[level-membership-for-neurosurgery-category]
CHAPTER 227 Selective Dorsal Rhizotomy for Spastic Cerebral Palsy
Cerebral palsy (CP) is the leading cause of developmental disability in children, with more than 500,000 individuals affected in the United States alone.1,2 CP is diagnosed in 1 in 500 live births, and this incidence continues to increase because of improvements in the care and survival of very-low-birth-weight infants.1 There have been significant advances in both medical and surgical treatment of CP spasticity in recent years. From the introduction of injectable botulinum toxin A (Botox) to the implantation of pump devices for chronic intrathecal baclofen administration, patients now have a number of pharmacologic tools to treat spasticity on a long-term basis. Permanent reductions in CP spasticity, however, can be achieved only with selective dorsal rhizotomy (SDR). Currently, a large volume of literature is available on SDR as a therapeutic modality for CP. This chapter attempts to synthesize the reported information on SDR, as well as our own experience with the procedure.
Pathogenesis of Spastic Cerebral Palsy
Spastic CP is the most common subtype of cerebral palsy. Spastic diplegia and spastic quadriplegia affect approximately 60% of patients, but spasticity may also occur with dyskinesia or ataxia, or both, in mixed subtypes of CP. Thus, in total, spasticity afflicts 80% to 90% of all patients with CP.3,4 A distinctive epidemiologic feature of spastic CP—and spastic diplegia in particular—is its correlation with premature birth and low birth weight.5–7 This relationship does not exist with other subtypes of the disease, such as athetoid or dyskinetic CP.
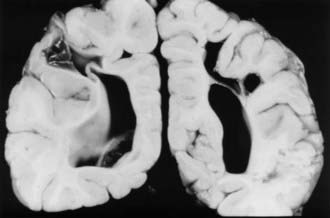
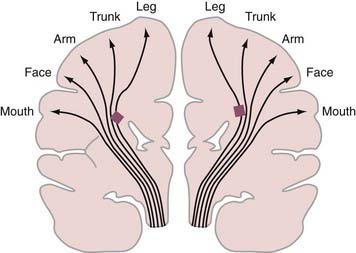
Periventricular leukomalacia (PVL) is the most common finding on brain magnetic resonance imaging (MRI) in children with spastic CP.8,9 Histologically, PVL is characterized by ischemic necrosis of the periventricular and subcortical white matter (Fig. 227-1), areas that are predisposed to hypoperfusion during hypotension.10 Reperfusion injury resulting from oxygen free radicals, cytokines, and excitatory amino acids such as glutamate probably mediate PVL.10 The high incidence of spastic diplegia in infants with PVL has led many to speculate that PVL is the neuropathologic basis for spastic CP.11 The neurological deficits in spastic CP may be explained in part by the topographic arrangements of projection fibers descending from the motor homunculus in the white matter. As seen in Figure 227-2, the leg fibers traverse the white matter closer to the ventricle than the arm fibers do. Thus, PVL is more likely to injure the leg fibers and spare the arm fibers. However, extensive leukomalacia will also injure other projection fibers and result in spastic quadriplegic CP.
Spinal cord ventral horn alpha motor neuron output is a primary determinant of muscle tone. The activity of alpha motor neurons is modulated by excitatory influences (e.g., Ia fibers from muscle spindles, descending input from the corticospinal and reticulospinal pathways) and inhibitory influences (e.g., descending GABAergic [γ-aminobutyric acid] pathways from the cerebellum and basal ganglia). Although the neural pathways involved in CP spasticity remain speculative, it is believed that spasticity results from an imbalance in these excitatory and inhibitory influences that creates hyperactive alpha and gamma neurons with exaggerated stretch reflex activity compounded by loss of inhibitory control (reviewed by Koman and colleagues12). Thus, excessive alpha motor neuron activity of the target muscles, coupled with decreased inhibition of antagonistic muscles, results in the co-contraction of agonist and antagonist muscle groups.13 Clinically, this results in a velocity-dependent, increased resistance to passive stretch.12
Deleterious Effects of Cerebral Palsy Spasticity
CP spasticity has harmful, long-term effects on growing children and thus requires optimal treatment at an early age. When untreated, inhibition of motor activity and limitation of voluntary movement result in decreased muscle and bone development, which ultimately causes deformities of the affected extremities.14 Long-lasting improvements in motor function are observed after SDR, thus supporting the argument for early intervention in the disease.15 Indeed, the spasticity-induced inhibition of motor activities does not abate if the spasticity is left untreated. Normally, longitudinal muscle growth proceeds as new myofilaments and sarcomeres are added to the ends of the muscle fibers.16 The greatest increase in sarcomere numbers occurs at an early age, with the increase taking place much more actively during development than after maturation. In parallel with the addition of sarcomeres, longitudinal muscle growth occurs rapidly during development. The increase in sarcomere number is not under neural control but is induced mainly by the amount of tension that the muscle is subjected to (i.e., repeated muscle stretch).17,18 Thus, when muscles are immobilized, there is a rapid decrease in the number of already existing sarcomeres, as well as a decrease in the number of new sarcomeres that should increase with normal development.19 Because CP spasticity in developing children limits muscle stretch in daily activities, it could cause loss of sarcomeres and inhibition of longitudinal muscle growth. In fact, the effect of spasticity on muscle growth was clearly shown in an experimental study in which longitudinal muscle growth was reduced by 45% in spastic mice as compared with control animals.20 The loss of sarcomeres accompanies a significant decrease in elasticity in the muscles21 and makes them resistant to passive stretch.22 Additionally, atrophy of type I and II muscle fibers compounds the slowed muscle growth.23 Recent work has implicated both shorter muscle (decreased sarcomere length, myocyte length, and fascicle length)24 and an increased extracellular elastic modulus25 in the clinical finding of stiffness. These pathologic events in the muscle cells cause abnormally shortened muscles, long tendons, and muscle contracture (i.e., increased resistance of the muscle to passive stretch in the absence of muscle contraction).22,26,27 The muscle contractures typically worsen as a child grows26 and produce various bone and joint deformities in the extremities.14
Treatment of Spastic Cerebral Palsy
Spasticity is commonly treated with oral medications, including the GABAB receptor agonist baclofen (Lioresal), benzodiazepines such as diazepam (Valium), the anticholinergic trihexyphenidyl, dantrolene, and tizanidine (Zanaflex).28,29 In general, oral medications are moderately effective in treating spasticity but frequently have significant adverse effects (sedation, confusion, dependence).
Intramuscular injections of alcohol and phenol for chemodenervation have been performed for many years. The introduction of intramuscular injection of Botox was a major advance in the treatment of CP spasticity, with predictable benefits and fewer adverse effects.29 Botox is injected directly into the affected muscle, and relaxation is generally seen within several days. Maximal benefit occurs approximately 4 weeks following injection, after which the effect declines and repeated injection is required in approximately 3 to 4 months.
Intrathecal Baclofen
Intrathecal administration of baclofen offers the benefit of delivering the medication directly into the central nervous system (CNS) without systemic side effects. Chronic intrathecal baclofen infusion (CIBI) with a surgically implanted pump device (SynchroMed Infusion System, Medtronic, Inc., Minneapolis, MN) has been shown to reduce spasticity and improve function,30–33 and its use has been indicated in nonambulatory or minimally ambulatory patients with spastic quadriparesis. The potential adverse effects with CIBI are significant and include infection, pump malfunction, and life-threatening withdrawal or overdose.
Stereotactic Intracranial Procedures
Stereotactic ablative procedures such as thalamotomy and dentatotomy have been used to treat CP patients with primarily unilateral dystonia and may offer some benefit in these patients.34–36 More recently, deep brain stimulation of the internal globus pallidus and thalamus have been performed in children with dystonia and tremor, respectively.37 The role of these procedures for CP spasticity has not been investigated.
