CHAPTER 20 Screening for Genetic Disease
Genetic disease affects individuals and their families dramatically but every person, and every couple having children, is at some risk of seeing a disorder with a genetic component suddenly appear. Our concepts and approaches to screening reflect the different burdens that these two realities impose. First, there is screening of individuals and couples known to be at significant or high risk because of a positive family history—sometimes referred to as targeted, or family, screening. This includes carrier, or heterozygote, screening, as well as presymptomatic testing. Second, there is the screening offered to the general population, who are at low risk—sometimes referred to as community genetics. Population screening involves the offer of genetic testing on an equitable basis to all relevant individuals in a defined population. Its primary objective is to enhance autonomy by enabling individuals to be better informed about genetic risks and reproductive options. A secondary goal is the prevention of morbidity resulting from genetic disease and alleviation of the suffering this would impose.
Screening Those at High Risk
Here we focus on the very wide range of general genetic disease as opposed to screening in the field of cancer genetics, which is addressed in Chapter 14. Prenatal screening is also covered in more detail in the next chapter. If it were easy to recognize carriers of autosomal and X-linked recessive disorders and persons who are heterozygous for autosomal dominant disorders that show reduced penetrance or a late age of onset, much doubt and uncertainty would be removed when providing information in genetic counseling. Increasingly, mutation analysis in genes that cause these disorders is indeed making the task easier. Where this is not possible, either because no gene test is available or the molecular pathology cannot be easily detected in a gene known to be associated with the disorder in question, a number of strategies and types of analysis is available to detect carriers for autosomal and X-linked recessive disorders, and for presymptomatic diagnosis of heterozygotes for autosomal dominant disorders.
Carrier Testing for Autosomal Recessive and X-Linked Disorders
In a number of autosomal recessive disorders, such as some of the inborn errors of metabolism (e.g., Tay-Sachs disease; p. 178) and the hemoglobinopathies (e.g., sickle cell disease; p. 159), carriers can be recognized with a high degree of certainty using biochemical or hematological techniques such that DNA analysis is not necessary. In other single-gene disorders, it is possible to detect or confirm carrier status by biochemical means in only a proportion of carriers; for example, mildly abnormal coagulation study results in a woman at risk of being a carrier for hemophilia (p. 309). A significant proportion of obligate carriers of hemophilia will have normal coagulation, however, so a normal result does not exclude a woman at risk from being a carrier.
There are several possible ways in which carriers of genetic diseases can be recognized.
Clinical Manifestations in Carriers
Occasionally, carriers for certain disorders can have mild clinical manifestations of the disease (Table 20.1), particularly with some of the X-linked disorders. These manifestations are usually so slight that they are apparent only on careful clinical examination. Such manifestations, even though minimal, are unmistakably pathological; for instance, the mosaic pattern of retinal pigmentation seen in manifesting female carriers of X-linked ocular albinism. Unfortunately, in most autosomal and X-linked recessive disorders there are either no manifestations at all in carriers, or they overlap with the variation seen in the general population. An example would be female carriers of hemophilia, who have a tendency to bruise easily—this is not a reliable sign of carrier status, as this is seen in a significant proportion of the general population, for different reasons. In X-linked adrenoleukodystrophy, a proportion of carrier females manifest neurological features, sometimes relatively late in life when the signs might easily be confused with the problems of aging. Thus clinical manifestations are helpful in detecting carriers only when they are unmistakably pathological, which is the exception rather than the rule with most single-gene disorders.
