Severe Sepsis and Multiple Organ Dysfunction
Introduction
The term sepsis is derived from the Greek word sepsin, which means “to make putrid.” The relationship between infection and sepsis has been recognized for many years. However, the precise mechanisms by which infection results in sepsis, severe sepsis, septic shock, or multiple organ dysfunction remain to be fully elucidated. Improvements in our understanding of this syndrome have led to the development of novel therapeutic strategies and have increased our appreciation for the complex interactions that exist in sepsis between pathogens and the host response to infection. Despite these advances, severe sepsis remains one of the most significant causes of morbidity and mortality in patients admitted to the intensive care unit (ICU).1 In this chapter we will discuss the current definitions, epidemiology, and pathogenesis of severe sepsis and multiple organ dysfunction. In addition, we will review current management options and discuss an approach to treatment based on pathophysiology.
Definitions
For many years the term sepsis was loosely applied in clinical practice and was used to describe a very heterogeneous patient population. In recognition of this problem, a consensus conference was convened to create standardized definitions and formulate a blueprint to guide future research in sepsis.2 The term systemic inflammatory response syndrome (SIRS) was introduced. SIRS can occur in response to a variety of severe clinical insults and is defined by the presence of two or more of the following conditions: (1) temperature > 38° C or < 36° C, (2) heart rate > 90 beats per minute, (3) respiratory rate > breaths per minute or a PaCO2 < 32 mm Hg, and (4) white blood cell count > 12,000 cells/mm3. Sepsis occurs when SIRS is caused by infection. Severe sepsis is sepsis with associated organ dysfunction, hypoperfusion, or hypotension. Hypoperfusion and perfusion abnormalities may include, but are not limited to, lactic acidosis, oliguria, or an acute alteration in mental status. Septic shock is defined by the presence of sepsis-induced hypotension (systolic blood pressure < 90 mm Hg or a reduction ≥ 40 mm Hg from baseline in the absence of other causes for hypotension), despite adequate fluid resuscitation along with the presence of perfusion abnormalities.2 The introduction of these definitions created a common language that was especially helpful in designing and defining populations for clinical trials.3 On the other hand, criticism of these definitions pointed out that they were too sensitive and were not useful when applied clinically to individual patients.4 In 2001 a second consensus conference with a broader representation was convened to revisit these definitions.5 The conference recommended keeping the 1992 definitions unchanged secondary to lack of new evidence to support new definitions. However, the consensus conference recommended expanding the diagnostic criteria for sepsis in an effort to enhance recognition at the bedside (Box 25.1). In addition, the Predisposition Insult infection Response Organ dysfunction (PIRO) system for staging sepsis was proposed. This staging system is still relatively new and further development and research will be needed prior to its implementation in clinical practice. Examples and possible measures for the future in each domain are shown in Table 25.1.
Table 25.1
The PIRO System for Staging Sepsis
| Domain | Present | Future |
| Predisposition | Premorbid conditions, age, and sex | Genetic polymorphism in components of the inflammatory response (e.g., TNF) |
| Insult infection | Culture and sensitivity of pathogens; identification of possible target for source control | Assays of specific microbial products and gene transcript profiles |
| Response | SIRS, other signs of sepsis, septic shock, C-reactive protein | Markers of activated inflammation or impaired host responsiveness |
| Organ dysfunction | Organ dysfunction as number of failing organs or composite scores | Measure of cellular response to insult-apoptosis, cytopathic hypoxia, cell stress |
SIRS, systemic inflammatory response syndrome; TNF, tumor necrosis factor.
Adapted from Levy M, Fink MP, Marshall JC, et al: 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med 2003;31:1250-1256.
Epidemiology
Severe sepsis constitutes a major health care problem.6–8 Estimates of the incidence of severe sepsis in the United States report that approximately 750,000 cases occur per year (3 cases per 1000 population).6 Almost 70% of these cases receive care in a high dependency unit (ICU, intermediate care unit, or coronary care unit).6 The incidence of severe sepsis and septic shock has increased over time both in North America and in Europe.6–8 The incidence of severe sepsis is projected to increase by 1.5% every year.6 These increases in incidence are attributed to an aging population with a growing number of patients with a compromised immune system, infected with resistant pathogens, and undergoing prolonged, high-risk surgical interventions.8 Severe sepsis is more frequent with increased age, in males, and in nonwhite patients.6,8 Before the mid-1980s, gram-negative bacteria were the most common pathogens responsible for severe sepsis. Over the years an increase in cases from gram-positive bacteria has been reported, and today gram-positive bacteria are the predominant pathogens in severe sepsis.8 The incidence of sepsis resulting from fungal organisms has increased substantially since the 1990s.8 The most common sites of infection include the respiratory system, the bloodstream, and the genitourinary tract.6,7,9
Although mortality for severe sepsis and septic shock has decreased over time, severe sepsis still kills one in four patients affected worldwide.6,7,10,11 Mortality increases with age in black men and with increased number of failing organs.8 Over time the hospital length of stay for patients with sepsis has decreased, and the number of discharges to nonacute medical care facilities has increased.8 In addition to causing high morbidity and mortality, severe sepsis has a significant economic impact. Estimates report an average cost per patient of $22,000, representing on annual impact to the health care system in excess of $16.5 billion in the United States alone.6
Pathophysiology
Severe sepsis is the result of complex interactions between infecting organisms and the host response. Important components of this host response in the early phases of sepsis include the immune system, activation of the inflammatory cascade, and alterations in hemostasis. In later stages of sepsis, organ failure, immunosuppression, and apoptosis play an important pathophysiologic role. Both characteristics of the infecting organism and of the host response influence the outcome of sepsis. Virulence factors, high burden of infection, and resistance to antibiotics are all organism characteristics associated with increased risk of severe sepsis. There is a growing body of literature suggesting that host responses might be influenced by genetic polymorphisms.12–17 This might explain why some patients develop severe sepsis to a particular pathogen and others do not. We will further discuss some of the relevant components of the host response in severe sepsis.
Role of the Immune System in the Early Phases of Sepsis
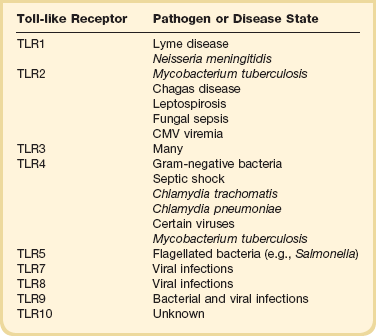
The immune response to infection takes place through the actions of two pathways: the innate immune system and the adaptive immune system. The goal of the innate immune system is to provide protection in the first minutes to hours after an infectious challenge. Although initially thought to be a nonspecific response, research has demonstrated that the innate immune system recognizes pathogens by means of pattern-recognition receptors (Toll-like receptors [TLRs], Table 25.2). Toll-like receptors bind to highly conserved structures on microorganisms, which are not easily altered by microbes to evade detection and are present on broad groups of organisms.18 Our current understanding of TLRs suggests that the immune cells use different TLRs to detect several features of an organism and based on the composite information gained generate a tailored response to the invading pathogen.18 Activation of Toll-like receptors by microorganisms stimulates signaling pathways that increase production of pro-inflammatory cytokines such as tumor necrosis factor (TNF-α), interleukin-1β (IL-1β), and nuclear factor-κB (NF-κB), as well as anti-inflammatory cytokines such as interleukin-10 (IL-10).18,19 Toll-like receptor activation also results in up-regulation of microbial killing mechanisms, such as the production of reactive nitrogen species.20 Toll-like receptors play a pivotal role in initiating the innate immune response and are important regulators of the adaptive immune response to infection. Recognition of these proteins and their functions expanded our understanding of the pathophysiology of sepsis and has provided a new target for therapeutic interventions.21
The adaptive immune system amplifies the response initiated by the innate immune system with a higher degree of specificity. In addition to their interactions with the innate immune system, microorganisms stimulate specific cell-mediated and humoral adaptive immune responses. Two types of lymphocytes, B cells and T cells, play an important role in the adaptive immune response. Adaptive immune responses (humoral and cellular) require days to develop. However, they are amnestic through the generation of memory T and B lymphocytes and, in the case of reexposure to the same pathogen, can elicit a faster response. CD4 T cells are divided into two types: type 1 helper T-cell (Th1) and type 2 helper T-cell (Th2). Factors such as type of organism, site of infection, and burden of infection influence the response elicited by T cells. In general, Th1 cells secrete pro-inflammatory cytokines (TNF-α and interleukin-1β) and Th2 cells secrete anti-inflammatory cytokines (interleukin-4 and interleukin-10).22 B lymphocyte cells are responsible for releasing immunoglobulins in response to microorganisms. These immunoglobulins bind to organism-specific antigens and enhance recognition and destruction by other immune cells (natural killer cells and neutrophils). Several other cell types are involved in the adaptive immune response to infection (Fig. 25.1).
