[level-membership-for-basic-science-category]
CHAPTER 26 Second Messengers
This chapter considers the remarkable variety of small molecules that carry signals inside living cells. These second messengers are chemically diverse, ranging from hydrophobic lipids confined to membrane bilayers, to an inorganic ion (Ca2+), to nucleotides (cyclic adenosine monophosphate [cAMP] and cyclic guanosine monophosphate [cGMP]), to a gas (nitric oxide). The messages that these molecules carry are encoded by their concentrations. In the simplest case, a rise or fall in the concentration of the second messenger conveys a signal from its source to its target. In other cases, the signal depends on the rate or frequency of the fluctuations in the concentration of the second messenger. The local concentration of a second messenger depends on the rate of production, the rate of diffusion from the site of production, and the rate of removal. Most second messengers are produced by enzymes that can switch on and off rapidly, allowing modulation of the concentration of second messengers on a millisecond time scale. In the case of Ca2+, the cytoplasmic concentration is determined by channels that release the ion from membrane-delimited stores and by pumps that remove it from cytoplasm.
The complexity of the signaling pathways is determined by the number of sources and the number of targets of each second messenger. Generally, multiple signal sources and multiple second messenger targets generate more complexity than one can fully appreciate. This chapter deals with this complexity only in passing. Chapter 27 considers a few model systems in which it is possible to understand how signals are integrated and transduced.
Cyclic Nucleotides
Two cyclic nucleoside monophosphates—adenosine 3′,5′-cyclic monophosphate (cAMP) and guanosine 3′,5′-cyclic monophosphate (cGMP)—are employed as second messengers (Fig. 26-1). Both act by binding reversibly to specific proteins. Enzymes that produce and degrade cyclic nucleotides determine the concentrations of these messengers available to bind targets. These enzymes turn over substrates rapidly, so they can amplify signals massively on a millisecond time scale, under the control of diverse signaling pathways (see Chapter 27). Cyclases make cyclic nucleotides in a single step from the corresponding nucleoside triphosphate, either adenosine triphosphate (ATP) or guano-sine triphosphate (GTP).
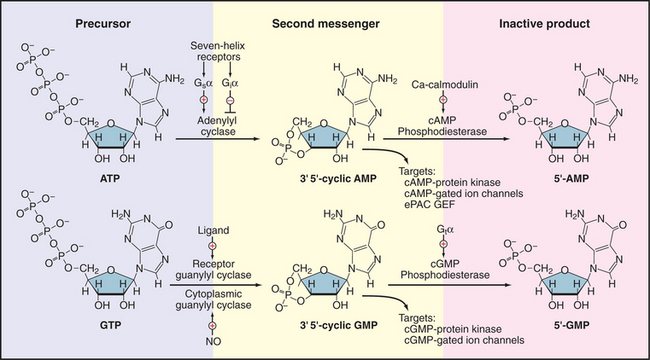
Figure 26-1 cyclic nucleotide metabolism. Synthesis and degradation of cAMP and cGMP, including regulatory inputs and targets. Gsα, Giα, and Gtα are trimeric GTPase a subunits (see Fig. 25-9). C A, calcium; NO, nitric oxide.
Cyclic nucleotides diffuse in the cytoplasm at about the same rate as in free solution, activating a modest repertoire of downstream targets, including protein kinases (see Figs. 25-3 and 25-4), cyclic nucleotide–gated ion channels (see Fig. 10-10), and, in the case of cAMP, one class of nucleotide exchange factors (Epac) for small GTPases (Rap1 and Rap2). The components of this system are quite ancient, since the protein domains that bind cAMP or cGMP are homologous to the cAMP-binding domain of CAP, a bacterial transcription factor. Enzymes called cyclic nucleotide phosphodiesterases degrade cAMP and cGMP to inactive nucleoside 5′-monophosphates. Eleven genes encode more than 40 different phosphodiesterases, which vary in their specificities for the two cyclic nucleotides, expression in various tissues, and localization to cellular compartments.
A family (10 human genes) of enzymes called adenylyl cyclases synthesize cAMP from ATP. Multiple transmembrane segments anchor most of these enzymes to the plasma membrane. Two homologous catalytic domains reside in the cytoplasm (C1 and C2 in Fig. 26-2). These cytoplasmic domains can be produced experimentally as soluble proteins separately from the transmembrane domains and can then be recombined to make a fully active enzyme. Both domains are necessary because the active site lies at the interface of the two domains. One adenylyl cyclase is a soluble enzyme that can concentrate in the nucleus. Generally, the concentration of adenylyl cyclases is very low relative to the trimeric G-proteins that regulate their activity.
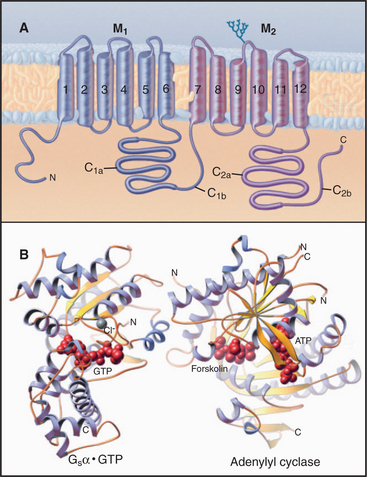
Multiple regulatory mechanisms act synergistically to regulate adenylyl cyclases. GTP-Gsα the GTPase subunit of a trimeric G-protein, activates many membrane-associated adenylyl cyclases by binding far from the active site (Fig. 26-2) and inducing a conformational change. Ca2+-calmodulin or protein kinase C (PKC) activates some adenylyl cyclases. GTP-Giα the GTPase subunit of another trimeric G-protein, or protein kinase A (PKA) inhibits some cyclases. Gβγ subunits of trimeric G-proteins activate some adenylyl cyclases but inhibit others. These diverse regulatory mechanisms allow adenylyl cyclases to integrate a variety of input signals. The diterpene forskolin from a Coleus plant binds to and activates adenylyl cyclases. Forskolin is useful experimentally for manipulating the cAMP concentration in cells and, on the basis of its ability to bind adenylyl cyclases, was essential in the initial purification of the enzyme by affinity chromatography. Regulation of a soluble form of adenylyl cyclase differs from that of all other isoforms. It is activated by bicarbonate, an essential step in sperm maturation.
In resting cells, the concentration of cAMP is so low, approximately 10−8M, that it does not bind its targets. Stimulation of appropriate receptors (such as the seven-helix β-adrenergic receptor, see Fig. 27-3) increases the cytoplasmic cAMP concentration more than 100-fold, enough to saturate the PKA regulatory subunits (Fig. 26-3). Dissociation of the regulatory (R) subunits frees the active PKA catalytic subunits to phosphorylate cytoplasmic and membrane substrates and to move into the nucleus to activate the transcription factor CREB (cyclic nucleotide regulatory element–binding protein) (see Fig. 15-22).
Outside the animal kingdom, cAMP has many functions. In bacteria, cAMP controls gene expression in response to nutritional conditions. The cellular slime mold Dictyostelium uses cAMP as an extracellular signal, acting through a seven-helix receptor, for its social interactions (see Fig. 38-12).
Guanylyl cyclases are dimeric enzymes similar to adenylyl cyclases. In fact, mutation of just two amino acid residues can convert a guanylyl cyclase to an adenylyl cyclase. Vertebrates express two types of guanylyl cyclases (see Fig. 24-9). A family of transmembrane receptors with cytoplasmic cyclase domains respond to ligand binding by producing cGMP. Cytoplasmic guanylyl cyclases are activated by nitric oxide and carbon monoxide, which bind a heme group in a regulatory domain (see the section titled “Nitric Oxide” later in this chapter).
Lipid-Derived Second Messengers
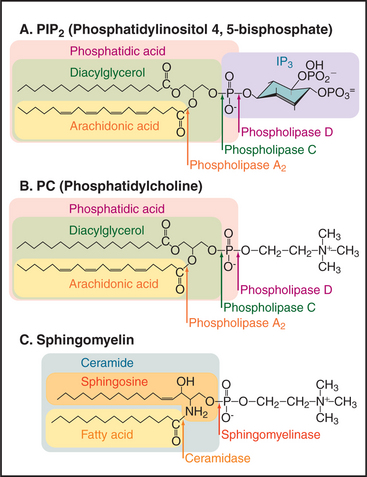
Phosphoglycerides (see Fig. 7-2) and sphingolipids (see Fig. 7-3) not only form cellular membranes but also participate in a wide range of signaling mechanisms. The long list of intracellular and extracellular second messengers derived from lipids will undoubtedly expand in the future. Three membrane lipids are the primary sources of these signaling molecules (Fig. 26-4):
Enzyme Reactions That Produce Lipid Second Messengers
Three types of enzyme reactions generate lipid second messengers:
Cells also use transferases that add or exchange head groups or fatty acids on membrane phosphoglycerides (see Fig. 7-2). These enzymes are essential for lipid biosynthesis but have not been implicated in signaling. For example, transferases add choline (as the nucleotide conjugate cytidine diphosphate [CDP]–choline) to DAG to make phosphatidylcholine). In the case of phosphatidylinositol, the head group is provided by dephosphorylation of IP3 to inositol, which is recombined enzymatically with a CDP conjugate of DAG to make phosphatidylinositol.
