CHAPTER 149 Scalp Tumors
Skin Tissue
Keratosis
AKs are regions of epithelial transformation that occur on portions of the skin that receive extensive sun or radiation exposure, such as the face and scalp (Fig. 149-1). Geographic regions with higher UV exposure have a higher prevalence of AKs. In Australia, the prevalence is estimated to be 40% to 50% in people older than 40 years as opposed to 11% to 26% in the United States.1 AKs are typically seen in fair-skinned people and other high-risk populations (including the elderly), in patients receiving immunosuppressive therapy and psoralen and ultraviolet A (PUVA) therapy, and in those with a history of arsenic exposure. Clinically, these lesions are red or darkened scaly raised patches or plaques with irregular borders. It has been estimated that approximately 8% of AKs progress to invasive squamous cell carcinoma (SCC),2 and this progression has been attributed to p53 mutations caused by ultraviolet B (UVB)-induced DNA damage. Current medical treatment options include topical agents such as 5-fluorouracil (5-FU), immunotherapy (5% imiquimod cream), diclofenac gel, and photodynamic therapy with 5-aminolevulinic acid.3,4 Surgical procedures such as cryosurgery, electrodesiccation and curettage, dermabrasion, shave excision, and carbon dioxide laser treatment are also effective.5
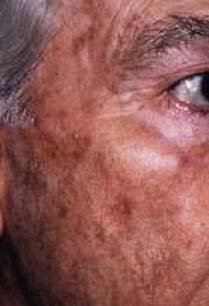
FIGURE 149-1 Elderly male patient with actinic keratosis of the left midfacial area, forehead, and scalp.
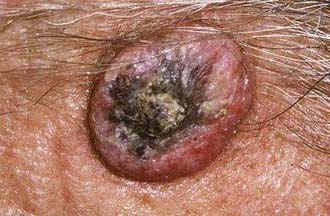
Keratoacanthomas are papillary lesions noted for their rapid growth (Fig. 149-2). These lesions are firm, skin-colored or reddish papules that may progress to dome-shaped nodules over a period of several weeks to months. In most cases, keratoacanthomas spontaneously regress but are often clinically confused with malignant lesions. UV light, chemical carcinogens, trauma, human papilloma virus (HPV), genetic factors, and immunocompromised status have been implicated as etiologic factors.6 Primary treatment is surgical excision, although radiation therapy and Mohs’ micrographic surgery (MMS) can be indicated in some cases.
Carcinoma
Basal Cell Carcinoma
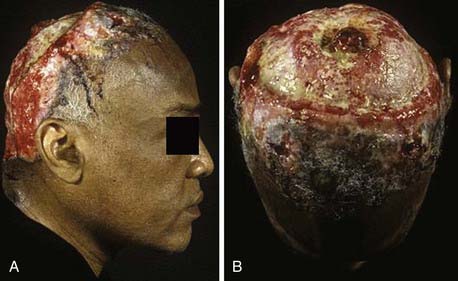
Basal cell carcinoma (BCC) is one of the most common and prevalent skin tumors in the white population, in whom the average lifetime risk is approximately 30%.7 Although it can be locally destructive, BCC rarely metastasizes and is associated with few deaths. These lesions arise in pluripotent basal cells of the epidermis and may grow to include the skin, vasculature, and periosteum. The disease may spread along the periosteum or fascia, which accounts for the high recurrence rates on the scalp,8 and in rare instances, the bone and dura may be involved. Cosmetic disfigurement is common and loss of vision may occur with orbital involvement (Fig. 149-3A and B). The main risk factor is UV exposure, specifically UVB radiation, which produces characteristic cytosine (C)-to-thymine (T) or CC-to-TT transition mutations in DNA.9 Fair-skinned individuals with blonde or red hair, light-colored eyes, high susceptibility to sunburn, and a history of solar dermatitis in childhood or adolescence are at the highest risk. Additional risk factors include the presence of immunosuppression, ingestion of arsenic acid, and exposure to ionizing radiation.7 Some familial syndromes predispose individuals to BCC, the most common being Gorlin’s syndrome (basal cell nevus syndrome [BCNS]).10
There are several variants of BCC, the most common of which is defined as nodular or nodular-ulcerative. This form of BCC is typically manifested as an elevated, light-colored, tan, or pink nodule (although in some instances it may be dark or black) with telangiectatic vessels and a pearly, rolled, well-circumscribed border. These lesions occur frequently on the head and neck and can ulcerate or become locally destructive if allowed to develop over time, although this form of the disease is generally well circumscribed. A second form of BCC, the pigmented type, is characterized by increased brown or black pigment in the lesion and is more common in darker skinned individuals. A third form is cystic BCC, characterized by translucent blue/gray cystic nodules that may mimic benign cystic lesions. A fourth form is the superficial multicentric type, which is more commonly found on the trunk and extremities and resembles a reddened, scaly patch of skin similar to that seen in psoriasis. This variant of BCC can be highly recurrent secondary to subclinically persistent radially oriented growth and has been linked to arsenic exposure.7 A fifth BCC subtype, the micronodular form, is characterized by yellow/white, small firm nodules and is very aggressive. The final subtype is the morpheaform variant, which appears as a scar-like, hardened plaque and is noted for aggressive behavior and high rates of recurrence. In addition to the aforementioned subtypes, BCC may be accompanied by squamous metaplasia. This group of BCC lesions appears to share some characteristics with SCC and is the form most likely to metastasize.
