[level-membership-for-cardiothoracic-surgery-category]
Chapter 4 Sedation, Analgesia, and Related Topics
In this chapter the indications, contraindications, and adverse effects of drugs used for sedation and analgesia in the intensive care unit (ICU) are reviewed. In addition, practical tools for the measurement of depth of sedation and quality of analgesia are outlined. The related topics of neuromuscular-blocking drugs and antiemetics are also discussed.
PHARMACOKINETIC CONSIDERATIONS
Duration of Effect
The elimination half-time is the time taken for the amount of drug in the body to decrease by 50%. This parameter is often quoted when describing the pharmacokinetic properties of a drug. However, elimination half-time only rarely reflects the duration of effect. When a drug is given intravenously, it is rapidly distributed to a central “virtual” compartment consisting of plasma, interstitial fluid, and organs with high blood flow (brain, heart, liver, kidneys). This central compartment is in equilibrium with the effect site and with the organs of elimination—the liver and the kidneys. Drugs with low lipid solubility, high ionization, and high protein binding tend to be confined to this central compartment and typically have a small steady-state volume of distribution (VSS; Fig. 4-1). Such drugs can be described using a one-compartment model (V1). For drugs that obey one-compartment kinetics, the duration of effect may be related to the elimination half-time. Examples include aminoglycosides and neuromuscular blocking drugs. However, highly lipid-soluble drugs, including most sedative-hypnotics and opioid analgesics, display multicompartment kinetics in which drugs are redistributed from the central compartment to one or two peripheral compartments (V2, V3; see Fig. 4-1).
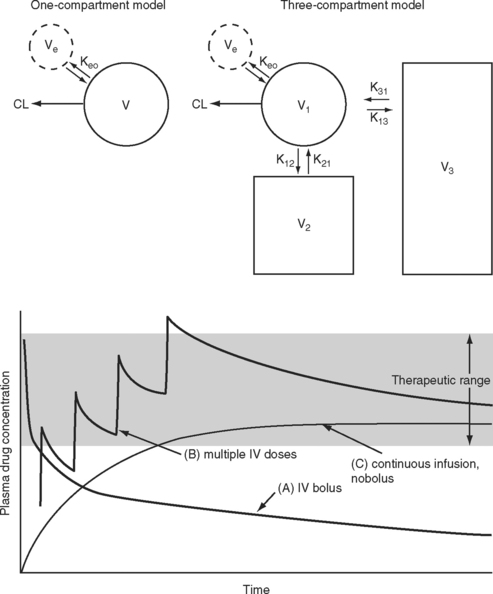
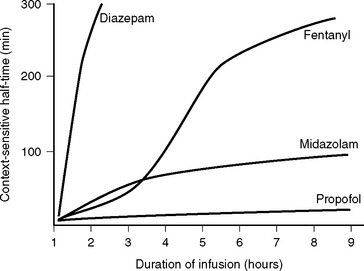
For drugs that display multicompartment kinetics, distribution and context-sensitive half-times are more useful concepts than elimination half-time. The distribution half-time is the time taken for the concentration within the central compartment to fall by 50%. Following a single intravenous dose, the distribution half-time determines the duration of effect of the drug (see Fig. 4-1). The context-sensitive half-time is the time taken for the effect-site concentration to fall by 50% following discontinuation of an intravenous infusion.1 Because a drug accumulates in the peripheral compartments over time, the context-sensitive half-time changes depending on the duration of infusion (Fig. 4-2). The context-sensitive half-time provides some indication of the duration of effect of the drug following both short- and long-term infusions (or repeated bolus doses). The percentage of decrease in concentration required for recovery from a drug’s effect is not necessarily 50%.
Onset of Action
The speed of onset of a drug depends on multiple factors; two that are of clinical importance for intravenously administered sedatives and analgesics are (1) the speed with which the drug is distributed within the central compartment and (2) the half-time for equilibration between the central and effect-site compartments (T1/2keo; see Fig. 4-1). Low cardiac output slows drug distribution within the central compartment and can greatly prolong the onset time. Thus, when administering potent sedative or analgesic medications to patients with low cardiac output, it is essential to give a small initial dose and wait a longer than normal time for the clinical effect to occur. Values for T1/2keo vary among drugs. For instance, the T1/2keo for morphine, fentanyl, and remifentanil are 17 minutes, 6.6 minutes, and 1.16 minutes, respectively. Therefore, morphine will have a slower onset of action than fentanyl and remifentanil.
Loading and Maintenance Doses
If a drug is administered by constant infusion or repeat doses it takes five (elimination) half-times to achieve 97% of the steady-state concentration (see Fig. 4-1). Thus, for a drug with a relatively long half-time, it is often useful to give a loading dose to rapidly increase the plasma concentration to within the therapeutic range. The loading dose (LD) depends on the volume of distribution (V) of the drug and the desired plasma concentration (CP):
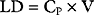
Note that loading dose does not depend on clearance; thus, the loading dose of some renally eliminated drugs, such as gentamicin, do not need to be reduced in patients with renal failure. However, for drugs that display multicompartment kinetics, it is important to be clear which loading dose is used in the calculation: V1 or VSS. A loading dose based on VSS will initially produce very high plasma levels because the drug will be delivered only to the central compartment volume (V1). To avoid this problem, multiple small loading doses based on V1 may be required. This is the approach that is recommended for amiodarone in Chapter 3.
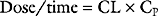
SEDATION
Sedation is part of a continuum of central nervous system (CNS) depression that ranges from anxiolysis through sedation, hypnosis (sleep), unconsciousness, and coma. Most sedative-hypnotic drugs produce anxiolysis at subhypnotic doses. Certain drugs, notably the benzodiazepines, also produce antegrade (i.e., following drug administration) amnesia at low doses. Some sedativehypnotics are anticonvulsants (e.g., benzodiazepines, barbiturates, and propofol). Anxiolysis is not the same as sedation. Antipsychotic drugs produce a state of outward calm but can increase feelings of anxiety and apprehension.
Indications for Sedation
Ventilated patients require sedation to tolerate endotracheal intubation and mechanical ventilation, facilitate nursing care, minimize the stress response, reduce oxygen consumption, diminish recall of unpleasant experiences, and prevent the development of posttraumatic stress disorder.2,3 Less commonly, sedation is indicated in extubated patients for the treatment of anxiety or delirium.
Mechanical ventilation, particularly using lungprotective strategies with long inspiratory times and permissive hypercapnia, is poorly tolerated by nonsedated patients and can result in ventilator dysynchrony (Chapter 29) and the sensation of dyspnea. Distressed patients may become tachycardic and hypertensive, which can exacerbate or provoke myocardial ischemia and bleeding. Such patients may also self-extubate or pull out their intravascular lines and surgical drains. A critically unwell patient commonly benefits from deep sedation, occasionally accompanied by neuromuscular blockade, during the acute phase of an illness. However, most patients do not require paralysis, only a level of sedation sufficient to allow tolerance of endotracheal intubation. Sedation of agitated patients should be commenced only after providing adequate analgesia and treating reversible physiologic causes.3
Adverse Effects of Sedation
Excessive sedation contributes to hypotension and delays awakening, needlessly prolonging the duration of mechanical ventilation.4 Sedation may also mask the development of intracranial, intrathoracic, or intraabdominal complications. The reduction in sympathetic tone that follows the administration of sedative and analgesic drugs can cause important hypotension. Hypotension is particularly marked in patients with high levels of endogenous catecholamines such as those that occur in the settings of hypovolemia and acute heart failure. Following prolonged administration of some sedatives (and opioid analgesics), tolerance may develop such that increased doses are required to elicit the same clinical effect. Abrupt discontinuation of certain sedatives, notably benzodiazepines, in a patient who has developed tolerance, may provoke a withdrawal syndrome (see discussion under subsequent heading Benzodiazepines). For these reasons, the need for sedation should be evaluated on an on-going basis and the depth of sedation regularly assessed.
