[level-membership-for-dermatology-category]
Selected Hereditary Diseases
Dawn H. Siegel, Kari L. Martin, Jennifer L. Hand
Approach to the child with a genetic skin condition
To effectively care for an affected newborn and provide information for anxious parents, an organized diagnostic approach is essential. The physical exam requires special attention to ectodermal involvement by assessing the hair, teeth, nails, palms and soles of the feet. Moving beyond the skin, a child should be examined for dysmorphic features, associated major or minor congenital anomalies and accompanying illness. Results of a newborn hearing screen and pediatric ophthalmology exam are often valuable. Finally, a detailed family history including ethnic background and miscarriage history is beneficial. After gathering clinical information, superb resources are available online to consider diagnostic possibilities. One example is Online Mendelian Inheritance in Man (OMIM; http://www.ncbi.nlm.nih.gov/omim).
OMIM is maintained by the National Center for Biotechnology Information (NCBI) and can be searched using a list of the clinical features in an affected patient. Each disorder is assigned a number based on broad categories such as inheritance pattern. ‘GeneTests’ (http://www.ncbi.nlm.nih.gov/sites/GeneTests) and a separate database, ‘GeneReviews’1 offers a frequently updated list of internationally available DNA-based genetic tests and concise, yet comprehensive overviews of selected disorders. For effective coordination of testing and advising the patient about the complexities of genetic testing and reproductive options, the GeneTests site provides a database of genetics services that is searchable by country and province or state. Increasingly, individual mutations that cause familial genetic disorders are recognized to affect important regulatory pathways. This chapter highlights pathways that are particularly important to growth and development of the skin.
Disorders of the RAS-MAPK pathway (RASopathies)
Several genetic skin disorders are caused by mutations in genes in the RAS-MAPK signaling pathway (Fig. 29.1) and have been coined the ‘RASopathies’.2 The RASopathies include neurofibromatosis-1 (NF-1), NF-like syndrome (Legius syndrome), Noonan syndrome, Noonan with multiple lentigines (formerly LEOPARD syndrome), cardiofaciocutaneous syndrome, Costello syndrome, capillary malformation–arteriovenous malformation (see Chapter 22), and hereditary gingival fibromatosis (Table 29.1). Neurofibromatosis and Legius syndrome are also discussed in Chapter 24.
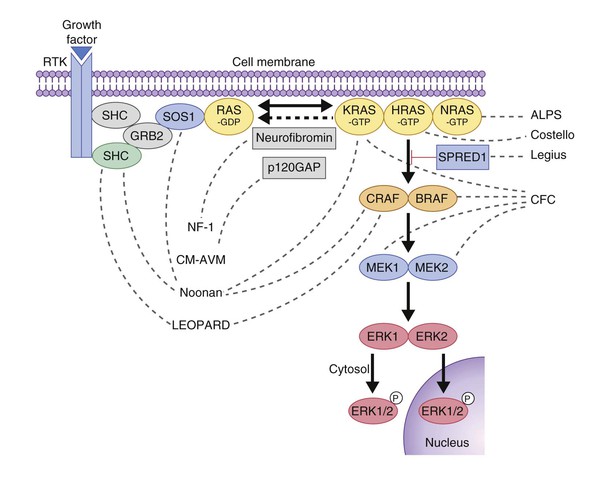
TABLE 29.1
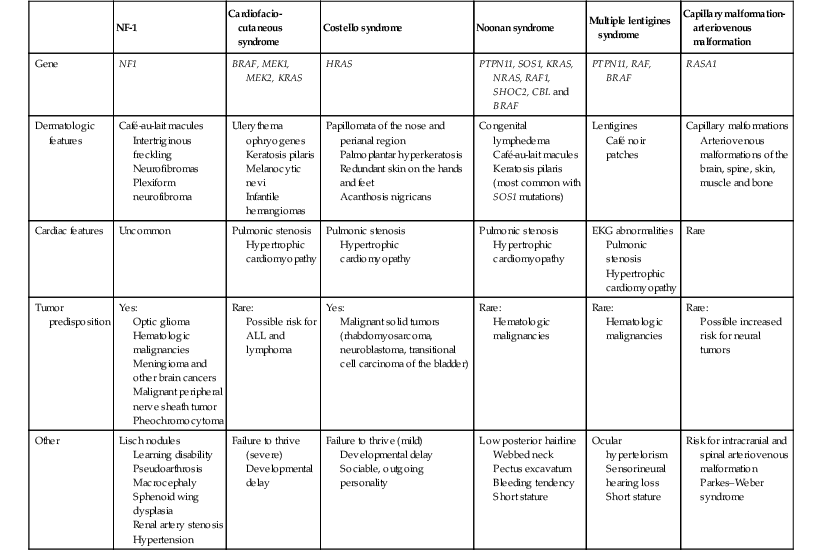
Comparison of the clinical features of the RASopathies
| NF-1 | Cardiofacio-cutaneous syndrome | Costello syndrome | Noonan syndrome | Multiple lentigines syndrome | Capillary malformation- arteriovenous malformation | |
| Gene | NF1 | BRAF, MEK1, MEK2, KRAS | HRAS | PTPN11, SOS1, KRAS, NRAS, RAF1, SHOC2, CBL and BRAF | PTPN11, RAF, BRAF | RASA1 |
| Dermatologic features | Café-au-lait macules Intertriginous freckling Neurofibromas Plexiform neurofibroma |
Ulerythema ophryogenes Keratosis pilaris Melanocytic nevi Infantile hemangiomas |
Papillomata of the nose and perianal region Palmoplantar hyperkeratosis Redundant skin on the hands and feet Acanthosis nigricans |
Congenital lymphedema Café-au-lait macules Keratosis pilaris (most common with SOS1 mutations) |
Lentigines Café noir patches |
Capillary malformations Arteriovenous malformations of the brain, spine, skin, muscle and bone |
| Cardiac features | Uncommon | Pulmonic stenosis Hypertrophic cardiomyopathy |
Pulmonic stenosis Hypertrophic cardiomyopathy |
Pulmonic stenosis Hypertrophic cardiomyopathy |
EKG abnormalities Pulmonic stenosis Hypertrophic cardiomyopathy |
Rare |
| Tumor predisposition | Yes: Optic glioma Hematologic malignancies Meningioma and other brain cancers Malignant peripheral nerve sheath tumor Pheochromocytoma |
Rare: Possible risk for ALL and lymphoma |
Yes: Malignant solid tumors (rhabdomyosarcoma, neuroblastoma, transitional cell carcinoma of the bladder) |
Rare: Hematologic malignancies |
Rare: Hematologic malignancies |
Rare: Possible increased risk for neural tumors |
| Other | Lisch nodules Learning disability Pseudoarthrosis Macrocephaly Sphenoid wing dysplasia Renal artery stenosis Hypertension |
Failure to thrive (severe) Developmental delay |
Failure to thrive (mild) Developmental delay Sociable, outgoing personality |
Low posterior hairline Webbed neck Pectus excavatum Bleeding tendency Short stature |
Ocular hypertelorism Sensorineural hearing loss Short stature |
Risk for intracranial and spinal arteriovenous malformation Parkes–Weber syndrome |
Neurofibromatosis type 1
Neurofibromatosis 1 (NF-1, MIM #162200) is a multisystem disorder characterized by age-related abnormalities of tissue proliferation. NF-1 is one of the most common autosomal dominant genetic conditions with an incidence of approximately 1 in 3000 individuals.3,4 Consensus criteria for the diagnosis of NF-1 were established at the 1988 NIH conference to be used as a guideline for clinical diagnosis (Box 29.1).5 In 95% of affected individuals, a diagnosis can be made by age 11 through the use of clinical evaluation alone.6 However, NF-1 can be a difficult condition to diagnose in some infants due to the high incidence of sporadic mutations, variability of clinical expression, and age-related penetrance of individual clinical manifestations. Hence, anticipatory guidance counseling should be provided in both established and suspected cases of NF-1.
Cutaneous findings
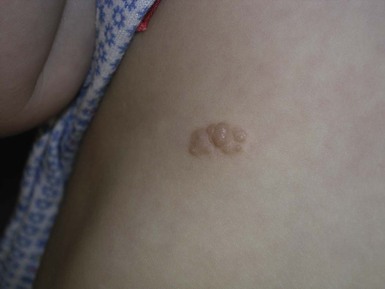
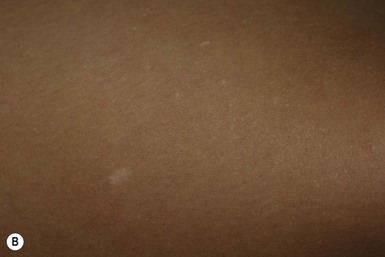
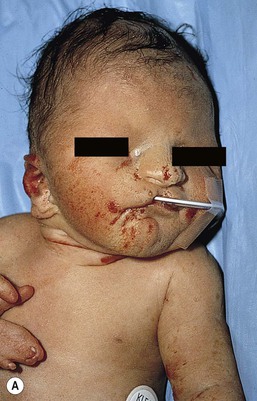
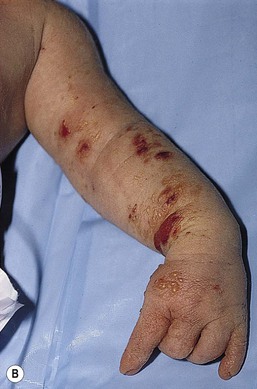
Café-au-lait macules (CALM) are light to dark brown sharply defined oval macules and patches which can occur on almost any skin surface (Fig. 29.2A). Infants with ≥6 café-au-lait macules >5 mm in diameter that are not confined to a single segmental region, should be evaluated and managed as though they have NF-1, even without other signs of NF-1, because of the high likelihood that they will develop other diagnostic signs with time. At puberty, if other features are lacking, the approach and diagnosis can be reconsidered.
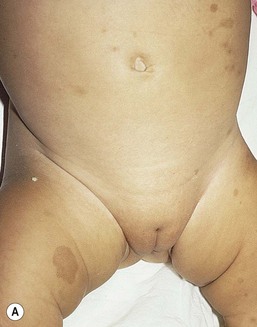
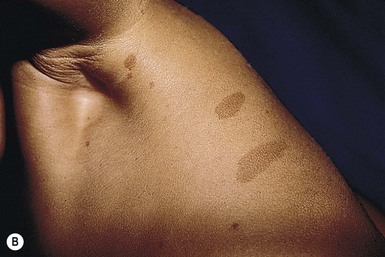
Intertriginous freckling of the axillary and inguinal regions may occasionally be present in infancy, but most often present between 2–10 years of age (Fig. 29.2B). Although freckling on the neck and trunk is common in NF-1, it is not accepted as a diagnostic criterion.
Peripheral neurofibromas are infrequent in early childhood NF-1, occurring in only 14% of children less than 10 years of age.6 Often the first sign of dermal neurofibromas are 3–6 mm, light blue, minimally raised papules which are most easily detected with side lighting. It has been suggested that a subset of children with large deletions of the NF-1 gene typically present with multiple neurofibromas in early childhood.7
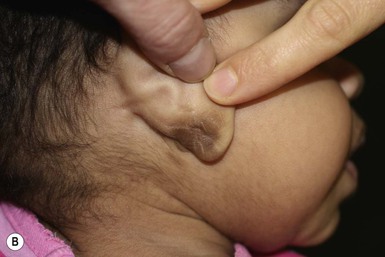
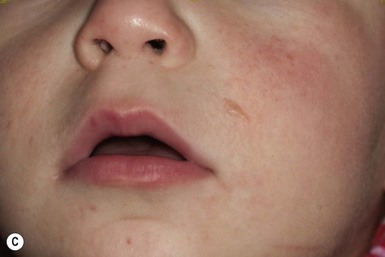
Plexiform neurofibromas may be apparent at birth or soon thereafter; however because they are often internal, may be difficult to detect. They occur in at least 25% of affected individuals (Fig. 29.2C). Most have overlying hyperpigmentation; some – but not all – have increased vascularity resembling a vascular anomaly or hypertrichosis resembling a congenital melanocytic nevus. Plexiform neurofibromas may grow rapidly and interdigitate with and surround normal structures. Radiologic imaging and neurosurgical consultation should be considered if lesions are extensive or close to major nerve bundles.
Extracutaneous findings
Optic gliomas are astrocytomas arising anywhere along the optic pathway. These lesions tend to arise in infancy or early childhood. Half of the tumors are symptomatic, causing loss of visual acuity, decreased field of vision, proptosis, or interference with the hypothalamopituitary axis. Symptomatic optic gliomas are often diagnosed by 6 years of age.
Lisch nodules are pigmented iris hamartomas that rarely present in infants, and only 20% of individuals under 5 years of age with NF-1 have Lisch nodules. They are best seen on slit-lamp examination and do not result in functional disability. Congenital glaucoma occurs in less than 0.05% of individuals with NF-1 and may present with an ipsilateral neurofibroma of the eyelid.
Relatively short stature and large head size are frequent findings and more common than the more diagnostic skeletal changes, pseudoarthrosis and sphenoid wing dysplasia. Pseudoarthrosis represents the failure of union after fracture. It is always unilateral and most commonly presents in the tibia as anterolateral bowing. Ultimately, pseudoarthrosis can progress to severe deformity. Sphenoid wing dysplasia is a unilateral defect of the orbit present in approximately 5% of individuals with NF-1 and results in a change in orbit structure. Approximately half of those cases with sphenoid wing dysplasia develop an ipsilateral temporal–orbital plexiform neurofibroma, and half of individuals presenting with a temporal–orbital tumor also have an underlying plexiform neurofibroma. Learning disabilities occur in approximately 50% of children with NF-1. The main learning difficulties include reading, deficits in perceptual skills (visuospatial) and executive functioning. Attention issues are also common.8
Etiology and pathogenesis
NF-1 is due to an autosomal dominant mutation localized to chromosome 17 that results in defects in neurofibromin, a tumor suppressor protein that stimulates hydrolysis of guanosine triphosphate (GTP) bound to ras.9
Differential diagnosis and diagnosis
Café-au-lait macules may be found in many other conditions, including segmental pigmentary disorder and McCune–Albright syndrome (see Chapter 24). Additional differential diagnoses for multiple café-au-lait macules are shown in Box 29.2. Genetic testing for NF-1 is available from several commercial and research laboratories (see: www.genetests.org, for specific information). Decisions regarding whether laboratory-based NF testing is appropriate are best made in conjunction with a geneticist and genetic counselor.
Treatment and care
Patients with neurofibromatosis require age-related anticipatory guidance counseling and regular follow-up with a pediatrician, ophthalmologist and geneticist. Additional specialists can be included, based on symptoms or complications and may include orthopedics, neurology, dermatology, neuropsychology, and neurosurgery (Box 29.3). Ophthalmologists and neurologists should evaluate for optic nerve pathway tumors and glaucoma. Anterolateral tibial bowing when present require prompt orthopedic evaluation, because the critical time for fracture and poor healing is infancy to early childhood.6 Periodic evaluation for scoliosis is also necessary. Regular physical examinations should include careful measurement of blood pressure because of a higher incidence of hypertension secondary to renovascular disease, vasoactive secreting tumors, and coarctation of the aorta.6 Head circumference should be monitored because of the risk of hydrocephalus and the occurrence of macrocephaly without hydrocephalus. Careful developmental assessment is a key part of management, as the risk of neurologic abnormalities and learning disabilities is increased.
Dermal neurofibromas are benign and should only be excised if they are symptomatic or disfiguring. Plexiform neurofibromas require close evaluation. Skin should be palpated carefully as plexiform neurofibromas may remain below the surface. If there are symptoms or neurologic abnormalities on clinical exam, MRI scans should be obtained to determine the extent of plexiform neurofibromas. Serial imaging or volumetric MRI may be necessary to assess potential growth. Pain and/or growth may herald malignant transformation, but this is exceedingly rare in infancy. Neurologic compromise may result from perineural extension, however, and neurology and/or neurosurgery may need to be consulted for plexiform lesions near neurovascular structures (such as the neck, axilla and spinal area).6 Orbitotemporal neurofibromas may be better managed by numerous surgical procedures over time; plastic surgery should be involved early for management of these tumors.
Leguis syndrome
Legius syndrome (MIM #611431) was first recognized in 2007 as an NF-1-like syndrome in families with multiple café-au-lait macules, but negative NF-1 gene testing.10 The phenotype is milder than NF-1 and seems to lack the propensity for tumor development. The clinical features include a mild NF-like phenotype and 50% of these patients meet diagnostic criteria for NF-1. Of the diagnostic criteria for NF-1 (Box 29.1), the features that have been reported in Legius include >6 CALM >5 mm, intertriginous freckling, positive family history, macrocephaly, short stature and learning disabilities.11 Neurofibromas, plexiform neurofibromas, bone dysplasia and scoliosis have not been associated with Legius syndrome. Legius syndrome is caused by loss of function (LOF) mutations in the SPRED1 gene.10 Like neurofibromin, SPRED1 is a negative regulator of the RAS-MAPK pathway (Fig. 29.1).11
Noonan syndrome
Noonan syndrome (MIM #163950) is an autosomal dominant multisystem disorder characterized by congenital lymphedema, broad or webbed neck, low posterior hairline, short stature, and cardiac malformations (Box 29.4).
Cutaneous findings
The neonate with Noonan syndrome is unlikely to have skin manifestations other than nuchal webbing and peripheral lymphedema that suggest the diagnosis. Keratosis pilaris atrophicans faciei (ulerythema ophryogenes), characterized by horny, whitish, hemispherical, or acuminate papules at the opening of pilosebaceous follicles, is generally noted in older children, but may manifest in the external third of the eyebrows by a few months after birth. Some children with Noonan syndrome have multiple CALM and/or lentigines.
