a result of smooth muscle contraction, include vomiting, diarrhea, cramping, and abdominal pain.
Hypotension and reflex tachycardia may develop quickly. This occurs in response to massive vasodilation and rapid loss of circulating volume. Jugular veins appear flat as right ventricular end-diastolic volume is decreased. The eventual outcome is circulatory failure and ineffective tissue perfusion.68,70 The patient’s level of consciousness may deteriorate to unresponsiveness.
Assessment of the hemodynamic parameters of a patient in anaphylactic shock reveals a decreased CO and CI. Venous vasodilation and massive volume loss lead to a decrease in preload, which results in a decline in the RAP and PAOP. Vasodilation of the arterial system results in a decrease in the afterload of the heart, as evidenced by a decrease in the SVR. Box 35-11 outlines the clinical criteria for diagnosing anaphylaxis.
Medical Management
Epinephrine is the first-line treatment of choice for anaphylaxis and should be administered when initial signs and symptoms occur.65,67 It promotes bronchodilation, vasoconstriction, and increased myocardial contractility and inhibits further release of biochemical mediators. In mild cases of anaphylaxis, 0.2 to 0.5 mg (0.3 to 0.5 mL) of a 1 : 1000 dilution of epinephrine is administered by intramuscular injection into the anterolateral thigh and repeated every 5 to 15 minutes until anaphylaxis is resolved.65–68,70–71 Subcutaneous injection is no longer recommended.71 For anaphylactic shock with hypotension, epinephrine is administered intravenously. The intravenous dose is 0.05 to 0.1 mg (1 mL) of a 1 : 10,000 dilution administered over 5 minutes.70 If hypotension persists, a continuous infusion of epinephrine is recommended, administered at 1 to 4 mcg/min with titration up to 15 mcg/min as needed.67,70 Patients receiving beta-blockers may have a limited response to epinephrine. Intravenous glucagon administered as a 20 to 30 mcg/kg bolus over 5 minutes followed by continuous infusion at 5 to 15 mcg/minute is recommended to treat bronchospasm and hypotension in these patients.66–67,71
Rapid volume replacement with crystalloid or colloid solutions is also used for patients with hypotension.66–67,71 Up to 1 L in 5 to 10 minutes is suggested if needed to restore perfusion.68 Vasopressors may be necessary to reverse the vasodilation and increase blood pressure.65–66,68,71
Several medications are used as second-line adjunctive therapy. Inhaled beta-adrenergic agents are used to treat bronchospasm unresponsive to epinephrine.67–68,70 Diphenhydramine (Benadryl) given 1 to 2 mg/kg (25 to 50 mg) by a slow intravenous is used to block histamine response.65,67,71 Ranitidine, given in conjunction with diphenhydramine at a dose of 1mL/kg intravenously over 10 to 15 minutes, has been found helpful.65,67 Corticosteroids are not effective in the immediate treatment of acute anaphylaxis,67,73 but may be given with the goal of preventing a prolonged or delayed reaction.66,70–71
Nursing Management
The patient in anaphylactic shock may have any number of nursing diagnoses, depending on the progression of the process (Box 35-12). Nursing interventions include administering epinephrine, facilitating ventilation, administering volume replacement, providing comfort and emotional support, maintaining surveillance for recurrent reactions and preventing and maintaining surveillance for complications.
 Box 35-12 Nursing Diagnoses
Box 35-12 Nursing Diagnoses
Anaphylactic Shock
• Deficient Fluid Volume related to relative loss, p. 1132
• Decreased Cardiac Output related to alterations in preload, p. 1128
• Decreased Cardiac Output related to alterations in afterload, p. 1128
• Ineffective Breathing Pattern related to decreased lung expansion, p. 1149
• Impaired Gas Exchange related to ventilation/perfusion mismatching or intrapulmonary shunting, p. 1144
• Imbalanced Nutrition: Less Than Body Requirements related to increased metabolic demands or lack of exogenous nutrients, p. 1143
• Ineffective Coping related to situational crisis and personal vulnerability, p. 1150
• Compromised Family Coping related to a critically ill family member, p. 1127
Neurogenic Shock
Description
Neurogenic shock, another type of distributive shock, is the result of the loss or suppression of sympathetic tone. The lack of sympathetic tone leads to decreased tissue perfusion and initiation of the general shock response (Fig. 35-5). Neurogenic shock is the most uncommon form of shock.