Selective Dorsal Rhizotomy
SDR has been shown in several controlled trials to reduce spasticity and increase range of motion.15,38–41 In conjunction with physical therapy, SDR produces significant improvements in gross motor function and gait.15,41 SDR also decreases the rate of subsequent orthopedic surgeries.42
Indications
Indications for SDR vary among medical centers. Our current indications for SDR are summarized in Table 227-1. Primary beneficiaries of SDR are children with spastic diplegia secondary to premature birth; however, children with spastic quadriplegic CP can also benefit from SDR. In spastic hemiplegic CP, spasticity is not a predominant cause of the motor impairment, and reduction of spasticity may not greatly improve motor function (see also Table 227-2). The optimal age for children to undergo SDR is 2 to 6 years. Because of the significant deleterious effects of spasticity outlined earlier, early treatment is recommended to reduce the chance of severe orthopedic deformities of the lower extremities. SDR is not considered for children younger than 2 years because approximately 30% of children in whom CP is diagnosed at the age of 1 year later become free of symptoms.43 Adolescents and adults younger than 40 years can also enjoy good functional gains with SDR.45 The risk associated with dorsal rhizotomy in adolescents and adults is no greater than that in young children when performed through a single-level laminectomy. It is possible that reduction of spasticity will lessen the impact of aging on the physical stress caused by CP, such as abnormal stress on bones and muscles, wear and tear on joints, and increasing joint and muscle pain.
TABLE 227-1 Indications for Selective Dorsal Rhizotomy for Spastic Cerebral Palsy
TABLE 227-2 Contraindications to Selective Dorsal Rhizotomy for Spastic Cerebral Palsy
Contraindications
Table 227-2 provides a partial list of contraindications to SDR. Such contraindications include a history of severe congenital hydrocephalus or severe neonatal CNS infection such as intrauterine encephalitis (e.g., toxoplasmosis or cytomegalovirus infection) or bacterial meningitis. Likewise, patients with severe head trauma or hypoxic encephalopathy may not be suitable candidates for SDR. Although SDR is contraindicated in patients with extensive neuronal migration disorders, those with limited schizencephaly involving the sensory motor area, a rare cause of spastic diplegia, may achieve a reduction in spasticity and functional improvements after SDR.
Patients with damage to the basal ganglia deserve special consideration because rigidity and dystonia may coexist with spasticity in this setting. Of particular concern is severe basal ganglia damage in children younger than 5 years. Dystonia may develop anytime in the first 5 years of life.46 As noted earlier, patients with mixed types of CP spasticity (i.e., those with components of dystonia or athetosis) do not enjoy the same degree of functional improvement after SDR. Thus, in the setting of basal ganglia damage it is best to wait until the age of 5, when the predominance of dystonia can more reliably be ascertained. After the age of 5, however, SDR may be considered even in the presence of demonstrable basal ganglia damage if spasticity is the primary clinical symptom. Likewise, athetosis and ataxia are also relative contraindications. Similar to dystonia, if spasticity is the predominant feature in patients with concomitant athetosis, SDR may be of benefit. In these cases, SDR reduces spasticity postoperatively, but the athetosis and dystonia remain unchanged.
Preoperative Evaluation
Motor strength can be determined by testing individual muscles. In young children, past motor milestones, speed of movement, and the ability to isolate joint movements are reliable indices of motor strength. If the motor milestone is close to normal and, for instance, the child can sit alone by the age of 2 years, the child most likely has adequate motor strength and will walk in the future. Crawling or walking in a walker and making rapid transitions between positions are also signs of relatively good motor function. As a rule, the greater the motor impairment, the slower the movement. In our experience, a neurological sign that can be an excellent predictor of gait outcome in children younger than 3 to 4 years is isolated joint movements in the lower extremities (see “Gait” in the section “Outcome”).47
Surgical Considerations and Operative Technique
Between 1987 and 1991, I (T.S.P.) performed dorsal rhizotomies on 173 patients through a laminectomy at the L2-S2 levels, as described by Peacock and colleagues.48 In 1991, however, the potential risk for late spinal deformities after the procedure led to the development of a dorsal rhizotomy procedure through an L1-2 or L1 laminectomy.49 Aside from minor technical refinements,50 the surgical technique described here has been performed between 1991 and 2008 on 1719 children and adults, with a single instance of cerebrospinal fluid (CSF) leakage and no other postoperative complications. Please refer to Tables 227-3 and 227-4.
TABLE 227-3 Subtypes of Spastic Cerebral Palsy Treated by Selective Dorsal Rhizotomy
| SPASTIC CEREBRAL PALSY SUBTYPES | NO. OF PATIENTS | PERCENT |
|---|---|---|
| Diplegia | 1358 | 78 |
| Triplegia or quadriplegia | 356 | 21 |
| Hemiplegia | 5 | <1 |
| Total | 1719 | 100 |
Data from St. Louis Children’s Hospital, 1987 to 2008.
TABLE 227-4 Prerhizotomy Ambulatory Function in Patients Who Underwent Selective Dorsal Rhizotomy for Spastic Cerebral Palsy
| AMBULATION | NO. OF PATIENTS | PERCENT |
|---|---|---|
| Walking with a walker | 719 | 42 |
| Walking with crutches or a cane | 124 | 7 |
| Independent walking | 557 | 32 |
| Some locomotion | 180 | 11 |
| Crawling | 117 | 7 |
| No independent mobility | 22 | 1 |
| Total | 1719 | 100 |
Data from St. Louis Children’s Hospital, 1987 to 2008.
Operative Exposure of the Conus Medullaris
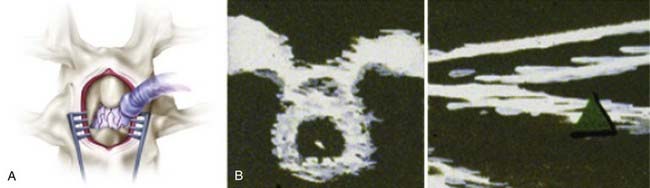
An incision is made in accordance with the localization just described. Electrocautery is used to expose the spinous process and lamina. In children, ultrasound is again used to examine the intradural structures through the exposed interlaminar space. In adults, a keyhole laminotomy may be performed to improve visualization with ultrasound. The conus is easily distinguished from the cauda equina with ultrasound (Fig. 227-3). Sagittal views show the conus as a hypodense triangle tapering caudally, whereas the cauda equina appears hyperdense. On axial views, the conus appears as a hypodense circular structure centered within the dural sac. A narrow gap between the dorsal and ventral spinal roots, lateral to the conus, can also be appreciated on the axial view. In general, axial views localize the conus more reliably than do sagittal views. A single-level laminectomy is performed to visualize 5 to 10 mm of the caudal conus and provide adequate exposure to safely separate the dorsal roots from the ventral roots later in the operation. The laminectomy may be extended to include a portion of the lamina above or below to achieve this exposure. Finally, a Midas Rex craniotome with a B5 attachment (Midas Rex Pneumatic Tools, Inc., Fort Worth, TX) is used to extend the laminectomy to the medial margin of the facet. The location of the conus is confirmed with ultrasound one final time before opening the dura. A midline durotomy is then made sharply, and the dural edges are tacked back with sutures.
Separation of the Dorsal Roots
Saline irrigation is not used after the dura is opened because it alters the EMG responses. An operating microscope is then brought into the field and used during EMG testing and sectioning of the dorsal root fascicles. The operating table is rotated slightly away from the surgeon as the contralateral spinal roots are dissected. The arachnoid is removed by sharp dissection, and the filum terminale is identified. Next, the L1 root is identified at the neural foramen, and the dorsal root is separated from the ventral root (Fig. 227-4). The L2 ventral and dorsal roots are traced back to the conus until the cleft between the ventral and dorsal roots is identified. Then the L2 and adjacent dorsal roots are gently retracted medially, and a cotton pad is placed over the ventral roots. The L1 root is left untouched at this point. Next, the conus and filum terminale are examined, and the S2-5 sacral roots exiting the conus are identified medially. The S2 dorsal root can be bulky, especially in patients with a postfixed lumbosacral plexus, but there is always an abrupt and marked decrease in its size. The individual S3-5 spinal roots appear as thin threads. The dorsal and ventral roots at this level are close together without intervening space, so all of the S3-5 spinal roots are left intact. The lower sacral roots can best be identified by gently lifting the dorsal roots from the entry zone on the dorsal aspect of the conus. Whenever the surgeon is unsure of the exact identification of the S3-5 spinal roots, it is prudent to spare the S2 dorsal root. It should be emphasized that direct electrical stimulation of the dorsal roots does not help identify the sacral nerve roots.51
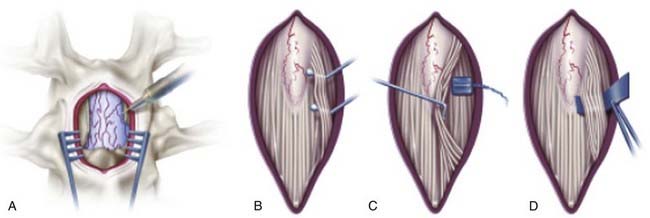
 -inch cotton pad until a 5-mm Silastic sheet can be passed between them.