The molecular genetics of BCC involves two pathways: the Hedgehog (HH) signaling pathway (via the PTCH1 gene) and p53 gene mutations. HH is a 45-kD protein that has a role in the differentiation of various tissues during development, including the neural tube, skin, and hair follicles. Binding of HH to the transmembrane receptor PTCH1 starts a cascade of events leading to cell proliferation. Mutations of PTCH1 are linked to chromosome 9q22 and are found in Gorlin’s syndrome (BCNS) and 30% of sporadic BCC cases.7 Mutations of the p53 tumor suppressor gene, which resides on chromosome 17p, are frequent (>50%) in sporadic BCC. These mutations allow cells that have sustained UVB damage to propagate.10
Squamous Cell Carcinoma
Like BCC, SCC of the skin is a frequently occurring and highly curable disease despite its greater probability of metastasis. The lifetime risk of SCC developing in a child born in 1994 in the United States is 7% to 11%.11 Lesions arise in the keratinizing cells of the epidermis and infiltrate locally but may spread to lymph nodes and metastasize. Risk factors include increasing age, male sex, fair skin, living close to the equator, chronic scarring conditions, exposure to sunlight or radiation, chronic immunosuppression, exposure to arsenic and organic hydrocarbons, and the presence of HPV.12 Clinically, SCC appears as scaly plaques that may be crusted or ulcerated, are frequently light or even whitish in color, and are not associated with telangiectasia or rolled, pearly borders (Fig. 149-4).
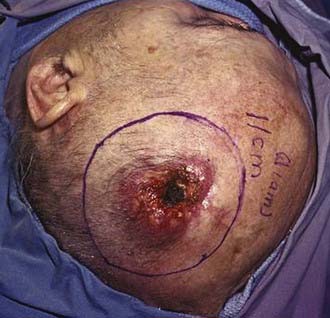
When compared with BCC, the pattern and degree of structural changes in the genome in SCC are more diverse and involve many more chromosomal regions (13q, 9p, 17p, 17q, 3p).13 Loss of heterozygosity at 17p is associated with p53 UV-induced mutations (C to T or CC to TT), which leads to an abnormal apoptotic response.14 Progression from AK to invasive SCC has been suggested to correlate with deletion of the 9p21 region harboring the CDKN2A gene.15 However, the association between the accumulation of genetic changes and progression to malignancy is not fully understood.
Staging and Treatment
BCCs and SCCs are staged by the TNM (tumor, node, metastasis) staging system of the American Joint Committee on Cancer (AJCC) and graded according to depth of invasion, proportion of differentiated cells, and degree of atypia.16 The primary goal of treatment is complete excision of the lesion. The extent of tumor involvement should be carefully determined beforehand with imaging, if appropriate. On the scalp, fixed lesions should be assessed for bony as well as possible dural or intracranial involvement.