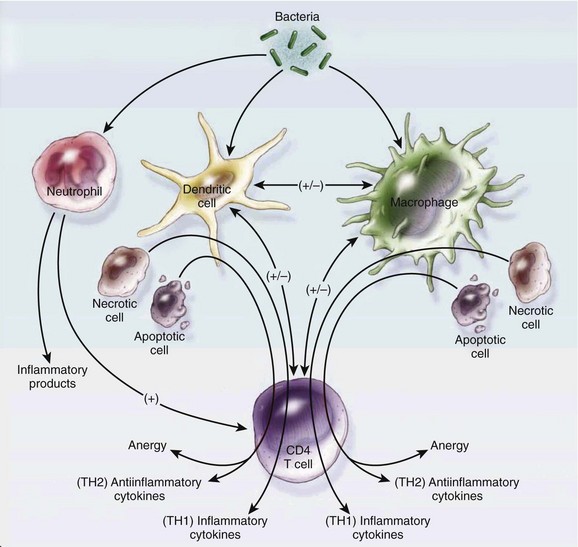
Role of Inflammation
For many years the prevailing theory has been that sepsis is the result of an uncontrolled inflammatory response.1,2 This paradigm was based on extensive animal experimentation with models of inflammation that may not necessarily reflect human disease. Animal models of sepsis that utilized large doses of endotoxin or bacteria created a “cytokine storm” that when blocked resulted in improvements in mortality. However, in human sepsis most patients have a complex host response that includes activation of both pro-inflammatory and anti-inflammatory cascades. Early death from overwhelming inflammation is not the norm, and most patients who die develop complications related to immunosuppression, apoptosis, and multiorgan failure later in the course of the disease. These differences may partially explain why so many anti-inflammatory compounds worked in animal models yet failed to improve mortality in human clinical trials. The interplay between pro-inflammatory cytokines, anti-inflammatory cytokines, and cytokine inhibitors is a dynamic process that influences the host response to sepsis. Pro-inflammatory cytokines such as TNF-α and IL-1β increase early in sepsis and have overlapping and synergistic effects in further stimulating the inflammatory cascade.23 Pro-inflammatory cytokines activate monocytes, macrophages, and neutrophils; stimulate neutrophil margination; and increase gluconeogenesis. In addition, pro-inflammatory cytokines have an important role in the development of clinical abnormalities such as fever, hypotension, capillary leakage with decreased intravascular volume, and myocardial depression.23 More recently, pro-inflammatory cytokines such as macrophage migration inhibitory factor (MIF) and high mobility group 1 protein (HMG-1) have received attention as downstream mediators of inflammation and potential therapeutic targets.24–27 The role of anti-inflammatory cytokines in sepsis is still not fully understood. Current understanding suggests that sepsis-induced multiorgan failure and death may be caused in part by a shift to an anti-inflammatory phenotype and by apoptosis of key immune cells.28,29 This shift is driven in part by increased levels of anti-inflammatory cytokines and results from a shift in helper T-cell populations (from Th1 to Th2).30 Inflammation plays an important role in the host response to sepsis. It is now apparent that simple therapeutic strategies that block specific pro-inflammatory cytokines are insufficient to modulate this response.31,32 As our understanding of the intricate relationship between pro-inflammatory and anti-inflammatory responses increases, we might become more successful in modulating these to improve patients’ outcomes.
Alterations of Hemostasis
Another important factor in the pathophysiology of sepsis is the alteration of the hemostatic balance. In sepsis this balance is altered by an increase in procoagulant factors paired with a decrease in anticoagulant factors (Fig. 25.2). Under normal conditions the intraluminal vascular surface has anticoagulant properties. During sepsis, stimulation from cytokines promotes expression of tissue factor on endothelial cells, monocytes, and neutrophils.33,34 Tissue factor triggers the extrinsic coagulation pathway by activating factor VII. Activation of the extrinsic pathway leads to the formation of thrombin. The intrinsic pathway is triggered by activation of factor XI and leads to amplification of the coagulation cascade with further formation of thrombin. Excessive coagulation is normally counterbalanced by several anticoagulant factors. Anticoagulant factors such as antithrombin III, activated protein C, protein S, and tissue factor pathway inhibitor are decreased in sepsis.35 These circumstances push the hemostatic balance toward the procoagulant state. Activation of the coagulation cascade leads to a consumption of coagulation factors. The clinical expression of this phenomenon is disseminated intravascular coagulation (DIC). Disseminated intravascular coagulation is characterized by a consumptive coagulopathy, which can result in an increased risk of bleeding but more commonly in sepsis causes damage by increasing the risk of thrombosis. In sepsis the excessive formation of fibrin from thrombin compounded by the suppression of fibrinolysis and the impairment of anticoagulant pathways leads to widespread formation of microthrombi. It has been proposed that these microthrombi lead to microcirculatory alterations and play an integral role in the pathogenesis of organ failure.36,37
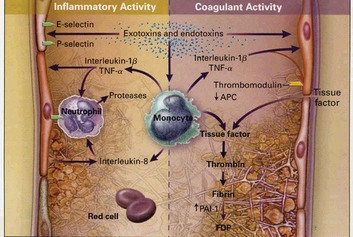
Management
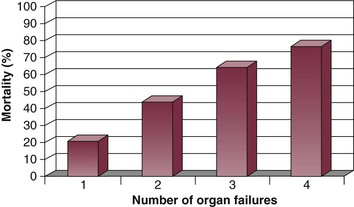
Severe sepsis is a medical emergency. When one considers its morbidity and the relationship between number of organ failures and mortality (Fig. 25.3), it makes sense to treat patients emergently and institute therapies that can prevent the progression of organ failure and improve outcomes in a time-sensitive fashion. Several therapies for severe sepsis have a potential time-sensitive effect on outcome (e.g., when instituted early have a higher likelihood of improving outcomes than when instituted with time delays) (Box 25.2). Although severe sepsis is associated with a higher mortality than other diseases considered medical emergencies, such as trauma, acute ischemic stroke, and acute myocardial infarction, it is still not treated with the same degree of urgency. This may be secondary to difficulties in recognizing severe sepsis early and a lack of understanding its consequences and their therapeutic implications by physicians outside the intensive care unit.
Recognizing these problems, the Society of Critical Care Medicine (SCCM), the European Society of Intensive Care Medicine (ESICM), and the International Sepsis Forum (ISF) created the Surviving Sepsis Campaign (SSC). The SSC conglomerates experts in the field of sepsis from around the world and currently counts with the endorsement of 29 international medical societies and the Institute for Healthcare Improvement (www.ihi.gov). The campaign has aimed to improve standards of patient care, secure funding for research, and ultimately reduce the mortality of severe sepsis worldwide. To achieve these goals, the SCC has published evidence-based practice guidelines and consensus recommendations for the management of patients with severe sepsis.38,39,39a These guidelines were first published in 2004 and revised in 2008, and a second revision for 2012 is currently in press. To increase the impact of these clinical guidelines at the bedside, the SCC created the sepsis bundles.40 The sepsis resuscitation bundle should be implemented over the first 6 hours after recognition of a patient with severe sepsis, and the sepsis management bundle should be implemented over the first 24 hours of admission to the hospital. A number of nonrandomized studies have shown that compliance with the bundles and application of their clinical recommendations in the form of protocols can improve patient outcomes.41–43 More important, the publication of phase 2 of the SSC international performance improvement program demonstrated the significant impact compliance with the sepsis bundles has on reducing mortality in severe sepsis.44 This large prospective study evaluated the implementation of a multifaceted intervention to facilitate compliance with selected guideline recommendations in the intensive care unit, emergency department, and wards of hospitals from around the world. Data from 15,022 subjects at 165 sites were analyzed to determine the compliance with bundle targets and association with hospital mortality. Compliance with the entire resuscitation bundle increased from 10.9% to 31.3% by the end of 2 years (p < 0.0001). Compliance with the entire management bundle started at 18.4% and increased to 36.1% by the end of 2 years (p = 0.008). This increase in compliance was associated with a decrease in unadjusted hospital mortality from 37% to 30.8% (p = .001). The adjusted odds ratio for mortality improved the longer the site participated in the SSC.44 Based on emerging data and recently published studies, the last revision of the guidelines recommends dropping the management bundle and dividing the resuscitation bundle into two parts:39a
The complete new bundles are shown in Box 25.3 and Box 25.4. The optimal treatment of severe sepsis is a dynamic and constantly evolving process. We will discuss current treatment recommendations based on up-to-date clinical data. However, as new research emerges it is likely that new therapies will be described, and treatment recommendations presented in this chapter may need to be modified.
Like in other medical emergencies the first priority in treating patients with severe sepsis should be assessing and optimizing the “ABCs”: airway, breathing, and circulation. In conjunction with initial stabilization of physiologic abnormalities, one should initiate appropriate diagnostic interventions to assess potential sources of infection and severity of organ dysfunction. Therapeutic interventions for severe sepsis should be implemented quickly and in conjunction. For the sake of discussion we will approach the treatment of severe sepsis based on pathophysiologic abnormalities produced by the syndrome (Fig. 25.4). We will discuss in further detail management of the infectious insult, hemodynamic optimization, modulation of the host response, and finally supportive therapies.
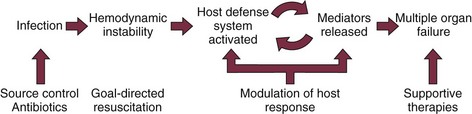
Figure 25.4 Approach to treatment of severe sepsis.
Infection Management
Severe sepsis is initiated by an infectious insult. Therefore, infection management constitutes one of the cornerstones of treatment in these patients. Infection management consists of source control and the administration of appropriate empiric antimicrobials that are effective against presumed causative pathogens. Administration of appropriate antibiotics is a time-sensitive intervention. Administration of antibiotics is often delayed, and this can result in worse outcomes.45 Delays in appropriate antibiotic administration are much more likely to result from system failures (e.g., order not written by physician, delay from pharmacy, etc.) than from bacteriologic resistance.46 Current guidelines recommend that appropriate antibiotics be administered to patients with severe sepsis within 1 hour of diagnosis.39 Results from a retrospective study in a large group of septic shock patients suggests that every hour appropriate antibiotics are delayed after the onset of hypotension, the odds ratio for mortality increases in a stepwise manner.47 Additional studies have shown increased mortality with delays in appropriate antibiotic administration.43,44,48,49 One study done in patients in the emergency department showed that if antibiotics were given prior to the onset of shock, mortality was significantly decreased.50 However, among patients who received antibiotics after shock recognition, mortality did not change with hourly delays in antibiotic administration.50 The goal should be to administer appropriate antibiotics as soon as possible in patients with severe sepsis. To accomplish this objective, hospitals must examine their particular dynamics and devise systems to optimize antibiotic administration.