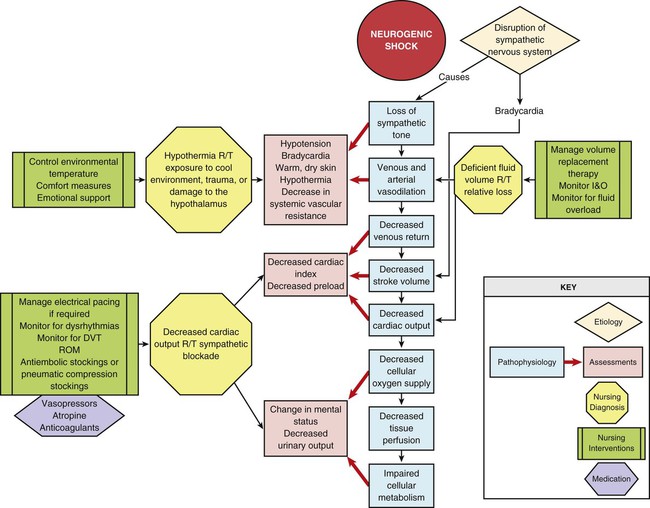
Etiology
Neurogenic shock can be caused by anything that disrupts the SNS. The problem can occur as the result of interrupted impulse transmission or blockage of sympathetic outflow from the vasomotor center in the brain.74–75 The most common cause is spinal cord injury (SCI). Neurogenic shock may mistakenly be referred to as spinal shock. The latter condition refers to loss of neurologic activity below the level of SCI, but it does not necessarily involve ineffective tissue perfusion.74–76
Pathophysiology
Loss of sympathetic tone results in massive peripheral vasodilation, inhibition of the baroreceptor response, and impaired thermoregulation. Arterial vasodilation leads to a decrease in SVR and a fall in blood pressure. Venous vasodilation leads to relative hypovolemia and pooling of blood in the venous circuit. The decreased venous return results in a decrease in end-diastolic volume or preload, causing a decrease in SV and CO. The fall in blood pressure and CO leads to inadequate or ineffective tissue perfusion. Loss of sympathetic tone and inhibition of the baroreceptor response result in bradycardia.74–75,77–78 The slow heart rate worsens CO, which further compromises tissue perfusion. Impaired thermoregulation occurs because of loss of vasomotor tone in the cutaneous blood vessels that dilate and constrict to maintain body temperature. The patient becomes poikilothermic, or dependent on the environment for temperature regulation.
Assessment and Diagnosis
The patient in neurogenic shock characteristically presents with hypotension, bradycardia, and warm, dry skin.74–75,78–79 The decreased blood pressure results from massive peripheral vasodilation. The decreased heart rate is caused by inhibition of the baroreceptor response and unopposed parasympathetic control of the heart.74–75,78 Consensus on the specific blood pressure and heart rate thresholds for the diagnosis of neurogenic shock has not been established.74–75 Hypothermia develops from uncontrolled peripheral heat loss. The warm, dry skin occurs as a consequence of pooling of blood in the extremities and loss of vasomotor control in surface vessels of the skin that control heat loss.
Assessment of the hemodynamic parameters of a patient in neurogenic shock reveals a decreased CO and CI. Venous vasodilation leads to a decrease in preload, which results in a decline in the RAP and PAOP. Vasodilation of the arterial system causes a decrease in the afterload of the heart, as evidenced by a decrease in the SVR.75,78
Medical Management
Hypovolemia is treated with careful fluid resuscitation. The minimal amount of fluid is administered to ensure adequate tissue perfusion. Volume replacement is initiated for systolic blood pressure lower than 90 mm Hg or evidence of inadequate tissue perfusion. Base deficit or lactate levels are recommended to guide fluid resuscitation in patients with SCI.77 The patient is carefully observed for evidence of fluid overload. Vasopressors are used as necessary to maintain blood pressure and organ perfusion.74,76–79 Higher than typical MAPs are commonly needed for patients with acute SCI to prevent cord ischemia, but optimal pressures have not been determined.74,77 Bradycardia associated with neurogenic shock rarely requires specific treatment, but atropine, intravenous infusion of a beta-adrenergic agent, or electrical pacing can be used when necessary.78,80 Hypothermia is treated with warming measures and environmental temperature regulation.
Nursing Management
The patient in neurogenic shock may have any number of nursing diagnoses, depending on the progression of the process (Box 35-13). Nursing interventions include treating hypovolemia and maintaining tissue perfusion, maintaining normothermia, monitoring for and treating dysrhythmias, providing comfort and emotional support, and preventing and maintaining surveillance for complications.
 Box 35-13 Nursing Diagnoses
Box 35-13 Nursing Diagnoses
Neurogenic Shock
• Deficient Fluid Volume related to relative loss, p. 1132
• Decreased Cardiac Output related to sympathetic blockade, p. 1128
• Hypothermia related to exposure to cold environment, trauma, or damage to the hypothalamus, p. 1142
• Imbalanced Nutrition: Less Than Body Requirements related to increased metabolic demands or lack of exogenous nutrients, p. 1143
• Anxiety related to threat to biologic, psychologic, or social integrity, p. 1125
• Compromised Family Coping related to a critically ill family member, p. 1127
Severe Sepsis and Septic Shock
Description
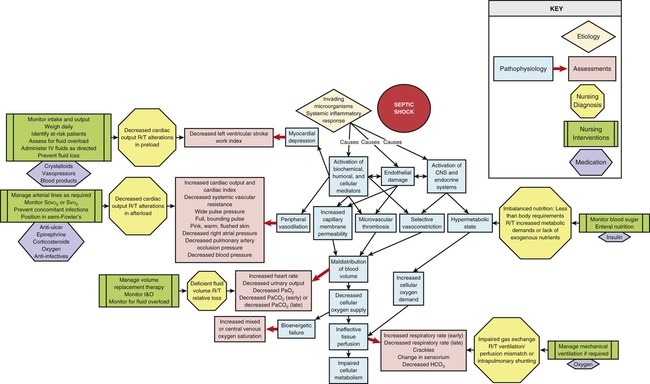
Sepsis occurs when microorganisms invade the body and initiate a systemic inflammatory response. This host response often results in perfusion abnormalities with organ dysfunction (severe sepsis) and eventually hypotension (septic shock). The primary mechanism of this type of shock is the maldistribution of blood flow to the tissues (Fig. 35-6).4 Severe sepsis is estimated to result in more than 700,000 hospitalizations annually in the United States, with an estimated hospital mortality rate of 23% to 50%.81–84 It is the leading cause of in-hospital death and the eleventh leading cause of all deaths in the United States.82,85
Specific terms are used to describe the continuum of conditions that the patient with an infection may experience. In 1991 at the American College of Chest Physicians/Society of Critical Care Medicine (ACCP/SCCM) Consensus Conference, definitions were developed to describe and differentiate these conditions (Box 35-14).86 These definitions were clarified and reinforced in subsequent conferences in 2001, 2004, and 2008.17,87–88 This discussion focuses on severe sepsis and septic shock.