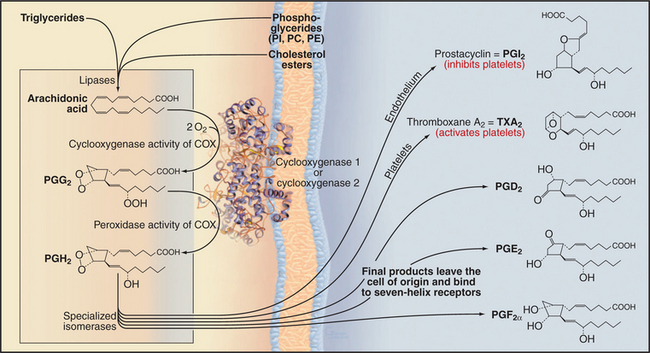
Sequential action of two or more enzymes produces some lipid second messengers. For example, cells can make DAG in two steps from phosphatidylcholine: PLD first makes phosphatidic acid, which is dephosphorylated by phosphatidic acid phosphatase. PLD followed by PLA2 makes lysophosphatidic acid from phosphatidylcholine. PLA2 also initiates the production of a huge family of fatty acid derivatives called eicosanoids by cleaving the fatty acid arachidonic acid from phosphatidylcholine. Cyclooxygenases and lipoxygenases then modify arachidonic acid to make prostaglandins, thromboxanes, and leukotrienes (Figs. 26-9 and 26-10), which leave the cell to interact with cell surface receptors.
Targets of Lipid Second Messengers
Downstream targets of the lipid second messengers are diverse but can be generalized to some extent into three categories (Fig. 26-5):
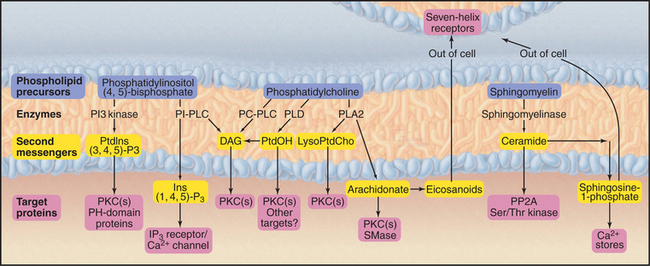
Lipid second messengers participate in many processes that are considered elsewhere in the book. For example, regulated secretion (see Chapter 21) in response to an agonist binding a seven-helix or tyrosine kinase receptor requires a transient Ca2+ signal in the cytoplasm. The Ca2+ is released from the membrane stores by IP3, which is produced by the action of a PI-PLC on PIP2. IP3-mediated Ca2+ release from the endoplasmic reticulum also controls smooth muscle contraction (see Fig. 39-21).
Protein Kinase C
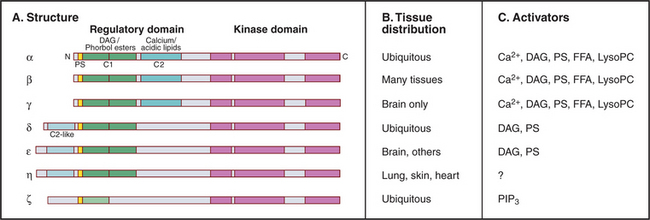
Many lipid second messengers, including DAG, PIP3, arachidonic acid, phosphatidic acid, and lysophosphatidylcholine activate one or more of the 10 protein kinase C (PKC) isozymes expressed by vertebrate cells (Fig. 26-6). These multiple PKC isozymes provide a selective response to various lipid second messengers. Some, but not all, PKC isozymes also require Ca2+ for activation. Sphingosine may inhibit some PKC isozymes.
PKCs have a protein kinase catalytic domain (see Fig. 25-3) and N-terminal regulatory domains. A pseudosubstrate sequence binds intramolecularly to the active site and inhibits activity by blocking substrate binding. Pseudosubstrates have alanine at the phosphorylation site instead of serine as substrates have. During apoptosis, caspases cleave off this regulatory domain (see Fig. 46-10) producing constitutively active PKC isoforms.
PKCs can provide either positive or negative feedback to the signaling pathways that turn them on. PKC activates PLD and PLA2 and provides positive feedback, because those enzymes produce more DAG to sustain the activation of PKC. On the other hand, PKC provides negative feedback when it phosphorylates and inhibits both growth factor receptors and PI-PLCγ1. PKC also phosphorylates and inhibits PI-PLCβ, generating negative feedback after activation of seven-helix receptors. Negative feedback makes both of these signaling events transient.
Phosphoinositide Signaling Pathways
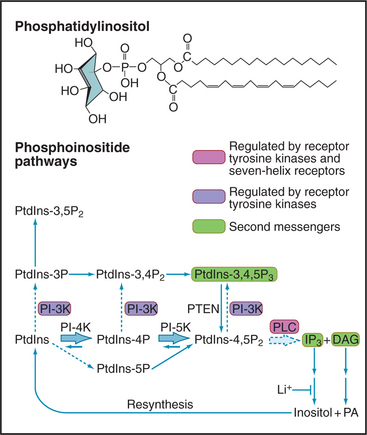
Although minor in terms of mass in biological membranes, phosphoinositides are major players in signal-ing (Fig. 26-7). The parent compound, phosphatidyl-inositol (PI), is a phosphoglyceride with a cyclohexanol head group called inositol. Specific lipid kinases phosphorylate the 4 and 5 hydroxyl groups of phosphatidylinositol to form PI(4-)P and PI(4,5-)P2, usually referred to simply as PIP and PIP2. A specific phosphatase can remove the D5 phosphate. Inactivation of the single copy of this gene on the X-chromosome causes Lowe’s syndrome with cataracts, renal failure, and mental retardation.
PIP2 is a substrate for a family of receptor-controlled PI-PLCs that cleave off the phosphorylated head group, producing two potent second messengers: IP3 and DAG. Water-soluble IP3 activates Ca2+ release channels in the endoplasmic reticulum (Fig. 26-12), and lipid-soluble DAG activates PKC. In contrast to Ca2+, which diffuses slowly and acts locally, IP3 diffuses rapidly through the cytoplasm, triggering Ca2+ release. DAG is confined to membranes but diffuses laterally to bind and activate PKC. Enzymes inactivate both IP3 and DAG. DAG is inactivated by phosphorylation to make phosphatidic acid, a second messenger in its own right but also an intermediate in the resynthesis of phosphoinositides. IP3 is dephosphorylated to inositol, which is inactive as a second messenger. LiCl inhibits the final step: dephosphorylation of inositol-1-phosphate. Remarkably, Li+ is clinically useful as a treatment for bipolar disorder, presumably by interfering with phosphoinositide signaling in the brain. In some tissues, IP3 is inactivated by phosphorylation to inositol 1,3,4,5-tetrakisphosphate (IP4).
Vertebrates use 10 classes of PI-PLCs to provide tissue-specific coupling of various receptors to the production of IP3 and DAG. PI-PLCγ1, expressed in the brain, is activated by tyrosine phosphorylation when its SH2 domains bind a tyrosine-phosphorylated receptor tyrosine kinase (Fig. 26-12). The trimeric G-protein Gqα activates another isozyme, PI-PLCβ.
Receptor tyrosine kinases activate another family of lipid kinases, PI-3 kinases, which phosphorylate the 3-hydroxyl group of PI, PIP, and PIP2. The products are PI(3-)P, PI(3,4-)P2, and PI(3,4,5-)P3. PI(3,4,5-)P3 is not a substrate for the PI-PLCs that produce IP3 and DAG, but it activates some PKC isozymes and binds specifically to certain pleckstrin homology domains (see Fig. 25-11), bringing enzymes such as protein kinase B (PKB/Akt) to the plasma membrane. Association with the membrane leads to phosphorylation and activation of PKB as part of insulin signaling (see Fig. 27-7). A phosphatase that removes the D3 phosphate from PIP3 is a tumor suppressor called PTEN (phosphatase and tensin homolog deleted on chromosome 10). Loss of PTEN function in tumors allows PIP3 to build up, activating PKB and growth promoting downstream pathways in many tumors. A steroid-like molecule called wortmannin inhibits PI-3 kinase relatively specifically and is used to investigate the physiological roles of PIP3.