Assessment of Sedation
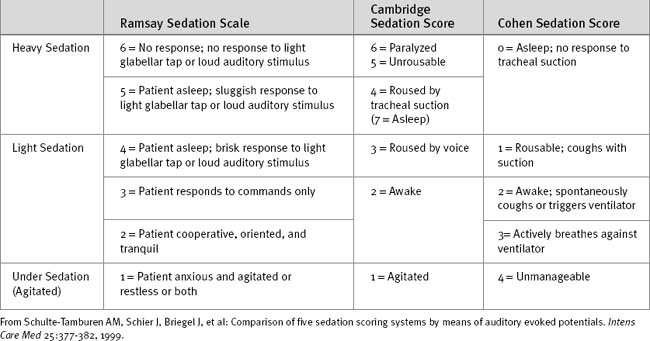
If the clinical state allows, sedation should be stopped each day until the patient shows signs of awakening. Sedation can then be restarted if still indicated. A number of sedation scoring systems have been developed to quantify the depth of sedation and allow sedative drugs to be titrated to effect. Some commonly used scoring systems are shown in Table 4-1. Although primarily a system for monitoring neurologic function after trauma, the Glasgow Coma Scale (Table 4-2) may also be used to monitor sedation, although much information is lost in patients who are intubated and cannot respond verbally. One option in ventilated patients is to revise the Glasgow Coma Scale score to a maximum of 10, with the annotation that the patient is intubated.
| Verbal Response | Motor Response | Eye Response |
|---|---|---|
| 5 = Appropriate | 6 = Obeys commands | 4 = Opens spontaneously |
| 4 = Disorientated | 5 = Localizes to pain | 3 = Opens on command |
| 3 = Unconnected words | 4 = Withdraws from pain | 2 = Opens with pain |
| 2 = Sounds only | 3 = Abnormal flexion to pain | 1 = No response |
| 1 = Nothing | 2 = Extends to pain | |
| 1 = No response | Total = V + M + E = 3-15 |
The Bispectral Index (BIS) monitor, a highly processed electroencephalogram, is widely used to assess depth of anesthesia during surgery and has been used on a limited basis to monitor depth of sedation in the ICU.5,6 A potential problem with this monitor in the ICU environment is that BIS recordings are increased—implying a reduced depth of sedation—by electromyographic activity.7 Thus, BIS recordings tend to be lower in paralyzed patients than in nonparalyzed patients for an equivalent depth of sedation. This phenomenon could potentially lead to paralyzed patients receiving inadequate sedation, resulting in unpleasant awareness.
Sedative Drugs
Propofol
The onset of action following a bolus dose usually occurs within 30 seconds. Propofol has a distribution half-time of 2 to 4 minutes, which results in an offset of effect of 5 to 10 minutes following a bolus dose. There is minimal residual sedation. Propofol has a relatively stable context-sensitive half-time (see Fig. 4-2), and awakening is rapid even after prolonged infusion. In one study of cardiac surgery patients, extubation occurred after a mean time of 7.6 minutes after cessation of propofol infusion (mean dose of 82.8 mg/hr) following 17 hours of continuous sedation.8 The corresponding extubation time for patients given midazolam (mean dose 2.3 mg/hr) was 125 minutes. This rapid offset of clinical effect following prolonged infusion occurs because propofol has high hepatic and extrahepatic clearance (pharmacokinetic effect) and because subhypnotic concentrations of propofol cause minimal sedation (pharmacodynamic effect).
The main side effects of propofol relate to cardiac and respiratory depression. Hypotension due to vasodilation tends to be more marked than with other sedatives. Bolus doses must be used with extreme caution because as little as 20 mg can cause profound hypotension in critically unwell patients. Respiratory depression and apnea are also common, particularly following bolus doses. In extubated patients, equipment for bag-mask ventilation and endotracheal intubation should be immediately available. Doses in excess of 5 mg/kg/hr for prolonged periods have been associated with propofol infusion syndrome. This syndrome is characterized by metabolic acidosis and progressive hemodynamic collapse, and it is potentially fatal.9 Prolonged infusions may result in hyperlipidemia resulting from the intralipid emulsion.
Midazolam
Midazolam can be given enterally or parenterally and has an oral bioavailability of about 50%. For sedation in the ICU, midazolam is given by intermittent intravenous bolus or by continuous infusion. The usual dose range is 2 to 10 mg/hr, but much higher doses are occasionally required. Following a single intravenous dose, midazolam has a rapid onset of action and a short duration of effect. The distribution half-time is about 8 minutes. Bolus doses should be administered slowly (1 mg/min) and titrated to effect because the peak effect may be delayed for several minutes in patients with low cardiac output. Following prolonged infusion the context-sensitive half-time is increased (see Fig. 4-2), which results in a greatly prolonged duration of effect.
Midazolam undergoes hepatic metabolism by hydroxylation—by the cytochrome P-450 (CYP) 3A4 enzyme system—and conjugation. The 1-hydroxy metabolite is pharmacologically active and can contribute to the clinical effect. Drugs that inhibit the CYP3A4 enzyme system (Table 4-3) can prolong the effect of midazolam.
Table 4-3 Selected Substrates, Inhibitors, and Inducers of the CYP3A4 and 2D6 Hepatic Enzyme Systems
| CYP3A4 | ||
|---|---|---|
| Substrates | Inhibitors | Inducers |
| Calcium channel blockers | Antiarrhythmics | Rifamycins |
| Diltiazem | Amiodarone | Rifabutin |
| Felodipine | Calcium channel blockers | Rifampin |
| Verapamil | Diltiazem | Rifapentine |
| Benzodiazepines | Verapamil | Anticonvulsants |
| Midazolam | Nicardipine | Carbamazepine |
| Alprazolam | Azole antifungals | Phenobarbital |
| Immunosuppressives | Itraconazole | Phenytoin |
| Cyclosporine | Ketoconazole | Others |
| Tacrolimus | Voriconazole | St. |
| Sirolimus | Macrolide antibiotics | Anti-HIV agents |
| Statins | Erythromycin | |
| Atorvastatin | Clarithromycin | |
| Lovastatin | Troleandomycin | |
| Macrolide antibiotics | Others | |
| Erythromycin | Grapefruit juice | |
| Clarithromycin | Anti-HIV agents | |
| Others | Metoclopramide | |
| Losartan | ||
| Sildenafil | ||
| Anti-HIV agents | ||
| CYP2D6 | ||
| Substrates | Inhibitors | |
| β blockers | Antidepressants and antipsychotics | |
| Alprenolol | Chlorpromazine | |
| Bufuralol | Haloperidol | |
| Carvedilol | Fluoxetine | |
| Metoprolol | Paroxetine | |
| Propranolol | Clomipramine | |
| Timolol | Doxepin | |
| Antiarrhythmics | Antiarrhythmics | |
| Flecainide | Quinidine | |
| Mexiletine | Amiodarone | |
| Propafenone | Antihistamines | |
| Antipsychotics | H2 antagonists (ranitidine) | |
| Haloperidol | H1 receptor antagonists | |
| Antidepressants | ||
| Fluoxetine | ||
| Paroxetine | ||
| Venlafaxine | ||
| Some tricyclic antidepressants | ||
| Opioids | ||
| Codeine | ||
| Dextromethorphan | ||
| Tramadol | ||
Substrate drugs’metabolisms or inhibitors of the relevant enzyme system. Two are enhanced or inhibited other important CYPenzyme systems are 2C9, which is involved in the metabolism of warfarin, and 2C19, which is involved in the metabolism of the proton pump inhibitors (omeprazole, pantoprazole, etc.)
(Modified from Wilkinson GR: Drug metabolism and variability among patients in drug response. N Engl J Med 352:2211, 2005.)48 HIV, human immunodeficiency virus.
Diazepam
Diazepam may be administered enterally and parenterally and has an oral bioavailability of 100%. Oral or rectal diazepam, in a dose of 5 to 20 mg, is used to treat anxiety and alcohol withdrawal and for night sedation. Intravenously, diazepam is used as boluses of 2.5 to 10 mg as a sedative or anticonvulsant or for acute alcohol withdrawal. Following an intravenous bolus dose, the onset of effect is rapid, similar to that of midazolam. However, the duration of effect is longer because of the relatively long distribution half-time of about 1 hour. Also, the context-sensitive half-time of diazepam is greatly increased following prolonged administration (see Fig. 4-2). Diazepam has several active metabolites that also prolong the clinical effect.