Etiology
Sepsis is caused by a wide variety of microorganisms, including gram-negative and gram-positive aerobes, anaerobes, fungi, and viruses. The source of these microorganisms varies. The respiratory system is the most common site of infection producing severe sepsis and septic shock, followed by the genitourinary and gastrointestinal systems.82,83 Gram-positive bacteria are the predominant cause of sepsis.83,89 Sepsis and septic shock are associated with a wide variety of intrinsic and extrinsic precipitating factors (Box 35-15). All of these factors interfere directly or indirectly with the body’s anatomic and physiologic defense mechanisms. Several of the intrinsic factors are not modifiable or are very difficult to control. Several of the extrinsic factors may be required for diagnosis and management. All critically ill patients are therefore at risk for septic shock.
Pathophysiology
The syndrome encompassing severe sepsis and septic shock is a complex systemic response that is initiated when a microorganism enters the body and stimulates the inflammatory/immune system. In a host-pathogen interaction, both the invading organism and injured tissue release intracellular proteins activating neutrophils, monocytes, lymphocytes, macrophages, mast cells, and platelets as well as numerous plasma enzyme cascades (complement, kinin/kallikrein, coagulation, and fibrinolytic factors). When this reaction is localized, infection is contained and eradicated. But when the magnitude of the infectious insult is great or the patient is physiologically unable to generate an effective host response, containment fails.90 The result is a systemic release of the pathogen, activated cells, and mediators, including cytokines, which initiate a chain of complex interactions leading to an uncontrolled SIRS.90–94
With systemic activation, a variety of physiologic and pathophysiologic events occur that affect clotting, the distribution of blood flow to the tissues and organs, capillary membrane permeability, and the metabolic state of the body. Subsequently, a systemic imbalance between cellular oxygen supply and demand develops that results in cellular hypoxia, damage, hibernation, and death.5,91,93–94
Hallmarks of severe sepsis are endothelial damage and coagulation dysfunction.91–92,95–96 Tissue factor (TF) is released from endothelial cells and monocytes in response to stimulation by inflammatory cytokines.91,95–97 Release of TF initiates the coagulation cascade, producing widespread microvascular thrombosis and further stimulation of the systemic inflammatory pathways.91–92,95–97 Diffuse endothelial damage impairs endogenous anticlotting mechanisms.91–92,95,97 Mediator-induced suppression of fibrinolysis slows clot breakdown. The result is DIC with eventual consumption of coagulation factors, bleeding, and hemorrhage.92,96–97
Significant alterations in cardiovascular hemodynamics are caused by the activation of inflammatory cytokines and endothelial damage.90–92 Massive peripheral vasodilation results in the development of relative hypovolemia. Increased capillary permeability produces a loss of intravascular volume to the interstitium, which accentuates the reduction in preload and CO. These changes, coupled with the microvascular thrombosis, produce maldistribution of circulating blood volume, decreased tissue perfusion, and inadequate oxygen delivery to the cells. Microcirculatory shunting is a key feature of this distributive shock.91,98 Impaired ventricular contractility results from cytokine activity.5,91–92,96
Activation of the central nervous and endocrine systems also occurs as part of the response to invading microorganisms. This activation leads to stimulation of the SNS and the release of ACTH. These events trigger the release of epinephrine, norepinephrine, glucocorticoids, aldosterone, glucagon, renin, and growth hormone, resulting in the development of a hypermetabolic state and contributing to vasoconstriction of the renal, pulmonary, and splanchnic beds. Selective vasoconstriction in the splanchnic bed may contribute to hypoperfusion of the gastrointestinal mucosal barrier, an area particularly vulnerable to the effects of inflammatory cytokines.92,99 The resulting gut injury propagates the inflammatory response.2,99
Several metabolic alterations occur as a result of CNS, endocrine system, and cytokine activation. The hypermetabolic state increases energy expenditure and oxygen demand, and it contributes to cellular hypoxia. Lactic acid is produced as a result of increased metabolic lactate production and hypoxic anaerobic metabolism. Glucocorticoids, ACTH, epinephrine, glucagon, and growth hormone are all catabolic hormones that are released as part of this response. In conjunction with the inflammatory cytokines, these hormones stimulate catabolism of protein stores in the visceral organs and skeletal muscles to fuel glucose production in the liver, hyperglycemia, and insulin resistance.94,100 The cytokines also stimulate the use of fats for energy production (lipolysis).94,100–101
Metabolic derangements in severe sepsis and septic shock include an inability of the cells to use oxygen even if blood flow is adequate. Mitochondrial dysfunction is thought to be the underlying mechanism.91,94,101–102 This bioenergetic failure plays an important role in the development of tissue ischemia and multiple organ dysfunction.91,94,101–102 These complex and interrelated pathophysiologic changes produce a pathologic imbalance between cellular oxygen demand and cellular oxygen supply and consumption.
The exaggerated systemic inflammatory response associated with severe sepsis and septic shock results in cell death via both ischemic necrosis and, to a large degree, apoptosis. Apoptosis is a programmed cell death or cellular suicide mediated by caspase-3, a cysteine protease, and affecting endothelial, gastrointestinal epithelial, and immune cells in particular.91–92,94,96 Apoptosis of immune cells results in immunosuppression and secondary infection as well as release of immune cell toxins. Both ischemic necrosis and apoptosis stimulate further inflammation, propagating an ongoing cycle of infection, tissue injury, and SIRS.90,96 If unabated, this situation ultimately results in MODS and death.
