[level-membership-for-pediatrics-category]
Chapter 586 Seizures in Childhood
A seizure is a transient occurrence of signs and/or symptoms resulting from abnormal excessive or synchronous neuronal activity in the brain. The International Classification of Epileptic Seizures divides epileptic seizures into 2 large categories: In focal (partial) seizures, the first clinical and electroencephalographic (EEG) changes suggest initial activation of a system of neurons limited to part of one cerebral hemisphere; in generalized seizures, the first clinical and EEG changes indicate synchronous involvement of all of both hemispheres (Table 586-1). Approximately 30% of patients who have a first afebrile seizure have later epilepsy; the risk is about 20% if neurologic exam, EEG, and neuroimaging are normal. Febrile seizures are a special category. Acute symptomatic seizures occur secondary to an acute problem affecting brain excitability such as electrolyte imbalance or meningitis. Most children with these types of seizures do well, but sometimes such seizures signify major structural, inflammatory, or metabolic disorders of the brain, such as meningitis, encephalitis, acute stroke, or brain tumor; the prognosis depends on the underlying disorder, including its reversibility or treatability and the likelihood of developing epilepsy from it. Unprovoked seizure is not an acute symptomatic seizure. Remote symptomatic seizure is thought to be secondary to a distant brain injury such as an old stroke.
Table 586-1 TYPES OF EPILEPTIC SEIZURES
SELF-LIMITED SEIZURE TYPES
Focal Seizures
Generalized Seizures
CONTINUOUS SEIZURE TYPES
Generalized Status Epilepticus
Focal Status Epilepticus
PRECIPITATING STIMULI FOR REFLEX SEIZURES
From International League Against Epilepsy: Epileptic seizure types and precipitating stimuli for reflex seizures (website), May 13, 2009. http://www.ilae-epilepsy.org/Visitors/Centre/ctf/seizure_types.cfm. Accessed October 8, 2010.
An epileptic syndrome is a disorder that manifests one or more specific seizure types and has a specific age of onset and a specific prognosis. Several types of epileptic syndromes can be distinguished (Tables 586-2 to 586-4). This classification has to be distinguished from the classification of epileptic seizures that refers to single events rather than to clinical syndromes. In general, seizure type is the primary determinant of the type of medications the patient is likely to respond to, and the epilepsy syndrome determines the type of prognosis one could expect. An epileptic encephalopathy is an epilepsy syndrome in which the severe EEG abnormality is thought to result in cognitive and other impairments in the patient. Idiopathic epilepsy is an epilepsy syndrome that is genetic or presumed genetic and in which there is no underlying disorder affecting development or other neurologic function (e.g., petit mal epilepsy). Symptomatic epilepsy is an epilepsy syndrome caused by an underlying brain disorder (e.g., epilepsy secondary to tuberous sclerosis). A cryptogenic epilepsy (also termed presumed symptomatic epilepsy) is an epilepsy syndrome in which there is a presumed underlying brain disorder causing the epilepsy and affecting neurologic function, but the underlying disorder is not known.
Table 586-2 CLASSIFICATION FOR EPILEPSY SYNDROMES WITH AN INDICATION OF AGE OF ONSET, DURATION OF ACTIVE EPILEPSY, PROGNOSIS, AND THERAPEUTIC OPTIONS
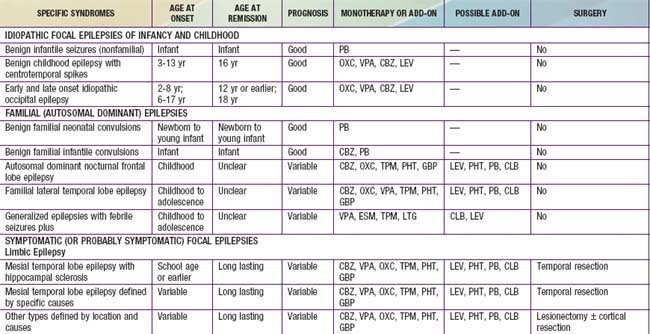
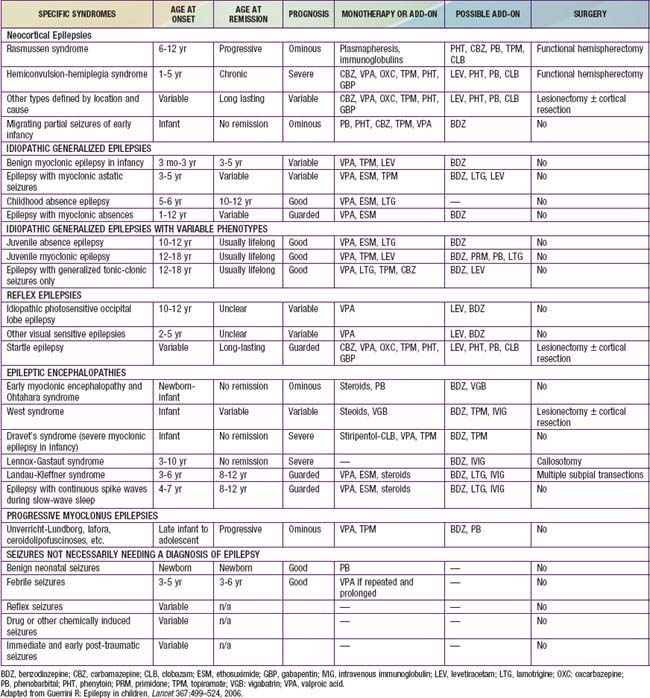
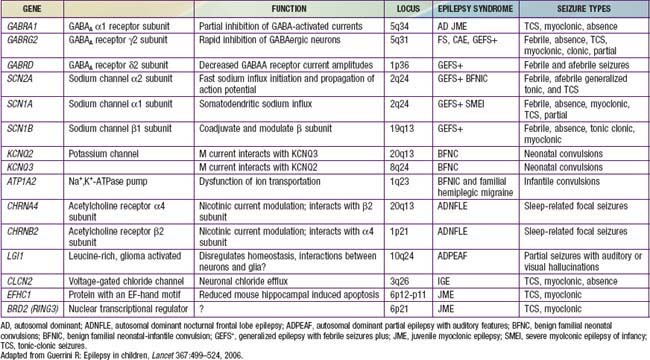
Table 586-4 CHILDHOOD EPILEPTIC SYNDROMES WITH GENERALLY GOOD PROGNOSIS
| SYNDROME | COMMENT |
|---|---|
| Benign neonatal familial convulsions | Dominant, may be severe and resistant during a few days Febrile or afebrile seizures (benign) occur later in a minority |
| Infantile familial convulsions | Dominant, seizures often in clusters (overlap with benign partial complex epilepsy of infancy) |
| Febrile convulsions plus syndromes (see Table 586-2) | In some families, febrile and afebrile convulsions occur in different members, GEFS+ The old dichotomy between febrile convulsions or epilepsy does not always hold |
| Benign myoclonic epilepsy of infancy | Often seizures during sleep, one rare variety with reflex myoclonic seizures (touch, noise) |
| Partial idiopathic epilepsy with rolandic spikes | Seizures with falling asleep or on awakening; focal sharp waves with centrotemporal location on EEG; genetic |
| Idiopathic occipital partial epilepsy | Early childhood form with seizures during sleep and ictal vomiting; can occur as status epilepticus Later forms with migrainous symptoms; not always benign |
| Petit mal absence epilepsy | Cases with absences only, some have generalized seizures. 60-80% full remission In most cases, absences disappear on therapy but there are resistant cases (unpredictable) |
| Juvenile myoclonic epilepsy | Adolescence onset, with early morning myoclonic seizures and generalized seizures during sleep; often history of absences in childhood |
EEG, electroencephalogram; GEFS+, generalized epilepsy with febrile seizures plus.
From Deonna T: Management of epilepsy, Arch Dis Child 90:5–9, 2005.
586.1 Febrile Seizures
Between 2% and 5% of neurologically healthy infants and children experience at least 1, usually simple, febrile seizure. Simple febrile seizures do not have an increased risk of mortality even though they are concerning to the parents. Complex febrile seizures may have an approximately 2-fold long-term increase in mortality, as compared to the general population over the subsequent 2 yr, probably secondary to coexisting pathology. There are no long-term adverse effects of having ≥1 simple febrile seizures. Specifically, recurrent simple febrile seizures do not damage the brain. Compared with age-matched controls, patients with febrile seizures do not have any increase in incidence of abnormalities of behavior, scholastic performance, neurocognitive function, or attention. Children who develop later epilepsy might experience such difficulties. Febrile seizures recur in approximately 30% of those experiencing a first episode, in 50% after 2 or more episodes, and in 50% of infants <1 yr old at febrile seizure onset. Several factors affect recurrence risk (Table 586-5). Although about 15% of children with epilepsy have had febrile seizures, only 2-7% of children who experience febrile seizures proceed to develop epilepsy later in life. There are several predictors of epilepsy after febrile seizures (Table 586-6).
Table 586-5 RISK FACTORS FOR RECURRENCE OF FEBRILE SEIZURES
MAJOR
MINOR
Having no risk factors carries a recurrence risk of about 12%; 1 risk factor, 25-50%; 2 risk factors, 50-59%; 3 or more, 73-100%.
Modified from Mikati MA, Rahi A: Febrile seizures: from molecular biology to clinical practice, Neurosciences 10:14–22, 2004.
Table 586-6 RISK FACTORS FOR OCCURRENCE OF SUBSEQUENT EPILEPSY
| RISK FACTOR | RISK FOR SUBSEQUENT EPILEPSY |
|---|---|
| Simple febrile seizure | 1% |
| Neurodevelopmental abnormalities | 33% |
| Focal complex febrile seizure | 29% |
| Family history of epilepsy | 18% |
| Fever <1 hr before febrile seizure | 11% |
| Complex febrile seizure, any type | 6% |
| Recurrent febrile seizures | 4% |
Modified from Mikati MA, Rahi A: Febrile seizures: from molecular biology to clinical practice, Neurosciences 10:14–22, 2004.
Work-Up
The general approach the patient with febrile seizures is delineated in Figure 586-1. Each child who presents with a febrile seizure requires a detailed history and a thorough general and neurologic examination. These are the cornerstones of the evaluation. Febrile seizures often occur in the context of otitis media, roseola and human herpesvirus 6 (HHV6) infection, shigella, or similar infections, making the evaluation more demanding. Several investigations need to be considered.
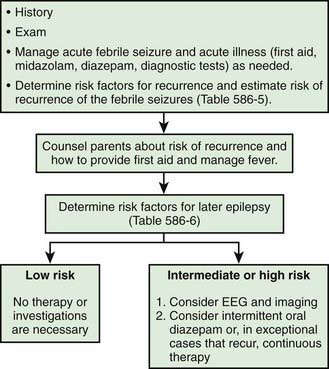
Figure 586-1 Management of febrile seizures.
