Vascular Malformations
Sheilagh M. Maguiness, Maria C. Garzon
Introduction to vascular malformations
In 1982, Mulliken and Glowacki1 proposed a biologic classification of vascular birthmarks that has become widely accepted. It was modified slightly in 1996 by the International Society for the Study of Vascular Anomalies (ISSVA).2 Two major groups of vascular birthmarks are recognized: vascular malformations, which are composed of dysplastic, malformed vessels; and vascular tumors, which demonstrate cellular hyperplasia. The distinction between malformations and tumors is emphasized by their varying histologic appearance, cellular markers, and natural history (see Chapter 21 for a full review of vascular tumors).3,4 The ISSVA classification of vascular anomalies allows for more precise diagnoses and helps avoid the perpetuation of out-dated terminology such as ‘cavernous hemangioma,’ which has been previously used to describe both tumors and malformations (Table 22.1).3
TABLE 22.1
ISSVA Classification of vascular anomalies
| Vascular tumors | Vascular malformations |
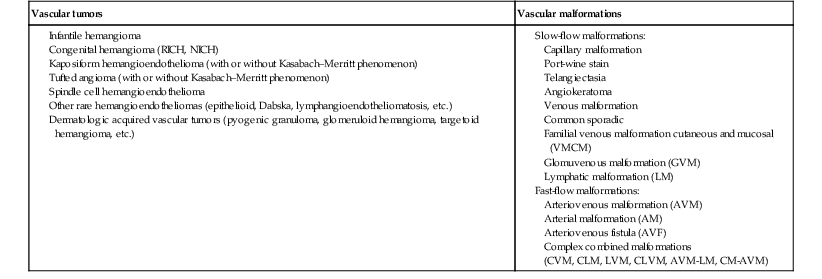
(Mulliken and Glowacki 1982; The International Society for the Study of Vascular Anomalies, ISSVA 1996, 2003)
Adapted from Enjolras O, Wassef M, Chapot R. Color atlas of vascular tumors and vascular malformations. New York, NY: Cambridge University Press; 2007.
Vascular malformations are subcategorized according to flow characteristics and predominant anomalous channels. They can occur alone or in combination: slow-flow malformations (capillary, C; venous, V; lymphatic, L) or fast-flow malformations (arteriovenous malformation, AVM; and arteriovenous fistula, AVF). Vascular malformations are often localized and circumscribed lesions, but can also present in a segmental/regional pattern or in a multifocal, disseminated form. Vascular malformations can also be observed in the setting of various syndromes, many of which arise due to known genetic mutations. In several cases, the discovery of the genetic mutation has advanced our understanding of the pathogenesis of the underlying conditions. A number of complex and/or combined vascular malformations also exist: CVM, CLVM, LVM, AVM, CAVM, CLAVM, and so forth, with some of them known by eponyms. In this chapter, we discuss a variety of these sometimes eponymous syndromes associated with vascular malformations. We have grouped them according to their predominant malformation but recognize these categories are not always rigid and significant overlap exists.
Capillary malformations
Nevus simplex (salmon patch, fading capillary stain)
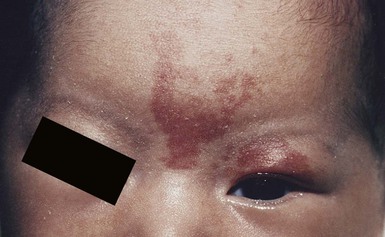
A salmon patch is a capillary malformation (CM), also known as an ‘angel kiss’ when located on the forehead or eyelids, and ‘stork bite’ when located on the nape of the neck. It is present in nearly half of all newborns and affects males and females equally. It has a characteristic predilection for the midline, with the most common locations being the nape, upper glabella, nose, and upper lip (Fig. 22.1). The occiput and lower back may also be affected.5 If a salmon patch involves the upper eyelid and does not occur concomitantly with the classic V-shaped patch in the middle of the forehead, it may be difficult to differentiate from a partial V1 port-wine stain or a hemangioma precursor. Occasionally, widespread involvement of nevus simplex at many different sites can exist – the term ‘nevus simplex complex’ has recently been proposed to describe such cases in an attempt to prevent confusion with port-wine stains.6 Nevus simplex usually disappears within 1 or 2 years, but some persist, particularly those at the nape.7
A nevus simplex localized to the midline sacral region has sometimes been referred to as a ‘butterfly-shaped mark’. Involvement at this site is most often seen in infants with multiple salmon patches elsewhere. In most cases, these are benign and not associated with underlying spinal dysraphism. However, prospective studies with MR imaging are lacking, so it is not possible to exclude this risk completely. In the absence of any other concerning signs, a nevus simplex in this location is usually innocent.
Although nevus simplex usually do not have underlying associations, rarely they are a manifestation of certain syndromes, such as Beckwith–Wiedemann, macrocephaly–capillary malformation (MCM/MCAP), or Nova syndromes (see below).
Capillary malformations
The terms port-wine stain (PWS) and capillary malformation (CM) are used interchangeably. CMs are almost always evident at birth and found in up to 0.3% of newborns.8 They are mostly sporadic in origin. Histopathologically, they are comprised of normal capillaries within the superficial dermis without evidence of proliferation. CMs can be observed in the setting of several different syndromes discussed later in this chapter.
Pathogenesis
The etiology of CM is not fully understood, however recent reports of mutations in GNAQ in both syndromic and non-syndromic CMs point to an underlying genetic basis.9 Another familial form of CM occurring with AVM has been reported in the CM-AVM syndrome.
Cutaneous findings
PWS are pink or red patches that typically grow proportionately with the child’s somatic growth, and persist throughout the patient’s life. They consist of ectatic dermal capillaries and may occur anywhere on the body (Fig. 22.2A). PWS may appear to lighten over the first 3–6 months of life, but this should not be taken as a sign of resolution. Rather, this physiologic lightening is due to the decrease in blood hemoglobin concentration corresponding to the change from fetal to adult type hemoglobin (Fig 22.2B).10 Capillary malformations can occur anywhere on the body. They may or may not be associated with overgrowth of the affected area (Fig. 22.2). Port-wine stains on the face tend to follow a dermatomal pattern, respecting the trigeminal nerve V1-V3 distribution or comprising multiple dermatomes (Fig. 22.3). The natural history of port-wine stains over a lifetime is often one of gradual darkening from pink-red to a crimson or deep purple hue. Skin thickening and soft tissue and/or bony hypertrophy may also develop. Eczematous changes can occur in PWS and salmon patches, either with or without treatment. Nodular vascular lesions (pyogenic granulomas) may appear within PWS during childhood or adult life and may require surgical intervention.11 The association of PWS with pigmentary anomalies such as extensive Mongolian spots, nevus spilus, or nevoid hyperpigmentation is a feature of phakomatosis pigmentovascularis (see further discussion of this entity below, Fig. 22.4, Table 22.2).
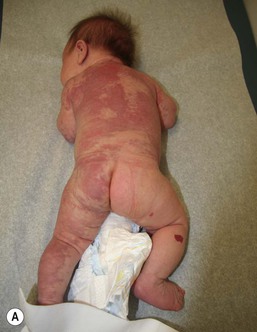
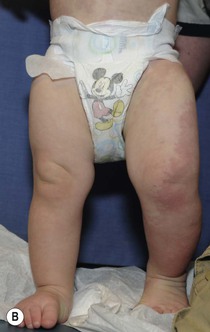
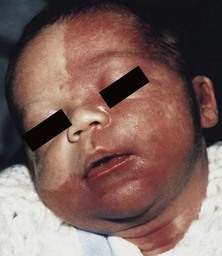
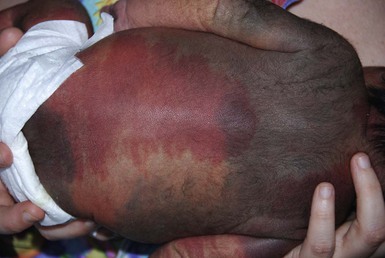
TABLE 22.2
Classification of phakomatosis pigmentovascularis
| Traditional classification | Alternate (Happle) classification | Vascular anomaly | Pigment anomaly | Other |
| I | – | CM | Epidermal nevus | – |
| II | Cesioflammea | CM | Dermal melanosis (Mongolian spots, Nevus of Ota) | ± Nevus anemicus |
| III | Spilorosea | CM | Nevus spilus | ± Nevus anemicus |
| IV | Unclassifiable | CM | Dermal melanosis (Mongolian spots, Nevus of Ota) Nevus spilus |
± Nevus anemicus |
| V | Cesiomarmorata | CMTC | Dermal melanosis | – |
Extracutaneous findings
Progressive soft tissue and bony overgrowth may occur during childhood, especially with V2 PWS, and subtle changes are sometimes noted even in the neonatal period.12 Asymmetric maxillary hypertrophy associated with distortion of the facial features requires multidisciplinary management with orthodontic follow-up and treatment. Some patients require procedures to correct skeletal overgrowth in late childhood. Gingival hypertrophy may also develop. Port-wine stains are also a manifestation of several different syndromes with other extracutaneous features discussed later in the chapter (see ‘Syndromes associated with vascular malformations’, below).