Assessment and Diagnosis
Effective treatment of severe sepsis and septic shock depends on timely recognition. The diagnosis of severe sepsis is based on the identification of three conditions: known or suspected infection, two or more of the clinical indications of the systemic inflammatory response, and evidence of at least one organ dysfunction. Clinical indications of systemic inflammatory response and sepsis were included in the original ACCP/SCCM consensus definitions and are listed in Box 35-14. The second consensus conference expanded this list to facilitate prompt clinical recognition (Box 35-16).88
Signs of individual organ dysfunction are discussed later in the chapter. The two most common organs to demonstrate dysfunction in severe sepsis are the cardiovascular system and the lungs. The patient with persistent hypotension requiring vasopressor therapy despite adequate volume resuscitation is demonstrating cardiovascular dysfunction. Pulmonary dysfunction is manifested by a Pao2/Fio2 (fraction of oxygen in inspired air) ratio of less than 300, indicating ARDS.17,103 Signs indicating septic shock are hypotension despite adequate fluid resuscitation and the presence of perfusion abnormalities such as lactic acidosis, oliguria, or acute change in mentation.
The patient in severe sepsis or septic shock may present with a variety of clinical manifestations that may change dynamically as the condition progresses (Box 35-17). During the initial stage, massive vasodilation occurs in the venous and arterial beds. Dilation of the venous system leads to a decrease in venous return to the heart, which results in a decrease in the preload of the right and left ventricles. This is evidenced by a decline in the RAP and PAOP. Dilation of the arterial system results in a decrease in the afterload of the heart, as evidenced by a decrease in the SVR. The patient’s skin becomes pink, warm, and flushed as a result of the massive vasodilation. Myocardial contractility is decreased, as evidenced by a decline in the left ventricular stroke work index (LVSWI) and ejection fraction.
In the lungs, ventilation/perfusion mismatching develops as a result of pulmonary vasoconstriction and the formation of pulmonary microemboli. Hypoxemia occurs, and the respiratory rate increases to compensate for the lack of oxygen. Crackles develop as increased pulmonary capillary membrane permeability leads to pulmonary edema.103
The level of consciousness starts to change as a result of decreased cerebral perfusion, immune mediator activation, hyperthermia, and lactic acidosis. This septic encephalopathy is demonstrated by acute onset of impaired cognitive functioning, or delirium, which may fluctuate during its course.104 The patient may appear disoriented, confused, combative, or lethargic.
ABG values initially reveal hypocarbia, hypoxemia, and metabolic acidosis. This is demonstrated by a low Pao2, low Paco2, and low HCO3− level, respectively. The respiratory alkalosis is caused by the patient’s increased respiratory rate. As pathologic pulmonary changes progress and the patient becomes fatigued, the effectiveness of respirations decreases and the Paco2 increases, resulting in respiratory acidosis. The metabolic acidosis is the result of a lack of oxygen to the cells and the development of lactic acidemia. Serum lactate levels increase above 2 mmol/L because of anaerobic metabolism. The mixed venous oxygen saturation (Svo2) may increase because of microcirculatory shunting or decrease because of inadequate oxygen delivery.91,98 The white blood cell (WBC) count is elevated as part of the immune response to the invading microorganisms. The WBC differential count reveals an increase in immature neutrophils (shift to the left). This occurs because the body has to mobilize increasing numbers of WBCs to fight the infection. An elevated procalcitonin level is a valuable biomarker of significant bacterial infection and procalcitonin levels have been used in clinical trials to guide antibiotic therapy with positive outcomes.91,105 Serum glucose levels increase as part of the hypermetabolic response and the development of insulin resistance. The patient’s temperature is elevated in response to pyrogens released from the invading microorganisms, immune mediator activation, and increased metabolic activity. Urine output declines because of decreased perfusion of the kidneys. As impaired tissue perfusion develops, a variety of other clinical manifestations appear that indicate the development of MODS.
Medical Management
Treatment of the patient in severe sepsis or septic shock requires a multifaceted approach. The goals of treatment are to reverse the pathophysiologic responses, control the infection, and promote metabolic support. This approach includes supporting the cardiovascular system and enhancing tissue perfusion, identifying and treating the infection, limiting the systemic inflammatory response, restoring metabolic balance, and initiating nutritional therapy. Dysfunction of the individual organ systems must be prevented. Early treatment reduces mortality.17,106–107 Guidelines for the management of severe sepsis and septic shock have been developed and updated under the auspices of the Surviving Sepsis Campaign (SSC), an international effort of more than 11 organizations to improve patient outcomes.17,87 From these guidelines, a group (“bundle”) of selected interventions was identified as having the most impact on patient outcomes (Box 35-18). The sepsis resuscitation bundle should be implemented within the first 6 hours, and the sepsis management bundle should be implemented within the first 24 hours. More information regarding these interventions is available at the SSC website (http://www.survivingsepsis.org).