(Modified from Mikati MA, Rahi A: Febrile seizures: from molecular biology to clinical practice, Neurosciences 10:14–22, 2004.)
Treatment
In general, antiepileptic therapy, continuous or intermittent, is not recommended for children with one or more simple febrile seizures. Parents should be counseled about the relative risks of recurrence of febrile seizures and recurrence of epilepsy, educated on how to handle a seizure acutely, and given emotional support. If the seizure lasts for >5 min, then acute treatment with diazepam, lorazepam, or midazolam is needed (see Chapter 586.8 for acute management of seizures and status epilepticus). Rectal diazepam is often prescribed to be given at the time of recurrence of febrile seizure lasting >5 min (see Table 586-12 for dosing). Alternatively, buccal or intranasal midazolam may be used and is often preferred by parents. Intravenous benzodiazepines, phenobarbital, phenytoin, or valproate may be needed in the case of febrile status epilepticus. If the parents are very anxious concerning their child’s seizures, intermittent oral diazepam can be given during febrile illnesses (0.33 mg/kg every 8 hr during fever) to help reduce the risk of seizures in children known to have had febrile seizures with previous illnesses. Intermittent oral nitrazepam, clobazam, and clonazepam (0.1 mg/kg/day) have also been used. Other therapies have included intermittent diazepam prophylaxis (0.5 mg/kg administered as a rectal suppository every 8 hr), phenobarbital (4-5 mg/kg/day in 1 or 2 divided doses), and valproate (20-30 mg/kg/day in 2 or 3 divided doses). In the vast majority of cases it is not justified to use these medications owing to the risk of side effects and lack of demonstrated long-term benefits, even if the recurrence rate of febrile seizures is expected to be decreased by these drugs. Other antiepileptic drugs (AEDs) have not been shown to be effective.
American Academy of Pediatrics. Febrile seizures: clinical practice guideline for the long-term management of the child with simple febrile seizures. Pediatrics. 2008:1281-1286.
Berkovic SF, Harkin L, McMahon JM, et al. De-novo mutations of the sodium channel gene SCN1A in alleged vaccine encephalopathy: a retrospective study. Lancet Neurol. 2006;5:488-492.
Chen SY, Tsai CN, Lai MW, et al. Norovirus infection as a cause of diarrhea-associated benign infantile seizures. Clin Infect Dis. 2009;48:849-855.
Guerrini R. Epilepsy in children. Lancet. 2006;367:499-524.
Hartfield DS, Tan J, Yager JY, et al. The association between iron deficiency and febrile seizures in childhood. Clin Pediatr. 2009;48:420-426.
Kimia A, Ben-Joseph EP, Rudloe T, et al. Yield of lumbar puncture among children who present with their first complex febrile seizure. Pediatrics. 2010;126:62-69.
Kimia A, Capraro AJ, Hummel D, et al. Utility of lumbar puncture for first simple febrile seizure among children 6 to 18 months of age. Pediatrics. 2009;123:6-12.
Nørgaard M, Ehrenstein V, Mahon BE, et al. Febrile seizures and cognitive function in young adult life: a prevalence study in Danish conscripts. J Pediatr. 2009;155:404-409.
Provenzale JM, Barboriak DP, VanLandingham K, et al. Hippocampus MRI signal hyperintensity after febrile status epilepticus is predictive of subsequent mesial temporal sclerosis. AJR Am J Roentgenol. 2008;190:976-983.
Sadleir LG, Scheffer IE. Febrile seizures. BMJ. 2007;334:307-311.
Strengell T, Uhari M, Tarkka R, et al. Antipyretic agents for preventing recurrences of febrile seizures. Arch Pediatr Adolesc Med. 2009;163:799-804.
Vestergaard M, Pedersen MG, Ostergaard JR, et al. Death in children with febrile seizures: a population-based cohort study. Lancet. 2008;372:457-463.
586.2 Unprovoked Seizures
History and Examination
The history should also include details of the seizure manifestations, particularly those that occurred at its initial onset. These could give clues to the type and brain localization of the seizure. One should question whether there were other previous signs or symptoms that might signify the occurrence of seizures that the parents overlooked or did not report. In some instances, if the events have been going on for a time and there is a question about their nature (e.g., sleep myoclonus versus seizures), then the family can video record the patient and make the video available to the health care provider. Having the parents imitate the seizure can also be helpful. Seizure patterns (e.g., clustering), precipitating conditions (e.g., sleep or sleep deprivation, television, visual patterns, mental activity, stress), exacerbating conditions (e.g., menstrual cycle, medications), frequency, duration, time of occurrence, and other characteristics need to be carefully documented. Parents often overlook, do not report, or underreport absence, complex partial, or myoclonic seizures. A history of personality change or symptoms of increased intracranial pressure can suggest an intracranial tumor. Similarly, a history of cognitive regression can suggest a degenerative or metabolic disease. Certain medications such as stimulants or antihistamines can precipitate seizures. A history of prenatal or perinatal distress or of developmental delay can suggest etiologic congenital or perinatal brain dysfunction. Details of the spells can suggest nonepileptic paroxysmal disorders that mimic seizures (Chapter 587).
Differential Diagnosis
The various types of seizures, as classified by the International League Against Epilepsy (ILAE), are enumerated in Table 586-1. Some seizures might begin with an aura. Auras are sensory experiences reported by the patient and not observed externally. These can take the form of visual (e.g., flashing lights or seeing colors or complex visual hallucinations), somatosensory (tingling), olfactory, auditory, vestibular, or experiential (e.g., déjà vu, déjà vécu feelings) sensations, depending upon the precise localization of the origin of the seizures.
Absence seizures are generalized seizures consisting of staring, unresponsiveness, and eye flutter lasting usually for few seconds. Typical absences are associated with 3 Hz spike–and–slow wave discharges and with petit mal epilepsy, which has a good prognosis. Atypical absences are associated with 1-2 Hz spike–and–slow wave discharges, with head atony and myoclonus during the seizures and with Lennox-Gastaut syndrome, which has a poor prognosis. Seizure type together with the other nonseizure clinical manifestations helps determine the type of epilepsy syndrome with which a particular patient is afflicted (Table 586-7; Chapters 586.3 and 586.4).
Table 586-7 SELECTED EPILEPSY SYNDROMES BY AGE OF ONSET
NEONATAL PERIOD
INFANCY
CHILDHOOD
ADOLESCENCE
AGE-RELATED (AGE OF ONSET LESS SPECIFIC)
SEIZURE DISORDERS THAT ARE NOT TRADITIONALLY GIVEN THE DIAGNOSIS OF EPILEPSY
EPILEPTIC ENCEPHALOPATHIES
OTHER SECONDARY GENERALIZED EPILEPSIES
Lists from International League Against Epilepsy: Table 1: genetic and developmental epilepsy syndromes by age of onset (website). http://www.ilae.org/Visitors/Centre/ctf/CTFtable1.cfm. Accessed October 26, 2010; and International League Against Epilepsy: Table 2. epileptic encephalopathies and other forms of secondary generalized epilepsies (website). http://www.ilae.org/Visitors/Centre/ctf/CTFtable2.cfm. Accessed October 26, 2010.
Guidelines on the evaluation and treatment of a first unprovoked nonfebrile seizure include a careful history and physical examination and brain imaging by head CT or MRI. Emergency head CT in the child presenting with a first unprovoked nonfebrile seizure is often useful for acute management of the patient. Laboratory studies are recommended in specific clinical situations: Spinal tap is considered in patients with suspected meningitis or encephalitis, in children without brain swelling or papilledema, and in children in whom a history of intracranial bleeding is suspected without evidence of such on head CT. In the second of these, examination of the CSF for xanthochromia is essential. CSF tests can also confirm with the appropriate clinical setup the diagnosis of glucose transporter deficiency, cerebral folate deficiency, pyridoxine dependency, pyridoxal dependency, mitochondrial disorders, nonketotic hyperglycemia, and neurotransmitter deficiencies. Electrocardiography (ECG) to rule out long QT or other cardiac dysrhythmias and other tests directed at disorders that could mimic seizures may be needed (Chapter 587).
Approach to the Patient and Additional Testing
The approach to the patient with epilepsy is based on the diagnostic scheme proposed by the ILAE Task Force on Classification and Terminology, presented in Table 586-8. This emphasizes the total approach to the patient, including identification, if possible, of the underlying etiology of the epilepsy and the impairments that result from it. The impairments are very often just as important as, if not more important than, the seizures themselves. There are now many epilepsy syndromes that have been associated with specific gene mutations (see Table 586-2). Different mutations of the same gene can result in different epilepsy syndromes, and mutations of different genes can cause the same epilepsy syndrome phenotype. The clinical use of gene testing in the diagnosis and management of childhood epilepsy has been limited to patients manifesting specific underlying malformational, metabolic, or degenerative disorders, patients with severe named epilepsy syndromes (such as West and Dravet syndromes and progressive myoclonic epilepsies), and, rarely, patients with familial syndromes (see Table 586-2).
Table 586-8 PROPOSED DIAGNOSTIC SCHEME FOR PEOPLE WITH EPILEPTIC SEIZURES AND WITH EPILEPSY
From International League Against Epilepsy: Table 2: proposed diagnostic scheme for people with epileptic seizures and with epilepsy (website). http://www.ilae-epilepsy.org/Visitors/Centre/ctf/table2.cfm. Accessed October 26, 2010.
MR spectroscopy is performed for lactate and creatine peaks.
Deprez L, Jansen A, De Jonghe P. Genetics of epilepsy syndromes starting in the first year of life. Neurology. 2009;72:273-281.
Engel JJr. Report of the ILAE classification core group. Epilepsia. 2006;47:1558-1568.
Guerrini R. Epilepsy in children. Lancet. 2006;367:499-524.
Harden CL, Huff JS, Schwartz TH, et al. Reassessment: neuroimaging in the emergency patient presenting with seizure (an evidence-based review): report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2007;69:1772-1780.
Hirtz D, Ashwal S, Berg A, et al. Practice parameter: evaluating a first nonfebrile seizure in children: report of the quality standards subcommittee of the American Academy of Neurology, the Child Neurology Society, and the American Epilepsy Society. Neurology. 2000;55:616-623.
Krumholz A, Wiebe S, Gronseth G, et al. Practice parameter: evaluating an apparent unprovoked first seizure in adults (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 2007;69:1996-2007.
Lagae L. What’s new in genetics in childhood epilepsy. Eur J Pediatr. 2008;167(7):715-722.
586.3 Partial Seizures and Related Epilepsy Syndromes
Secondary Generalized Seizures
EEG in patients with partial seizures usually shows focal spikes or sharp waves in the lobe where the seizure originates. A sleep-deprived EEG with recording during sleep increases the diagnostic yield and is advisable in all patients whenever possible (Fig. 586-2). Despite that, about 15% of children with epilepsy initially have normal EEGs because the discharges are relatively infrequent or the focus is deep. If repeating the test does not detect paroxysmal findings, then 24-hour video EEG monitoring may be helpful and can allow visualization of the clinical events and the corresponding EEG tracing.