Table 20.1 Clinical and Biochemical Abnormalities Used in Carrier Detection of X-Linked Disorders
| Disorder | Abnormality |
|---|---|
| Clinical | |
| Ocular albinism | Mosaic retinal pigmentary pattern |
| Retinitis pigmentosa | Mosaic retinal pigmentation, abnormal electroretinographic findings |
| Anhidrotic ectodermal dysplasia | Sweat pore counts reduced, dental anomalies |
| Lowe syndrome | Lens opacities |
| Alport syndrome | Hematuria |
| Biochemical | |
| Hemophilia A | Reduced factor VIII activity : antigen ratio |
| Hemophilia B | Reduced levels of factor IX |
| G6PD deficiency | Erythrocyte G6PD activity reduced |
| Lesch-Nyhan syndrome fibroblasts | Reduced hypoxanthine-guanine phosphoribosyl transferase activity in skin |
| Hunter syndrome | Reduced sulfoiduronate sulfatase activity in skin fibroblasts |
| Vitamin D–resistant rickets | Serum phosphate level reduced |
| Duchenne muscular dystrophy | Raised serum creatine kinase level |
| Becker muscular dystrophy | Raised serum creatine kinase level |
| Fabry disease | Reduced α-galactosidase activity in hair root follicles |
G6PD, Glucose 6-phosphate dehydrogenase.
Biochemical Abnormalities in Carriers
By far the most important approach to determining the carrier status for autosomal recessive and X-linked disorders has been the demonstration of detectable biochemical abnormalities in carriers of certain diseases. In some disorders, the biochemical abnormality seen is a direct product of the gene and the carrier status can be tested for with confidence. For example, in carriers of Tay-Sachs disease the range of enzyme activity (hexosaminidase) is intermediate between levels found in normal and affected people. In many inborn errors of metabolism, however, the enzyme activity levels in carriers overlap with those in the normal range, so that it is not possible to distinguish reliably between heterozygote carriers and those who are homozygous normal.
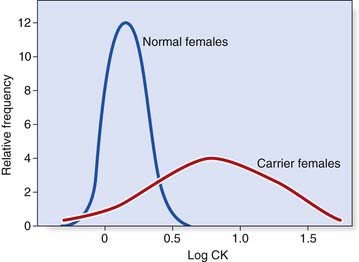
In many single-gene disorders, the biochemical abnormality used in the diagnosis of the disorder in the affected individual is not a direct result of action of the gene product but the consequence of a secondary or downstream process. Such abnormalities are further from the primary action of the gene and, consequently, are usually even less likely to be useful in identifying carriers. For example, in Duchenne muscular dystrophy (DMD) there is an increased permeability of the muscle membrane, resulting in the escape of muscle enzymes into the blood. A grossly raised serum creatine kinase (CK) level often confirms the diagnosis of DMD in a boy presenting with features of the disorder (p. 307). Obligate female carriers of DMD have, on average, serum CK levels that are increased compared with those of the general female population (Figure 20.1). There is, however, a substantial overlap of CK values between normal and obligate carrier females. Nevertheless, this information can be used in conjunction with pedigree risk information (p. 344) and the results of linked DNA markers (p. 345) to help calculate the likelihood of a woman being a carrier for this disorder.
There is another reason for difficulty with carrier testing in the case of X-linked recessive disorders. Random inactivation of the X chromosome in females (p. 103) means that many, often the majority, of female carriers of X-linked disorders cannot be detected reliably by biochemical methods. An exception to this involves analysis of individual clones to look for evidence of two populations of cells, as with peripheral blood lymphocytes in female carriers of some of the X-linked immunodeficiency syndromes (p. 204). In a clinical setting, this is referred to as ‘X-inactivation studies’.
Linkage between a Disease Locus and a Polymorphic Marker
DNA Polymorphic Markers
The advent of recombinant DNA technology revolutionized the approach to carrier detection. Relatively seldom, nowadays, do conventional biochemical and blood group polymorphisms have a role because there are few that are sufficiently informative to be of practical clinical value. The large number of different types of DNA sequence variants (p. 17) in the human genome means that, if sufficient numbers of families are available, linkage of any disease with a polymorphic DNA marker is possible, providing the disease is not genetically heterogeneous (see locus heterogeneity below). After this is achieved, the information can be applied to smaller families. The demonstration of linkage between a DNA sequence variant and a disease locus overcomes the need to identify a biochemical defect or protein marker and the necessity for it to be expressed in accessible tissues. In addition, use of markers at the DNA level also overcomes the difficulties that occur in carrier detection due to X-inactivation for women at risk for X-linked disorders (p. 103).