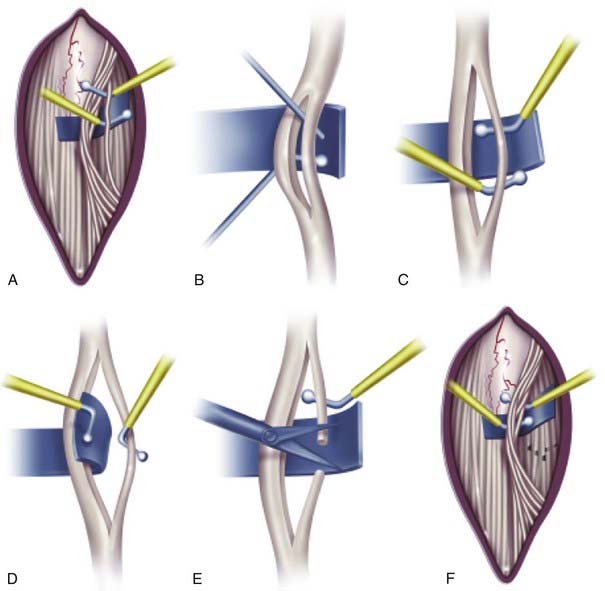
-inch cotton pad until a 5-mm Silastic sheet can be passed between them.Once the L2-S2 dorsal roots are identified, a 5-mm-wide blue Silastic sheet is placed around all the dorsal roots and distant from the conus (Fig. 227-5A). The Silastic sheet keeps the L2-S2 dorsal roots safely separate from the ventral and lower sacral roots during the rest of the operation. Before starting EMG testing, to ensure that no ventral root or lower sacral root is over the Silastic material, the surgeon again examines three structures: the L2 foraminal exit, the cleft lateral to the conus between the ventral and dorsal roots, and the S3-5 roots.
Identification of Individual Dorsal Roots
A shortcoming of this particular dorsal rhizotomy procedure is difficulty identifying individual dorsal roots with certainty. However, precise identification of the dorsal roots is not critical for SDR for the following reasons: (1) the dorsal roots project to multiple segments in the spinal cord,52 (2) all major muscles of the lower extremity in children with spastic CP receive motor innervation from several segments,53 and (3) somatotopic organization occurs in the spinal cord and brain after deafferentation.54
Electromyographic Examination and Sectioning of the Dorsal Roots
Whether intraoperative EMG testing helps in determining the dorsal root fibers to be cut is a topic of persisting controversy. One of us (T.S.P.) continues to use intraoperative EMG testing because there is no conclusive evidence definitely invalidating it and such testing helps identify spinal root levels during the operation. After the innervation of a dorsal root is determined by EMG responses to electrical stimulation of the whole spinal root, the root is sharply subdivided with a Scheer needle (Storz, St. Louis, MO) into three to seven smaller rootlet fascicles of equal size (Fig. 227-5B). Each rootlet fascicle is suspended over the two hooks of the rhizotomy probes. Single constant square-wave pulses 0.1 msec in duration are applied to the rootlet at a rate of 0.5 Hz. The stimulus intensity is increased stepwise until a reflex response appears from the ipsilateral muscles. After the reflex threshold is determined, a 50-Hz train of tetanic stimulation for 1 second is applied to the rootlet (Fig. 227-5C). The reflex response is then graded according to the criteria detailed in Table 227-5. Our experience has been that the great majority of rootlets produce 1+ to 4+ responses. Thus, we base our decision to section a given rootlet on the number of rootlets producing sustained responses at that level and the intensity of the responses. Rootlets that produce a response of 0 are left intact. Rootlets producing 3+ and 4+ responses are cut, and those producing 1+ and 2+ responses are sometimes spared. If only 1+ and 2+ responses are detected, the rootlets with the most active responses are cut. At least one rootlet is left, irrespective of the EMG responses, to avoid postoperative sensory loss. The dorsal rootlets spared from sectioning are placed behind the Silastic sheet and thus kept apart from the rootlets yet to be tested (Fig. 227-5D to F). The procedure is carried out in sequence on the remaining L3-S2 dorsal roots. Using the criteria presented in Table 227-5, I (T.S.P.) section 50% to 75% of the rootlets examined. Finally, the L1 dorsal root is identified at the neural foramen, and half of the dorsal root is cut without EMG testing. In our experience, EMG testing of the L1 root is unreliable. Sectioning of the L1 dorsal root is necessary to further reduce spasticity in the hip flexors, especially in patients with a large L1 root associated with a prefixed lumbosacral plexus. Once the L1 rhizotomy is complete, the entire procedure is performed in an identical manner on the contralateral side.
TABLE 227-5 Criteria for Grading Electromyographic Responses during Selective Dorsal Rhizotomy For Spastic Cerebral Palsy
| GRADE | ELECTROMYOGRAPHIC RESPONSES |
|---|---|
| 0 | Unsustained or single discharge in response to a train of stimuli |
| 1+ | Sustained discharges from muscles innervated through the segment stimulated in the ipsilateral lower extremity |
| 2+ | Sustained discharges from muscles innervated through the segment stimulated and immediately adjacent segments |
| 3+ | Sustained discharges from segmentally innervated muscles, as well as muscles innervated through segments distant to the segment stimulated |
| 4+ | Sustained discharges from contralateral muscles with or without sustained discharges from the ipsilateral muscles |
Postoperative Course and Complications
Reduction of spasticity is apparent immediately after SDR. Patients typically complain of numbness, tingling, and a feeling of heaviness in the lower extremities for 5 to 10 days postoperatively. Voluntary urination is regained within 72 hours. Most patients who were independent walkers preoperatively walk with assistance by the 5th postoperative day and resume full independent walking by the 14th day. Patients who walked with aids preoperatively take a slower postoperative course; it often takes more than 6 weeks for their motor performance to reach preoperative levels. Recovery of motor performance is faster after this variation of dorsal rhizotomy than after the operation introduced by Peacock and colleagues.48 We surmise that the rapid recovery can be attributed to the lack of traction on the ventral spinal roots and consequent neurapraxic injury during the former operation.
Potential immediate postoperative complications after SDR include CSF leakage, meningitis, neurogenic bladder, sensory loss, ileus, bronchospasm, pneumonia, urinary tract infection, and sexual dysfunction.55 To date, in none of our patients has a neurological complication developed; however, bronchospasm, pneumonia, and urinary tract infections have developed in a small number of patients, and one adult patient experienced a CSF leak.
Other investigators have reported rates of bladder dysfunction after SDR in the range of 1% to 24%.55–57 These rates have been attributed to an increase in the tendency to include the S2 root in the rhizotomy. To this end, pudendal afferent mapping has been performed and has shown that genital sensation may be carried to some degree by these fibers.58 Huang and associates suggested that these afferents may contribute to sexual function and bowel and bladder continence. Although we do include S2 in most cases of SDR, we have not observed new-onset sexual dysfunction after SDR, even on direct questioning at follow-up clinic visits.
Spinal deformities (i.e., spondylosis, spondylolisthesis),59 lumbar spinal stenosis,60 scoliosis,61 and lumbar hyperlordosis62 have been reported in patients who underwent SDR through a multilevel lumbosacral laminectomy or laminotomy. Peter and coworkers found that isthmic spondylolysis or grade I spondylolisthesis developed in 20% of 99 patients after five-level lumbosacral laminectomies for SDR.59 In 19 of their patients, the isthmic defects involved the L3-4, L4-5, or L5-S1 levels, and 6 patients had spondylolisthesis. Golan and coauthors reported that 19% of their patients had spondylolisthesis, 17% had hyperlordosis, and 44% had at least mild scoliosis.63 In the latter study, hyperlordosis was less likely to develop in younger patients, whereas spondylolisthesis was more likely to develop in ambulatory children. Spinal stenosis occurred in 2 of 130 patients who underwent SDR for spastic diplegia 4 years after L1-S2 laminectomy and 2 years after L2-S2 laminotomy.60 Lumbar hyperlordosis occurred in 2 nonambulatory children with spastic quadriplegia who had significant trunk weakness.62 Osteoplastic laminotomy may reduce the risk for these observed spinal deformities; however, convincing evidence for this assertion remains to be demonstrated. In fact, one study of SDR with laminoplasty reported that 12% of patients had spondylolisthesis (1 requiring fusion) and that scoliosis developed in 16% postoperatively.64 These and other reported spinal deformities after SDR through an extensive lumbosacral laminectomy highlight the importance of a limited laminectomy approach for SDR.