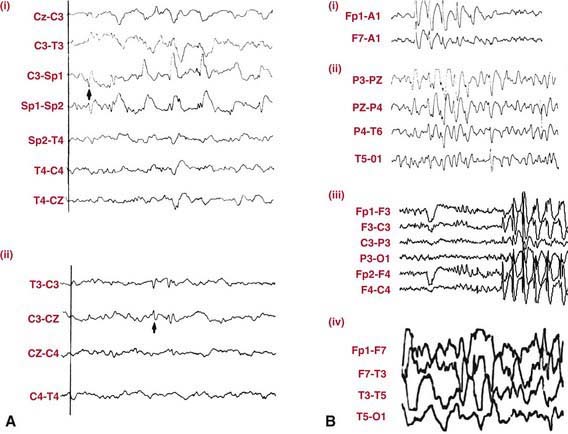
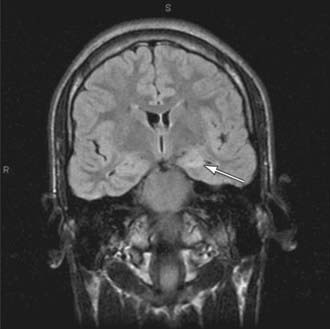
Brain imaging is critical in patients with focal seizures. In general, MRI is preferable to CT and can show pathologies such as changes due to previous strokes or hypoxic injury, malformations, medial temporal sclerosis, arteriovenous malformations, or tumors (Fig. 586-3).
Benign Epilepsy Syndromes with Partial Seizures
In infants, several less-common benign infantile familial convulsion syndromes have been reported. For some of these, the corresponding gene mutation and its function are known (see Tables 586-2 and 586-4), but for others, the genetic underpinnings are yet to be determined. Specific syndromes include benign infantile familial convulsions with parieto-occipital foci linked to chromosomal loci 19q and 2q, benign familial infantile convulsions with associated choreoathetosis linked to chromosomal locus 16p12-q12, and benign infantile familial convulsions with hemiplegic migraine linked to chromosome 1. A number of benign infantile nonfamilial syndromes have been reported, including complex partial seizures with temporal foci, secondary generalized tonic-clonic seizures with variable foci, tonic seizures with midline foci, and partial seizures in association with mild gastroenteritis. All of these have a good prognosis and respond to treatment promptly, often necessitating only short-term (e.g., 6 mo), if any, therapy. Nocturnal autosomal dominant frontal lobe epilepsy has been linked to acetylcholine-receptor gene mutations and manifests with nocturnal seizures with dystonic posturing that respond promptly to carbamazepine. Several other less-frequent familial benign epilepsy syndromes with different localizations have also been described, some of which occur exclusively or predominantly in adults (see Table 586-2).
586.4 Generalized Seizures and Related Epilepsy Syndromes
Generalized Motor Seizure
The most common generalized motor seizures are generalized tonic-clonic seizures that can be either primarily generalized (bilateral) or secondarily generalized (as described in Chapter 586.3) from a unilateral focus. If there is no partial component then the seizure usually starts with loss of consciousness and at times with a sudden cry, upward rolling of the eyes, and a generalized tonic contraction with falling, apnea, and cyanosis. In some, a clonic or myoclonic component precedes the tonic stiffening. The tonic phase is followed by a clonic phase that, as the seizure progresses, shows slowing of the rhythmic contractions until the seizure stops usually 1-2 min later. Incontinence and a postictal period often follow. The latter usually lasts for 30 min to several hours with semicoma or obtundation and postictal sleepiness, ataxia, hyper- or hyporeflexia, and headaches. There is a risk of aspiration and injury. First aid measures include positioning the patient on his or her side, clearing the mouth if it is open, loosening tight clothes or jewelry, and gently extending the head and, if possible, insertion of an airway by a trained professional. The mouth should not be forced open with a foreign object (this could dislodge teeth, causing aspiration) or with a finger in the mouth (this could result in serious injury to the examiner’s finger). Many patients have single idiopathic generalized tonic-clonic seizures that may be associated with intercurrent illness or with a cause that cannot be ascertained (Chapter 586.2). Generalized tonic, atonic, and astatic seizures often occur in severe pediatric epilepsies. Generalized myoclonic seizures can occur in either benign or difficult-to-control epilepsies.
Benign Generalized Epilepsies
Petit mal epilepsy typically starts in mid-childhood, and most patients outgrow it before adulthood. Approximately 25% of patients also develop generalized tonic-clonic seizures, half before and half after the onset of absences. Benign myoclonic epilepsy of infancy consists of the onset of myoclonic and other seizures during the 1st yr of life, with generalized 3 Hz spike–and–slow wave discharges. Often it is initially difficult to distinguish this type from more-severe syndromes, but follow-up clarifies the diagnosis. Febrile seizures plus syndrome manifests febrile seizures and multiple types of generalized seizures in multiple family members, and at times different individuals within the same family manifest different generalized and febrile seizure types (Chapter 586.1).
Juvenile myoclonic epilepsy (Janz syndrome) is the most common generalized epilepsy in young adults, accounting for 5% of all epilepsies. It has been linked to mutations in many genes including CACNB4; CLNC2; EJM2, 3, and 4; GABRA1; GABRD; and Myoclonin1/EFHC1 (see Table 586-2). Typically, it starts in early adolescence with one or more of the following manifestations: myoclonic jerks in the morning, often causing the patient to drop things; generalized tonic clonic or clonic-tonic-clonic seizures upon awakening; and juvenile absences. Sleep deprivation, alcohol (in older patients), and photic stimulation or, rarely, certain cognitive activities can act as precipitants. The EEG usually shows generalized 4-5 Hz polyspike–and–slow wave discharges. There are other forms of generalized epilepsies such as photoparoxysmal epilepsy, in which occipital, generalized tonic clonic, absence or myoclonic generalized seizures are precipitated by photic stimuli such as flipping through TV channels and viewing video games. Other forms of reflex (i.e., stimulus-provoked) epilepsy can occur; associated seizures are usually generalized, although some may be focal (see Table 586-1).
Severe Generalized Epilepsies
Severe generalized epilepsies are associated with intractable seizures and developmental delay. Early myoclonic infantile encephalopathy (EMIE) starts during the first 2 mo of life with severe myoclonic seizures and burst suppression pattern on EEG. It is usually caused by inborn errors of metabolism. Early epileptic infantile encephalopathy (EEIE, Ohtahara syndrome) has similar age of onset and EEG but manifests tonic seizures and is usually caused by brain malformations or syntaxin binding protein 1 mutations. Severe myoclonic epilepsy of infancy (Dravet syndrome) starts as focal febrile status epilepticus and later manifests myoclonic and other seizure types (Chapter 586.1).
West syndrome starts between the ages of 2 and 12 mo and consists of a triad of infantile spasms that usually occur in clusters (particularly in drowsiness or upon arousal), developmental regression, and a typical EEG picture called hypsarrhythmia (see Fig. 586-2); hypsarrhythmia is a high-voltage, slow, chaotic background with multifocal spikes. Patients with cryptogenic (sometimes called idiopathic) disease have normal development before onset, and symptomatic patients have preceding developmental delay owing to perinatal encephalopathies, malformations, underlying metabolic disorders, or other etiologies (Chapter 586.2). In boys, West syndrome can also be caused by ARX gene mutations (often associated with ambiguous genitalia). Recognizing West syndrome, especially in cryptogenic cases, is a medical emergency because diagnosis delayed for 3 wk or longer can affect long-term prognosis. The spasms are often overlooked by parents and by physicians, being mistaken for startles due to colic or for other benign paroxysmal syndromes (Chapter 587).
Many patients start with Ohtahara syndrome, develop West syndrome, and then progress to Lennox-Gastaut syndrome. Lennox-Gastaut syndrome typically starts between the age of 2 and 10 yr and consists of a triad of developmental delay, multiple seizure types that as a rule include atypical absences, and myoclonic, astatic, and tonic seizures. The tonic seizures occur either in wakefulness (causing falls and injuries) or also, typically, in sleep. The third component is the EEG findings (see Fig. 586-2): 1-2 Hz spike–and-slow waves, polyspike bursts in sleep, and a slow background in wakefulness. Patients commonly have myoclonic, atonic, and other seizure types, and most are left with long-term mental retardation and intractable seizures despite multiple therapies. Myoclonic astatic epilepsy is a syndrome similar to but milder than Lennox-Gastaut syndrome that usually does not have tonic seizures or polyspike bursts in sleep. The prognosis is more favorable than that for Lennox Gastaut syndrome.
 yr. It is often confused with autism, in that both conditions are associated with a loss of language function. Landau-Kleffner syndrome is characterized by loss of language skills in a previously normal child. At least 70% have an associated seizure disorder. The aphasia may be primarily receptive or expressive, and auditory agnosia may be so severe that the child is oblivious to everyday sounds. Hearing is normal, but behavioral problems, including irritability and poor attention span, are particularly common.
yr. It is often confused with autism, in that both conditions are associated with a loss of language function. Landau-Kleffner syndrome is characterized by loss of language skills in a previously normal child. At least 70% have an associated seizure disorder. The aphasia may be primarily receptive or expressive, and auditory agnosia may be so severe that the child is oblivious to everyday sounds. Hearing is normal, but behavioral problems, including irritability and poor attention span, are particularly common.586.5 Mechanisms of Seizures
Bockenhauer D, Feather S, Stanescu HC, et al. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med. 2009;360:1960-1970.
Caleo M. Epilepsy: synapses stuck in childhood. Nature Med. 2009;15:1126-1127.
Camfield C, Camfield P. Preventable and unpreventable causes of childhood-onset epilepsy plus mental retardation. Pediatrics. 2007;120:e52-e55.
Caraballo RH, Capovilla G, Vigevano F, et al. The spectrum of benign myoclonus of early infancy: clinical and neurophysiologic features in 102 patients. Epilepsia. 2009;50:1176-1183.
Christensen J, Pedersen MG, Pedersen CB, et al. Long-term risk of epilepsy after traumatic brain injury in children and young adults: a population-based cohort study. Lancet. 2009;373:1105-1110.
Cope DW, Di Giovanni G, Fyson SJ, et al. Enhanced tonic GABAA inhibition in typical absence epilepsy. Nature Med. 2009;15:1392-1398.
Dichter MA. Emerging concepts in the pathogenesis of epilepsy and epileptogenesis. Arch Neurol. 2009;66(4):443-447.
Helbig I, Mefford HC, Sharp AJ, et al. 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy. Nat Genet. 2009;41:160-162.
Johnson JN, Hofman N, Haglund CM, et al. Identification of a possible pathogenic link between congenital long QT syndrome and epilepsy. Neurology. 2009;72:224-231.