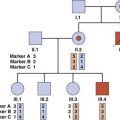
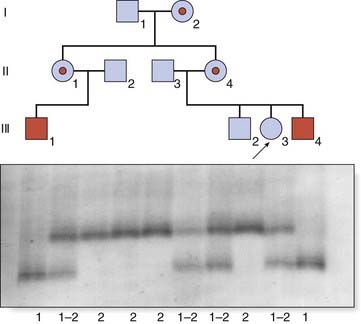
Linked polymorphic DNA markers were frequently used in determining the carrier status of females in families where DMD has occurred; nowadays, direct gene sequencing is more likely to be used. An example is shown in Figure 20.2; individual III3 wants to know whether she is a carrier and therefore at risk for having sons affected with DMD. Analysis of the pedigree reveals that her mother, II4, along with her sister, II1, and their mother, I2, are all obligate carriers of DMD. The family is informative for a polymorphic CA dinucleotide repeat (p. 69) in the closely flanking region 5′ to the dystrophin gene known as Dys 5′ II, which can be demonstrated by polymerase chain reaction (PCR) (p. 56). The mutation in the dystrophin gene in the family is segregating with allele 1 and, because individual III3 has inherited this allele from her mother, she is likely to be a carrier. Linked polymorphic DNA markers can be used for prenatal diagnosis to predict whether a male fetus is likely to be affected with DMD, even without knowing the specific dystrophin gene mutation in the affected male(s) (p. 308).
Potential Pitfalls with Linked Polymorphic DNA Markers
Recombination
The first potential pitfall is the chance of recombination occurring between the polymorphic DNA marker and the disease locus to which linkage has been shown. The risk of a recombination can be minimized, in most instances, by the identification of either intragenic or closely linked markers on either side of the disease locus—termed flanking markers. In some instances, for example the dystrophin locus, there is a ‘hotspot’ for recombination (p. 308). Even with closely flanking or intragenic markers there appears to be a minimal chance of approximately 12% that recombination will occur in any meiosis in a female. The uncertainty introduced by this possibility needs to be accounted for when combining the results of linked polymorphic DNA markers with pedigree risks and the results of CK testing for women at risk of being carriers of DMD (p. 308).
Polymorphic Variation
Another problem encountered in the use of linked DNA markers is whether the family possesses the necessary variation in a linked marker to be what is known as informative. This is increasingly rare due to the availability of large numbers of different DNA sequence variants in the human genome, for example tandem repeat sequences such as CA dinucleotide repeats (p. 69), and single nucleotide polymorphisms (p. 67). It has become less problematic as specific mutation analysis becomes available for an increasing number of single-gene disorders.
Presymptomatic Diagnosis of Autosomal Dominant Disorders
Many autosomal dominant single-gene disorders either have a delayed age of onset (p. 341) or exhibit reduced penetrance (p. 340). The results of clinical examination, specialist investigations, biochemical studies, and family DNA studies can enable the genetic status of the person at risk to be determined before the onset of symptoms or signs. This is known as presymptomatic, or predictive, testing.
Clinical Examination
In some dominantly inherited disorders, simple clinical means can be used for presymptomatic diagnosis, taking into account possible pleiotropic effects of a gene (p. 109). For example, individuals with neurofibromatosis type I (NF1) can have a variety of clinical features (p. 298). It is not unusual to examine an apparently unaffected relative of someone with NF1, who has had no medical problems, only to discover that they have sufficient numbers of café-au-lait spots or cutaneous neurofibromas to confirm that they are affected. However, NF1 is a relatively rare example of a dominantly inherited disorder that is virtually 100% penetrant by the age of 5 or 6 years, with visible external features. With many other disorders, clinical examination is less helpful.