The patient in severe sepsis or septic shock requires immediate resuscitation of the hypoperfused state. Specific interventions are aimed at increasing cellular oxygen supply and decreasing cellular oxygen demand. These treatments include administration of fluids, vasopressors, and positive inotropic agents. Early goal-directed therapy during the first 6 hours of resuscitation improves survival84,106 and is recommended in the SSC guidelines.17 This therapy includes aggressive fluid resuscitation to augment intravascular volume and increase preload until a CVP of 8 to 12 mm Hg (12 to 15 mm Hg in mechanically ventilated patients) is achieved. Crystalloids are the initial fluid of choice.17 A fluid challenge for hypovolemia should be initiated with at least 1000 mL of crystalloids or 300 to 500 mL of colloids over 30 minutes. Vasopressors should be administered as necessary to maintain a MAP of at least 65 mm Hg. These agents reverse the massive peripheral vasodilation and increase SVR. Norepinephrine and dopamine have both been recommended as first-choice agents, but recent evidence strongly indicates superiority of norepinephrine in lowering mortality rates and a higher risk of dysrhythmias when dopamine is used.108–109 Epinephrine is recommended as an alternative agent if response to norepinephrine is poor.17 Arterial line placement is recommended for any patient requiring vasopressor therapy. Intermittent or continuous monitoring of central venous or mixed venous oxygen saturation (Scvo2 or Svo2) allows evaluation of the effectiveness of oxygen delivery. If the Scvo2 is less than 70% or the Svo2 is less than 65% after the CVP goal is achieved, administration of packed red cells to achieve a hematocrit of at least 30% or inotropic stimulation with dobutamine (administered to a maximum of 20 mcg/kg/minute) to counteract myocardial depression and maintain adequate CO is recommended to obtain this goal.17,106 The dobutamine infusion should be reduced or discontinued if a tachycardia greater than 120 beats/minute develops.106
Intubation and mechanical ventilatory support are usually required to optimize oxygenation and ventilation for the patient in severe sepsis or septic shock. Ventilation with lower than traditional tidal volumes (6 versus 12 mL/kg) in patients with ARDS decreases mortality.110 SSC guidelines recommend the goals of 6 mL/kg of predicted body weight and plateau pressures no more than 30 cm H2O for patients with severe sepsis or septic shock with ARDS.17 Increased Paco2 may result from this therapy and is acceptable if tolerated as evidenced by hemodynamic stability. Ventilator settings should include positive end-expiratory pressure (PEEP) and be adjusted to provide the patient with a Pao2 greater than 70 mm Hg. Patients receiving mechanical ventilation should be maintained in a semirecumbent position with the head of the bed raised to 45 degrees to decrease the incidence of ventilator-associated pneumonia.17 Prone positioning should be considered in the septic patient with ARDS requiring high levels of oxygen.17 Sedation protocols using intermittent bolus or continuous infusion using a standardized sedation scale and specific goals are recommended for all patients requiring mechanical ventilation. Daily interruption of sedative infusions to allow wakefulness and re-evaluation of sedation needs reduce duration of mechanical ventilation and is recommended.17 Neuromuscular blocking agents should be avoided, if possible, to prevent prolonged blockade after discontinuation.17
A key measure in the treatment of septic shock is finding and eradicating the cause of the infection. At least two blood cultures plus urine, sputum, and wound cultures should be obtained to find the location of the infection before antibiotic therapy is initiated.17 Antibiotic therapy should be started within 1 hour of recognition of severe sepsis without delay for cultures.17 Each hour of delay is associated with a substantial drop in the survival rate.107 If the microorganism is unknown, anti-infective therapy with one or more agents known to be effective against likely pathogens should be initiated, with daily reassessment of the regimen. Combination therapy is recommended for known or suspected Pseudomonas infection and for neutropenic patients but should be limited to less than 3 to 5 days.17 A specific source of infection should be established within 6 hours of presentation.17 Surgical intervention to débride infected or necrotic tissue or to drain abscesses may be necessary to facilitate removal of the septic source.17 Intravascular devices that may be the source of the infection should be removed after establishment of alternative vascular access.
Only one medication has been brought to market specifically for the treatment of severe sepsis and septic shock, recombinant human activated protein C (rhAPC), marketed as drotrecogin alfa [activated] under the trade name Xigris. This medication was recommended only for patients at high risk for death.17 However, a Cochrane review found that none of the results of subsequent randomized clinical trials supported the beneficial findings in mortality demonstrated in the original trials used for drug approval both in the United States and Europe.111 Following failure to show a survival benefit in a major clinical trial launched to reassess the medication, Xigris was voluntarily withdrawn from the market by the manufacturer in October 2011.112–113 Studies of numerous other medications thought to block or alter the effects of immune mediators have failed to demonstrate effectiveness or have been associated with unacceptable adverse effects.90,96
Low-dose intravenous corticosteroids reduce mortality in a select group of catecholamine-dependent septic shock patients.114–115 Therefore, intravenous hydrocortisone is recommended only for the patient in septic shock who remains hypotensive despite adequate fluid resuscitation and vasopressor therapy.17,116 High doses of 300 mg/day or more may be harmful and should not be used.17,116 Steroid therapy should be weaned when vasopressors are no longer required.