Dexmedetomidine
Dexmedetomidine is a highly selective α2 receptor agonist (Chapter 3) that is approved for short-term (<24 hours) sedation in the ICU. It is administered as a loading dose of 1 μg/kg over 10 to 20 minutes followed by a continuous infusion of 0.2-0.7 μg/kg/hr. Dexmedetomidine has a short duration of action (minutes) due to rapid redistribution from the central compartment and is therefore easily titratable.
Stimulation of α2 receptors in the brain causes reduced sympathetic nervous system activity and sedation; stimulation of α2 receptors in the spinal cord results in analgesia. Patients receiving dexmedetomidine appear calm and sleepy but usually can easily be roused. The drug causes minimal respiratory depression, and the infusion need not be stopped prior to extubation. Requirements for supplemental opioid analgesia are greatly reduced with dexmedetomidine compared to propofol.10
Rapid intravenous administration of dexmedetomidine can cause transient hypertension due to weak peripheral α1 receptor agonist activity. This effect may be avoided by administering the loading dose slowly. During the infusion, modest hypotension and bradycardia may occur, particularly at higher doses. Hypotension is slightly greater with dexmedetomidine than with propofol.10
ANTIPSYCHOTIC DRUGS
Side Effects
The side effects of antipsychotics include sedation (central antihistamine and anticholinergic effects), peripheral anticholinergic effects (dry mouth, blurred vision, constipation, urinary retention), postural hypotension (α receptor blockade), and extrapyramidal motor effects. Extrapyramidal effects may be acute or chronic; acute effects include dystonic reactions (manifest as painful muscle spasms involving primarily the face and neck) and motor restlessness. Antipsychotics should be avoided in patients with Parkinson disease. The risks for respiratory depression and airway obstruction are lower than with the benzodiazepines and propofol, but they can still occur. A number of antipsychotic drugs (including thioridazine, haloperidol, and droperidol) are associated with QT prolongation and torsades de pointes ventricular tachycardia.11 This has led to a “black-box warning” for droperidol that has greatly diminished its use. However, the risk for torsades de pointes with droperidol has probably been overstated.12 Nevertheless, this class of drugs should be avoided in patients with QT prolongation and in those with other risk factors for torsades de pointes (Chapter 21). The QT interval should be monitored during treatment, and if it lengthens by >25%, treatment should be discontinued.
Antipsychotics
Risperidone
Risperidone is an atypical antipsychotic that is used mainly to treat psychotic disorders such as schizophrenia. However, evidence of its effectiveness in treating acute delirium is emerging.13 Risperidone is generally not available for parenteral use so is not useful for the acute control of aggressive patients. The starting dose is 0.25 to 0.5 mg twice daily, which may be increased to a maximum of 8 mg per day.13 Extrapyramidal side effects are fewer than with haloperidol but can occur. Risperidone is not associated with torsades de pointes ventricular tachycardia.11
ANALGESIC DRUGS
Pain following cardiac surgery arises from the sternotomy incision, from pleural irritation caused by chest drains, from osteoarticular trauma caused by retraction of the thoracic cage, and from sites from which bypass conduits have been obtained (saphenous vein, radial artery). Occult rib fractures are also common. In one study, 44 rib fractures were identified on radionuclide bone scans following median sternotomy in 24 patients.14 Pain is maximal on the first and second postoperative days, with the greatest intensity in the sternal, substernal, and parasternal regions.15 Pain scores are significantly higher in younger patients (<60 years) than in older patients.15 Severe pain also limits deep breathing and forceful coughing, which contributes to sputum retention and atelectasis and increases the risk for pneumonia. Some investigators have shown a reduction in postoperative pain16 and improved pulmonary function17 with the use of minimally invasive surgical techniques rather than conventional midline sternotomy. However, these benefits have not been consistent findings.18,19 Postoperative pain varies dramatically from patient to patient and also changes over time. Thus, analgesic regimes must be individualized and drug therapy titrated to effect.
Pain Assessment
In awake patients, pain may be subjectively graded on a numeric rating scale of 0 to 10, where 0 means no pain and 10 means the worst pain imaginable. Pain scores should be recorded at rest and with coughing every few hours for the first two days following surgery. The goal is to maintain pain scores of 3/10 at rest and 5/10 with coughing. The degree of sedation and the presence of any nausea or vomiting should be assessed at the same time as the pain scores. In extubated patients receiving opioids, sedation may be rated on a 4-point scale where A = alert, V = rousable to voice, P = rousable to pain, and U = unrousable. The assessment of regional nerve blocks is discussed in Chapter 12.
In ventilated or confused patients who are unable to verbalize, pain is not easy to assess. It may be very difficult to distinguish inadequate analgesia from inadequate sedation. Doctors, and to a lesser extent nurses, tend to underestimate pain in ICU patients.20 Various physiologic parameters, such as tachycardia, hypertension, and sweating, can indicate pain. Additionally, behavioral responses such as face grimacing, eye shutting, and fist clenching may be indicative of pain.21 If there is doubt, analgesics should be administered in preference to sedatives.
Analgesic Regimens
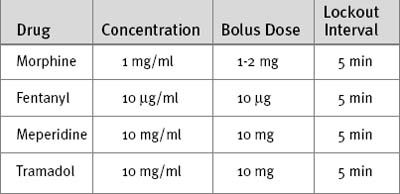
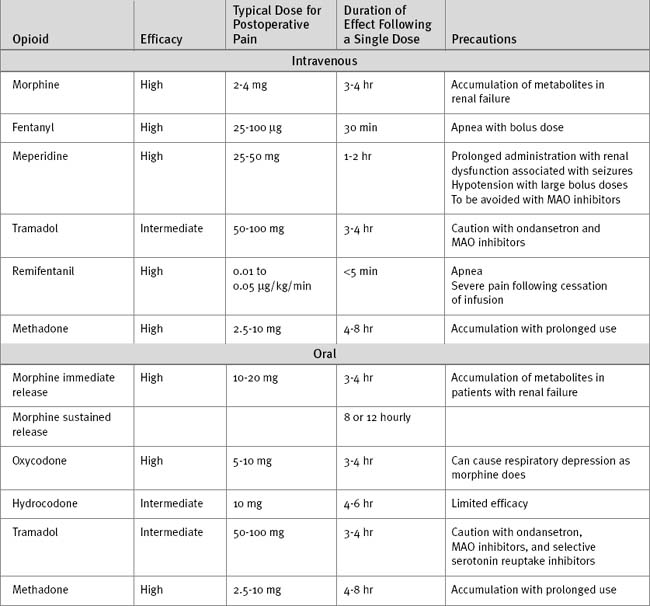
While patients are sedated and intubated, an opioid is administered intravenously, usually by intermittent bolus and on the basis of the patient’s physiologic and behavioral responses. Once the patient is awake and extubated, intravenous opioid analgesia may be administered via a patient controlled analgesia (PCA) system (Table 4-4). PCA provides enhanced analgesia and increased patient satisfaction, and it may improve pulmonary function over that found with the use of conventional intravenous opioid techniques.22,23 An alternative to PCA, once patients are able to take medications enterally, is the combination of a long- and a short-acting oral opioid (Table 4-5). The long-acting agent is taken morning and night for 2 to 3 days following surgery; the short-acting agent is taken for breakthrough pain or prior to planned activities such as physical therapy. A full agonist should not be combined with a weak agonist (see Strong and Weak Opioids in subsequent material). After the first few days following surgery, acetaminophen, either alone or in combination with a weak opioid, usually provides adequate analgesia. A simple measure that helps with pain following sternotomy is the use of a rolled towel or small pillow that the patient can clutch over the wound during repositioning and coughing.