In tuberous sclerosis (TSC) a number of body systems may be involved and the external manifestations, such as the facial rash of angiokeratoma (Chapter 7; see Figure 7.5, A, p. 111) may not be present. Similarly, seizures and learning difficulties are not inevitable. In autosomal dominant polycystic kidney disease, which is extremely variable and may have a delayed age of onset, there may be no suspicion of the condition from routine examination, and hypertension may be borderline without raising suspicions of an underlying problem. Reaching a diagnosis in Marfan syndrome (p. 300) can be notoriously difficult because of the variable features and overlap with other joint hypermobility disorders, even though very detailed diagnostic criteria have been established.
Specialist Investigation
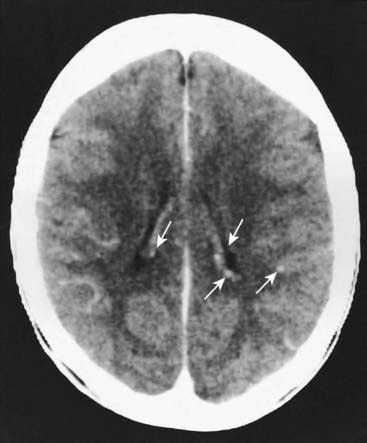
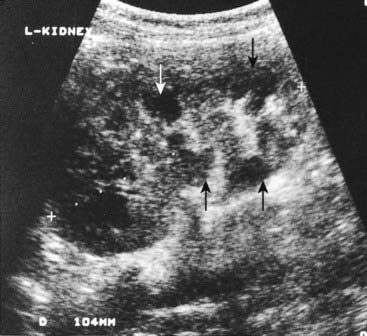
In conditions where clinical assessment leaves diagnostic doubt or ambiguity, special investigations of relevant body systems can serve to clarify status and presymptomatic diagnosis. In TSC imaging, studies of the brain by computed tomography to look for intracranial calcification (Figure 20.3) is more or less routine, as well renal ultrasonography to identify the cysts known as angiomyolipoma(ta) (Figure 20.4). Use of these relatively non-invasive tests in relatives of persons with TSC can often detect evidence of the condition in asymptomatic persons.
Biochemical Tests
For a number of autosomal dominant disorders, biochemical tests can determine whether or not a person at risk has inherited a gene. Examples include the use of serum cholesterol levels in those at risk for familial hypercholesterolemia (p. 175), currently more straightforward than genetic testing, and assay of the appropriate urinary porphyrins or the specific enzyme deficiency in the various dominant porphyrias (p. 179).
Linked DNA Markers
Linked DNA polymorphic markers can be used in presymptomatic diagnosis of dominantly inherited disorders but the principles and pitfalls discussed earlier apply. A common difficulty in dominantly inherited conditions is that of informativity, because often a key individual is deceased or unavailable for some other reason. Despite this, the availability of linked DNA markers has found widespread use in presymptomatic or predictive testing for a number of single-gene disorders inherited in an autosomal dominant manner. Increasingly, however, direct mutation analysis is possible and has replaced the use of linked markers—for example, in Huntington disease (HD). In NF1, TSC, and Marfan syndrome, mutation analysis is expensive and used more discerningly because it is not guaranteed to identify the causative mutation even where there is confidence about the diagnosis. Judicious use of linked DNA markers can be very helpful. Box 20.1 lists some of the more common conditions in which DNA analysis is regularly used to offer presymptomatic diagnosis, but there are of course many more.
Ethical Considerations in Carrier Detection and Predictive Testing
Medically, there are often advantages in being able to determine the carrier status of a person at risk an autosomal or X-linked recessive disorder, mainly relating to a couple being able to make an informed choice when having children. For some individuals and couples, however, the knowledge that there is a significant risk of having an affected child may present options and choices that they would rather not have. The attendant risk, and the awareness that prenatal diagnosis is available, may create a sense of guilt whichever decision is taken—either to have a child knowing it could be affected, or to have prenatal testing and possible termination of pregnancy. The latter option is especially difficult when the prognosis of the disease in question cannot be stated with any certainty because of variability or reduced penetrance, or if there is hope that treatment may be developed in time to help the child. Because of these difficulties and potential dilemmas it is normal practice to suggest that information is passed on within families, rather than by professionals. In general this approach works well, but professional dilemmas can arise if family members refuse to communicate with one another when the disease in question carries significant morbidity and the risk may be high, particularly with X-linked conditions.
