[level-membership-for-anesthesiology-category]
Chapter 61 Severe sepsis
The incidence of sepsis has increased steadily over the last three decades, at least in part because of ageing western populations.1 Nearly 15% of patients in intensive care have severe sepsis and two-thirds of them have septic shock.2,3 Despite our increased understanding, improved support and more powerful antibiotic therapy, severe sepsis is consistently reported as a leading cause of death in non-cardiac intensive care units (ICUs). Mortality remains high3,4 and severe sepsis claims far more lives than the common diseases of western countries such as acute myocardial infarction, stroke and trauma.1,4
DEFINITION
In 1991 the American College of Chest Physicians and Society of Critical Care Medicine convened a consensus conference with the goal of providing a uniform definition for sepsis and its sequelae, using common clinical findings such as alteration in body temperature, tachycardia, tachypnoea and abnormalities in the white blood cell count that were identifiable at the bedside or early in the clinical process.5 They proposed to differentiate systemic inflammation from the inflammatory response itself, and advocated a model in which sepsis is defined as infection in association with a state of systemically activated inflammation (Table 61.1). To differentiate illness severity further, severe sepsis was defined as sepsis with organ dysfunction and septic shock as sepsis with haemodynamic collapse.5
Table 61.1 Sepsis definitions from the 1991 American College of Chest Physician and Society of Critical Care Medicine Consensus Conference5
| Systemic inflammatory response syndrome (SIRS) | At least two among the following: |
These consensus definitions delineate gradations of mortality risk, but they do not adequately stratify patients into homogeneous groups with respect to the underlying pathophysiology or potential to respond to therapy. They define concepts, but do not identify patients with a single disease.6,7 For these reasons, a new consensus conference held in 2001 presented a new framework of consensus definitions and a staging system. This system, called PIRO (Table 61.2), includes domains for predisposition (premorbid illness), insult/infection (site, bacteriology of infection, severity of other insults such as trauma), response (hypotension, severity of illness measures such as such as Acute Physiology, Age and Chronic Health Evaluation – APACHE), and organ dysfunction (aggregate organ dysfunction scores such as multiple-organ dysfunction syndrome and Sequential Organ Failure Assessment – SOFA).6,7
Table 61.2 The PIRO sepsis staging system6
| Predisposition | Previous illness with reduced probability of short-term survival |
| Age | |
| Genetic polymorphisms in components of inflammatory response | |
| Insult/infection | Culture and sensitivity of infecting pathogens |
| Disease amenable to source control | |
| Gene transcript profiles | |
| Response | Systemic inflammatory response syndrome (SIRS) |
| Sepsis | |
| Severe sepsis | |
| Septic shock | |
| Markers of activated inflammation (C-reactive protein, procalcitonin, interleukin-6) | |
| Markers of impaired host responsiveness (human leukocyte antigen (HLA)-DR) | |
| Detection of therapy target (protein C, tumour necrosis factor, platelet-activating factor) | |
| Organ dysfunction | Number of failing organs |
| Composite scores |
PATHOGENESIS
Severe sepsis and septic shock evolve from a systemic inflammatory and coagulation response to a documented infection. Gram-negative bacilli (mainly Escherichia coli, Klebsiella spp. and Pseudomonas aeruginosa) and Gram-positive cocci (mainly staphylococci and streptococci) are the pathogens most commonly associated with the development of sepsis, although fungi, viruses and parasites cause the syndrome. Fungi, mostly Candida, account for only 5% of all cases of severe sepsis.7,8
The pathophysiology of bacterial sepsis is initiated by the outer-membrane components of both Gram-negative organisms (lipopolysaccharide, lipid A, flagellin and peptidoglycan) and Gram-positive organisms (teichoic acid, lipoteichoic acid, peptidoglycan). These outer-membrane components and other cell wall components are able to bind to the CD14 receptor, a protein anchored in the outer leaflet of the surface of monocytes. Recently, some coreceptors called Toll-like receptors were identified and it was demonstrated that bacterial components also interact with them. Ten members of the Toll-like receptor family have been identified, each of which shows a degree of specificity for pathogenic microorganisms and cellular products (e.g. TLR2 for peptidoglycan, lipoteichoic acid or TLR4 for lipopolysaccharide).7,8
Binding of Toll-like receptors activates intracellular signalling pathways that trigger transcription factors, such as nuclear factor-κB, which in turn control the expression of immune response genes, resulting in the release of cytokines (Figure 61.1). They secrete either cytokines with inflammatory properties (including tumour necrosis factor-α (TNF-α), interleukin-1 (IL-1), IL-2, IL-6) or cytokines with anti-inflammatory properties (IL-4 and IL-10).7,8