Niehusmann P, Dalman J, Rudlowski C, et al. Diagnostic value of N-methyl-D-aspartate receptor antibodies in women with new-onset epilepsy. Arch Neurol. 2009;6:458-464.
Reid CA, Berkovic SF, Petrou S. Mechanisms of human inherited epilepsies. Prog Neurobiol. 2009;87(1):41-57.
Shorvon S, Neligan A. Risk of epilepsy after head trauma. Lancet. 2009;373:1060-1061.
Tanaka M, Olsen RW, Medina MT, et al. Hyperglycosylation and reduced GABA currents of mutated GABRB3 polypeptide in remitting childhood absence epilepsy. Am J Hum Genet. 2008;82:1249-1261.
586.6 Treatment of Seizures and Epilepsy
Deciding on Long-Term Therapy
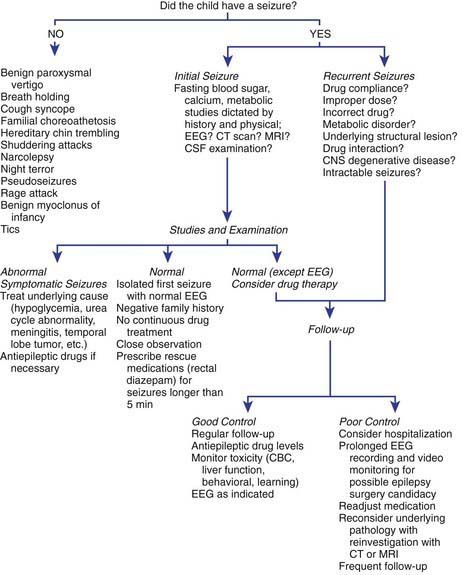
After a first seizure, if the risk of recurrence is low and the patient has normal neurodevelopmental status, EEG, and MRI (risk about 20%), then treatment is usually not started. If the patient has abnormal EEG, MRI, development, and/or neurologic exam and/or a positive family history of epilepsy, then the risk is higher and often treatment is started. Other considerations are also important, such as motor vehicle driving status and type of employment in older patients or the parents’ ability to deal with recurrences or AED drug therapy in children. The decision is therefore always individualized. All aspects of this decision-making process should be discussed with the family. An overview of the approach to the treatment of seizures and epilepsy is presented in Figure 586-4.
Counseling
An important part of the management of a patient with epilepsy is educating the family and the child about the disease, its management, and the limitations it might impose and how to deal with them. It is important to establish a successful therapeutic alliance. Some restrictions on driving (in adolescents) and on swimming are usually necessary (Table 586-9). In most states, the physician is not required to report the epileptic patient to the motor vehicle registry; this is the responsibility of the patient. Also in most states, a seizure-free period of 6 mo, and in some states longer, is required before driving is allowed. Often swimming in rivers, lakes, or sea, and underwater diving are prohibited but swimming in swimming pools may be allowable. When swimming, patients with epilepsy, even if the epilepsy is under excellent control, should be under the continuous supervision of an observer who is aware of their condition and capable of lifeguard-level rescue.
Table 586-9 SPORTS AND SPECIAL CONSIDERATIONS FOR THE CHILD WITH EPILEPSY*
| SPORTS TYPE | SPECIAL CONSIDERATIONS |
|---|---|
| Body contact sports | If there are more than occasional seizures, physician evaluation of benefits and risks of participation should be made based on the child’s condition |
| Noncontact sports | Anxiety and fatigue can cause a problem in some children Individualization based on clinical history must be the rule |
| Gymnastics | A fall can result if the child experiences a sudden seizure, especially with trampolines, parallel bars, and rope climbing, which therefore should be avoided Individual consideration remains the basic determinant |
| Swimming | The child should always be under supervision, and competitive underwater swimming should be discouraged |
* Specific advice should be individualized depending on the patient’s clinical condition. Many patients actually have fewer seizures when they are active than when they are idle.
Based on Committee on Children with Handicaps: The epileptic child and competitive school athletics, Pediatrics 42:700–702, 1968.
The American Academy of Pediatrics recommends that the physician, parents, and child jointly evaluate the risk of involvement in athletic activities. To participate in athletics, proper medical management, good seizure control, and proper supervision are crucial to avoid significant risks. Any activity where a seizure might cause a dangerous fall should be avoided; these activities include rope climbing, use of the parallel bars, and high diving. Participation in collision or contact sports depends on the patient’s condition. Epileptic children should not automatically be banned from participating in hockey, baseball, basketball, football, or wrestling. Rather, individual consideration should be based on the child’s specific case (see Table 586-9).
Choice of Drug According to Seizure Type and Epilepsy Syndrome
Drug therapy should be based on the type of seizure and the epilepsy syndrome. In general, the drugs of first choice for focal seizures and epilepsies are oxcarbazepine and carbamazepine; for absence seizures, ethosuximide; for juvenile myoclonic epilepsy, valproate and lamotrigine; for Lennox-Gastaut syndrome, valproate, topiramate, lamotrigine, and, most recently, as add on, rufinamide; and for infantile spasms, adrenocorticotropic hormone (ACTH). Lamotrigine has been shown to be effective for partial seizures, and valproate has been shown to be effective for generalized and unclassified epilepsies. There is significant controversy about these choices, and therapy should always be individualized (see the next section and Table 586-10).
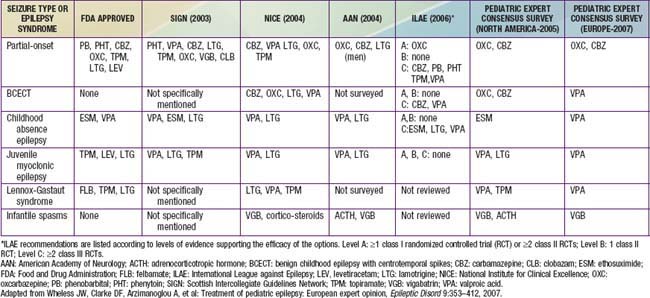
Partial and secondary generalized tonic and clonic seizures can be treated with oxcarbazepine, carbamazepine, phenobarbital, topiramate, valproic acid, lamotrigine, clobazam, clonazepam, or levetiracetam (see Table 586-8). Oxcarbazepine, levetiracetam, carbamazepine (USA) or valproate (Europe) are often used first. One study favored lamotrigine as initial monotherapy for partial seizures and valproate for generalized seizures. Almost any of these medications has been used as first or second choice depending on the individualization of the therapy.
Choice of Drug: Other Considerations
Comparative effectiveness and potential for paradoxical seizure aggravation by some AEDs (e.g., precipitation of absence seizures and myoclonic seizures by carbamazepine and tiagabine) must be considered (see Table 586-10).
The presence of comorbid conditions: For example, the presence of migraine in a patient with epilepsy can lead to the choice of a medication that is effective against both conditions such as valproate or topiramate. In an obese patient, a medication such as valproate might be avoided, and a medication that decreases appetite such as topiramate might be used instead. In adolescent girls of child-bearing potential, enzyme-inducing AEDs should be avoided because they can interfere with birth control pills; other AEDs, particularly valproate, can increase risks for fetal malformations (see Table 586-9).
Teratogenic profiles: Some AEDs, including valproate and to a lesser extent carbamazepine, phenobarbital, and phenytoin, are associated with teratogenic effects (Table 586-11).
Table 586-11 TERATOGENESIS AND PERINATAL OUTCOMES OF ANTIEPILEPTIC DRUGS
| FINDING | RECOMMENDATION | LEVEL OF EVIDENCE |
|---|---|---|
| VPA as part of polytherapy and possibly monotherapy probably contributes to the development of major congenital malformations and adverse cognitive outcome | If possible, avoidance of valproate polytherapy during the first trimester of pregnancy should be considered so as to decrease the risk of major congenital malformations and adverse cognitive outcome | B |
| AED polytherapy, as compared to monotherapy, regimens probably contribute to the development of major congenital malformations and to adverse cognitive outcomes | If possible, avoidance of AED polytherapy during the first trimester of pregnancy should be considered to decrease the risk of major congenital malformations and adverse cognitive outcome | B |
| Monotherapy exposure to phenytoin or phenobarbital possibly increases the likelihood of adverse cognitive outcomes | If possible, avoidance of phenytoin and phenobarbital during pregnancy may be considered to prevent adverse cognitive outcomes | C |
| Neonates of women with epilepsy taking AEDs probably have an increased risk of being small for gestational age and possibly have an increased risk of a 1-min Apgar score of <7 | Pregnancy risk stratification should reflect that the offspring of women with epilepsy taking AEDs are probably at increased risk for being small for gestational age (level B) and possibly at increased risk of 1-min Apgar scores of <7 | C |
Levels of recommendation: A: strongest recommendation; based on Class 1 data, B and C: lower levels of recommendations.
Types of malformations: Prior studies had reported the occurrence of spina bifida with valproate and carbamazepine therapy, and of cardiac malformation and cleft palate after carbamazepine phenytoin and phenobarbital exposure. There is variability from study to study. However, in general the relative incidence of major malformations of about 10% for valproate monotherapy, higher with valproate polytherapy, and in the range of 5% for monotherapy with the other above three AEDs and higher with polytherapy.
FDA categories: Valproate, phenobarbital, carbamazepine, and phenytoin are classified by the FDA as category D. Ethosuximide, felbamate, gabapentin, lamotrigine, levetiracetam, oxcarbazepine, tiagabine, topiramate, and zonisamide are category C. Category C: Animal studies have shown an adverse effect and there are no adequate and well-controlled studies in pregnant women or no animal studies have been conducted and there are no adequate and well-controlled studies in pregnant women. Category D: Studies, adequate well-controlled or observational, in pregnant women have demonstrated a risk to the fetus. However, the benefits of therapy might outweigh the potential risk.
AED, antiepileptic drug; VPA, valproate.
Data from Harden CI, Meador KJ, Pennell PB, et al: Practice parameter update: management issues for women with epilepsy—focus on pregnancy (an evidence-based review): teratogenesis and perinatal outcomes. Report of the Quality Standards Subcommittee and Therapeutics and Technology Subcommittee of the American Academy of Neurology and American Epilepsy Society, Neurology 73(2):133–141, 2009.
Some of these considerations can be addressed by resorting to expert opinion surveys (see Table 586-10) or to guidelines developed by concerned societies such as the ILAE, National Institute for Clinical Excellence (NICE) in England, Scottish Intercollegiate Guidelines Network (SIGN), or the American Academy of Neurology (AAN). Some guidelines are totally evidence based (AAN, ILAE), and others (NICE, SIGN) incorporate other considerations as well. However, no guideline is able to incorporate all the considerations relevant to each patient. Thus, the process of choosing an AED involves incorporating the evidence from randomized controlled trials, guidelines, expert opinion surveys, and all of the other considerations inherent in individualizing therapy and in tailoring therapy to the patient’s specific condition.