Management
The gradual thickening and nodularity of PWS provide a medical rationale for treatment during infancy and childhood.11 The flashlamp-pumped pulsed dye laser (PDL) is the gold standard treatment for PWS and poses a very low risk of scarring, even in young infants. It has been used for longer than 15 years and advances such as the development of a cooling system have improved efficacy. Most PDL units have a wavelength of 595 nm. Settings of 0.45–1.5 ms pulse duration, 6–10 J/cm2 fluences, and 7–10 mm spot size along with the coolant system (dynamic cooling device) are used in the majority of cases. Although only 15–20% of PWS clear completely with PDL, the majority of treated lesions lighten significantly.13,14 Response to laser treatment varies by area and skin type: outcomes are better on the face and neck, albeit with less improvement in V2 than other facial sites, and are also better in patients with type I–II skin.14 The extremities do not respond as well.13–15 Controversy exists regarding the age at which treatment should begin. Some authors have noted a better therapeutic response with fewer treatments when treatment was initiated in early infancy,16 but others have found no difference.17 Treating earlier (in infancy or early childhood) can be helpful in reducing stigmatization and may help prevent thickening of the skin.18 Treating early in infancy may enable the physician to treat with topical anesthesia, as small infants can be swaddled and held during a brief treatment and local anesthetic can be applied prior to the procedure. In older children with large stains, general anesthesia may be required. In most cases, re-darkening occurs with time and touch-up treatments are often needed in later childhood or teen years.19
In treatment-resistant CM, other laser modalities have recently been studied in an attempt to decrease thickness, color, and nodularity. These include the long-pulsed neodymium : yttrium-aluminum-garnet (Nd : YAG) laser, the combined PDL and Nd : YAG laser, and the alexandrite laser.20–24
Other novel therapies show some initial promise, including intense pulsed light (IPL), photodynamic therapy (PDT), and application of topical anti-angiogenic factors (imiquimod and rapamycin) in conjunction with laser ablation.25–35
In cases where there is soft tissue or bony overgrowth, or in syndromic CM, a multidisciplinary approach to management is often necessary. Patients may require orthodontic work, maxillofacial reconstruction or soft tissue debulking. Ideally, follow-up in a multidisciplinary vascular anomalies center is beneficial.
Telangiectasia
Though not true CMs, telangiectasias can present in infancy or early childhood and often persist. They are usually composed of small, punctate linear vessels distributed in either a segmental, unilateral nevoid, or diffuse pattern. A variety of telangiectatic skin lesions have been described. Most are absent at birth, often developing in childhood. A pale halo of vasoconstriction may surround small telangiectasias. Toddlers and young children sometimes develop so-called spider angiomas comprised of a brightly erythematous central punctum with radiating ‘spider-like’ telangiectasias. They are usually located on sun-exposed areas. Risk factors include fair skin and a history of minor skin injury at the site. These may disappear spontaneously over years, or persist. Fine telangiectasias are occasionally present on the cheeks of normal young children, but extensive telangiectasias in the photo-distribution should prompt consideration of conditions with photosensitivity such as Rothmund–Thomson syndrome. Persistent telangiectasias can also be seen as a sequela of neonatal lupus. The facial telangiectasias associated with hereditary hemorrhagic telangiectasia (HHT) and ataxia–telangiectasia usually present later in life.
Venous malformations
Venous malformations (VM) are slow-flow vascular malformations that are usually evident at birth. They may involve skin, mucosa, subcutaneous tissues, muscle and rarely bone. They are composed of ill-defined venous channels with irregularly attenuated walls, focally lacking smooth muscle cells, permeating the skin and adnexal structures. Glomuvenous malformation (GVM) is a subtype of VM composed of anomalous venous channels lined by glomus cells, which are actin positive and do not lack smooth muscle. Glomus cells have a characteristic appearance on histopathology.
Pathogenesis
VMs are sporadic in the majority of cases, but, familial cases of both VM and (more often) GVM occur. The genetic basis for familial VMs (VMCM syndrome, see below) and GVM are now known. The mutated gene in familial VMCM syndrome maps to chromosome 9p17; and is an activating mutation in the kinase domain of the receptor tyrosine kinase Tie2.36 Sporadic Tie2 mutations are also found in 50% of sporadic VMs.37 GVM can be either a sporadic or an autosomal dominant condition, linked to mutations in the glomulin gene (mapping to 1p21–p22). Unlike VMs, far more cases of GVM are familial (approx. 64%).38,39
Cutaneous findings
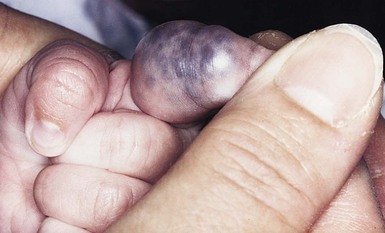
At birth, the majority of VM are subtle, bluish, ill-defined, compressible plaques or nodules. Rarely, a bulky venous mass is observed in the neonatal period (Fig. 22.5). VMs may be diffuse or localized. Affected skin and mucous membranes are typically blue with normal skin temperature, without a thrill or bruit. Lesions swell when dependent or with crying. In many cases, the blue-hued cutaneous component heralds involvement of deeper structures. Local venous thromboses can lead to the formation of phleboliths, which can be tender when palpated and are visible on radiographs as round calcifications.
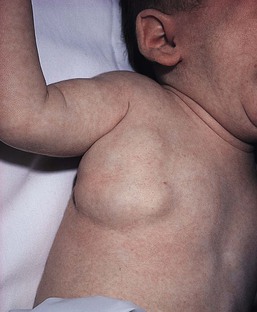
Glomuvenous malformations (GVM; formerly called ‘glomangioma’) may be solitary or multiple, and may be localized or involve a larger territory of skin (Fig. 22.6). They are often bluish to purple, cobblestoned or plaque-like in appearance. During childhood, they typically acquire a deeper blue hue and thicken, and become tender when palpated. Congenital plaque-like GVMs are usually pink at birth, with noticeable thickening and change in color to blue-purple during childhood. These plaque-like GVMs may arise sporadically, or occur as a manifestation of autosomal dominant GVM. Other family members may have smaller, blue vascular lesions scattered over the skin, increasing in number with age.38
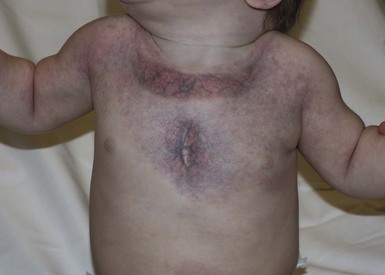
Extracutaneous findings
In addition to skin and mucosal involvement, VM can also involve deeper soft tissues, muscles, joints, and in severe cases, visceral sites such as the abdomen and pelvis. A majority of patients with extensive VMs develop a chronic localized intravascular coagulopathy (LIC), which is distinct from Kasabach–Merritt phenomenon (KMP). VM associated LIC can result in either thrombosis (with pain and phlebolith formation) or bleeding, and persists throughout life. It differs from KMP because the primary process is one of ongoing clotting, with consumption of clotting factors, low fibrinogen and elevated d-dimers, but without the marked thrombocytopenia of KMP.40,41 Large and intramuscular VMs are more likely to have associated VM-LIC. Pain is a common complaint either due to phlebolith formation or often in larger extremity lesions.42 ‘Blue rubber bleb nevus’ syndrome, the association of multiple cutaneous venous malformations with gastrointestinal and other internal lesions, is discussed later in this chapter. Some patients with VM experience worsening of symptoms during puberty or in pregnancy. In contrast to VMs, GVMs are typically localized to the skin and soft tissues without mucosal or intramuscular involvement. GVMs are not associated with LIC.