Continuous infusion of intravenous insulin is recommended by SSC guidelines when blood glucose level exceeds 180 mg/dL with a goal blood glucose level of approximately 150 mg/dL.17,117 Glucose levels should be monitored every 1 to 2 hours until stable and then every 4 hours. Low glucose levels measured by capillary testing may be inaccurate in this population.17 Platelets should be administered when counts are less than 5000/mm3 and red blood cell transfusions are recommended when the hemoglobin level is less than 7.0 g/dL to obtain a target value of 7 to 9 g/dL.17 Stress ulcer prophylaxis using H2-receptor antagonists or proton pump inhibitors and DVT prophylaxis are recommended for all patients with severe sepsis or septic shock. The SCCM guidelines recommend against the use of sodium bicarbonate for lactic acidemia if the pH is equal to or greater than 7.15.17 Low-dose dopamine infusion for renal protection is not beneficial and should not be used.17
The initiation of nutritional therapy is critical in the management of the patient in severe sepsis or septic shock. The goal is to improve the patient’s overall nutritional status, enhance immune function, and promote wound healing. A daily caloric intake of 20 to 30 kcal/kg of usual body weight is recommended for critically ill patients. The enteral route is strongly preferred. When compared to enteral nutrition, parenteral nutrition or a combination of both methods has been associated with higher mortality in patients with severe sepsis or septic shock.118
Sufficient protein needs to be provided because of the metabolic derangements that develop in the hypermetabolic state. A range of 1.2 to 2.0 g/kg actual body weight per day is recommended for patients with a body mass index (BMI) less than 30, with progressively larger amounts for those with higher BMI.38 Specific nutritional therapies to reduce the inflammatory and hypermetabolic responses associated with sepsis, such as antioxidant supplementation and feeding with long-chain n-3 polyunsaturated fatty acids, are the source of much debate and evaluation.100,119 Glutamine is considered by some to be an essential amino acid in critically ill patients and has the most empirical support,91,119 but others may actually worsen outcomes. Current recommendations stress caution with any immune-modulating formulations in patients with severe sepsis.38
Nursing Management
Nursing recommendations to complement the SSC guidelines have recently been published and focus on infection prevention and transmission, early recognition and treatment of severe sepsis and septic shock and its progression, and supportive nursing care.120 Prevention of severe sepsis and septic shock is one of the primary responsibilities of the nurse in the critical care area. These measures include the identification of patients at risk and reduction of their risk factors, including exposure to invading microorganisms. Hand washing, aseptic technique, and an understanding of evidence-based practice to reduce nosocomial infection in critically ill patients are essential components of preventive nursing care. Early identification allows for early treatment and decreases mortality. Figure 35-7 is a simple screening tool for identifying patients with severe sepsis. Subsequent continual observation to detect subtle changes that indicate the progression of the septic process is vitally important, as is evidence-based practice to prevent further infection. Immunosuppression is common as sepsis progresses,96 and recent research has found that a resurgence in opportunistic infection in patients with severe sepsis and septic shock occurs in the later stages, more than 2 weeks after initial diagnosis and treatment.121 Evidence-based practice to prevent complications of critical illness and prolonged bed rest are essential to prevent further compromise and negative short- and long-term outcomes. Ongoing mortality and impaired quality of life persist in the months and years after hospital discharge for survivors of sepsis.122

The patient in septic shock may have any number of nursing diagnoses, depending on the progression of the process (Box 35-19). Nursing interventions include early identification of sepsis syndrome; administering prescribed fluids, medications, and nutrition; providing comfort and emotional support; and preventing and maintaining surveillance for complications.
 Box 35-19 Nursing Diagnoses
Box 35-19 Nursing Diagnoses
Septic Shock
• Deficient Fluid Volume related to relative loss, p. 1132
• Decreased Cardiac Output related to alterations in preload, p. 1128
• Decreased Cardiac Output related to alterations in afterload, p. 1128
• Decreased Cardiac Output related to alterations in contractility, p. 1128
• Impaired Gas Exchange related to ventilation/perfusion mismatching or intrapulmonary shunting, p. 1144
• Imbalanced Nutrition: Less Than Body Requirements related to increased metabolic demands or lack of exogenous nutrients, p. 1143
• Anxiety related to threat to biologic, psychologic, or social integrity, p. 1125
• Compromised Family Coping related to a critically ill family member, p. 1127
Evidence-based guidelines for the management of the patient with severe sepsis or septic shock are listed in the Box 35-20.
Box 35-20
Evidence-Based Practice
Severe Sepsis and Septic Shock Management Guidelines

1. Protocolized, quantitative resuscitation of patients with sepsis-induced tissue hypoperfusion. Goals during first 6 hrs of resuscitation:
a) Central venous pressure 8–12 mm Hg
b) Mean arterial pressure (MAP) ≥ 65 mm Hg
c) Urine output ≥ 0.5 mL/kg/hr
d) Central venous (superior vena cava) or mixed venous oxygen saturation 70% or 65%, respectively (grade 1C).
2. In patients with elevated lactate levels, targeting resuscitation to normalize lactate (grade 2C).
B Screening for Sepsis and Performance Improvement
1. Routine screening of potentially infected seriously ill patients for severe sepsis to allow earlier implementation of therapy (grade 1C).
2. Hospital–based performance improvement efforts in severe sepsis (UG).
1. Cultures before antimicrobial therapy if delay is <45 min in the start of antimicrobial(s) (grade 1C). ≥2 sets of blood cultures (aerobic and anaerobic) with at least 1 drawn percutaneously and 1 drawn through each vascular access device, unless the device was inserted <48 hrs (grade 1C).
2. 1,3 beta-D-glucan assay (grade 2B), mannan and anti-mannan antibody assays (2C), if available and invasive candidiasis is in differential diagnosis.
3. Imaging studies to confirm a potential source of infection (UG).
1. Goal: IV antimicrobials within first hour of septic shock (grade 1B) and severe sepsis without septic shock (grade 1C).
2a. Initial empiric anti-infective therapy of drug(s) active against all likely pathogens and that penetrate in adequately into sepsis source tissues (grade 1B).
2b. Antimicrobial regimen reassessed daily for potential deescalation (grade 1B).
3. Low procalcitonin levels or similar biomarkers assist clinicians in discontinuation of therapy in patients who initially appeared septic, but have no subsequent evidence of infection (grade 2C).