Studies in patients with sepsis have reported an incidence of positive blood cultures in the range of 20% to 50%.8,51–53 Considering the growing need for broad-spectrum empirical regimens and the need to narrow down antimicrobial regimens in order to decrease resistance, obtaining blood cultures prior to the administration of antibiotics is essential. In most cases, one must start antibiotics without bacteriologic confirmation of the causative pathogen. Studies have demonstrated that the appropriateness of initial antibiotic therapy has a significant impact on patient outcomes.54,55 In one prospective cohort study of critically ill patients, inadequate initial antibiotic therapy was associated with a statistically significant increase in all-cause and infection-related hospital mortality.56 Factors associated with administration of inadequate antibiotics included prior administration of antibiotics, bloodstream infections, increasing acute physiology and chronic health evaluation (APACHE II) scores, and decreasing age.56 If one considers the detrimental effect on mortality, it is apparent that in patients with severe sepsis, one cannot afford to miss potential causative organisms when empirically selecting an antimicrobial regimen. The choice of antibiotics should be based on the following factors:
• Probable pathogens based on clinical diagnosis and source of infection (pneumonia, bloodstream infection, abdominal source, etc.)
• Site where infection was acquired (community versus hospital acquired)
• Results obtained from diagnostic tests such as Gram stains
• Resistance patterns of local and hospital bacterial flora
• Patient comorbidities, drug allergies, and previous antibiotic exposure
Initial empiric anti-infective therapy should include one or more drugs that have activity against likely pathogens and that penetrate the presumptive site of infection in adequate concentrations. Recently used anti-infective drugs in a particular patient should be avoided as the likelihood of resistance increases. Clinicians should also consider whether candidemia is a likely pathogen based on the presence of predisposing risk factors. When indicated, empirical antifungal therapy should take into account local flora and previous exposure of the patient to azole drugs. Recent Infectious Disease Society of America (IDSA) guidelines recommend either fluconazole or an echinocandin.57 Empiric use of echinocandin is preferred in critically ill patients, those exposed to azole drugs, and in settings where infection with C. glabrata is suspected or documented. Initial antibiotic therapy for severe sepsis should be broad in spectrum and progressively narrowed as microbiologic data become available. In culture-negative patients, the de-escalation of antibiotics may become challenging. In these cases, clinical evolution can be used to guide decisions. A detailed discussion of specific antibiotic regimens is beyond the scope of this chapter; the reader is referred to other chapters in the textbook and to the synopsis in Table 25.3.
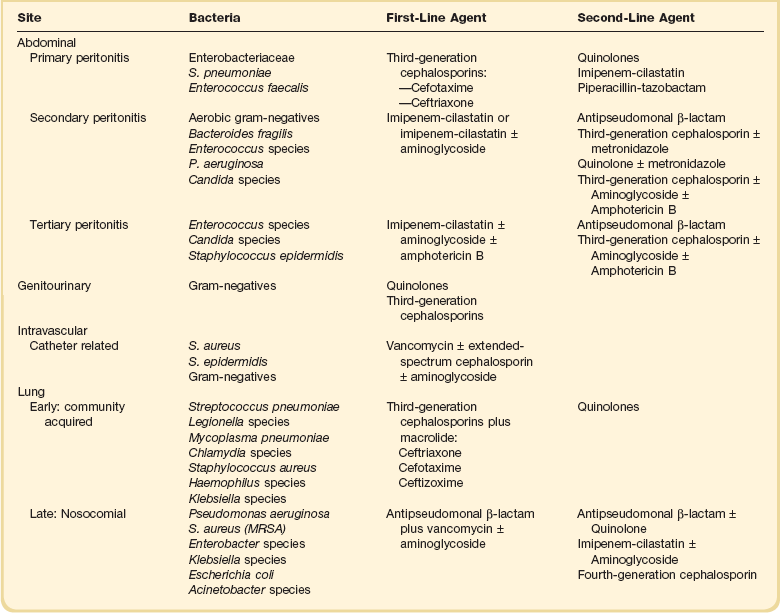
The term source control refers to measures implemented to control the source of infection. Source control interventions can be divided into three broad categories: (1) drainage of an abscess, (2) debridement/drainage/incision of infected tissue, and (3) removal of an infected foreign body.58 Attention to identifying potential sources amenable to source control measures should be part of the initial evaluation of patients with sepsis. The timing of intervention depends on several factors. When source interventions are simple, such as removal of an infected central venous catheter, they should be implemented immediately. In cases of unstable patients were surgery might be required, delaying source control while optimizing hemodynamic status may be appropriate. Finally, in cases such as necrotizing fasciitis, in which delays carry a significant risk of increasing mortality, one must proceed to surgery as early as possible. Examples of specific source control measures in patients with sepsis are shown on Table 25.4.

Hemodynamic Optimization
Severe sepsis is associated with a host of hemodynamic abnormalities. These abnormalities can ultimately lead to sepsis-induced tissue hypoperfusion if not addressed early and aggressively. The hemodynamic profile of severe sepsis and septic shock is initially characterized by components of hypovolemic, cardiogenic, and distributive shock.59 In the initial phases of resuscitation, addressing the hypovolemic component is most important. Early in sepsis, increased capillary leak and increased venous capacitance will result in effective hypovolemia with decreased venous return to the heart. Low intravascular volume paired with sepsis-induced myocardial depression will result in a decrease in stroke volume (SV). Administration of intravascular fluids can alter this early phase of sepsis characterized by hypovolemia, tachycardia, and depressed cardiac output. Initial steps in hemodynamic optimization for patients with severe sepsis should include evaluation for signs of sepsis-induced tissue hypoperfusion. Signs of global hypoperfusion such as hypotension, tachycardia, oliguria, delayed capillary refill, altered mentation, increased blood lactate, and low mixed venous oxygen saturation are helpful when present to establish tissue hypoperfusion. However, these signs are not always sensitive and they must be complemented with assessment of indices of regional hypoperfusion. Patients with severe sepsis should have good venous access. Central venous access is preferred as it can also be used for hemodynamic monitoring.
The importance of early intervention in patients with sepsis-induced tissue hypoperfusion has been highlighted by the results of an early goal-directed therapy (EGDT) clinical trial by Rivers and colleagues.60 In this study, patients with sepsis-induced hypoperfusion (lactate > 4 mmol or hypotension after fluids) were randomized to receive either standard resuscitation or an early goal-directed (EGDT) protocol during the first 6 hours of admission to the emergency department. In both groups, end points of resuscitation included central venous pressure (CVP) ≥ 8 to 12 mm Hg, mean arterial pressure (MAP) ≥ 65 mm Hg, and urine output ≥ 0.5 mL/kg/hr. To achieve these goals, patients were treated with intravenous crystalloids and vasopressors. The EGDT group had as an additional end point, central venous oxygen saturation (ScvO2) ≥ 70%, which was continuously measured from a subclavian or jugular central venous catheter. ScvO2 was used as an index for oxygen delivery. If ScvO2 was < 70% after reaching targets for CVP and MAP, patients received packed red blood cells for a hematocrit ≤ 30, or dobutamine infusion if the hematocrit was ≥ 30. Patients in the EGDT group received more fluids, dobutamine, and transfusions in the first 24 hours. In-hospital mortality was significantly lower in the EGDT group when compared to the standard therapy group (30.5% versus 46.5%, respectively, [p = 0.009]). Observational studies published after the Rivers study have demonstrated a strong association between improved clinical outcomes and maintenance of MAP ≥ 65 mm Hg as well as central venous oxygen saturation (ScvO2) of ≥ 70%.61 Furthermore, several more recent studies have showed improved outcomes with the use of protocolized quantitative resuscitation in severe sepsis and sepsis-induced tissue hypoperfusion.42,62–65 Although the specific merits of each individual intervention within a quantitative resuscitation or EGDT protocol can be discussed, the results of these studies strongly support early intervention with predefined hemodynamic settings and protocolized care.
As stated before, the initial step in optimizing hemodynamics in patients with severe sepsis is aggressive fluid resuscitation. Although experts agree on the value of early and aggressive volume replacement, controversy persists over the optimal type of fluid. This debate revolves around the use of crystalloids (saline, Ringer’s lactate) versus colloids (albumin, hydroxyethyl starches). A large meta-analysis evaluated data from 56 trials and found no difference in mortality between crystalloids and colloids when used for initial fluid resuscitation.66 Three randomized studies did not find a difference in mortality when starches (heta-, hexa-, or penta-) where compared to other fluids. However, these studies did report a significant increase in acute kidney injury with the use of starches.67–69 The Saline versus Albumin Fluid Evaluation (SAFE) study prospectively randomized 7000 critically ill patients to receive 4% albumin or 0.9% saline for fluid resuscitation.70 There were no significant differences between groups in mortality and other secondary outcomes. A subgroup analysis conducted in patients with sepsis revealed a trend toward improved outcomes in patients treated with albumin, although this difference did not achieve statistical significance. We believe that achieving end points of resuscitation is more important than the type of fluid utilized. In North America consideration for cost differences has made crystalloids the initial fluid of choice for resuscitating patients with severe sepsis. However, based on emerging data it seems appropriate to add albumin to the initial fluid resuscitation regimen in severe sepsis and septic shock.
Patients with severe sepsis may present with significant intravascular volume depletion. Aggressive fluid boluses are usually required to restore tissue perfusion. It is recommended that patients receive at least 20 to 30 mL/kg of crystalloid initially.39,71 This may be supplemented with more fluids based on markers of perfusion in repeated boluses of 300 to 500 mL.71 Current guidelines recommend achieving the following hemodynamic end points of resuscitation during the first 6 hours of treatment: CVP ≥ 8 to 12 mm Hg, mean arterial pressure (MAP) ≥ 65 mm Hg, urine output ≥ 0.5 mL/kg/h, and central venous oxygen saturation (ScvO2) ≥ 70%.39,72 For further discussion on the pathophysiology and treatment of hemodynamic abnormalities in sepsis, the reader is referred to Chapter 23.