Diagnosis
The diagnosis is usually established on the basis of clinical features. Imaging modalities including ultrasonography, Doppler, MRI, and CT scans are useful for evaluating the extent of involvement.43 The finding of associated LIC and specifically elevated d-dimer levels, can help confirm the diagnosis of VM in uncertain cases.44 Apart from helping to confirm the diagnosis, the decision to image early in infancy depends on whether functional problems are present or early treatments are planned. Many VMs, however, especially larger ones, eventually do require imaging studies, and MRI with contrast is the best study to delineate disease. A number of radiologic classification schema have been proposed and may predict response to treatment, with smaller and more localized lesions being more amenable to sclerotherapy.45 In individuals with craniofacial VMs, it is advisable to image the brain: developmental venous anomalies (DVA) in the brain are more common in patients with craniofacial VMs than in the general population (25% vs 0.5%).46 DVAs are uncommon trajectories of the brain’s venous drainage and pose little risk of cerebral hemorrhage, but documenting their presence can help avoid misdiagnosis of a more worrisome condition later in life. Histopathology can also help to confirm the diagnosis when uncertainty is present and/or to distinguish between VM and GVM.
Differential diagnosis
Extensive VMs in a leg or an arm and adjacent trunk must be differentiated from Klippel–Trenaunay and other overgrowth syndromes, as management and prognosis is variable (see Table 22.4). Sinus pericranii should be considered in the differential diagnosis of a VM located on the central forehead. This presents as a bluish, nonpulsating mass that is usually congenital and quickly expands when the patient puts their head in a dependent position or with crying. Sinus pericranii represents a direct communication between superficial veins and intracranial venous sinuses through a bony defect. It is best imaged using CT with bone windows.
Course
Venous malformations tend to enlarge and become more symptomatic with time. Their clinical course and complications depend on anatomic location, with differing problems in the craniofacial area, trunk, and limbs.41 The cheek, lip and tongue are common locations for craniofacial VMs, and there is sometimes deep extension to the temporal and orbital areas. Swelling is noted with dependency and activity. Over time, the VM may progressively distort the facial features and mold the underlying developing bones, which can result in deformities such as an open bite or enlargement of the orbit. Extensive retropharyngeal involvement can result in obstructive sleep apnea, occasionally even in young children. VMs located on the trunk and limbs may involve skin, skeletal muscles, joints, and bones, often causing pain. During infancy and early childhood, the skin component of the VM expands and becomes deep blue. However, the deeper component may remain undiagnosed until it becomes symptomatic. Swelling, functional impairment, and limited joint motion occur when the child becomes older and more active, especially if playing sports. VM-LIC associated with large and intramuscular VM can manifest as early as the neonatal period.
Management
Due to the heterogeneity of VMs and the clinical and radiologic diagnostic overlap with other vascular anomalies, multidisciplinary management is recommended. VMs are reported to comprise up to 40% of referrals to vascular anomalies centers.47 VMs can be treated with many different modalities including sclerotherapy, excision, endovenous laser ablation, cutaneous laser ablation or a combination of these modalities. Sclerotherapy has emerged as the mainstay of treatment for VMs, followed by surgical excision. The decision regarding when to treat is usually based on whether there is significant functional impairment or disfigurement. Sclerotherapy involves the introduction of an endothelial-cell cidal sclerosant into the vascular spaces of the malformation.48
Several different sclerosants can be used, including ethanol or sodium tetradecyl sulfate. Complications of sclerotherapy include tissue necrosis, nerve palsies, hemoglobinuria and oliguria.49 Surgical management can be most helpful in small or localized lesions. A combination of sclerotherapy followed by surgical debulking is often used in large or complex cases. In craniofacial lesions, therapy is aimed at preventing distortion of facial anatomy, limiting bone deformity, open bite or shift of the dental midline, lip expansion, and displacement of the lip commissures.50,51 MRI and ultrasound features may be especially helpful in determining optimal management.51 Multiple treatments are often required over years. Laser or radiofrequency ablation are occasionally helpful. Conservative therapy with compression should be initiated early on and is even encouraged from infancy in diffuse extremity VMs. Compression increases comfort, limits swelling, and improves coagulopathy. Compression is less useful for intramuscular VMs and may exacerbate pain.
Evaluation of large VMs includes assessment for, and possibly treatment of, VM-LIC. d-dimer levels should be obtained periodically to assess the extent of coagulopathy. Treatment with low-dose aspirin can be a helpful adjunct to conservative therapy in some cases. Low molecular weight heparin (LMWH) has been found to be useful in patients with chronic VM-LIC during episodes of exacerbation (thrombosis, pain) or before and after planned procedures such as sclerotherapy or surgical excision, to prevent thrombosis and/or control intraoperative hemorrhage.52 In patients with a history of thromboembolic events or in those with high risk, large and/or combined malformations, placement of an inferior or superior vena cava filter may help prevent pulmonary embolus.53
Lymphatic malformations
Lymphatic malformations (LMs) are slow-flow vascular anomalies known in much of the literature as ‘lymphangioma’. They can be macrocystic, microcystic, combined or very rarely generalized. LMs arise in a sporadic manner and can also be syndromic.
Pathogenesis
The etiology of LMs is still unknown. LMs arise in a sporadic manner and no associated genetic basis has yet been discovered. This may suggest that a genetic cause is likely related to somatic mutations, which, if occurring within the germline, would be incompatible with life.54 This is in contrast to primary lymphedema, where several genetic mutations leading to specific lymphedema syndromes have been discovered and have improved our understanding of the molecular pathways involved in lymphangiogenesis.55 In primary lymphedema, defective lymphatic drainage leads to accumulation of lymphatic fluid in the interstitial space (Fig. 22.7).
Several forms of lymphedema are known, including familial congenital lymphedema, Nonne–Milroy syndrome, Meige disease, and Hennekam syndrome, summarized in Table 22.3.
TABLE 22.3
Hereditary lymphedema syndromes
| Syndrome | Genetic basis | Clinical features |
| Primary congenital lymphedema (Nonne–Milroy disease) | Autosomal dominant Mutations in VEGFR3 |
Congenital lymphedema of lower limbs, mainly below knees. Slow to progress. Can be associated with hydrocele, enlarged leg veins and recurrent cellulitis |
| Pubertal onset lymphedema (Meige disease, lymphedema distichiasis syndrome) | Mutations in FOXC2 | Pubertal onset, most common type of primary lymphedema. Can be associated with distichiasis, ptosis, yellow nails, syndactyly, cleft palate and cardiac septal defects |
| Hypotrichosis–Lymphedema–Telangiectasia syndrome | Autosomal dominant and recessive forms reported mutations in SOX18 | Lymphedema, sparse hair, cutaneous telangiectasias |
| Hennekam syndrome (generalized lymphatic dysplasia, GLD) | Autosomal recessive Mutations in CCBE1 in some cases |
Lymphedema, intestinal lymphangiectasia, protein-losing enteropathy, developmental delay, multiorgan involvement |
Cutaneous findings
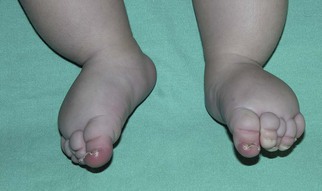
Macrocystic LMs are usually visible at birth and are commonly diagnosed by prenatal ultrasound investigation. They occur more commonly in the neck and axilla, where they are often referred to as cystic hygroma (Fig. 22.8).56 The detection in utero of some huge LMs of the neck, axilla, and thoracic area may lead to a discussion about terminating the pregnancy, as the prognosis is poor. Microcystic LMs can infiltrate diffusely throughout the dermis. Clear or hemorrhagic vesicles (so-called ‘lymphangioma circumscriptum’), which may intermittently leak lymphatic fluid, may be visible on the surface of the lesion. Severe combined LMs may also occur on the trunk and limbs.57
Extracutaneous findings
Cervicofacial LMs involving the tongue and floor of the mouth will interfere with normal development of the jaw and create an open bite deformity. The most severe forms of combined micro/macrocystic LMs, which are more common on the head and neck, can cause life-threatening airway disease and other functional problems in the neonate (Fig. 22.9). Intraoral involvement also causes drainage of serosanguineous lymphatic fluid, aggressive caries, and loss of teeth. Involvement of the mandible with ensuing overgrowth is present in about 40% of these patients.58,59 Extra- and intraconal orbital LM occurs in association with eyelid LM; this uncommon location causes severe complications including disfigurement, bleeding, infection, proptosis, and visual loss.60 Severe cervicofacial LMs, usually the combined micro/macrocystic type, involving the hypopharynx and larynx, the tongue and the floor of the mouth, can cause airway and esophageal obstruction requiring nasogastric tube feeding and emergency tracheotomy in the newborn.61 Of 31 cases with such severe involvement, 58% required tracheostomy in infancy and one-third could not be decannulated.59 Visceral LMs, intrathoracic or abdominal, are less common, representing about one-tenth of cases. LMs are more susceptible to bacterial infections, and recurrent cellulitis can be a complication.