Extracutaneous findings
Neonates have a characteristic facial appearance consisting of a tall forehead, low posterior hairline, hypertelorism, downslanting palpebral fissures, epicanthal folds, short and broad, depressed nasal root, deeply grooved philtrum and micrognathia. The chest shape is unique with superior pectus carinatum and inferior pectus excavatum. Feeding difficulties, gastroesophageal reflux and failure to thrive are frequent problems in infancy. Unilateral or bilateral cryptorchidism are common in boys. Infants are at risk for transient monocytosis, thrombocytopenia and myeloproliferative disorder.12 Coagulation defects occur in approximately one-third of patients with Noonan syndrome and may present with easy bruising or prolonged bleeding times. There is a slightly increased risk of hematologic malignancy (most commonly juvenile myelomonocytic leukemia, JMML) compared with the general population. The classic congenital heart defect in Noonan syndrome is pulmonic stenosis.
Etiology and pathogenesis
The incidence of Noonan syndrome is approximately 1 in 1000–2500 live births.13 Noonan syndrome may be caused by a mutation in one of several genes in the RAS-MAPK signaling pathway, including PTPN11, SOS1, KRAS, NRAS, RAF1, SHOC2, CBL, and BRAF.14 PTPN11 and CBL mutations have a higher rate of bleeding diathesis and JMML. Patients with SOS1 mutations have a lower rate of intellectual disability. SHOC2 mutations have been associated with a Noonan phenotype with loose anagen hair.15 PTPN11 mutations are also the genetic basis of Noonan with multiple lentigines (formerly known as LEOPARD syndrome;16 see Chapter 24).
Differential diagnosis
In the neonatal period, the RASopathies can be very difficult to distinguish as they share the features of congenital heart defects, severe feeding difficulties and developmental delay.17 Later in infancy, facial and ectodermal features become more distinct for each of the RASopathies. Cardiofaciocutaneous syndrome and Costello syndrome are described in more detail below.
Treatment and care
Patients with Noonan syndrome should be monitored for growth deficiency and developmental delay. Electrocardiogram and echocardiography should be performed at the time of diagnosis and repeated as indicated based on findings. A renal ultrasound is recommended at diagnosis. Coagulopathy work-up should be performed if the patient has a history of either easy bruising or prolonged bleeding.14
Cardiofaciocutaneous syndrome
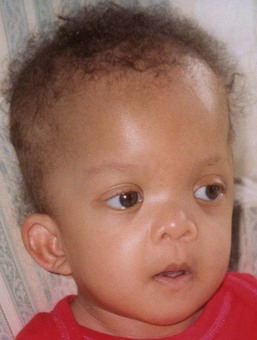
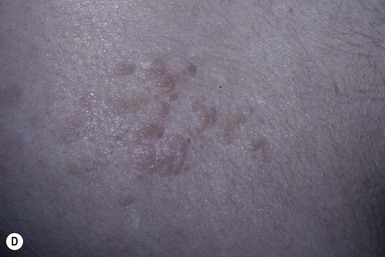
Cardiofaciocutaneous (CFC) syndrome (MIM #115150) is characterized by short stature, congenital heart defects, intellectual disability, ectodermal abnormalities and a characteristic coarse facial appearance (Table 29.2). Numerous cutaneous findings have been reported in CFC. In 2010, Siegel and colleagues18 evaluated the cutaneous manifestations in 61 mutation-positive individuals with CFC syndrome. All had dermatologic findings. One of the striking features identified in this study was a high number of melanocytic nevi. In the study, 23% of participants had over 50 nevi and 36% of those patients reported over 100 nevi. The amount of nevi increased with age and were not a prominent feature in infancy. Keratosis pilaris and ulerythema ophryogenes were very common and affected the majority of individuals in childhood and adolescence (Fig. 29.3). Infantile hemangiomas occurred at a greater frequency when compared with the general population. Additional features in CFC include macrocephaly, characteristic facial appearance, growth retardation, cardiac defects, neurologic impairment, gastrointestinal dysfunction, and ocular abnormalities.
TABLE29.2
Dermatologic features found in cardiofaciocutaneous syndrome compared with Costello syndrome
| Percentage of cases | ||
| Cardiofaciocutaneous | Costello | |
| Curly hair | 93.4% | 95.7% |
| Eyebrow density | 90.0% sparse | 47.8% thick |
| Keratosis pilaris | 80.3% | 32.6% |
| Infantile hemangioma | 26.2% | 10.9% |
| Papillomas | 4.9% | 71.7% |
| More than 50 nevi | 23.0% | 4.0% |
| Palmoplantar keratoderma | 36.1% | 76.1% |
(Adapted from Siegel DH, Mann JA, Krol AL, Rauen KA. Dermatological phenotype in Costello syndrome: consequences of Ras dysregulation in development. Br J Dermatol 2012; 166(3):601–607.)
CFC syndrome is caused by mutations in BRAF, MEK1 or MEK2.19,20 These mutations were discovered in part because of the similarity of the phenotypic features of Noonan and Costello syndromes, both of which were known to have mutations involving the RAS-MAPK pathway. Given insights into genetics and pathogenesis, it is not surprising that the main differential diagnoses for CFC syndrome include Noonan and Costello syndromes.
Costello syndrome
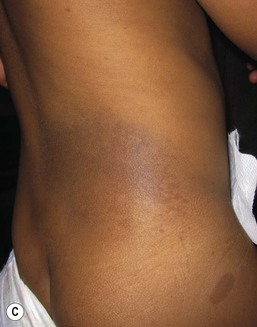
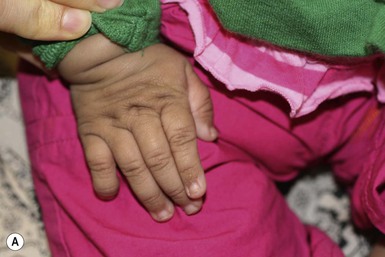
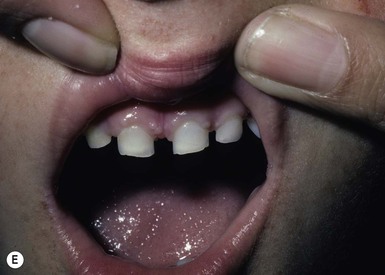
Costello syndrome (CS) (MIM #218040) is a rare, autosomal dominant, multiple congenital anomaly syndrome associated with failure to thrive, developmental delay, and an increased risk of malignancy.21 The features of Costello syndrome in the neonatal and infantile period include macrosomia, severe feeding problems, developmental delay, coarse facial features, gingival hyperplasia, osteopenia, hypertrophic cardiomyopathy and atrial arrhythmias.22 The most common malignancies include rhabdomyosarcoma, neuroblastoma and transitional cell cancer of the bladder. CS is caused by mutations in the HRAS gene, at 11p15.5, leading to constitutive activation of the RAS-MAPK pathway.21 The cutaneous findings include curly hair, papillomas on the perinasal, perioral and perianal skin, palmoplantar keratoderma, unusual body odor and heat intolerance. The skin on hands is loose and redundant (Fig. 29.4A). Acanthosis nigricans has been reported in 37% of the cases (Fig. 29.4B).23 In the neonatal period, it can be difficult to distinguish Costello, CFC and Noonan syndromes, as all three conditions can manifest with neonatal macrosomia, coarse facial features, failure to thrive and developmental delay.17
Capillary malformation-arteriovenous malformation (CM-AVM) (MIM #608354)
CM-AVM is an autosomal dominant disorder characterized by multiple, small, oval capillary malformations on the face, body and limbs, which are associated with arteriovenous malformations in about 30% of cases. The AVMs can occur in the brain, spine, muscle or skin. CM-AVM is caused by mutations in the RASA1 gene.24 See Chapter 22 for a more in-depth discussion about this condition.
Neurofibromatosis type 2
Neurofibromatosis type 2 (NF-2) (MIM #101000) is an autosomal dominant condition characterized by a high burden of schwannoma and meningioma tumor development.
Presentation in infancy is rare, although cutaneous schwannomas have been reported.25 Cutaneous features which may be present in infancy or early childhood include CALM and cutaneous schwannomas (Fig. 29.5). The age range at the onset of NF-2-related symptoms is 5–55 years. The initial symptoms that lead to the diagnosis include hearing loss, visual disturbance, and enlarging subcutaneous mass. The incidence of NF-2 is difficult to predict due to a high rate of mosaicism, but is estimated at about 1 : 25 000.26 NF-2 is caused by mutations in the merlin (also called schwannomin) tumor suppressor gene on chromosome 22q12.27 The differential diagnosis for NF-2 includes NF-1 and schwannomatosis.
Disorders of the PI3K-AKT/mTOR pathway and overgrowth syndromes
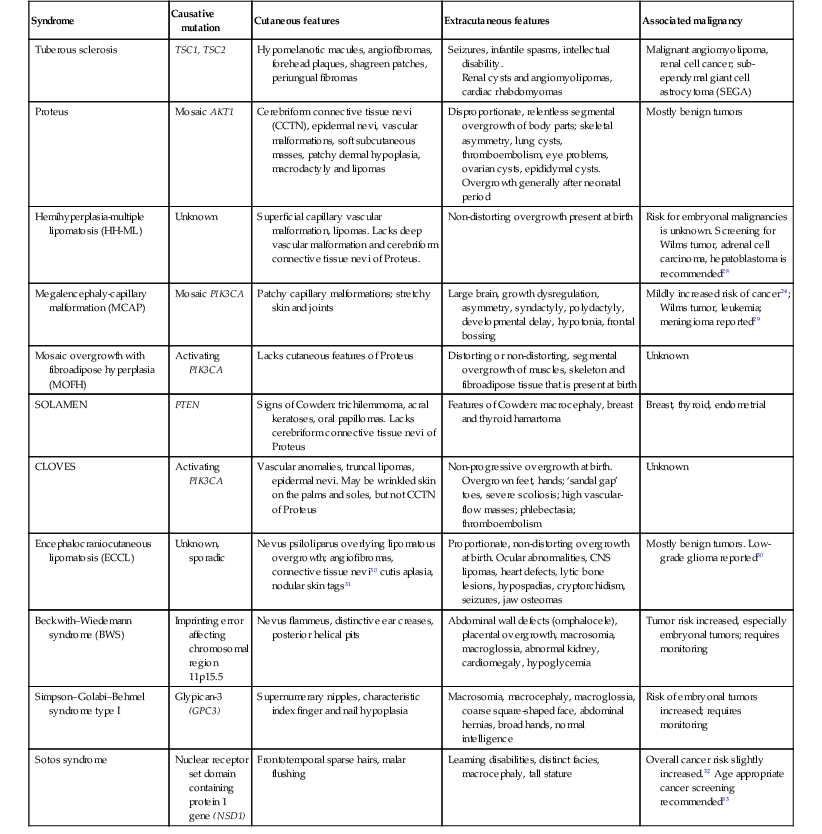
Overgrowth syndromes and human cancer share disruption of similar critical regulatory pathways (Table 29.3). Intensive cancer research, therefore, has improved our understanding of these rare but important disorders. The phosphatidylinositol 3-kinase (PI3K)-AKT pathway, in particular, critically guides cell growth and metabolism (Fig. 29.6). The PI3K enzymes are a family of highly conserved enzymes that regulate cell growth, migration and survival and disruption in the embryonic period typically impacts vascular, limb or brain development.29 Mutations cause recognizable patterns of increased cell number, hypertrophy, increased interstitium, or a combination of these.34 Typical neonatal clues of an overgrowth syndrome are abnormally increased body length, macrosomia, and macrocephaly, dysregulated growth of a body part or asymmetry (Fig. 29.7). Almost all overgrowth syndromes are associated with neoplasms, especially solid tumors.
TABLE 29.3
Overgrowth syndromes and associated clinical features
| Syndrome | Causative mutation | Cutaneous features | Extracutaneous features | Associated malignancy |
| Tuberous sclerosis | TSC1, TSC2 | Hypomelanotic macules, angiofibromas, forehead plaques, shagreen patches, periungual fibromas | Seizures, infantile spasms, intellectual disability. Renal cysts and angiomyolipomas, cardiac rhabdomyomas |
Malignant angiomyolipoma, renal cell cancer; sub-ependymal giant cell astrocytoma (SEGA) |
| Proteus | Mosaic AKT1 | Cerebriform connective tissue nevi (CCTN), epidermal nevi, vascular malformations, soft subcutaneous masses, patchy dermal hypoplasia, macrodactyly and lipomas | Disproportionate, relentless segmental overgrowth of body parts; skeletal asymmetry, lung cysts, thromboembolism, eye problems, ovarian cysts, epididymal cysts. Overgrowth generally after neonatal period |
Mostly benign tumors |
| Hemihyperplasia-multiple lipomatosis (HH-ML) | Unknown | Superficial capillary vascular malformation, lipomas. Lacks deep vascular malformation and cerebriform connective tissue nevi of Proteus. | Non-distorting overgrowth present at birth | Risk for embryonal malignancies is unknown. Screening for Wilms tumor, adrenal cell carcinoma, hepatoblastoma is recommended28 |
| Megalencephaly-capillary malformation (MCAP) | Mosaic PIK3CA | Patchy capillary malformations; stretchy skin and joints | Large brain, growth dysregulation, asymmetry, syndactyly, polydactyly, developmental delay, hypotonia, frontal bossing | Mildly increased risk of cancer24; Wilms tumor, leukemia; meningioma reported29 |
| Mosaic overgrowth with fibroadipose hyperplasia (MOFH) | Activating PIK3CA | Lacks cutaneous features of Proteus | Distorting or non-distorting, segmental overgrowth of muscles, skeleton and fibroadipose tissue that is present at birth | Unknown |
| SOLAMEN | PTEN | Signs of Cowden: trichilemmoma, acral keratoses, oral papillomas. Lacks cerebriform connective tissue nevi of Proteus | Features of Cowden: macrocephaly, breast and thyroid hamartoma | Breast, thyroid, endometrial |
| CLOVES | Activating PIK3CA | Vascular anomalies, truncal lipomas, epidermal nevi. May be wrinkled skin on the palms and soles, but not CCTN of Proteus | Non-progressive overgrowth at birth. Overgrown feet, hands; ‘sandal gap’ toes, severe scoliosis; high vascular-flow masses; phlebectasia; thromboembolism |
Unknown |
| Encephalocraniocutaneous lipomatosis (ECCL) | Unknown, sporadic | Nevus psiloliparus overlying lipomatous overgrowth; angiofibromas, connective tissue nevi30 cutis aplasia, nodular skin tags31 | Proportionate, non-distorting overgrowth at birth. Ocular abnormalities, CNS lipomas, heart defects, lytic bone lesions, hypospadias, cryptorchidism, seizures, jaw osteomas | Mostly benign tumors. Low-grade glioma reported30 |
| Beckwith–Wiedemann syndrome (BWS) | Imprinting error affecting chromosomal region 11p15.5 | Nevus flammeus, distinctive ear creases, posterior helical pits | Abdominal wall defects (omphalocele), placental overgrowth, macrosomia, macroglossia, abnormal kidney, cardiomegaly, hypoglycemia | Tumor risk increased, especially embryonal tumors; requires monitoring |
| Simpson–Golabi–Behmel syndrome type I | Glypican-3 (GPC3) | Supernumerary nipples, characteristic index finger and nail hypoplasia | Macrosomia, macrocephaly, macroglossia, coarse square-shaped face, abdominal hernias, broad hands, normal intelligence | Risk of embryonal tumors increased; requires monitoring |
| Sotos syndrome | Nuclear receptor set domain containing protein 1 gene (NSD1) | Frontotemporal sparse hairs, malar flushing | Learning disabilities, distinct facies, macrocephaly, tall stature | Overall cancer risk slightly increased.32 Age appropriate cancer screening recommended33 |
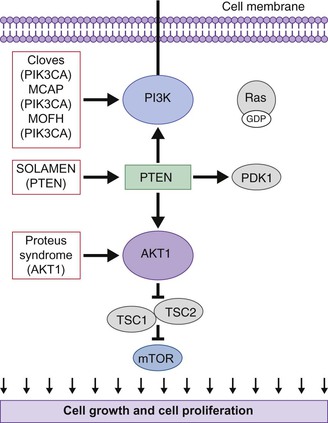
PI3K-AKT activity is considered so essential that disruptive germline mutations are often presumed fatal. For example, an activating PI3K pathway mutation appears only in the mosaic form. In addition to impacting embryonic development, the PI3K-AKT pathway participates in normal cellular function by regulating glucose metabolism and apoptosis.35 Therefore, mutations affecting this pathway may cause congenital anomalies as well as ongoing complications. Improved understanding of the PI3K pathway, especially by revealing therapeutic targets for inhibitory drugs like rapamycin, gives hope for future trials and treatment.36
Tuberous sclerosis
Tuberous sclerosis (TSC, MIM #191100) is a multisystem disorder characterized by tumors and hamartomas affecting the skin, brain, heart, kidneys and lungs, often in association with seizures and developmental delay (Box 29.5). TSC is discussed in further detail in Chapter 23.
Cutaneous findings
There is a very high prevalence of cutaneous findings in TSC.37 Hypomelanotic macules are usually present at birth or appear within the first few months of life and ultimately are present in approximately 90% of TSC patients. Classically, they present as an ‘ash leaf macule,’ an oval area with reduction in pigment (Fig. 29.8A). These areas can also be irregular in outline and shape, or very small and guttate (confetti-like) (Fig. 29.8B). In fair-skinned infants, a Wood’s lamp examination may be necessary for detection. They can also occur on the scalp, with lightening of the hair within the patch. A single, hypopigmented macule in an infant, without other features of TSC, should not cause concern,38 but multiple lesions (≥3) should lead to further investigation.

Classic facial angiofibromas typically appear after 4 years of age, however fibrous plaques resembling coalescence of angiofibromas may be present over the scalp, face, or neck at birth or appear shortly thereafter, as firm slightly raised patches or plaques that are commonly erythematous (Fig. 29.8C). Shagreen patches – firm, palpable thickened dermal plaques – can occur in the lumbar region as a roughened area of erythematous skin with a rubbery consistency or develop in other areas of the torso (Fig. 29.8D). They range in size from a few millimeters to 15 cm in diameter, and generally appear by adolescence but are infrequent in young infants. Periungual fibromas are similarly uncommon in the first decade. Gingival fibromas are a less common feature, but occasionally develop in young children (Fig. 29.8E).