Phosphatidylcholine Signaling Pathways
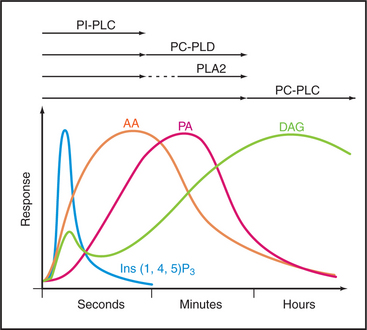
Phosphatidylcholine is not only a major structural lipid of the plasma membrane but also an important source of DAG and a large family of other signaling molecules (Fig. 26-5). In response to agonist stimulation, cells produce two waves of DAG (Fig. 26-8). Within seconds, PI-PLCs activated by seven-helix or tyrosine kinase receptors produce the first wave of DAG from PIP2. Then, over a period of minutes, a second wave of DAG is derived from phosphatidylcholine, either directly by a PC-PLC or in two steps by PLD (to remove choline) and a phosphatidic acid phosphatase (to remove the phosphate from phosphatidic acid). The first wave of DAG may contribute to the second wave as PKC activates one PLD isoform. Ca2+ produced in the first wave may also activate PLD.
Lipid-Derived Second Messengers for Intercellular Communication
Eicosanoids (eicosa is Greek for “20”) are a diverse family of metabolites derived from the 20-carbon fatty acid arachidonic acid, including prostaglandins, thromboxanes, leukotrienes, and lipoxins (Figs. 26-9 and 26-10). Depending on the particular receptor and G-protein, eicosanoids selectively activate or inhibit the synthesis of cAMP, release Ca2+, activate PKC, or regulate ion channels. Biological consequences are diverse, depending on the specific eicosanoid and target cell. Eicosanoids are important medically as mediators of inflammation.
Prostaglandins and thromboxanes are synthesized from arachidonic acid by pairs of enzymes, the first being generic and the second being specific for a particular product (Fig. 26-9). Typically, differentiated cells express primarily one second-step enzyme and thus produce just one of these local hormones.
Physiological responses to each eicosanoid depend on selective expression of specific seven-helix receptors. For example, receptors for prostaglandin F2a prepare the uterus, but not other organs of pregnant mammals, for delivery. Receptors for counteracting eicosanoids control the response of platelets to blood vessel damage (see Fig. 30-14). Normally, endothelial cells lining blood vessels produce prostaglandin I2 (prostacyclin), which inhibits interaction of platelets with the vessel wall and promotes blood flow by relaxing smooth muscle cells in the wall of the vessel. In response to damage, the blood-clotting protein thrombin activates platelets to aggregate and plug gaps in the endothelium. Activated platelets produce thromboxane A2, which diffuses locally to promote irreversible platelet aggregation and blood vessel constriction by triggering production of IP3, release of Ca2+, and contraction of smooth muscle (see Fig. 39-21). This secondary response mediated by positive feedback through thromboxane A2 minimizes blood loss but can contribute to pathological formation of clots in vital blood vessels, resulting in heart attacks and strokes. Tissue injury also provokes synthesis of prostaglandins E2 and I2, which mediate inflammation locally by dilating blood vessels, sensitizing pain receptors, and causing fever.
Nonsteroidal anti-inflammatory drugs, including aspirin and ibuprofen, target both cyclooxygenase isozymes. Most of these drugs competitively inhibit arachidonic acid binding to both enzymes, but aspirin covalently and irreversibly acetylates a serine residue in their active sites. Either way, these drugs inhibit the synthesis of all prostaglandins and thromboxanes. Aspirin reduces the incidence of heart attacks and strokes by inhibiting the synthesis of thromboxane A2 by platelets, which reduces platelet aggregation and pathological clotting in blood vessels. Low doses of aspirin are selective and effective for platelets, as platelets have only COX-I and lack the capacity for protein synthesis to replace inactivated enzyme. Aspirin also protects against colon cancer. High doses of aspirin and other nonsteroidal anti-inflammatory drugs reduce inflammation, pain, and fever by inhibiting the synthesis of other eicosanoids, but this is not without side effects, including gastrointestinal bleeding. Drugs that selectively inhibit COX-2 relieve inflammation without the gastrointestinal side effects caused by inhibition of COX-1. However, they inhibit the synthesis of prostaglandin I2 by endothelium. The lack of prostaglandin I2 to inhibit platelet aggregation might increase the risk of heart attacks and strokes.
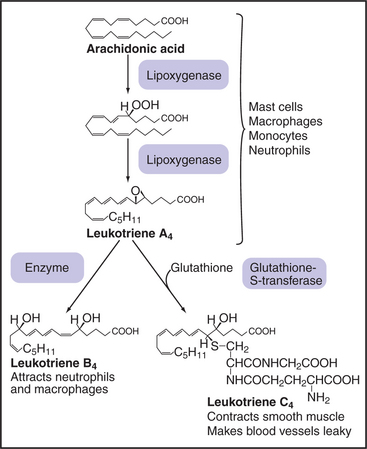
Macrophages and white blood cells produce an enzyme called 5-lipoxygenase that synthesizes leukotrienes and lipoxins by addition of oxygen to specific double bonds of arachidonic acid (Fig. 26-10). The first biologically active product, leukotriene A4, can be modified by addition and subsequent trimming of glutathione to yield a variety of active leukotrienes. Leukotrienes mediate inflammatory reactions by constricting blood vessels, allowing plasma to leak from small vessels and attracting white blood cells into connective tissue. These effects, together with constriction of the respiratory tract, contribute to the symptoms of asthma. Drugs that inhibit 5-lipoxygenase and leukotriene receptors are being tested in treatment of asthma and other inflammatory diseases, including atherosclerosis. Lipoxins are another family of mediators that act on blood vessels. They are formed by the addition of oxygen to both the C-5 and C-15 positions of arachidonic acid.
Phosphatidylcholine is also the starting point for production of two other water-soluble, intercellular, second messengers: lysophosphatidic acid (LPA) and platelet-activating factor (PAF). Activated platelets and injured fibroblasts produce LPA from phosphatidylcholine by the action of the phospholipases PLD and PLA2 (Fig. 26-4). A variety of cells synthesize PAF in two steps, using PLA2 to remove the C2 fatty acid from phosphatidylcholine molecules with an ether-bonded fatty alcohol (rather than an ester-bonded fatty acid) on the C1 position and a second enzyme to acetylate the C2 hydroxyl group. LPA and PAF escape cells and stimulate target cells by binding to seven-helix receptors. Depending on their signaling hardware, cells respond to LPA in different ways: Activation of the PLC/IP3 pathway releases intracellular Ca2+ in some cells; activation of a mitogen-activated protein (MAP) kinase pathway (see Fig. 27-5) stimulates some cells to divide; and activation of Rho-family small GTPases stimulates formation of actin bundles in cultured cells (see Fig. 33-20). As is expected from its name, PAF activates platelets, but it also modifies the behavior of other blood cells, inhibits heart contractions, and stimulates contraction of the uterus.
Endocannabinoids are a family of fatty acid amides that activate the seven-helix receptors that also respond to δ9-tetrahydrocannabinol, the active ingredient in marijuana. Many tissues, including the brain, synthesize the classic endocannabinoid, N-arachidonoyl-ethanolamine, from ethanolamine and arachidonic acid. Brain cannabinoid receptors are found exclusively on the axonal side of synapses. Stimulation of the postsynaptic neuron results in Ca2+ entry, activating the enzymes that synthesize endocannabinoids. When these lipid second messengers diffuse from the postsynaptic cell back to the presynaptic terminal, they bind class 1 cannabinoid receptors, which suppress transmitter release by blocking Ca2+ entry. Some of these presynaptic terminals secrete the inhibitory neurotransmitter GABA, so the endocannabinoid increases the activity of the postsynaptic neuron, including those that suppress the sensation of pain. The functions of cannabinoid receptors in other tissues are less well understood. Another lipid amide, oleamide (cis-9,10-octadecenoamide), induces sleep in mammals, most likely through GABA receptors (see Fig. 10-12). An enzyme called fatty acid amide hydrolase degrades all of these signaling molecules.
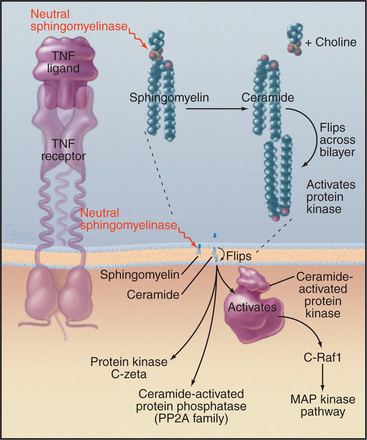
Sphingomyelin/Ceramide Signaling Pathways
Activation of plasma membrane receptors for tumor necrosis factor (TNF) and interleukin-2 (IL-2) stimulates formation of the lipid-soluble second messenger ceramide from sphingomyelin (Fig. 26-11). Sphingomyelin is concentrated in the external leaflet of the plasma membrane (Fig. 26-4). Ligand binding to TNF or IL-2 receptors activates a plasma membrane sphingomyelinase (a specialized PLC) that removes phosphorylcholine from sphingomyelin, producing ceramide. Ceramide flips across the lipid bilayer to the cytoplasmic surface and activates a proline-directed, serine/threonine protein kinase associated with the plasma membrane. Target proteins have the sequence X-Ser/Thr-Pro-X and include epidermal growth factor (EGF) receptor and Raf kinase. Downstream events include activation of MAP kinase, which activates transcription factors and other effectors (see Fig. 27-6). Ceramide also activates a protein phosphatases 1 and 2A.