Diagnosis
The diagnosis is established either clinically or using CT, MRI, or ultrasound. Fluid aspiration and analysis may be helpful because a number of neonatal growths, including some malignant tumors, can present with large cyst-like swellings.62 Histologically, lymphatic vessels or cysts have thin walls and lumina appear empty. Several positive immunohistochemical markers of lymphatic vessels have been identified, including LYVE-1, VEGFR-3 and D2-40, and can help to differentiate LM from other vascular anomalies.
Treatment
Lymphatic malformations are benign and treatment is usually directed at managing complications or attempting to restore anatomy. Conservative measures include observation, prophylactic antibiotics when infection is a concern, and in the case of primary lymphedema, compression. Sclerotherapy has emerged as an important treatment for LMs, and can be done early, even in the neonatal period. A variety of sclerosing agents have been used, including doxycycline, ethanol, bleomycin, OK-432 and STS. Macrocystic LMs respond very well to sclerotherapy. Surgical resection is another therapeutic option for macrocystic LMs, as well as for microcystic and combined types, but recurrences and complications such as seroma or chronic lymphatic leakage may occur.58–60,63,64 Radiofrequency ablation can be helpful in the management of LMs, most commonly for the lips and tongue to help decrease chronic leakage and bleeding.65,66
A multidisciplinary approach is required for complex LMs.59
Arteriovenous malformations
Arteriovenous malformations (AVMs) are fast-flow anomalies that most commonly arise in the head and neck area. Macular erythema mimicking a PWS, with increased local warmth, and subtle skin thickening can be clues to the diagnosis.
Pathogenesis
Arteriovenous malformations arise due to errors in vascular development, which occur during embryogenesis. AVMs represent direct shunting from the arterial to the venous system due to the absence of an intervening capillary bed. Recently, the genetic basis for familial types of AVM has been discovered. Mutations in RASA1 have been found in individuals with AVM of the extremity (Parkes Weber syndrome) and in CM AVM syndrome (see below for further discussion on Parkes Weber syndrome), vein of Galen aneurysmal malformation, and other fast-flow vascular anomalies caused by RASA1 mutations.9 Other mutations associated with AVMs or AVM variants include endoglin, which gives rise to hereditary hemorrhagic telangiectasia syndrome (HHT) and PTEN as seen in high flow lesions of the PTEN hamartoma tumor syndrome (see below).
Cutaneous findings
Most commonly, AVMs present as a pink vascular stain on the skin, mimicking port-wine stain. They are often located on the head and neck, the nose and central face/lip are common locations as is the ear (Fig. 22.10). As mentioned above, lesions are warm and may have a thrill or a bruit. AVMs may develop pyogenic granulomas within them that bleed and may need to be excised or treated with laser ablation.67 Any patient who presents with small, multifocal lesions that resemble PWS but demonstrate high flow on Doppler examination should receive a workup for CM AVM syndrome (see below). Lesions may become thicker, more nodular and darker in color with time. AVMs often progress during puberty or with trauma.
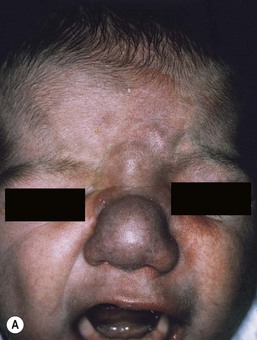
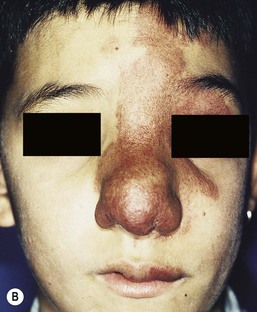
Extracutaneous findings
AVMs can be life-threatening lesions that worsen over time. Complications include disfigurement, pain, bony erosion, hemorrhage, and even death. Large AVMs may have associated high-output heart failure at birth or later in life, requiring intervention.
Diagnosis
Most AVMs can be diagnosed based on their natural history and physical examination characteristics. They may not be obvious at birth, most commonly lesions mimic port-wine stain, but unlike PWS, AVMs expand through childhood. In general, AVMs are warm, a bruit may be audible, and a thrill may be palpable. An actual vascular mass with tense draining veins, a thrill, and a bruit is uncommon for AVM in a newborn, and may represent a congenital hemangioma (see Chapter 21). The Schobinger Classification of AVMs divides the presentation into four stages based on severity and associated findings: Stage I, lesions are quiescent; Stage II, AVMs have expanded and a thrill or bruit is palpable; Stage III, AVM destruction occurs and lesions can lead to ulceration, bleeding and pain; and Stage IV AVMs are associated with cardiac compromise.68
Color Doppler ultrasonography as well as MR with MR angiography may be helpful in diagnosis, as well as delineating the extent of disease. Angiography is sometimes done to aid in diagnosis or in preparation for embolization or resection. In contrast to VMs, AVMs are not known to be associated with LIC.
Management
Early on, conservative observation may be the best course for Stage I and II AVMs, as interventions may lead to enlargement or worsening of the lesions secondary to trauma. In more advanced malformations, where there are complications or functional compromise, embolization is usually the first-line treatment. Embolization can also be done prior to a planned surgical resection.69 Embolization involves the delivery of an embolic agent, usually ethanol, onyx, polyvinyl alcohol or coils, into the AVM in an attempt to block the blood flow and reduce shunting.70 Embolization is considered a palliative measure, as there is no cure for AVMs. Surgical management of AVMs should be performed with caution as exacerbation or recurrences are common and any resulting deformity may not be cosmetically acceptable.70
Combined malformations
Vascular malformations occur most commonly as described above, manifesting as CM, VM, LM, or AVM. However, any combination of malformed vessels may occur together and a number of combined malformations are well recognized. In the past, many of these disorders were named eponymously, which sometimes leads to confusion in the diagnosis of these rare entities. Classification based on the vessel type predominant in the malformation may be a better way to distinguish them. Nonetheless, many of the eponyms persist and are widely used. Many of the combined vascular malformations are associated with overgrowth of the affected areas of the body (Table 22.4). Most commonly, capillary-lymphatic-venous malformation (CLVM) is described in association with various conditions including Klippel–Trenaunay and CLOVES. Combined fast-flow malformations such as capillary arteriovenous malformation (CAVM) and capillary arteriovenous fistulae (CAVF) are also described and are the main presenting feature of Parkes Weber syndrome. Some CAVM patients have underlying RASA1 mutations or CM-AVM syndrome (see below).
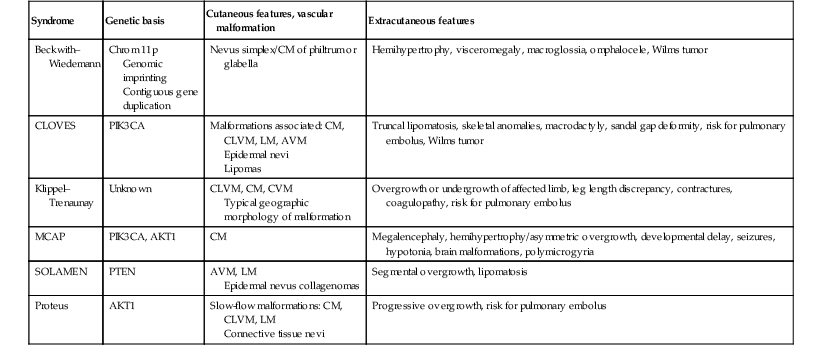
TABLE 22.4
Vascular malformation syndromes associated with overgrowth
| Syndrome | Genetic basis | Cutaneous features, vascular malformation | Extracutaneous features |
| Beckwith–Wiedemann | Chrom 11p Genomic imprinting Contiguous gene duplication |
Nevus simplex/CM of philtrum or glabella | Hemihypertrophy, visceromegaly, macroglossia, omphalocele, Wilms tumor |
| CLOVES | PIK3CA | Malformations associated: CM, CLVM, LM, AVM Epidermal nevi Lipomas |
Truncal lipomatosis, skeletal anomalies, macrodactyly, sandal gap deformity, risk for pulmonary embolus, Wilms tumor |
| Klippel–Trenaunay | Unknown | CLVM, CM, CVM Typical geographic morphology of malformation |
Overgrowth or undergrowth of affected limb, leg length discrepancy, contractures, coagulopathy, risk for pulmonary embolus |
| MCAP | PIK3CA, AKT1 | CM | Megalencephaly, hemihypertrophy/asymmetric overgrowth, developmental delay, seizures, hypotonia, brain malformations, polymicrogyria |
| SOLAMEN | PTEN | AVM, LM Epidermal nevus collagenomas |
Segmental overgrowth, lipomatosis |
| Proteus | AKT1 | Slow-flow malformations: CM, CLVM, LM Connective tissue nevi |
Progressive overgrowth, risk for pulmonary embolus |
Syndromes associated with vascular malformations
The syndromes described below are associated with various vascular anomalies. We have elected to group them based on the most prominent or characteristic cutaneous vascular anomaly as this is often the first presenting sign of the disorder. Most of these malformations are evident at birth or become apparent in infancy or early childhood. In some cases, the genetic basis for the syndrome is known.