Modulation of the Host Response
Over the years, research efforts in severe sepsis have been heavily involved with therapies targeted at modulating the host response. Several pathways and mechanisms have been studied in clinical trials (Table 25.5). Unfortunately, very little success has been found in these endeavors. Initial attempts were aimed at blunting the inflammatory response with nonspecific agents such as high-dose glucocorticoids and ibuprofen.73–75 Another unsuccessful strategy involved the use of antibodies directed at endotoxin in patients with gram-negative sepsis.76–81 However, the area that received the greatest attention was modulation of the inflammatory cascade by targeting specific pro-inflammatory cytokines such as tumor necrosis factor (TNF-α) and interleukin-1β (IL-1β). Multiple clinical trials enrolling thousands of patients tested compounds directed at specific pro-inflammatory cytokines, among them TNF monoclonal antibody, interleukin-1 receptor antagonist, and soluble TNF receptor.82–89 Unfortunately, none of these compounds improved survival of patients with severe sepsis in randomized studies. The failure of these therapies led to a reappraisal of the pathophysiology, potential therapeutic targets, and clinical trial design in severe sepsis. As the role of the coagulation cascade and its crosstalk with inflammation in sepsis was recognized, a series of new clinical trials took place. Three large trials studied the effects of anticoagulants in severe sepsis (Box 25.5).
Table 25.5
Pathways and Mediators of Sepsis, Potential Treatments, and Results of Randomized, Controlled Trials (RCTs)*
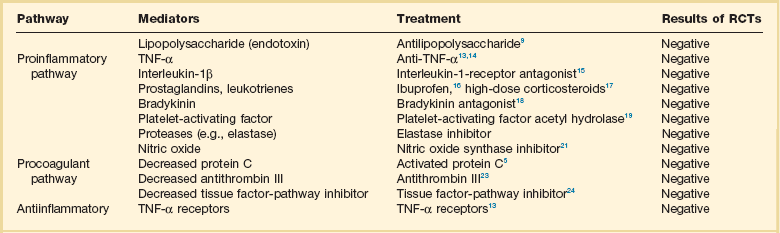
*Studies cited in table may be found in the complete list of references for this chapter provided online.
Adapted from Russell, JA: Management of sepsis. N Engl J Med 2006; 355:1699-1713.
Antithrombin III (AT III) is a progressive inhibitor of thrombin and factor Xa.90 Studies showed that AT III supplementation attenuated the systemic inflammatory response in patients with severe sepsis.91 A large (n = 2314) multicenter, double-blinded, placebo-controlled trial evaluated the safety and efficacy of AT III in adult patients with severe sepsis.92 At 28 days there was no difference in mortality between the treatment group and the placebo group (38.9% versus 38.7%, respectively). Patients who received AT III had a higher risk of bleeding (relative risk > 1.7). A subgroup analysis of patients not receiving concomitant heparin showed a trend (statistically nonsignificant) toward reduced mortality at 28 and 90 days with AT III. This specific subgroup of patients with severe sepsis may warrant further investigations. Tissue factor pathway inhibitor (TFPI) has been show to modulate the extrinsic pathway in preclinical models of severe sepsis. Recombinant human TFPI (tifacogin) was evaluated in clinical trials of patients with severe sepsis. Although the results of a phase II trial suggested a trend toward improved mortality, a large phase III trial, the OPTIMIST (Optimized Phase III Tifacogin in Multicenter International Sepsis Trial) study failed to show a mortality benefit in patients treated with this compound.93,94 Patients treated with tifacogin had a higher risk of bleeding complications irrespective of their baseline international normalized ratios (INRs).93 Studies evaluating different dosing regimens and the application of this drug in patients with pneumonia are still being conducted. Finally, recombinant human activated protein C (rhAPC) was evaluated in clinical trials. A landmark study, the protein C worldwide evaluation in severe sepsis (PROWESS) trial, demonstrated improved 28-day survival in patients with severe sepsis treated with rhAPC.95
The phase III randomized, double-blind, placebo-controlled, multicenter international study, PROWESS trial evaluated the efficacy of drotrecogin alfa (activated) in patients with severe sepsis.95 This study enrolled 1690 patients and was terminated early after an interim safety analysis found a significant reduction in mortality in the treatment group compared to placebo (24.7% versus 30.8%, respectively [p = 0.005], relative and absolute risk reductions of 19.4% and 6.1%, respectively). Patients treated with drotrecogin alfa (activated) showed a trend toward a higher incidence of bleeding (3.5% versus 2%; p = 0.06). Subgroup analysis demonstrated that patients at higher risk of death as measured by APACHE II scores (APACHE II ≥ 25) and number of organ failures (two or more organ failures) had an increased benefit from the drug. Effects on mortality seemed to be lost in low-severity patients. Based on this study, the Food and Drug Administration (FDA) approved the use of drotrecogin alfa (activated) in adult patients with severe sepsis with a high risk of death. Angus and colleagues reported, after long-term follow-up, that those patients who were treated with drotrecogin alfa (activated) had an increased median survival (9 months) compared to patients treated with placebo.96 Once again, beneficial effects of the drug seemed to be greatest in patients with a higher severity of disease. Two additional phase IV studies were published post the FDA’s approval of the drug. The ENHANCE trial was a single-arm open-label study that enrolled 2375 patients with severe sepsis.97 ENHANCE evaluated the use of drotrecogin alfa (activated) in a more routine clinical setting beyond the restrictions of a controlled randomized study. In this study, the effect of the drug on mortality was similar to PROWESS (25.3% in ENHANCE versus 24.7% in PROWESS). The risk of bleeding during infusion was higher when compared to PROWESS (3.6% versus 2.4%). However, the higher rate of postinfusion bleeding observed in the ENHANCE population (3.2% versus 1.2%) suggests a higher incidence of background bleeding. A second phase IV trial, the ADDRESS study, was a randomized, blinded, placebo-controlled trial that evaluated the efficacy of the drug in severe sepsis in patients judged prospectively by the enrolling clinician to have a low risk of death (APACHE II score < 25 or single organ failure based on regulation requirements in countries of study entry).98 This study enrolled 2646 patients and found that treatment with the drug offered no mortality benefit when compared to placebo in a low-risk-of-death population. Serious bleeding events were similar to those reported in PROWESS. There was significant criticism from several academic experts on the approval of drotrecogin alfa (activated) based on one single positive prospective randomized trial that was terminated prematurely because of positive results. Additionally critics argued that the drug was associated with a higher risk of bleeding when used in the clinical setting and that it was approved for a population that had not been prospectively evaluated in randomized trials (based on the post hoc analysis of the APACHE II quartiles). The publication of the PROWESS SHOCK trial, showing no benefit of drotrecogin alfa (activated) in patients with septic shock (mortality 26.4% in patients given drotrecogin alfa [activated] versus 24.2% in patients receiving placebo) led to the worldwide withdrawal of the drug from the market.99 The choice to withdraw was a voluntary decision by Eli Lilly and Company and was likely heavily influenced by business calculations. Drotrecogin alfa (activated) has important biologic effects that could be helpful in modulating the host response in severe sepsis. It is very probable that we do not know how best to select patients who would benefit from this drug. In the future, we should consider ways to improve our process in drug development from clinical trial design to regulatory approval. Furthermore, it seems that more sophisticated selection of patients will be instrumental in future studies evaluating new agents designed to modulate the host response to sepsis.100
Supportive Therapies
As with other critical illnesses, patients with severe sepsis require various supportive therapies (see Box 25.5). These therapies are general supportive measures that prevent complications associated with critical illness. Improvement in these therapies over the years probably plays a role in the historical decrease in mortality observed in several disease processes such as severe sepsis and acute respiratory distress syndrome (ARDS). Patients with severe sepsis often present with tachypnea and hypoxemia. Mechanical ventilation is often utilized for support. Studies in patients with ARDS have demonstrated that ventilation strategies utilizing low tidal volume (6 mL/kg) are associated with significantly lower mortality than ventilation with more traditional tidal volumes (12 mL/kg).101 This is most likely due to a decrease in ventilator-induced lung injury. Several meta-analyses have suggest decreased mortality in patients with established ARDS who are treated with a volume and pressure limited ventilator strategy.102,103 Current guidelines recommend the use of a protective lung strategy (low tidal volume; inspiratory plateau pressure < 30 cm H2O) in mechanically ventilated patients with severe sepsis.39 Goals for oxygen saturation should be an SaO2 ≥ 90%. This can be achieved by increasing the fraction of inspired oxygen (FiO2) and/or application of positive end-expiratory pressure (PEEP). In patients with sepsis-induced ARDS who do not have evidence of tissue hypoperfusion, a conservative approach to fluid management is recommended.104 Patients on mechanical ventilation who are clinically improving should be evaluated on a daily basis for weaning from mechanical ventilation. Patients with severe sepsis on mechanical ventilation should be managed with appropriate sedatives and analgesics. For a detailed discussion, the reader is referred to Chapter 19.
