Chapter 154 Scleroderma and Raynaud Phenomenon
Classification
Localized scleroderma is distinct from systemic scleroderma and rarely progresses to systemic disease. Within the category of LS there are several subtypes that are differentiated by both the distribution of the lesions and the depth of involvement (Table 154-1). Up to 15% of children have a combination of 2 or more subtypes.
Table 154-1 CLASSIFICATION OF PEDIATRIC SCLERODERMA (MORPHEA)
LOCALIZED SCLERODERMA
Plaque Morphea
Generalized Morphea
Bullous Morphea
Bullous lesions that can occur with any of the subtypes of morphea
Linear Scleroderma
Linear lesions can extend through the dermis, subcutaneous tissue, and muscle to underlying bone; more likely unilateral
Deep Morphea
SYSTEMIC SCLEROSIS
Diffuse
Limited
Clinical Manifestations
Localized Scleroderma
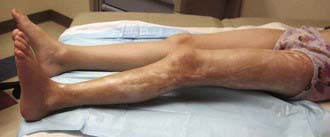
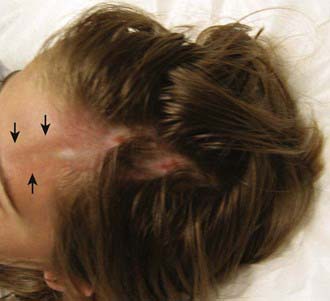
The onset of scleroderma is generally insidious, and manifestations vary according to disease subtype. The initial skin manifestations of localized disease usually include erythema or a bluish hue seen around an area of waxy induration; subtle erythema may be the only presenting sign (Fig. 154-1). Early edema and erythema are followed by indurated, hypopigmented or hyperpigmented, atrophic lesions (Fig. 154-2). Linear scleroderma varies in size from a few centimeters to the entire length of the extremity, with varying depth. Patients sometimes present with arthralgias, synovitis, or flexion contractures (Fig. 154-3). Children also experience limb length discrepancies as a result of growth impairment due to involvement of muscle and bone. Children with en coup de sabre (Fig. 154-4) may have symptoms unique to central nervous system (CNS) involvement, such as seizures, hemifacial atrophy, ipsilateral uveitis, and learning/behavioral changes.
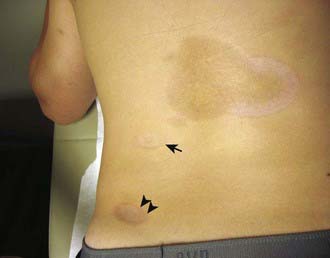
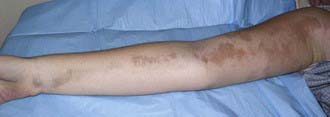
Figure 154-2 Inactive linear scleroderma demonstrating hyperpigmented lesion with areas of normal skin (skip lesions).
Systemic Scleroderma
The skin manifestations of SSc include an early phase of edema that spreads proximally from the dorsum of the hands and fingers and includes the face. An eventual decrease in edema is followed by induration and fibrosis of skin, ultimately resulting in loss of subcutaneous fat, sweat glands, and hair follicles. Later, atrophic skin becomes shiny and waxy in appearance. As lesions spread proximally, flexion contractures develop at the elbows, hips, and knees associated with secondary muscle weakness and atrophy. In the face, this process results in a small oral stoma with decreased mouth aperture. Skin ulceration over pressure points, such as the elbows, may be associated with subcutaneous calcifications. Severe Raynaud phenomenon causes ulceration of the fingertips with subsequent loss of tissue pulp and tapered fingers (sclerodactyly) (Fig. 154-5). Resorption of the distal tufts of the distal phalanges may occur (acro-osteolysis). Hyperpigmented postinflammatory changes surrounded by atrophic depigmentation gives a salt-and-pepper appearance. Over a period of years, remodeling of lesions sometimes results in focal improvement in skin thickening.