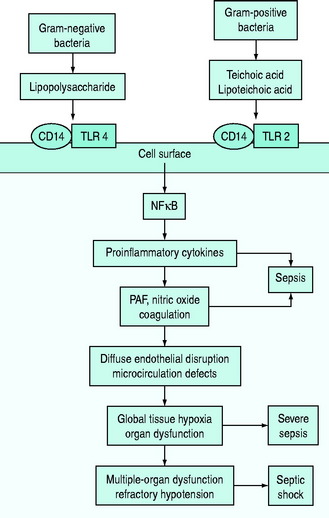
A network of inflammatory mediators activates leukocytes, promotes leukocyte–vascular endothelium adhesion and induces endothelial damage.6 This endothelial damage, in turn, leads to tissue factor expression and activation of the tissue factor-dependent clotting cascade with subsequent formation of thrombin, so that microaggregates of fibrin, platelets, neutrophils and red blood cells impair capillary blood flow, thereby decreasing oxygen and nutrient delivery.6–8
In particular, early inflammatory cytokines increase the expression of the enzyme-inducible nitric oxide synthase (iNOS) in endothelial cells; increased synthesis of the potent vasodilator nitric oxide leads to a decrease in systemic vascular resistance characteristic of shock. Inflammatory cytokines such as TNF also contribute to disruption of the tight junctions between endothelial cells, resulting in increased permeability to plasma proteins and fluid with generalised tissue oedema.7,9 IL-6 alters hepatocyte protein synthesis, inducing the synthesis of acute-phase reactants and promoting anaemia. The acute-phase response also results in downregulation of the production of albumin and anticoagulant proteins such as protein C.7,9 Microcirculatory dysfunction plays a key role in the development of organ dysfunction in septic patients.10 As a result of the vicious cycle of inflammation and coagulation, cardiovascular insufficiency (due to the myocardial-depressant effect of TNF, vasodilation and capillary leak) and multiple-organ failure occur, often leading to death (see Figure 61.1).
DIAGNOSIS
The initial presentation of severe sepsis and septic shock is often non-specific and its severity is cryptic. Diagnosis requires the presence of a presumed or known site of infection, evidence of a systemic inflammatory response syndrome and acute sepsis-associated organ dysfunction (Table 61.3).
Table 61.3 Diagnosis of sepsis-associated organ dysfunctions
| Cardiovascular | Systolic arterial blood pressure < 90 mmHg |
| Decrease in systolic blood pressure > 40 mmHg | |
| Mean arterial blood pressure < 70 mmHg | |
| Decreased capillary refill or mottling | |
| Respiratory | PaO2/Fio2 < 300 |
| Renal | Creatinine increase > 60 μµmol/l from baseline |
| Creatinine increase > 60 μµmol/l within last 24 hours | |
| Urine output < 0.5 ml/kg per hour for 2 hours despite fluid resuscitation | |
| Coagulation | Activated partial thromboplastin time > 60 seconds |
| International normalised ratio > 1.5 | |
| Platelets < 100 000/μµl | |
| Liver | Bilirubin > 70 mmol/l |
| Acid–base | Lactate > 2.1 mmol/l |
Laboratory and invasive haemodynamic measurements include:
CARDIOVASCULAR FAILURE
Cardiovascular failure is caused by an inadequate supply or inappropriate use of metabolic substrate. There is increasing evidence that sepsis is accompanied by a hypermetabolic state, with enhanced glycolysis and hyperlactataemia that do not necessarily indicate tissue hypoxia.11 Hypotension is frequently the result of low systemic vascular resistance. Shock is typically defined as a systolic pressure lower than 90 mmHg that is unresponsive to fluids or that requires vasoactive drugs.
LIVER DYSFUNCTION
The liver is a mechanical and immunologic filter for portal blood and may be a major source of cytokines. An increase in aminotransferase and bilirubin levels is common,12 but hepatic failure is rare.
COAGULATION ABNORMALITIES
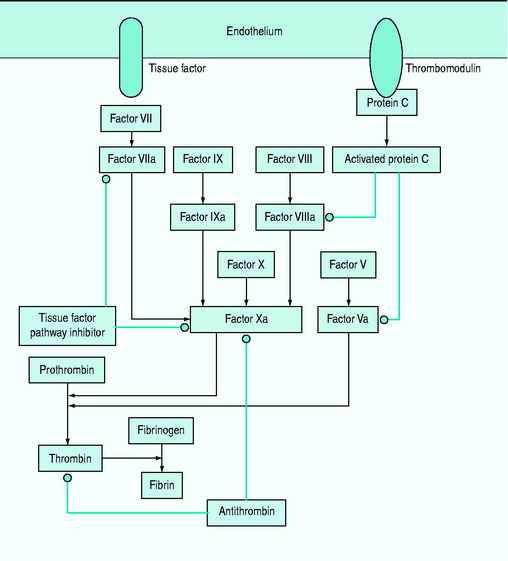
Subclinical coagulopathy, with a mild elevation of the prothrombin or partial thromboplastin time, or a moderate reduction in platelet count or an increase in plasma fibrinogen degradation and D-dimer levels may occur. Coagulopathy is caused by deficiencies of the coagulation system proteins (antithrombin III, protein C, tissue factor pathway inhibitor and the kinin system) (Figure 61.2). Simultaneous activation of coagulation by inflammatory cytokines and reduced production of anticoagulant proteins contribute to disseminated intravascular coagulation.12
SEPSIS-ASSOCIATED ENCEPHALOPATHY
A diffuse cerebral dysfunction is often present in sepsis and may ensue even before signs of other organ failure. It is better defined as ‘sepsis-associated encephalopathy’ (SAE), in order to stress the absence of direct infection of the central nervous system. The main sign of SAE is an altered mental status. Electroencephalography is the most sensitive diagnostic test, and allows the grading of the severity of cerebral dysfunction that is related to outcome. SAE is potentially reversible, but always worsens the prognosis. The pathophysiology of SAE is still not completely understood, and it is probably multifactorial. Indeed, brain dysfunction in sepsis may be related to microorganism toxin activity, to the effects of inflammatory mediators, to metabolic alterations and to abnormalities in cerebral circulation.13,14
TREATMENT
Timely and proper care of the patient with severe sepsis is essential and includes: (1) treatment of the infection with source control and antimicrobial agents; (2) supportive measures such as fluid therapy and vasoactive agents; and (3) adjuvant therapies (Table 61.4).