Extracutaneous findings
Seizures occur in more than 60–80% of patients with TSC.39 Conversely 4–50% of infants with infantile spasms have TSC.39,40 TSC patients with early onset of seizures (<2 years of age) or infantile spasms have an elevated risk for intellectual disability. The most common lesions in the brain include tubers, subependymal nodules and subependymal giant-call astrocytomas (SEGA). Renal cysts, angiomyolipomas, and cardiac rhabdomyomas are findings in newborns and infants that suggest TSC. Cardiac rhabdomyomas are often discovered on routine antenatal ultrasound; 30–50% of infants with TSC have cardiac rhabdomyomas, and 80–90% of infants with these lesions have TSC.41 They are rarely symptomatic and typically regress spontaneously.42 An expert panel at the Tuberous Sclerosis Consensus Conference4 recommended a baseline electrocardiogram both at the time of diagnosis and prior to surgery, as cardiac rhabdomyomas can be associated with pre-excitation and arrhythmias on the electrocardiogram.43 Angiomyolipomas are the most common renal manifestation of TSC.
Etiology and pathogenesis
TSC may be caused by an autosomal dominant mutation in the TSC1 gene (encoding hamartin) or the TSC2 gene (encoding tuberin). The two proteins form a complex that is involved with the phosphoinositide-3-kinase (PI3K) signaling pathway, which regulates cell growth and proliferation (Fig. 29.6).44
Differential diagnosis
The differential diagnosis for the hypopigmented macules seen in the neonatal period includes vitiligo, nevus depigmentosus, nevus anemicus, and piebaldism. A single lesion, or several lesions occurring along the lines of Blaschko suggests the diagnosis of nevoid hypopigmentation rather than TSC (see Chapter 23). Connective tissue nevi may be sporadic and have also been associated with Buschke–Ollendorff or Proteus syndrome. Angiofibromas have been described in MEN145 and Birt–Hogg–Dubé syndrome, and also as an isolated autosomal dominant disorder.
Treatment and care
Consensus recommendations for screening and evaluation of TSC were proposed in 1998 and updated in 2000 (Box 29.6).43,46 The Scottish Clinical Genetics Service and the UK Tuberous Sclerosis Association have also created clinical guidelines. Neurology and/or neurosurgery should be consulted for management of seizures, brain tumors, and shunting of obstructive hydrocephalus. Ophthalmology should also examine patients to assist in confirming the diagnosis. Renal ultrasound or renal MRI are important to screen angiomyolipomas. This may also help to identify patients with coexisting polycystic kidney disease due to a contiguous gene deletion syndrome involving the TSC2 and PKD1 genes. Echocardiography and electrocardiogram are recommended at diagnosis for confirmation (to detect cardiac rhabdomyomas and arrhythmias) and to screen for aortic aneurysms.47
Pulsed dye laser has been recommended for flat erythematous angiofibromas, and both Potassium titanyl phosphate (KTP) laser and carbon dioxide laser are used to treat more elevated lesions, but are rarely indicated in infants.48 Fibrous forehead plaques are generally left untreated, but may also be treated with lasers or surgery. Several case reports have recently described the utility of topical rapamycin for the treatment of angiofibromas. This approach holds promise, but further study is needed to establish the safety, efficacy and dosing guidelines.49,50
Proteus syndrome
Proteus syndrome (MIM #176920) is a very rare condition characterized by dramatic segmental or mosaic overgrowth. Common complications include skeletal asymmetry, characteristic overgrowth of the palms or soles referred to as ‘cerebriform connective tissue nevi’ (CCTN),51 linear epidermal nevi, deep or superficial vascular malformations, dysregulated adipose tissue (formerly referred to as lipomas) and tumor predisposition (see also Chapter 22).
Cutaneous findings
Almost all individuals with Proteus syndrome have a dermatologic manifestation. Over 40% of affected neonates will demonstrate at least some evidence of the disease at birth, such as epidermal nevi or vascular malformations.52 The three main cutaneous findings are epidermal nevi, vascular malformations, and soft subcutaneous masses. Epidermal nevi are usually linear and verrucous, but may be macular and hyper- or hypopigmented. Malformations may be venous, capillary, and/or lymphatic.53 The cerebriform connective tissue nevus, when on the sole of the foot, is caused by hyperplasia of cutaneous and subcutaneous tissues and considered virtually pathognomonic.54 The tissue is very firm; similar lesions may also occur on the hands, perinasal area, or near the canthus. Prominent cutaneous venous structures may occur as a result of patchy dermal hypoplasia. Macrodactyly and adipose overgrowth may also be observed.
Extracutaneous findings
Overgrowth in Proteus syndrome is disproportionate, asymmetric, progressive, distorting, and persistent (see Fig. 29.7). Overgrowth usually presents between 6 and 18 months of age and can occur in areas that were completely normal at birth. Overgrowth affects most tissues including bones, cartilage, muscle, and connective tissues. At least some overgrowth and asymmetry is present at birth in 17.5% of cases.52 Orifices may be affected, causing respiratory obstruction, conductive hearing loss, or gastric outlet obstruction. Cystic degeneration of the lungs may lead to pneumonia.55 Affected individuals are predisposed to deadly deep venous thrombosis and pulmonary embolism.56 The central nervous system is commonly affected by hemimegalencephaly (unilateral enlargement of the brain), but most patients are asymptomatic.57 Eye complications are common and range from strabismus to epibulbar hamartomas. Ovarian cystadenomas and cystic lesions of the epididymis are also common.
Etiology and pathogenesis
The cause of Proteus syndrome is a de novo post-zygotic activating mutation in the AKT1 oncogene. Proteus features are an example of somatic mosaicism that is lethal in the non-mosaic state.51 Because Proteus is a mosaic disorder, biopsy of affected tissue is required to make a genetic diagnosis.
Differential diagnosis
Proteus syndrome has been overdiagnosed,58 prompting the creation of diagnostic criteria (Box 29.7) to separate Proteus syndrome from other overgrowth syndromes, many of which share asymmetric hypertrophy as a feature but almost always to a less severe degree.52 Proteus stands apart by having fairly rapid postnatal progression, relentless deforming overgrowth and a poor prognosis.58
Hemihyperplasia-multiple lipomatosis (HH-ML).
This is most commonly misdiagnosed as Proteus syndrome. HH-ML includes superficial capillary vascular malformation (similar to port-wine stain), but lacks progressive, distorting overgrowth, deep vascular malformations and cerebriform connective tissue nevi on the palms or soles.59 In non-distorting overgrowth that characterizes HH-ML, a bone is normally shaped and larger than expected, but without growths or odd edges. In HH-ML, non-distorting overgrowth is present at birth,59 whereas, the distinctive distorting overgrowth that characterizes Proteus may not be apparent until age 2 or 3.52 Lipomas may recur after surgical removal. Therefore, removal of symptomatic lipomas only is recommended.
Megalencephaly-capillary malformation (MCAP).
Sometimes also referred to as M-CM, it has previously been termed macrocephaly-capillary malformation and macrocephaly-cutis marmorata telangiectatica congenital. MCAP sometimes includes markedly large brain size with variable cortical malformation, growth dysregulation with variable asymmetry, patchy capillary malformations frequently on the philtrum, upper lip and nose as well as the limbs and trunk, and distal limb abnormalities such as syndactyly and polydactyly, and mild connective tissue dysplasia (hyperextensible joints and skin).29,60 Congenital Chiari I malformation had been associated with MCAP, however, the term ‘cerebellar tonsil herniation’ (CTH) is now preferred to describe acquired herniation caused by cerebellar overgrowth that occurs in up to 70% of MCAP cases.61 With capillary vascular malformation and asymmetric overgrowth, children often meet the criteria for Klippel–Trenaunay syndrome. Affected children typically have developmental delay (85%) and neonatal hypotonia (68%). Frontal bossing and prominent nevus simplex are additional facial features.60 There is also a mildly increased risk of cancer (about 3%).29 MCAP has been associated with postzygotic mutations in the PIK3CA gene.29
Mosaic overgrowth with fibroadipose hyperplasia (MOFH).
This is an example of an overgrowth syndrome defined by its newly identified causative mutation, an activating mutation in the PIK3CA gene.62 Affected children have segmental overgrowth that affects the muscles, skeleton, and fibroadipose tissue without cutaneous features of Proteus syndrome. Both distorting and non-distorting overgrowth are reported. The features of MOFH often begin at birth, in contrast to Proteus syndrome which generally appears later.62
SOLAMEN syndrome.
Segmental overgrowth, lipomatosis, arteriovenous malformation and epidermal nevus (SOLAMEN) is a ‘Proteus-like’ syndrome,63 now known to be a mosaic form of Cowden disease. Affected individuals carry a germline PTEN mutation and a mosaic second hit to the PTEN gene on the opposite allele in affected tissues.63 Absence of the cerebriform connective tissue nevi on the palms and soles distinguishes SOLAMEN syndrome. In addition, characteristic features of Cowden disease such as macrocephaly, breast and thyroid hamartomas and skin changes are likely to be present.63
CLOVES syndrome (MIM #612918).
Congenital lipomatosis, overgrowth, vascular malformations, epidermal nevi and skeletal anomalies (CLOVES) describes another subset of patients, most of whom were initially misdiagnosed with Proteus syndrome.58 In CLOVES syndrome, overgrowth is present at birth with truncal vascular anomalies, truncal lipomatous masses and overgrown feet with acral or musculoskeletal anomalies. In contrast to Proteus, the overgrowth is not progressive.64 Patients may have wrinkled skin on the palms and soles, but lack the firm rubbery CCTN of Proteus syndrome. Since CLOVES syndrome was suspected to be a mosaic disorder at the outset, massive DNA sequencing of affected tissues revealed causative activating mutations in PIK3CA.65 Severe scoliosis, large truncal masses, high flow vascular lesions, phlebectasia with thromboembolism and orthopedic problems of the hands and feed may all require early, aggressive intervention in affected individuals.64 Prognosis seems better compared to Proteus syndrome but this condition can still be very severe, even life-threatening.66
Encephalocraniocutaneous lipomatosis (ECCL).
The so-called ‘nevus psiloliparus’ is the cutaneous hallmark, with a localized non-scarring patch of alopecia on the scalp which may or may not overlie a fatty tissue mass. ECCL, while once thought to be a localized form of Proteus syndrome, does not meet the diagnostic criteria of Proteus, lacking disproportionate growth31 as a salient feature (see also Chapter 31).
Differential diagnosis of Proteus and similar disorders
Maffucci syndrome consists of multiple enchondromatosis (which may be confused with hyperostosis) and vascular malformations.67 Klippel–Trenaunay (MIM #149000) syndrome consists of vascular malformations with overgrowth, typically in the same segment. Axillary freckling and neurofibromas help distinguish neurofibromatosis.
Management
Management of these syndromes depends on the specific disease; if overgrowth is severe – as in Proteus syndrome – management is often very difficult. A multidisciplinary approach best addresses all aspects of the disease adequately. Ongoing ophthalmology, neurology, and developmental assessment exams are recommended.52 Caregivers should be informed of the risk of pulmonary embolism and stroke so that healthcare providers consider it if an emergency arises.52 Orthopedic surgery should be consulted before functional deficits appear and before the patient becomes too debilitated to undergo surgery. Cancer surveillance is an important part of management as dictated by clinical symptoms; routine imaging is not recommended.68
Isolated hemihyperplasia places individuals at increased risk (5.9%) for embryonal malignancies such as Wilms tumor, adrenal cell carcinoma and hepatoblastoma.28 A study of 260 children with hemihyperplasia69 reported that the risk in truly isolated idiopathic hemihyperplasia is lower (1.2%) and in syndrome-related hemihyperplasia higher (10%) than previously reported. The risk of malignancy in specific syndromes with hemihyperplasia, including HH-ML and excluding Klippel–Trenaunay, is not clear. Until further information becomes available, screening with abdominal ultrasound every 3–6 months until age 8, checking of blood alpha fetoprotein level every 3 months until age 4 and referral to a geneticist for identifying associated syndromes is recommended.28 Individuals with SOLAMEN syndrome have a risk of malignancies associated with Cowden (PTEN hamartoma) syndrome.
Additional overgrowth syndromes
Of the generalized overgrowth syndromes, Beckwith–Wiedemann Syndrome (BWS) (MIM #130650) is the most common.70 Cutaneous features include prominent nevus simplex, distinctive anterior ear creases, and posterior helical pits. Other characteristic features are abdominal wall defects (omphalocele), placental overgrowth, macrosomia, macroglossia, kidney abnormalities, cardiomegaly, and hypoglycemia. As the phenotype is variable, mild cases are likely to be missed.70 Mosaicism may lead to asymmetric hypertrophy of a limb or body part. BWS patients have an increased risk of Wilms tumor and hepatoblastoma.
Beckwith–Wiedemann is a disorder of imprinting, an epigenetic process using methylation to silence either the maternal or paternal allele in a gene pair. Imprinting affects only specific regions of the genome, especially areas with genes impacting fetal growth.71 Beckwith–Wiedemann syndrome affects chromosomal region 11p15.5. This region contains an imprinting center that regulates several adjacent genes. Since 15% of cases are inherited, siblings are considered at risk for the disorder and require screening for complications. Monitoring for neonatal hypoglycemia, for example, may prevent neurologic injury. Affected children may benefit from tongue reduction surgery and speech therapy. Surveillance for abdominal embryonal tumor (described above) with ultrasound and blood testing and ongoing screening for renal abnormalities is recommended.
Simpson–golabi–behmel syndrome type I
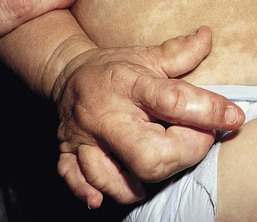
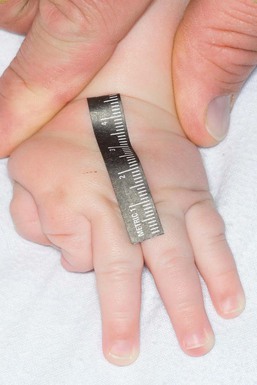
Simpson–Golabi–Behmel syndrome type I is an overgrowth syndrome with cutaneous features including supernumerary nipples72 and dysplastic fingernails. The index fingers are affected with hypoplasia and nail abnormality and this characteristic feature may help in early diagnosis (Fig. 29.9).72 Extracutaneous features are macrosomia, macrocephaly, macroglossia, coarse square-shaped face, abdominal hernias, broad hands, and normal intelligence. The X-linked recessive disorder is73 caused by a mutation in glypican-3 (GPC3) gene. Risk of embryonal tumors is increased and requires monitoring.
Sotos syndrome
This is an overgrowth syndrome with cutaneous features including frontotemporal sparse hairs and malar flushing.74 Other identifying features are learning disabilities, distinct facial appearance, and macrocephaly along with tall stature. The inheritance pattern is autosomal dominant caused by a mutation in the nuclear receptor set domain containing protein 1 gene (NSD1).74
Ectodermal dysplasias
There are over 150 rare syndromes whose primary features involve alterations in two or more of the structures that derive from the embryonic ectoderm, which include developmental defects in hair, teeth, nails, sweat glands, and the lens of the eye. These are referred to as ‘ectodermal dysplasias’ (ED). The National Foundation for Ectodermal Dysplasia is an excellent resource for families and provides information to professionals caring for children with a wide variety of ED types (www.nfed.org).
Hypohidrotic ectodermal dysplasia
Hypohidrotric ectodermal dysplasia (HED, Christ–Siemens–Touraine syndrome (MIM #305100))75 is most often an X-linked recessive condition and is the most common form of ED encountered by clinicians.
Cutaneous findings
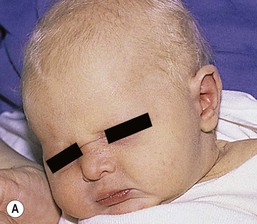
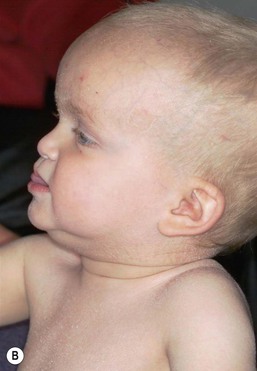
Patients with hypohidrotic ectodermal dysplasia often present at birth with a collodion membrane or marked scaling of the skin, which may be misconstrued as a marker of congenital ichthyosis or of postmaturity.76 After membrane shedding, the skin is soft, thin, and light-colored with fine periorbital wrinkling (Fig. 29.10A,B). Even in the neonatal period, the hair tends to be absent or sparse, short, and blond. The nails are usually normal. Patients have an increased frequency of atopic dermatitis, particularly periorbital dermatitis.
In cases without a positive family history, repeated bouts of unexplained fevers most often bring infants with HED to medical attention. As the infants produce little to no sweat, they cannot make the appropriate physiologic response to increased environmental temperature, resulting in core temperature elevation. Diminished or absent sweat pores may be appreciated both clinically and histologically.
Extracutaneous findings
Most infants with HED have a typical facies, easily recognizable to educated family members and/or physicians, characterized by a square forehead with frontal bossing, a flattened nasal bridge with prominent nostrils, wide cheekbones with flat malar ridges, a relatively thick, everted lower lip, prominent chin, and small, pointed, low-lying ears. Lacrimal and mucous glands are hypoplastic, leading to reduced tearing, chronic thick nasal discharge, and impacted cerumen with an increased frequency of otitis media and respiratory tract infections. As the teeth are not present in the neonatal period, the typical peg-shaped or missing teeth cannot be used to aid in diagnosis, but dental X-rays can demonstrate these findings, even in young infants.