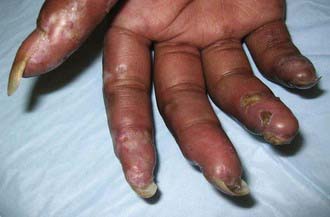
Raynaud Phenomenon
Raynaud phenomenon (RP) is the most frequent initial symptom in pediatric systemic sclerosis, present in 70% of affected children months to years before other manifestations. Raynaud phenomenon refers to the classic triphasic sequence of blanching, cyanosis, and erythema of the digits induced by cold exposure and/or emotional stress. Raynaud phenomenon is most commonly independent of an underlying rheumatic disease (Raynaud disease), but it can be a consequence of other diseases as well as scleroderma, such as systemic lupus erythematosus and mixed connective tissue disease (Table 154-2). The color changes are brought about by (1) initial arterial vasoconstriction, resulting in hypoperfusion and pallor (blanching), (2) venous stasis (cyanosis), and (3) reflex vasodilatation caused by the factors released from the ischemic phase (erythema). The color change is classically reproduced by immersing the hands in iced water and reversed by warming. During the blanching phase, there is inadequate tissue perfusion in the affected area, associated with pain and paresthesias and resulting in ischemic damage only when associated with a rheumatic disease. The blanching usually affects the distal fingers but may also involve thumbs, toes, ears, and tip of the nose. The affected area is usually well demarcated and uniformly white.
Table 154-2 CLASSIFICATION OF RAYNAUD PHENOMENON
From Firestein GS, Budd RC, Harris ED Jr, et al, editors: Kelley’s textbook of rheumatology, ed 8, vol II, Philadelphia, 2009, Saunders/Elsevier.
Diagnosis
The diagnosis of localized scleroderma is based on the distribution and depth of characteristic lesions. Biopsy is helpful to confirm the diagnosis. Classification criteria for juvenile systemic sclerosis were recently devised, reflecting differences in presentation and course compared with adult onset disease. The new classification requires proximal sclerosis/induration of the skin as well as the presence of 2 of 20 minor criteria (Table 154-3).
Table 154-3 PROVISIONAL CRITERIA FOR THE CLASSIFICATION OF JUVENILE SYSTEMIC SCLEROSIS (SSC)
MAJOR CRITERION (REQUIRED)
Proximal skin sclerosis/induration of the skin
MINOR CRITERIA (AT LEAST 2 REQUIRED)
From Zulian F, Woo P, Athreya BH, et al: The Pediatric Rheumatology European Society/American College of Rheumatology/European League against Rheumatism provisional classification criteria for juvenile systemic sclerosis, Arthritis Rheum 57:203–212, 2007.
Charles C, Clements P, Furst DE. Systemic sclerosis: hypothesis-driven treatment strategies. Lancet. 2006;367:1683-1690.
Fitch PG, Rettig P, Burnham JM, et al. Treatment of pediatric localized scleroderma with methotrexate. J Rheumatol. 2006;33:609.
Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. N Engl J Med. 2009;360:1989-2002.
Gu YS, Kong J, Cheema GS, et al. The immunobiology of systemic sclerosis. Semin Arthritis Rheum. 2008;38:132-160.
Hudson M, Fritzler MJ, Baron M, et al. Systemic sclerosis: establishing diagnostic criteria. Medicine. 2010;89(3):159-165.
Kreuter A, Gambichler T, Breuckmann F, et al. Pulsed high-dose corticosteroids combined with low-dose methotrexate in severe localized scleroderma. Arch Dermatol. 2005;141:847.
Laxer RM, Zulian F. Localized scleroderma. Curr Opin Rheumatol. 2006;18:606-613.
Martini G, Foeldvari I, Russo R, et al. Systemic sclerosis in childhood: clinical and immunologic features of 153 patients in an international database. Arthritis Rheum. 2006;54:3971-3978.
Nigrovic PA, Fuhlbrigge RC, Sundel RP. Raynaud’s phenomenon in children: a retrospective review of 123 patients. Pediatrics. 2003;111:715-721.
Peterson LS, Nelson AM, Su WPD, et al. The epidemiology of morphea (localized scleroderma) in Olmsted county 1960–1993. J Rheumatol. 1997;24:73-80.
Scalapino K, Arkachaisri T, Lucas M, et al. Childhood onset systemic sclerosis: classification, clinical and serologic features, and survival in comparison with adult onset disease. J Rheumatol. 2006;33:1004-1013.
Tashkin DP, Elashoff R, Clements PJ, et al. Cyclophosphamide versus placebo in scleroderma lung disease. N Engl J Med. 2006;354:2655-2666.
Uziel Y, Feldman BM, Krafchik BR, et al. Methotrexate and corticosteroid therapy for pediatric localized scleroderma. J Pediatr. 2000;136:91.
Zulian F, Martini G. Childhood systemic sclerosis. Curr Opin Rheumatol. 2007;19:592-597.
Zulian F, Vallongo C, Woo P, et al. Localized scleroderma in childhood is not just a skin disease. Arthritis Rheum. 2005;52:2873-2881.