In those at risk for late-onset autosomal dominant disorders, many of which have neurological features, there can in some instances be a clear advantage in presymptomatic diagnosis. For example, in those at risk for familial adenomatous polyposis (p. 221), colonoscopy looking for the presence of colonic polyps can be offered as a regular screening procedure to those who have been shown to be at high risk of developing colonic cancer by molecular studies. Conversely, individuals who have been shown to have not inherited a mutation in the APC gene do not need to be screened.
In contrast, for persons at risk for HD, in which there is not yet any effective treatment to delay the onset or progression of the disorder, the benefit of predictive testing is not immediately obvious. The same is true for familial forms of Alzheimer disease, motor neurone disease, CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy), and the spinocerebellar ataxias. Although choice is often considered to be of paramount importance in genetic counseling for those at risk for inherited disorders, it is important to remember that those considering presymptomatic or predictive testing should proceed only if they can give truly informed consent and are free from coercion from any outside influence. It is possible that employers, life insurance companies, and society in general will put indirect, and on occasion direct, pressure on those at high at risk to be tested (p. 366). Indeed, there are examples in which individuals at risk of HD have received prejudicial treatment in relation to employment, and higher than average insurance premiums can be expected on the basis of the family history alone. Here again, the very knowledge itself may torment and frustrate an individual who wants to move on in their life but, once acquired, the knowledge and awareness cannot be removed.
A significant problem raised by predictive testing for late-onset disorders is that it can, in theory, be used for children and minors. This issue can be very contentious, with parents sometimes arguing that it is their right to know the status of their child(ren). However, this conflicts with the high ideal of upholding the principle of individual autonomy wherever possible. Presymptomatic testing of children is therefore usually discouraged unless an early medical intervention or screening is beneficial for the disorder in question. The latter is certainly true for a number of the familial cancer conditions, but also applies to Marfan syndrome and autosomal dominant polycystic kidney disease, among others. The issue of genetic testing of children is addressed more fully in Chapter 24 (p. 365).
Population Screening
One definition of population screening is: ‘The systematic application of a test or inquiry, to identify individuals at sufficient risk of a specific disorder to warrant further investigation or treatment, amongst persons who have not sought medical attention on account of symptoms of that disorder’. Neonatal screening for phenylketonuria is the paradigm of a good screening program and has been available for more than 40 years, with screening for congenital hypothyroidism not far behind. In the United Kingdom, since 1996, population screening has been overseen by the UK National Screening Committee, which advises the government. Under their auspices a National Programme Centre was established in 2002. The current, nationally managed, screening programs are listed in Box 20.2. The implementation of a screening program is a huge logistical exercise requiring financial, expertise, and technology resources, as well as setting up practical mechanisms to introduce the program and monitor outcomes and quality assurance.
Criteria for a Screening Program
These can be considered under the headings of the disease, the test, and the practical aspects of the program (Box 20.3). These criteria apply equally to prenatal screening, which is addressed in the next chapter.
The Disease
To justify the applied effort and resources allocated to screening, the disease should be sufficiently common and have potentially serious effects that are amenable to prevention or amelioration. This may involve early treatment, as in phenylketonuria diagnosed in the neonatal period (p. 167), or the offer of termination of pregnancy for disorders that cannot be treated effectively and are associated with serious morbidity and/or mortality.
The Test
The test should be accurate and reliable with high sensitivity and specificity. Sensitivity refers to the proportion of cases that are detected. A measure of sensitivity can be made by determining the proportion of false-negative results (i.e., how many cases are missed). Thus, if a test detects only 70 of 100 cases, it shows a sensitivity of 70%. Specificity refers to the extent to which the test detects only affected individuals. If unaffected people test positive, these are referred to as false positives. Thus, if 10 of 100 unaffected individuals have a false-positive test result, the test shows a specificity of 90%. Table 20.2 explains this further. Of great interest too is the positive predictive value of a screening test, which is the proportion of positive tests that are true positives; this is illustrated in Table 20.3.