In some centers, regional analgesia (Chapter 12), often involving epidural or intrathecal opioids, is used for the management of pain following cardiac surgery. The advantages of regional analgesia include improved postoperative pain relief and the potential for earlier extubation.24,25 Countered against this is the fact that for most patients the pain following sternotomy is of moderate, rather than severe, intensity15 and is typically less than the pain experienced following a lateral thoracotomy or upper abdominal incision. Furthermore, despite the good safety record noted in reported series, many clinicians are concerned about the potential for the formation of epidural hematoma in patients in whom the epidural space has been instrumented and who then receive heparin shortly thereafter.
Analgesics: Opioids
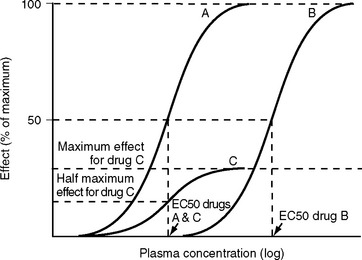
Strong and Weak Opioids: Efficacy Versus Potency
Efficacy describes the maximum clinical effect of a drug; potency describes the plasma concentration, and therefore the dose, required for a particular clinical effect—usually the EC50, the plasma concentration at which 50% of a drug’s maximal effect occurs (Fig. 4-3). Strong opioids, such as morphine and fentanyl, have high efficacy and are termed full agonists or simply agonists. However, fentanyl has a greater affinity for the μ receptor than does morphine, and it is able to produce a similar clinical effect at a lower dose; thus, fentanyl is more potent than morphine. A drug that produces a submaximal effect, irrespective of the dose, is called a partial agonist (see Fig. 4-3). Propoxyphene, oxycodone, and buprenorphine are examples of partial agonists at opioid receptors. Such drugs are also called weak opioids. Weak opioids may have high (e.g., buprenorphine) or low (e.g., codeine) potency.
When an opioid antagonist drug such as naloxone binds to an opioid receptor it exerts no pharmacologic effect (zero efficacy). An antagonist drug functions by preventing access to the receptor population by an agonist. Thus naloxone, which is highly potent, can antagonize the effect of morphine or fentanyl. Similarly, a potent partial agonist such as buprenorphine can potentially antagonize the effect of a less potent strong opioid such as morphine. For this reason, strong and weak opioids should not be used together. The characteristics of opioids that are commonly used in the ICU are shown in Table 4-5.
Clinical Effects
Opioids reduce the sensation of pain in a dose-dependent manner but they do not alter the sensory threshold of pain. Thus, patients report improved analgesia but are usually still aware of their pain. Opioids cause drowsiness and mental clouding and can also cause unpleasant dreams and hallucinations. They diminish the sensation of dyspnea and suppress cough. Other effects include respiratory depression, nausea and vomiting, miosis (small pupils), reduced gastrointestinal motility, urinary retention, and inhibition of the secretion of antidiuretic hormone. The occurrence and severity of these effects are dependent primarily on the efficacy and dose of the opioid rather than on the specific characteristics of a particular agent. However, a patient may tolerate one agent better than another. Therefore, if a particular drug causes unpleasant side effects, such as hallucinations or nausea, it is worth trying another agent.
Respiratory depression is a feature of all strong opioids, is dose-dependent, and can be fatal. Life-threatening respiratory depression is a particular risk when strong opioids are administered by continuous infusion to extubated patients, but respiratory depression is highly unlikely when the same drug is administered by PCA.26 Severe thoracic or abdominal pain can cause chest splinting and respiratory insufficiency. In this situation, administration of a strong opioid can actually improve ventilation and gas exchange.
Fentanyl
Fentanyl is a synthetic strong opioid with a potency 60 to 80 times greater than that of morphine. It is available for intravenous, transmucosal, and transdermal use. Following an intravenous dose, the onset of effect occurs within 60 seconds; this is faster than the onset of morphine because of the shorter effect-site half-time of fentanyl (see Speed of Onset in earlier material). At doses less than about 500 μg, the duration of action of fentanyl is relatively short (<60 min) because of redistribution; the distribution half-time of fentanyl is 13 minutes. Despite having a shorter duration of effect, fentanyl actually has a longer elimination half-time than morphine: 3.6 versus 3.0 hours. At higher doses or following prolonged infusion (see Fig. 4-2), the duration of action of fentanyl is greatly increased.
Tramadol
Tramadol is a partial agonist at μ opioid receptors but also exerts its analgesic effect through inhibition of serotonin and norepinephrine uptake within the CNS. It is less efficacious than morphine for the management of severe pain but has only a limited capacity to cause respiratory depression. The incidence and severity of nausea are similar to or worse than those that occur with morphine. Tramadol is usually combined with a nonopioid analgesic for the treatment of moderate pain. Additionally, there are some limited data that indicate that the combination of tramadol and morphine provides superior analgesia to the analgesia of morphine alone for the treatment of severe pain.27 Although the combination of a full and a partial agonist might seem counterproductive, the effect of tramadol on serotonin and norepinephrine reuptake may explain the benefit in this case. When tramadol is combined with the antiemetic ondansetron, a mutual reduction in efficacy occurs.28 Both drugs have been associated with the serotonin syndrome (see later discussion). Tramadol is available for enteral and parenteral use (see Table 4-5) and has an oral bioavailability of nearly 100%. Part of the analgesic activity of tramadol derives from the active metabolite mono-O-desmethyltramadol, which is dependent on the CYP2D6 enzyme system. Inhibition of CYP2D6 (see Table 4-3) greatly reduces the analgesic efficacy of tramadol. CYP2D6 is deficient in 5% to 10% of Caucasians.
Oxycodone
Oxycodone is a strong opioid with a duration of action that is similar to that of morphine. Like morphine, oxycodone is available in a sustained-release formulation. Oxycodone has a side-effect profile similar to that of morphine, including the potential for life-threatening respiratory depression. Codeine, hydrocodone, and propoxyphene are weak opioids that are commonly formulated with acetaminophen. The analgesic effect of codeine comes almost entirely from its metabolism to morphine by CYP2D6 (see Tramadol, earlier, and Table 4-3). Codeine is also useful to suppress cough and, because it has a powerful constipating effect, for treating diarrhea.
Naloxone
Naloxone is a highly potent opioid receptor antagonist that is used to reverse the effect of opioid-induced respiratory depression or, when combined with flumazenil, to rule out a pharmacologic cause of unexplained coma. Intravenously, naloxone may be given in 0.1 mg increments, titrated to effect. The duration of effect of a single dose of 0.4 mg naloxone is only 30 to 45 minutes, which may be far less than the opioid agonist that is being reversed. Naloxone should be used with caution because it can precipitate severe pain or acute withdrawal in patients who are opioid tolerant. Tachycardia, hypertension, and acute pulmonary edema may occur.
Analgesics: Acetaminophen (Paracetamol)
Acetaminophen is an analgesic and antipyretic drug that has almost no antiinflammatory effects. Its mechanism of action is unknown, but it is thought to act centrally through inhibition of the enzymes cyclooxygenase (COX) type 3 (constitutive) and COX-2b (inducible) (see NSAIDs below). At therapeutic doses it has almost no side effects, provides effective analgesia for mild to moderate pain, and has an opioid-sparing effect for severe pain.29 Acetaminophen has been available for oral and rectal use for many years, and recently an intravenous formulation has become available. Intravenous acetaminophen is likely to replace the acetaminophen prodrug, propacetamol, which is currently available for intravenous use in some countries. The oral bioavailability of acetaminophen is 100%, and the dose, whether oral, rectal, or intravenous, is 500 to 1000 mg 6 hourly. Overdose with as little as 10 g of acetaminophen can cause fatal hepatotoxicity. Acetaminophen should be withheld in patients with hepatocellular dysfunction.
Analgesics: Nonsteroidal Antiinflammatory Drugs
NSAIDs inhibit COX, an enzyme necessary for the synthesis of prostaglandins and thromboxane. Two major forms of the enzyme are found in the periphery: COX-2, the inducible form, is involved in inflammation. COX-1, the constitutive form, is found in various sites throughout the body, including blood vessels, the stomach, and the kidney. Many of the prostaglandins formed under the influence of COX-1 have protective functions such as renal vasodilation (Chapter 1) and protection of the gastric mucosa. NSAIDs inhibit both forms of the enzyme.