| Monitoring of heart rate, blood pressure, pulse oximetry and urine output |
| Investigation of the infection source |
| Laboratory tests for organ dysfunction and global tissue hypoxia (lactate) |
| Empiric broad-spectrum antibiotics |
| Central venous access and early goal-directed therapy in patients with lactate > 4 mmol/l or systolic arterial pressure < 90 mmHg even after 20–40 ml/kg fluid challenge |
| Source control (abscess drainage, tissue debridement, prothesis removal) |
| Fluid therapy, vasoactive and inotrope drugs |
| Consider adjuvant therapies |
| Additional general treatment components |
During the past few years, several randomised controlled trials in patients with severe sepsis and septic shock have demonstrated significant reductions in mortality rates with new applied therapies. Concurrently, antimicrobial resistance to several agents has emerged and changed considerations about empirical therapy. Advances in imaging and non-invasive interventional techniques have also led to new diagnostic and therapeutic strategies for early source control. Some of the new approaches to management of severe sepsis appear to be time-dependent, suggesting a ‘golden hour’ and a ‘silver day’ perspective on the management of this syndrome.15
SURVIVING SEPSIS CAMPAIGN
In order to improve the standard of care offered to patients with sepsis, the Society of Critical Care Medicine, the European Intensive Care Society and the International Sepsis Forum launched the Surviving Sepsis Campaign (SSC) with a founding statement that became known as the ‘Barcelona declaration’.16 All aspects of management of the patient with severe sepsis were covered and recommendations or bundles, depending on the available level of evidence, were developed for each category16 (Table 61.5).
Table 61.5 Issues considered by the Surviving Sepsis Campaign Guidelines16
| Diagnosis |
| Intial resuscitation |
| Antibiotic therapy |
| Fluid therapy |
| Vasopressors |
| Inotropic therapy |
| Steroids |
| Activated protein C |
| Blood product administration |
| Mechanical ventilation |
| Sedation/analgesia |
| Glucose control |
| Renal replacement |
| Bicarbonate therapy |
| Deep-vein thrombosis prophylaxis |
| Stress ulcer prophylaxis |
| Consideration regarding limitation of life support |
However, at the moment, there is no evidence that their beneficial effects are synergistic.17,18
SOURCE CONTROL OF INFECTION
Adequate source control of infection is as important as appropriate antimicrobial therapy. All patients presenting with the clinical syndrome of sepsis should be evaluated for the possible presence of a focus of infection amenable to treatment by source control measures. A source of infection should be rapidly sought by integration of the clinical history, physical examination and results of focused diagnostic tests and imaging examinations.7 Advances in diagnostic or interventional radiology and surgical procedure have revolutionised the management of many intra-abdominal infections: Specifically, the diagnostic impact of abdominal computed tomography (CT) scan has been significant in patients with multiple potential causes of their acute abdominal pain or sepsis of unknown origin.15
Source control includes removal of infected foreign bodies (such as intravascular catheters, vascular draft); incision and drainage (open or percutaneous) of abscesses or fluid collections; the debridement of infected necrotic tissue and the definitive management of anatomical derangements that permit ongoing microbial contamination. For patients with necrotising fasciitis mortality and extent of tissue loss are directly related to the rapidity of surgical intervention.7
ANTIMICROBIAL THERAPY
Observational studies suggest a significant reduction in mortality when antibiotics are administered within 4 and 8 hours of hospital admission.19 Current SSC recommendations are to administer antibiotics within 1 hour of sepsis diagnosis.16 Specific antibiotic strategies may be found in Chapter 64.