| Disease Status | ||
|---|---|---|
| Affected | Unaffected | |
| Screening test result | ||
| Positive | a (true positive) | b (false positive) |
| Negative | c (false negative) | d (true negative) |
| Sensitivity: a/(a + c) − proportion of true positives | ||
| Specificity: d/(d + b) − proportion of true negatives | ||

Neonatal Screening
Newborn screening programs have been introduced on a widespread basis for phenylketonuria, galactosemia, and congenital hypothyroidism. In all of these disorders early treatment can dramatically prevent the development of learning disability. To this group have been added hemoglobinopathies, cystic fibrosis, and medium-chain acyl-CoA dehydrogenase more recently, where knowledge of the condition and intervention is important but less dramatic in its benefits. Screening for several other disorders is carried out more selectively in different centers (Table 20.4). In the United States, for example, all 50 states have a legal duty to provide screening to all newborns, at least for the first three conditions mentioned. In some states, (West Virginia, Montana, and South Dakota), screening is limited to these three conditions but at the other extreme up to 30 diseases are screened for (North Carolina and Oregon). In the United Kingdom, consideration is being given to screening for Pompe disease, maple syrup urine disease, tyrosinemia, congenital adrenal hyperplasia, isovaleric academia, glutaric aciduria type 1, and homocystinuria.
Table 20.4 Conditions for Which Neonatal Screening Can Be Undertaken
| Disorder | Test/Method |
|---|---|
| Widely Applied | |
| Phenylketonuria | Guthrie testa or automated fluorometric assay |
| Congenital hypothyroidism | Thyroxine or thyroid-stimulating hormone |
| Other Inborn Errors | |
| Biotinidase deficiency | Specific enzyme assay |
| Galactosemia | Modified Guthrie test |
| Homocystinuria | Modified Guthrie test |
| Maple syrup urine disease | Modified Guthrie test |
| Tyrosinemia | Modified Guthrie test |
| Miscellaneous | |
| Congenital adrenal hyperplasia | 17-Hydroxyprogesterone assay |
| Cystic fibrosis | Immunoreactive trypsin and DNA analysis |
| Duchenne muscular dystrophy | Creatine kinase |
| Sickle-cell disease | Hemoglobin electrophoresis |
a The Guthrie test is based on reversal of bacterial growth inhibition by a high level of phenylalanine.
For most of these conditions, the rationale is to prevent subsequent morbidity and in most countries is either mandatory or pursued under the auspices of implied consent. The Netherlands, by contrast, have adopted a more explicit consent process, whereas the screening remains highly recommended. The importance of adhering to the principle of screening for a disorder that needs to be treated early is illustrated by the Swedish experience of neonatal screening for α1-antitrypsin deficiency. In this condition neonatal complications occur in up to 10%, but for most cases the morbidity is seen in adult life, and the main message on diagnosing the disorder is avoidance of smoking. Between 1972 and 1974, 200,000 newborns were screened and follow-up studies showed that considerable anxiety was generated when the information was conveyed to parents, who perceived their children to be at risk of a serious, life-threatening disorder. The case of newborn screening for Duchenne muscular dystrophy also deviates from the screening paradigm because, thus far, no early intervention is helpful. Here, the parents (or mother) can be counseled before having more children and, in the wider family, identification of female carriers (of reproductive age) may be possible. However, here again, parental reaction has not been uniformly favorable.
Phenylketonuria
Routine biochemical screening of newborn infants for phenylketonuria was recommended by the Ministry of Health in the United Kingdom in 1969, after it had been shown that a low-phenylalanine diet could prevent the severe learning disabilities that previously had been a hallmark of this condition (p. 167). The screening test, which is sometimes known as the Guthrie test, is carried out on a small sample of blood obtained by heel-prick at age 7 days. An abnormal test result is further investigated by repeat analysis of phenylalanine levels in a venous blood sample. A low-phenylalanine diet is extremely effective in preventing learning disabilities, and, although it is not particularly palatable, most affected children can be persuaded to adhere to it until early adult life when it can be relaxed. However, because high phenylalanine levels are toxic to the developing brain, a woman with phenylketonuria who is contemplating pregnancy should adhere to a strict low-phenylalanine diet both before and during pregnancy (p. 261).