NSAIDs have analgesic, antipyretic, antiplatelet, and antiinflammatory effects and may be used as supplements to acetaminophen and opioids for the treatment of pain following cardiac surgery.30,31 However, NSAIDs are associated with a number of adverse effects, including renal dysfunction and gastrointestinal bleeding. NSAID-induced renal dysfunction is more likely in patients with preexisting renal dysfunction, impaired ventricular function, or hemodynamic instability. The risk for gastrointestinal bleeding is greatest in patients taking anticoagulant medications in those with histories of peptic ulcer disease. NSAIDs can provoke bronchospasm in patients with asthma and potentially can increase postoperative bleeding. For these reasons, the routine use of NSAIDs in cardiac surgery patients is controversial.32,33
NSAIDs should be avoided in the following circumstances:
If a decision is made to use an NSAID, it may be prudent to wait until the first postoperative creatinine level has been confirmed as normal before administering the first dose. There is some evidence that the concomitant use of ibuprofen reduces the antiplatelet effects of aspirin,34 suggesting that an alternative agent should be used following cardiac surgery. In a recent large-cohort study, the combined use of NSAIDs (including ibuprofen) and aspirin was not associated with an increased risk for myocardial infarction compared to the use of aspirin alone.35
Analgesics: COX-2 Inhibitors
Drugs with specific activity against COX-2 act as analgesics and cause less gastrointestinal side effects than the NSAIDs. COX-2 inhibitors have been associated with an increased risk for myocardial infarction, stroke, and sternal wound infection. A number have been withdrawn from the market. They should be avoided in cardiac surgery patients.36,37
NEUROMUSCULAR-BLOCKING DRUGS
Neuromuscular-blocking drugs are used in the ICU to facilitate tracheal intubation and to provide paralysis of skeletal muscles in patients who are mechanically ventilated. Neuromuscular-blocking drugs can be extremely useful for patients in the acute phase of a critical illness characterized by severe cardiac or respiratory insufficiency. Neuromuscular-blocking drugs are also sometimes indicated following routine cardiac surgery in patients who develop ventilator dysynchrony or shivering. The majority of patients who are mechanically ventilated do not require muscular paralysis, and these drugs should be reserved for patients with specific indications and should be discontinued as soon as possible. Inappropriate use of neuromuscular-blocking drugs delays extubation, can exacerbate postextubation respiratory insufficiency, and predisposes patients to the development of critical-illness polymyoneuropathy (Chapter 37).38 Furthermore, the use of paralyzing drugs in the absence of sufficient sedation and analgesia results in unpleasant awareness.
Mechanism of Action
Assessment of Neuromuscular Function
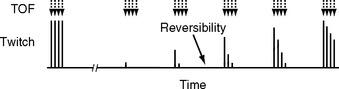
One way of gauging neuromuscular function by using a nerve stimulator is to assess the strength of thumb adduction in response to a train-of-four stimulation of the ulnar nerve. The nerve stimulator is connected to surface electrodes on the distal ulnar side of the forearm. One electrode is placed 1 cm proximal to the wrist crease and the other electrode is placed 2 to 3 cm farther up on the forearm. The nerve stimulator is then set to deliver four brief impulses at 60 to 80 mA, 0.5 seconds apart. The normal response to train-of-four stimulation is four strong twitches of the thumb, each of equal intensity, whereas complete neuromuscular blockade fails to elicit any response. As recovery of function occurs, the first of the four stimuli elicits a weak twitch; then progressively, the second, third, and fourth twitches return (Fig. 4-4). Once four twitches have returned, residual neuromuscular blockade is identified by the presence of fade, where the intensity of the first twitch is greater than that of the last twitch. Patients should not be extubated until there are four strong twitches with minimal fade. Residual neuromuscular blockade resulting from nondepolarizing agents can be reversed with neostigmine, as described later.
Nondepolarizing Neuromuscular-blocking Drugs
Rocuronium
Rocuronium is a steroid-based neuromuscular-blocking drug that has a faster onset and shorter duration of action than pancuronium. Following a dose of 1 mg/kg, optimal intubating conditions are achieved within 60 seconds, and muscular paralysis lasts for about 45 minutes. Rocuronium is hepatically metabolized and is eliminated unchanged into the bile. The rapid onset of action of rocuronium means it is suitable for use in a (modified) rapid-sequence intubation (Chapter 40) as an alternative to suxamethonium, but its greater cost and shorter duration of action than pancuronium mean that it is unsuitable for patients who require repeat doses over a number of days.
Reversal of Nondepolarizing Muscle Relaxants
Suxamethonium (succinylcholine)
Suxamethonium is a depolarizing neuromuscular-blocking drug that consists of two acetylcholine molecules joined together. At a dose of 1 to 1.5 mg/kg, suxamethonium causes extremely rapid muscular paralysis, and optimal intubating conditions are obtained within 30 to 60 seconds. Paralysis is preceded by a brief period of intense muscle fasiculation and rigidity. The drug is rapidly metabolized by plasma cholinesterase, which results in a duration of effect of 5 to 10 minutes following a standard dose. Suxamethonium is indicated primarily for rapid-sequence intubation (Chapter 40) because intubating conditions are obtained with sufficient speed that a period of bag-mask ventilation is not required. However, the use of suxamethonium is limited by a range of adverse effects, some of which are life-threatening.
In normal subjects, suxamethonium causes a small rise in serum potassium of about 0.5 mmol/l. However, in certain disease states (Table 4-6), a massive rise in serum potassium sufficient to cause life-threatening ventricular arrhythmias can occur. Two conditions that are associated with suxamethonium hyperkalemia are of particular concern in the ICU: stroke and critical illness polymyoneuropathy. The risk of suxamethonium hyperkalemia evolves over the first few days following a stroke and resolves over several months. An alternative to suxamethonium should also be considered in patients with prolonged immobility.
Table 4-6 Conditions in which Suxamethonium is Associated with Massive Hyperkalemia
| Stroke |
| Paraplegia |
| Muscular dystrophy |
| Critical illness polymyoneuropathy |
| Prolonged and severe intraabdominal infections |
| Head injury |
| Trauma |
| Burns |
| Prolonged immobility |
ANTIEMETICS
Nausea and vomiting are common following cardiac surgery, occurring in 30% to 50% of patients.39,40 The incidence and severity are greatest on the first postoperative day and then usually settle. However, in some patients nausea and vomiting persist for days, precluding oral intake and delaying recovery. The incidence is higher in females and younger patients and in those with histories of motion sickness or postoperative nausea and vomiting. Routine gastric decompression with a nasogastric tube does not reduce the incidence.41
Dopaminergic Antagonists
The antipsychotic medications described earlier all have antiemetic properties. Prochlorperazine (10 to 25 mg rectally, 8 hourly), droperidol (0.5 to 1 mg intravenously, 8 hourly), and metoclopramide (10 mg orally or intravenously, 6 hourly) are widely used as antiemetics. In addition to an antagonist effect at central dopamine receptors, metoclopramide is also a gastrointestinal prokinetic, which may contribute to its antiemetic effect. Metoclopramide, at the dose described, has a very low incidence of side effects but is less effective than droperidol and ondansetron.42
Serotonin Antagonists
Antagonists of the 5-hydroxytryptamine type 3 receptor (5-HT3), such as ondansetron and granisetron, are useful antiemetics and have very few side effects; headache and transient elevations in liver function tests occasionally occur.43 For the treatment of established nausea and vomiting, 4 mg of ondansetron is as effective as 8 mg.44 The dose may be repeated 8 hourly. Combined use of a 5-HT3 receptor antagonist and tramadol should be avoided (see Tramadol, earlier).
SEROTONIN SYNDROME
Serotonin syndrome is a state of heightened CNS activity manifested by agitation, autonomic hyperactivity, hyperreflexia, and delirium.47 It occasionally occurs as a side effect of drugs that potentiate serotonin activity, particularly when multiple serotoninergic drugs are used in combination. The syndrome should always be considered in patients who are being treated with a selective serotonin reuptake inhibitor antidepressant and who develop symptoms of CNS excitation. Commonly used drugs that are associated with the syndrome are listed in Table 4-7.