EARLY HAEMODYNAMIC RESUSCITATION (FIRST 6 HOURS)
Haemodynamic resuscitation to normal physiological parameters has been trialled as early goal-directed therapy (EGDT), which revealed a statistically significant mortality reduction of 16.5%.20 EGDT aims to restore the balance between oxygen supply and demand within the first 6 hours. The strategy involves obtaining adequate oxygen delivery by optimisation of intravascular volume (preload) with the use of: (1) central venous pressure (CVP) monitoring; (2) blood pressure (afterload) with mean arterial pressure monitoring; (3) contractility monitoring to avoid tachycardia; and (d) restoration of the balance between systemic oxygen delivery and oxygen demand to resolve global tissue hypoxia (guided by ScvO2).15,21,22 A low ScvO2 (< 70%), coupled with an elevated lactate level, suggests a mismatch between systemic oxygen delivery and oxygen consumption of the tissues. When a low ScvO2 is identified, therapies to augment the components of oxygen delivery are recommended to restore the balance between systemic oxygen delivery and consumption (oxygen-carrying capacity, arterial oxygen saturation and cardiac output). ScvO2 can be measured intermittently from venous gas samples taken from the distal port of a standard central venous catheter.23 At the moment, EGDT appears to decrease ICU pulmonary artery catheter utilisation.23,24 In any case, the pulmonary artery catheter remains effective as a measurement site, although outcome benefit from its use remains to be demonstrated.23,24
Resuscitation goals proposed in SSC16 include: central venous pressure of 8–12 mmHg, mean arterial pressure ≥ 65 mmHg, urine output ≥ 0.5 ml/kg per hour, central venous (superior vena cava) oxygen saturation ≥ 70% or mixed venous ≥ 65%.
FLUID THERAPY
Initial resuscitation efforts should incorporate intravenous fluid therapy. The first goal of volume therapy of sepsis is repletion of the patient’s intravascular volume. The selection of crystalloid versus colloid solutions has been vigorously debated. At the moment no randomised controlled trial or systemic review has definitively shown a benefit in the critically ill septic patient for the use of colloid or crystalloid fluid.25,26 A larger volume of crystalloids (2–3 times) is usually required. A smaller volume of colloid will produce a similar improvement in volume status, but these fluids are more expensive and may alter blood coagulation. Intravenous fluid therapy should begin with 1000 ml crystalloid fluid challenge or 500 ml colloids over 30 min.16 Rapid and repeated 500-ml boluses of either crystalloid or colloid fluid may be necessary up to an initial resuscitation volume of 20–40 ml/kg body weight.15,21
VASOACTIVE AGENTS AND INOTROPES
Organ perfusion cannot be maintained with fluids alone. Vasoactive agents (Table 61.6) should be administered when hypotension is persistent or mean arterial blood pressure is less than 65 mmHg, after a crystalloid challenge of 20–40 ml/kg.15,21 However, existing evidence does not clearly support the superiority of one vasopressor over another.27 Both noradrenaline (norepinephrine) and dopamine have been advocated as first-line vasopressor agents in cases of sepsis,15,21 although some prefer norepinephrine.28 Norepinephrine and phenylephrine, agents with more alpha-agonist effects, may be preferable for patients with tachycardia or ischaemic cardiac diseases. The combination of noradrenaline plus low-dose dobutamine has been shown to prevent gastric mucosal perfusion in comparison with adrenaline (epinephrine) or dopamine.28,29
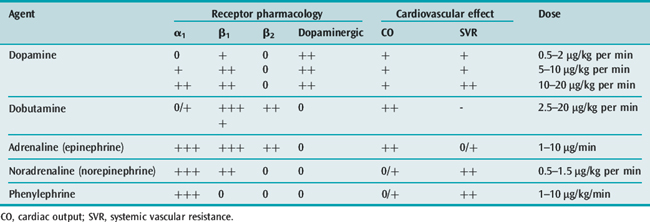
Inotropic support with dobutamine may treat the myocardial depression, improving contractility.15 When hypotension persists, a vasopressin deficiency may be considered. Exogenously administered vasopressin in physiologic replacement doses may act synergistically with other vasopressor agents.18,23 The SSC16 recommends vasopressin infusion for refractory shock and in limited doses.