Sphingosine and sphingosine-1-phosphate are produced from sphingomyelin by a succession of enzymatic reactions (Fig. 26-4). Ceramidase removes the fatty acid, and sphingomyelinase removes the phosphorylcholine head group to form sphingosine. Then sphingosine kinase adds phosphate to C1 to make sphingosine-1-phosphate. Growth factor signaling pathways might regulate this lipid kinase. Sphingosine-1-phosphate escapes from cells and activates seven-helix receptors on the same or other cells. Cells respond by releasing Ca2+, which influences motility and growth, as well as smooth muscle contraction. The biological effects of sphingosine are incompletely defined but include regulation of PKC.
Cross Talk
Interaction (cross talk) among lipid second messenger pathways is thought to integrate signals from different agonists. Consequently, it is difficult to define distinct linear pathways from an agonist to an individual effector through lipid second messengers. The following are some examples of cross talk using enzymes defined in Figure 26-4:
Calcium
Overview of Calcium Regulation
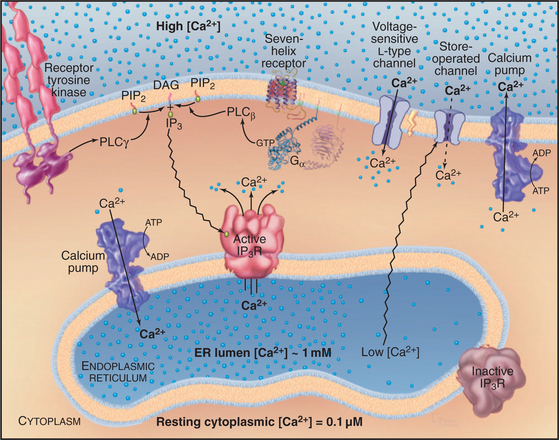
In contrast to all other second messengers, which are synthesized and metabolized, Ca2+ levels are controlled by release into and removal from the cytoplasm (Fig. 26-12). ATP-driven Ca2+ pumps of the plasma membrane and endoplasmic reticulum keep cytoplasmic Ca2+ levels low (about 0.1μM in resting cells) and generate a 10,000-fold concentration gradient across these membranes. A remarkable variety of stimuli, operating through many different receptors (Table 26-1), open Ca 2+ channels in the plasma membrane or endoplasmic reticulum, allowing a concentrated puff of Ca2+, called a Ca2+ transient or “spark,” to enter the cytoplasm. Peak cytoplasmic Ca2+ concentrations in stimulated cells are in the micromolar range. The durations of Ca2+ signals range from milliseconds to minutes depending on the nature of the stimulus, the type of release channel, the rate of Ca2+ pumping out of cytoplasm, and the rates of binding and dissociation on target proteins. Ca2+ levels can rise and fall locally, flood the whole cytoplasm, or travel in waves across a cell.

Ca2+ signals work locally in cytoplasm. Although Ca2+ is a small ion with a large diffusion coefficient in water, its diffusion through cytoplasm is very slow, owing to efficient sequestering mechanisms and abundant Ca2+-binding proteins, estimated to be 300μM. Thus, only about 1 in 100 Ca2+ ions is free to diffuse in cytoplasm; the other 99 are bound. At a concentration of 0.1μM in the cytoplasm of resting cell, the half-time for a free Ca2+ion is about 30μs, and its range of diffusion is only about 0.1μm. Thus, although Ca2+ pours through release channels at a rate of 106 ions per second, it does not spread but acts locally. Ca2+, when bound to a protein messenger such as calmodulin, has a wider range of about 5μm.
Cellular responses to Ca2+ signals depend on the available repertoire of Ca2+-sensitive proteins and effector systems (Appendix 26-1). Some Ca2+-binding proteins respond directly, such as Ca2+-activated ion channels, whereas others initiate a cascade of downstream reactions. For example, Ca2+ binding to calmodulin has no direct consequence, but Ca2+-calmodulin regulates more than a dozen proteins, including protein kinases and adenylyl cyclase.
Removal of Ca2+ from Cytoplasm
The best-characterized Ca2+ storage compartment is the sarcoplasmic reticulum of striated muscle (see Fig. 39-10), a specialized smooth endoplasmic reticulum with remarkably high concentrations of the SERCA Ca-ATPase pump and ryanodine receptor Ca2+ channels (Table 26-2). These proteins give sarcoplasmic reticulum a great capacity to handle the millisecond Ca2+ transients that control muscle contraction (see Fig. 39-15). Other cells use similar mechanisms but are endowed more modestly with these Ca2+-handling proteins. Mitochondria sequester Ca2+, using carriers driven by the electrochemical potential across the inner membrane. Although their Ca2+ content is high, mitochondria do not participate in signal transduction by regulated release of Ca2+ into cytoplasm.

Pumps
ATP-driven Ca2+ pumps of the plasma membrane and endoplasmic reticulum remove Ca2+ from cytoplasm (see Fig. 8-7). These P-type pumps move Ca2+ out of cytoplasm against a concentration gradient of about 104, hydrolyzing one ATP for every two Ca2+ transferred from the cytoplasm out of the cell or into the endoplasmic reticulum storage compartment. Cytoplasmic Ca2+ activates Ca2+ pumps until the Ca2+ in the cytoplasm falls to about 0.1μM, the resting level. Three different genes and alternative splicing produce at least five different Ca2+-ATPase pumps. In the heart, the activity of the Ca2+-ATPase is modulated by phospholamban, a 6-kD integral membrane protein of the sarcoplasmic reticulum. Phosphorylation by PKA and calmodulin-activated (CaM) kinase dissociates phospholamban from the Ca2+ pump, stimulating its activity.
Sequestering Proteins
Proteins in the lumen of the endoplasmic reticulum bind much of the Ca2+, but their low affinity ensures that bound and free Ca2+ are in a rapid equilibrium, providing free Ca2+ for release when membrane channels open. Calsequestrin is a major Ca2+-binding protein of the sarcoplasmic reticulum. In nonmuscle cells, the endoplasmic reticulum lumen sometimes contains calsequestrin, but more commonly, it contains calreticulin, a 47-kD protein with a low affinity (Kd = 250μM) but high capacity (25 moles) for Ca2+. Calreticulin is also a chaperone for protein folding in the endoplasmic reticulum (see Fig. 20-10).
Refilling Endoplasmic Reticulum
Repeated stimulation can deplete Ca2+ from intracel-lular stores, because some released Ca2+ is pumped out of the cell by plasma membrane pumps. Endoplasmic reticulum stores can also be depleted experimentally by thapsigargin, a lactone isolated from plants that inhibits most known endoplasmic reticulum Ca2+ pumps. In either case, cells replenish the stores by admitting extracellular Ca2+ through low-conductance channels, most likely but not yet definitely proven to be members of the Trp family (see Fig. 10-9). This is called store-operated Ca2+ entry. Some Trp channels respond to DAG or arachidonic acid, while others respond to decreased levels of Ca2+ in the endoplasmic reticulum, yet the physiological stimuli for refilling the endoplasmic reticulum in most cell types are still unclear.
Calcium-Release Channels
Voltage-gated and agonist-gated channels in the plasma membrane (Table 26-3 and Fig. 26-12) admit Ca2+ into the cytoplasm from outside. Chapter 10 explains how the membrane potential or agonists open these channels. Voltage-gated channels are essential for rapid responses in excitable cells such as those of muscles and neurons. Owing to rapid inactivation by negative feedback from the released Ca2+, most of these channels produce brief, self-limited Ca2+ pulses.
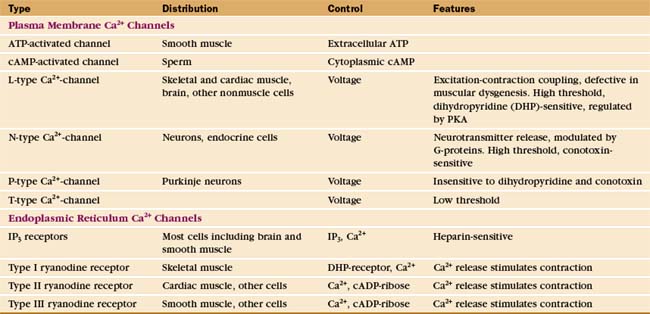
Inositol 1,4,5-Trisphosphate Receptor Ca2+ Channels
Numerous signal transduction pathways generate IP3 (Fig. 26-7), which activates IP3 receptors to release Ca2+ from the endoplasmic reticulum in animal cells. Plants and fungi appear to lack IP3 receptors.