A variety of treatment options for BCC and SCC are currently available. Medical treatments include topical chemotherapy (5-FU), topical immune response modifiers (imiquimod), and photodynamic therapy. These treatment options are indicated for premalignant and in situ lesions. Systemic retinoids are effective in reducing the number of new SCCs but have not been shown to be beneficial in treating existing SCC or reducing the risk for recurrence after treatment.17 Emerging evidence from animal and human studies suggests that regular use of nonsteroidal anti-inflammatory drugs may protect against the development of both AK and SCC,18 but this is not currently standard therapy. Surgical options include electrodesiccation and curettage, surgical excision, MMS, and cryosurgery (contraindicated for scalp tumors). Radiation treatment is indicated for patients who cannot undergo surgery (such as the elderly or those with multiple comorbid conditions) and as an adjunct in metastatic or high-risk SCC. Radiotherapy is not generally recommended for younger patients, however, because of the long-term consequences associated with exposure to radiation. Systemic chemotherapy, such as capecitabine and oral 5-FU combined with interferon, has been used to treat metastatic cutaneous SCC.19 As well as histology, selection of treatment depends on the size and anatomic location of the lesion. Certain risk factors are associated with higher rates of recurrence or metastasis, including size, depth, location, perineural invasion, and immunosuppression. The 5-year survival rate for patients with metastatic cutaneous SCC is 34%.20
By virtue of its sequential microscopic examination of tumor invasion, MMS has high cure rates (>90%) in experienced hands and is generally selected for BCC or SCC lesions in which therapy failure rates are high (e.g., scalp lesions), for large tumors (>2 cm), for tumors in locations where tissue conservation is important for cosmetic reasons, or for BCC lesions of the morpheaform or sclerotic subtype.21,22 Rowe and colleagues noted that local recurrences are less frequent when SCC is treated by MMS than when treated by all non-Mohs’ modalities.20 This local recurrence rate differential in favor of MMS was observed in primary SCC of the skin and lip (3.1% versus 10.9%), for locally recurrent SCC (10% versus 23.3%), for poorly differentiated SCC (32.6% versus 53.6%), and for SCC with perineural involvement (0% versus 47%).
In general, electrodesiccation with curettage is a less preferable alternative for scalp tumors than either MMS or simple surgical excision because of reported recurrence rates as high as 50%. Complex or extensive lesions, including those that invade bone or dura, may require extensive surgical resection, often accompanied by skin-bone-dural grafts or flaps. In general, for low-risk lesions (<2 cm in maximal diameter), surgical excision should include a 4-mm margin of normal tissue. However, for larger lesions (>2 cm in maximal diameter), a 1-cm margin is preferable.23 Despite the availability of successful treatment regimens for the two diseases, prevention of BCC and SCC remains the ultimate clinical goal, given the reduction in risk associated with the use of sunscreen and avoidance of sunlight exposure.
Melanocytic Nevus
Melanocytic nevi are benign tumors of melanocytes and nevus cells that produce the pigment melanin. These nevi are of neural crest origin and are noted in approximately 1% of newborns.24 Typically, these lesions are categorized as (1) common acquired; (2) relatively rare subtypes, such as congenital giant (“hairy”), blue, compound Spitz’, and Ota’s; or (3) dysplastic nevi (Clark’s nevi). Histologically, nevi are defined as intradermal, junctional (located at the dermoepidermal junction within the epidermis), compound (in the dermis, with associated intraepidermal nevus cell nests), large pigmented, and dysplastic.
Common acquired nevi, often termed moles, are characterized by a light to dark brown color and may be slightly elevated or have hair. Histology and clinical course vary by body location. Nevi located on the palms or soles tend to be compound or junctional and have a low probability of “neurotization,” a process by which mature dermal melanocytes undergo differentiation into spherical structures resembling tactile nerve endings.25 Nevi on the face and scalp are more likely to undergo neurotization and change to pale lesions or disappear entirely via the formation of halo nevi.24 In general, common acquired nevi are benign lesions smaller than 1 cm in diameter, are evenly pigmented, and require no treatment, although in some instances they may become atypical and require excision. Any nevus that grows rapidly in size, changes color, bleeds, or becomes ulcerated should undergo biopsy and be treated.