Etiology and pathogenesis
Most patients are male and carry the X-linked form; however, mutations in several individual genes with autosomal dominant and recessive inheritance patterns may lead to identical developmental abnormalities of the hair and glands. All of the involved genes are within the ectodysplasin signaling pathway (Fig. 29.11) and include ectodysplasin-A1 (EDA-A1), Eda-A1 receptor (EDAR), and EDAR associated death domain (EDARADD).77,78 Female carriers of ectodysplasin mutations show varied manifestations including more subtle clinical findings overall, or distinct manifestations such as hypohidrosis following the lines of Blaschko.
Differential diagnosis
Hypohidrosis may be found in another group of ectodermal dysplasias resulting from autosomal dominant mutations in p63 (Rapp–Hodgkin syndrome, AEC syndrome, EEC syndrome, limb–mammary syndrome, or ADULT syndrome), but that group has associated facial clefting and split-hand/foot malformations.79,80
Treatment and care
Temperature must be carefully regulated with cool baths, air conditioning, light clothing, spray-mist bottles to dampen clothing during activities, and avoidance of warm environments (Box 29.8).81 Some families find commercially available cooling suits to be helpful. Reduced glandular secretion may be treated with lubricating eye drops and nasal irrigation. Treatment of recurrent otitis media, respiratory infections, atopic dermatitis, and asthma should be individualized. Dental manifestations should be managed early.
Hypohidrotic ectodermal dysplasia with immunodeficiency
Cutaneous findings
Patients with hypohidrotic ectodermal dysplasia with immunodeficiency (HED-ID) have similar findings to patients with HED although they may be less clinically obvious. They have hypohidrosis along with conical and peg-shaped teeth. They also have pale and dry or mildly scaly skin with periorbital darkening, although typically less noticeable than in patients with HED. They may also have extensive seborrheic dermatitis and intertrigo.
Extracutaneous findings
The extracutaneous findings are what help separate this disorder from HED. Patients are prone to severe and sometimes fatal infections. This can include infection with either Gram-positive or Gram-negative bacteria leading to skin and soft tissue abscesses, acute otitis media, pneumonia, osteomyelitis, gastrointestinal infections and sepsis.
Etiology and pathogenesis/management
HED-ID (MIM #300291) is most often inherited in an X-linked manner and is due to mutations in IKBKG (inhibitor of kappa light polypeptide gene enhancer in B-cells, kinase gamma).82 Autosomal dominant forms also exist and are due to mutations in IκBα. Both mutations result in impaired NF-κB signaling (Fig. 29.11).83 Somatic mosaic mutations in NEMO can also occur which leads to a milder phenotype.83,84
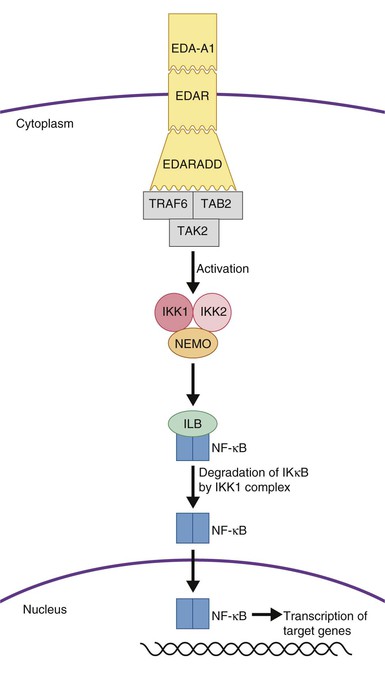
Defective NF-κB signaling is responsible for both defective ectodermal appendage development as well as the immunodeficiency in these patients. Antibody response is poor, natural killer cells are ineffective and T cells lack development and maturation which accounts for the wide variety of infectious diseases patients with HED-ID encounter.
The ED components are managed similar to those in HED; manifestations of the immunodeficiency require a multidisciplinary approach with immunologists and infectious disease specialists.
Incontinentia pigmenti
Cutaneous findings
Incontinentia pigmenti (IP, MIM #308300)85 is an X-linked multisystem disorder with characteristic cutaneous manifestations. Classically, the skin changes occur in four stages: vesicular, verrucous, hyperpigmented, and atrophic. A patient may not develop all stages and several stages may overlap.
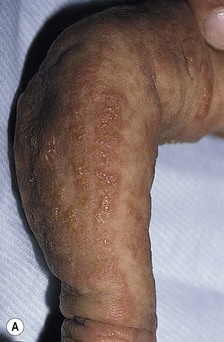
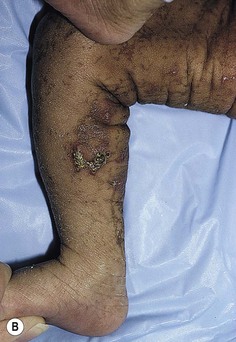
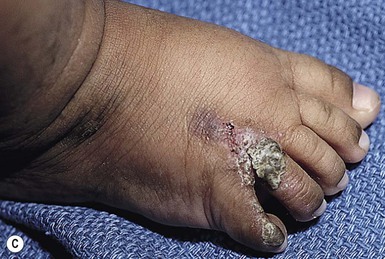
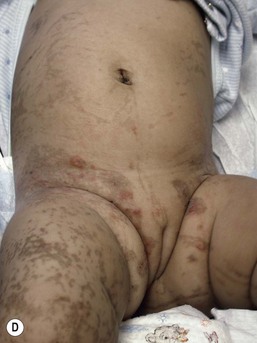
In the newborn period, affected infants develop small, clustered blisters on an erythematous base, scattered along the lines of Blaschko (Fig. 29.12A). This stage usually resolves by 4–6 months of age, but milder, short-lived eruptions may continue during the first year of life or longer, sometimes associated with an acute febrile illness. The second phase occurs as warty, hyperkeratotic, linear lesions (Fig. 29.12B,C), typically resolving by 6 months.85 The presence and extent of the third, hyperpigmented, stage is highly variable (Fig. 29.12D) and is often unrelated to the distribution of the previous stages. By the age of 16 months, most pigmented lesions will have faded.86 The last hypopigmented/atrophic stage becomes apparent with resolution of the lesions from the first three stages and demonstrates loss of hair and sweat glands. Alopecia and nail dystrophy are common.
The histopathology of IP is stage specific. Blisters show intercellular edema and intraepidermal vesicles filled with eosinophils, along with dyskeratotic keratinocytes. Patients may have peripheral eosinophilia or leukocytosis as well. Warty lesions show hyperkeratosis, papillomatosis, and mild dyskeratosis. In areas of hyperpigmentation, pigment-laden melanophages are evident in the dermis and focal dyskeratosis.
Extracutaneous findings
The most characteristic ocular finding is retinal vascular proliferation,87 which can result in bleeding, fibrosis, retinal detachment,88 and in 10% of patients, enough scarring to produce permanent blindness.86 All neonates with IP need prompt and periodic evaluation by an experienced ophthalmologist. The magnitude of risk of central nervous system abnormalities is controversial but probably lower than previously believed, with current estimates varying from 10% to 30%.85,89 Most neurologic features present in the neonatal period and include seizures, encephalopathy, encephalitis and ischemic stroke.90
Etiology and pathogenesis
IP results from mutations in the X chromosomal gene IKBKG (previously known as NEMO, NF-κB essential modulator),91 which is involved in immune, inflammatory, and apoptotic pathways, as discussed above.91 The mutation is believed to be lethal in affected 46,XY males, but male cases have been reported in the setting of XXY genotype, and other cases in males are presumably due to somatic mosaicism or half-chromatid mutations.
Differential diagnosis
In the newborn, IP must be differentiated from other causes of blistering, including infections (bacteria and herpes simplex virus), erythema toxicum, and epidermolysis bullosa (see Chapters 10 and 11). The warty phase of IP is unique, but may be confused with a linear epidermal nevus. Linear and whorled nevoid hypermelanosis may appear identical to stage 3 of IP. Although history helps to distinguish between the two conditions, a biopsy may be necessary, as stage 3 IP has occurred as late as 15 months of age without any antecedent skin changes.
Treatment and care
The skin changes of IP do not require any treatment other than wound care for blisters to prevent secondary infection. A baseline eye examination and close follow up by an ophthalmologist and a full neurological assessment with anticipatory evaluation for the possibility of neurologic deficits are appropriate. Dental evaluation should be considered after teeth erupt.
Hidrotic ectodermal dysplasia
Also known as Clouston syndrome, hidrotic ectodermal dysplasia (MIM #129500) is an autosomal dominant form of hidrotic or ‘sweating’ ectodermal dysplasia characterized by nail dystrophy, hyperkeratosis of the palms and soles, and hair defects.92,93 Affected newborns may have milky-white-appearing nails and dry skin, or may display no clinical signs. Chronic paronychial infections frequently develop. Hair may be normal during infancy and childhood, and the diagnosis may not be recognized until abnormal sparse, fine, and brittle hair is detected or progressive nail dystrophy develops. The terminal phalanges may be tufted. Individuals show normal sweating, facies, and dentition.
Autosomal dominant mutations in connexin 30 (GJB6) cause the disorder.94
The diagnosis of hidrotic ectodermal dysplasia is unlikely to be made in the newborn period in the absence of a positive family history. Nail dystrophy may be the only manifestation in one-third of patients. Swollen, tufted terminal phalanges may aid in diagnosis. One should differentiate hidrotic ectodermal dysplasia from pachyonychia congenita and other palmoplantar keratodermas (see Chapters 19 and 32).
Keratolytics and surgical debridement may be effective for keratoderma. Paronychial infections can be treated with antibiotics or anti-candidal agents as appropriate.
Ectodermal dysplasias due to p63 mutations
Several forms of ectodermal dysplasia with overlapping clinical features are due to mutations in p63, a transcription factor that is structurally related to p53, localized to gene locus 3q27 (Fig. 29.13). These include: Rapp–Hodgkin syndrome; ectrodactyly, ectodermal dysplasia, and cleft lip/palate syndrome; ankyloblepharon–ectodermal defects–cleft lip/palate syndrome; acrodermato–ungual–lacrimal–tooth syndrome; limb–mammary syndrome; and nonsyndromic split-hand/foot malformation.95,96 p63 is involved in epidermal development, differentiation and homeostasis. By encoding two N-termini and multiple C-termini, many different isoforms can be produced, each of which has differing functions within keratinocytes.97,98 For example, the ΔNp63 isoform controls expression of basal layer keratins and then, upon receiving a differentiation stimulus, can change its transcriptional activity and activate genes required for cell cycle exit (IKKα).99
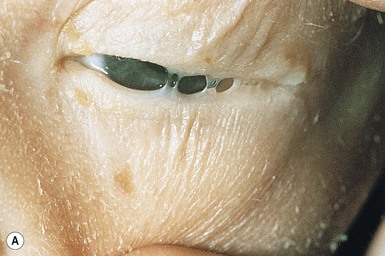
Ankyloblepharon–ectodermal dysplasia–clefting (AEC) syndrome
This syndrome (AEC, Hay–Wells syndrome, MIM #106260) is an uncommon form of ectodermal dysplasia with autosomal dominant inheritance.100
Cutaneous findings
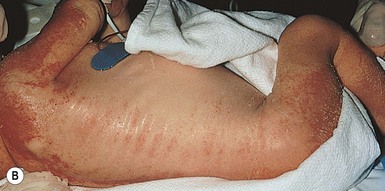
The upper and lower eyelids of the newborn have fine strands of skin between them (so-called ankyloblepharon), which are pieces of tissue that can be thick or thin, may tear spontaneously, or require surgical lysis (Fig. 29.14A). These are a cardinal feature of this condition, but are not mandatory for diagnosis. During the newborn period, the rest of the skin is erythematous and fissured with a collodion membrane appearance, resembling bullous congenital ichthyosiform erythroderma with peeling, erythema, and erosions (Fig. 29.14B) (see also Chapters 10 and 19). After the membrane has shed over the first few weeks, the underlying skin is dry and thin. Recurrent scalp infections, erosions, and granulation tissue may have their onset in early infancy and are present in two-thirds to three-quarters of older infants, children, and adults with this condition.101 The hair is sparse and coarse. Sweating is usually not significantly affected. Nail dystrophy is variable. The nails can be thickened and malformed, thin or absent.
Extracutaneous findings
Cleft palate, with or without cleft lip, is the third major sign of AEC syndrome, occurring in 80% of affected newborns. The reported hypodontia associated with the condition may reflect the degree of severity of the clefting, rather than a primary ectodermal defect.
A few males with AEC have had hypospadias. External ear malformations are described in some patients. Supernumerary nipples and ectopic breast tissue occur in a minority of cases. There may be tear duct abnormalities and recurrent lid inflammation.
Differential diagnosis
Other diagnoses to be considered when presented with an infant with cleft palate and a collodion membrane include EEC (ectodermal dysplasia, ectrodactyly, cleft lip/palate syndrome), distinguished by its limb involvement, and Rapp–Hodgkin syndrome. Not surprisingly, there is considerable overlap between Rapp–Hodgkin and AEC syndromes, as they are allelic. The presence of ankyloblepharon can distinguish between the two, being a key characteristic of AEC, but now that the genetics of the conditions are appreciated, a true distinction may not be absolutely necessary.
Treatment and care
Treatment is limited to surgical management of eyelid involvement and oral facial clefting. The use of light emollients may speed the shedding of dry, cracking neonatal skin. Careful handling of the scalp and prompt attention to secondary bacterial infection may decrease long-term complications. Patients will require ongoing ocular hygiene.
Ectrodactyly–ectodermal dysplasia–clefting syndrome (EEC)
Cutaneous findings
Cutaneous findings in EEC syndrome are mild. Fair skin and fine, sparse, light-colored hair are common features. Nails overlying abnormal, and occasionally normal, phalanges are dystrophic. Skin may be dry with occasional hypohidrosis.
Extracutaneous findings
Ectrodactyly (abnormal development of the median rays of the hands and feet) serves to distinguish this disorder from other ectodermal dysplasias and occurs in 80–100% of affected individuals. Depending on the series, cleft palate with or without cleft lip occurs in 70–100% of patients.102,103 Lacrimal duct hypoplasia or atresia is seen in over 90% of affected individuals, leading to excessive tearing, conjunctivitis, and blepharitis.104–106 Genitourinary malformations affect over one-third of patients. Chronic otitis media with secondary hearing loss occurs in 50%.107
Etiology and pathogenesis
Three forms of EEC have been described, EEC1 (MIM #129900), EEC2 (MIM #602077), and EEC3 (MIM #604292). Most individuals harbor mutations in p63, a tumor-suppressor gene (see above).108
Differential diagnosis
The diagnosis of EEC is self-evident when all three features (ectrodactyly, ectodermal dysplasia and clefting) are present. In the absence of limb defects, other ectodermal dysplasias associated with oral facial clefting, including Hay–Wells, Rapp–Hodgkin, and limb–mammary syndromes, need to be considered. Other diseases with limb defects include odontotrichomelic syndrome, which presents with severe absence deformities of the limbs, and Adams–Oliver syndrome (aplasia cutis congenita with limb defects), which has neither clefting nor ectodermal defects other than localized absence of skin.
Treatment and care
Treatment is mainly surgical. Consultations may include plastic surgery for cleft palate, orthopedics for limb repair, ENT for otitis media and hearing loss, ophthalmology for symptoms related to lacrimal duct hypoplasia, and urology for possible genitourinary malformations.
Disorders with skin laxity and redundant skin
Soft, hyperelastic skin, lax skin, or redundant skin, with or without bruising, fragility, or abnormal healing, is seen in a variety of related and distinct inherited disorders. Most are clinically evident in infancy or early childhood, but a correct diagnosis may be delayed until later in childhood.
Cutis laxa
Cutaneous findings
Cutis laxa is a term that encompasses the clinical finding of loose, inelastic skin that droops rather than stretches (Fig. 29.15), and does not spring back to place on release of tension. Three forms with different inheritance patterns have been described (Table 29.4). Loss of skin elasticity is progressive, and sagging may not be evident in the newborn, although in autosomal recessive disease flaccid skin is often evident at birth.109 Autosomal dominant cutis laxa patients tend to develop skin laxity in later childhood.110
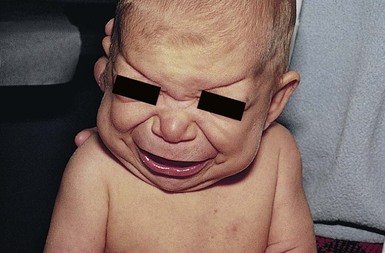
TABLE 29.4
Disorders to consider in the differential diagnosis of cutis laxa
| Disease name | Gene symbol | MIM # | Inheritance | Clinical findings | |||
| Cutis laxa | Emphysema | Aneurysms | Developmental delay | ||||
| ALDH18A1-related cutis laxa | ALDH18A1 | 612652 | AR | + | − | − | ++ |
| FBLN5-related cutis laxa | FBLN5 | 219100 | AR | +++ | +++ | − | − |
| EFEMP2-related cutis laxa | EFEMP2 (FBLN4) | 219100 | AR | ++ | ++ | +++ | − |
| Autosomal recessive cutis laxa type 2A | ATP6V0A2 | 278250 219200 |
AR | ++ | − | − | ++ |
| Autosomal dominant cutis laxa | ELN or FBLN5 | 123700 | AD | + | + | + | − |
| Gerodermia osteodysplastica | GORAB | 231070 | AR | + + | − | − | − |
| De Barsy syndrome (PYCR1-related progeroid syndrome) | PYCR1 | 219150 | AR | + | − | − | +++ |
| Autosomal recessive cutis laxa type 2B | PYCR1 | 612940 | AR | + | − | _ | +++ |
| LTBP4-related cutis laxa | LTBP4 | 613177 | AR | + | ++ | + | + |
| RIN2-related cutis laxa | RIN2 | 613075 | AR | + | − | − | ± |
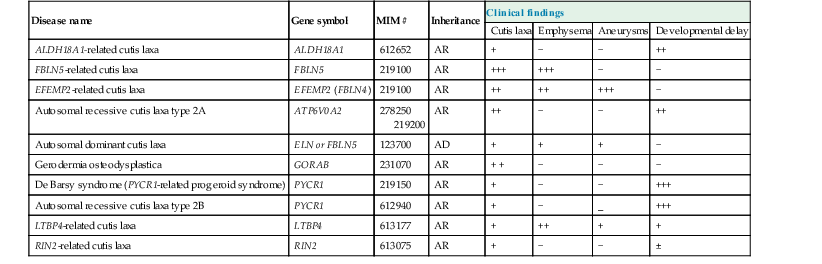
(Reproduced with permission from Van Maldergem L, Dobyns W, Kornak U. ATP6V0A2-Related Cutis Laxa. 2009 Mar 19 [Updated 2011 May 10]. In: Pagon RA, Bird TD, Dolan CR, et al. editors. GeneReviews™ [Internet]. Seattle (WA): University of Washington, Seattle; 1993. Available from: http://www.ncbi.nlm.nih.gov/books/NBK5200/)
Extracutaneous findings
Cutis laxa may present in the newborn with skin changes, joint laxity, and abdominal and inguinal hernias. Pulmonary emphysema may develop either during childhood or later in the course of the disease.109 In X-linked cutis laxa, bladder and gastrointestinal diverticuli can occur. Many newborns present with congenital dislocation of the hip. The characteristic long-bone and occipital exostoses (occipital horns) occur over time. Autosomal recessive disease is often more severe than the other forms, with flaccid skin evident at birth, early-onset emphysema, and vascular abnormalities such as aortic aneurysms or pulmonary artery/valve stenosis.109 Death from complications related to emphysema may occur early in infancy. The autosomal dominant form typically has fewer internal manifestations and a normal associated lifespan.110
Etiology and pathogenesis
The inherited forms of cutis laxa are detailed in Table 29.4.