Syndromes associated with capillary malformations
Sturge–Weber syndrome
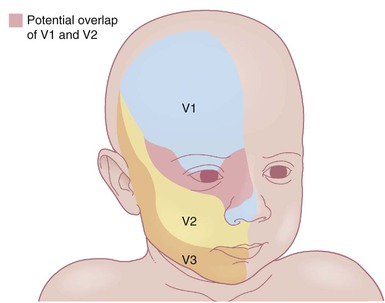
The classic triad in Sturge–Weber syndrome (SWS) includes the association of a facial port-wine stain, invariably involving V1 (although it may be more extensive) (Fig. 22.2), ipsilateral eye abnormalities (choroidal vascular anomalies, increased ocular pressure, buphthalmos, and glaucoma), and leptomeningeal and brain abnormalities (leptomeningeal vascular malformation, calcifications, cerebral atrophy, enlarged choroid plexus, and developmental venous anomalies in the brain). The risk of SWS with V1 PWS alone is variably reported but is approximately 10%. The risk increases to ≥25% with either bilateral V1 or concurrent V1, V2, and V3 involvement. Patients with V2 or V3 PWS alone without involvement of the V1 skin are not at risk for SWS. However, individual anatomic variations in the distribution of V1 and V2 at the internal or external canthus of the eye may pose difficulties in determining whether a port-wine stain involves V1, with its associated risk of SWS (Fig. 22.11). The possibility of SWS should be considered in any infant with a PWS that includes the V1 distribution.71
SWS can cause significant medical and ophthalmologic problems. Consequences of intracranial vascular anomalies include seizures, headaches (including migraines), spastic hemiparesis, visual field defects, cognitive impairment and behavioral disorders including attention deficit disorder. Transient ischemic attacks and strokes may also occur in some patients. An increased prevalence of growth hormone deficiency and hypothyroidism is described in patients with SWS, so at-risk individuals need to be assessed for these potential complications.72 Potential visual loss via acute or chronic glaucoma requires ongoing ophthalmologic follow-up throughout a patient’s lifetime. Even an individual who has a V1 PWS without CNS findings should have periodic ophthalmologic evaluations throughout their lifetime.
The pathogenesis of SWS has recently been attributed to somatic activating mutations in GNAQ.9 The three mesectodermal tissues involved (the nasofrontal skin known as V1 skin, the ocular choroid, and the leptomeninges) have a common origin in the anterior neural primordium and a somatic mutation arising during development has been hypothesized. Neuroimaging consisting of MRI with gadolinium enhancement may be helpful in making an early diagnosis, but can be normal in some cases. Early subtle changes on standard MRI can include an enlarged choroid plexus or a pattern of local accelerated myelination. Typical neuroimaging changes include visualization of the pial vascular malformation, cerebral atrophy, and calcifications of the leptomeninges, the abnormal cortex and the underlying white matter.73 Newer MRI modalities such as susceptibility-weighted imaging may prove useful in detecting abnormalities earlier in life. In most patients, the first seizures in SWS occur before 2 years of age but may arise later in life. The progressive nature of SWS has been demonstrated using functional neuroimaging tools, with hyperperfusion noted prior to the development of seizures, followed by hypoperfusion with decreased glucose utilization after the onset of seizures.74,75 Newer noninvasive tools including quantitative electroencephalography and transcranial Doppler are under investigation and may prove promising for the evaluation and management of children with SWS.72
Careful follow-up is recommended for newborns with an at-risk V1 PWS. Although controversial, some pediatric neurologists believe that prophylactic anti-seizure medications or low dose aspirin regimens are worth considering for at-risk infants.76,77 Therapeutic management of SWS (Box 22.1) is often characterized by a lifelong struggle to preserve vision, motor and psychomotor development, and to ameliorate disfigurement.
Macrocephaly capillary malformation syndrome (megalencephaly capillary malformation polymicrogyria syndrome)
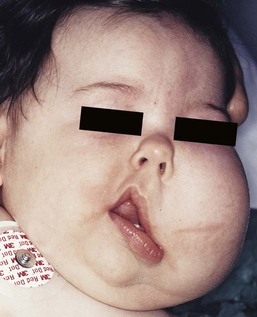
Macrocephaly capillary malformation syndrome (MCM/MCAP) was formerly known as macrocephaly CMTC. However, the most commonly associated vascular anomalies are a persistent nevus simplex or typical CM/PWS (not CMTC) and the diagnostic criteria for this condition were revised to reflect this.78,79 MCM/MCAP is associated with megalencephaly, developmental delay, brain and body asymmetry, capillary malformations, digital anomalies (syndactyly, polydactyly), and brain malformations; characteristically, polymicrogyria. Other features include seizures, developmental delay, hydrocephalus, and joint laxity. Recently, the genetic basis for this condition has been identified. Affected individuals were found to harbor mutations in the AKT3, PIK3CA or PIK3R2 genes.80 MCM/MCAP is a heterogeneous overgrowth syndrome, most commonly patients present with macrocephaly and cutaneous CMs. The associated capillary stain is most commonly located on the central face (philtrum and glabella) but can be seen on any area of the body. As in all overgrowth syndromes, there is a possibility of increased risk for Wilms tumor – many experts recommend serial abdominal ultrasounds in infancy and early childhood.
Beckwith–Wiedemann syndrome
Beckwith–Wiedemann syndrome (BWS) is a pediatric overgrowth disorder that carries an increased risk for malignancy, specifically Wilms tumor. BWS can present antenatally with visceromegaly on prenatal ultrasonography. It is associated with persistent nevus simplex or capillary malformation of the mid-forehead. Other findings include overgrowth of tissues and organs, macroglossia, and abdominal wall defects, usually omphalocele. High birthweight, hemihypertrophy, and neonatal hypoglycemia are also reported. Intelligence is usually not impaired.
The genetic basis of BWS is complex and several different modes of inheritance are possible. Inheritance can be via autosomal dominant, contiguous gene duplication or genomic imprinting. Chromosome 11p is implicated in several of these inheritance modes.81
Nova syndrome
Nova syndrome is a familial disorder in which a congenital glabellar capillary stain occurs in association with neurologic malformations, including Dandy–Walker malformation, hydrocephalus, cerebellar vermis agenesis, and mega cisterna magna.
Phakomatosis pigmentovascularis
Phakomatosis pigmentovascularis (PPV) is a term used to describe the association of cutaneous pigmentary and vascular anomalies. Five types with two subtypes per group are classically described. An alternate classification uses descriptive terminology to highlight the cutaneous features (Table 22.2, Fig. 22.4). Capillary malformations are described in types I–IV and cutis marmorata telangiectatica congenita in type V. The pigmentary anomalies include blue-gray macules/patches (dermal melanocytosis), nevus spilus and epidermal nevi which are darkly pigmented. Nevus anemicus is also reported. PPV has been described in association with other vascular anomalies including Klippel–Trenaunay syndrome and Sturge–Weber syndrome.82,83
Syndromes associated with venous malformations
Venous malformations cutaneous and mucosal (VMCM), glomuvenous malformations and blue rubber bleb nevus syndrome (Bean syndrome)
Somatic and familial venous malformations arise due to mutations in the Tie2 gene, a tyrosine kinase receptor involved in angiogenesis.37 In the familial VMCM syndrome, the malformations are typically small and superficial, located mainly in the skin and mucosa, though they can be found invading skeletal muscle and have been reported both in the GI tract and the brain. Since the lesions are usually small and asymptomatic, family members may be unaware of the syndrome, even though it is inherited in an autosomal dominant pattern. Lesions can be present early on and individuals can acquire more VMs with time. This is due to the mode of inheritance, whereby a second mutation within the Tie2 gene leads to the development of more lesions, analogous to Knudson’s two-hit hypothesis theory postulated for retinoblastoma.84
Familial glomuvenous malformations are usually inherited in an autosomal dominant manner, but can also be sporadic. Mutations in the glomulin gene have been identified in affected patients. As described above, GVM often resemble VM clinically. They can be small or large/segmental in their appearance (Fig. 22.6). They are bluish to purple, cobblestoned in appearance and often painful on palpation. There is great heterogeneity among affected family members, which suggests that a single mutation in the glomulin gene is not enough to produce the lesion, thus as in VMCM, a second post-zygotic mutation in the unaffected allele is required for the lesion to develop.