4a. Combination therapy for neutropenic patients with severe sepsis (grade 2B) and for patients with difficult-to-treat, multidrugresistant bacterial pathogens (grade 2B). For patients with severe infections associated with respiratory failure and septic shock, combination therapy with an extended spectrum beta-lactam and either an aminoglycoside or a fluoroquinolone (for P. aeruginosa bacteremia) (grade 2B). A combination of beta-lactam and macrolide for patients with septic shock from bacteremic Streptococcus pneumoniae infections (grade 2B).
4b. Combination therapy should not be administered for >3–5 days. De-escalation to most appropriate single therapy should be performed as soon as the susceptibility profile is known (grade 2B).
5. Duration of therapy typically 7–10 days; longer courses may be appropriate in patients who have a slow clinical response, undrainable foci of infection, bacteremia with S. aureus; and some fungal and viral infections or immunologic deficiencies (grade 2C).
6. Antiviral therapy initiated as early as possible in patients with severe sepsis or septic shock of viral origin (grade 2C).
7. Antimicrobial agents should not be used in patients with severe inflammatory states determined to be of noninfectious cause (UG).
1. Specific anatomical diagnosis and emergent source control should be diagnosed or excluded as rapidly as possible; source control intervention should be undertaken <12 hr after diagnosis, if feasible (grade 1C).
2. When source is potentially infected peripancreatic necrosis, definitive intervention is best delayed until after adequate demarcation of viable tissues (grade 2B).
3. For source control in severely septic patients, effective interventions associated with the least physiologic insult should be used (UG).
4. If IV access devices are possible sources, they should be removed promptly after other vascular access has been established (UG).
1a. Selective oral and digestive decontamination should be considered to reduce the incidence. of ventilator-associated pneumonia; this can be instituted in settings where it is effective (grade 2B).
1b. Oral chlorhexidine gluconate be used to reduce risk of ventilator-associated pneumonia in ICU patients with severe sepsis (grade 2B).
G Fluid Therapy of Severe Sepsis
1. Crystalloids initially in resuscitation of severe sepsis and septic shock (grade 1B).
2. Against hydroxyethyl starches for resuscitation of severe sepsis and septic shock (grade 1B).
3. Albumin in resuscitation of severe sepsis and septic shock when patients require substantial crystalloids (grade 2C).
4. Initial fluid challenge (to a minimum of 30 mL/kg of crystalloids) in patients with sepsis-induced tissue hypoperfusion with suspicion of hypovolemia. More rapid administration and greater amounts may be needed in some (grade 1C).
5. Fluid challenge technique be applied wherein fluid administration is continued if there is hemodynamic improvement based on dynamic or static variables (UG).
1. Vasopressor therapy initially to target mean arterial pressure (MAP) of 65 mm Hg (grade 1C).
2. Norepinephrine (NE) as first choice vasopressor (grade 1B).
3. Epinephrine (added to and potentially substituted for norepinephrine) when additional agent is needed to maintain adequate BP (grade 2B).
4. Vasopressin 0.03 units/minute can be added to NE to raise MAP or decrease NE dosage (UG).
5. Low dose vasopressin not recommended as the single initial vasopressor for treatment of sepsis-induced hypotension. Vasopressin doses >0.03-0.04 units/minute should be reserved for salvage therapy (UG).
6. Dopamine as an alternative to norepinephrine only in highly selected patients (grade 2C).
7. Phenylephrine not recommended in the treatment of septic shock except where (a) norepinephrine is associated with serious arrhythmias, (b) cardiac output is high and BP persistently low or (c) as salvage therapy when combined inotrope/vasopressor drugs and low dose vasopressin have failed to achieve MAP target (grade 1C).
8. Low-dose dopamine should not be used for renal protection (grade 1A).
9. All patients requiring vasopressors have an arterial catheter placed as soon as practical (UG).
1. A trial of dobutamine infusion up to 20 mcg/kg/min be administered or added to vasopressor (if in use) in the presence of (a) myocardial dysfunction, or (b) ongoing signs of hypoperfusion, despite adequate intravascular volume and adequate MAP (grade 1C).
2. Not using a strategy to increase cardiac index to predetermined supranormal levels (grade 1B).
1. Do not use IV hydrocortisone to treat adults if adequate fluid resuscitation and vasopressor therapy are able to restore hemodynamic stability. Otherwise, IV hydrocortisone alone at a dose of 200 mg per day (grade 2C) is suggested.
2. Do not use ACTH stimulation test to identify adults who should receive hydrocortisone (grade 2B).
3. If used, hydrocortisone tapered when vasopressors no longer required (grade 2D).
4. Do not use corticosteroids for treatment of sepsis without shock (grade 1D).
5. When hydrocortisone is given, use continuous flow (grade 2D).
K Blood Product Administration
1. Once tissue hypoperfusion has resolved (in absence of extenuating circumstances), red blood cell transfusion is recommended only when Hgb decreases to <7.0 g/dL to target a Hgb concentration of 7.0 –9.0 g/dL in adults (grade 1B).
2. Do not use erythropoietin to treat anemia associated with severe sepsis (grade 1B).
3. Do not use fresh frozen plasma to correct laboratory clotting abnormalities in the absence of bleeding or planned invasive procedures (grade 2D).
4. Do not use antithrombin for treatment of severe sepsis and septic shock (grade 1B).
5. In patients with severe sepsis, administer platelets prophylactically when <10,000/mm3 (10 x 109/L) in absence of apparent bleeding. Prophylactic platelet transfusion suggested when <20,000/mm3 (20 x 109/L) if patient has significant risk of bleeding. Higher platelet counts (≥50,000/mm3 [50 x 109/L]) advised for active bleeding, surgery, or invasive procedures (grade 2D).