Dysplastic nevi (Clark’s nevi) are atypical melanocytic nevi that are precursor or marker lesions for the development of melanoma,25,26 although the probability that any one dysplastic nevus will become a melanoma is small. Clinically, these lesions are flat, thinly papular, and relatively broad. Frequently, such lesions exhibit target-like or “fried-egg” morphology with a central papular zone and a macular surrounding area with differing pigmentation. They occur on both sun-exposed and non–sun-exposed regions of the body. These lesions may be present at birth or be recognized later in life. Families that share similar patterns or numbers of dysplastic nevi, sometimes in association with melanoma, have been identified.25,26 Among sporadic cases, the extent to which the syndrome of dysplastic nevi is congenital or acquired is a topic of much debate. An important clinical fact is that risk for melanoma is increased in portions of the skin without dysplastic nevi, as well as in areas marked by such nevi. Thus, careful skin examination of the entire body should be performed regularly in these patients. Any suspicious lesion should undergo biopsy given the risk for melanoma.
Melanoma
Melanoma is one of the few cancers that increased in both incidence and mortality in the United States between 1973 and 1999.27 Analysis of U.S. Surveillance, Epidemiology, and End Results (SEER) data from this period demonstrated a disproportionate burden of melanoma deaths in middle-aged and older white men. Although melanoma mortality rates have fallen 39% in women and 29% in men 20 to 44 years old over this period, they have increased 66% in men 45 to 64 years old and 157% in older men (≥65 years old).27 The current lifetime risk in the United States is 1 in 68.28 Cutaneous melanomas (which make up >90% of all melanomas) arise from epidermal melanocytes, cells that are derived from the neural crest and produce the photoprotective pigment melanin. During embryogenesis, these cells migrate to the epidermis and come to rest in the basal layer of the epidermis. Although the exact mechanism by which melanoma develops remains unclear, it probably involves a multistep process of progressive mutations that (1) alter cell proliferation, differentiation, and death and (2) have an effect on susceptibility to the carcinogenic effects of UV radiation.29
Epidemiologic data suggest that several factors are highly correlated with risk for melanoma. In particular, excessive exposure to UV radiation plays a key role in the development of melanoma, with individuals having light-colored eyes and fair skin being at greatest risk for development of the disease. More specifically, the presence of a blistering sunburn in childhood increases the risk significantly.30 Interestingly, most melanomas occur on areas of the body exposed intermittently, but probably intensely to the sun, such as the back on white men and the legs on white women, with relative sparing of areas more frequently exposed to the sun such as the scalp, face, and upper extremities. In African American, Hispanic, and Asian individuals, the most common site is the plantar surface of the foot, followed by subungual, palmar, and mucosal sites.31 In addition to sun exposure, the presence of nevi, both dysplastic and nondysplastic, has been correlated with an increased risk for melanoma,32,33 with possession of large numbers of clinically recognized dysplastic nevi being associated with up to a 12-fold increase in risk.32 Immunosuppressed status and a family history of melanoma are also risk factors.
Much active research has gone into identifying the molecular pathways involved in the pathogenesis of melanoma. Familial melanoma accounts for some 10% of all melanoma cases, and approximately 40% of such patients have germline mutations in the gene CDKN2A.34 This is a tumor suppressor gene that normally inhibits tumor formation by limiting cell growth. Loss-of-heterozygosity analysis has led to identification of the CDKN2A locus on chromosome 9p21 for familial melanoma. This locus encodes two proteins: p16INKA, which inhibits cyclin-dependent kinase (CDK) 4 and 6, enzymes involved in cell cycle regulation, and p14ARF, which stabilizes p53.35 Penetrance of CDKN2A mutations varies by geographic location, which suggests that common factors may modify both geographic variation and mutation penetrance.36
Polymorphisms in the BRAF proto-oncogene have been demonstrated in up to 60% of melanoma tumors.37 This gene is one of three serine/threonine kinases regulated by binding to RAS, an oncogene that has been implicated in approximately 15% of all cancers. When mutated, the protein has increased kinase activity. The estimated proportion of risk for melanoma attributable to variants in BRAF is 1.6%, as opposed to 0.2% attributable to CDK2NA variants.38
More recently, several low-penetrance risk alleles have been identified. Variants in the melanocortin-1 receptor (MC1R) gene are associated with red hair39 and a twofold to fourfold increase in melanoma risk.40 Melanoma was also overrepresented in families with the BRCA2 mutation, who have a relative risk of 2.58.41 A genome-wide association study using samples from two Australian case-control studies identified a new melanoma risk locus on chromosome 20q11.22 that was able to be replicated in two further samples (combined P < 1 × 10−15).42 This chromosome region contains the gene ASIP, which has been associated with both cutaneous melanoma and BCC.