Differential diagnosis
Ehlers–Danlos syndrome (EDS) has similar skin laxity, but has the elastic recoil lacking in cutis laxa. Unlike in EDS, vascular fragility and problems with wound healing are lacking. Cutis laxa may also present as a component of other genetic disorders. Acquired cutis laxa is often late onset and associated with drug exposure to penicillin, d-penicillamine, or isoniazid. Alternatively, acquired disease may develop from crops of well-demarcated inflammatory plaques in association with fever, malaise, and peripheral neutrophilia or eosinophilia.111 This form, which has sometimes been called ‘Marshall syndrome’ has been reported in infancy.112 Areas of loose wrinkled skin are also evident in the so-called ‘prune belly syndrome’ where lax skin is seen in association with renal anomalies and hypoplastic abdominal musculature (Fig. 29.16).113
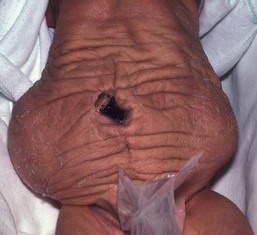
The diagnosis is confirmed by elastin staining (Verhoeff–van Gieson, orcein) of a skin biopsy. X-linked disease has abnormally large collagen fibrils with normal elastic fibers, whereas autosomal disease has reduced elastin, an abnormal dense amorphous component, and variation in collagen fibril diameter with collagen ‘flowers.’ Low serum ceruloplasmin or copper levels may also help in the diagnosis of affected boys.109
Treatment and care
All patients should have a complete physical examination for associated abnormalities, chest radiography to screen for emphysema and cardiomyopathy, and echocardiography for potential cardiac valve involvement (Box 29.9). Pulmonary function tests should be considered for early detection of emphysema. As solar elastosis aggravates cutaneous disease, sun protection should be emphasized.114 Early parenteral copper-histidine replacement may prolong life and delay the onset of symptoms in patients with the X-linked form.115
De barsy syndrome
This rare condition is characterized by very lax, wrinkled skin and a progeroid facies with thin hair, thin skin, and lack of subcutaneous fat, all present at birth without progression over time. A prominent vascular pattern is probably due to the thin dermis. Affected infants also have intrauterine growth retardation and poor postnatal growth. The hands are held in fists, whereas other joints are typically lax. Mental retardation is present; choreoathetosis develops over time. Eye findings include cataracts, strabismus, and myopia. De Barsy syndrome has an autosomal recessive inheritance pattern and is associated with mutations in the P5CS and PYCR1 genes.116
Ehlers–danlos syndromes
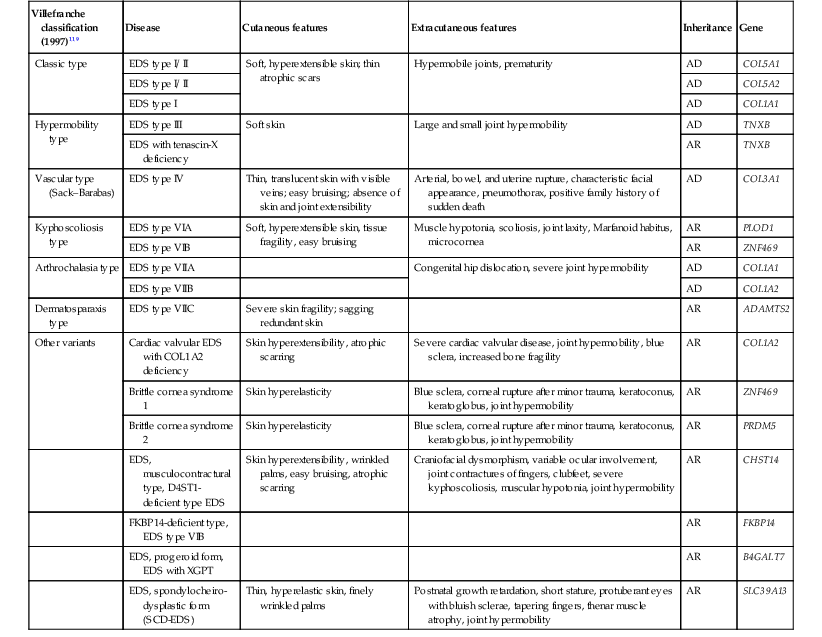
The Ehlers–Danlos syndromes (EDS) are a group of inherited connective tissue disorders characterized by articular hypermobility, skin extensibility, and tissue fragility. A significant number of individuals with all types of EDS have cardiac abnormalities.117 They were originally classified as 11 separate disorders,118 however, as the genetic mutations underlying this group of disorders are becoming elucidated, the classification has been updated (Table 29.5).119–121
TABLE 29.5
Clinical features, inheritance patterns, and biochemical defects of the Ehlers–Danlos syndromes120,121
| Villefranche classification (1997)119 | Disease | Cutaneous features | Extracutaneous features | Inheritance | Gene |
| Classic type | EDS type I/ II | Soft, hyperextensible skin; thin atrophic scars | Hypermobile joints, prematurity | AD | COL5A1 |
| EDS type I/ II | AD | COL5A2 | |||
| EDS type I | AD | COL1A1 | |||
| Hypermobility type | EDS type III | Soft skin | Large and small joint hypermobility | AD | TNXB |
| EDS with tenascin-X deficiency | AR | TNXB | |||
| Vascular type (Sack–Barabas) | EDS type IV | Thin, translucent skin with visible veins; easy bruising; absence of skin and joint extensibility | Arterial, bowel, and uterine rupture, characteristic facial appearance, pneumothorax, positive family history of sudden death | AD | COL3A1 |
| Kyphoscoliosis type | EDS type VIA | Soft, hyperextensible skin, tissue fragility, easy bruising | Muscle hypotonia, scoliosis, joint laxity, Marfanoid habitus, microcornea | AR | PLOD1 |
| EDS type VIB | AR | ZNF469 | |||
| Arthrochalasia type | EDS type VIIA | Congenital hip dislocation, severe joint hypermobility | AD | COL1A1 | |
| EDS type VIIB | AD | COL1A2 | |||
| Dermatosparaxis type | EDS type VIIC | Severe skin fragility; sagging redundant skin | AR | ADAMTS2 | |
| Other variants | Cardiac valvular EDS with COL1A2 deficiency | Skin hyperextensibility, atrophic scarring | Severe cardiac valvular disease, joint hypermobility, blue sclera, increased bone fragility | AR | COL1A2 |
| Brittle cornea syndrome 1 | Skin hyperelasticity | Blue sclera, corneal rupture after minor trauma, keratoconus, keratoglobus, joint hypermobility | AR | ZNF469 | |
| Brittle cornea syndrome 2 | Skin hyperelasticity | Blue sclera, corneal rupture after minor trauma, keratoconus, keratoglobus, joint hypermobility | AR | PRDM5 | |
| EDS, musculocontractural type, D4ST1-deficient type EDS | Skin hyperextensibility, wrinkled palms, easy bruising, atrophic scarring | Craniofacial dysmorphism, variable ocular involvement, joint contractures of fingers, clubfeet, severe kyphoscoliosis, muscular hypotonia, joint hypermobility | AR | CHST14 | |
| FKBP14-deficient type, EDS type VIB | AR | FKBP14 | |||
| EDS, progeroid form, EDS with XGPT | AR | B4GALT7 | |||
| EDS, spondylocheiro-dysplastic form (SCD-EDS) | Thin, hyperelastic skin, finely wrinkled palms | Postnatal growth retardation, short stature, protuberant eyes with bluish sclerae, tapering fingers, thenar muscle atrophy, joint hypermobility | AR | SLC39A13 |
Cutaneous findings
Babies with the classic type of EDS have soft, velvety, extensible skin that feels doughy and is very fragile with bruising and splitting secondary to minimal trauma. Wounds are slow to heal, quickly dehisce, and often resolve with cigarette paper-like atrophic scars. Patients with kyphoscoliosis-type and arthrochalasia-type EDS have less prominent skin fragility, bruisability, and dermal hyperextensibility than those with classic EDS.117 Patients with the periodontal form have similar skin findings to those in classic EDS, but additionally have excessive wrinkling of the palms and soles. Over time, the skin becomes hyperpigmented and markedly atrophic. In dermatosparaxis-type EDS a patient’s skin is characterized by extreme fragility and is sagging, redundant, and not in-elastic.
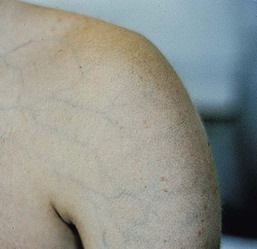
Rather than having the extensible and doughy skin seen in classic EDS, the skin in vascular-type EDS is thin and translucent with a visible vascular pattern (Fig. 29.17). It is not fragile and heals normally, but bruising is still common. Patients have a typical facial appearance, characterized by a thin nose, thin lips, and prominent eyes. Unless the disorder is already known to be in the family, none of these features is likely to be appreciated in the newborn or young infant.
Extracutaneous findings
Of the neonates with EDS, 40% are delivered prematurely.122 All forms of EDS have joint hypermobility with joint dislocations; congenital dislocation of the hip occurs in the classic, vascular, and arthrochalasia types. Although the prevalence is unknown, cardiac and aortic abnormalities seem to be much more common than in the general population. This is most evident in patients with the vascular type of EDS, who are at high risk for rupture of medium-sized arteries, aorta, and bowel, either spontaneously or following minor trauma. Special precautions should be taken when performing surgery on these patients.
Etiology and pathogenesis
The pathogenesis and inheritance patterns of EDS are detailed in Table 29.5.
Differential diagnosis
Cutis laxa also has hyperextensible skin, but it is loose and sagging, whereas the skin in EDS is elastic and recoils. Although patients with Marfan syndrome may also have mild to moderate joint hypermobility, the skin in newborns with Marfan syndrome does not show distinctive findings in this age group as it may with EDS.
Treatment and care
Treatment of EDS is mostly supportive and preventative (Box 29.10). Large doses of ascorbic acid (a cofactor of lysyl hydroxylase), 2–4 g/day, have been used in patients with kyphoscoliosis-type EDS, with clinical response.123 Specific therapies are not routinely used in other subtypes, although some physicians recommend high-dose ascorbic acid for all subtypes.124
A cardiovascular examination should be performed on all patients. Those with evidence of abnormalities or any patient with the vascular form of EDS should have an echocardiogram (Box 29.10). Alternatively, some recommend echocardiography at diagnosis for all patients, to assess aortic size.117
Because of tissue friability, surgical procedures are difficult, with dehiscence and wound breakdown being common. Although scars will still spread over time, a combination of adhesive tape, skin closure surgical adhesive tape, absorbable running subcuticular suturing, increased density of sutures, or cyanoacrylate adhesives may facilitate healing. Pseudotumor formation may be prevented by applying pressure bandages to hematomas. Parents should be instructed on how to protect the infant from trauma.
Mosaic disorders
The understanding of the genetics of mosaicism and its impact on skin development is increasing. In general, asymmetric and/or linear skin changes may be attributed to mosaicism, which can be the result of distinct cell lines or X inactivation. Female carriers of X-linked disorders such as incontinentia pigmenti express pigment changes along the lines of Blaschko. Children mosaic for chromosomal aneuploidy may have hyperpigmentation or hypopigmentation along the lines of Blaschko, or in other patterns, such as a phylloid (leaf-like) or checkerboard appearance. These cutaneous patterns represent the endpoint of cell migration during embryogenesis.
Focal dermal hypoplasia of goltz
Cutaneous findings
Focal dermal hypoplasia (FDH, MIM #305600) is characterized by congenital linear or reticulated atrophic, hypo- or hyperpigmented lesions following the lines of Blaschko, often with prominent telangiectasias (Fig. 29.18A).125,126 Fat herniations, hyperkeratosis, aplasia cutis, and scarring with pinpoint, pore-like depressions may occur within these lesions (Fig. 29.18B).127 The skin lesions are usually regarded as essential for the diagnosis, but cases without skin involvement have been reported.128 Biopsy of affected areas reveals dermal hypoplasia with upward extension of subcutaneous fat almost to normal epidermis.
Other common findings include raspberry-like papillomas, which are commonly found on the perineum, vulva, anus, or lips. These vascular papillomas usually develop over time, but may be present at birth.129 Nails are often dystrophic, and hair may be sparse and brittle.
Extracutaneous findings
After skin disease, skeletal abnormalities constitute the second most constant manifestation of FDH and include limb reduction, asymmetric growth, small stature, and split-hand/split-foot malformations (‘lobster-claw’ deformity).127 Vertical striations in the metaphyses of long bones (osteopathia striata) may be seen in many patients and are a useful index for diagnosis.130,131 Ocular abnormalities affect approximately 40% of patients and range from strabismus to microphthalmia.127 Teeth are commonly malformed or absent. Less commonly described findings include clavicular anomalies, abdominal wall defects, renal anomalies, and congenital heart defects.127,132,133
Etiology and pathogenesis
FDH is X-linked dominant and presumed lethal in hemizygous males. It is due to mutations in the PORCN gene which encodes a protein important in Wnt signaling.134,135 Lyonization of the X chromosome may explain the cutaneous Blaschko linear distribution. Rare cases in males may arise from somatic mosaicism and half-chromatid mutations.133
Differential diagnosis
The diagnosis of FDH is clinical, with histologic and radiographic corroboration (see above). Skin lesions of FDH do not blister and are static in comparison with IP. There is no epiphyseal stippling in FDH as in Conradi–Hunermann–Happle syndrome (X-linked dominant chondrodysplasia punctata), which also has linear ichthyosis and follicular atrophoderma as opposed to the telangiectases, hypo-/hyperpigmentation, and/or dermal atrophy of FDH. Distal extremities are not involved in MIDAS (microphthalmia, dermal aplasia, and sclerocornea) syndrome, and herniations of fatty tissue are not found. The initial skin changes of linear porokeratosis and porokeratotic eccrine and ostial dermal duct nevus may also resemble FDH, but evolution over time and histopathology can help to distinguish these conditions.
Treatment and care
After a complete physical examination looking for possible systemic manifestations, the management of FDH is mainly symptomatic. Infants should be evaluated by dermatology, ophthalmology, and orthopedic surgery as needed because of the common occurrence of skeletal, cutaneous, and ophthalmologic disease. Other anomalies should be investigated in symptomatic individuals.
Epidermal nevus syndrome
The sporadic association of epidermal nevi (see also Chapter 26) with developmental abnormalities in other organ systems constitutes epidermal nevus syndrome (ENS, MIM #163200).
Epidermal nevi vary according to their predominant component and include nevus sebaceus (sebaceous glands), nevus comedonicus (hair follicles) and verrucous nevus (keratinocytes). The mosaic genetic variations underlying these conditions are beginning to become clear. Somatic mosaic mutations in RAS have been identified in keratinocytic and sebaceous epidermal nevi.136,137 Multiple other mucocutaneous findings may occur simultaneously with the epidermal nevus. Central nervous, ocular, and musculoskeletal systems are predominantly affected, however, a wide range of abnormalities may occur within each of these systems, and occasionally other organ systems may be involved. ENS occurs sporadically and should be considered in any patient with extensive epidermal nevi or epidermal nevi associated with systemic abnormalities. The possibility that an epidermal nevus is a component of another syndrome, such as Proteus, CHILD (congenital hemidysplasia with ichthyosiform nevus and limb defects), or phakomatosis pigmentovascularis, should be considered. Patients suspected of having ENS should have a thorough physical evaluation with special attention to the musculoskeletal, neurologic, ocular, and cardiovascular systems. Depending on the findings of the physical examination, management should be multidisciplinary.