Patients with severe sepsis should receive prophylaxis for the development of deep vein thrombosis (DVT). In the absence of contraindications, patients should receive pharmacologic DVT prophylaxis. Treatment with low-dose unfractioned heparin (UFH), adjusted-dose UFH, or low-molecular-weight heparin (LMWH) is recommended.105 Treatment for DVT prophylaxis with UFH or LMWH is not contraindicated during infusion of drotrecogin alfa (activated). Stress ulcer prophylaxis is recommended for all patients with severe sepsis. Histamine-2 receptor antagonists are more effective than sucralfate in decreasing bleeding risk and transfusion requirements.106 Proton pump inhibitors have not been assessed in a direct comparison with histamine-2 receptor antagonists but do demonstrate equivalency and ability to increase gastric pH.105
Severe sepsis is a catabolic state. Metabolic alterations in patients with severe sepsis include breakdown of proteins, carbohydrates, and lipids; negative nitrogen balance; and hyperglycemia with insulin resistance. As with other critically ill patients, those with severe sepsis require adequate nutritional support. Enteral nutrition offers several advantages including lower cost, preservation of gastric mucosa integrity, decreased incidence of infections, and avoidance of parenteral nutritional catheters and their potential complications.107 In patients who cannot tolerate enteral nutrition, parenteral nutrition should be utilized.108 Immunomodulation through nutritional supplements has been proposed in patients with severe sepsis but remains experimental at this point.
Hyperglycemia and insulin resistance are commonly present in patients with severe sepsis. This phenomenon is a common feature of the metabolic response to critical illness and stress and has been described after major surgery, in trauma, acute myocardial infarction, and several other disease states. Furthermore, there is a growing body of literature suggesting that hyperglycemia related to critical illness is associated with poor outcomes.109–113 Proposed mechanisms for this deleterious effect include impaired neutrophil function, increased risk of infection, poor wound healing, and procoagulant state as a consequence of hyperglycemia.114 Treatment of critical illness–related hyperglycemia with insulin has been proposed to modulate these effects and improve patient outcomes. Van den Berghe and associates studied the effects of tight glycemic control on outcomes in a population of mechanically ventilated surgical critical care patients.115 In this study, patients were randomized to receive intensive insulin therapy (target blood glucose 80-110 mg/dL) or standard therapy (target blood glucose 180-200 mg/dL). Patients treated with the intensive insulin regimen had significant improvements in overall ICU mortality rates (4.6% versus 8.0%). This benefit in mortality was more pronounced among patients who stayed in the ICU longer than 5 days (10.6% versus 20.2%, p = 0.005). In addition, intensive insulin therapy was associated with a 46% reduction in bloodstream infections, a 44% reduction in the incidence of critical illness polyneuropathy, a 41% decrease in the need for renal replacement therapy, and a 50% reduction in number of transfused units of packed red blood cells. The same group reported the results of a similar study in medical intensive care unit patients.116 In this clinical trial, intensive insulin was not associated with improved mortality when compared to standard therapy. Intensive insulin therapy was associated with decreased mortality in patients who remained in the ICU > 3 days, but it was associated with increased mortality in those who remained in the ICU < 3 days. Prospective identification of these patient groups was difficult. Studies published more recently have not found the same benefit in mortality with intensive insulin therapy.69,117–119 The NICE-SUGAR study, a large randomized trial with > 6000 patients, found that intensive glucose control increased mortality among adults in the ICU.120 In this study, a blood glucose target of 180 mg or less/dL resulted in lower mortality than did a target of 81 to 108 mg per deciliter.120 Two questions remain germane to the glycemic control issue: First, what is the downside of tight glycemic control? Second, what level of glucose should we target? The biggest downside to tight glycemic control probably relates to the risk of hypoglycemia and the morbidity/mortality this could cause in critically ill patients. In both studies by Van den Berghe and colleagues, hypoglycemia was more common in the intensive insulin group than in the standard group (surgical study: 5.2% versus 0.7% and medical study: 18.7% versus 3.1%, respectively).115,116 In subsequent studies, the incidence of hypoglycemia was consistently higher. As an example in the NICE-SUGAR study, severe hypoglycemia (blood glucose level, ≤40 mg/dL) was reported in 6.8% of the patients in the intensive-control group and in 0.5% of the patients in the conventional-control group (p < 0.001).120 Concerns for the effects of hypoglycemia on ICU patients are well founded. However, it does not appear that short-term hypoglycemia that is quickly recognized and treated carries deleterious consequences.121 There is no clear answer with respect to what glucose level we should target in patients with severe sepsis. It is important to note that the studies that showed benefit with tight glycemic control compared intensive insulin therapy to high controls (180-200 mg/dL), whereas those that did not demonstrate benefit compared intensive insulin therapy to moderate controls (108-180 mg/dL). Considering the current available evidence, it is recommended that glucose be kept < 180 mg/dL in patients with severe sepsis. To minimize the risk of hypoglycemia, patients on intensive insulin regimens should have frequent blood glucose monitoring.
Multiple Organ Dysfunction
Multiple organ dysfunction is a common complication of sepsis. Multiple organ dysfunction syndrome (MODS) occurs when two or more organ systems fail sequentially or at the same time in a patient with sepsis. Various organs, such as the brain, heart, lung, kidney, and liver, can be affected in patients with severe sepsis. Often these organs are distant from the site of primary insult, and development of organ failure occurs as a response to complicated interactions and pathophysiologic events. Metabolic and hematologic dysfunctions are also common with severe sepsis and MODS. MODS significantly contributes to higher mortality. Studies have shown that mortality in patients with severe sepsis increases in parallel with increases in number and severity of organ failures.122,123 Russell and associates evaluated the pattern of organ dysfunction in early sepsis and its relationship with mortality.124 In this study, clinically significant pulmonary dysfunction, although common early in sepsis, was not associated with 30-day mortality. Early dysfunction of other organs and, particularly, worsening neurologic, coagulation, and renal dysfunction over the first 3 days were associated with significantly higher 30-day mortality.
Recognition of early organ dysfunction is important because it is likely that early intervention can affect outcomes. The clinical manifestations of MODS for individual organs are summarized in Figure 25.5. The cornerstones of treatment for MODS are based on appropriate treatment for the underlying cause (sepsis) and on early organ-specific support interventions. As discussed previously, early implementation of therapies directed at control of infection, hemodynamic support, and modulation of the host response are key to improving organ dysfunction and patient outcomes. Perhaps the single most important aspect relates to early and aggressive hemodynamic support. As demonstrated in the study by Rivers and colleagues, goal-directed interventions instituted in the first 6 hours of presentation to the hospital have a tremendous impact on long-term organ function and survival.60 We further discuss some salient features of the pathophysiology of MODS and the use of scoring systems. For a more detailed discussion on organ-specific supportive therapies, the reader is referred to other chapters in this textbook.
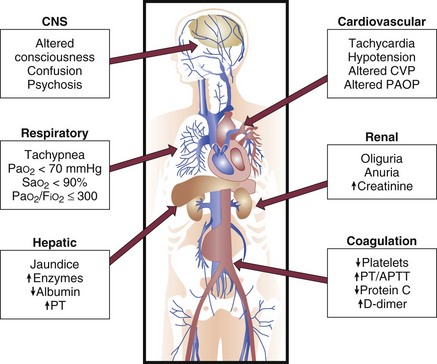
Pathophysiology of Multiple Organ Dysfunction Syndrome in Sepsis
In early unresuscitated sepsis, tissue hypoperfusion is to a great extent driven by decreased intravascular volume and the resulting drop in cardiac output (hypovolemic shock).54 Despite aggressive volume resuscitation, however, many patients still show evidence of tissue hypoperfusion, probably secondary to vasodilation and maldistribution of blood flow (distributive shock).71 Furthermore, a subset of patients with normalized macrovascular hemodynamic parameters (e.g., blood pressure, CVP, and cardiac output) can still show evidence of sepsis-induced tissue hypoperfusion.125 New technology has allowed investigators to evaluate the microcirculatory flow in patients with severe sepsis.126 Redistribution of capillary blood flow has been demonstrated in both animal models and clinical sepsis.36,37 The importance of this finding has been highlighted by studies demonstrating that functional (impaired blood flow) and structural (shunting, redistribution) abnormalities in microcirculation are associated with death and organ failure in patients with severe sepsis and septic shock.37,127
In the early phases of sepsis, decreased oxygen delivery (DO2) can result in tissue hypoperfusion. However, in late sepsis there is evidence of impaired tissue oxygen utilization even after optimization of DO2.128 The inability of cells to use oxygen in the face of adequate DO2 in sepsis has been termed cytopathic hypoxia.128 Development of cytopathic hypoxia is closely linked to mitochondrial dysfunction. The inability of the mitochondria to use oxygen to produce energy in the form of adenosine triphosphate leads to impaired cellular function. Proposed mechanisms that result in cytopathic hypoxia in sepsis include diminished delivery of pyruvate into the mitochondria, inhibition of mitochondrial enzymes, and activation of poly-(adenosine phosphate-ribosyl) polymerase (PARP).129 The exact mechanisms leading to organ failure in sepsis remain unidentified. Organ failure in sepsis is reversible in patients who survive. Furthermore, in patients who do not survive, there is no histopathologic evidence of tissue damage.28 Hotchkiss and associates have described extensive lymphocyte apoptosis in sepsis and have proposed this mechanism as an important driver of the impaired immune response seen in late sepsis and MODS.130,131 Finally, multiple organ failure has been hypothesized to be an adaptive metabolic response to overwhelming inflammation in sepsis.132 The hypothesis holds that multiple organ failure induced by sepsis is primarily a functional abnormality that serves as a protective reactive mechanism and that the decline of organ function is triggered by a decrease in mitochondrial activity. This decrease in mitochondrial activity leads to a reduction in cell metabolism and occurs as a consequence of humoral and mediator-induced changes.118
Organ Dysfunction Scoring Systems
Severity of illness scoring systems have been developed and applied in the ICU to describe patient populations. These scoring systems have been useful in predicting expected mortality and comparing different patient populations. The use of general outcome prediction models, such as the APACHE, the Mortality Probability Model (MPM), and the second Simplified Acute Physiologic Score (SAPS), is discussed in detail in Chapter 74. Therefore, this discussion is limited to scoring systems used to specifically assess organ dysfunction.