Initiating and Monitoring Therapy
In nonemergency situations or when loading is not necessary, the maintenance dose of the chosen AED is started (Table 586-12). With some medications (e.g., carbamazepine and topiramate), in many cases even smaller doses are initially started then gradually increased up to the maintenance dose to build tolerance to adverse effects such as sedation. For example, the starting dose of carbamazepine is usually 5-10 mg/kg/day. Increments of 5 mg/kg/day can be added every 3 days until a therapeutic level is achieved and a therapeutic response is established or until unacceptable adverse effects occur. With other medications such as zonisamide, phenobarbital, phenytoin, or valproate, starting at the maintenance dose is usually tolerated. With some, such as levetiracetam and gabapentin, either approach can be used. Patients should be counseled about potential adverse effects, and these should be monitored during follow-up visits (Table 586-13).
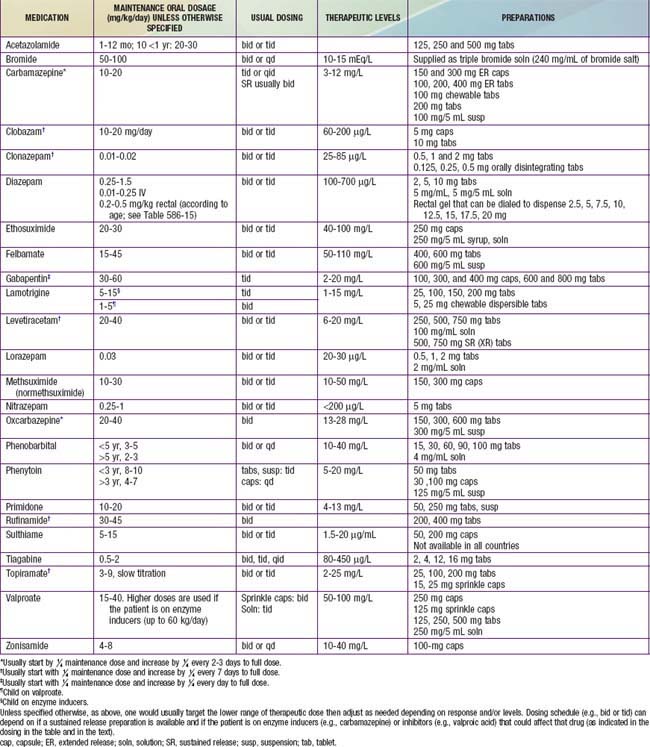
Table 586-13 SOME COMMON ADVERSE EFFECTS OF ANTIEPILEPTIC DRUGS
| ANTIEPILEPTIC DRUG | SIDE EFFECT(S) |
|---|---|
| Acetazolamide | Nuisance: Dizziness, polyuria, electrolyte imbalance Serious: Stevens-Johnson syndrome |
| Benzodiazepines | Nuisance: Dose-related neurotoxicity (drowsiness, sedation, ataxia), hyperactivity, drooling, increased secretions Serious: Apnea |
| Bromide | Nuisance: Irritability, spurious hyperchloremia (falsely high chloride owing to bromide) Serious: Psychosis, rash, toxicity developing slowly owing to the very long half-life |
| Carbamazepine | Nuisance: Tics, transient leukopenia; hyponatremia, weight gain, nausea; dizziness Serious: Stevens-Johnson syndrome, agranulocytosis, aplastic anemia, liver toxicity |
| Felbamate | Nuisance: Anorexia, vomiting, insomnia, hyperactivity, dizziness Serious: Major risks for liver and hematologic toxicity requiring close monitoring (1 : 500) |
| Gabapentin | In children: Acute onset of aggression, hyperactivity In adults: Euphoria and behavioral disinhibiting, weight gain |
| Lamotrigine | Nuisance: CNS side effects: headache, ataxia, dizziness, tremor, but usually less than other AEDs Serious: Stevens-Johnson syndrome, rarely liver toxicity |
| Levetiracetam | CNS adverse events: Somnolence, asthenia, dizziness, but usually less than other AEDs In adults: Depressive mood; in children behavioral symptoms are common |
| Oxcarbazepine | Somnolence, headache, dizziness, nausea, apathy, rash, hypertrichosis, gingival hypertrophy, hyponatremia |
| Phenobarbital and other barbiturates | Neurotoxicity, insomnia, hyperactivity, signs of distractibility, fluctuation of mood, aggressive outbursts Serious: Liver toxicity, Stevens-Johnson syndrome |
| Phenytoin and other hydantoins | Nuisance: Gingival hyperplasia, coarsening of the facies, hirsutism, cerebellovestibular symptoms (nystagmus and ataxia) Serious: Stevens-Johnson syndrome, liver toxicity |
| Primidone | Nuisance: CNS toxicity (dizziness, slurred speech, giddiness, drowsiness, depression) Serious: Liver toxicity, Stevens-Johnson syndrome |
| Rufinamide | Nuisance: Somnolence, vomiting Serious: Contraindicated in familial short QT interval |
| Succinimides | Nuisance: Nausea, abdominal discomfort, anorexia, hiccups Serious: Stevens-Johnson syndrome, drug-induced lupus |
| Tiagabine | Nuisance: Dizziness, somnolence, asthenia, headache and tremor, precipitation of absence or myoclonic seizures Serious: Precipitation of nonconvulsive status epilepticus |
| Topiramate | Nuisance: Cognitive dysfunction; weight loss; renal calculi; hypohydrosis, fever Serious: precipitation of glaucoma |
| Valproic acid | Nuisance: Weight gain; hyperammonemia tremor, alopecia, menstrual irregularities Serious: Hepatic and pancreatic toxicity |
| Vigabatrin | Nuisance: Hyperactivity Serious: Irreversible visual field deficits, retinopathy |
| Zonisamide | Fatigue, dizziness, anorexia, psychomotor slowing, ataxia, rarely hallucinations, hypohydrosis and fever |
Essentially all AEDs can cause CNS toxicity and potentially rashes and serious allergic reactions. Lacosamide has recently been approved as add-on therapy for partial seizures for patients ≥17 yr of age and requires a baseline EEG before starting it.
AED, antiepileptic drug; CNS, central nervous system; EEG, electroencephalogram.
Additional Treatment
Patients with drug resistance (at times also referred to as intractable or refractory) epilepsy warrant a careful diagnostic reevaluation to look for degenerative, metabolic or inflammatory underlying disorders (e.g., mitochondrial disease, Rasmussen’s encephalitis, Chapter 586.2). Treatable metabolic disorders that can manifest as intractable epilepsy include pyridoxine-dependent and pyridoxal-responsive epilepsy, folinic acid–responsive seizures (recently demonstrated to be the same disorder as pyridoxine-dependent epilepsy), cerebral folate deficiency, neurotransmitter disorders, biotinidase deficiency, glucose transporter 1 deficiency (responds to the ketogenic diet), serine synthesis defects, creatine deficiency syndromes, and untreated phenylketonuria. Often patients who do not respond to antiepileptic drugs are candidates for steroids, intravenous gamma globulin, or the ketogenic diet.
Discontinuation of Therapy
The decision to attempt AED withdrawal must be assessed mutually among the clinician, the parents, and the child. Risk factors should be identified and precautionary measures should be anticipated in case of seizure relapse. The patient and family should be counseled fully on what to expect, what precautions to take (including cessation of driving for a period of time), and what to do in case of relapse. A prescription for rectal diazepam to be given at the time of seizures that might occur during and after tapering may be warranted (see Table 586-12 for dosing).
Anderson M. Benzodiazepines for prolonged seizures. Arch Dis Child. 2010;95:183-189.
Arana A, Wentworth CE, Ayuso-Mateos JL, et al. Suicide-related events in patients treated with antiepileptic drugs. N Engl J Med. 2010;363:542-551.
Bonnett LJ, Tudur-Smith C, Williamson PR, et al. Risk of recurrence after a first seizure and implications for driving: further analysis of the multicentre study of early epilepsy and single seizures. BMJ. 2010;341:1260-1261.
Camfield P, Camfield C. When is it safe to discontinue AED treatment? Epilepsia. 2008;49(Suppl 9):25-28.
Chen P, Lin JJ, Lu CS, et al. Carbamazepine-induced toxic effects and HLA-B*1502 screening in Taiwan. N Engl J Med. 2011;364(12):1126-1133.
Cross JH, Jayakar P, Nordli D, et al. Proposed criteria for referral and evaluation of children for epilepsy surgery: recommendations of the Subcommission for and the International Bureau for Epilepsy (IBE). International League against Epilepsy, Subcommission for Pediatric Epilepsy Surgery, Commissions of Neurosurgery and Pediatrics. Epilepsia. 2006;46:470-472.
Cross JH, Mclellan A, Neal EG, et al. The ketogenic diet in childhood epilepsy: where are we now? Arch Dis Child. 2010;95:550-553.
Desguerre I, Nabbout R, Dulac O. The management of infantile spasms. Arch Dis Child. 2008;93:462-463.
Engel JJr. Surgical treatment for epilepsy. JAMA. 2008;300:2548-2550.
Fisher R, Salanova V, Witt T, et al. Electrical stimulation of the anterior nucleus of thalamus for treatment of refractory epilepsy. Epilepsia. 2010;51:899-908.
French JA, Pedley TA. Initial management of epilepsy. N Engl J Med. 2008;359:166-176.
Glauser TA, Cnaan A, Shinnar S, et al. Ethosuximide, valproic acid, and lamotringine in childhood absence epilepsy. N Engl J Med. 2010;362:790-798.
Glauser T, Ben-Menachem E, Bourgeois B, et al. ILAE treatment guidelines: evidence-based analysis of antiepileptic drug efficacy and effectiveness as initial monotherapy for epileptic seizures and syndromes. Epilepsia. 2006;47:1094-1120.
Harden CL, Hopp J, Ting TY, et al. Practice parameter update: management issues for women with epilepsy—focus on pregnancy (an evidence-based review): obstetrical complications and change in seizure frequency. Report of the Quality Standards Subcommittee and Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and American Epilepsy Society. Neurology. 2009;73:126-132.
Harden CI, Meador KJ, Pennell PB, et al. Practice parameter update: management issues for women with epilepsy—focus on pregnancy (an evidence -based review): teratogenesis and perinatal outcomes. Report of the Quality Standards Subcommittee and Therapeutics and Technology Subcommittee of the American Academy of Neurology and American Epilepsy Society. Neurology. 2009;73(2):133-141.
Holmes GL, Zhao Q. Choosing the correct antiepileptic drugs: from animal studies to the clinic. Pediatr Neurol. 2008;38(3):151-162.
Holsti M, Dudley N, Schunk J, et al. Intranasal midazolam vs rectal diazepam for the home treatment of acute seizures in pediatric patients with epilepsy. Arch Pediatr Adolesc Med. 2010;164:747-753.