ADJUVANT THERAPIES
ACTIVATED PROTEIN C (APC)
During sepsis the APC pathway serves as a major system for controlling thrombosis, inhibiting thrombin formation, limiting inflammatory responses and potentially decreasing endothelial cell apoptosis. Recombinant APC (rhAPC) could also improve microcirculatory blood flow in septic patients, as recently measured in vivo.10 There are concerns about the effects of rhAPC, although it has been shown to yield a 6.1% absolute reduction in 28-day all-cause mortality when administered to a heterogeneous group of patients with severe sepsis (Protein C Worldwide Evaluation in Severe Sepsis (PROWESS) trial).31 This was supported by the open-label extended evaluation of recombinant human protein C (ENHANCE study).32 However, a second large randomised controlled trial that compared rhAPC with placebo in patients with less severe sepsis was terminated early, when there appeared to be no mortality benefit for this patient population (Administration of Drotrecogin alfa (activated) in Early Stage Severe Sepsis (ADDRESS) trial).33
The SSC recommends rhAPC in patients at high risk of death (APACHE II ≥ 25), sepsis-induced multiple-organ failure, septic shock or sepsis-induced acute respiratory distress syndrome (ARDS) and with no absolute contraindication related to bleeding risk or relative contraindication that outweighs the potential benefit of rhAPC.16 This may not seem the most reliable method for choosing patients because the use of this drug has not been validated in this context, and there are numerous variables in the calculation of the APACHE II score, including age.18 Further studies are necessary to optimise the use of this drug; many uncertainties remain and it must also be taken into account that the drug is expensive, carries an elevated risk of bleeding and that its introduction was accompanied by more than the usual amount of scientific debate.7,34,35
STEROIDS
Steroid administration in severe sepsis and septic shock remains controversial. The SSC recommends the use of intravenous corticosteroids in patients with septic shock who, despite adequate fluid replacement, require vasopressor therapy to maintain blood pressure.16
The use of large doses of methylprednisolone has not been shown to be beneficial.36 Recently, the concept of inadequate adrenal reserve, or relative adrenal insufficiency (RAI), has led to the consideration of administering moderate doses of steroids. The administration of low doses of hydrocortisone (200–300 mg/day for 7 days) plus fludrocortisone (50 μg orally per day for 7 days) to patients with septic shock decreased their requirements for vasopressors and lowered their mortality rate.37–39 In other studies RAI was diagnosed using the 250-μg adrenocorticotrophin test to identify patients who would benefit more from steroid administration.37–39
Recently, in a multicentre, randomised, double-blind, placebo-controlled trial on about 500 patients, hydrocortisone did not improve survival or reversal of septic shock, either overall or in patients who did not have a response to corticotrophin.40
TIGHT BLOOD GLUCOSE CONTROL
When conservative glycaemic control was compared with tight control in a multicentre randomised clinical trial, tight control led to a significant reduction in mortality and improved morbidity at 1 year.41 Other advantages included shorter intensive care stay, a reduced risk of renal failure and a much lower incidence of critical-illness polyneuropathy. The study involved cardiac surgery patients. A recent trial by the same group in medical critically ill patients showed no significant difference.42 As it is not clear if slightly less stringent control has the same advantages, it is necessary to carry out meticulous monitoring of blood glucose in order to prevent the risk of hypoglycaemia, particularly in unstable septic patients.
PROTECTIVE LUNG STRATEGIES
High tidal volumes coupled with high plateau pressures should be avoided in those patients who develop acute lung injury (ALI) or ARDS as a consequence of severe sepsis.43 Mechanical ventilation with large tidal volume can stretch lung tissue to excess (volume trauma), exacerbating the inflammatory response and leading to ALI. In one study patients with ALI or ARDS were randomised to receive ventilation with tidal volumes of 12 ml/kg predicted body weight (control) or 6 ml/kg predicted body weight (treatment), with plateau pressure maintained at < 30 cm H2O. An absolute reduction of 9.9% in 28-day mortality was demonstrated in the low tidal volume group,44 with fewer days on ventilatory support and lower levels of circulating IL-6.
EXTRACORPOREAL BLOOD PURIFICATION TECHNIQUES
These techniques may hold promise for the treatment of sepsis.45 At the moment definitive studies have yet to be performed. Dialysis does improve survival among patients with sepsis who have established renal insufficiency. It is not certain that dialysis, plasmapheresis or plasma exchange will improve outcomes in patients with sepsis who do not have renal failure. Studies in animals and humans examining this issue have been inconclusive. There is also a potential for harm, since this therapy could remove beneficial compounds or molecules as well.46
ADDITIONAL GENERAL TREATMENT COMPONENTS
Additional components in the care of severe sepsis include:
OUTCOME
The overall hospital mortality rate for patients with severe sepsis is high. Since this disease is reversible, prompt recognition and resuscitation are key to optimising outcome. Studies focused on novel targets, mechanism of action and combination therapy may also improve outcome.48
1 Finfer S, Bellomo R, Lipman J, et al. Adult population incidence of severe sepsis in Australian and New Zealand intensive care units. Intens Care Med. 2004;30:527-529.
2 Martin GS, Mannino DM, Eaton S, et al. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546-1554.
3 Brun Buisson C. Epidemiology of severe sepsis. Presse Med. 2006;35:513-520.
4 Angus DC, Linde-Zwirble WT, Lidicker J, et al. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome and associated costs of care. Crit Care Med. 2001;29:1303-1310.
5 Bone RC, Balk RA, Cerra F, et al. Definition for sepsis and organ failure and guidelines for use of innovative therapies in sepsis: American College of Chest Physicians/Society of Critical Care Medicine. Chest. 1992;101:1644-1655.
6 Levy MM, Fink M, Marshall JC, et al. SCCM/ESICM/ACCP/ATS/SIS international sepsis definitions conference. Crit Care Med. 2003;34:1250-1256.
7 Marshall JC. Sepsis: current status, future prospects. Curr Opin Crit Care. 2004;10:250-264.
8 Bochud PY, Calandra T. Pathogenesis of sepsis: new concepts and implications for future treatment. Br Med J. 2003;326:262-266.