It is recognized that the risk of death for patients with severe sepsis is directly related to the number of dysfunctional organs. Organ dysfunction scoring systems have been developed as a tool for the clinician to characterize the severity of illness and follow the clinical evolution of patients with sepsis. The more commonly used systems are the Sequential Organ Failure Assessment (SOFA), the Logistic Organ Dysfunction System, and the Multiple Organ Dysfunction Score. Perhaps the most commonly used is the SOFA score (Table 25.6). It was initially described by Vincent and colleagues to assess the incidence of organ dysfunction in critically ill patients.123 Using the SOFA scores in patients with severe sepsis, these researchers found that mortality rates were lowest in patients without organ dysfunction (9%) and rose progressively with the number of organ dysfunctions (one organ, 22%; two organs, 38%; three organs, 69%; ≥ four organs, 83%).123 The type of organ dysfunction also affects mortality. Hebert and coworkers106 used logistic aggression analysis of the results of a simple multiple system organ failure score to determine the odds ratio for death for specific organ system dysfunctions. This study showed that the adjusted odds ratios (OR) for covariates most predictive of mortality were hematologic (OR = 6.2), neurologic (OR = 4.4), hepatic (OR = 3.4), cardiovascular (OR = 2.6), and age (OR = 1.05). It is important to remember that there are caveats when employing organ dysfunction scores for the management of individual patients with severe sepsis. Most important, organ dysfunction is not a static process and it changes over time. Levy and colleagues reported that changes in SOFA score over the first 24 hours were associated with outcomes in patients with severe sepsis. Improvement in cardiovascular, renal, or respiratory failure over the first 24 hours was associated with lower mortality.133 On the other hand, worsening SOFA scores for these organ systems were associated with higher mortality (an approximately 60% mortality rate). Finally, how these scores change over time in response to therapeutic interventions is probably of greater value than an initial organ dysfunction score.
Table 25.6
The Sequential Organ Failure Assessment Score

Adapted from Vincent JL, de Mendonca A, Cantraine F, et al: Use of the SOFA score to assess the incidence of organ dysfunction/failure in intensive care units: Results of a multicenter, prospective study. Working group on “sepsis-related problems” of the European Society of Intensive Care Medicine. Crit Care Med 1998;26:1793-1800.
References
1. Stone, R. Search for sepsis drugs goes on despite past failures. Science. 1994; 264:365–367.
2. Bone, RC, Balk, RA, Cerra, FB, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest. 1992; 101:1644–1655.
3. Trzeciak, S, Zanotti-Cavazzoni, S, Parrillo, JE, Dellinger, RP. Inclusion criteria for clinical trials in sepsis: Did the American College of Chest Physicians/Society of Critical Care Medicine consensus conference definitions of sepsis have an impact? Chest. 2005; 127:242–245.
4. Vincent, JL. Dear SIRS, I’m sorry to say that I don’t like you. Crit Care Med. 1997; 25:372–374.
5. Levy, MM, Fink, MP, Marshall, JC, et al. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med. 2003; 31:1250–1256.
6. Angus, DC, Linde-Zwirble, WT, Lidicker, J, et al. Epidemiology of severe sepsis in the United States: Analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001; 29:1303–1310.
7. Annane, D, Aegerter, P, Jars-Guincestre, MC, Guidet, B. Current epidemiology of septic shock: The CUB-Rea Network. Am J Respir Crit Care Med. 2003; 168:165–172.
8. Martin, GS, Mannino, DM, Eaton, S, Moss, M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003; 348:1546–1554.
9. Sands, KE, Bates, DW, Lanken, PN, et al. Epidemiology of sepsis syndrome in 8 academic medical centers. JAMA. 1997; 278:234–2340.
10. Friedman, G, Silva, E, Vincent, JL. Has the mortality of septic shock changed with time. Crit Care Med. 1998; 26:2078–2086.
11. Dombrovskiy, VY, Martin, AA, Sunderram, J, Paz, HL. Rapid increase in hospitalization and mortality rates for severe sepsis in the United States: A trend analysis from 1993 to 2003. Crit Care Med. 2007; 35:1244–1250.
12. Arcaroli, J, Fessler, MB, Abraham, E. Genetic polymorphisms and sepsis. Shock. 2005; 24:300–312.
13. Arcaroli, J, Silva, E, Maloney, JP, et al. Variant IRAK-1 haplotype is associated with increased nuclear factor-kappaB activation and worse outcomes in sepsis. Am J Respir Crit Care Med. 2006; 173:1335–1341.
14. Barber, RC, Aragaki, CC, Rivera-Chavez, FA, et al. TLR4 and TNF-alpha polymorphisms are associated with an increased risk for severe sepsis following burn injury. J Med Genet. 2004; 41:808–813.
15. Freeman, BD, Buchman, TG. Gene in a haystack: Tumor necrosis factor polymorphisms and outcome in sepsis. Crit Care Med. 2000; 28:3090–3091.
16. Gordon, AC, Lagan, AL, Aganna, E, et al. TNF and TNFR polymorphisms in severe sepsis and septic shock: A prospective multicentre study. Genes and immunity. 2004; 5:631–640.
17. Lin, MT, Albertson, TE. Genomic polymorphisms in sepsis. Crit Care Med. 2004; 32:569–579.
18. Underhill, DM, Ozinsky, A. Toll-like receptors: key mediators of microbe detection. Current opinion in immunology. 2002; 14:103–110.
19. Brower, RG, Lanken, PN, MacIntyre, N, et al. Higher versus lower positive end-expiratory pressures in patients with the acute respiratory distress syndrome. N Engl J Med. 2004; 351:327–336.
20. Brightbill, HD, Libraty, DH, Krutzik, SR, et al. Host defense mechanisms triggered by microbial lipoproteins through toll-like receptors. Science. 1999; 285:732–736.
21. Modlin, RL, Brightbill, HD, Godowski, PJ. The toll of innate immunity on microbial pathogens. N Engl J Med. 1999; 340:1834–1835.
22. Abbas, AK, Murphy, KM, Sher, A. Functional diversity of helper T lymphocytes. Nature. 1996; 383:787–793.
23. Zanotti, S, Kumar, A, Kumar, A. Cytokine modulation in sepsis and septic shock. Expert Opin Investig Drugs. 2002; 11:1061–1075.
24. Wang, H, Yang, H, Tracey, KJ. Extracellular role of HMGB1 in inflammation and sepsis. J Intern Med. 2004; 255:320–331.
25. Czura, CJ, Tracey, KJ. Targeting high mobility group box 1 as a late-acting mediator of inflammation. Crit Care Med. 2003; 31:S46–S50.
26. Yang, H, Ochani, M, Li, J, et al. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci U S A. 2004; 101:296–301.
27. Calandra, T, Echtenacher, B, Roy, DL, et al. Protection from septic shock by neutralization of macrophage migration inhibitory factor. Nat Med. 2000; 6:164–170.
28. Hotchkiss, RS, Karl, IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003; 348:138–150.
29. Remick, DG. Pathophysiology of sepsis. Am J Pathol. 2007; 170:1435–1444.
30. Gogos, CA, Drosou, E, Bassaris, HP, Skoutelis, A. Pro- versus anti-inflammatory cytokine profile in patients with severe sepsis: A marker for prognosis and future therapeutic options. J Infect Dis. 2000; 181:176–180.
31. Li, J, Carr, B, Goyal, M, Gaieski, DF. Sepsis: The inflammatory foundation of pathophysiology and therapy. Hosp Pract. 2011; 39:99–112.
32. Okazaki Y, Matsukawa A: Pathophysiology of sepsis and recent patents on the diagnosis, treatment and prophylaxis for sepsis: Recent patents on inflammation & allergy drug discovery. 2009;3:26–32.
33. Esmon, CT, Taylor, FB, Jr., Snow, TR. Inflammation and coagulation: Linked processes potentially regulated through a common pathway mediated by protein C. Thromb Haemost. 1991; 66:160–165.
34. Taylor, FB, Jr., Kinasewitz, GT, Lupu, F. Pathophysiology, staging and therapy of severe sepsis in baboon models. J Cell Mol Med. 2012; 16:672–862.
35. Fourrier, F, Chopin, C, Goudemand, J, et al. Septic shock, multiple organ failure, and disseminated intravascular coagulation: Compared patterns of antithrombin III, protein C, and protein S deficiencies. Chest. 1992; 101:816–823.
36. Lam, C, Tyml, K, Martin, C, Sibbald, W. Microvascular perfusion is impaired in a rat model of normotensive sepsis. J Clin Invest. 1994; 94:2077–2083.
37. Sakr, Y, Dubois, MJ, De Backer, D, et al. Persistent microcirculatory alterations are associated with organ failure and death in patients with septic shock. Crit Care Med. 2004; 32:1825–1831.
38. Dellinger, RP, Carlet, JM, Masur, H, et al. Surviving Sepsis Campaign guidelines for management of severe sepsis and septic shock. Crit Care Med. 2004; 32:858–873.
39. Dellinger, RP, Levy, MM, Carlet, JM, et al. Surviving Sepsis Campaign: International guidelines for management of severe sepsis and septic shock: 2008. Crit Care Med. 2008; 36:296–327.
39a. Dellinger, RP, et al, Surviving sepsis campaign: International guidelines for management of severe sepsis and septic shock, 2012. Crit Care Med 2013 Feb; 41:580. http://dx.doi.org/10.1097/CCM.0b013e31827e83af
40. Levy, MM, Pronovost, PJ, Dellinger, RP, et al. Sepsis change bundles: Converting guidelines into meaningful change in behavior and clinical outcome. Crit Care Med. 2004; 32:S595–S597.
41. Gao, F, Melody, T, Daniels, DF, et al. The impact of compliance with 6-hour and 24-hour sepsis bundles on hospital mortality in patients with severe sepsis: A prospective observational study. Crit Care. 2005; 9:R764–R770.