Congenital Hypothyroidism
Screening for congenital hypothyroidism was first introduced in the United States in 1974 and is now undertaken in most parts of the developed world. The test is based on assay of either thyroxine or thyroid-stimulating hormone. This disorder is particularly suitable for screening as it is relatively common, with an incidence of approximately 1 in 4000, and treatment with lifelong thyroxine replacement is extremely effective in preventing the severe developmental problems associated with the classic picture of ‘cretinism’. The most common cause of congenital hypothyroidism is absence of the thyroid gland rather than an inborn error of metabolism (p. 167). Congenital absence of the thyroid gland is usually not caused by genetic factors but on rare occasion is part of a wider syndrome.
Sickle Cell Disease and Thalassemia
Newborn screening based on hemoglobin electrophoresis is undertaken in many countries with a significant Afro-Caribbean community. As with cystic fibrosis, it is hoped that early prophylaxis will reduce morbidity and mortality, thereby improving the long-term outlook. In the case of sickle cell disease, treatment involves the use of oral penicillin to reduce the risk of pneumococcal infection resulting from immune deficiency secondary to splenic infarction (p. 152). Even in Western countries with good medical facilities, a significant proportion of sickle cell homozygotes, possibly as many as 15%, die as a result of infection in early childhood. In the case of thalassemia, early diagnosis makes it possible to optimize transfusion regimens and iron-chelation therapy from an early stage. Neonatal screening programs for both of these hemoglobinopathies were implemented in the United Kingdom in 2005, and antenatal screening (the mother, followed by the father if necessary) is under way in some areas of high risk. In some low-risk areas, there is a preference for antenatal screening to be targeted to high-risk couples after completion of an ethnicity questionnaire (p. 164).
Population Carrier Screening
Widespread screening for carriers of autosomal recessive disorders in high-incidence populations was first introduced for the hemoglobinopathies (see Chapter 10) and has been extended to several other disorders (Table 20.5). The rationale behind these programs is that carrier detection can be supported by genetic counseling so that carrier couples can be forewarned of the 1 in 4 risk that each of their children could be affected. The example of Tay-Sachs disease in orthodox Jewish communities has been discussed previously (pp. 313–314); this does not amount to ‘population’ screening.
Table 20.5 Autosomal Recessive Disorders Suitable for Population Carrier Screening
| Disorder | Ethnic Group or Community | Test |
|---|---|---|
| α-Thalassemia electrophoresis | China and eastern Asia | Mean corpuscular hemoglobin and hemoglobin |
| β-Thalassemia electrophoresis | Indian subcontinent and Mediterranean countries | Mean corpuscular hemoglobin and hemoglobin |
| Sickle cell disease | Afro-Caribbean | Sickle test and hemoglobin electrophoresis |
| Cystic fibrosis | Western European whites | Common mutation analysis |
| Tay-Sachs disease | Ashkenazi Jews | Hexosaminidase A |
Thalassemia
α- and β-thalassemia are caused by abnormal globin chain synthesis because of mutations involving the α- and β-globin genes or their promoter regions (p. 160). Both disorders show autosomal recessive inheritance and are extremely common in certain parts of the world, notably South-East Asia (α-thalassemia), Cyprus and the Mediterranean region, Italy, and the Indian subcontinent (β-thalassemia).
Sickle Cell Disease
In contrast to the Cypriot response to β-thalassemia screening, early attempts to introduce sickle cell carrier detection in the black population of North America were disastrous. Information pamphlets tended to confuse the sickle cell carrier state, or trait, which is usually harmless, with the homozygous disease, which conveys significant morbidity (p. 159). Several US states passed legislation making sickle cell screening in black people mandatory, and carriers suffered discrimination by employers and insurance companies. It is not surprising that public criticism was aroused, leading to abandonment of the screening programs and amendment of the ill-conceived legislation.