9 Opal SM, Esmon CT. Bench-to-bedside review: functional relationships between coagulation and the innate immune response and their respective roles in the pathogenesis of sepsis. Crit Care. 2003;7:23-38.
10 Hoffmann JN, Fertmann JM, Jauch KW. Microcirculatory disorders in sepsis and transplantation: therapy with natural coagulatory inhibitors antithrombin and activated protein C. Curr Opin Crit Care. 2006;12:426-430.
11 Levy B. Lactate and shock state: the metabolic view. Curr Opin Crit Care. 2006;1:315-321.
12 Marshall JC. Inflammation, coagulopathy, and the pathogenesis of the multiple organ dysfunction syndrome. Crit Care Med. 2001;29:S106.
13 Gren R, Scott LK, Minagar A, et al. Sepsis associated encephalopathy (SAE): a review. Front Biosci. 2004;9:1637-1641.
14 Consales G, De Gaudio AR. Sepsis associated encephalopathy. Minerva Anestesiol. 2005;71:39-52.
15 Nguyen BN, Rivers EP, Abraham FM, et al. Severe sepsis and septic shock: review of the literature and emergency department management guidelines. Ann Emerg Med. 2006;48:1-54.
16 Dellinger RP, Levy M, Carlet J. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2008. Intens Care Med. 2008;34:17-60.
17 Poulton B. Advances in the management of sepsis: the randomised controlled trials behind the Surving Sepsis campaign recommendations. Int J Antimicrob Agents. 2006;27:97-101.
18 Vincent JL. Is the current management of severe sepsis and septic shock really evidence based? Plos Med. 2006;3:346-350.
19 Houck PM, Bratzler DW. Administration of first hospital antibiotics for community-acquired pneumonia: does timeliness affect outcomes? Curr Opin Infect Dis. 2005;18:151-156.
20 Rivers E, Nguyen B, Havstad S, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001;345:1368-1377.
21 Rivers EP, McIntyre L, Morro DC, et al. Early and innovative interventions for severe sepsis and septic shock: taking advantage of a window of opportunity. CMAJ. 2005;173:1054-1065.
22 Hollenberg SM, Ahrens TS, Annane D, et al. Practice parameters for hemodynamic support of sepsis in adult patient: 2004 update. Crit Care Med. 2004;32:1928-1948.
23 Trzeciak S, Dellinger RP, Abate NL, et al. Translating research to clinical practice: a 1-year experience with implementing early goal-directed therapy for septic shock in the emergency department. Chest. 2006;129:225-232.
24 Harvey S, Harrison DA, Singer M, et al. Assessment of the clinical effectiveness of pulmonary artery catheters in management of patients in intensive care. Lancet. 2005;366:472-477.
25 Finfer S, Bellomo R, Boyce N, et al. A comparison of albumin and saline for fluid resuscitation in the intensive care unit. N Engl J Med. 2004;350:2247-2256.
26 Boldt J. Do plasma substitutes have additional properties beyond correcting volume deficits? Shock. 2006;25:103-116.
27 Sessler CN, Perry JC, Varney KL. Management of severe sepsis and septic shock. Curr Opin Crit Care. 2004;10:354-363.
28 Leone M, Vallet B, Teboul J, et al. Survey of the use of catecholamines by French physicians. Intens Care Med. 2004;30:984-988.
29 Zhou SX, Qiu HB, Huang YZ, et al. Effects of norepinephrine, epinephrine, and epinephrine – dobutamine on systemic and gastric mucosal oxygenation in septic shock. Acta Pharmacol Sin. 2002;23:654-658.
30 Vincent JL, Baron JF, Reinhart K, et al. Anemia and blood transfusion in critically ill patients. JAMA. 2002;288:1499-1507.
31 Bernard GR, Vincent JL, Laterre PF, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699-709.
32 Vincent JL, Bernard GR, Beale R, et al. Drotrecogin alfa (activated) treatment in severe sepsis from the global open-label trial ENHANCE. Crit Care Med. 2005;33:2266-2277.
33 Abraham E, Laterre PF, Garg R, et al. Administration of Drotrecogin alfa (activated) in Early Stage Severe Sepsis (ADDRESS) study group: drotrecogin alfa (activated) for adults with severe sepsis and a low risk of death. N Engl J Med. 2005;353:1332-1341.
34 Warren HS, Suffredini AF, Eicchacker PQ, et al. Risks and benefits of activated protein C treatment for severe sepsis. N Engl J Med. 2002;347:1027-1030.
35 Eichacker PQ, Natanson C. Increasing evidence that the risks of rhAPC may outweigh its benefits. Intens Care Med. 2007;33:396-399.
36 Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138-150.
37 Briegel J, Forst H, Haller M, et al. Stress doses of hydrocortisone reverse hyperdynamic septic shock: a prospective, randomized, double-blind, single-center study. Crit Care Med. 1999;27:723-732.