IP3 receptors are tetramers of giant 313-kD polypeptides with multiple domains mostly in the cytoplasm (Fig. 26-13). Six transmembrane segments near the C-terminus form a tetrameric Ca2+ channel similar to other cation channels, including a P-loop between segments 5 and 6 facing the lumen of the ER (see Fig. 10-3). The pore is large and nonselective, but Ca2+ is the main ion crossing the open channel, owing to its large concentration gradient between the ER lumen and the cytoplasm. IP3 binds with submicromolar affinity between two domains near the N-terminus. Specificity for IP3 is provided by a network of hydrogen bonds between basic residues of the two domains and all three phosphates as well as the hydroxyls of IP3. Ca2+ binds to the IP3-binding domains and several other sites along the polypeptide. The organization of the approximately 1500 residues between the IP3 binding domains and the channel domain is not known.
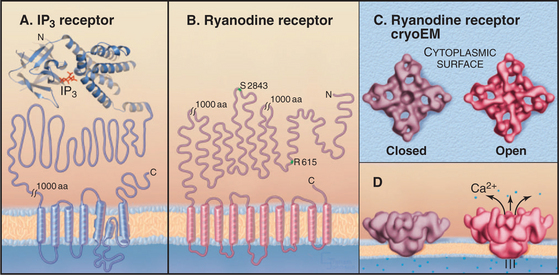
Cytoplasmic IP3 and Ca2+ cooperate to open and close these channels, with IP3 setting the sensitivity of the channel to Ca2+ (Fig. 26-14). A tetrameric IP3 receptor has four highly selective binding sites for inositol 1,4,5-triphosphate. IP3 must occupy at least two, and perhaps as many as four, of these sites to open the channel. Channels respond rapidly, because binding and dissociation of both ligands occurs quickly (k+ = 33μM−1s−1, k– = 6s−1 for IP3). High concentrations of Ca2+ in the endoplasmic reticulum lumen sensitize receptors to IP3. Phosphorylation by PKA, PKC, and CaM kinase can raise or lower the sensitivity to IP3.
Ryanodine Receptor Ca2+ Channels
Ryanodine receptors are homotetramers of 565-kD subunits with a massive cytoplasmic domain and a cation channel domain near the C-terminus (Fig. 26-13B), an architecture similar to that of IP3 receptors. Three ryanodine receptor genes encode proteins that are about 60% identical and expressed in different cells (Table 26-3). Ryanodine receptors are the sole release channels in striated muscles. In smooth muscle and nonmuscle cells, ryanodine receptors augment IP3 receptor Ca2+-release channels.
Cyclic adenosine diphosphate (cADP)–ribose sets the Ca2+ sensitivity of ryanodine receptors, just as IP3 does for its receptor. At low concentrations of cADP-ribose, high levels of cytoplasmic Ca2+ are required to open the channel, whereas at high concentrations of cADP-ribose, even resting Ca2+ concentrations open the channel. A single enzymatic step produces cADP-ribose from the metabolite nicotinamide adenine dinucleotide, NAD+. cGMP regulates ADP-ribosyl cyclase, presumably through a cGMP-dependent protein kinase. cADP-ribose has been implicated in the Ca2+ transient that triggers secretion of insulin from pancreatic b cells in response to glucose. In fertilization of echinoderm eggs, cADP-ribose releases Ca2+ through endoplasmic reticulum ryanodine receptors in parallel with IP3-mediated Ca2+ release. Vertebrate eggs depend entirely on the IP3 release mechanism (Fig. 26-15).
Physiological and pharmacological agents can stimulate or inhibit ryanodine receptor activity. Phosphorylation by PKA increases ryanodine receptor channel activity and may contribute to the effects of β-adrenergic receptor stimulation on the heart (see Fig. 11-12). Caffeine activates Ca2+ release by ryanodine receptors and is used to stimulate sperm in fertility tests. Numerous agents suppress the spontaneous release of Ca2+ by ryanodine receptors in heart and skeletal muscle: FKBP (the protein that binds the immunosuppressant drug FK506), micromolar ryanodine, the local anesthetic procaine, and calmodulin.
Calcium Dynamics in Cells
Methods to visualize Ca2+ concentrations inside living cells revealed an amazing temporal and spatial complexity of Ca2+ signals. The original experimental Ca2+ sensor was a jellyfish protein called aequorin, which emits light when it binds Ca2+ (see Fig. 39-16). An evolving series of Ca2+-sensitive fluorescent dyes (Fura-2, calcium green, calcium red, Fluo-3) have largely replaced aequorin and made possible the observa-tions in the following paragraphs. Ca2+ biosensors, constructed by fusing calmodulin to variants of green fluorescent protein, offer advantages, including targeting to particular cellular compartments and the ability to adjust the response range by mutating the Ca2+-binding site.
Voltage-dependent Ca2+ channels in excitable cells such as neurons and striated muscles respond rapidly (<1μs) to an action potential to admit extracellular Ca2+. At chemical synapses, this produces a brief, locally confined Ca2+ pulse that triggers the release of synaptic vesicles (see Fig. 11-8). In striated muscles, each action potential causes a transient, global increase in cytoplasmic Ca2+ lasting tens of milliseconds (see Fig. 39-15). The details differ in skeletal and cardiac muscle, but the elementary events are similar. Voltage-sensitive Ca2+ channels in T tubules (invaginations of the plasma membrane; see Fig. 39-10) activate one or a few ryanodine receptor channels in the adjacent endoplasmic reticulum through direct physical interaction in skeletal muscle and through Ca2+ release in the heart. Ca2+ released by these ryanodine receptors triggers Ca2+ release from nearby ryanodine receptors, generating a local pulse of Ca2+ called a Ca2+ spark. Because T tubules penetrate throughout the muscle cytoplasm, thousands of these sparks are produced simultaneously, yielding a transient global rise in Ca2+. The brief duration of the excitatory signal and the robust Ca2+ pumping activity of the endoplasmic reticulum limit the duration of the signal.
Nonexcitable cells generally rely on slower, receptor-mediated production of IP3 to produce transient changes in cytoplasmic Ca2+. For example, activation of a frog egg at one point by entry of a sperm or artificial activation by pricking with a needle (Fig. 26-15) results in a self-propagating wave of cytoplasmic Ca2+ that spreads slowly around the cell. This produces a wave of se-cretion and activation of downstream effectors, such as PKC.
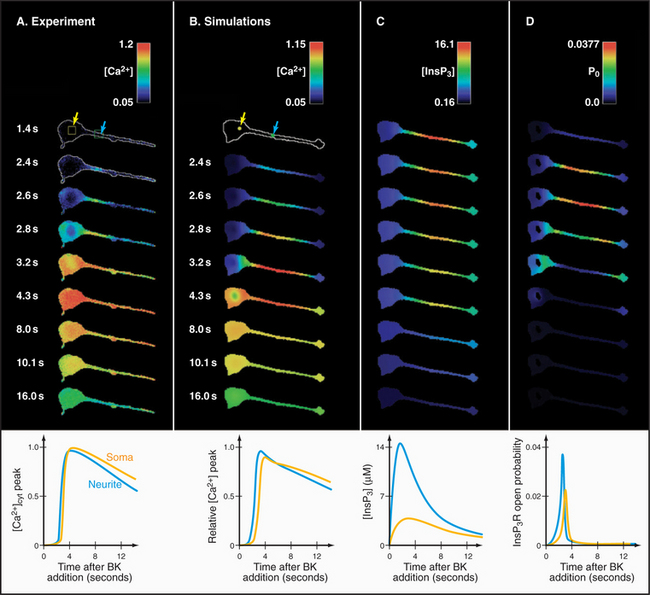
Many cells with type I and II IP3 receptors respond to constant agonist stimulation with transient bursts of cytoplasmic Ca2+ (Fig. 26-16). The agonist concentration sets the level of cytoplasmic IP3 (and, possibly, cADP-ribose), which determines the sensitivity of release channels to cytoplasmic Ca2+. A region of a cell with a high density of the most sensitive channels then initiates the release of Ca2+ at a focus. The resulting Ca2+ transient may spread through the cytoplasm as a planar or spiral wave that is driven locally by Ca2+-induced Ca2+ release from either ryanodine receptors or IP3 receptors. Such transients are self-limited locally, because cytoplasmic Ca2+ concentrations exceeding 0.5μM inhibit both IP3 receptors and ryanodine receptors. This negative feedback closes release channels and allows Ca2+ pumps to clear the cytoplasm of Ca2+. Release channels recover slowly from this negative feedback, creating an interval between Ca2+ transients. In this way, the cell decodes the concentration of agonist as a function of the frequency of Ca2+ pulses. Colliding waves annihilate each other, owing to negative feedback by high concentrations of Ca2+ or local depletion of Ca2+ stores.