42. Micek, ST, Roubinian, N, Heuring, T, et al. Before-after study of a standardized hospital order set for the management of septic shock. Crit Care Med. 2006; 34:2707–2713.
43. Ferrer, R, Artigas, A, Suarez, D, et al. Effectiveness of treatments for severe sepsis: A prospective, multicenter, observational study. Am J Respir Crit Care Med. 2009; 180:861–866.
44. Levy, MM, Dellinger, RP, Townsend, SR, et al. The Surviving Sepsis Campaign: Results of an international guideline-based performance improvement program targeting severe sepsis. Crit Care Med. 2010; 38:367–374.
45. Pittet, D, Thievent, B, Wenzel, RP, et al. Bedside prediction of mortality from bacteremic sepsis: A dynamic analysis of ICU patients. Am J Respir Crit Care Med. 1996; 153:684–693.
46. Iregui, M, Ward, S, Sherman, G, et al. Clinical importance of delays in the initiation of appropriate antibiotic treatment for ventilator-associated pneumonia. Chest. 2002; 122:262–268.
47. Kumar, A, Roberts, D, Wood, KE, et al. Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Crit Care Med. 2006; 34:1589–1596.
48. Castellanos-Ortega, A, Suberviola, B, Garcia-Astudillo, LA, et al. Impact of the Surviving Sepsis Campaign protocols on hospital length of stay and mortality in septic shock patients: Results of a three-year follow-up quasi-experimental study. Crit Care Med. 2010; 38:1036–1043.
49. Puskarich, MA, Trzeciak, S, Shapiro, NI, et al. Association between timing of antibiotic administration and mortality from septic shock in patients treated with a quantitative resuscitation protocol. Crit Care Med. 2011; 39:2066–2071.
50. Puri, N, Puri, V, Dellinger, RP. History of technology in the intensive care unit. Crit Care Clin. 2009; 25:185–200.
51. Blot, F, Schmidt, E, Nitenberg, G, et al. Earlier positivity of central-venous- versus peripheral-blood cultures is highly predictive of catheter-related sepsis. J Clin Microbiol. 1998; 36:105–109.
52. Leibovici, L, Greenshtain, S, Cohen, O, et al. Bacteremia in febrile patients: A clinical model for diagnosis. Arch Intern Med. 1991; 151:1801–1806.
53. Weinstein, MP, Reller, LB, Murphy, JR, Lichtenstein, KA. The clinical significance of positive blood cultures: A comprehensive analysis of 500 episodes of bacteremia and fungemia in adults. I. Laboratory and epidemiologic observations. Rev Infect Dis. 1983; 5:35–53.
54. Ibrahim, EH, Sherman, G, Ward, S, et al. The influence of inadequate antimicrobial treatment of bloodstream infections on patient outcomes in the ICU setting. Chest. 2000; 118:146–155.
55. Leibovici, L, Shraga, I, Drucker, M, et al. The benefit of appropriate empirical antibiotic treatment in patients with bloodstream infection. J Intern Med. 1998; 244:379–386.
56. Kollef, MH, Sherman, G, Ward, S, Fraser, VJ. Inadequate antimicrobial treatment of infections: A risk factor for hospital mortality among critically ill patients. Chest. 1999; 115:462–474.
57. Pappas, PG, Kauffman, CA, Andes, D, et al. Clinical practice guidelines for the management of candidiasis: 2009 update by the Infectious Diseases Society of America. Clin Infect Dis. 2009; 48:503–535.
58. Jimenez, MF, Marshall, JC. Source control in the management of sepsis. Intensive Care Med. 2001; 27(Suppl 1):S49–S62.
59. Parrillo, JE, Parker, MM, Natanson, C, et al. Septic shock in humans. Advances in the understanding of pathogenesis, cardiovascular dysfunction, and therapy. Ann Intern Med. 1990; 113:227–242.
60. Rivers, E, Nguyen, B, Havstad, S, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001; 345:1368–1377.
61. Varpula, M, Tallgren, M, Saukkonen, K, et al. Hemodynamic variables related to outcome in septic shock. Intensive Care Med. 2005; 31:1066–1071.
62. Kortgen, A, Niederprum, P, Bauer, M. Implementation of an evidence-based “standard operating procedure” and outcome in septic shock. Crit Care Med. 2006; 34:943–949.
63. Nguyen, HB, Corbett, SW, Steele, R, et al. Implementation of a bundle of quality indicators for the early management of severe sepsis and septic shock is associated with decreased mortality. Crit Care Med. 2007; 35:1105–1112.
64. Sebat, F, Johnson, D, Musthafa, AA, et al. A multidisciplinary community hospital program for early and rapid resuscitation of shock in nontrauma patients. Chest. 2005; 127:1729–1743.
65. Shapiro, NI, Howell, MD, Talmor, D, et al. Implementation and outcomes of the Multiple Urgent Sepsis Therapies (MUST) protocol. Crit Care Med. 2006; 34:1025–1032.
66. Perel, P, Roberts, I. Colloids versus crystalloids for fluid resuscitation in critically ill patients. Cochrane Database Syst Rev. 2011.
67. Schortgen, F, Lacherade, JC, Bruneel, F, et al. Effects of hydroxyethylstarch and gelatin on renal function in severe sepsis: A multicentre randomised study. Lancet. 2001; 357:911–916.
68. McIntyre, LA, Fergusson, D, Cook, DJ, et al. Fluid resuscitation in the management of early septic shock (FINESS): A randomized controlled feasibility trial. Can J Anaesth. 2008; 55:819–826.
69. Brunkhorst, FM, Engel, C, Bloos, F, et al. Intensive insulin therapy and pentastarch resuscitation in severe sepsis. N Engl J Med. 2008; 358:125–139.
70. Finfer, S, Bellomo, R, Boyce, N, et al. A comparison of albumin and saline for fluid resuscitation in the intensive care unit. N Engl J Med. 2004; 350:2247–2256.
71. Dellinger, RP. Cardiovascular management of septic shock. Crit Care Med. 2003; 31:946–955.
72. Hollenberg, SM, Ahrens, TS, Annane, D, et al. Practice parameters for hemodynamic support of sepsis in adult patients: 2004 update. Crit Care Med. 32, 2004. [1928–1248].
73. Bernard, GR, Wheeler, AP, Russell, JA, et al. The effects of ibuprofen on the physiology and survival of patients with sepsis. The Ibuprofen in Sepsis Study Group. N Engl J Med. 1997; 336:912–918.
74. Bone, RC, Fisher, CJ, Jr., Clemmer, TP, et al. A controlled clinical trial of high-dose methylprednisolone in the treatment of severe sepsis and septic shock. N Engl J Med. 1987; 317:653–658.
75. Sprung, CL, Caralis, PV, Marcial, EH, et al. The effects of high-dose corticosteroids in patients with septic shock: A prospective, controlled study. N Engl J Med. 1984; 311:1137–1143.
76. Bigatello, LM, Greene, RE, Sprung, CL, et al. HA-1A in septic patients with ARDS: Results from the pivotal trial. Intensive Care Med. 1994; 20:328–334.
77. Derkx, B, Wittes, J, McCloskey, R. Randomized, placebo-controlled trial of HA-1A, a human monoclonal antibody to endotoxin, in children with meningococcal septic shock. European Pediatric Meningococcal Septic Shock Trial Study Group. Clin Infect Dis. 1999; 28:770–777.
78. McCloskey, RV, Straube, RC, Sanders, C, et al. Treatment of septic shock with human monoclonal antibody HA-1A: A randomized, double-blind, placebo-controlled trial. CHESS Trial Study Group. Ann Intern Med. 1994; 121:1–5.
79. Smith, CR, Straube, RC, Ziegler, EJ. HA-1A. A human monoclonal antibody for the treatment of gram-negative sepsis. Infect Dis Clin North Am. 1992; 6:253–266.
80. Wortel, CH, von der Mohlen, MA, van Deventer, SJ, et al. Effectiveness of a human monoclonal anti-endotoxin antibody (HA-1A) in gram-negative sepsis: Relationship to endotoxin and cytokine levels. J Infect Dis. 1992; 166:1367–1374.
81. Ziegler, EJ, Fisher, CJ, Jr., Sprung, CL, et al. Treatment of gram-negative bacteremia and septic shock with HA-1A human monoclonal antibody against endotoxin: A randomized, double-blind, placebo-controlled trial. The HA-1A Sepsis Study Group. N Engl J Med. 1991; 324:429–436.
82. Abraham, E, Anzueto, A, Gutierrez, G, et al. Double-blind randomised controlled trial of monoclonal antibody to human tumour necrosis factor in treatment of septic shock. NORASEPT II Study Group. Lancet. 1998; 351:929–933.
83. Abraham, E, Laterre, PF, Garbino, J, et al. Lenercept (p55 tumor necrosis factor receptor fusion protein) in severe sepsis and early septic shock: A randomized, double-blind, placebo-controlled, multicenter phase III trial with 1,342 patients. Crit Care Med. 2001; 29:503–510.
84. Abraham, E, Wunderink, R, Silverman, H, et al. Efficacy and safety of monoclonal antibody to human tumor necrosis factor alpha in patients with sepsis syndrome: A randomized, controlled, double-blind, multicenter clinical trial. TNF-alpha MAb Sepsis Study Group. JAMA. 1995; 273:934–941.
85. Dhainaut, JF, Yan, SB, Margolis, BD, et al. Drotrecogin alfa (activated) (recombinant human activated protein C) reduces host coagulopathy response in patients with severe sepsis. Thromb Haemost. 2003; 90:642–653.
86. Fein, AM, Bernard, GR, Criner, GJ, et al. Treatment of severe systemic inflammatory response syndrome and sepsis with a novel bradykinin antagonist, deltibant (CP-0127): Results of a randomized, double-blind, placebo-controlled trial. CP-0127 SIRS and Sepsis Study Group. JAMA. 1997; 277:482–487.