MIDAS syndrome
Microphthalmia, dermal aplasia, and sclerocornea (MIM #309801) are the hallmarks of MIDAS or MLS (microphthalmia and linear skin defects). Dermal aplasia typically presents as atrophic linear scars on the face, scalp, and neck (Fig. 29.19).138 The skin shows irregular, linear, erythematous areas of atrophic skin similar in appearance to fresh lesions of aplasia cutis congenita. Eye defects are usually bilateral and include microphthalmia, corneal opacities, and orbital cysts. Congenital heart disease and neurologic abnormalities may be found.139
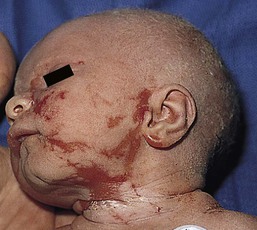
MIDAS syndrome is an X-linked dominant disorder caused in some cases by a deletion in HCCS, which encodes mitochondrial holocytochrome c–type synthase.140 This hampers oxidative phosphorylation and regulation of apoptosis which results in the clinical phenotype.
In contrast to FDH, aplastic skin lesions are limited to the upper half of the body. Fat herniation and other manifestations of FDH, such as papillomas, clefting of the hands or feet, syndactyly, and coloboma, do not occur in MIDAS syndrome.
Patients should have complete physical and ophthalmic examinations, with treatment individualized to the patient.
Chromosomal disorders
Turner syndrome
Turner syndrome (Ullrich–Turner syndrome) occurs in approximately 1 in 3000 females and is caused by absence or partial deletion of one of the X chromosomes. Lymphedema, neck webbing, short stature, gonadal dysfunction, and cardiac malformations are the characteristic features (Box 29.11).
Cutaneous findings
Lymphatic abnormalities are a common presenting manifestation of Turner syndrome (TS). Fetal edema, viewed on ultrasound, can lead to the prenatal diagnosis. Any infant presenting with congenital lymphedema of the extremities or neck requires a karyotype analysis. Lymphatic defects result in many clinical manifestations, including pterygium coli (webbing of the neck), redundant neck folds, and a low hairline over the nape (Fig. 29.20).141 Nails may become hypoplastic and concave, with an increased insertion angle secondary to lymphedema.142 Cutis verticis gyrata or congenital areas of fixed skin folds are also presumed to be caused by in utero entrapment and fixation of edematous skin.143 Acral edema usually resolves by 2 years of age, but may persist or recur in childhood in a minority of patients. No sequelae of lymphedema, such as cellulitis or elephantiasis nostras verrucosa, have been reported.141
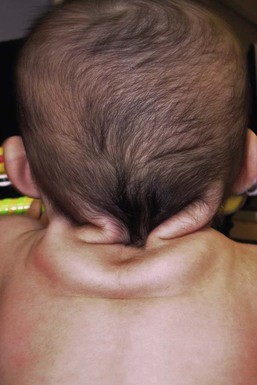
Benign melanocytic nevi are increased in number in TS, with one study showing the average number of nevi to be 115,144 versus 20–40 per person in the general population.145 The nevi are not dysplastic clinically or histologically, and patients do not appear to have an increased risk of developing malignant melanoma.146 Other cutaneous stigmata including alopecia areata, café-au-lait macules, a tendency to form keloids, psoriasis, and vitiligo are of questionable increased frequency.141 A subset of patients who are mosaics for their chromosome alterations present with pigmentary alterations along the lines of Blaschko.
Extracutaneous findings
The physical phenotype of patients with TS is highly variable, with many patients appearing physically normal except for short stature.147 Nevertheless, several clinical features related to bone deformities may be found, including a square-shaped ‘shield’ chest with widely spaced nipples, cubitus valgus, chondrodysplasia of the distal radial epiphysis (Madelung’s deformity), and brachymetacarpalia/tarsalia.141 Additional features include micrognathia, downward displacement of the outer corner of the eyes and epicanthic folds, low-set ears, and a high arched palate.
Etiology and pathogenesis
Turner syndrome is a sporadic disease of females characterized by the absence of all or part of the second X chromosome. TS is often diagnosed incidentally by amniocentesis or chorionic villous sampling performed for unrelated reasons.148 However, many infants are diagnosed prenatally based on the ultrasonic finding of fetal edema or an abnormal marker screen. Other ultrasonic findings may include nuchal translucency, cystic hygroma, coarctation of the aorta, renal anomalies, growth retardation, and fetal hydrops.149 Up to one-third of affected girls are diagnosed at birth based on puffy hands and feet or redundant nuchal skin.148 Another third have edema at birth, but it is commonly overlooked until adolescence or adulthood.
Treatment and care
Baseline evaluation of patients diagnosed with Turner syndrome should include physical examination, echocardiography, renal ultrasound, thyroid function tests, and a hearing screen. Patients should also be monitored for ovarian failure, growth issues, and psychosocial issues.148,151
Trisomy 21
In 1959, Down syndrome (MIM #190685) was found by Lejeune and colleagues152 to be caused by the presence of three copies of chromosome 21. In most instances, trisomy 21 is sporadic, the risk increasing with increased maternal age. The chromosome has been sequenced and is estimated to contain over 200 genes.153 Despite considerable effort to determine which features, if any, of trisomy 21 are due to dosage effects of specific genes, none have so far been identified.
There are many phenotypic changes associated with trisomy 21 (Box 29.12). One feature relevant to evaluation in the newborn period is a pustular eruption which can occur in the setting of a transient neonatal leukemoid reaction, which occurs in as many as 10% of newborns with trisomy 21.154 Most cases resolve spontaneously; however, children with trisomy 21 are also at risk for the development of acute leukemia, both ALL and AML (see Chapter 10). Other cutaneous associations in trisomy 21 include elastosis perforans serpiginosa, multiple syringomas, alopecia areata, milia-like idiopathic calcinosis cutis, and crusted scabies, and psoriasis but these rarely if ever occur in the neonatal period or early infancy.155,156
Trisomy 18
Trisomy 18 (Edwards syndrome) occurs in approximately 1 in 3600–8500 live births. It is fatal by 1 year of age in 90% of cases. Characteristic features (Box 29.13) include overriding fingers, nail hypoplasia, short hallux, and distinctive facial appearance. Neurologic features include cerebellar hypoplasia and agenesis of the corpus collosum.157 Skin involvement may include redundancy and hirsutism of the forehead and back. Cutis marmorata and scalp defects have also been reported. G-banded karyotype or aneuploidy fluorescent in situ hybridization analysis can be used to confirm the diagnosis. The differential diagnosis includes fetal akinesia sequence, which is characterized by polyhydramnios, characteristic facial features, joint contractures, and overriding fingers.
Trisomy 13
Trisomy 13 (Patau syndrome) was first reported in 1960 and is a cause of multiple congenital anomalies occurring in approximately 1 in 10 000–20 000 live births (Box 29.14). Orofacial clefts, holoprosencephaly, microphthalmia, and postaxial polydactyly are the cardinal features, with parieto-occipital aplasia cutis of the scalp also being a diagnostic feature. Minor findings include hemangioma on the forehead, anterior cowlick, loose skin, and abnormal ears.157
Miscellaneous disorders
Cerebello-trigeminal dermal dysplasia
Cerebello-trigeminal dermal dysplasia (Gomez–Lopez–Hernandez syndrome, MIM #601853)158 is a rare condition characterized by bilateral localized scalp alopecia and cerebellar malformation (rhombencephalosynapsis) with variable trigeminal nerve anesthesia.159 Alopecia appears on the scalp at birth in a band-like pattern, most often symmetrically in the parieto-occipital region. The alopecia might be mild and easily hidden, asymmetric or unilateral.159 In addition, facial scars may develop later in life, secondary to self-injury from trigeminal anesthesia. Patients may have retardation of motor and mental development. Anesthesia may occur in the trigeminal distribution and cornea leading to corneal opacities. Variably, the skull is asymmetric, with midfacial hypoplasia and low-set ears. Fifth finger clinodactyly, corneal opacities, and short stature have been associated.158 Cerebellar alterations can be detected clinically with characteristic structural abnormalities seen on both CT and MRI.158 Expressivity is highly variable with mild cases easily missed.160 Failure of migration and multiplication from a specific ectodermal region have been hypothesized to cause this syndrome.161 All cases have been sporadic with more affected boys than girls.160,159 The differential diagnosis includes healed areas of aplasia cutis congenita; however, other signs of disease become evident in patients with cerebello-trigeminal dermal dysplasia as the individuals age. Neurology consultation for evaluation and treatment of motor development delay, neuropsychiatric evaluation and ophthalmology evaluation for corneal abnormalities is recommended.
Familial dysautonomia
Familial dysautonomia (Riley–Day syndrome, hereditary sensory and autonomic neuropathy type III; MIM #223900) is an autosomal recessive condition seen almost exclusively in Ashkenazi Jews (Box 29.15). The USA has the largest prevalence of familial dysautonomia in the world related to a founder mutation.162 Due to population-based genetic screening for at-risk couples, and use of reproductive technology such as pre-implantation genetic diagnosis, the number of newborns with familial dysautonomia has steadily dropped.163,164 Familial dysautonomia causes progressive systemic sensory and autonomic dysfunction. Excessive drooling and sweating, and blood pressure dysregulation are common. Survival is reduced, and almost one-fifth of those affected die in infancy or childhood.
Cutaneous findings
Absent tearing, a cardinal feature of Riley–Day syndrome, is usually not appreciated in neonates, as overflow tearing does not develop until 2–3 months of age in normal infants. Affected babies may have striking blotching and mottling of the skin. This usually appears one month after birth and within the first year, as do bouts of hyperpyrexia. Emotional excitement often precipitates the appearance of red blotches. The hands and feet may be cool and mottled from vasoconstriction, or swollen and red from vasodilation. In familial dysautonomia, indifference or insensitivity to pain results in progressive self-mutilation, burns, and ulcers. Absent fungiform papillae on the tip of the tongue are an important diagnostic feature and the associated absence of taste buds leads to decreased ability to taste, especially sweetness. Insensitivity to pain causes oro-dental-self-mutilation of the tongue, lips and cheeks or loss of teeth. Compulsive biting in those areas may cause tumor-like masses called Riga–Fede disease (see Fig. 30.14), or ulcers. In toddlers, injuries to the tongue from emerging teeth are common.
Extracutaneous findings
Although usually not diagnosed until several years of age, generalized signs appear within the newborn period in over 80% of patients.165 These signs include breech birth in one-quarter, prematurity, intrauterine growth retardation, hypotonia, respiratory insufficiency, and poor feeding with swallowing difficulty and aspiration, developmental delay, short stature, scoliosis and corneal disease. Unexplained episodes called ‘dysautonomic crises’ of fever, labile blood pressure, tachycardia, aspiration pneumonia, and vomiting are typical. Deep tendon reflexes are invariably absent. Dysmorphic facial features are not associated, but facial asymmetry and a straightened mouth develop due to abnormal tone and molding of the facial bones.163
Etiology and pathogenesis
The causative gene for familial dysautonomia syndrome is the IKBKAP (I-kappa-B kinase complex associated protein) gene on chromosome 9q31. Disease expression is variable even though 99.5% of affected children have the same homozygous mutations.166 The cause is decreased survival of certain sensory, sympathetic, and parasympathetic nerve cells.
Differential diagnosis
In an infant with unexplained hyperpyrexia, abnormal sweating, and failure to thrive, the primary differential diagnosis is hypohidrotic ectodermal dysplasia versus familial dysautonomia syndrome. Injection of histamine (1 : 10 000) does not elicit a flare response in familial dysautonomia. The flare response can discriminate between the two conditions, as can the presence of teeth on a Panorex examination of the jaw. Further, in familial dysautonomia, the pupil is hypersensitive to parasympathomimetic drops like methacholine and pilocarpine. Upon exposure, the pupil almost always shrinks in familial dysautonomia, but not in a normal eye. The astute physician, aware of the feature of random blotching and mottling of the skin, may be able to make the diagnosis of Riley–Day syndrome in an infant whose multiplicity of problems have eluded correct diagnosis.
Treatment and care
Due to poor coordination, breast-feeding is difficult and delivery of proper nutrition might require a nasogastric tube or possible gastrostomy tube in the first year.163 Use of artificial tears, and avoidance of aspiration are necessary. Referral to neurology and orthopedics for specialized care is recommended. The involvement of multiple subspecialists in addition to close follow-up with the primary care provider is important.
Coffin–siris syndrome
A heterogenous disorder, hypoplastic or absent fifth digits or nails are the most consistent feature of Coffin–Siris syndrome (MIM #135900).167 Hypertrichosis, thick eyebrows and long eyelashes (see Chapter 31) with sparse temporal and frontal scalp hair characterize the most common ‘classic/Type A’, but thin ‘penciled’ eyebrows characterize the more rare ‘Type B’ variant (Box 29.16).167 Developmental delay and mental retardation are necessary to make the diagnosis. The facial features of Coffin–Siris fall into two groups with coarse facial features, thick eyebrows and thick lips characterizing ‘Type A’ and thin eyebrows and thin upper lips characterizing ‘Type B’. Short stature and delayed bone age are also characteristic. Delayed dentition has been reported.168 Work-up should include hand radiographs to assess for hypoplastic or absent phalanges on the fifth digit, renal ultrasound, ophthalmologic evaluation, hearing screen and cardiac evaluation to rule out cardiac malformations.
Whole exome sequencing has shown de novo autosomal dominant mutations in the SWItch/Sucrose (SWI/SNF) complex, which regulates chromatin structure and allows appropriate gene expression.169 Since most cases are sporadic with two unaffected parents, an autosomal dominant inheritance mechanism is indistinguishable from autosomal recessive. Therefore, with an undetected mutation, parents had received an estimated recurrence risk of 10%.170 With an identified de novo mutation in a child using DNA technology, the recurrence risk is reduced to 1–2%.170 The differential diagnosis includes mosaic trisomy 9, brachymorphism-onychodysplasia-dysphalangism, deafness-onychodystrophy-osteodystrophy-mental retardation,167 fetal hydantoin syndrome, fetal alcohol syndrome, and Cornelia de Lange syndrome. Patients with Coffin–Siris syndrome have been reported to have frequent upper respiratory and ear infections. Younger infants may have feeding problems. Anesthesia in children may be complicated by apnea.171
Mitochondrial disorders
Mitochondrial enzyme abnormalities are characterized by an unexplained association of symptoms with multiple involved organs that have no common embryologic origin or biologic function. An increasing number of involved organs and systems, appearing during the course of disease, is characteristic. In recent years, the causative mutations for many mitochondrial disorders have been described. Mitochondrial disorders have a heterogeneous group of findings, such as poor growth, vomiting, developmental delay, renal tubular dysfunction, and seizures. Therefore, the diagnosis is often delayed for many months. Over the past several years, cutaneous findings have begun to be recognized in mitochondrial diseases. The most common cutaneous findings in patients with mitochondrial disease include lipomas, alopecia, mottled pigmentation or poikiloderma, erythematous patches, hypertrichosis, and acrocyanosis.172,173 The alopecia seems to be related to hair shaft abnormalities, fractures (trichoschisis), twists (pili torti), and trichorrhexis nodosa. The mottled or reticulated pigmentation is most commonly in sun-exposed areas, such as the neck, dorsal arms, and forehead. Hypertrichosis has frequently been noted in Leigh syndrome. Neurologic features may include seizures, developmental delay, myoclonus, and ataxia. Skeletal muscle is involved, resulting in myopathy and hypotonia. Sideroblastic anemia and pancytopenia can be hematologic complications. The endocrine manifestations include hypoparathyroidism, growth deficiency, and diabetes mellitus. Cardiomyopathy and conduction defects are the cardiac manifestations. In addition, sensorineural deafness and lactic acidosis may also be present.174 Mitochondria are ubiquitous and are the main organelle responsible for producing energy in cells. Several different mutations in the mitochondrial DNA (mtDNA) have been identified resulting in a variety of diseases, including Leigh syndrome (subacute necrotizing encephalopathy) and MELAS (mitochondrial encephalomyopathy, acidosis, and stroke-like episodes), and NARP (neurogenic muscle weakness, ataxia, and retinitis pigmentosa). Mitochondrial DNA mutations can be either inherited or acquired. Acquired mutations may be related to ultraviolet-induced damage and may play a role in aging.175 Evaluation should include plasma lactate, pyruvate and ketone body levels, head CT or MRI, and muscle biopsy for histology and for genetic analysis of the mitochondrial DNA. Treatment is supportive.
Access the full reference list at ExpertConsult.com 
[/level-membership-for-dermatology-category][not-level-membership-for-dermatology-category]
Selected Hereditary Diseases
Dawn H. Siegel, Kari L. Martin, Jennifer L. Hand
Approach to the child with a genetic skin condition
To effectively care for an affected newborn and provide information for anxious parents, an organized diagnostic approach is essential. The physical exam requires special attention to ectodermal involvement by assessing the hair, teeth, nails, palms and soles of the feet. Moving beyond the skin, a child should be examined for dysmorphic features, associated major or minor congenital anomalies and accompanying illness. Results of a newborn hearing screen and pediatric ophthalmology exam are often valuable. Finally, a detailed family history including ethnic background and miscarriage history is beneficial. After gathering clinical information, superb resources are available online to consider diagnostic possibilities. One example is Online Mendelian Inheritance in Man (OMIM; http://www.ncbi.nlm.nih.gov/omim).
OMIM is maintained by the National Center for Biotechnology Information (NCBI) and can be searched using a list of the clinical features in an affected patient. Each disorder is assigned a number based on broad categories such as inheritance pattern. ‘GeneTests’ (http://www.ncbi.nlm.nih.gov/sites/GeneTests) and a separate database, ‘GeneReviews’1 offers a frequently updated list of internationally available DNA-based genetic tests and concise, yet comprehensive overviews of selected disorders. For effective coordination of testing and advising the patient about the complexities of genetic testing and reproductive options, the GeneTests site provides a database of genetics services that is searchable by country and province or state. Increasingly, individual mutations that cause familial genetic disorders are recognized to affect important regulatory pathways. This chapter highlights pathways that are particularly important to growth and development of the skin.