Blue rubber bleb nevus syndrome (BRBNS) is a rare congenital disorder in which patients present with multifocal venous malformations in the skin, soft tissue, and gastrointestinal tract.85 The associated VMs are typically small black-blue papules and skin-colored subcutaneous nodules that often involve the palms and soles. GI bleeding occurs in infancy or early childhood and can lead to chronic anemia necessitating transfusion. Lesions are commonly observed in the small bowel and colon. There is much confusion in the literature regarding the diagnosis of BRBNS, as other familial VM syndromes such as VMCM and GVM can present with similar-appearing lesions. BRBNS is less common than the other familial VM syndromes, and it is thought that GI involvement and associated bleeding are unique to BRBNS. Surgical management of patients with chronic GI bleeding and resultant anemia has been reported as a successful intervention.86 The differential diagnosis of BRBNS in the newborn period also includes multifocal infantile hemangiomas and multifocal lymphangioendotheliomatosis (see Chapter 21).
Maffucci syndrome
Maffucci syndrome (MS) is a rare sporadic syndrome characterized by vascular lesions of the skin and multiple bone tumors. This form of enchondromatosis is associated with spindle cell hemangioma, begins in childhood and worsens with maturity. Congenital forms occur and disease presents in 25% of cases by the first year of life.87 The skin lesions clinically resemble venous malformations, are nodular, develop slowly, and are rare in infancy. Although they have features of slow-flow venous anomalies – phleboliths, hypersignal on signal-enhanced T2 sequences with MRI – histologic examination reveals a spindle cell hemangioma, in addition to malformed venous channels.4 Enchondromas are benign cartilage-forming tumors within the medullary cavity of the bone. These tumors are identical to those present in another form of multiple enchondromatosis, Ollier disease. They involve both the metaphyses and the diaphyses, and may cause bony distortion, fragility, and shortening of an affected limb. The hands and feet are involved in 90% of patients. Cranial enchondromas result in severe neuro-ophthalmologic consequences. Over time, enchondromas may develop malignant transformation. Somatic mosaic IDH1 and IDH2 mutations are associated with the lesions in both Ollier disease and MS.88
Cutis marmorata telangiectatica congenita
Cutis marmorata telangiectatica congenita (CMTC) is a form of vascular malformation with a distinctive reticulated pattern. Most cases are sporadic. A female predominance is reported in some cases series, while others show no difference in incidence among genders.89,90 CMTC can be confined to a small area, have a regional distribution, or more diffuse skin involvement. At birth, a reticulated purple network is noted. The skin is streaked with linear and patchy vascular lesions intermingled with telangiectasia. Within affected areas there are often focal areas of atrophy and/or ulceration, even during the neonatal period (Fig. 22.12). These changes are often most prominent over the limbs. This conspicuous atrophic reticulate pattern differs from physiologic cutis marmorata, a normal finding in newborns, in that the pattern is coarse and less regular. CMTC may be associated with port-wine stains, which can become more apparent with maturity as the reticulate lesions fade. CMTC may improve with age, but rarely disappears completely with areas of atrophy often persisting. Ulcerations may continue to arise during infancy and childhood, particularly in areas overlying the joints, resulting in scaly areas of scarring.
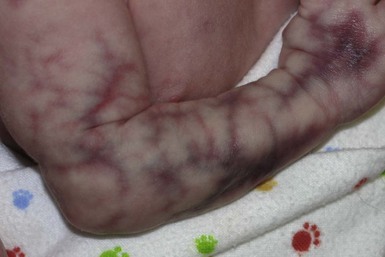
Localized or regional CMTC with involvement of one or two areas of skin (most often the extremities) is more common than the diffuse type, but because of its generally benign course is likely less frequently reported in the literature. A difference in limb girth is common, and the affected limb may have a thinner, pseudo-‘athletic’ appearance compared with the normal extremity as a result of having less fat or diminished muscles and bones. Subsequent growth is usually proportional to the original degree of limb asymmetry.91 Multiple abnormalities have been reported in association with CMTC, however, care should be taken when reviewing older case series, since cases of macrocephaly CM syndrome associated with a reticulated capillary malformation and not true CMTC, may have been included. The most frequently described associated anomaly in many case series is body asymmetry. Other reported associated anomalies include musculoskeletal anomalies, vascular abnormalities including co-existing capillary malformations/PWS, arterial stenosis and cardiac defects. CMTC, syndactyly, cardiac defects and alopecia are features of Adams Oliver syndrome.92 Less frequently reported anomalies include brain and spinal cord defects, glaucoma and other ocular anomalies, imperforate anus, abnormal genitalia, dystrophic teeth, congenital hypothyroidism, stenosing tendonitis, and others.89,91,93–96,90
The differential diagnosis of CMTC includes macrocephaly CM syndrome, which is discussed above, and persistent true cutis marmorata. Neonatal lupus can be associated with extensive livedo reticularis, telangiectasia, and atrophic striae, and mimics CMTC.97 Less generalized PWS with a blotchy reticulated quality are also sometimes mistaken for CMTC. The residual, persistent, reticulate vascular lesions of CMTC respond poorly to pulsed dye laser treatments and can be associated with a greater risk of scarring than is usually associated with this mode of therapy. Associated port-wine stains, however, are amenable to PDL therapy. Management of extracutaneous associated abnormalities is directed at specific signs or symptoms. Infants with CMTC located in a distribution similar to the port-wine stain of Sturge–Weber syndrome are at higher risk for CNS and ophthalmologic complications and require evaluation.
Verrucous hemangioma and angiokeratoma circumscriptum
Verrucous hemangioma (VH) is a rare congenital vascular anomaly which has historically been classified as a vascular malformation but shows immunohistochemical features of both a vascular tumor and vascular malformation.98,99 VH presents in infancy and is typically located on the lower extremities but may arise at other sites (Fig. 22.13). It may be a single lesion or grouped. Initially, there are blue to purple soft compressible lesions that develop overlying hyperkeratosis and bleeding over time. In its early stages VH may mimic an infantile hemangioma, venous malformation or combined venous lymphatic malformation. The lesions persist and do not involute. As hyperkeratosis develops, the lesion may mimic angiokeratoma, and capillary-lymphatic–venous malformation or even a pigmented/melanocytic lesion.
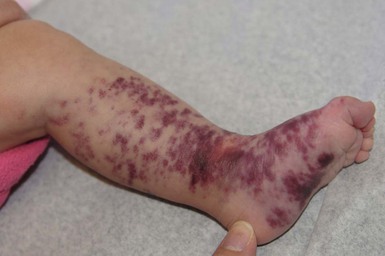
Angiokeratoma circumscriptum is another rare congenital vascular anomaly which often occurs on the lower extremities. At birth, it may present as pink-red patches and become increasingly hyperkeratotic with age.99–101 Histopathologic evaluation of VH reveals verrucous hyperplasia of the epidermis overlying a vascular lesion composed of small sized vessels separated by fibrous and adipose tissue. There is extension into the subcutis and in some cases the fascia. This is particularly important when undertaking removal, since wide excision is required in order to prevent recurrence. These features help to differentiate them from angiokeratoma, which may be congenital or appear later in childhood and are usually small and superficial. Immunohistochemical analysis can reveal focal immunoreactivity for GLUT1 (a marker that is typically seen in infantile hemangiomas)99 and negative for staining for lymphatic differentiation and Wilms Tumor 1 transcription factor (WT1). WT1 is typically positive in vascular tumors and negative in vascular malformations.102,103 The diagnosis of this rare vascular anomaly is best established with clinicopathologic correlation.