Ca2+ Targets
The diversity of targets for Ca2+ (Appendix 26-1) emphasizes the widespread and complex effects of this second messenger. The response to a Ca2+ signal depends on the available targets, as well as modulat-ing effects of parallel signaling pathways. Some proteins bind Ca2+ directly, including Ca2+-activated plasma membrane channels for K+ and Cl−, troponin-C in striated muscles, synaptotagmin (a Ca2+-sensing synaptic vesicle protein), and calpain (a Ca2+-activated protease). Parvalbumin and calbindin D28K buffer the cytoplasmic Ca2+ concentration. Ca2+ activates many proteins indirectly by first binding and activat-ing calmodulin or S100 proteins. Some calmodulin targets, such as CaM kinase II, modify multiple sub-strate proteins, greatly amplifying the effect of Ca2+. A small protein called “regulator of calmodulin signaling” inhibits calmodulin, provided that this regulator is phosphorylated by PKA.
Nitric Oxide
The free radical gas nitric oxide (NO˙) provides cells with a unique way to transmit signals. It diffuses rapidly through membranes, allowing the signal to spread from cell to cell rather than being confined to the cell of origin like most second messengers. Nitric oxide has long been known as a mildly toxic air pollutant, so it was easy to accept the finding that macrophages produce nitric oxide to the kill microorganisms and tumor cells. On the other hand, it was surprising to learn in the late 1980s that nitric oxide is a diffusible messenger: Endothelial cells lining blood vessels make nitric oxide that relaxes smooth muscle in the walls of arteries, and some neurons in the central and peripheral nervous systems produce nitric oxide as an unconventional neurotransmitter. Carbon monoxide (CO) might be another gaseous intercellular second messenger under some circumstances.
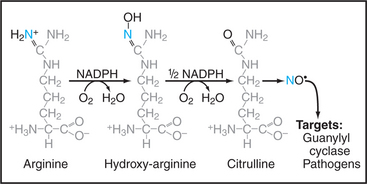
Enzymes called nitric oxide synthases produce nitric oxide by converting l-arginine and molecular oxygen into citrulline and nitric oxide (Fig. 26-17). NADPH provides reducing equivalents for the reaction. Nitric oxide synthase (NOS) is really two enzymes in one. The N-terminal oxidase domain has a heme group that participates directly in oxidation of arginine. The C-terminal reductase domain supplies electrons to the oxidase domain. NOS depends on an extraordinary number of cofactors to handle the electron transfers that are required to produce nitric oxide: heme and tetrahydrobiopterin bound to the oxidase domain, and flavin adenine dinucleotide, flavin mononucleotide, and NADPH in the reductase domain. Some gram-positive bacteria have an NOS with only the N-terminal oxidase domain that is presumably the evolutionary ancestor of eukaryotic NOS. These enzymes catalyze nitration of substrates rather than production of nitric oxide.
The main target for the low concentrations of nitric oxide used for signaling is soluble guanylyl cyclase, the cytoplasmic enzyme that makes cGMP (see Fig. 24-9). Nitric oxide binds reversibly to iron in the heme group of guanylyl cyclase, causing a conformational change that activates the enzyme. Nitric oxide also reacts with cysteine side chains of proteins (S-nitrosylation). This covalent posttranslational modification can modify the activity of sensitive proteins such as NSF (N-ethylmaleimide-sensitive factor), a protein that is required for membrane trafficking (see Fig. 21-12).
Nitric oxide from three different sources regulates blood flow and blood pressure in response to local physiological demands. During exercise, the repeated release of Ca2+ that stimulates skeletal muscle contraction (see Fig. 39-16) also binds calmodulin and activates NOS to produce nitric oxide. Nitric oxide diffuses out of the skeletal muscle and into smooth muscle cells surrounding nearby blood vessels. Nitric oxide relaxes smooth muscle by activating cGMP production and lowering the internal Ca2+ level through incompletely understood mechanisms. This increases local blood flow to supply oxygen and nutrients. Endothelial cells lining the inside of blood vessels also use nitric oxide to regulate vascular smooth muscle. Mechanical shear stress from blood flow continuously stimulates endothelial cell phosphatidylinositol-3 kinase to produce PIP3, which stimulates PKB/Akt to phosphorylate and activate NOS3. This provides a sustained, long-term signal to relax vascular smooth muscle. Acutely, hormones such as acetylcholine and bradykinin can stimulate endothelial cells to produce nitric oxide by causing Ca2+ release from endoplasmic reticulum. Mice that lack NOS3 have high blood pressure.
Mono- and di-NG-methylated arginines strongly inhibit NOS and therefore have been used experimentally to reveal many biological functions of nitric oxide. Inhibition of NOS compromises killing of bacteria by macrophages. Similarly, inhibition of endothelial nitric oxide production causes vascular smooth muscle cells to contract, raising blood pressure and demonstrating a constitutive role for nitric oxide in relaxing these cells. Nitric oxide produced by autonomic nerves causes erection of the penis by stimulating the production of cGMP and relaxing vascular smooth muscle cells. The drug sildenafil (Viagra) is used to treat erectile dysfunction, as it inhibits the phosphodiesterase that breaks down cGMP. Sildenafil and related drugs bind in the active site, excluding cGMP. Other autonomic nerves use nitric oxide to control the smooth muscle cells in the walls of the intestines. Nitric oxide is the active metabolite of nitroglycerin, a drug that is widely used to relieve pain (angina pectoris) associated with compromised blood flow in the heart. It dilates coronary arteries and improves circulation. In addition to regulating cerebral blood flow, nitric oxide produced by nerve cells in the brain may contribute to certain types of learning by reinforcing the release of neurotransmitters (see Fig. 11-10).
ACKNOWLEDGMENTS
Thanks go to Barbara Ehrlich and Paul Insel for their suggestions on revisions to this chapter.
Baillie GS, Scott JD, Houslay MD. Compartmentalisation of phosphodiesterases and protein kinase A: Opposites attract. FEBS Lett. 2005;579:3264-3270.
Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signaling. Nat Rev Mol Cell Biol. 2000;1:11-21.
Bolotina VM, Csutora P. CIF and other mysteries of the store-operated Ca2+-entry pathway. Trends Biochem Sci. 2005;30:378-387.
Bos JL. EPAC: A new cAMP target and new avenues in cAMP research. Nat Rev Mol Cell Biol. 2003;4:733-738.
Bredt DS. Nitric oxide signaling specificity: The heart of the problem. J Cell Sci. 2003;116:9-15.
De Caterina R, Zampolli A. From asthma to atherosclerosis: 5-lipoxygenase, leukotrienes and inflammation. New Engl J Med. 2004;350:4-7.
Futerman AH, Hannun YA. The complex life of simple sphingolipids. EMBO Rep. 2004;5:777-782.
Garavito RM, Mulichak AM. The structure of mammalian cyclooxygenases. Annu Rev Biophys Biomol Struct. 2003;32:183-206.
Geiger M, Wrulich OA, Jenny M, et al. Defining the human targets of phorbol ester and diacylglycerol. Curr Opin Mol Ther. 2003;5:631-641.
Guatimosim S, Dilly K, Santana LF, et al. Local Ca2+ signaling and EC coupling in heart. J Mol Cardiol. 2002;34:941-950.
Guse AH. Second messenger function and the structure-activity relationship of cyclic adenosine diphosphoribose (cADPR). FEBS J. 2005;272:4590-4597.
Ikura M. Calcium binding and conformational response in EF-hand proteins. Trends Biochem Sci. 1996;21:14-17.
McKinney MK, Cravatt BF. Structure and function of fatty acid amide hydrolase. Annu Rev Biochem. 2005;74:411-432.
McLaughlin S, Wang J, Gambhir A, Murray D. PIP2 and proteins. Interactions, organization, and information flow. Annu Rev Biophys Biomol Struct. 2002;31:151-175.
Meldolesi J, Pozzan T. The endoplasmic reticulum Ca2+ store. A view from the lumen. Trends Biochem Sci. 1998;23:10-14.
Niggli E, Egger M. Calcium quarks. Front Biosci. 2002;7:1288-1297.
Patrono C, Garcia Rodriguez LA, Landolfi R, Baigent C. Low-dose aspirin for the prevention of atherthrombosis. New Engl J Med. 2005;353:2373-2383.
Patterson RL, Boehning D, Snyder SH. Inositol 1,4,5-trisphosphate receptors as signal integrators. Annu Rev Biochem. 2004;73:437-465.
Radmark O, Samuelsson B. Regulation of 5-lipoxygenase enzyme activity. Biochem Biophys Res Comm. 2005;338:102-110.
Rhee SG. Regulation of phosphoinositide-specific phospholipase C. Annu Rev Biochem. 2001;70:281-312.
Rosen H, Goetzl EJ. Sphingosine 1-phosphate and its receptors: An autocrine and paracrine network. Nature Rev Immunol. 2005;5:560-570.
Schopfer FJ, Baker PRS, Freeman BA. NO-dependent protein nitration: A cell signaling event or an oxidative inflammatory response? Trends Biochem Sci. 2003;28:646-654.
Sulis ML, Parsons R. PTEN: From pathology to biology. Trends Cell Biol. 2003;13:478-483.