Cystic Fibrosis
Initial studies of attitudes to cystic fibrosis carrier detection yielded quite divergent results. A casual, written invitation generates a poor take-up response of around 10%, whereas personal contact during early pregnancy, whether mediated through general practice or the antenatal clinic, results in uptake rates of more than 80%. Studies have been undertaken to explore attitudes to cystic fibrosis screening among specific groups, such as school leavers and women in early pregnancy.
Positive and Negative Aspects of Population Screening
Well-planned population screening enhances informed choice and offers the prospect of a significant reduction in the incidence of disabling genetic disorders. These potential advantages have to be weighed against the potential disadvantages that can arise from the overenthusiastic pursuit of a poorly planned or ill-judged screening program (Box 20.4). Experience to date indicates that in relatively small, well-informed groups, such as the Greek Cypriots and American Ashkenazi Jews, community screening is welcomed. When screening is offered to larger populations the outcome is less certain.
Genetic Registers
Regional genetic centres maintain a genetic register of families and individuals who are either affected by, or at risk of developing, a serious hereditary disorder. The main difference with a conventional digital medical record is the facility to link biological relatives, some affected by a disorder, some unaffected, and others at risk. This makes it possible to manage, support, and inform the family (Box 20.5), as well as schedule review, screening, and the offer of appropriate and timely genetic tests. Ideally, patients should be aware that their information is held in this way and the genetics unit is responsible for security and confidentiality. Unlike other medical records, the destruction of these records at a given time after patients’ death is strongly resisted by geneticists because the information may be crucially important to the generations that follow.
Box 20.5
Roles of a Genetic Register
Genetic registers are especially appropriate for conditions that are relatively common, have potentially serious effects, convey high risks to other family members, and in which complications can be treated or prevented (Box 20.6). They are particularly valuable for conditions that show a delayed age of onset or those for which unaffected carriers could be at high risk of having seriously affected children.
Well-organized registers can facilitate the coordination of a multidisciplinary approach to the management of families with familial cancer-predisposing syndromes (p. 229). This often involves the interpretation of molecular investigations and the organization of hospital appointments and screening (see Table 14.9, p. 228). This is an important part of patient care and registers will continue to be necessary as more genetic health information is generated in the future.
Axworthy D, Brock DJH, Bobrow M, Marteau TM. 1996 Psychological impact of population-based carrier testing for cystic fibrosis: 3-year follow-up. Lancet. 1996;347:1443-1446.
A review of the impact of carrier testing for cystic fibrosis on over 700 individuals.
Baily MA, Murray TH, editors. Ethics and newborn genetic screening: new technologies, new challenges. Baltimore: Johns Hopkins University Press, 2009.
Brock DJH, Rodeck CH, Ferguson-Smith MA, editors. Prenatal diagnosis and screening. Edinburgh: Churchill Livingstone, 1992.
A huge multiauthor textbook with excellent chapters on all aspects of genetic screening.
Cunningham GC, Tompkinson DG. Cost and effectiveness of the California triple marker prenatal screening program. Genet Med. 1999;1:199-206.
Harper PS. Practical genetic counselling, 6th ed. London: Arnold; 2004.
Marteau T, Richards M, editors. The troubled helix. Cambridge: Cambridge University Press, 1996.
Perspectives on the social and psychological implications of genetic testing and screening.
Modell B, Modell M. Towards a healthy baby. Oxford: Oxford University Press; 1992.
A clearly written and easily understood guide to genetic counseling and community genetics.
Nuffield Council on Bioethics. Genetic screening: ethical issues. London: Nuffield Council on Bioethics; 1993.
Pauli R, Motulsky AG. Risk counselling in autosomal dominant disorders with undetermined penetrance. J Med Genet. 1981;18:340-343.
Pembrey ME, Davies KE, Winter RM, et al. Clinical use of DNA markers linked to the gene for Duchenne muscular dystrophy. Arch Dis Child. 1984;59:208-216.
Elements