Outcome
Spasticity
Reduction of spasticity is the principal goal of SDR. It can be achieved in nearly all patients with spastic diplegia15,38,39,41,65,66 and in many patients with spastic quadriplegia.67–69 The marked reduction in spasticity is apparent shortly after the operation as decreased muscle tone, decreased knee and ankle jerks, and absence of ankle clonus. The reduction in spasticity can be quantified over time,65 and although muscle tone may increase to a minor degree months to years postoperatively, it remains reduced from the preoperative level.40,70,71
Strength
Motor weakness of varying degrees is invariably present in patients with CP. When spasticity is reduced or eliminated, the motor weakness underlying the spasticity surfaces, thus giving the impression that SDR produces weakness. In quantitative strength measurements, our group and others have found that strength in CP patients, although less than that in able-bodied children, had increased significantly by 6 months postoperatively.65 Indeed, with aggressive postoperative therapy, strength improves greatly within the first year,15,69 but further gains have been noted successively over a 5-year period.70–72
Gross Motor Function
Three controlled studies evaluated the effect of SDR on gross motor function measure (GMFM) scores.15,39,41 The GMFM assesses several gross motor functions in CP patients, including lie/roll, sit, crawl/kneel, stand, walk/run/jump.73 The three studies included a group receiving physical therapy alone and another group undergoing SDR combined with physical therapy. Two studies found the SDR group to have significantly higher GMFM scores at 9 and 12 months than the group receiving physical therapy alone.15,41 In contrast, one study found no beneficial effect of SDR on GMFM scores assessed at 12 and 24 months postoperatively.39 All three studies are limited in scope because quality of gait and other motor performance, efficiency of gait, quality of life, and deformities of the lower extremities were not examined. Moreover, patient follow-up was too short to evaluate the long-term effects of reduced spasticity on, for example, joint or extremity deformities or rates of subsequent orthopedic surgery.
A meta-analysis addressed the discordance among the clinical trials investigating physical therapy alone versus SDR plus physical therapy.38 This study showed a statistically significant reduction in spasticity, as well as an increase in GMFM scores, in the group that received SDR plus physical therapy when compared with physical therapy alone. These findings appear to be durable inasmuch as one study found improvement in GMFM scores more than 5 years postoperatively.71
Gait
Walking is an element of gross motor function, but gait outcome deserves a separate discussion because it is a primary concern to many parents of children with CP. A frequently asked question is what would a child’s level of walking be after SDR. To answer the question, one should take into consideration the diagnosis of CP, the level of motor development, and the ability to dorsiflex the foot. Nearly all hemiplegics walk independently, and 87% of diplegics walk with or without assistive devices.74 No quadriplegics can walk independently; they are either nonambulatory or walk with assistive devices.75 Regarding motor development for prediction of gait outcome, the child should sit and stand before taking steps. Thus, children who can sit alone at 2 years of age will most likely walk either independently or with aids.74 Sitting ability is a predictor of walking, but it does not specifically predict independent walking. One can use the child’s ability to dorsiflex as a predictor of independent walking after SDR47; in our experience, virtually all children who can dorsiflex the foot bilaterally without associated simultaneous movements of other joints, either before or after SDR, become independent walkers. If the dorsiflexion is unilateral and occurs without associated movements of other joints, there is a high probability of the child becoming an independent walker; however, the child’s walking will be significantly abnormal because of the more involved extremity.47 If dorsiflexion of the foot is bilateral but associated with other joint movements, household-independent walking can be achieved. If dorsiflexion of the foot is lacking bilaterally, independent walking after SDR is not possible, and a walker or crutches will be needed for assistance. The predictive value of foot dorsiflexion stems from the fact that active foot movements are most vulnerable to cerebral lesions, and hence retention of the ability to perform dorsiflexion of the foot indicates a relatively mild injury to the motor area.
Computerized gait analysis has been used to assess gait outcome after SDR.76–78 Gait analysis quantitatively assesses gait parameters in children with CP, including cadence (steps per minute), velocity (m/s), stride length (m), pelvic tilt, hip extension and abduction, and knee and ankle range of movement. Studies have demonstrated that SDR results in significantly improved range of movement in the lower limbs, increased stride length, and increased gait velocity, as well as improvements in other gait parameters.40,65,79–82 The improvements in gait pattern have been maintained for longer than 10 years postoperatively.83,84
Orthopedic Surgery
Because spasticity is a cause of muscle contractures and deformities, one would anticipate a decreased rate of orthopedic surgery after SDR in patients with spastic CP. We have shown that SDR, when performed between 2 and 4 years of age, reduces the need for orthopedic surgery for heel cord, hamstring, and adductor release.42 Subsequent reports from our group85 and others71,86,87 have confirmed the reduction in orthopedic surgeries after SDR. It is rare to encounter any need for late orthopedic surgery in diplegic children who walk unaided after SDR at 2 to 4 years of age. In contrast, children who walk with assistive devices or cannot walk at all often need orthopedic surgery.88 With the exception of spastic quadriplegics,89 the rate of orthopedic surgery after SDR is highest in spastic diplegic children with more severe involvement. Delayed SDR in independent ambulators and limited muscle stretching in assisted ambulators or nonambulators may necessitate subsequent orthopedic surgery.43 These assisted ambulators or nonamubulatory patients in particular require close orthopedic follow-up after SDR.
Hip Deformities
Hip subluxation and dislocation are common problems in children with spastic CP and occur in 3% to 59% of patients.90,91 The cause of the hip deformities is multifactorial, but spasticity in the hip adductor and iliopsoas muscles, combined with lack of normal weight bearing, plays an important role in their development. By decreasing spasticity, SDR may decrease the incidence of hip subluxation in children with CP.72,92 We examined changes in lateral hip migration in spastic diplegic and quadriplegic children undergoing SDR. First, 67 diplegics between 2 and 11 years of age at the time of surgery were monitored for 6 to 46 months postoperatively. Of all hips examined radiographically, 75% remained unchanged, 17% improved, and 7% worsened.93 Next, 45 quadriplegics were examined in a similar manner. In these more involved children, 80% of the hips remained unchanged, 9% improved, and 11% worsened.68 The results contradict a suggestion in a previous report94 and indicate that SDR prevents progressive hip subluxation in the majority of spastic diplegic and quadriplegic children who undergo the operation.
Because of the high incidence of hip abnormalities in children with CP, however, it is essential to obtain hip radiographs at the initial evaluation. The severity of the hip abnormalities influences the decision regarding timing and performance of the operation. It is also important to monitor the hips after SDR to avoid delayed orthopedic interventions in the event of progressive hip subluxation secondary to a dysplastic acetabulum.95 Postrhizotomy follow-up is especially important in children younger than 5 years who have not yet completed hip maturation.96
Cognition and Language
Some children who appear developmentally delayed before SDR show marked improvements in their attention, language, temperament, mood, and social interactions after the operation.97,98 The improvements may result in part from alleviation of spasticity-associated physical discomfort,99 elevated mood, or the intensified therapy after SDR. The improvements may also be related to the suprasegmental effects of SDR on neural circuits beyond the lumbosacral segments.69 Although the concept of suprasegmental effects of SDR remains to be substantiated, positive changes in cognitive performance after SDR have been demonstrated in a neuropsychological investigation.97 Groups of children with spastic diplegia and able-bodied children were tested with visual attention tasks and Woodcock-Johnson psychoeducational battery measures (vocabulary, spatial relationships, quantitative concepts, phonemic blending, memory for sentences, and visual-auditory–associated learning). The children with spastic diplegia received either SDR plus therapy or therapy alone. In a 6-month interval, those who underwent SDR made more improvements in visual attention tasks and visual-auditory–associated learning than did those receiving therapy only. In addition to cognitive improvements, patients have been shown to make functional gains in self-care and social interactions.100
Upper Limb Function
Positive effects of SDR on upper extremity function have been demonstrated in several studies.98,101–103 Through proposed suprasegmental effects, gains in upper extremity function enable increased independence in performing activities of daily living102 and fine upper extremity and trunk control.101 In one study, significant improvements in grasping, hand-eye coordination, and manual dexterity were seen as long as 5 years postoperatively.103 Spastic quadriplegic children who have some upper extremity movement may show significant improvement in range of motion after SDR. In contrast, children who have either severe spastic quadriplegia with little upper extremity movement or mild spastic quadriplegia with full range of motion are less likely show upper extremity improvement.