Table 4-7 Drugs that Are Associated with the Serotonin Syndrome
| Selective serotonin reuptake inhibitors |
| Monoamine oxidase inhibitors |
| Tricyclic antidepressants |
| Analgesics: meperidine and tramadol |
| Antiemetics: metoclopramide, ondansetron, granisetron |
| Antibiotics: linezolid, ritonavir |
| Others: valproate, lithium, sumatriptan (for migraine), sibutramine |
From Boyer EW, Shannon M: The serotonin syndrome. N Engl J Med 352:1112-1120, 2005.49
1 Hughes MA, Glass PS, Jacobs JR. Context-sensitive half-time in multicompartment pharmacokinetic models for intravenous anesthetic drugs. Anesthesiology. 1992;76:334-341.
2 Ostermann ME, Keenan SP, Seiferling RA, et al. Sedation in the intensive care unit: a systematic review. JAMA. 2000;283:1451-1459.
3 Jacobi J, Fraser GL, Coursin DB, et al. Clinical practice guidelines for the sustained use of sedatives and analgesics in the critically ill adult. Crit Care Med. 2002;30:119-141.
4 Kollef MH, Levy NT, Ahrens TS, et al. The use of continuous IV sedation is associated with prolongation of mechanical ventilation. Chest. 1998;114:541-548.
5 Mondello E, Siliotti R, Noto G, et al. Bispectral index in ICU: correlation with Ramsay score on assessment of sedation level. J Clin Monitor Comput. 2002;17:271-277.
6 Berkenbosch JW, Fichter CR, Tobias JD. The correlation of the bispectral index monitor with clinical sedation scores during mechanical ventilation in the pediatric intensive care unit. Anesth Analges. 2002;94:506-511.
7 Vivien B, Di Maria S, Ouattara A, et al. Overestimation of Bispectral Index in sedated intensive care unit patients revealed by administration of muscle relaxant. Anesthesiology. 2003;99:9-17.
8 McMurray TJ, Collier PS, Carson IW, et al. Propofol sedation after open heart surgery: a clinical and pharmacokinetic study. Anaesthesia. 1990;45:322-326.
9 Cremer OL, Moons KG, Bouman EA, et al. Long-term propofol infusion and cardiac failure in adult head-injured patients. Lancet. 2001;357:117-118.
10 Herr DL, Sum-Ping ST, England M. ICU sedation after coronary artery bypass graft surgery: dexmedetomidine-based versus propofol-based sedation regimens. J Cardiothorac Vasc Anesth. 2003;17:576-584.
11 Glassman AH, Bigger JTJ. Antipsychotic drugs: prolonged QTc interval, torsades de pointes, and sudden death. Am J Psychiatr. 2001;158:1774-1782.
12 Shale JH, Mastin WD, Shale CM. A review of the safety and efficacy of droperidol for the rapid sedation of severely agitated and violent patients. J Clin Psychiatr. 2003;64:500-505.
13 Schwartz TL, Masand PS. The role of atypical antipsychotics in the treatment of delirium. Psychosomatics. 2002;43:171-174.
14 Greenwald LV, Baisden CE, Symbas PN. Rib fractures in coronary bypass patients: radionuclide detection. Radiology. 1983;148:553-554.
15 Mueller XM, Tinguely F, Tevaearai HT, et al. Pain location, distribution, and intensity after cardiac surgery. Chest. 2000;118:391-396.
16 Izzat MB, Yim APC, El-Zufari MH, et al. Upper T mini-sternotomy for aortic valve operations. Chest. 1998;114:291-294.
17 Lichtenberg A, Hagl C, Harringer W, et al. Effects of minimal invasive coronary artery bypass on pulmonary function and postoperative pain. Ann Thorac Surg. 2002;70:461-465.
18 Bauer M, Pasic M, Ewert R, et al. Ministernotomy versus complete sternotomy for coronary bypass operations: no difference in postoperative pulmonary function. J Thorac Cardiovasc Surg. 2001;121:702-707.
19 Diegeler A, Walther T, Metz S, et al. Comparison of MIDCAP versus conventional CABG surgery regarding pain and quality of life. Heart Surg Forum. 1999;2:290-295.
20 Whipple JK, Lewis KS, Quebbeman EJ, et al. Analysis of pain management in critically ill patients. Pharmacotherapy. 1995;15:592-599.
21 Puntillo KA, Morris AB, Thompson CL, et al. Pain behaviors observed during six common procedures: Results from Thunder Project II. Crit Care Med. 2004;32:421-427.
22 Boldt J, Thaler E, Lehmann A, et al. Pain management in cardiac surgery patients: comparison between standard therapy and patient-controlled analgesia regimen. J Cardiothorac Vasc Anesth. 1998;12:654-658.
23 Gust R, Pecher S, Gust A, et al. Effect of patient-controlled analgesia on pulmonary complications after coronary artery bypass grafting. Crit Care Med. 1999;27:2218-2223.
24 Priestley MC, Cope L, Halliwell R, et al. Thoracic epidural anesthesia for cardiac surgery: the effects on tracheal intubation time and length of hospital stay. Anesth Analges. 2002;94:275-282.
25 Stenseth R, Bjella L, Berg EM, et al. Effects of thoracic epidural analgesia on pulmonary function after coronary artery bypass surgery. Eur J Cardiothorac Surg. 1996;10:859-865.
26 Schug SA, Torrie JJ. Safety assessment of postoperative pain management by an acute pain service. Pain. 1993;55:387-391.
27 Webb AR, Leong S, Burn SJ, et al. The addition of a tramadol infusion to morphine patient-controlled analgesia after abdominal surgery: a double-blinded, placebo-controlled randomized trial. Anesth Analges. 2002;95:1713-1718.
28 Hammonds B, Sidebotham DA, Anderson BJ. Aspects of tramadol and ondansetron interactions. Acute Pain. 2003;5:31-34.
29 Schug SA, Sidebotham DA, McGuinnety M, et al. Acetaminophen as an adjunct to morphine by patient-controlled analgesia in the management of acute postoperative pain. Anesth Analges. 1998;87:368-372.
30 Hynninen MS, Cheng DC, Hossain I, et al. Non-steroidal anti-inflammatory drugs in treatment of postoperative pain after cardiac surgery. Can J Anaesth. 2000;47:1182-1187.
31 Rapanos T, Murphy P, Szalai JP, et al. Rectal indomethacin reduces postoperative pain and morphine use after cardiac surgery. Can J Anaesth. 1999;46:725-730.
32 Griffin M. Con: nonsteroidal anti-inflammatory drugs should not be routinely administered for postoperative analgesia after cardiac surgery. J Cardiothorac Vasc Anesth. 2000;14:735-738.
33 Ralley FE, Day FJ, Cheng DC. Pro: nonsteroidal anti-inflammatory drugs should be routinely administered for postoperative analgesia after cardiac surgery. J Cardiothorac Vasc Anesthes. 2000;14:731-734.
34 Catella-Lawson F, Reilly MP, Kapoor SC, et al. Cyclooxygenase inhibitors and the antiplatelet effects of aspirin. N Engl J Med. 2001;345:1809-1817.
35 Garcia Rodriguez LA, Varas-Lorenzo C, Maguire A, et al. Nonsteroidal antiinflammatory drugs and the risk of myocardial infarction in the general population. Circulation. 2004;109:3000-3006.
36 Fitzgerald GA. Coxibs and cardiovascular disease. N Engl J Med. 2004;351:1709-1711.
37 Nussmeier NA, Whelton AA, Brown MT, et al. Complications of the COX-2 inhibitors parecoxib and valdecoxib after cardiac surgery. N Engl J Med. 2005;352:1081-1091.
38 Garnacho-Montero J, Madrazo-Osuna J, Garcia-Garmendia JL, et al. Critical illness polyneuropathy: risk factors and clinical consequences: a cohort study in septic patients. Intens Care Med. 2001;27:1288-1296.
39 Grebenik CR, Allman C. Nausea and vomiting after cardiac surgery. Br J Anaesth. 1996;77:356-359.
40 Woodward DK, Sherry KM, Harrison D. Antiemetic prophylaxis in cardiac surgery: comparison of metoclopramide and ondansetron. Br J Anaesth. 1999;83:933-935.