N Mechanical Ventilation of Sepsis-Induced Acute Respiratory Distress Syndrome (ARDS)
1. Target tidal volume of 6 mL/kg predicted body weight in patients with sepsis-induced ARDS (grade 1A vs. 12 mL/kg).
2. Measure plateau pressures in patients with ARDS. Initial upper limit goal in a passively inflated lung is ≤30 cm H2O (grade 1B).
3. Apply positive end-expiratory pressure (PEEP) to avoid alveolar collapse at end expiration (grade 1B).
4. Use strategies based on higher levels of PEEP for patients with sepsis- induced moderate or severe ARDS (grade 2C).
5. Use recruitment maneuvers in sepsis patients with severe refractory hypoxemia (grade 2C).
6. Use prone positioning in sepsis-induced ARDS patients with a Pao2/Fio2 ratio ≤100 mm Hg (grade 2B).
7. Maintain mechanically ventilated sepsis patients with head of bed elevated 30-45° to limit aspiration risk and prevent development of ventilator-associated pneumonia (grade 1B).
8. Use noninvasive mask ventilation (NIV) in that minority of sepsis-induced ARDS patients in whom the benefits of NIV have been carefully considered (grade 2B).
9. Put a weaning protocol in place. Mechanically ventilated patients with severe sepsis should undergo spontaneous breathing trials regularly to evaluate ability to discontinue mechanical ventilation when they: a) are arousable; b) are hemodynamically stable (without vasopressor agents); c) have no new potentially serious conditions; d) have low ventilator and end-expiratory pressure requirements; and e) have low Fio2 requirements that can be met with a face mask or nasal cannula. If spontaneous breathing trial is successful, consider extubation (grade 1A).
10. Do not routinely use pulmonary artery catheter for patients with sepsis-induced ARDS (grade 1A).
11. Use a conservative fluid strategy for patients with established sepsis-induced ARDS who do not have evidence of tissue hypoperfusion (grade 1C).
12. In the absence of specific indications, do not use beta 2-agonists for treatment of sepsis-induced ARDS (grade 1B).
O Sedation, Analgesia, and Neuromuscular Blockade in Sepsis
1. Continuous or intermittent sedation should be minimized in mechanically ventilated sepsis patients, targeting specific titration endpoints (grade 1B).
2. Avoid neuromuscular blocking agents (NMBAs) if possible in septic patients without ARDS due to risk of prolonged neuromuscular blockade following discontinuation. If NMBAs must be maintained, use either intermittent bolus as required or continuous infusion with train-of-four monitoring (grade 1C).
3. Use a short course of NMBA ≤48 hours for patients with early sepsis-induced ARDS and Pao2/Fio2 < 150 mm Hg (grade 2C).
1. A protocolized approach to management in ICU patients with severe sepsis commencing insulin dosing when 2 consecutive blood glucose levels are >180 mg/dL. Approach should target an upper blood glucose ≤180 mg/dL (grade 1A).
2. Blood glucose values should be monitored every 1–2 hrs until glucose values and insulin infusion rates stabilize, then every 4 hr, thereafter (grade 1C).
3. Glucose levels obtained with point-of-care testing of capillary blood should be interpreted with caution (UG).
1. Continuous renal replacement therapies and intermittent hemodialysis are equivalent in severe sepsis and acute renal failure (grade 2B).
2. Use continuous therapies to facilitate management of fluid balance in hemodynamically unstable septic patients (grade 2D).
1. Do not use sodium bicarbonate therapy for improving hemodynamics or reducing vasopressor requirements in patients with hypoperfusion-induced lactic acidemia with pH ≥7.15 (grade 2B).
S Deep Vein Thrombosis Prophylaxis
1. Patients with severe sepsis should receive daily pharmacoprophylaxis against venous thromboembolism (grade 1B). Accomplish with daily subcutaneous low-molecular weight heparin (LMWH) (grade 1B vs. twice daily UFH, grade 2C vs. three times daily UFH). If creatinine clearance <30 mL/min, use dalteparin (grade 1A) or another form of LMWH that has a low degree of renal metabolism (grade 2C) or UFH (grade 1A).
2. Treat patients with severe sepsis with combination of pharmacologic therapy and intermittent pneumatic compression devices if possible (grade 2C).
3. Do not use pharmacoprophylaxis in septic patients who have a contraindication for heparin use (grade 1B).Use mechanical prophylactic treatment (grade 2C), unless contraindicated. When risk decreases, start pharmacoprophylaxis (grade 2C).
1. Give stress ulcer prophylaxis using H2 blocker or proton pump inhibitor to patients with severe sepsis/septic shock who have bleeding risk factors (grade 1B).
2. If used, proton pump inhibitors preferred over H2RA (grade 2D)
3. Patients without risk factors do not receive prophylaxis (grade 2B).
1. Administer oral or enteral feedings rather than either complete fasting or provision of only IV glucose within the first 48 hours after diagnosis of severe sepsis/septic shock (grade 2C).
2. Avoid mandatory full caloric feeding in first week. Suggest low dose feeding, advancing only as tolerated (grade 2B).
3. Use IV glucose and enteral nutrition rather than total parenteral nutrition alone or parenteral nutrition in conjunction with enteral feeding in the first 7 days after diagnosis of severe sepsis/septic shock (grade 2B).
4. Use nutrition with no specific immunomodulating supplementation in patients with severe sepsis (grade 2C).
Data from Dellinger RP, Levy MM, Rhodes A, et al. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med. 2013:41(2):580.