Bladder Function
Because of the loss of descending inhibitory input, patients with CP exhibit increased intravesical pressure consistent with spasticity,104 and urodynamic studies of incontinent children reveal uninhibited contractions of the bladder, detrusor-sphincter dyssynergia, a small-capacity bladder, a hypertonic bladder, or any combination of these problems.105 More than a third of patients experience dysfunctional voiding symptoms such as dribbling, stress incontinence, enuresis, urgency, and frequency.106 Sweetser and associates reported improvement in incontinence and urgency in spastic diplegics after SDR but found no beneficial effect in spastic quadriplegics after SDR.107 Anecdotally, we and others72 have also seen young spastic diplegic children who completed potty training within a few months of SDR.
Craft S, Park TS, White DA, et al. Changes in cognitive performance in children with spastic diplegic cerebral palsy following selective dorsal rhizotomy. Pediatr Neurosurg. 1995;23:68-74.
Crawford K, Karol LA, Herring JA. Severe lumbar lordosis after dorsal rhizotomy. J Pediatr Orthop. 1996;16:336-339.
Damiano DL, Laws E, Carmines DV, et al. Relationship of spasticity to knee angular velocity and motion during gait in cerebral palsy. Gait Posture. 2006;23:1-8.
Damiano DL, Martellotta TL, Sullivan DJ, et al. Muscle force production and functional performance in spastic cerebral palsy: relationship of cocontraction. Arch Phys Med Rehabil. 2000;81:895-900.
de la Tour EH, Tabary JC, Tabary C, et al. The respective roles of muscle length and muscle tension in sarcomere number adaptation of guinea-pig soleus muscle. J Physiol (Paris). 1979;75:589-592.
Decter RM, Bauer SB, Khoshbin S, et al. Urodynamic assessment of children with cerebral palsy. J Urol. 1987;138:1110-1112.
1 Clark SL, Hankins GD. Temporal and demographic trends in cerebral palsy—fact and fiction. Am J Obstet Gynecol. 2003;188:628-633.
2 Kuban KC, Leviton A. Cerebral palsy. N Engl J Med. 1994;330:188-195.
3 Grether JK, Cummins SK, Nelson KB. The California Cerebral Palsy Project. Paediatr Perinat Epidemiol. 1992;6:339-351.
4 Taft LT. Cerebral palsy. Pediatr Rev. 1995;16:411-418.
5 Atkinson S, Stanley FJ. Spastic diplegia among children of low and normal birthweight. Dev Med Child Neurol. 1983;25:693-708.
6 Ellenberg JH, Nelson KB. Cluster of perinatal events identifying infants at high risk for death or disability. J Pediatr. 1988;113:546-552.
7 Krageloh-Mann I, Hagberg G, Meisner C, et al. Bilateral spastic cerebral palsy—a comparative study between southwest Germany and western Sweden. II: Epidemiology. Dev Med Child Neurol. 1994;36:473-483.
8 Okumura A, Kato T, Kuno K, et al. MRI findings in patients with spastic cerebral palsy. II: Correlation with type of cerebral palsy. Dev Med Child Neurol. 1997;39:369-372.
9 Park TS, Phillips LH, Torner JC. Magnetic resonance imaging in selective dorsal rhizotomy for spastic cerebral palsy. In: Park TS, Phillips LH, Peacock WJ, editors. Neurosurgery: State of the Art Reviews: Management of Spasticity in Cerebral Palsy and Spinal Cord Injury. Philadelphia: Hanley & Belfus; 1989:485-495.
10 Folkerth RD. Periventricular leukomalacia: overview and recent findings. Pediatr Dev Pathol. 2006;9:3-13.
11 Folkerth RD. Neuropathologic substrate of cerebral palsy. J Child Neurol. 2005;20:940-949.
12 Koman LA, Smith BP, Shilt JS. Cerebral palsy. Lancet. 2004;363:1619-1631.
13 Damiano DL, Martellotta TL, Sullivan DJ, et al. Muscle force production and functional performance in spastic cerebral palsy: relationship of cocontraction. Arch Phys Med Rehabil. 2000;81:895-900.
14 Morrell DS, Pearson JM, Sauser DD. Progressive bone and joint abnormalities of the spine and lower extremities in cerebral palsy. Radiographics. 2002;22:257-268.
15 Steinbok P, Reiner AM, Beauchamp R, et al. A randomized clinical trial to compare selective posterior rhizotomy plus physiotherapy with physiotherapy alone in children with spastic diplegic cerebral palsy. Dev Med Child Neurol. 1997;39:178-184.
16 Williams PE, Goldspink G. Longitudinal growth of striated muscle fibres. J Cell Sci. 1971;9:751-767.
17 de la Tour EH, Tabary JC, Tabary C, et al. The respective roles of muscle length and muscle tension in sarcomere number adaptation of guinea-pig soleus muscle. J Physiol (Paris). 1979;75:589-592.
18 Goldspink G, Tabary C, Tabary JC, et al. Effect of denervation on the adaptation of sarcomere number and muscle extensibility to the functional length of the muscle. J Physiol. 1974;236:733-742.
19 Williams PE, Goldspink G. The effect of immobilization on the longitudinal growth of striated muscle fibres. J Anat. 1973;116:45-55.
20 Ziv I, Blackburn N, Rang M, et al. Muscle growth in normal and spastic mice. Dev Med Child Neurol. 1984;26:94-99.
21 Tabary JC, Tardieu C, Tardieu G, et al. Experimental rapid sarcomere loss with concomitant hypoextensibility. Muscle Nerve. 1981;4:198-203.
22 Tardieu C, Huet de la Tour E, Bret MD, et al. Muscle hypoextensibility in children with cerebral palsy: I. Clinical and experimental observations. Arch Phys Med Rehabil. 1982;63:97-102.
23 Castle ME, Reyman TA, Schneider M. Pathology of spastic muscle in cerebral palsy. Clin Orthop Relat Res. 1979;142:223-232.
24 Mohagheghi AA, Khan T, Meadows TH, et al. In vivo gastrocnemius muscle fascicle length in children with and without diplegic cerebral palsy. Dev Med Child Neurol. 2008;50:44-50.
25 Friden J, Lieber RL. Spastic muscle cells are shorter and stiffer than normal cells. Muscle Nerve. 2003;27:157-164.
26 Hoffer MM, Knoebel RT, Roberts R. Contractures in cerebral palsy. Clin Orthop Relat Res. 1987;219:70-77.
27 Rose J, Haskell WL, Gamble JG, et al. Muscle pathology and clinical measures of disability in children with cerebral palsy. J Orthop Res. 1994;12:758-768.
28 Sanger TD, Bastian A, Brunstrom J, et al. Prospective open-label clinical trial of trihexyphenidyl in children with secondary dystonia due to cerebral palsy. J Child Neurol. 2007;22:530-537.
29 Tilton AH. Therapeutic interventions for tone abnormalities in cerebral palsy. NeuroRx. 2006;3:217-224.
30 Albright AL. Intrathecal baclofen for childhood hypertonia. Childs Nerv Syst. 2007;23:971-979.
31 Albright AL, Barron WB, Fasick MP, et al. Continuous intrathecal baclofen infusion for spasticity of cerebral origin. JAMA. 1993;270:2475-2477.
32 Gilmartin R, Bruce D, Storrs BB, et al. Intrathecal baclofen for management of spastic cerebral palsy: multicenter trial. J Child Neurol. 2000;15:71-77.
33 Van Schaeybroeck P, Nuttin B, Lagae L, et al. Intrathecal baclofen for intractable cerebral spasticity: a prospective placebo-controlled, double-blind study. Neurosurgery. 2000;46:603-609.
34 Broggi G, Angelini L, Bono R, et al. Long term results of stereotactic thalamotomy for cerebral palsy. Neurosurgery. 1983;12:195-202.
35 Gornall P, Hitchcock E, Kirkland IS. Stereotaxic neurosurgery in the management of cerebral palsy. Dev Med Child Neurol. 1975;17:279-286.
36 Trejos H, Araya R. Stereotactic surgery for cerebral palsy. Stereotact Funct Neurosurg. 1990;54-55:130-135.
37 Albright AL. Neurosurgical treatment of spasticity and other pediatric movement disorders. J Child Neurol. 2003;18(suppl 1):S67-S78.
38 McLaughlin J, Bjornson K, Temkin N, et al. Selective dorsal rhizotomy: meta-analysis of three randomized controlled trials. Dev Med Child Neurol. 2002;44:17-25.