Kirmani BF, Crisp ED, Kayani S, Rajab H. Role of intravenous levetiracetam in acute seizure management of children. Pediatr Neurol. 2009;41:37-39.
Langfitt JT, Holloway RG, McDermott MP, et al. Health care costs decline after successful epilepsy surgery. Neurology. 2007;68:1290-1298.
Macleod S, Appleton RE. The new antiepileptic drugs. Arch Dis Child Educ Pract Ed. 2007;92:ep182-ep188.
Mann MW, Pons G. Various pharmacogenetic aspects of antiepileptic drug therapy: a review. CNS Drugs. 2007;21:143-164.
Marson AG, Al-Kharusi AM, Alwaidth M, et al. The SANAD study of effectiveness of valproate, lamotrigine, or topiramate for generalized and unclassifiable epilepsy: an unblinded randomized controlled trial. Lancet. 2007;369:1016-1026.
Marson AG, Al-Kharusi AM, Alwaidth M, et al. The SANAD study of effectiveness of carbamazepine, gabapentin, oxcarbazepine, or topiramate for treatment of partial epilepsy: an unblinded randomized controlled trial. Lancet. 2007;369:1000-1014.
McCormack M, Alfirevic A, Bourgeois S, et al. HLA-A*3101 and carbamazepine-induced hypersensitivity reactions in Europeans. N Engl J Med. 2011;364(12):1134-1143.
McMullan J, Sasson C, Panciolo A, Silbergleit R. Midazolam versus diazepam for the treatment of status epilepticus in children and young adults: a meta-analysis. Acad Emerg Med. 2010;17:575-582.
McTague A, Appleton R. Treatment of difficult epilepsy. Arch Dis Child. 2011;96:200-204.
The Medical Letter. Vigabatrin (Sabril) for epilepsy. Med Lett. 2010;52:14-16.
The Medical Letter: Lacosamide for epilepsy, Med Lett 51:50–52, 2009.
The Medical Letter. Valproic acid delayed release (Stavzor). Med Lett. 2009;51:27-28.
Morse RP. Dravet syndrome: inroads into understanding epileptic encephaopathies. J Pediatr. 2011;158(3):354-359.
Obeid M, Wyllie E, Rahi AC, Mikati MA. Approach to pediatric epilepsy surgery: state of the art. Part I: general principles and presurgical workup. Eur J Paediatr Neurol. 2009;13(2):102-114.
Obeid M, Wyllie E, Rahi AC, Mikati MA. Approach to pediatric epilepsy surgery: state of the art. Part II: approach to specific epilepsy syndromes and etiologies. Eur J Paediatr Neurol. 2009;13(2):115-127.
Partikian A, Mitchell WG. Neurodevelopmental and epilepsy outcomes in a North American cohort of patients with infantile spasms. J Child Neurol. 2010;25:423-428.
Patorno E, Bohn RL, Wahl PM, et al. Anticonvulsant medications and the risk of suicide, attempted suicide, or violent death. JAMA. 2010;303:1401-1408.
Sadleir LG, Scheffer IE. Optimizing electroencephalographic studies for epilepsy diagnosis in children with new-onset seizures. Arch Neurol. 2010;67(11):1345-1349.
Scottish Intercollegiate Guidelines Network. Diagnosis and management of epilepsies in children and young people: A national clinical guideline. Edinburgh: Scottish Intercollegiate Guidelines Network; 2007.
Sillanpää M, Shinnar S. Long-term mortality in childhood-onset epilepsy. N Engl J Med. 2010;363(26):2522-2529.
Wheless JW, Clarke DF, Arzimanoglou A, et al. Treatment of pediatric epilepsy: European expert opinion. Epileptic Disord. 2007;9:353-412.
Yoong M, Chin RFM, Scott RC. Management of convulsive status epilepticus in children. Arch Dis Child Educ Pract Ed. 2009;94:1-9.
Zupanc ML, dos Santos Rubio EJ, Werner RR, et al. Epilepsy surgery outcomes: quality of life and seizure control at Children’s Hospital of Wisconsin. Pediatr Neurol. 2010;42:12-20.
Zupec-Kania B, Zupanc ML. Long-term management of the ketogenic diet: seizure monitoring, nutrition, and supplementation. Epilepsia. 2008;49(Suppl 8):23-26.
586.7 Neonatal Seizures
Types of Neonatal Seizures
Etiology
Causes of neonatal seizures are shown in Table 586-14.
Table 586-14 CAUSES OF NEONATAL SEIZURES
AGES 1-4 DAYS
AGES 4-14 DAYS
AGES 2-8 WK
From Kliegman RM, Greenbaum LA, Lye PS: Practical strategies in pediatric diagnosis and therapy, ed 2, Philadelphia, 2004, Elsevier, p 681.
Metabolic Disturbances
Neonatal seizures can also result from disturbances in amino acid or organic acid metabolism. These are usually associated with acidosis and/or hyperammonemia. However, even in the absence of these findings, if a cause of the seizures is not immediately evident, then ruling out metabolic causes requires a full metabolic work-up (Chapter 586.2) including examination of serum amino acids, acyl carnitine profile, lactate, pyruvate, and ammonia, examination of urine for amino acids and organic acids, and examination of CSF for glucose, protein, cells, amino acids, very long chain fatty acids (for neonatal adrenoleukodystropy and Zellweger syndrome), lactate, pyruvate, and perhaps other tests. This is because many inborn errors of metabolism such as nonketotic hyperglycinemia can manifest with neonatal seizures (often mistaken initially for hiccups) and can be detected only by performing these tests. Definitive diagnosis of nonketotic hyperglycinemia, for example, requires measuring the ratio of CSF glycine to plasma glycine.
Pyridoxine and pyridoxal dependency, which are malfunctions of pyridoxine metabolism, can cause severe seizures. These seizures, which are often multifocal clonic, usually start during the first hours of life. Mental retardation is often associated if therapy is delayed (Chapter 586.6).
Neonatal Seizure Syndromes
Seizure syndromes include benign idiopathic neonatal seizures (fifth day fits), which are usually apneic and focal motor seizures that start around the fifth day of life. Interictal EEG shows a distinctive pattern called theta pointu alternant (runs sharp 4-7 Hz activity), and ictal EEG shows multifocal electrographic seizures. Patients have a good response to medications and a good prognosis. Autosomal dominant benign familial neonatal seizures have onset at 2-4 days of age and usually remit at 2-15 wk of age. The seizures consist of ocular deviation, tonic posturing, clonic jerks, and, at times, motor automatisms. Interictal EEG is usually normal. These have been shown to be due to mutations in the KCNQ2 and KCNQ3 genes. Approximately 16% of patients develop later epilepsy. Early myoclonic encephalopathy and early infantile epileptic encephalopathy (Ohtahara syndrome) are discussed in Chapter 586.4.
Treatment
Lawrence R, Mathur A, Tich SNT, et al. A pilot study of continuous limited-channel aEEG in term infants with encephalopathy. J Pediatr. 2009;154:835-841.
Murray DM, Boylan GB, Ali I, et al. Defining the gap between electrographic seizure burden, clinical expression and staff recognition of neonatal seizures. Arch Dis Child Fetal Neonatal Ed. 2008;93:F187-F191.
Silverstein FS, Jensen FE, Inder T, et al. Improving the treatment of neonatal seizures: National Institute of Neurological Disorders and Stroke workshop report. J Pediatr. 2008;153:12-15.
Silverstein FS, Jensen FE. Neonatal seizures. Ann Neurol. 2007;62:112-120.
Thibeault-Eybalin MP, Lortie A, Carmant L. Neonatal seizures: do they damage the brain? Pediatr Neurol. 2009;40:175-180.
586.8 Status Epilepticus
Etiology
Etiologies include new-onset epilepsy of any type, drug intoxication (e.g., tricyclic antidepressants) in children and drug and alcohol abuse in adolescents, drug withdrawal or overdose in patients on AEDs, hypoglycemia, electrolyte imbalance (hypocalcemia, hyponatremia, hypomagnesemia), acute head trauma, encephalitis, meningitis, ischemic (arterial or venous) stroke, intracranial hemorrhage, pyridoxine, folinic acid and pyridoxal phosphate dependency, inborn errors of metabolism (Chapter 586.2) such as nonketotic hyperglycinemia in neonates and mitochondrial encephalopathy with lactic acidosis (MELAS) in children and adolescents, hypoxic-ischemic injury (e.g., after cardiac arrest), systemic conditions (such as hypertensive encephalopathy, renal or hepatic encephalopathy), brain tumors, and any other disorders that can cause epilepsy (such as brain malformations, neurodegenerative disorders, different types of progressive myoclonic epilepsy, storage diseases).
A rare condition called hemiconvulsion, hemiplegia, epilepsy (HHE) syndrome consists of prolonged febrile status epilepticus presumably due to focal acute encephalitis with resultant atrophy in the involved hemisphere, contralateral hemiplegia, and chronic epilepsy and needs to be suspected early on to attempt to control the seizures as early as possible. A somewhat similar condition in older children presenting as fever-induced refractory epileptic encephalopathy (FIRES) has been reported. Rasmussen encephalitis often causes epilepsia partialis continua (Chapter 586.3) and sometimes convulsive status epilepticus. Several types of infections are more likely to cause encephalitis with status epilepticus such as herpes simplex (complex partial and convulsive status), Bartonella (particularly nonconvulsive status), Epstein-Barr virus, and mycoplasma (postinfections encephalomyelitis with any type of status epilepticus). Postinfectious encephalitis and acute disseminated encephalomyelitis are common causes of status epilepticus including refractory status epilepticus.
Therapy
The initial therapy usually involves intravenous lorazepam, which is at least as effective as intravenous diazepam but has fewer side effects (Table 586-15). In infants, a trial of pyridoxine is often warranted. If intravenous access is not available, buccal midazolam or intranasal lorazepam are 2 effective options. With all options, respiratory depression is a potential side effect for which the patient should be monitored and managed as needed. Nasal midazolam and rectal diazepam have also been used, but there is less evidence to support their use in status epilepticus as compared to the other options.
Table 586-15 DOSES OF COMMONLY USED ANTIEPILEPTIC DRUGS IN STATUS EPILEPTICUS
| DRUG | ROUTE | DOSAGE (mg/kg) |
|---|---|---|
| Lorazepam | Intravenous | 0.05-0.1 |
| Intranasal | 0.1 | |
| Midazolam | Intravenous | 0.2 loading 0.08-0.23/hr maintenance |
| Intramuscular | 0.1-0.5 | |
| Intranasal | 0.2-0.3 | |
| Buccal | 0.2-0.5 | |
| Diazepam | Intravenous | 0.2-0.5 |
| Rectal | 2-5 yr: 0.5 | |
| 6-11 yr: 0.3 | ||
| ≥12 yr: 0.2 | ||
| Phosphenytoin | Intravenous | 15-20 PE, then 3-6/24 hr |
| Paraldehyde | Intramuscular | 0.2 mL/kg |
| Rectal | 0.4 mL/kg + same volume of olive oil | |
| Phenobarbital | 5-20 | |
| Pentobarbital coma | 13.0, then 1-5/hr | |
| Propofol | 1 (bolus), then 1-15/hr (infusion) | |
| Thiopental | 5/1st hour, then 1-2/hr | |
| Valproate | Intravenous | Loading: 25, then 30-60/24 hr |
Abend NS, Dlugos DJ. Treatment of refractory status epilepticus: literature review and a proposed protocol. Pediatr Neurol. 2008;38:377-390.