87. Fisher, CJ, Jr., Agosti, JM, Opal, SM, et al. Treatment of septic shock with the tumor necrosis factor receptor:Fc fusion protein. The Soluble TNF Receptor Sepsis Study Group. N Engl J Med. 1996; 334:1697–1702.
88. Fisher, CJ, Jr., Dhainaut, JF, Opal, SM, et al. Recombinant human interleukin 1 receptor antagonist in the treatment of patients with sepsis syndrome. Results from a randomized, double-blind, placebo-controlled trial. Phase III rhIL-1ra Sepsis Syndrome Study Group. JAMA. 1994; 271:1836–1843.
89. Opal, S, Laterre, PF, Abraham, E, et al. Recombinant human platelet-activating factor acetylhydrolase for treatment of severe sepsis: Results of a phase III, multicenter, randomized, double-blind, placebo-controlled, clinical trial. Crit Care Med. 2004; 32:332–341.
90. Inthorn, D, Hoffmann, JN, Hartl, WH, et al. Antithrombin III supplementation in severe sepsis: Beneficial effects on organ dysfunction. Shock. 1997; 8:328–334.
91. Inthorn, D, Hoffmann, JN, Hartl, WH, et al. Effect of antithrombin III supplementation on inflammatory response in patients with severe sepsis. Shock. 1998; 10:90–96.
92. Warren, BL, Eid, A, Singer, P, et al. Caring for the critically ill patient. High-dose antithrombin III in severe sepsis: A randomized controlled trial. JAMA. 2001; 286:1869–1878.
93. Abraham, E, Reinhart, K, Opal, S, et al. Efficacy and safety of tifacogin (recombinant tissue factor pathway inhibitor) in severe sepsis: A randomized controlled trial. JAMA. 2003; 290:238–247.
94. Abraham, E, Reinhart, K, Svoboda, P, et al. Assessment of the safety of recombinant tissue factor pathway inhibitor in patients with severe sepsis: A multicenter, randomized, placebo-controlled, single-blind, dose escalation study. Crit Care Med. 2001; 29:2081–2089.
95. Bernard, GR, Vincent, JL, Laterre, PF, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001; 344:699–709.
96. Angus, DC, Laterre, PF, Elterbrand, J. The effects of drotrecogin alfa (activated) on long term survival after severe sepsis. Chest. 2002; 122:51S.
97. Bernard, GR, Margolis, BD, Shanies, HM, et al. Extended evaluation of recombinant human activated protein C United States Trial (ENHANCE US): A single-arm, phase 3B, multicenter study of drotrecogin alfa (activated) in severe sepsis. Chest. 2004; 125:2206–2216.
98. Abraham, E, Laterre, PF, Garg, R, et al. Drotrecogin alfa (activated) for adults with severe sepsis and a low risk of death. N Engl J Med. 2005; 353:1332–1341.
99. Ranieri, VM, Thompson, BT, Barie, PS, et al. Drotrecogin alfa (activated) in adults with septic shock. N Engl J Med. 2012; 366:2055–2064.
100. Angus, DC. Drotrecogin alfa (activated) … a sad final fizzle to a roller-coaster party. Crit Care. 2012; 16:107.
101. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N Engl J Med. 2000; 342:1301–1308.
102. Putensen, C, Theuerkauf, N, Zinserling, J, et al. Meta-analysis: Ventilation strategies and outcomes of the acute respiratory distress syndrome and acute lung injury. Ann Intern Med. 2009; 151:566–576.
103. Burns, KE, Adhikari, NK, Slutsky, AS, et al. Pressure and volume limited ventilation for the ventilatory management of patients with acute lung injury: A systematic review and meta-analysis. PLoS ONE. 2011; 6:e14623.
104. National Heart, L, Wiedemann, HP, et al. Comparison of two fluid-management strategies in acute lung injury. Blood Institute Acute Respiratory Distress Syndrome Clinical Trials N. N Engl J Med. 2006; 354:2564–2575.
105. Trzeciak, S, Dellinger, RP. Other supportive therapies in sepsis: An evidence-based review. Crit Care Med. 2004; 32:S571–S577.
106. Cook, D, Guyatt, G, Marshall, J, et al. A comparison of sucralfate and ranitidine for the prevention of upper gastrointestinal bleeding in patients requiring mechanical ventilation. Canadian Critical Care Trials Group. N Engl J Med. 1998; 338:791–797.
107. Heyland, DK. Nutritional support in the critically ill patients: A critical review of the evidence. Crit Care Clin. 1998; 14:423–440.
108. Heyland, DK, MacDonald, S, Keefe, L, Drover, JW. Total parenteral nutrition in the critically ill patient: A meta-analysis. JAMA. 1998; 280:2013–2019.
109. Finney, SJ, Zekveld, C, Elia, A, Evans, TW. Glucose control and mortality in critically ill patients. JAMA. 2003; 290:2041–2047.
110. Malmberg, K, Norhammar, A, Wedel, H, Ryden, L. Glycometabolic state at admission: Important risk marker of mortality in conventionally treated patients with diabetes mellitus and acute myocardial infarction: Long-term results from the Diabetes and Insulin-Glucose Infusion in Acute Myocardial Infarction (DIGAMI) study. Circulation. 1999; 99:2626–2632.
111. Michaud, LJ, Rivara, FP, Longstreth, WT, Jr., Grady, MS. Elevated initial blood glucose levels and poor outcome following severe brain injuries in children. J Trauma. 1991; 31:1356–1362.
112. Norhammar, AM, Ryden, L, Malmberg, K. Admission plasma glucose: Independent risk factor for long-term prognosis after myocardial infarction even in nondiabetic patients. Diabetes Care. 1999; 22:1827–1831.
113. Zindrou, D, Taylor, KM, Bagger, JP. Admission plasma glucose: An independent risk factor in nondiabetic women after coronary artery bypass grafting. Diabetes Care. 2001; 24:1634–1639.
114. Nylen, ES, Muller, B. Endocrine changes in critical illness. J Intensive Care Med. 2004; 19:67–82.
115. van den Berghe, G, Wouters, P, Weekers, F, et al. Intensive insulin therapy in the critically ill patients. N Engl J Med. 2001; 345:1359–1367.
116. Van den Berghe, G, Wilmer, A, Hermans, G, et al. Intensive insulin therapy in the medical ICU. N Engl J Med. 2006; 354:449–461.
117. Arabi, YM, Dabbagh, OC, Tamim, HM, et al. Intensive versus conventional insulin therapy: A randomized controlled trial in medical and surgical critically ill patients. Crit Care Med. 2008; 36:3190–3197.
118. Investigators, CS, Annane, D, Cariou, A, et al. Corticosteroid treatment and intensive insulin therapy for septic shock in adults: A randomized controlled trial. JAMA. 2010; 303:341–348.
119. Preiser, JC, Devos, P, Ruiz-Santana, S, et al. A prospective randomised multi-centre controlled trial on tight glucose control by intensive insulin therapy in adult intensive care units: The Glucontrol study. Intensive Care Med. 2009; 35:1738–1748.
120. Investigators, N-SS, Finfer, S, Chittock, DR, et al. Intensive versus conventional glucose control in critically ill patients. N Engl J Med. 2009; 360:1283–1297.
121. Vriesendorp, TM, DeVries, JH, van Santen, S, et al. Evaluation of short-term consequences of hypoglycemia in an intensive care unit. Crit Care Med. 2006; 34:2714–2718.
122. Hebert, PC, Drummond, AJ, Singer, J, et al. A simple multiple system organ failure scoring system predicts mortality of patients who have sepsis syndrome. Chest. 1993; 104:230–235.
123. Vincent, JL, de Mendonca, A, Cantraine, F, et al. Use of the SOFA score to assess the incidence of organ dysfunction/failure in intensive care units: Results of a multicenter, prospective study. Working group on “sepsis-related problems” of the European Society of Intensive Care Medicine. Crit Care Med. 1998; 26:1793–1800.
124. Russell, JA, Singer, J, Bernard, GR, et al. Changing pattern of organ dysfunction in early human sepsis is related to mortality. Crit Care Med. 2000; 28:3405–3411.
125. Zanotti Cavazzoni, SL, Dellinger, RP. Hemodynamic optimization of sepsis-induced tissue hypoperfusion. Crit Care. 2006; 10(Suppl 3):S2.
126. Ince, C. The microcirculation is the motor of sepsis. Crit Care. 2005; 9(Suppl 4):S13–S19.
127. Trzeciak, S, Dellinger, RP, Parrillo, JE, et al. Early microcirculatory perfusion derangements in patients with severe sepsis and septic shock: Relationship to hemodynamics, oxygen transport, and survival. Ann Emerg Med. 2007; 49:88–98.
128. Fink, MP. Bench-to-bedside review: Cytopathic hypoxia. Crit Care. 2002; 6:491–499.
129. Fink, MP. Cytopathic hypoxia: Is oxygen use impaired in sepsis as a result of an acquired intrinsic derangement in cellular respiration? Crit Care Clin. 2002; 18:165–175.
130. Hotchkiss, RS, Swanson, PE, Freeman, BD, et al. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med. 1999; 27:1230–1251.
131. Hotchkiss, RS, Tinsley, KW, Swanson, PE, et al. Sepsis-induced apoptosis causes progressive profound depletion of B and CD4+ T lymphocytes in humans. J Immunol. 2001; 166:6952–6963.
132. Singer, M, De Santis, V, Vitale, D, Jeffcoate, W. Multiorgan failure is an adaptive, endocrine-mediated, metabolic response to overwhelming systemic inflammation. Lancet. 2004; 364:545–548.
133. Levy, MM, Macias, WL, Vincent, JL, et al. Early changes in organ function predict eventual survival in severe sepsis. Crit Care Med. 2005; 33:2194–2201.