41 Burlacu CL, Healy D, Buggy DJ, et al. Continuous gastric decompression for postoperative nausea and vomiting after coronary revascularization surgery. Anesth Analges. 2005;100:321-326.
42 Domino KB, Anderson EA, Polissar NL, et al. Comparative efficacy and safety of ondansetron, droperidol, and metoclopramide for preventing postoperative nausea and vomiting: a meta-analysis. Anesth Analges. 1999;88:1370-1379.
43 Tramer MR, Reynolds DJ, Moore RA, et al. Efficacy, dose-response, and safety of ondansetron in prevention of postoperative nausea and vomiting: a quantitative systematic review of randomized placebo-controlled trials. Anesthesiology. 1997;87:1277-1289.
44 Tramer MR, Moore RA, Reynolds DJ, et al. A quantitative systematic review of ondansetron in treatment of established postoperative nausea and vomiting. BMJ. 1997;314:1088-1092.
45 Henzi I, Walder B, Tramer MR. Dexamethasone for the prevention of postoperative nausea and vomiting: a quantitative systematic review. Anesthes Analges. 2000;90:186-194.
46 Lee Y, Lai HY, Lin PC, et al. A dose ranging study of dexamethasone for preventing patient-controlled analgesia-related nausea and vomiting: a comparison of droperidol with saline. Anesth Analges. 2004;98:1066-1071.
47 Schulte-Tamburen AM, Scheier J, Briegel J, et al. Comparison of five sedation scoring systems by means of auditory evoked potentials. Intens Care Med. 1999;25:377-382.
48 Wilkinson GR. Drug metabolism and variability among patients in drug response. N Engl J Med. 2005;352:2211-2221.
49 Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med. 2005;352:1112-1120.
[/level-membership-for-cardiothoracic-surgery-category][not-level-membership-for-cardiothoracic-surgery-category]
Chapter 4 Sedation, Analgesia, and Related Topics
In this chapter the indications, contraindications, and adverse effects of drugs used for sedation and analgesia in the intensive care unit (ICU) are reviewed. In addition, practical tools for the measurement of depth of sedation and quality of analgesia are outlined. The related topics of neuromuscular-blocking drugs and antiemetics are also discussed.
PHARMACOKINETIC CONSIDERATIONS
Duration of Effect
The elimination half-time is the time taken for the amount of drug in the body to decrease by 50%. This parameter is often quoted when describing the pharmacokinetic properties of a drug. However, elimination half-time only rarely reflects the duration of effect. When a drug is given intravenously, it is rapidly distributed to a central “virtual” compartment consisting of plasma, interstitial fluid, and organs with high blood flow (brain, heart, liver, kidneys). This central compartment is in equilibrium with the effect site and with the organs of elimination—the liver and the kidneys. Drugs with low lipid solubility, high ionization, and high protein binding tend to be confined to this central compartment and typically have a small steady-state volume of distribution (VSS; Fig. 4-1). Such drugs can be described using a one-compartment model (V1). For drugs that obey one-compartment kinetics, the duration of effect may be related to the elimination half-time. Examples include aminoglycosides and neuromuscular blocking drugs. However, highly lipid-soluble drugs, including most sedative-hypnotics and opioid analgesics, display multicompartment kinetics in which drugs are redistributed from the central compartment to one or two peripheral compartments (V2, V3; see Fig. 4-1).
For drugs that display multicompartment kinetics, distribution and context-sensitive half-times are more useful concepts than elimination half-time. The distribution half-time is the time taken for the concentration within the central compartment to fall by 50%. Following a single intravenous dose, the distribution half-time determines the duration of effect of the drug (see Fig. 4-1). The context-sensitive half-time is the time taken for the effect-site concentration to fall by 50% following discontinuation of an intravenous infusion.1 Because a drug accumulates in the peripheral compartments over time, the context-sensitive half-time changes depending on the duration of infusion (Fig. 4-2). The context-sensitive half-time provides some indication of the duration of effect of the drug following both short- and long-term infusions (or repeated bolus doses). The percentage of decrease in concentration required for recovery from a drug’s effect is not necessarily 50%.
Onset of Action
The speed of onset of a drug depends on multiple factors; two that are of clinical importance for intravenously administered sedatives and analgesics are (1) the speed with which the drug is distributed within the central compartment and (2) the half-time for equilibration between the central and effect-site compartments (T1/2keo; see Fig. 4-1). Low cardiac output slows drug distribution within the central compartment and can greatly prolong the onset time. Thus, when administering potent sedative or analgesic medications to patients with low cardiac output, it is essential to give a small initial dose and wait a longer than normal time for the clinical effect to occur. Values for T1/2keo vary among drugs. For instance, the T1/2keo for morphine, fentanyl, and remifentanil are 17 minutes, 6.6 minutes, and 1.16 minutes, respectively. Therefore, morphine will have a slower onset of action than fentanyl and remifentanil.
Loading and Maintenance Doses
If a drug is administered by constant infusion or repeat doses it takes five (elimination) half-times to achieve 97% of the steady-state concentration (see Fig. 4-1). Thus, for a drug with a relatively long half-time, it is often useful to give a loading dose to rapidly increase the plasma concentration to within the therapeutic range. The loading dose (LD) depends on the volume of distribution (V) of the drug and the desired plasma concentration (CP):
Note that loading dose does not depend on clearance; thus, the loading dose of some renally eliminated drugs, such as gentamicin, do not need to be reduced in patients with renal failure. However, for drugs that display multicompartment kinetics, it is important to be clear which loading dose is used in the calculation: V1 or VSS. A loading dose based on VSS will initially produce very high plasma levels because the drug will be delivered only to the central compartment volume (V1). To avoid this problem, multiple small loading doses based on V1 may be required. This is the approach that is recommended for amiodarone in Chapter 3.
SEDATION
Sedation is part of a continuum of central nervous system (CNS) depression that ranges from anxiolysis through sedation, hypnosis (sleep), unconsciousness, and coma. Most sedative-hypnotic drugs produce anxiolysis at subhypnotic doses. Certain drugs, notably the benzodiazepines, also produce antegrade (i.e., following drug administration) amnesia at low doses. Some sedativehypnotics are anticonvulsants (e.g., benzodiazepines, barbiturates, and propofol). Anxiolysis is not the same as sedation. Antipsychotic drugs produce a state of outward calm but can increase feelings of anxiety and apprehension.
Indications for Sedation
Ventilated patients require sedation to tolerate endotracheal intubation and mechanical ventilation, facilitate nursing care, minimize the stress response, reduce oxygen consumption, diminish recall of unpleasant experiences, and prevent the development of posttraumatic stress disorder.2,3 Less commonly, sedation is indicated in extubated patients for the treatment of anxiety or delirium.
Mechanical ventilation, particularly using lungprotective strategies with long inspiratory times and permissive hypercapnia, is poorly tolerated by nonsedated patients and can result in ventilator dysynchrony (Chapter 29) and the sensation of dyspnea. Distressed patients may become tachycardic and hypertensive, which can exacerbate or provoke myocardial ischemia and bleeding. Such patients may also self-extubate or pull out their intravascular lines and surgical drains. A critically unwell patient commonly benefits from deep sedation, occasionally accompanied by neuromuscular blockade, during the acute phase of an illness. However, most patients do not require paralysis, only a level of sedation sufficient to allow tolerance of endotracheal intubation. Sedation of agitated patients should be commenced only after providing adequate analgesia and treating reversible physiologic causes.3
Adverse Effects of Sedation
Excessive sedation contributes to hypotension and delays awakening, needlessly prolonging the duration of mechanical ventilation.4 Sedation may also mask the development of intracranial, intrathoracic, or intraabdominal complications. The reduction in sympathetic tone that follows the administration of sedative and analgesic drugs can cause important hypotension. Hypotension is particularly marked in patients with high levels of endogenous catecholamines such as those that occur in the settings of hypovolemia and acute heart failure. Following prolonged administration of some sedatives (and opioid analgesics), tolerance may develop such that increased doses are required to elicit the same clinical effect. Abrupt discontinuation of certain sedatives, notably benzodiazepines, in a patient who has developed tolerance, may provoke a withdrawal syndrome (see discussion under subsequent heading Benzodiazepines). For these reasons, the need for sedation should be evaluated on an on-going basis and the depth of sedation regularly assessed.