Syndromes associated with arteriovenous malformations
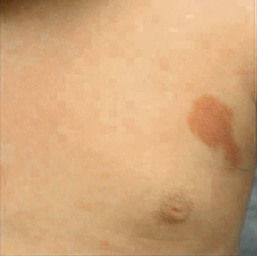
Capillary malformation-arteriovenous malformation syndrome
The capillary malformation-arteriovenous malformation (CM-AVM) syndrome104 was first described in 2003. It is characterized by the combination of congenital and acquired capillary malformations and an AVM of the soft tissue or central nervous system. Infants are often born with multiple pink patches, which may mimic a hemangioma precursor, or a larger CM (Fig. 22.14). The pink patches often increase in number with maturity. CM-AVM is inherited as an autosomal dominant trait, with wide expressivity. Some affected family members may have symptomatic AVMs, whereas others exhibit only small, pink patches, however the true incidence of underlying AVM is not fully understood because not all individuals have undergone complete evaluation. In the families that have been evaluated, approximately two-thirds of individuals will have the AVM located in the soft tissue and one-third in the central nervous system. In many patients, the AVM will underlie the largest CM. Parkes Weber syndrome (see below) is one of the ways CM-AVM will present and may do so in the neonatal period with an enlarged limb, overlying CM and a underlying fast-flow AVM or AVF, or vein of Galen malformation which can lead to cardiac compromise. The disease is caused by mutations of RASA1. Genetic testing is available and evaluation of the index patient and family members is recommended, as these skin lesions may be a marker for CNS AVM risk.9,105
Wyburn–Mason, Bonnet–Dechaume–Blanc, and Brégeat syndromes
These rare syndromes are characterized by arteriovenous malformations in the craniofacial area with fast-flow vascular anomalies in the skin (midline or hemifacial), orbit, retina, and brain (thalamus, hypothalamus and optic chiasm). These eponyms are now considered to be synonymous. In infancy, the cutaneous AVM commonly mimics a facial port-wine stain, although it is usually fainter and less well-demarcated than PWS (Fig. 22.10). It is warm on palpation and is sometimes associated with an abnormally increased skin thickness at birth. MR is a helpful noninvasive tool that may be used in infants for the detection of the enlarged tortuous vessels and AV shunting. These findings are more clearly delineated later in life with conventional arteriography. Lesions slowly enlarge over years and may cause distortion of facial features, visual loss, and cerebral hemorrhage.106
Cobb syndrome
Cobb syndrome (cutaneomeningospinal angiomatosis) is the association of a dermatomal skin vascular malformation (trunk and arm or leg), a fast-flow intramedullary spinal AVM, and a vertebral vascular anomaly in the same segment. This metameric angiomatosis is the truncal counterpart of the syndromic cephalic AVMs. In infancy, this syndrome may be undiagnosed because the cutaneous vascular signs are subtle or are diagnosed as skin capillary malformation. Patients may present with neurologic symptoms in childhood or later in life. Hyperpigmentation has also been described overlying the spinal AVM.107,108 The diagnosis is often established later, when an abnormal vertebra is incidentally imaged or if neurologic symptoms of spinal cord compression occur. The existing literature on Cobb syndrome in infants needs to be interpreted with caution, given that some case reports of Cobb syndrome in infants actually represent segmental infantile hemangiomas associated with intraspinal hemangiomas or tethered cord.
Hereditary hemorrhagic telangiectasia
Also known as Osler–Weber–Rendu syndrome, hereditary hemorrhagic telangiectasia (HHT), is an autosomal dominant disorder with various phenotypes corresponding to at least three distinct genotypes.109 Genetic testing makes early diagnosis available to relatives in a given affected family. HHT is characterized by skin and mucosal telangiectasia, spontaneous epistaxis and a risk of arteriovenous malformations in lungs, brain, and liver.110 Telangiectasia of the skin, lips, and mouth are not visible during infancy and mucosal and visceral hemorrhages are typically uncommon during this period. However, it is important to note that there are reports of infants of parents with known HHT presenting with intracranial hemorrhage in the neonatal period.111
Ataxia–telangiectasia
This is a rare autosomal recessive disease characterized by progressive cerebellar ataxia, telangiectasia, elevated α-fetoprotein levels, B- and T-cell immunodeficiency with sinopulmonary infections, cancer susceptibility (especially hematologic malignancies and breast cancer), sensitivity to ionizing radiation and radiomimetic drugs, and premature aging.112 The onset of telangiectasia in the newborn period is rare, however ocular telangiectasia may be noted in children as young as 1 year of age. In a group of 48 patients, the median age of onset of gait abnormalities was 15 months, and 72 months for telangiectasias. The median age of diagnosis was 78 months, shortly after the appearance of telangiectasia in two-thirds of patients.113 The mutated gene (ATM) maps to 11q22–23, and more than 400 mutations have been documented in affected individuals.
Syndromes associated with lymphatic malformations
Gorham syndrome
This sporadic syndrome of progressive bony destruction is associated with a lymphatic malformation. Involvement of the skin and subcutaneous tissue is variable. Soft tissue masses as well as purple plaques have been described in association with the destructive bony lesions.114 The cause of the extensive bone destruction is not clearly understood. Lesions usually become obvious in childhood and the course is variable with some patients experiencing a period of stabilization after a period of osteolysis and other with more aggressive proliferation. Visceral life-threatening lymphatic anomalies, including pleural effusions, and gastrointestinal tract involvement, may develop in association with the bone-destructive process. Management of this disorder is challenging and includes supportive care. Surgical management and radiation therapy have been described but have been of limited effectiveness. More recently, medical therapies including anti-angiogenic agents such as interferon-alpha, bevacizumab and sirolimus are under investigation.115–117
Hennekam syndrome
Hennekam syndrome, also known as generalized lymphatic dysplasia, is a rare diffuse lymphatic anomaly that is typically characterized by lymphedema of the extremities, which may be progressive and asymmetric, intestinal lymphangiectasia resulting in protein-losing enteropathy, developmental delay and abnormal fascias. Involvement of the pleura, pericardium, kidney and thyroid is reported.118–120 Autosomal recessive inheritance is suspected and in some cases it has been linked to mutations in the CCBE1 gene.121 The expansion of the phenotype raises the question of more than one gene defect.
Hereditary cholestasis with lymphedema (Aagenaes syndrome)
Hereditary cholestasis with lymphedema is an autosomal recessive disease that occurs mostly in infants of Norwegian ancestry. Significant leg lymphedema due to lymph vessel hypoplasia that is congenital or develops later in life, requires lifelong treatment. Cholestasis and obstructive jaundice are present at birth and may improve in adulthood, but in childhood, they may be lethal. Children have severe bleeding if vitamin K supplementation is not provided. They also complain of itching, and have growth retardation.
Syndromes associated with either combined malformations or overgrowth
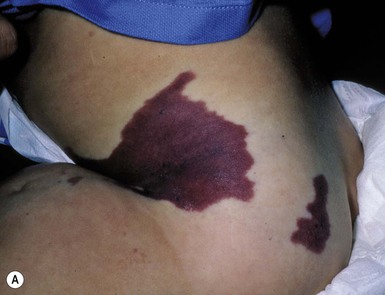
Klippel–Trenaunay syndrome
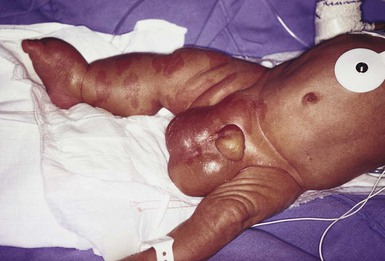
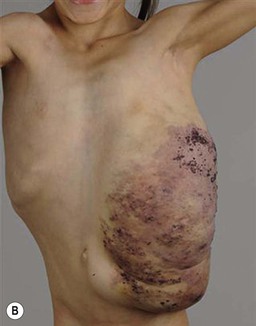
Klippel and Trenaunay described the association of a port-wine stain (CM), venous anomaly and limb overgrowth in 1900. Since the first description, it has become well recognized that this group of patients are part of a heterogenous spectrum and not all patients described in the literature have the same features. Over the last few years, there has been an attempt to refine the classification of this disorder with some authors restricting the designation of true Klippel–Trenaunay syndrome (KTS) to individuals with a combined capillary lymphatic-venous malformation, while others divide KTS into simple and complex forms based upon the presence or absence of lymphatic anomalies, significant venous anomalies and a geographic stain.122–124 Regardless of the classification, the heterogeneity of features in patients diagnosed with KTS highlights the importance of recognizing early clues that might predict more significant overgrowth and complications. It is important to note that limb capillary malformations may occur without significant venous anomalies or overgrowth and these patients have a good prognosis. However, in the newborn period, it might be difficult for a less experienced clinician to predict which infants are more likely to have a milder course, therefore it is essential to reassess neonates and infants periodically for signs of more extensive involvement. The presence at birth of a sharply demarcated geographic stain on the external lateral aspect of the affected extremity, mainly the thigh, is predictive of associated lymphatic anomalies and a poorer prognosis. If there is an associated lymphatic anomaly, small hemorrhagic ‘blebs’, which are actually lymphatic vesicles are often visible from birth or develop during early infancy (Fig. 22.15).125 Bony hypertrophy can increase progressively, resulting in a limb length discrepancy. Some infants have a milder presentation and course, with a large PWS involving a limb, soft tissue hypertrophy, but minimal to absent venous or lymphatic venous disease. These patients often have proportionate limb growth and a less morbid course than more severe cases of KTS. Recently, some authors have distinguished these patients from those with ‘true’ KTS. However, it becomes particularly important to follow young infants with limb PWS because venous anomalies may not become apparent until later in childhood.