Disorders of the RAS-MAPK pathway (RASopathies)
Several genetic skin disorders are caused by mutations in genes in the RAS-MAPK signaling pathway (Fig. 29.1) and have been coined the ‘RASopathies’.2 The RASopathies include neurofibromatosis-1 (NF-1), NF-like syndrome (Legius syndrome), Noonan syndrome, Noonan with multiple lentigines (formerly LEOPARD syndrome), cardiofaciocutaneous syndrome, Costello syndrome, capillary malformation–arteriovenous malformation (see Chapter 22), and hereditary gingival fibromatosis (Table 29.1). Neurofibromatosis and Legius syndrome are also discussed in Chapter 24.
TABLE 29.1
Comparison of the clinical features of the RASopathies
| NF-1 | Cardiofacio-cutaneous syndrome | Costello syndrome | Noonan syndrome | Multiple lentigines syndrome | Capillary malformation- arteriovenous malformation | |
| Gene | NF1 | BRAF, MEK1, MEK2, KRAS | HRAS | PTPN11, SOS1, KRAS, NRAS, RAF1, SHOC2, CBL and BRAF | PTPN11, RAF, BRAF | RASA1 |
| Dermatologic features | Café-au-lait macules Intertriginous freckling Neurofibromas Plexiform neurofibroma |
Ulerythema ophryogenes Keratosis pilaris Melanocytic nevi Infantile hemangiomas |
Papillomata of the nose and perianal region Palmoplantar hyperkeratosis Redundant skin on the hands and feet Acanthosis nigricans |
Congenital lymphedema Café-au-lait macules Keratosis pilaris (most common with SOS1 mutations) |
Lentigines Café noir patches |
Capillary malformations Arteriovenous malformations of the brain, spine, skin, muscle and bone |
| Cardiac features | Uncommon | Pulmonic stenosis Hypertrophic cardiomyopathy |
Pulmonic stenosis Hypertrophic cardiomyopathy |
Pulmonic stenosis Hypertrophic cardiomyopathy |
EKG abnormalities Pulmonic stenosis Hypertrophic cardiomyopathy |
Rare |
| Tumor predisposition | Yes: Optic glioma Hematologic malignancies Meningioma and other brain cancers Malignant peripheral nerve sheath tumor Pheochromocytoma |
Rare: Possible risk for ALL and lymphoma |
Yes: Malignant solid tumors (rhabdomyosarcoma, neuroblastoma, transitional cell carcinoma of the bladder) |
Rare: Hematologic malignancies |
Rare: Hematologic malignancies |
Rare: Possible increased risk for neural tumors |
| Other | Lisch nodules Learning disability Pseudoarthrosis Macrocephaly Sphenoid wing dysplasia Renal artery stenosis Hypertension |
Failure to thrive (severe) Developmental delay |
Failure to thrive (mild) Developmental delay Sociable, outgoing personality |
Low posterior hairline Webbed neck Pectus excavatum Bleeding tendency Short stature |
Ocular hypertelorism Sensorineural hearing loss Short stature |
Risk for intracranial and spinal arteriovenous malformation Parkes–Weber syndrome |
Neurofibromatosis type 1
Neurofibromatosis 1 (NF-1, MIM #162200) is a multisystem disorder characterized by age-related abnormalities of tissue proliferation. NF-1 is one of the most common autosomal dominant genetic conditions with an incidence of approximately 1 in 3000 individuals.3,4 Consensus criteria for the diagnosis of NF-1 were established at the 1988 NIH conference to be used as a guideline for clinical diagnosis (Box 29.1).5 In 95% of affected individuals, a diagnosis can be made by age 11 through the use of clinical evaluation alone.6 However, NF-1 can be a difficult condition to diagnose in some infants due to the high incidence of sporadic mutations, variability of clinical expression, and age-related penetrance of individual clinical manifestations. Hence, anticipatory guidance counseling should be provided in both established and suspected cases of NF-1.
Cutaneous findings
Café-au-lait macules (CALM) are light to dark brown sharply defined oval macules and patches which can occur on almost any skin surface (Fig. 29.2A). Infants with ≥6 café-au-lait macules >5 mm in diameter that are not confined to a single segmental region, should be evaluated and managed as though they have NF-1, even without other signs of NF-1, because of the high likelihood that they will develop other diagnostic signs with time. At puberty, if other features are lacking, the approach and diagnosis can be reconsidered.
Intertriginous freckling of the axillary and inguinal regions may occasionally be present in infancy, but most often present between 2–10 years of age (Fig. 29.2B). Although freckling on the neck and trunk is common in NF-1, it is not accepted as a diagnostic criterion.
Peripheral neurofibromas are infrequent in early childhood NF-1, occurring in only 14% of children less than 10 years of age.6 Often the first sign of dermal neurofibromas are 3–6 mm, light blue, minimally raised papules which are most easily detected with side lighting. It has been suggested that a subset of children with large deletions of the NF-1 gene typically present with multiple neurofibromas in early childhood.7
Plexiform neurofibromas may be apparent at birth or soon thereafter; however because they are often internal, may be difficult to detect. They occur in at least 25% of affected individuals (Fig. 29.2C). Most have overlying hyperpigmentation; some – but not all – have increased vascularity resembling a vascular anomaly or hypertrichosis resembling a congenital melanocytic nevus. Plexiform neurofibromas may grow rapidly and interdigitate with and surround normal structures. Radiologic imaging and neurosurgical consultation should be considered if lesions are extensive or close to major nerve bundles.
Extracutaneous findings
Optic gliomas are astrocytomas arising anywhere along the optic pathway. These lesions tend to arise in infancy or early childhood. Half of the tumors are symptomatic, causing loss of visual acuity, decreased field of vision, proptosis, or interference with the hypothalamopituitary axis. Symptomatic optic gliomas are often diagnosed by 6 years of age.
Lisch nodules are pigmented iris hamartomas that rarely present in infants, and only 20% of individuals under 5 years of age with NF-1 have Lisch nodules. They are best seen on slit-lamp examination and do not result in functional disability. Congenital glaucoma occurs in less than 0.05% of individuals with NF-1 and may present with an ipsilateral neurofibroma of the eyelid.
Relatively short stature and large head size are frequent findings and more common than the more diagnostic skeletal changes, pseudoarthrosis and sphenoid wing dysplasia. Pseudoarthrosis represents the failure of union after fracture. It is always unilateral and most commonly presents in the tibia as anterolateral bowing. Ultimately, pseudoarthrosis can progress to severe deformity. Sphenoid wing dysplasia is a unilateral defect of the orbit present in approximately 5% of individuals with NF-1 and results in a change in orbit structure. Approximately half of those cases with sphenoid wing dysplasia develop an ipsilateral temporal–orbital plexiform neurofibroma, and half of individuals presenting with a temporal–orbital tumor also have an underlying plexiform neurofibroma. Learning disabilities occur in approximately 50% of children with NF-1. The main learning difficulties include reading, deficits in perceptual skills (visuospatial) and executive functioning. Attention issues are also common.8
Etiology and pathogenesis
NF-1 is due to an autosomal dominant mutation localized to chromosome 17 that results in defects in neurofibromin, a tumor suppressor protein that stimulates hydrolysis of guanosine triphosphate (GTP) bound to ras.9
Differential diagnosis and diagnosis
Café-au-lait macules may be found in many other conditions, including segmental pigmentary disorder and McCune–Albright syndrome (see Chapter 24). Additional differential diagnoses for multiple café-au-lait macules are shown in Box 29.2. Genetic testing for NF-1 is available from several commercial and research laboratories (see: www.genetests.org, for specific information). Decisions regarding whether laboratory-based NF testing is appropriate are best made in conjunction with a geneticist and genetic counselor.
Treatment and care
Patients with neurofibromatosis require age-related anticipatory guidance counseling and regular follow-up with a pediatrician, ophthalmologist and geneticist. Additional specialists can be included, based on symptoms or complications and may include orthopedics, neurology, dermatology, neuropsychology, and neurosurgery (Box 29.3). Ophthalmologists and neurologists should evaluate for optic nerve pathway tumors and glaucoma. Anterolateral tibial bowing when present require prompt orthopedic evaluation, because the critical time for fracture and poor healing is infancy to early childhood.6 Periodic evaluation for scoliosis is also necessary. Regular physical examinations should include careful measurement of blood pressure because of a higher incidence of hypertension secondary to renovascular disease, vasoactive secreting tumors, and coarctation of the aorta.6 Head circumference should be monitored because of the risk of hydrocephalus and the occurrence of macrocephaly without hydrocephalus. Careful developmental assessment is a key part of management, as the risk of neurologic abnormalities and learning disabilities is increased.
Dermal neurofibromas are benign and should only be excised if they are symptomatic or disfiguring. Plexiform neurofibromas require close evaluation. Skin should be palpated carefully as plexiform neurofibromas may remain below the surface. If there are symptoms or neurologic abnormalities on clinical exam, MRI scans should be obtained to determine the extent of plexiform neurofibromas. Serial imaging or volumetric MRI may be necessary to assess potential growth. Pain and/or growth may herald malignant transformation, but this is exceedingly rare in infancy. Neurologic compromise may result from perineural extension, however, and neurology and/or neurosurgery may need to be consulted for plexiform lesions near neurovascular structures (such as the neck, axilla and spinal area).6 Orbitotemporal neurofibromas may be better managed by numerous surgical procedures over time; plastic surgery should be involved early for management of these tumors.
Leguis syndrome
Legius syndrome (MIM #611431) was first recognized in 2007 as an NF-1-like syndrome in families with multiple café-au-lait macules, but negative NF-1 gene testing.10 The phenotype is milder than NF-1 and seems to lack the propensity for tumor development. The clinical features include a mild NF-like phenotype and 50% of these patients meet diagnostic criteria for NF-1. Of the diagnostic criteria for NF-1 (Box 29.1), the features that have been reported in Legius include >6 CALM >5 mm, intertriginous freckling, positive family history, macrocephaly, short stature and learning disabilities.11 Neurofibromas, plexiform neurofibromas, bone dysplasia and scoliosis have not been associated with Legius syndrome. Legius syndrome is caused by loss of function (LOF) mutations in the SPRED1 gene.10 Like neurofibromin, SPRED1 is a negative regulator of the RAS-MAPK pathway (Fig. 29.1).11
Noonan syndrome
Noonan syndrome (MIM #163950) is an autosomal dominant multisystem disorder characterized by congenital lymphedema, broad or webbed neck, low posterior hairline, short stature, and cardiac malformations (Box 29.4).
Cutaneous findings
The neonate with Noonan syndrome is unlikely to have skin manifestations other than nuchal webbing and peripheral lymphedema that suggest the diagnosis. Keratosis pilaris atrophicans faciei (ulerythema ophryogenes), characterized by horny, whitish, hemispherical, or acuminate papules at the opening of pilosebaceous follicles, is generally noted in older children, but may manifest in the external third of the eyebrows by a few months after birth. Some children with Noonan syndrome have multiple CALM and/or lentigines.
Extracutaneous findings
Neonates have a characteristic facial appearance consisting of a tall forehead, low posterior hairline, hypertelorism, downslanting palpebral fissures, epicanthal folds, short and broad, depressed nasal root, deeply grooved philtrum and micrognathia. The chest shape is unique with superior pectus carinatum and inferior pectus excavatum. Feeding difficulties, gastroesophageal reflux and failure to thrive are frequent problems in infancy. Unilateral or bilateral cryptorchidism are common in boys. Infants are at risk for transient monocytosis, thrombocytopenia and myeloproliferative disorder.12 Coagulation defects occur in approximately one-third of patients with Noonan syndrome and may present with easy bruising or prolonged bleeding times. There is a slightly increased risk of hematologic malignancy (most commonly juvenile myelomonocytic leukemia, JMML) compared with the general population. The classic congenital heart defect in Noonan syndrome is pulmonic stenosis.
Etiology and pathogenesis
The incidence of Noonan syndrome is approximately 1 in 1000–2500 live births.13 Noonan syndrome may be caused by a mutation in one of several genes in the RAS-MAPK signaling pathway, including PTPN11, SOS1, KRAS, NRAS, RAF1, SHOC2, CBL, and BRAF.14 PTPN11 and CBL mutations have a higher rate of bleeding diathesis and JMML. Patients with SOS1 mutations have a lower rate of intellectual disability. SHOC2 mutations have been associated with a Noonan phenotype with loose anagen hair.15 PTPN11 mutations are also the genetic basis of Noonan with multiple lentigines (formerly known as LEOPARD syndrome;16 see Chapter 24).
Differential diagnosis
In the neonatal period, the RASopathies can be very difficult to distinguish as they share the features of congenital heart defects, severe feeding difficulties and developmental delay.17 Later in infancy, facial and ectodermal features become more distinct for each of the RASopathies. Cardiofaciocutaneous syndrome and Costello syndrome are described in more detail below.
Treatment and care
Patients with Noonan syndrome should be monitored for growth deficiency and developmental delay. Electrocardiogram and echocardiography should be performed at the time of diagnosis and repeated as indicated based on findings. A renal ultrasound is recommended at diagnosis. Coagulopathy work-up should be performed if the patient has a history of either easy bruising or prolonged bleeding.14
Cardiofaciocutaneous syndrome
Cardiofaciocutaneous (CFC) syndrome (MIM #115150) is characterized by short stature, congenital heart defects, intellectual disability, ectodermal abnormalities and a characteristic coarse facial appearance (Table 29.2). Numerous cutaneous findings have been reported in CFC. In 2010, Siegel and colleagues18 evaluated the cutaneous manifestations in 61 mutation-positive individuals with CFC syndrome. All had dermatologic findings. One of the striking features identified in this study was a high number of melanocytic nevi. In the study, 23% of participants had over 50 nevi and 36% of those patients reported over 100 nevi. The amount of nevi increased with age and were not a prominent feature in infancy. Keratosis pilaris and ulerythema ophryogenes were very common and affected the majority of individuals in childhood and adolescence (Fig. 29.3). Infantile hemangiomas occurred at a greater frequency when compared with the general population. Additional features in CFC include macrocephaly, characteristic facial appearance, growth retardation, cardiac defects, neurologic impairment, gastrointestinal dysfunction, and ocular abnormalities.
TABLE29.2
Dermatologic features found in cardiofaciocutaneous syndrome compared with Costello syndrome
| Percentage of cases | ||
| Cardiofaciocutaneous | Costello | |
| Curly hair | 93.4% | 95.7% |
| Eyebrow density | 90.0% sparse | 47.8% thick |
| Keratosis pilaris | 80.3% | 32.6% |
| Infantile hemangioma | 26.2% | 10.9% |
| Papillomas | 4.9% | 71.7% |
| More than 50 nevi | 23.0% | 4.0% |
| Palmoplantar keratoderma | 36.1% | 76.1% |
(Adapted from Siegel DH, Mann JA, Krol AL, Rauen KA. Dermatological phenotype in Costello syndrome: consequences of Ras dysregulation in development. Br J Dermatol 2012; 166(3):601–607.)
CFC syndrome is caused by mutations in BRAF, MEK1 or MEK2.19,20 These mutations were discovered in part because of the similarity of the phenotypic features of Noonan and Costello syndromes, both of which were known to have mutations involving the RAS-MAPK pathway. Given insights into genetics and pathogenesis, it is not surprising that the main differential diagnoses for CFC syndrome include Noonan and Costello syndromes.
Costello syndrome
Costello syndrome (CS) (MIM #218040) is a rare, autosomal dominant, multiple congenital anomaly syndrome associated with failure to thrive, developmental delay, and an increased risk of malignancy.21 The features of Costello syndrome in the neonatal and infantile period include macrosomia, severe feeding problems, developmental delay, coarse facial features, gingival hyperplasia, osteopenia, hypertrophic cardiomyopathy and atrial arrhythmias.22 The most common malignancies include rhabdomyosarcoma, neuroblastoma and transitional cell cancer of the bladder. CS is caused by mutations in the HRAS gene, at 11p15.5, leading to constitutive activation of the RAS-MAPK pathway.21 The cutaneous findings include curly hair, papillomas on the perinasal, perioral and perianal skin, palmoplantar keratoderma, unusual body odor and heat intolerance. The skin on hands is loose and redundant (Fig. 29.4A). Acanthosis nigricans has been reported in 37% of the cases (Fig. 29.4B).23 In the neonatal period, it can be difficult to distinguish Costello, CFC and Noonan syndromes, as all three conditions can manifest with neonatal macrosomia, coarse facial features, failure to thrive and developmental delay.17
Capillary malformation-arteriovenous malformation (CM-AVM) (MIM #608354)
CM-AVM is an autosomal dominant disorder characterized by multiple, small, oval capillary malformations on the face, body and limbs, which are associated with arteriovenous malformations in about 30% of cases. The AVMs can occur in the brain, spine, muscle or skin. CM-AVM is caused by mutations in the RASA1 gene.24 See Chapter 22 for a more in-depth discussion about this condition.
Neurofibromatosis type 2
Neurofibromatosis type 2 (NF-2) (MIM #101000) is an autosomal dominant condition characterized by a high burden of schwannoma and meningioma tumor development.
Presentation in infancy is rare, although cutaneous schwannomas have been reported.25 Cutaneous features which may be present in infancy or early childhood include CALM and cutaneous schwannomas (Fig. 29.5). The age range at the onset of NF-2-related symptoms is 5–55 years. The initial symptoms that lead to the diagnosis include hearing loss, visual disturbance, and enlarging subcutaneous mass. The incidence of NF-2 is difficult to predict due to a high rate of mosaicism, but is estimated at about 1 : 25 000.26 NF-2 is caused by mutations in the merlin (also called schwannomin) tumor suppressor gene on chromosome 22q12.27 The differential diagnosis for NF-2 includes NF-1 and schwannomatosis.