Taylor CW, da Fonseca PCA, Morris EP. IP3 receptors: The search for structure. Trends Biochem Sci. 2004;29:210-219.
Toyoshima C, Inesi G. Structural basis of ion pumping by Ca2+-ATPase of the sarcoplasmic reticulum. Annu Rev Biochem. 2004;73:269-292.
Vanhaesebroeck B, Leevers SJ, Ahmadi K, et al. Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem. 2001;70:535-602.
Vetter SW, Leclerc E. Novel aspects of calmodulin target recognition and activation. Eur J Biochem. 2003;270:404-414.
Wehrens SHT, Marks AR. Altered function and regulation of cardiac ryanodine receptors in cardiac disease. Trends Biochem Sci. 2003;28:671-678.
Wilson RI, Nicoll RA. Endocannaboid signaling in the brain. Science. 2002;296:678-682.
Wolfe MM, Lichtenstein DR, Singh G. Gastrointestinal toxicity of nonsteroidal antiinflammatory drugs. N Engl J Med. 1999;340:1888-1899.
Yamasaki M, Churchill GC, Galione A. Calcium signalling by nicotinic acid dinucleotide phosphate (NAADP). FEBS J. 2005;272:4598-4906.
Zaccolo M, Magalhães P, Pozzan T. Compartmentalisation of cAMP and Ca2+ signals. Curr. Opin Cell Biol. 2002;14:160-166.
APPENDIX 26-1 Examples of Ca2+ Regulated Proteins
| Protein | Binding Site | Function |
|---|---|---|
| First-Order Proteins That Bind Ca2+ Directly | ||
| Membrane Proteins | ||
| Annexins | Novel | Promote membrane interactions |
| Ca2+ -activated Cl− channels | Novel | Participate in secretion |
| Ca2+ -activated K+ channels | Novel | Control membrane excitability |
| IP3 receptor | Novel | Ca2+ -release channel; activated and inhibited by Ca2+ |
| Ryanodine receptor | Novel | Ca2+ -release channel, activated and inhibited by Ca2+ |
| Synaptotagmin | Novel | A synaptic vesicle Ca2+ sensor |
| Enzymes | ||
| Calmodulin-domain protein kinases | a EF hand | Plant protein kinases |
| Calpain | EF hand | Ca2+ -dependent protease |
| Protein kinase C, some isozymes | C2 domain | Multifunctional protein kinases activated by Ca2+ |
| Cytoskeletal Proteins | ||
| α-Actinin (some isoforms) | EF hand | Actin filament cross-linking protein |
| Centrin/caltractin | EF hand | Ca2+-sensitive contractile fibers |
| Gelsolin, villin | Novel | Actin filament severing and capping proteins |
| Molluscan myosin light chains | EF hand | Regulate muscle contraction; activated by Ca2+ |
| Troponin C | EF hand | Ca2+ -activated regulator striated muscle contraction |
| Calcium-Binding Proteins | ||
| Calmodulin | EF hand | Ca2+ -activated regulator of many proteins |
| Calbindin-D28K | EF hand | Cytoplasmic Ca2+ buffer |
| Calretinin | EF hand | Activates guanylyl cyclase |
| Parvalbumin | EF hand | Cytoplasmic Ca2+ buffer |
| S100 proteins (18 human isoforms) | EF hand | Diverse regulatory functions; some isoforms may be secreted |
| S100 calbindin-D9K | EF hand | Cytoplasmic Ca2+ buffer |
| Recoverin | EF hand | Regulates visual phototransduction |
| Second-Order Proteins Activated by Ca2+-Calmodulin | ||
| Membrane Proteins | ||
| Adenylyl cyclase (some isoforms) | Produces cAMP | |
| Ca2+ -dependent Na+ channels | Na+ currents | |
| cGMP-gated cation channels | Phototransduction | |
| Plasma membrane Ca2+ -ATPase pumps | Clears cytoplasm of Ca2+ | |
| Enzymes | ||
| Calcineurin | Protein phosphatase 2B | |
| CaM kinase (several isozymes) | Multifunctional protein kinase | |
| cAMP phosphodiesterase | Degrades cAMP | |
| IP3 kinase | Phosphorylates IP3 | |
| Myosin light chain kinase | Activates smooth muscle and cytoplasmic myosin | |
| NAD kinase | Phosphorylates NAD | |
| Nitric oxide synthetase | Makes nitric oxide | |
| Phosphorylase kinase | Phosphorylates phosphorylase | |
| Cytoskeletal Proteins | ||
| MARCKS | Actin filament cross-linking protein | |
a EF hand is the abbreviation for the Ca2+ binding site in calmodulin consisting of α-helices E and F.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
CHAPTER 26 Second Messengers
This chapter considers the remarkable variety of small molecules that carry signals inside living cells. These second messengers are chemically diverse, ranging from hydrophobic lipids confined to membrane bilayers, to an inorganic ion (Ca2+), to nucleotides (cyclic adenosine monophosphate [cAMP] and cyclic guanosine monophosphate [cGMP]), to a gas (nitric oxide). The messages that these molecules carry are encoded by their concentrations. In the simplest case, a rise or fall in the concentration of the second messenger conveys a signal from its source to its target. In other cases, the signal depends on the rate or frequency of the fluctuations in the concentration of the second messenger. The local concentration of a second messenger depends on the rate of production, the rate of diffusion from the site of production, and the rate of removal. Most second messengers are produced by enzymes that can switch on and off rapidly, allowing modulation of the concentration of second messengers on a millisecond time scale. In the case of Ca2+, the cytoplasmic concentration is determined by channels that release the ion from membrane-delimited stores and by pumps that remove it from cytoplasm.
The complexity of the signaling pathways is determined by the number of sources and the number of targets of each second messenger. Generally, multiple signal sources and multiple second messenger targets generate more complexity than one can fully appreciate. This chapter deals with this complexity only in passing. Chapter 27 considers a few model systems in which it is possible to understand how signals are integrated and transduced.
Cyclic Nucleotides
Two cyclic nucleoside monophosphates—adenosine 3′,5′-cyclic monophosphate (cAMP) and guanosine 3′,5′-cyclic monophosphate (cGMP)—are employed as second messengers (Fig. 26-1). Both act by binding reversibly to specific proteins. Enzymes that produce and degrade cyclic nucleotides determine the concentrations of these messengers available to bind targets. These enzymes turn over substrates rapidly, so they can amplify signals massively on a millisecond time scale, under the control of diverse signaling pathways (see Chapter 27). Cyclases make cyclic nucleotides in a single step from the corresponding nucleoside triphosphate, either adenosine triphosphate (ATP) or guano-sine triphosphate (GTP).
Figure 26-1 cyclic nucleotide metabolism. Synthesis and degradation of cAMP and cGMP, including regulatory inputs and targets. Gsα, Giα, and Gtα are trimeric GTPase a subunits (see Fig. 25-9). C A, calcium; NO, nitric oxide.
Cyclic nucleotides diffuse in the cytoplasm at about the same rate as in free solution, activating a modest repertoire of downstream targets, including protein kinases (see Figs. 25-3 and 25-4), cyclic nucleotide–gated ion channels (see Fig. 10-10), and, in the case of cAMP, one class of nucleotide exchange factors (Epac) for small GTPases (Rap1 and Rap2). The components of this system are quite ancient, since the protein domains that bind cAMP or cGMP are homologous to the cAMP-binding domain of CAP, a bacterial transcription factor. Enzymes called cyclic nucleotide phosphodiesterases degrade cAMP and cGMP to inactive nucleoside 5′-monophosphates. Eleven genes encode more than 40 different phosphodiesterases, which vary in their specificities for the two cyclic nucleotides, expression in various tissues, and localization to cellular compartments.
A family (10 human genes) of enzymes called adenylyl cyclases synthesize cAMP from ATP. Multiple transmembrane segments anchor most of these enzymes to the plasma membrane. Two homologous catalytic domains reside in the cytoplasm (C1 and C2 in Fig. 26-2). These cytoplasmic domains can be produced experimentally as soluble proteins separately from the transmembrane domains and can then be recombined to make a fully active enzyme. Both domains are necessary because the active site lies at the interface of the two domains. One adenylyl cyclase is a soluble enzyme that can concentrate in the nucleus. Generally, the concentration of adenylyl cyclases is very low relative to the trimeric G-proteins that regulate their activity.
Multiple regulatory mechanisms act synergistically to regulate adenylyl cyclases. GTP-Gsα the GTPase subunit of a trimeric G-protein, activates many membrane-associated adenylyl cyclases by binding far from the active site (Fig. 26-2) and inducing a conformational change. Ca2+-calmodulin or protein kinase C (PKC) activates some adenylyl cyclases. GTP-Giα the GTPase subunit of another trimeric G-protein, or protein kinase A (PKA) inhibits some cyclases. Gβγ subunits of trimeric G-proteins activate some adenylyl cyclases but inhibit others. These diverse regulatory mechanisms allow adenylyl cyclases to integrate a variety of input signals. The diterpene forskolin from a Coleus plant binds to and activates adenylyl cyclases. Forskolin is useful experimentally for manipulating the cAMP concentration in cells and, on the basis of its ability to bind adenylyl cyclases, was essential in the initial purification of the enzyme by affinity chromatography. Regulation of a soluble form of adenylyl cyclase differs from that of all other isoforms. It is activated by bicarbonate, an essential step in sperm maturation.