39 McLaughlin JF, Bjornson KF, Astley SJ, et al. Selective dorsal rhizotomy: efficacy and safety in an investigator-masked randomized clinical trial. Dev Med Child Neurol. 1998;40:220-232.
40 Steinbok P. Outcomes after selective dorsal rhizotomy for spastic cerebral palsy. Childs Nerv Syst. 2001;17:1-18.
41 Wright FV, Sheil EM, Drake JM, et al. Evaluation of selective dorsal rhizotomy for the reduction of spasticity in cerebral palsy: a randomized controlled trial. Dev Med Child Neurol. 1998;40:239-247.
42 Chicoine MR, Park TS, Kaufman BA. Selective dorsal rhizotomy and rates of orthopedic surgery in children with spastic cerebral palsy. J Neurosurg. 1997;86:34-39.
43 Arens LJ, Peacock WJ, Peter J. Selective posterior rhizotomy: a long-term follow-up study. Childs Nerv Syst. 1989;5:148-152.
44 Reference deleted in proofs.
45 Peter JC, Arens LJ. Selective posterior lumbosacral rhizotomy in teenagers and young adults with spastic cerebral palsy. Br J Neurosurg. 1994;8:135-139.
46 Burke RE, Fahn S, Gold AP. Delayed-onset dystonia in patients with static encephalopathy. J Neurol Neurosurg Psychiatry. 1980;43:789-797.
47 Chicoine MR, Park TS, Vogler GP, et al. Predictors of ability to walk after selective dorsal rhizotomy in children with cerebral palsy. Neurosurgery. 1996;38:711-714.
48 Peacock WJ, Arens LJ, Berman B. Cerebral palsy spasticity. Selective posterior rhizotomy. Pediatr Neurosci. 1987;13:61-66.
49 Park TS. Selective dorsal rhizotomy for the spasticity of cerebral palsy. In: Neurosurgery Operative Atlas. Park Ridge, IL: American Association of Neurological Surgeons Publications Committee; 1995.
50 Park TS, Johnston JM. Surgical techniques of selective dorsal rhizotomy for spastic cerebral palsy. Technical note. Neurosurg Focus. 2006;21(2):e7.
51 Ojemann JG, Park TS, Komanetsky R, et al. Lack of specificity in electrophysiological identification of lower sacral roots during selective dorsal rhizotomy. J Neurosurg. 1997;86:28-33.
52 Imai Y, Kusama T. Distribution of the dorsal root fibers in the cat. An experimental study with the Nauta method. Brain Res. 1969;13:338-359.
53 Phillips LH2nd, Park TS. Electrophysiologic mapping of the segmental anatomy of the muscles of the lower extremity. Muscle Nerve. 1991;14:1213-1218.
54 Basbaum AI, Wall PD. Chronic changes in the response of cells in adult cat dorsal horn following partial deafferentation: the appearance of responding cells in a previously non-responsive region. Brain Res. 1976;116:181-204.
55 Abbott R. Complications with selective posterior rhizotomy. Pediatr Neurosurg. 1992;18:43-47.
56 Fasano VA, Broggi G, Barolat-Romana G, et al. Surgical treatment of spasticity in cerebral palsy. Childs Brain. 1978;4:289-305.
57 Laitinen LV, Nilsson S, Fugl-Meyer AR. Selective posterior rhizotomy for treatment of spasticity. J Neurosurg. 1983;58:895-899.
58 Huang JC, Deletis V, Vodusek DB, et al. Preservation of pudendal afferents in sacral rhizotomies. Neurosurgery. 1997;41:411-415.
59 Peter JC, Hoffman EB, Arens LJ. Spondylolysis and spondylolisthesis after five-level lumbosacral laminectomy for selective posterior rhizotomy in cerebral palsy. Childs Nerv Syst. 1993;9:285-287.
60 Gooch JL, Walker ML. Spinal stenosis after total lumbar laminectomy for selective dorsal rhizotomy. Pediatr Neurosurg. 1996;25:28-30.
61 Li Z, Zhu J, Liu X. Deformity of lumbar spine after selective dorsal rhizotomy for spastic cerebral palsy. Microsurgery. 2008;28:10-12.
62 Crawford K, Karol LA, Herring JA. Severe lumbar lordosis after dorsal rhizotomy. J Pediatr Orthop. 1996;16:336-339.
63 Golan JD, Hall JA, O’Gorman G, et al. Spinal deformities following selective dorsal rhizotomy. J Neurosurg. 2007;106:441-449.
64 Spiegel DA, Loder RT, Alley KA, et al. Spinal deformity following selective dorsal rhizotomy. J Pediatr Orthop. 2004;24:30-36.
65 Engsberg JR, Olree KS, Ross SA, et al. Spasticity and strength changes as a function of selective dorsal rhizotomy. J Neurosurg. 1998;88:1020-1026.
66 Peacock WJ, Staudt LA. Functional outcomes following selective posterior rhizotomy in children with cerebral palsy. J Neurosurg. 1991;74:380-385.
67 Gormley MEJr, Krach LE, Piccini L. Spasticity management in the child with spastic quadriplegia. Eur J Neurol. 2001;8(suppl 5):127-135.
68 Heim RC, Park TS, Vogler GP, et al. Changes in hip migration after selective dorsal rhizotomy for spastic quadriplegia in cerebral palsy. J Neurosurg. 1995;82:567-571.
69 Steinbok P. Selective dorsal rhizotomy for spastic cerebral palsy: a review. Childs Nerv Syst. 2007;23:981-990.
70 Gul SM, Steinbok P, McLeod K. Long-term outcome after selective posterior rhizotomy in children with spastic cerebral palsy. Pediatr Neurosurg. 1999;31:84-95.
71 Mittal S, Farmer JP, Al-Atassi B, et al. Long-term functional outcome after selective posterior rhizotomy. J Neurosurg. 2002;97:315-325.
72 Farmer JP, Sabbagh AJ. Selective dorsal rhizotomies in the treatment of spasticity related to cerebral palsy. Childs Nerv Syst. 2007;23:991-1002.
73 Russell DJ, Rosenbaum PL, Cadman DT, et al. The gross motor function measure: a means to evaluate the effects of physical therapy. Dev Med Child Neurol. 1989;31:341-352.
74 Molnar GE, Gordon SU. Cerebral palsy: predictive value of selected clinical signs for early prognostication of motor function. Arch Phys Med Rehabil. 1976;57:153-158.
75 Watt JM, Robertson CM, Grace MG. Early prognosis for ambulation of neonatal intensive care survivors with cerebral palsy. Dev Med Child Neurol. 1989;31:766-773.
76 Boscarino LF, Ounpuu S, Davis RB3rd, et al. Effects of selective dorsal rhizotomy on gait in children with cerebral palsy. J Pediatr Orthop. 1993;13:174-179.
77 Cahan LD, Adams JM, Perry J, et al. Instrumented gait analysis after selective dorsal rhizotomy. Dev Med Child Neurol. 1990;32:1037-1043.
78 Thomas SS, Aiona MD, Pierce R, et al. Gait changes in children with spastic diplegia after selective dorsal rhizotomy. J Pediatr Orthop. 1996;16:747-752.
79 Abel MF, Damiano DL, Gilgannon M, et al. Biomechanical changes in gait following selective dorsal rhizotomy. J Neurosurg. 2005;102:157-162.
80 Damiano DL, Laws E, Carmines DV, et al. Relationship of spasticity to knee angular velocity and motion during gait in cerebral palsy. Gait Posture. 2006;23:1-8.
81 Granata KP, Padua DA, Abel MF. Repeatability of surface EMG during gait in children. Gait Posture. 2005;22:346-350.
82 Wong AM, Pei YC, Lui TN, et al. Comparison between botulinum toxin type A injection and selective posterior rhizotomy in improving gait performance in children with cerebral palsy. J Neurosurg. 2005;102:385-389.
83 Langerak NG, Lamberts RP, Fieggen AG, et al. A prospective gait analysis study in patients with diplegic cerebral palsy 20 years after selective dorsal rhizotomy. J Neurosurg Pediatr. 2008;1:180-186.
84 Subramanian N, Vaughan CL, Peter JC, et al. Gait before and 10 years after rhizotomy in children with cerebral palsy spasticity. J Neurosurg. 1998;88:1014-1019.
85 OBrien DF, Park TS. A review of orthopedic surgeries after selective dorsal rhizotomy. Neurosurg Focus. 2006;21(2):e2.