Appleton R, Macleod S, Martland T: Drug management for acute tonic-clonic convulsions including convulsive status epilepticus in children, Cochrane Database Syst Rev (3):CD001905, 2008.
Jackson A, Lieberman S, McKiernan C, et al. Management of convulsive status epilepticus in children, clinical practice guideline. Baystate Health System (2002). (website) http://libraryinfo.bhs.org/secure/pediepil.pdf Accessed October 8, 2010
Novorol CL, Chin RFM, Scott RC. Outcome of convulsive status epilepticus: a review. Arch Dis Child. 2007;92:948-951.
Riviello JJJ, Ashwal S, Hirtz D, et al. Practice parameter: diagnostic assessment of the child with status epilepticus (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2006;67:1542-1550.
Singh RK, Gaillard WD. Status epilepticus in children. Curr Neurol Neurosci Rep. 2009;9(2):137-144.
Sofou K, Kristjánsdóttir R, Papachatzakis NE, et al. Management of prolonged seizures and status epilepticus in childhood: a systematic review. J Child Neurol. 2009;24(8):918-926.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 586 Seizures in Childhood
A seizure is a transient occurrence of signs and/or symptoms resulting from abnormal excessive or synchronous neuronal activity in the brain. The International Classification of Epileptic Seizures divides epileptic seizures into 2 large categories: In focal (partial) seizures, the first clinical and electroencephalographic (EEG) changes suggest initial activation of a system of neurons limited to part of one cerebral hemisphere; in generalized seizures, the first clinical and EEG changes indicate synchronous involvement of all of both hemispheres (Table 586-1). Approximately 30% of patients who have a first afebrile seizure have later epilepsy; the risk is about 20% if neurologic exam, EEG, and neuroimaging are normal. Febrile seizures are a special category. Acute symptomatic seizures occur secondary to an acute problem affecting brain excitability such as electrolyte imbalance or meningitis. Most children with these types of seizures do well, but sometimes such seizures signify major structural, inflammatory, or metabolic disorders of the brain, such as meningitis, encephalitis, acute stroke, or brain tumor; the prognosis depends on the underlying disorder, including its reversibility or treatability and the likelihood of developing epilepsy from it. Unprovoked seizure is not an acute symptomatic seizure. Remote symptomatic seizure is thought to be secondary to a distant brain injury such as an old stroke.
Table 586-1 TYPES OF EPILEPTIC SEIZURES
SELF-LIMITED SEIZURE TYPES
Focal Seizures
Generalized Seizures
CONTINUOUS SEIZURE TYPES
Generalized Status Epilepticus
Focal Status Epilepticus
PRECIPITATING STIMULI FOR REFLEX SEIZURES
From International League Against Epilepsy: Epileptic seizure types and precipitating stimuli for reflex seizures (website), May 13, 2009. http://www.ilae-epilepsy.org/Visitors/Centre/ctf/seizure_types.cfm. Accessed October 8, 2010.
An epileptic syndrome is a disorder that manifests one or more specific seizure types and has a specific age of onset and a specific prognosis. Several types of epileptic syndromes can be distinguished (Tables 586-2 to 586-4). This classification has to be distinguished from the classification of epileptic seizures that refers to single events rather than to clinical syndromes. In general, seizure type is the primary determinant of the type of medications the patient is likely to respond to, and the epilepsy syndrome determines the type of prognosis one could expect. An epileptic encephalopathy is an epilepsy syndrome in which the severe EEG abnormality is thought to result in cognitive and other impairments in the patient. Idiopathic epilepsy is an epilepsy syndrome that is genetic or presumed genetic and in which there is no underlying disorder affecting development or other neurologic function (e.g., petit mal epilepsy). Symptomatic epilepsy is an epilepsy syndrome caused by an underlying brain disorder (e.g., epilepsy secondary to tuberous sclerosis). A cryptogenic epilepsy (also termed presumed symptomatic epilepsy) is an epilepsy syndrome in which there is a presumed underlying brain disorder causing the epilepsy and affecting neurologic function, but the underlying disorder is not known.
Table 586-2 CLASSIFICATION FOR EPILEPSY SYNDROMES WITH AN INDICATION OF AGE OF ONSET, DURATION OF ACTIVE EPILEPSY, PROGNOSIS, AND THERAPEUTIC OPTIONS
Table 586-4 CHILDHOOD EPILEPTIC SYNDROMES WITH GENERALLY GOOD PROGNOSIS
| SYNDROME | COMMENT |
|---|---|
| Benign neonatal familial convulsions | Dominant, may be severe and resistant during a few days Febrile or afebrile seizures (benign) occur later in a minority |
| Infantile familial convulsions | Dominant, seizures often in clusters (overlap with benign partial complex epilepsy of infancy) |
| Febrile convulsions plus syndromes (see Table 586-2) | In some families, febrile and afebrile convulsions occur in different members, GEFS+ The old dichotomy between febrile convulsions or epilepsy does not always hold |
| Benign myoclonic epilepsy of infancy | Often seizures during sleep, one rare variety with reflex myoclonic seizures (touch, noise) |
| Partial idiopathic epilepsy with rolandic spikes | Seizures with falling asleep or on awakening; focal sharp waves with centrotemporal location on EEG; genetic |
| Idiopathic occipital partial epilepsy | Early childhood form with seizures during sleep and ictal vomiting; can occur as status epilepticus Later forms with migrainous symptoms; not always benign |
| Petit mal absence epilepsy | Cases with absences only, some have generalized seizures. 60-80% full remission In most cases, absences disappear on therapy but there are resistant cases (unpredictable) |
| Juvenile myoclonic epilepsy | Adolescence onset, with early morning myoclonic seizures and generalized seizures during sleep; often history of absences in childhood |
EEG, electroencephalogram; GEFS+, generalized epilepsy with febrile seizures plus.
From Deonna T: Management of epilepsy, Arch Dis Child 90:5–9, 2005.
586.1 Febrile Seizures
Between 2% and 5% of neurologically healthy infants and children experience at least 1, usually simple, febrile seizure. Simple febrile seizures do not have an increased risk of mortality even though they are concerning to the parents. Complex febrile seizures may have an approximately 2-fold long-term increase in mortality, as compared to the general population over the subsequent 2 yr, probably secondary to coexisting pathology. There are no long-term adverse effects of having ≥1 simple febrile seizures. Specifically, recurrent simple febrile seizures do not damage the brain. Compared with age-matched controls, patients with febrile seizures do not have any increase in incidence of abnormalities of behavior, scholastic performance, neurocognitive function, or attention. Children who develop later epilepsy might experience such difficulties. Febrile seizures recur in approximately 30% of those experiencing a first episode, in 50% after 2 or more episodes, and in 50% of infants <1 yr old at febrile seizure onset. Several factors affect recurrence risk (Table 586-5). Although about 15% of children with epilepsy have had febrile seizures, only 2-7% of children who experience febrile seizures proceed to develop epilepsy later in life. There are several predictors of epilepsy after febrile seizures (Table 586-6).
Table 586-5 RISK FACTORS FOR RECURRENCE OF FEBRILE SEIZURES
MAJOR
MINOR
Having no risk factors carries a recurrence risk of about 12%; 1 risk factor, 25-50%; 2 risk factors, 50-59%; 3 or more, 73-100%.
Modified from Mikati MA, Rahi A: Febrile seizures: from molecular biology to clinical practice, Neurosciences 10:14–22, 2004.
Table 586-6 RISK FACTORS FOR OCCURRENCE OF SUBSEQUENT EPILEPSY
| RISK FACTOR | RISK FOR SUBSEQUENT EPILEPSY |
|---|---|
| Simple febrile seizure | 1% |
| Neurodevelopmental abnormalities | 33% |
| Focal complex febrile seizure | 29% |
| Family history of epilepsy | 18% |
| Fever <1 hr before febrile seizure | 11% |
| Complex febrile seizure, any type | 6% |
| Recurrent febrile seizures | 4% |
Modified from Mikati MA, Rahi A: Febrile seizures: from molecular biology to clinical practice, Neurosciences 10:14–22, 2004.
Work-Up
The general approach the patient with febrile seizures is delineated in Figure 586-1. Each child who presents with a febrile seizure requires a detailed history and a thorough general and neurologic examination. These are the cornerstones of the evaluation. Febrile seizures often occur in the context of otitis media, roseola and human herpesvirus 6 (HHV6) infection, shigella, or similar infections, making the evaluation more demanding. Several investigations need to be considered.
Figure 586-1 Management of febrile seizures.
(Modified from Mikati MA, Rahi A: Febrile seizures: from molecular biology to clinical practice, Neurosciences 10:14–22, 2004.)
Treatment
In general, antiepileptic therapy, continuous or intermittent, is not recommended for children with one or more simple febrile seizures. Parents should be counseled about the relative risks of recurrence of febrile seizures and recurrence of epilepsy, educated on how to handle a seizure acutely, and given emotional support. If the seizure lasts for >5 min, then acute treatment with diazepam, lorazepam, or midazolam is needed (see Chapter 586.8 for acute management of seizures and status epilepticus). Rectal diazepam is often prescribed to be given at the time of recurrence of febrile seizure lasting >5 min (see Table 586-12 for dosing). Alternatively, buccal or intranasal midazolam may be used and is often preferred by parents. Intravenous benzodiazepines, phenobarbital, phenytoin, or valproate may be needed in the case of febrile status epilepticus. If the parents are very anxious concerning their child’s seizures, intermittent oral diazepam can be given during febrile illnesses (0.33 mg/kg every 8 hr during fever) to help reduce the risk of seizures in children known to have had febrile seizures with previous illnesses. Intermittent oral nitrazepam, clobazam, and clonazepam (0.1 mg/kg/day) have also been used. Other therapies have included intermittent diazepam prophylaxis (0.5 mg/kg administered as a rectal suppository every 8 hr), phenobarbital (4-5 mg/kg/day in 1 or 2 divided doses), and valproate (20-30 mg/kg/day in 2 or 3 divided doses). In the vast majority of cases it is not justified to use these medications owing to the risk of side effects and lack of demonstrated long-term benefits, even if the recurrence rate of febrile seizures is expected to be decreased by these drugs. Other antiepileptic drugs (AEDs) have not been shown to be effective.