In resting cells, the concentration of cAMP is so low, approximately 10−8M, that it does not bind its targets. Stimulation of appropriate receptors (such as the seven-helix β-adrenergic receptor, see Fig. 27-3) increases the cytoplasmic cAMP concentration more than 100-fold, enough to saturate the PKA regulatory subunits (Fig. 26-3). Dissociation of the regulatory (R) subunits frees the active PKA catalytic subunits to phosphorylate cytoplasmic and membrane substrates and to move into the nucleus to activate the transcription factor CREB (cyclic nucleotide regulatory element–binding protein) (see Fig. 15-22).
Outside the animal kingdom, cAMP has many functions. In bacteria, cAMP controls gene expression in response to nutritional conditions. The cellular slime mold Dictyostelium uses cAMP as an extracellular signal, acting through a seven-helix receptor, for its social interactions (see Fig. 38-12).
Guanylyl cyclases are dimeric enzymes similar to adenylyl cyclases. In fact, mutation of just two amino acid residues can convert a guanylyl cyclase to an adenylyl cyclase. Vertebrates express two types of guanylyl cyclases (see Fig. 24-9). A family of transmembrane receptors with cytoplasmic cyclase domains respond to ligand binding by producing cGMP. Cytoplasmic guanylyl cyclases are activated by nitric oxide and carbon monoxide, which bind a heme group in a regulatory domain (see the section titled “Nitric Oxide” later in this chapter).
Lipid-Derived Second Messengers
Phosphoglycerides (see Fig. 7-2) and sphingolipids (see Fig. 7-3) not only form cellular membranes but also participate in a wide range of signaling mechanisms. The long list of intracellular and extracellular second messengers derived from lipids will undoubtedly expand in the future. Three membrane lipids are the primary sources of these signaling molecules (Fig. 26-4):
Enzyme Reactions That Produce Lipid Second Messengers
Three types of enzyme reactions generate lipid second messengers:
Cells also use transferases that add or exchange head groups or fatty acids on membrane phosphoglycerides (see Fig. 7-2). These enzymes are essential for lipid biosynthesis but have not been implicated in signaling. For example, transferases add choline (as the nucleotide conjugate cytidine diphosphate [CDP]–choline) to DAG to make phosphatidylcholine). In the case of phosphatidylinositol, the head group is provided by dephosphorylation of IP3 to inositol, which is recombined enzymatically with a CDP conjugate of DAG to make phosphatidylinositol.
Sequential action of two or more enzymes produces some lipid second messengers. For example, cells can make DAG in two steps from phosphatidylcholine: PLD first makes phosphatidic acid, which is dephosphorylated by phosphatidic acid phosphatase. PLD followed by PLA2 makes lysophosphatidic acid from phosphatidylcholine. PLA2 also initiates the production of a huge family of fatty acid derivatives called eicosanoids by cleaving the fatty acid arachidonic acid from phosphatidylcholine. Cyclooxygenases and lipoxygenases then modify arachidonic acid to make prostaglandins, thromboxanes, and leukotrienes (Figs. 26-9 and 26-10), which leave the cell to interact with cell surface receptors.
Targets of Lipid Second Messengers
Downstream targets of the lipid second messengers are diverse but can be generalized to some extent into three categories (Fig. 26-5):
Lipid second messengers participate in many processes that are considered elsewhere in the book. For example, regulated secretion (see Chapter 21) in response to an agonist binding a seven-helix or tyrosine kinase receptor requires a transient Ca2+ signal in the cytoplasm. The Ca2+ is released from the membrane stores by IP3, which is produced by the action of a PI-PLC on PIP2. IP3-mediated Ca2+ release from the endoplasmic reticulum also controls smooth muscle contraction (see Fig. 39-21).
Protein Kinase C
Many lipid second messengers, including DAG, PIP3, arachidonic acid, phosphatidic acid, and lysophosphatidylcholine activate one or more of the 10 protein kinase C (PKC) isozymes expressed by vertebrate cells (Fig. 26-6). These multiple PKC isozymes provide a selective response to various lipid second messengers. Some, but not all, PKC isozymes also require Ca2+ for activation. Sphingosine may inhibit some PKC isozymes.
PKCs have a protein kinase catalytic domain (see Fig. 25-3) and N-terminal regulatory domains. A pseudosubstrate sequence binds intramolecularly to the active site and inhibits activity by blocking substrate binding. Pseudosubstrates have alanine at the phosphorylation site instead of serine as substrates have. During apoptosis, caspases cleave off this regulatory domain (see Fig. 46-10) producing constitutively active PKC isoforms.
PKCs can provide either positive or negative feedback to the signaling pathways that turn them on. PKC activates PLD and PLA2 and provides positive feedback, because those enzymes produce more DAG to sustain the activation of PKC. On the other hand, PKC provides negative feedback when it phosphorylates and inhibits both growth factor receptors and PI-PLCγ1. PKC also phosphorylates and inhibits PI-PLCβ, generating negative feedback after activation of seven-helix receptors. Negative feedback makes both of these signaling events transient.
Phosphoinositide Signaling Pathways
Although minor in terms of mass in biological membranes, phosphoinositides are major players in signal-ing (Fig. 26-7). The parent compound, phosphatidyl-inositol (PI), is a phosphoglyceride with a cyclohexanol head group called inositol. Specific lipid kinases phosphorylate the 4 and 5 hydroxyl groups of phosphatidylinositol to form PI(4-)P and PI(4,5-)P2, usually referred to simply as PIP and PIP2. A specific phosphatase can remove the D5 phosphate. Inactivation of the single copy of this gene on the X-chromosome causes Lowe’s syndrome with cataracts, renal failure, and mental retardation.
PIP2 is a substrate for a family of receptor-controlled PI-PLCs that cleave off the phosphorylated head group, producing two potent second messengers: IP3 and DAG. Water-soluble IP3 activates Ca2+ release channels in the endoplasmic reticulum (Fig. 26-12), and lipid-soluble DAG activates PKC. In contrast to Ca2+, which diffuses slowly and acts locally, IP3 diffuses rapidly through the cytoplasm, triggering Ca2+ release. DAG is confined to membranes but diffuses laterally to bind and activate PKC. Enzymes inactivate both IP3 and DAG. DAG is inactivated by phosphorylation to make phosphatidic acid, a second messenger in its own right but also an intermediate in the resynthesis of phosphoinositides. IP3 is dephosphorylated to inositol, which is inactive as a second messenger. LiCl inhibits the final step: dephosphorylation of inositol-1-phosphate. Remarkably, Li+ is clinically useful as a treatment for bipolar disorder, presumably by interfering with phosphoinositide signaling in the brain. In some tissues, IP3 is inactivated by phosphorylation to inositol 1,3,4,5-tetrakisphosphate (IP4).
Vertebrates use 10 classes of PI-PLCs to provide tissue-specific coupling of various receptors to the production of IP3 and DAG. PI-PLCγ1, expressed in the brain, is activated by tyrosine phosphorylation when its SH2 domains bind a tyrosine-phosphorylated receptor tyrosine kinase (Fig. 26-12). The trimeric G-protein Gqα activates another isozyme, PI-PLCβ.
Receptor tyrosine kinases activate another family of lipid kinases, PI-3 kinases, which phosphorylate the 3-hydroxyl group of PI, PIP, and PIP2. The products are PI(3-)P, PI(3,4-)P2, and PI(3,4,5-)P3. PI(3,4,5-)P3 is not a substrate for the PI-PLCs that produce IP3 and DAG, but it activates some PKC isozymes and binds specifically to certain pleckstrin homology domains (see Fig. 25-11), bringing enzymes such as protein kinase B (PKB/Akt) to the plasma membrane. Association with the membrane leads to phosphorylation and activation of PKB as part of insulin signaling (see Fig. 27-7). A phosphatase that removes the D3 phosphate from PIP3 is a tumor suppressor called PTEN (phosphatase and tensin homolog deleted on chromosome 10). Loss of PTEN function in tumors allows PIP3 to build up, activating PKB and growth promoting downstream pathways in many tumors. A steroid-like molecule called wortmannin inhibits PI-3 kinase relatively specifically and is used to investigate the physiological roles of PIP3.
Phosphatidylcholine Signaling Pathways
Phosphatidylcholine is not only a major structural lipid of the plasma membrane but also an important source of DAG and a large family of other signaling molecules (Fig. 26-5
[/not-level-membership-for-basic-science-category]