86 Carroll KL, Moore KR, Stevens PM. Orthopedic procedures after rhizotomy. J Pediatr Orthop. 1998;18:69-74.
87 Kan P, Gooch J, Amini A, et al. Surgical treatment of spasticity in children: comparison of selective dorsal rhizotomy and intrathecal baclofen pump implantation. Childs Nerv Syst. 2008;24:239-243.
88 OBrien DF, Park TS, Puglisi JA, et al. Orthopedic surgery after selective dorsal rhizotomy for spastic diplegia in relation to ambulatory status and age. J Neurosurg. 2005;103:5-9.
89 OBrien DF, Park TS, Puglisi JA, et al. Effect of selective dorsal rhizotomy on need for orthopedic surgery for spastic quadriplegic cerebral palsy: long-term outcome analysis in relation to age. J Neurosurg. 2004;101:59-63.
90 Lonstein JE, Beck K. Hip dislocation and subluxation in cerebral palsy. J Pediatr Orthop. 1986;6:521-526.
91 Samilson RL, Tsou P, Aamoth G, et al. Dislocation and subluxation of the hip in cerebral palsy. Pathogenesis, natural history and management. J Bone Joint Surg Am. 1972;54:863-873.
92 Engsberg JR, Ross SA, Collins DR, et al. Effect of selective dorsal rhizotomy in the treatment of children with cerebral palsy. J Neurosurg. 2006;105:8-15.
93 Park TS, Vogler GP, Phillips LH2nd, et al. Effects of selective dorsal rhizotomy for spastic diplegia on hip migration in cerebral palsy. Pediatr Neurosurg. 1994;20:43-49.
94 Greene WB, Dietz FR, Goldberg MJ, et al. Rapid progression of hip subluxation in cerebral palsy after selective posterior rhizotomy. J Pediatr Orthop. 1991;11:494-497.
95 Vidal J, Deguillaume P, Vidal M. The anatomy of the dysplastic hip in cerebral palsy related to prognosis and treatment. Int Orthop. 1985;9:105-110.
96 Harris NH, Lloyd-Roberts GC, Gallien R. Acetabular development in congenital dislocation of the hip. With special reference to the indications for acetabuloplasty and pelvic or femoral realignment osteotomy. J Bone Joint Surg Br. 1975;57:46-52.
97 Craft S, Park TS, White DA, et al. Changes in cognitive performance in children with spastic diplegic cerebral palsy following selective dorsal rhizotomy. Pediatr Neurosurg. 1995;23:68-74.
98 Loewen P, Steinbok P, Holsti L, et al. Upper extremity performance and self-care skill changes in children with spastic cerebral palsy following selective posterior rhizotomy. Pediatr Neurosurg. 1998;29:191-198.
99 Roscigno CI. Addressing spasticity-related pain in children with spastic cerebral palsy. J Neurosci Nurs. 2002;34:123-133.
100 Buckon CE, Thomas SS, Piatt JHJr, et al. Selective dorsal rhizotomy versus orthopedic surgery: a multidimensional assessment of outcome efficacy. Arch Phys Med Rehabil. 2004;85:457-465.
101 Beck AJ, Gaskill SJ, Marlin AE. Improvement in upper extremity function and trunk control after selective posterior rhizotomy. Am J Occup Ther. 1993;47:704-707.
102 Kinghorn J. Upper extremity functional changes following selective posterior rhizotomy in children with cerebral palsy. Am J Occup Ther. 1992;46:502-507.
103 Mittal S, Farmer JP, Al-Atassi B, et al. Impact of selective posterior rhizotomy on fine motor skills. Long-term results using a validated evaluative measure. Pediatr Neurosurg. 2002;36:133-141.
104 Houle AM, Vernet O, Jednak R, et al. Bladder function before and after selective dorsal rhizotomy in children with cerebral palsy. J Urol. 1998;160:1088-1091.
105 Decter RM, Bauer SB, Khoshbin S, et al. Urodynamic assessment of children with cerebral palsy. J Urol. 1987;138:1110-1112.
106 McNeal DM, Hawtrey CE, Wolraich ML, et al. Symptomatic neurogenic bladder in a cerebral-palsied population. Dev Med Child Neurol. 1983;25:612-616.
107 Sweetser PM, Badell A, Schneider S, et al. Effects of sacral dorsal rhizotomy on bladder function in patients with spastic cerebral palsy. Neurourol Urodyn. 1995;14:57-64.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
CHAPTER 227 Selective Dorsal Rhizotomy for Spastic Cerebral Palsy
Cerebral palsy (CP) is the leading cause of developmental disability in children, with more than 500,000 individuals affected in the United States alone.1,2 CP is diagnosed in 1 in 500 live births, and this incidence continues to increase because of improvements in the care and survival of very-low-birth-weight infants.1 There have been significant advances in both medical and surgical treatment of CP spasticity in recent years. From the introduction of injectable botulinum toxin A (Botox) to the implantation of pump devices for chronic intrathecal baclofen administration, patients now have a number of pharmacologic tools to treat spasticity on a long-term basis. Permanent reductions in CP spasticity, however, can be achieved only with selective dorsal rhizotomy (SDR). Currently, a large volume of literature is available on SDR as a therapeutic modality for CP. This chapter attempts to synthesize the reported information on SDR, as well as our own experience with the procedure.
Pathogenesis of Spastic Cerebral Palsy
Spastic CP is the most common subtype of cerebral palsy. Spastic diplegia and spastic quadriplegia affect approximately 60% of patients, but spasticity may also occur with dyskinesia or ataxia, or both, in mixed subtypes of CP. Thus, in total, spasticity afflicts 80% to 90% of all patients with CP.3,4 A distinctive epidemiologic feature of spastic CP—and spastic diplegia in particular—is its correlation with premature birth and low birth weight.5–7 This relationship does not exist with other subtypes of the disease, such as athetoid or dyskinetic CP.
Periventricular leukomalacia (PVL) is the most common finding on brain magnetic resonance imaging (MRI) in children with spastic CP.8,9 Histologically, PVL is characterized by ischemic necrosis of the periventricular and subcortical white matter (Fig. 227-1), areas that are predisposed to hypoperfusion during hypotension.10 Reperfusion injury resulting from oxygen free radicals, cytokines, and excitatory amino acids such as glutamate probably mediate PVL.10 The high incidence of spastic diplegia in infants with PVL has led many to speculate that PVL is the neuropathologic basis for spastic CP.11 The neurological deficits in spastic CP may be explained in part by the topographic arrangements of projection fibers descending from the motor homunculus in the white matter. As seen in Figure 227-2, the leg fibers traverse the white matter closer to the ventricle than the arm fibers do. Thus, PVL is more likely to injure the leg fibers and spare the arm fibers. However, extensive leukomalacia will also injure other projection fibers and result in spastic quadriplegic CP.
Spinal cord ventral horn alpha motor neuron output is a primary determinant of muscle tone. The activity of alpha motor neurons is modulated by excitatory influences (e.g., Ia fibers from muscle spindles, descending input from the corticospinal and reticulospinal pathways) and inhibitory influences (e.g., descending GABAergic [γ-aminobutyric acid] pathways from the cerebellum and basal ganglia). Although the neural pathways involved in CP spasticity remain speculative, it is believed that spasticity results from an imbalance in these excitatory and inhibitory influences that creates hyperactive alpha and gamma neurons with exaggerated stretch reflex activity compounded by loss of inhibitory control (reviewed by Koman and colleagues12). Thus, excessive alpha motor neuron activity of the target muscles, coupled with decreased inhibition of antagonistic muscles, results in the co-contraction of agonist and antagonist muscle groups.13 Clinically, this results in a velocity-dependent, increased resistance to passive stretch.12
Deleterious Effects of Cerebral Palsy Spasticity
CP spasticity has harmful, long-term effects on growing children and thus requires optimal treatment at an early age. When untreated, inhibition of motor activity and limitation of voluntary movement result in decreased muscle and bone development, which ultimately causes deformities of the affected extremities.14 Long-lasting improvements in motor function are observed after SDR, thus supporting the argument for early intervention in the disease.15 Indeed, the spasticity-induced inhibition of motor activities does not abate if the spasticity is left untreated. Normally, longitudinal muscle growth proceeds as new myofilaments and sarcomeres are added to the ends of the muscle fibers.16 The greatest increase in sarcomere numbers occurs at an early age, with the increase taking place much more actively during development than after maturation. In parallel with the addition of sarcomeres, longitudinal muscle growth occurs rapidly during development. The increase in sarcomere number is not under neural control but is induced mainly by the amount of tension that the muscle is subjected to (i.e., repeated muscle stretch).17,18 Thus, when muscles are immobilized, there is a rapid decrease in the number of already existing sarcomeres, as well as a decrease in the number of new sarcomeres that should increase with normal development.19 Because CP spasticity in developing children limits muscle stretch in daily activities, it could cause loss of sarcomeres and inhibition of longitudinal muscle growth. In fact, the effect of spasticity on muscle growth was clearly shown in an experimental study in which longitudinal muscle growth was reduced by 45% in spastic mice as compared with control animals.20 The loss of sarcomeres accompanies a significant decrease in elasticity in the muscles21 and makes them resistant to passive stretch.22 Additionally, atrophy of type I and II muscle fibers compounds the slowed muscle growth.23 Recent work has implicated both shorter muscle (decreased sarcomere length, myocyte length, and fascicle length)24 and an increased extracellular elastic modulus25 in the clinical finding of stiffness. These pathologic events in the muscle cells cause abnormally shortened muscles, long tendons, and muscle contracture (i.e., increased resistance of the muscle to passive stretch in the absence of muscle contraction).22,26,27 The muscle contractures typically worsen as a child grows26 and produce various bone and joint deformities in the extremities.14
Treatment of Spastic Cerebral Palsy
Spasticity is commonly treated with oral medications, including the GABAB receptor agonist baclofen (Lioresal), benzodiazepines such as diazepam (Valium), the anticholinergic trihexyphenidyl, dantrolene, and tizanidine (Zanaflex).28,29 In general, oral medications are moderately effective in treating spasticity but frequently have significant adverse effects (sedation, confusion, dependence).