38 Annane D, Sebille V, Charpentier C, et al. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA. 2002;288:862-871.
39 Annane D, Bellissant E, Bollaert P, et al. Corticosteroids for treating severe sepsis and septic shock. Cochrane Database Syst Rev. 2004;1:CD002243.
40 Sprung CL, Annane D, Keh D, et al. Hydrocortisone therapy for patients with septic shock. N Engl J Med. 2008;358:111-124.
41 Van den Berghe G, Wouters P, Weekers F, et al. Intensive insulin therapy in the critically ill patient. N Engl J Med. 2001;345:1359-1367.
42 Van den Berghe G, Wilmer A, Hermans G, et al. Intensive insulin therapy in the medical ICU. N Engl J Med. 2006;354:449-461.
43 Frank JA, Matthay MA. Science review: mechanisms of ventilator-induced injury. Crit Care. 2003;7:233-241.
44 The Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med. 2000;342:1301-1308.
45 Ronco C, Brendolan A, Lonnemann G, et al. A pilot study of coupled plasma filtration with adsorption in septic shock. Crit Care Med. 2002;30:1250-1255.
46 Hotchkiss RS, Karl IE. Sepsis – theory and therapies. N Engl J Med. 2003;348:1600-1602.
47 De Jonge E. Effects of selective decontamination of digestive tract on mortality and antibiotic resistance in the intensive-care unit. Curr Opin Crit Care. 2005;11:144-149.
48 Russell JA. Management of sepsis. N Engl J Med. 2006;355:1699-1713.
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
Chapter 61 Severe sepsis
The incidence of sepsis has increased steadily over the last three decades, at least in part because of ageing western populations.1 Nearly 15% of patients in intensive care have severe sepsis and two-thirds of them have septic shock.2,3 Despite our increased understanding, improved support and more powerful antibiotic therapy, severe sepsis is consistently reported as a leading cause of death in non-cardiac intensive care units (ICUs). Mortality remains high3,4 and severe sepsis claims far more lives than the common diseases of western countries such as acute myocardial infarction, stroke and trauma.1,4
DEFINITION
In 1991 the American College of Chest Physicians and Society of Critical Care Medicine convened a consensus conference with the goal of providing a uniform definition for sepsis and its sequelae, using common clinical findings such as alteration in body temperature, tachycardia, tachypnoea and abnormalities in the white blood cell count that were identifiable at the bedside or early in the clinical process.5 They proposed to differentiate systemic inflammation from the inflammatory response itself, and advocated a model in which sepsis is defined as infection in association with a state of systemically activated inflammation (Table 61.1). To differentiate illness severity further, severe sepsis was defined as sepsis with organ dysfunction and septic shock as sepsis with haemodynamic collapse.5
Table 61.1 Sepsis definitions from the 1991 American College of Chest Physician and Society of Critical Care Medicine Consensus Conference5
| Systemic inflammatory response syndrome (SIRS) | At least two among the following: |
These consensus definitions delineate gradations of mortality risk, but they do not adequately stratify patients into homogeneous groups with respect to the underlying pathophysiology or potential to respond to therapy. They define concepts, but do not identify patients with a single disease.6,7 For these reasons, a new consensus conference held in 2001 presented a new framework of consensus definitions and a staging system. This system, called PIRO (Table 61.2), includes domains for predisposition (premorbid illness), insult/infection (site, bacteriology of infection, severity of other insults such as trauma), response (hypotension, severity of illness measures such as such as Acute Physiology, Age and Chronic Health Evaluation – APACHE), and organ dysfunction (aggregate organ dysfunction scores such as multiple-organ dysfunction syndrome and Sequential Organ Failure Assessment – SOFA).6,7
Table 61.2 The PIRO sepsis staging system6
| Predisposition | Previous illness with reduced probability of short-term survival |
| Age | |
| Genetic polymorphisms in components of inflammatory response | |
| Insult/infection | Culture and sensitivity of infecting pathogens |
| Disease amenable to source control | |
| Gene transcript profiles | |
| Response | Systemic inflammatory response syndrome (SIRS) |
| Sepsis | |
| Severe sepsis | |
| Septic shock | |
| Markers of activated inflammation (C-reactive protein, procalcitonin, interleukin-6) | |
| Markers of impaired host responsiveness (human leukocyte antigen (HLA)-DR) | |
| Detection of therapy target (protein C, tumour necrosis factor, platelet-activating factor) | |
| Organ dysfunction | Number of failing organs |
| Composite scores |
PATHOGENESIS
Severe sepsis and septic shock evolve from a systemic inflammatory and coagulation response to a documented infection. Gram-negative bacilli (mainly Escherichia coli, Klebsiella spp. and Pseudomonas aeruginosa) and Gram-positive cocci (mainly staphylococci and streptococci) are the pathogens most commonly associated with the development of sepsis, although fungi, viruses and parasites cause the syndrome. Fungi, mostly Candida, account for only 5% of all cases of severe sepsis.7,8