Assessment of Sedation
If the clinical state allows, sedation should be stopped each day until the patient shows signs of awakening. Sedation can then be restarted if still indicated. A number of sedation scoring systems have been developed to quantify the depth of sedation and allow sedative drugs to be titrated to effect. Some commonly used scoring systems are shown in Table 4-1. Although primarily a system for monitoring neurologic function after trauma, the Glasgow Coma Scale (Table 4-2) may also be used to monitor sedation, although much information is lost in patients who are intubated and cannot respond verbally. One option in ventilated patients is to revise the Glasgow Coma Scale score to a maximum of 10, with the annotation that the patient is intubated.
| Verbal Response | Motor Response | Eye Response |
|---|---|---|
| 5 = Appropriate | 6 = Obeys commands | 4 = Opens spontaneously |
| 4 = Disorientated | 5 = Localizes to pain | 3 = Opens on command |
| 3 = Unconnected words | 4 = Withdraws from pain | 2 = Opens with pain |
| 2 = Sounds only | 3 = Abnormal flexion to pain | 1 = No response |
| 1 = Nothing | 2 = Extends to pain | |
| 1 = No response | Total = V + M + E = 3-15 |
The Bispectral Index (BIS) monitor, a highly processed electroencephalogram, is widely used to assess depth of anesthesia during surgery and has been used on a limited basis to monitor depth of sedation in the ICU.5,6 A potential problem with this monitor in the ICU environment is that BIS recordings are increased—implying a reduced depth of sedation—by electromyographic activity.7 Thus, BIS recordings tend to be lower in paralyzed patients than in nonparalyzed patients for an equivalent depth of sedation. This phenomenon could potentially lead to paralyzed patients receiving inadequate sedation, resulting in unpleasant awareness.
Sedative Drugs
Propofol
The onset of action following a bolus dose usually occurs within 30 seconds. Propofol has a distribution half-time of 2 to 4 minutes, which results in an offset of effect of 5 to 10 minutes following a bolus dose. There is minimal residual sedation. Propofol has a relatively stable context-sensitive half-time (see Fig. 4-2), and awakening is rapid even after prolonged infusion. In one study of cardiac surgery patients, extubation occurred after a mean time of 7.6 minutes after cessation of propofol infusion (mean dose of 82.8 mg/hr) following 17 hours of continuous sedation.8 The corresponding extubation time for patients given midazolam (mean dose 2.3 mg/hr) was 125 minutes. This rapid offset of clinical effect following prolonged infusion occurs because propofol has high hepatic and extrahepatic clearance (pharmacokinetic effect) and because subhypnotic concentrations of propofol cause minimal sedation (pharmacodynamic effect).
The main side effects of propofol relate to cardiac and respiratory depression. Hypotension due to vasodilation tends to be more marked than with other sedatives. Bolus doses must be used with extreme caution because as little as 20 mg can cause profound hypotension in critically unwell patients. Respiratory depression and apnea are also common, particularly following bolus doses. In extubated patients, equipment for bag-mask ventilation and endotracheal intubation should be immediately available. Doses in excess of 5 mg/kg/hr for prolonged periods have been associated with propofol infusion syndrome. This syndrome is characterized by metabolic acidosis and progressive hemodynamic collapse, and it is potentially fatal.9 Prolonged infusions may result in hyperlipidemia resulting from the intralipid emulsion.
Midazolam
Midazolam can be given enterally or parenterally and has an oral bioavailability of about 50%. For sedation in the ICU, midazolam is given by intermittent intravenous bolus or by continuous infusion. The usual dose range is 2 to 10 mg/hr, but much higher doses are occasionally required. Following a single intravenous dose, midazolam has a rapid onset of action and a short duration of effect. The distribution half-time is about 8 minutes. Bolus doses should be administered slowly (1 mg/min) and titrated to effect because the peak effect may be delayed for several minutes in patients with low cardiac output. Following prolonged infusion the context-sensitive half-time is increased (see Fig. 4-2), which results in a greatly prolonged duration of effect.
Midazolam undergoes hepatic metabolism by hydroxylation—by the cytochrome P-450 (CYP) 3A4 enzyme system—and conjugation. The 1-hydroxy metabolite is pharmacologically active and can contribute to the clinical effect. Drugs that inhibit the CYP3A4 enzyme system (Table 4-3) can prolong the effect of midazolam.
Table 4-3 Selected Substrates, Inhibitors, and Inducers of the CYP3A4 and 2D6 Hepatic Enzyme Systems
| CYP3A4 | ||
|---|---|---|
| Substrates | Inhibitors | Inducers |
| Calcium channel blockers | Antiarrhythmics | Rifamycins |
| Diltiazem | Amiodarone | Rifabutin |
| Felodipine | Calcium channel blockers | Rifampin |
| Verapamil | Diltiazem | Rifapentine |
| Benzodiazepines | Verapamil | Anticonvulsants |
| Midazolam | Nicardipine | Carbamazepine |
| Alprazolam | Azole antifungals | Phenobarbital |
| Immunosuppressives | Itraconazole | Phenytoin |
| Cyclosporine | Ketoconazole | Others |
| Tacrolimus | Voriconazole | St. |
| Sirolimus | Macrolide antibiotics | Anti-HIV agents |
| Statins | Erythromycin | |
| Atorvastatin | Clarithromycin | |
| Lovastatin | Troleandomycin | |
| Macrolide antibiotics | Others | |
| Erythromycin | Grapefruit juice | |
| Clarithromycin | Anti-HIV agents | |
| Others | Metoclopramide | |
| Losartan | ||
| Sildenafil | ||
| Anti-HIV agents | ||
| CYP2D6 | ||
| Substrates | Inhibitors | |
| β blockers | Antidepressants and antipsychotics | |
| Alprenolol | Chlorpromazine | |
| Bufuralol | Haloperidol | |
| Carvedilol | Fluoxetine | |
| Metoprolol | Paroxetine | |
| Propranolol | Clomipramine | |
| Timolol | Doxepin | |
| Antiarrhythmics | Antiarrhythmics | |
| Flecainide | Quinidine | |
| Mexiletine | Amiodarone | |
| Propafenone | Antihistamines | |
| Antipsychotics | H2 antagonists (ranitidine) | |
| Haloperidol | H1 receptor antagonists | |
| Antidepressants | ||
| Fluoxetine | ||
| Paroxetine | ||
| Venlafaxine | ||
| Some tricyclic antidepressants | ||
| Opioids | ||
| Codeine | ||
| Dextromethorphan | ||
| Tramadol | ||
Substrate drugs’metabolisms or inhibitors of the relevant enzyme system. Two are enhanced or inhibited other important CYPenzyme systems are 2C9, which is involved in the metabolism of warfarin, and 2C19, which is involved in the metabolism of the proton pump inhibitors (omeprazole, pantoprazole, etc.)
(Modified from Wilkinson GR: Drug metabolism and variability among patients in drug response. N Engl J Med 352:2211, 2005.)48 HIV, human immunodeficiency virus.
Diazepam
Diazepam may be administered enterally and parenterally and has an oral bioavailability of 100%. Oral or rectal diazepam, in a dose of 5 to 20 mg, is used to treat anxiety and alcohol withdrawal and for night sedation. Intravenously, diazepam is used as boluses of 2.5 to 10 mg as a sedative or anticonvulsant or for acute alcohol withdrawal. Following an intravenous bolus dose, the onset of effect is rapid, similar to that of midazolam. However, the duration of effect is longer because of the relatively long distribution half-time of about 1 hour. Also, the context-sensitive half-time of diazepam is greatly increased following prolonged administration (see Fig. 4-2). Diazepam has several active metabolites that also prolong the clinical effect.
[/not-level-membership-for-cardiothoracic-surgery-category]