Intramuscular injections of alcohol and phenol for chemodenervation have been performed for many years. The introduction of intramuscular injection of Botox was a major advance in the treatment of CP spasticity, with predictable benefits and fewer adverse effects.29 Botox is injected directly into the affected muscle, and relaxation is generally seen within several days. Maximal benefit occurs approximately 4 weeks following injection, after which the effect declines and repeated injection is required in approximately 3 to 4 months.
Intrathecal Baclofen
Intrathecal administration of baclofen offers the benefit of delivering the medication directly into the central nervous system (CNS) without systemic side effects. Chronic intrathecal baclofen infusion (CIBI) with a surgically implanted pump device (SynchroMed Infusion System, Medtronic, Inc., Minneapolis, MN) has been shown to reduce spasticity and improve function,30–33 and its use has been indicated in nonambulatory or minimally ambulatory patients with spastic quadriparesis. The potential adverse effects with CIBI are significant and include infection, pump malfunction, and life-threatening withdrawal or overdose.
Stereotactic Intracranial Procedures
Stereotactic ablative procedures such as thalamotomy and dentatotomy have been used to treat CP patients with primarily unilateral dystonia and may offer some benefit in these patients.34–36 More recently, deep brain stimulation of the internal globus pallidus and thalamus have been performed in children with dystonia and tremor, respectively.37 The role of these procedures for CP spasticity has not been investigated.
Selective Dorsal Rhizotomy
SDR has been shown in several controlled trials to reduce spasticity and increase range of motion.15,38–41 In conjunction with physical therapy, SDR produces significant improvements in gross motor function and gait.15,41 SDR also decreases the rate of subsequent orthopedic surgeries.42
Indications
Indications for SDR vary among medical centers. Our current indications for SDR are summarized in Table 227-1. Primary beneficiaries of SDR are children with spastic diplegia secondary to premature birth; however, children with spastic quadriplegic CP can also benefit from SDR. In spastic hemiplegic CP, spasticity is not a predominant cause of the motor impairment, and reduction of spasticity may not greatly improve motor function (see also Table 227-2). The optimal age for children to undergo SDR is 2 to 6 years. Because of the significant deleterious effects of spasticity outlined earlier, early treatment is recommended to reduce the chance of severe orthopedic deformities of the lower extremities. SDR is not considered for children younger than 2 years because approximately 30% of children in whom CP is diagnosed at the age of 1 year later become free of symptoms.43 Adolescents and adults younger than 40 years can also enjoy good functional gains with SDR.45 The risk associated with dorsal rhizotomy in adolescents and adults is no greater than that in young children when performed through a single-level laminectomy. It is possible that reduction of spasticity will lessen the impact of aging on the physical stress caused by CP, such as abnormal stress on bones and muscles, wear and tear on joints, and increasing joint and muscle pain.
TABLE 227-1 Indications for Selective Dorsal Rhizotomy for Spastic Cerebral Palsy
TABLE 227-2 Contraindications to Selective Dorsal Rhizotomy for Spastic Cerebral Palsy
Contraindications
Table 227-2 provides a partial list of contraindications to SDR. Such contraindications include a history of severe congenital hydrocephalus or severe neonatal CNS infection such as intrauterine encephalitis (e.g., toxoplasmosis or cytomegalovirus infection) or bacterial meningitis. Likewise, patients with severe head trauma or hypoxic encephalopathy may not be suitable candidates for SDR. Although SDR is contraindicated in patients with extensive neuronal migration disorders, those with limited schizencephaly involving the sensory motor area, a rare cause of spastic diplegia, may achieve a reduction in spasticity and functional improvements after SDR.
Patients with damage to the basal ganglia deserve special consideration because rigidity and dystonia may coexist with spasticity in this setting. Of particular concern is severe basal ganglia damage in children younger than 5 years. Dystonia may develop anytime in the first 5 years of life.46 As noted earlier, patients with mixed types of CP spasticity (i.e., those with components of dystonia or athetosis) do not enjoy the same degree of functional improvement after SDR. Thus, in the setting of basal ganglia damage it is best to wait until the age of 5, when the predominance of dystonia can more reliably be ascertained. After the age of 5, however, SDR may be considered even in the presence of demonstrable basal ganglia damage if spasticity is the primary clinical symptom. Likewise, athetosis and ataxia are also relative contraindications. Similar to dystonia, if spasticity is the predominant feature in patients with concomitant athetosis, SDR may be of benefit. In these cases, SDR reduces spasticity postoperatively, but the athetosis and dystonia remain unchanged.
Preoperative Evaluation
Motor strength can be determined by testing individual muscles. In young children, past motor milestones, speed of movement, and the ability to isolate joint movements are reliable indices of motor strength. If the motor milestone is close to normal and, for instance, the child can sit alone by the age of 2 years, the child most likely has adequate motor strength and will walk in the future. Crawling or walking in a walker and making rapid transitions between positions are also signs of relatively good motor function. As a rule, the greater the motor impairment, the slower the movement. In our experience, a neurological sign that can be an excellent predictor of gait outcome in children younger than 3 to 4 years is isolated joint movements in the lower extremities (see “Gait” in the section “Outcome”).47
Surgical Considerations and Operative Technique
Between 1987 and 1991, I (T.S.P.) performed dorsal rhizotomies on 173 patients through a laminectomy at the L2-S2 levels, as described by Peacock and colleagues.48 In 1991, however, the potential risk for late spinal deformities after the procedure led to the development of a dorsal rhizotomy procedure through an L1-2 or L1 laminectomy.49 Aside from minor technical refinements,50 the surgical technique described here has been performed between 1991 and 2008 on 1719 children and adults, with a single instance of cerebrospinal fluid (CSF) leakage and no other postoperative complications. Please refer to Tables 227-3 and 227-4.
TABLE 227-3 Subtypes of Spastic Cerebral Palsy Treated by Selective Dorsal Rhizotomy
| SPASTIC CEREBRAL PALSY SUBTYPES | NO. OF PATIENTS | PERCENT |
|---|---|---|
| Diplegia | 1358 | 78 |
| Triplegia or quadriplegia | 356 | 21 |
| Hemiplegia | 5 | <1 |
| Total | 1719 | 100 |
Data from St. Louis Children’s Hospital, 1987 to 2008.
TABLE 227-4 Prerhizotomy Ambulatory Function in Patients Who Underwent Selective Dorsal Rhizotomy for Spastic Cerebral Palsy
| AMBULATION | NO. OF PATIENTS | PERCENT |
|---|---|---|
| Walking with a walker | 719 | 42 |
| Walking with crutches or a cane | 124 | 7 |
| Independent walking | 557 | 32 |
| Some locomotion | 180 | 11 |
| Crawling | 117 | 7 |
| No independent mobility | 22 | 1 |
| Total | 1719 | 100 |
Data from St. Louis Children’s Hospital, 1987 to 2008.