The pathophysiology of bacterial sepsis is initiated by the outer-membrane components of both Gram-negative organisms (lipopolysaccharide, lipid A, flagellin and peptidoglycan) and Gram-positive organisms (teichoic acid, lipoteichoic acid, peptidoglycan). These outer-membrane components and other cell wall components are able to bind to the CD14 receptor, a protein anchored in the outer leaflet of the surface of monocytes. Recently, some coreceptors called Toll-like receptors were identified and it was demonstrated that bacterial components also interact with them. Ten members of the Toll-like receptor family have been identified, each of which shows a degree of specificity for pathogenic microorganisms and cellular products (e.g. TLR2 for peptidoglycan, lipoteichoic acid or TLR4 for lipopolysaccharide).7,8
Binding of Toll-like receptors activates intracellular signalling pathways that trigger transcription factors, such as nuclear factor-κB, which in turn control the expression of immune response genes, resulting in the release of cytokines (Figure 61.1). They secrete either cytokines with inflammatory properties (including tumour necrosis factor-α (TNF-α), interleukin-1 (IL-1), IL-2, IL-6) or cytokines with anti-inflammatory properties (IL-4 and IL-10).7,8
A network of inflammatory mediators activates leukocytes, promotes leukocyte–vascular endothelium adhesion and induces endothelial damage.6 This endothelial damage, in turn, leads to tissue factor expression and activation of the tissue factor-dependent clotting cascade with subsequent formation of thrombin, so that microaggregates of fibrin, platelets, neutrophils and red blood cells impair capillary blood flow, thereby decreasing oxygen and nutrient delivery.6–8
In particular, early inflammatory cytokines increase the expression of the enzyme-inducible nitric oxide synthase (iNOS) in endothelial cells; increased synthesis of the potent vasodilator nitric oxide leads to a decrease in systemic vascular resistance characteristic of shock. Inflammatory cytokines such as TNF also contribute to disruption of the tight junctions between endothelial cells, resulting in increased permeability to plasma proteins and fluid with generalised tissue oedema.7,9 IL-6 alters hepatocyte protein synthesis, inducing the synthesis of acute-phase reactants and promoting anaemia. The acute-phase response also results in downregulation of the production of albumin and anticoagulant proteins such as protein C.7,9 Microcirculatory dysfunction plays a key role in the development of organ dysfunction in septic patients.10 As a result of the vicious cycle of inflammation and coagulation, cardiovascular insufficiency (due to the myocardial-depressant effect of TNF, vasodilation and capillary leak) and multiple-organ failure occur, often leading to death (see Figure 61.1).
DIAGNOSIS
The initial presentation of severe sepsis and septic shock is often non-specific and its severity is cryptic. Diagnosis requires the presence of a presumed or known site of infection, evidence of a systemic inflammatory response syndrome and acute sepsis-associated organ dysfunction (Table 61.3).
Table 61.3 Diagnosis of sepsis-associated organ dysfunctions
| Cardiovascular | Systolic arterial blood pressure < 90 mmHg |
| Decrease in systolic blood pressure > 40 mmHg | |
| Mean arterial blood pressure < 70 mmHg | |
| Decreased capillary refill or mottling | |
| Respiratory | PaO2/Fio2 < 300 |
| Renal | Creatinine increase > 60 μµmol/l from baseline |
| Creatinine increase > 60 μµmol/l within last 24 hours | |
| Urine output < 0.5 ml/kg per hour for 2 hours despite fluid resuscitation | |
| Coagulation | Activated partial thromboplastin time > 60 seconds |
| International normalised ratio > 1.5 | |
| Platelets < 100 000/μµl | |
| Liver | Bilirubin > 70 mmol/l |
| Acid–base | Lactate > 2.1 mmol/l |
Laboratory and invasive haemodynamic measurements include:
CARDIOVASCULAR FAILURE
Cardiovascular failure is caused by an inadequate supply or inappropriate use of metabolic substrate. There is increasing evidence that sepsis is accompanied by a hypermetabolic state, with enhanced glycolysis and hyperlactataemia that do not necessarily indicate tissue hypoxia.11 Hypotension is frequently the result of low systemic vascular resistance. Shock is typically defined as a systolic pressure lower than 90 mmHg that is unresponsive to fluids or that requires vasoactive drugs.
LIVER DYSFUNCTION
The liver is a mechanical and immunologic filter for portal blood and may be a major source of cytokines. An increase in aminotransferase and bilirubin levels is common,12 but hepatic failure is rare.
COAGULATION ABNORMALITIES
Subclinical coagulopathy, with a mild elevation of the prothrombin or partial thromboplastin time, or a moderate reduction in platelet count or an increase in plasma fibrinogen degradation and D-dimer levels may occur. Coagulopathy is caused by deficiencies of the coagulation system proteins (antithrombin III, protein C, tissue factor pathway inhibitor and the kinin system) (Figure 61.2). Simultaneous activation of coagulation by inflammatory cytokines and reduced production of anticoagulant proteins contribute to disseminated intravascular coagulation.12