Parkes Weber syndrome
Parkes Weber syndrome (PKWS) is the association of vascular stain, limb overgrowth (length and girth), and a fast-flow vascular anomaly with multiple arteriovenous shunts (Fig. 22.16). It is important to differentiate this disorder from KTS, as the prognosis is different. In rare instances, PKWS is complicated at birth by high-output cardiac failure. In the neonatal period, noninvasive assessment of PKWS is best done using ultrasound/color Doppler and MR angiography. Arteriography is usually not performed in the neonatal period, unless endovascular arterial embolization of the AVFs is mandatory to reduce the arterial overload accountable for congestive heart failure. Thus, KTS is a slow-flow vascular anomaly, whereas PKWS is a fast-flow vascular anomaly, but both can result in hypertrophy of the affected limb. Parkes Weber syndrome may occur as a manifestation of CM-AVM syndrome, therefore it is essential that a complete family history be obtained from affected individuals and genetic testing for RASA1 mutations is recommended.
Diagnosis.
These diagnoses are usually made clinically, but vascular imaging techniques help delineate the vascular defects. The differential diagnosis of KTS, especially severe cases, includes Proteus syndrome, PTEN hamartoma syndrome, MCAP and CLOVES syndrome (see below). Doppler ultrasound evaluation is helpful. MRI and MRA are helpful in more severe cases. Arteriography, phlebography, or lymphography are rarely needed during infancy and childhood. Routine screening for Wilms tumor, as advised in infants with congenital hemihypertrophy, is not necessary for individuals with true/classic KTS,126 with the exception of patients having true generalized hemihypertrophy and KTS, or features that would suggest another overgrowth syndrome.127
Management.
Therapeutic management includes close orthopedic follow-up of limb growth.128 If limb length discrepancy is significant after 1 year of age, radiographic studies may be appropriate, and a shoe lift or other orthopedic appliance may be used. Ultimately, during infancy, if capillary stains are extensive, the use of a laser may be impractical, and responses on the extremities are poorer than at other sites.15 Varicosities worsen over time, and later in life, varicose veins develop in KTS. AV fistulae in PKWS in selected patients, may require treatment. Ideally, patients with slow-flow vascular anomalies of the limb should use compressive stockings, but proper fitting is difficult in infants and young children who are undergoing rapid somatic growth. Low-grade clotting and consumption coagulopathy similar to that seen in venous malformations can be seen in KTS (see above). Deep vein thrombosis is rare and pulmonary embolism is an infrequent but life-threatening event.129 Long-term iatrogenic complications and a bad cosmetic outcome can result from overenthusiastic aggressive treatments early in life. Parents need educational information and support, both in the newborn period and over time. A multidisciplinary approach is important, especially in the more severe cases.
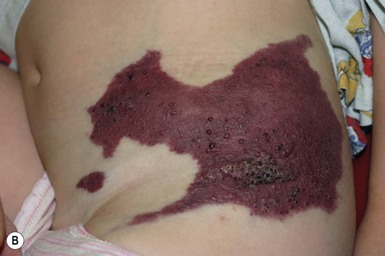
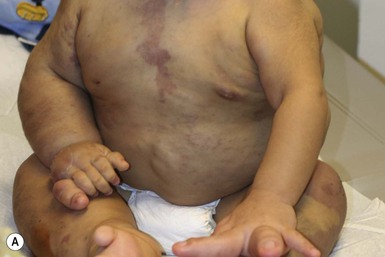
CLOVES syndrome
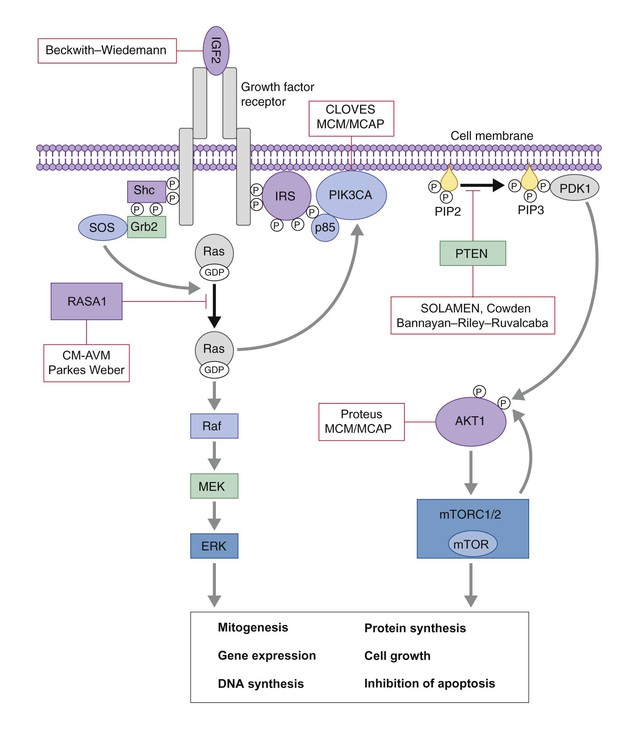
CLOVES syndrome refers to congenital lipomatous overgrowth, vascular malformations, epidermal nevi and skeletal anomalies/scoliosis. It is a newly described overgrowth syndrome resulting from post-zygotic activation mutations in the PIK3CA gene.130 In CLOVES, the large lipomatous masses are usually present on the trunk and lead to scoliosis of the spine. Other skeletal anomalies include macrodactyly and a widened space between the first and second toes, known as a sandal-gap deformity. Most patients with CLOVES syndrome have vascular malformations, most commonly CM or CLVM, however fast-flow lesions (spinal AVMs) are also described.131 In the past, patients with CLOVES were often misdiagnosed as having KT or Proteus syndrome, as there is considerable clinical overlap (Fig. 22.17). The identification of similar gene mutations in CLOVES, Proteus and MCAP (see above) suggests a common lineage, where each syndrome results from a mutation within the same molecular pathway (Fig. 22.18). Like many of the complex overgrowth syndromes, screening for Wilms tumor is also recommended.
Proteus syndrome
Proteus syndrome, first described by Wiedemann and colleagues,132 is characterized by asymmetric localized overgrowth of various body parts, affecting soft tissues and bones. It is caused by mutations in the AKT1 gene.133 The syndrome may be evident at birth, but a progressive course and mosaic distribution of the lesions are characteristic and necessary for diagnosis.134,135 The most characteristic features are the asymmetric, disproportionate growth with regional gigantism and cutaneous manifestations, including connective tissue nevus (cerebriform dermal thickening of soles and palms), epidermal nevi, lipomas, café-au-lait spots, and vascular malformations of the slow-flow type, such as extensive CLVMs.134,135 Visceral benign tumors, mainly lipomas, but tumors in the endocrine glands or CNS, and visceral vascular malformations are also observed. Intelligence is normal in most patients, but learning disabilities are present in one-third. Ophthalmologic and neurologic alterations or seizures have been reported. Surgical reconstruction is the primary treatment for these children. A multidisciplinary approach, including orthopedic management is essential because of discrepancies in limb and foot growth. Excision of lipomas or laser treatment of vascular lesions is sometimes indicated. The differential diagnosis for Proteus syndrome includes KTS, CLOVES, SOLAMEN and MCM/MCAP. Recent advances have led to the discovery of the specific genetic mutations of each of these conditions, where affected genes are involved in the same molecular signaling pathway (AKT1/PIK3CA). AKT1 is activated by loss of function mutations in PTEN, which helps to explain why patients with PTEN mutations (SOLAMEN, Cowden) have some overlapping yet distinct features (Table 22.3, Fig. 22.18).
PTEN hamartoma tumor syndrome
The PTEN gene encodes for a tumor suppressor phosphatase that has been implicated in familial cancer syndromes. The PTEN hamartoma tumor syndrome is the name given recently to this group of familial syndromes, which includes Cowden and Bannayan–Riley–Ruvalcaba syndromes (BRRS). Cowden syndrome is associated with a high risk for the development of cutaneous hamartomas and internal malignancies such as breast, thyroid and endometrial carcinoma. BRRS is an autosomal dominant disorder characterized by macrocephaly, developmental delay and penile lentigines. Both syndromes are due to mutations in the PTEN gene, which suggests a similar risk for the development of malignancy.136 In both syndromes, associated vascular malformations are frequently described and usually consist of high flow, intramuscular vascular anomalies associated with increased adipose tissue. These vascular malformations have been found to be quite specific to PTEN hamartoma tumor syndromes and should prompt investigation, workup and continued monitoring for early-onset malignancies in suspected cases.137 See Chapter 29 for further discussion on selected genetic overgrowth syndromes.
Access the full reference list at ExpertConsult.com 