Disorders of the PI3K-AKT/mTOR pathway and overgrowth syndromes
Overgrowth syndromes and human cancer share disruption of similar critical regulatory pathways (Table 29.3). Intensive cancer research, therefore, has improved our understanding of these rare but important disorders. The phosphatidylinositol 3-kinase (PI3K)-AKT pathway, in particular, critically guides cell growth and metabolism (Fig. 29.6). The PI3K enzymes are a family of highly conserved enzymes that regulate cell growth, migration and survival and disruption in the embryonic period typically impacts vascular, limb or brain development.29 Mutations cause recognizable patterns of increased cell number, hypertrophy, increased interstitium, or a combination of these.34 Typical neonatal clues of an overgrowth syndrome are abnormally increased body length, macrosomia, and macrocephaly, dysregulated growth of a body part or asymmetry (Fig. 29.7). Almost all overgrowth syndromes are associated with neoplasms, especially solid tumors.
TABLE 29.3
Overgrowth syndromes and associated clinical features
| Syndrome | Causative mutation | Cutaneous features | Extracutaneous features | Associated malignancy |
| Tuberous sclerosis | TSC1, TSC2 | Hypomelanotic macules, angiofibromas, forehead plaques, shagreen patches, periungual fibromas | Seizures, infantile spasms, intellectual disability. Renal cysts and angiomyolipomas, cardiac rhabdomyomas |
Malignant angiomyolipoma, renal cell cancer; sub-ependymal giant cell astrocytoma (SEGA) |
| Proteus | Mosaic AKT1 | Cerebriform connective tissue nevi (CCTN), epidermal nevi, vascular malformations, soft subcutaneous masses, patchy dermal hypoplasia, macrodactyly and lipomas | Disproportionate, relentless segmental overgrowth of body parts; skeletal asymmetry, lung cysts, thromboembolism, eye problems, ovarian cysts, epididymal cysts. Overgrowth generally after neonatal period |
Mostly benign tumors |
| Hemihyperplasia-multiple lipomatosis (HH-ML) | Unknown | Superficial capillary vascular malformation, lipomas. Lacks deep vascular malformation and cerebriform connective tissue nevi of Proteus. | Non-distorting overgrowth present at birth | Risk for embryonal malignancies is unknown. Screening for Wilms tumor, adrenal cell carcinoma, hepatoblastoma is recommended28 |
| Megalencephaly-capillary malformation (MCAP) | Mosaic PIK3CA | Patchy capillary malformations; stretchy skin and joints | Large brain, growth dysregulation, asymmetry, syndactyly, polydactyly, developmental delay, hypotonia, frontal bossing | Mildly increased risk of cancer24; Wilms tumor, leukemia; meningioma reported29 |
| Mosaic overgrowth with fibroadipose hyperplasia (MOFH) | Activating PIK3CA | Lacks cutaneous features of Proteus | Distorting or non-distorting, segmental overgrowth of muscles, skeleton and fibroadipose tissue that is present at birth | Unknown |
| SOLAMEN | PTEN | Signs of Cowden: trichilemmoma, acral keratoses, oral papillomas. Lacks cerebriform connective tissue nevi of Proteus | Features of Cowden: macrocephaly, breast and thyroid hamartoma | Breast, thyroid, endometrial |
| CLOVES | Activating PIK3CA | Vascular anomalies, truncal lipomas, epidermal nevi. May be wrinkled skin on the palms and soles, but not CCTN of Proteus | Non-progressive overgrowth at birth. Overgrown feet, hands; ‘sandal gap’ toes, severe scoliosis; high vascular-flow masses; phlebectasia; thromboembolism |
Unknown |
| Encephalocraniocutaneous lipomatosis (ECCL) | Unknown, sporadic | Nevus psiloliparus overlying lipomatous overgrowth; angiofibromas, connective tissue nevi30 cutis aplasia, nodular skin tags31 | Proportionate, non-distorting overgrowth at birth. Ocular abnormalities, CNS lipomas, heart defects, lytic bone lesions, hypospadias, cryptorchidism, seizures, jaw osteomas | Mostly benign tumors. Low-grade glioma reported30 |
| Beckwith–Wiedemann syndrome (BWS) | Imprinting error affecting chromosomal region 11p15.5 | Nevus flammeus, distinctive ear creases, posterior helical pits | Abdominal wall defects (omphalocele), placental overgrowth, macrosomia, macroglossia, abnormal kidney, cardiomegaly, hypoglycemia | Tumor risk increased, especially embryonal tumors; requires monitoring |
| Simpson–Golabi–Behmel syndrome type I | Glypican-3 (GPC3) | Supernumerary nipples, characteristic index finger and nail hypoplasia | Macrosomia, macrocephaly, macroglossia, coarse square-shaped face, abdominal hernias, broad hands, normal intelligence | Risk of embryonal tumors increased; requires monitoring |
| Sotos syndrome | Nuclear receptor set domain containing protein 1 gene (NSD1) | Frontotemporal sparse hairs, malar flushing | Learning disabilities, distinct facies, macrocephaly, tall stature | Overall cancer risk slightly increased.32 Age appropriate cancer screening recommended33 |
PI3K-AKT activity is considered so essential that disruptive germline mutations are often presumed fatal. For example, an activating PI3K pathway mutation appears only in the mosaic form. In addition to impacting embryonic development, the PI3K-AKT pathway participates in normal cellular function by regulating glucose metabolism and apoptosis.35 Therefore, mutations affecting this pathway may cause congenital anomalies as well as ongoing complications. Improved understanding of the PI3K pathway, especially by revealing therapeutic targets for inhibitory drugs like rapamycin, gives hope for future trials and treatment.36
Tuberous sclerosis
Tuberous sclerosis (TSC, MIM #191100) is a multisystem disorder characterized by tumors and hamartomas affecting the skin, brain, heart, kidneys and lungs, often in association with seizures and developmental delay (Box 29.5). TSC is discussed in further detail in Chapter 23.
Cutaneous findings
There is a very high prevalence of cutaneous findings in TSC.37 Hypomelanotic macules are usually present at birth or appear within the first few months of life and ultimately are present in approximately 90% of TSC patients. Classically, they present as an ‘ash leaf macule,’ an oval area with reduction in pigment (Fig. 29.8A). These areas can also be irregular in outline and shape, or very small and guttate (confetti-like) (Fig. 29.8B). In fair-skinned infants, a Wood’s lamp examination may be necessary for detection. They can also occur on the scalp, with lightening of the hair within the patch. A single, hypopigmented macule in an infant, without other features of TSC, should not cause concern,38 but multiple lesions (≥3) should lead to further investigation.
Classic facial angiofibromas typically appear after 4 years of age, however fibrous plaques resembling coalescence of angiofibromas may be present over the scalp, face, or neck at birth or appear shortly thereafter, as firm slightly raised patches or plaques that are commonly erythematous (Fig. 29.8C). Shagreen patches – firm, palpable thickened dermal plaques – can occur in the lumbar region as a roughened area of erythematous skin with a rubbery consistency or develop in other areas of the torso (Fig. 29.8D). They range in size from a few millimeters to 15 cm in diameter, and generally appear by adolescence but are infrequent in young infants. Periungual fibromas are similarly uncommon in the first decade. Gingival fibromas are a less common feature, but occasionally develop in young children (Fig. 29.8E).
Extracutaneous findings
Seizures occur in more than 60–80% of patients with TSC.39 Conversely 4–50% of infants with infantile spasms have TSC.39,40 TSC patients with early onset of seizures (<2 years of age) or infantile spasms have an elevated risk for intellectual disability. The most common lesions in the brain include tubers, subependymal nodules and subependymal giant-call astrocytomas (SEGA). Renal cysts, angiomyolipomas, and cardiac rhabdomyomas are findings in newborns and infants that suggest TSC. Cardiac rhabdomyomas are often discovered on routine antenatal ultrasound; 30–50% of infants with TSC have cardiac rhabdomyomas, and 80–90% of infants with these lesions have TSC.41 They are rarely symptomatic and typically regress spontaneously.42 An expert panel at the Tuberous Sclerosis Consensus Conference4 recommended a baseline electrocardiogram both at the time of diagnosis and prior to surgery, as cardiac rhabdomyomas can be associated with pre-excitation and arrhythmias on the electrocardiogram.43 Angiomyolipomas are the most common renal manifestation of TSC.
Etiology and pathogenesis
TSC may be caused by an autosomal dominant mutation in the TSC1 gene (encoding hamartin) or the TSC2 gene (encoding tuberin). The two proteins form a complex that is involved with the phosphoinositide-3-kinase (PI3K) signaling pathway, which regulates cell growth and proliferation (Fig. 29.6).44
Differential diagnosis
The differential diagnosis for the hypopigmented macules seen in the neonatal period includes vitiligo, nevus depigmentosus, nevus anemicus, and piebaldism. A single lesion, or several lesions occurring along the lines of Blaschko suggests the diagnosis of nevoid hypopigmentation rather than TSC (see Chapter 23). Connective tissue nevi may be sporadic and have also been associated with Buschke–Ollendorff or Proteus syndrome. Angiofibromas have been described in MEN145 and Birt–Hogg–Dubé syndrome, and also as an isolated autosomal dominant disorder.
Treatment and care
Consensus recommendations for screening and evaluation of TSC were proposed in 1998 and updated in 2000 (Box 29.6).43,46 The Scottish Clinical Genetics Service and the UK Tuberous Sclerosis Association have also created clinical guidelines. Neurology and/or neurosurgery should be consulted for management of seizures, brain tumors, and shunting of obstructive hydrocephalus. Ophthalmology should also examine patients to assist in confirming the diagnosis. Renal ultrasound or renal MRI are important to screen angiomyolipomas. This may also help to identify patients with coexisting polycystic kidney disease due to a contiguous gene deletion syndrome involving the TSC2 and PKD1 genes. Echocardiography and electrocardiogram are recommended at diagnosis for confirmation (to detect cardiac rhabdomyomas and arrhythmias) and to screen for aortic aneurysms.47
Pulsed dye laser has been recommended for flat erythematous angiofibromas, and both Potassium titanyl phosphate (KTP) laser and carbon dioxide laser are used to treat more elevated lesions, but are rarely indicated in infants.48 Fibrous forehead plaques are generally left untreated, but may also be treated with lasers or surgery. Several case reports have recently described the utility of topical rapamycin for the treatment of angiofibromas. This approach holds promise, but further study is needed to establish the safety, efficacy and dosing guidelines.49,50
Proteus syndrome
Proteus syndrome (MIM #176920) is a very rare condition characterized by dramatic segmental or mosaic overgrowth. Common complications include skeletal asymmetry, characteristic overgrowth of the palms or soles referred to as ‘cerebriform connective tissue nevi’ (CCTN),51 linear epidermal nevi, deep or superficial vascular malformations, dysregulated adipose tissue (formerly referred to as lipomas) and tumor predisposition (see also Chapter 22).
Cutaneous findings
Almost all individuals with Proteus syndrome have a dermatologic manifestation. Over 40% of affected neonates will demonstrate at least some evidence of the disease at birth, such as epidermal nevi or vascular malformations.52 The three main cutaneous findings are epidermal nevi, vascular malformations, and soft subcutaneous masses. Epidermal nevi are usually linear and verrucous, but may be macular and hyper- or hypopigmented. Malformations may be venous, capillary, and/or lymphatic.53 The cerebriform connective tissue nevus, when on the sole of the foot, is caused by hyperplasia of cutaneous and subcutaneous tissues and considered virtually pathognomonic.54 The tissue is very firm; similar lesions may also occur on the hands, perinasal area, or near the canthus. Prominent cutaneous venous structures may occur as a result of patchy dermal hypoplasia. Macrodactyly and adipose overgrowth may also be observed.
Extracutaneous findings
Overgrowth in Proteus syndrome is disproportionate, asymmetric, progressive, distorting, and persistent (see Fig. 29.7). Overgrowth usually presents between 6 and 18 months of age and can occur in areas that were completely normal at birth. Overgrowth affects most tissues including bones, cartilage, muscle, and connective tissues. At least some overgrowth and asymmetry is present at birth in 17.5% of cases.52 Orifices may be affected, causing respiratory obstruction, conductive hearing loss, or gastric outlet obstruction. Cystic degeneration of the lungs may lead to pneumonia.55 Affected individuals are predisposed to deadly deep venous thrombosis and pulmonary embolism.56 The central nervous system is commonly affected by hemimegalencephaly (unilateral enlargement of the brain), but most patients are asymptomatic.57 Eye complications are common and range from strabismus to epibulbar hamartomas. Ovarian cystadenomas and cystic lesions of the epididymis are also common.
Etiology and pathogenesis
The cause of Proteus syndrome is a de novo post-zygotic activating mutation in the AKT1 oncogene. Proteus features are an example of somatic mosaicism that is lethal in the non-mosaic state.51 Because Proteus is a mosaic disorder, biopsy of affected tissue is required to make a genetic diagnosis.
Differential diagnosis
Proteus syndrome has been overdiagnosed,58 prompting the creation of diagnostic criteria (Box 29.7) to separate Proteus syndrome from other overgrowth syndromes, many of which share asymmetric hypertrophy as a feature but almost always to a less severe degree.52 Proteus stands apart by having fairly rapid postnatal progression, relentless deforming overgrowth and a poor prognosis.58
Hemihyperplasia-multiple lipomatosis (HH-ML).
This is most commonly misdiagnosed as Proteus syndrome. HH-ML includes superficial capillary vascular malformation (similar to port-wine stain), but lacks progressive, distorting overgrowth, deep vascular malformations and cerebriform connective tissue nevi on the palms or soles.59 In non-distorting overgrowth that characterizes HH-ML, a bone is normally shaped and larger than expected, but without growths or odd edges. In HH-ML, non-distorting overgrowth is present at birth,59 whereas, the distinctive distorting overgrowth that characterizes Proteus may not be apparent until age 2 or 3.52 Lipomas may recur after surgical removal. Therefore, removal of symptomatic lipomas only is recommended.
Megalencephaly-capillary malformation (MCAP).
Sometimes also referred to as M-CM, it has previously been termed macrocephaly-capillary malformation and macrocephaly-cutis marmorata telangiectatica congenital. MCAP sometimes includes markedly large brain size with variable cortical malformation, growth dysregulation with variable asymmetry, patchy capillary malformations frequently on the philtrum, upper lip and nose as well as the limbs and trunk, and distal limb abnormalities such as syndactyly and polydactyly, and mild connective tissue dysplasia (hyperextensible joints and skin).29,60 Congenital Chiari I malformation had been associated with MCAP, however, the term ‘cerebellar tonsil herniation’ (CTH) is now preferred to describe acquired herniation caused by cerebellar overgrowth that occurs in up to 70% of MCAP cases.61 With capillary vascular malformation and asymmetric overgrowth, children often meet the criteria for Klippel–Trenaunay syndrome. Affected children typically have developmental delay (85%) and neonatal hypotonia (68%). Frontal bossing and prominent nevus simplex are additional facial features.60 There is also a mildly increased risk of cancer (about 3%).29 MCAP has been associated with postzygotic mutations in the PIK3CA gene.29
Mosaic overgrowth with fibroadipose hyperplasia (MOFH).
This is an example of an overgrowth syndrome defined by its newly identified causative mutation, an activating mutation in the PIK3CA gene.62 Affected children have segmental overgrowth that affects the muscles, skeleton, and fibroadipose tissue without cutaneous features of Proteus syndrome. Both distorting and non-distorting overgrowth are reported. The features of MOFH often begin at birth, in contrast to Proteus syndrome which generally appears later.62
SOLAMEN syndrome.
Segmental overgrowth, lipomatosis, arteriovenous malformation and epidermal nevus (SOLAMEN) is a ‘Proteus-like’ syndrome,63 now known to be a mosaic form of Cowden disease. Affected individuals carry a germline PTEN mutation and a mosaic second hit to the PTEN gene on the opposite allele in affected tissues.63 Absence of the cerebriform connective tissue nevi on the palms and soles distinguishes SOLAMEN syndrome. In addition, characteristic features of Cowden disease such as macrocephaly, breast and thyroid hamartomas and skin changes are likely to be present.63
CLOVES syndrome (MIM #612918).
Congenital lipomatosis, overgrowth, vascular malformations, epidermal nevi and skeletal anomalies (CLOVES) describes another subset of patients, most of whom were initially misdiagnosed with Proteus syndrome.58 In CLOVES syndrome, overgrowth is present at birth with truncal vascular anomalies, truncal lipomatous masses and overgrown feet with acral or musculoskeletal anomalies. In contrast to Proteus, the overgrowth is not progressive.64 Patients may have wrinkled skin on the palms and soles, but lack the firm rubbery CCTN of Proteus syndrome. Since CLOVES syndrome was suspected to be a mosaic disorder at the outset, massive DNA sequencing of affected tissues revealed causative activating mutations in PIK3CA.65 Severe scoliosis, large truncal masses, high flow vascular lesions, phlebectasia with thromboembolism and orthopedic problems of the hands and feed may all require early, aggressive intervention in affected individuals.64 Prognosis seems better compared to Proteus syndrome but this condition can still be very severe, even life-threatening.66
Encephalocraniocutaneous lipomatosis (ECCL).
The so-called ‘nevus psiloliparus’ is the cutaneous hallmark, with a localized non-scarring patch of alopecia on the scalp which may or may not overlie a fatty tissue mass. ECCL, while once thought to be a localized form of Proteus syndrome, does not meet the diagnostic criteria of Proteus, lacking disproportionate growth31 as a salient feature (see also Chapter 31).
Differential diagnosis of Proteus and similar disorders
Maffucci syndrome consists of multiple enchondromatosis (which may be confused with hyperostosis) and vascular malformations.67 Klippel–Trenaunay (MIM #149000) syndrome consists of vascular malformations with overgrowth, typically in the same segment. Axillary freckling and neurofibromas help distinguish neurofibromatosis.
Management
Management of these syndromes depends on the specific disease; if overgrowth is severe – as in Proteus syndrome – management is often very difficult. A multidisciplinary approach best addresses all aspects of the disease adequately. Ongoing ophthalmology, neurology, and developmental assessment exams are recommended.52 Caregivers should be informed of the risk of pulmonary embolism and stroke so that healthcare providers consider it if an emergency arises.52
[/not-level-membership-for-dermatology-category]