American Academy of Pediatrics. Febrile seizures: clinical practice guideline for the long-term management of the child with simple febrile seizures. Pediatrics. 2008:1281-1286.
Berkovic SF, Harkin L, McMahon JM, et al. De-novo mutations of the sodium channel gene SCN1A in alleged vaccine encephalopathy: a retrospective study. Lancet Neurol. 2006;5:488-492.
Chen SY, Tsai CN, Lai MW, et al. Norovirus infection as a cause of diarrhea-associated benign infantile seizures. Clin Infect Dis. 2009;48:849-855.
Guerrini R. Epilepsy in children. Lancet. 2006;367:499-524.
Hartfield DS, Tan J, Yager JY, et al. The association between iron deficiency and febrile seizures in childhood. Clin Pediatr. 2009;48:420-426.
Kimia A, Ben-Joseph EP, Rudloe T, et al. Yield of lumbar puncture among children who present with their first complex febrile seizure. Pediatrics. 2010;126:62-69.
Kimia A, Capraro AJ, Hummel D, et al. Utility of lumbar puncture for first simple febrile seizure among children 6 to 18 months of age. Pediatrics. 2009;123:6-12.
Nørgaard M, Ehrenstein V, Mahon BE, et al. Febrile seizures and cognitive function in young adult life: a prevalence study in Danish conscripts. J Pediatr. 2009;155:404-409.
Provenzale JM, Barboriak DP, VanLandingham K, et al. Hippocampus MRI signal hyperintensity after febrile status epilepticus is predictive of subsequent mesial temporal sclerosis. AJR Am J Roentgenol. 2008;190:976-983.
Sadleir LG, Scheffer IE. Febrile seizures. BMJ. 2007;334:307-311.
Strengell T, Uhari M, Tarkka R, et al. Antipyretic agents for preventing recurrences of febrile seizures. Arch Pediatr Adolesc Med. 2009;163:799-804.
Vestergaard M, Pedersen MG, Ostergaard JR, et al. Death in children with febrile seizures: a population-based cohort study. Lancet. 2008;372:457-463.
586.2 Unprovoked Seizures
History and Examination
The history should also include details of the seizure manifestations, particularly those that occurred at its initial onset. These could give clues to the type and brain localization of the seizure. One should question whether there were other previous signs or symptoms that might signify the occurrence of seizures that the parents overlooked or did not report. In some instances, if the events have been going on for a time and there is a question about their nature (e.g., sleep myoclonus versus seizures), then the family can video record the patient and make the video available to the health care provider. Having the parents imitate the seizure can also be helpful. Seizure patterns (e.g., clustering), precipitating conditions (e.g., sleep or sleep deprivation, television, visual patterns, mental activity, stress), exacerbating conditions (e.g., menstrual cycle, medications), frequency, duration, time of occurrence, and other characteristics need to be carefully documented. Parents often overlook, do not report, or underreport absence, complex partial, or myoclonic seizures. A history of personality change or symptoms of increased intracranial pressure can suggest an intracranial tumor. Similarly, a history of cognitive regression can suggest a degenerative or metabolic disease. Certain medications such as stimulants or antihistamines can precipitate seizures. A history of prenatal or perinatal distress or of developmental delay can suggest etiologic congenital or perinatal brain dysfunction. Details of the spells can suggest nonepileptic paroxysmal disorders that mimic seizures (Chapter 587).
Differential Diagnosis
The various types of seizures, as classified by the International League Against Epilepsy (ILAE), are enumerated in Table 586-1. Some seizures might begin with an aura. Auras are sensory experiences reported by the patient and not observed externally. These can take the form of visual (e.g., flashing lights or seeing colors or complex visual hallucinations), somatosensory (tingling), olfactory, auditory, vestibular, or experiential (e.g., déjà vu, déjà vécu feelings) sensations, depending upon the precise localization of the origin of the seizures.
Absence seizures are generalized seizures consisting of staring, unresponsiveness, and eye flutter lasting usually for few seconds. Typical absences are associated with 3 Hz spike–and–slow wave discharges and with petit mal epilepsy, which has a good prognosis. Atypical absences are associated with 1-2 Hz spike–and–slow wave discharges, with head atony and myoclonus during the seizures and with Lennox-Gastaut syndrome, which has a poor prognosis. Seizure type together with the other nonseizure clinical manifestations helps determine the type of epilepsy syndrome with which a particular patient is afflicted (Table 586-7; Chapters 586.3 and 586.4).
Table 586-7 SELECTED EPILEPSY SYNDROMES BY AGE OF ONSET
NEONATAL PERIOD
INFANCY
CHILDHOOD
ADOLESCENCE
AGE-RELATED (AGE OF ONSET LESS SPECIFIC)
SEIZURE DISORDERS THAT ARE NOT TRADITIONALLY GIVEN THE DIAGNOSIS OF EPILEPSY
EPILEPTIC ENCEPHALOPATHIES
OTHER SECONDARY GENERALIZED EPILEPSIES
Lists from International League Against Epilepsy: Table 1: genetic and developmental epilepsy syndromes by age of onset (website). http://www.ilae.org/Visitors/Centre/ctf/CTFtable1.cfm. Accessed October 26, 2010; and International League Against Epilepsy: Table 2. epileptic encephalopathies and other forms of secondary generalized epilepsies (website). http://www.ilae.org/Visitors/Centre/ctf/CTFtable2.cfm. Accessed October 26, 2010.
Guidelines on the evaluation and treatment of a first unprovoked nonfebrile seizure include a careful history and physical examination and brain imaging by head CT or MRI. Emergency head CT in the child presenting with a first unprovoked nonfebrile seizure is often useful for acute management of the patient. Laboratory studies are recommended in specific clinical situations: Spinal tap is considered in patients with suspected meningitis or encephalitis, in children without brain swelling or papilledema, and in children in whom a history of intracranial bleeding is suspected without evidence of such on head CT. In the second of these, examination of the CSF for xanthochromia is essential. CSF tests can also confirm with the appropriate clinical setup the diagnosis of glucose transporter deficiency, cerebral folate deficiency, pyridoxine dependency, pyridoxal dependency, mitochondrial disorders, nonketotic hyperglycemia, and neurotransmitter deficiencies. Electrocardiography (ECG) to rule out long QT or other cardiac dysrhythmias and other tests directed at disorders that could mimic seizures may be needed (Chapter 587).
Approach to the Patient and Additional Testing
The approach to the patient with epilepsy is based on the diagnostic scheme proposed by the ILAE Task Force on Classification and Terminology, presented in Table 586-8. This emphasizes the total approach to the patient, including identification, if possible, of the underlying etiology of the epilepsy and the impairments that result from it. The impairments are very often just as important as, if not more important than, the seizures themselves. There are now many epilepsy syndromes that have been associated with specific gene mutations (see Table 586-2). Different mutations of the same gene can result in different epilepsy syndromes, and mutations of different genes can cause the same epilepsy syndrome phenotype. The clinical use of gene testing in the diagnosis and management of childhood epilepsy has been limited to patients manifesting specific underlying malformational, metabolic, or degenerative disorders, patients with severe named epilepsy syndromes (such as West and Dravet syndromes and progressive myoclonic epilepsies), and, rarely, patients with familial syndromes (see Table 586-2).
Table 586-8 PROPOSED DIAGNOSTIC SCHEME FOR PEOPLE WITH EPILEPTIC SEIZURES AND WITH EPILEPSY
From International League Against Epilepsy: Table 2: proposed diagnostic scheme for people with epileptic seizures and with epilepsy (website). http://www.ilae-epilepsy.org/Visitors/Centre/ctf/table2.cfm. Accessed October 26, 2010.
MR spectroscopy is performed for lactate and creatine peaks.
Deprez L, Jansen A, De Jonghe P. Genetics of epilepsy syndromes starting in the first year of life. Neurology. 2009;72:273-281.
Engel JJr. Report of the ILAE classification core group. Epilepsia. 2006;47:1558-1568.
Guerrini R. Epilepsy in children. Lancet. 2006;367:499-524.
Harden CL, Huff JS, Schwartz TH, et al. Reassessment: neuroimaging in the emergency patient presenting with seizure (an evidence-based review): report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2007;69:1772-1780.
Hirtz D, Ashwal S, Berg A, et al. Practice parameter: evaluating a first nonfebrile seizure in children: report of the quality standards subcommittee of the American Academy of Neurology, the Child Neurology Society, and the American Epilepsy Society. Neurology. 2000;55:616-623.
Krumholz A, Wiebe S, Gronseth G, et al. Practice parameter: evaluating an apparent unprovoked first seizure in adults (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 2007;69:1996-2007.
Lagae L. What’s new in genetics in childhood epilepsy. Eur J Pediatr. 2008;167(7):715-722.
586.3 Partial Seizures and Related Epilepsy Syndromes
Secondary Generalized Seizures
EEG in patients with partial seizures usually shows focal spikes or sharp waves in the lobe where the seizure originates. A sleep-deprived EEG with recording during sleep increases the diagnostic yield and is advisable in all patients whenever possible (Fig. 586-2). Despite that, about 15% of children with epilepsy initially have normal EEGs because the discharges are relatively infrequent or the focus is deep. If repeating the test does not detect paroxysmal findings, then 24-hour video EEG monitoring may be helpful and can allow visualization of the clinical events and the corresponding EEG tracing.
Brain imaging is critical in patients with focal seizures. In general, MRI is preferable to CT and can show pathologies such as changes due to previous strokes or hypoxic injury, malformations, medial temporal sclerosis, arteriovenous malformations, or tumors (Fig. 586-3).
Benign Epilepsy Syndromes with Partial Seizures
In infants, several less-common benign infantile familial convulsion syndromes have been reported. For some of these, the corresponding gene mutation and its function are known (see Tables 586-2 and 586-4), but for others, the genetic underpinnings are yet to be determined. Specific syndromes include benign infantile familial convulsions with parieto-occipital foci linked to chromosomal loci 19q and 2q, benign familial infantile convulsions with associated choreoathetosis linked to chromosomal locus 16p12-q12, and benign infantile familial convulsions with hemiplegic migraine linked to chromosome 1. A number of benign infantile nonfamilial syndromes have been reported, including complex partial seizures with temporal foci, secondary generalized tonic-clonic seizures with variable foci, tonic seizures with midline foci, and partial seizures in association with mild gastroenteritis. All of these have a good prognosis and respond to treatment promptly, often necessitating only short-term (e.g., 6 mo), if any, therapy. Nocturnal autosomal dominant frontal lobe epilepsy has been linked to acetylcholine-receptor gene mutations and manifests with nocturnal seizures with dystonic posturing that respond promptly to carbamazepine. Several other less-frequent familial benign epilepsy syndromes with different localizations have also been described, some of which occur exclusively or predominantly in adults (see Table 586-2).