The clinical manifestation of scalp melanoma is generally an asymptomatic pigmented lesion with irregular borders. The lesion is frequently dark or black in color, although lighter colored melanomas do exist, and many melanomas arise from regions where no previous lesion was noted. A change in the color, texture, size, or shape of a lesion or preexisting mole is cause for concern, particularly if the mole becomes ulcerated or crusted or bleeds. During skin examination, care must be taken to fully examine the scalp because lesions may be hidden under the hair, particularly in individuals with thick hair or dark skin. With primary cutaneous melanoma isolated to the scalp, the 5-year survival rate was 86% for patients with lesions located in non–hair-bearing regions and 47% in those with lesions in hair-bearing regions.43 It is possible that delayed detection of lesions in hair-bearing areas played a role in this difference in survival.
In general, there are four primary patterns of melanoma: superficial spreading melanoma, nodular melanoma, lentigo maligna melanoma, and acral-lentiginous melanoma. Superficial spreading melanoma is the most common of the four and represents approximately 60% to 75% of all cases. This variant is characterized by horizontal growth along the dermoepidermal junction and usually has a fairly good prognosis if diagnosed early. It is generally larger than 6 mm in maximal diameter, has irregular asymmetric borders, and histologically is characterized by buckshot (pagetoid) scatter of atypical melanocytes within the epidermis. Nodular melanoma, which accounts for approximately 15% to 30% of all cases, is characterized by vertical growth and a blue or black color, although an unpigmented (amelanotic) variant exists that may be difficult to detect clinically. Lentigo maligna melanoma (5% to 10% of cases but increasing in the United States44) generally develops from an in situ lesion (Hutchinson’s freckle) that is usually large (>1 to 3 cm in diameter), is present for a minimum of 10 to 15 years, and demonstrates macular pigmentation ranging from dark brown to black, although hypopigmented (white) areas are common within lentigo maligna. Dermal invasion (progression to lentigo maligna melanoma) is characterized by the development of raised blue-black nodules within the in situ lesion. These lesions are frequently seen on the head and face of older individuals with a history of sunburn.
The final type, acral-lentiginous melanoma (2% to 8% of cases), is notable for its location on the palms and soles, as well as under the nail beds. This form of the disease constitutes most of the melanomas (29% to 72%) occurring in dark-skinned individuals and, because of delays in diagnosis, may be associated with a worse prognosis.31,45 It is characterized by atypical melanocytes along the dermoepidermal junction.
There are two other widely used methods of pathologic staging of melanoma: one developed by Breslow, which uses invasion thickness cutoff points of 0.75, 1.5, and 4 mm as measures of prognosis,46 and a second by Clark and colleagues, which defines five increasing levels of invasion: epidermis only, to dermis, to reticular dermis, through reticular dermis, and to subcutaneous tissue.47 In general, the Breslow method is thought to predict survival probability more accurately. The AJCC uses TNM staging based on invasion characteristics of the disease, as well as metastatic status.48 The initial classification system divided melanoma into four stages and incorporated tumor thickness and anatomic level of invasion for stages I and II (localized cutaneous disease), with a later recommendation to follow Breslow depth over Clark level when any discordance arose. Stage III disease involved the regional lymph nodes, and stage IV disease included distant skin, subcutaneous, nodal, visceral, skeletal, or central nervous system metastasis. However, revisions to this system in 2002 included the incorporation of histologic ulceration and the number of lymph nodes involved (instead of size) to better stratify metastatic risk and patient prognosis. In the revised staging system, the Clark level is included only in thin primary tumors (<1 mm in depth, stages IA and IB) because its prognostic value is minimal in thicker primary melanomas. Microscopic regional lymph node metastasis as detected by SLNB is differentiated from macroscopic nodal metastasis.49
Treatment of melanoma varies by stage, but surgical excision with clear margins remains a mainstay of treatment in early-stage disease. Currently, surgical margins of 5 mm are recommended for melanoma in situ, and margins of 1 cm are recommended for melanomas up to 1 mm in depth (low-risk primaries).50 In some settings, tissue sparing may be critical and Mohs’ margin–controlled excision may be appropriate. Randomized prospective studies show that 2-cm margins are appropriate for tumors of intermediate thickness (1 to 4 mm Breslow depth), although 1-cm margins have proved effective for tumors 1 to 2 mm in thickness.51,52 Margins of 2 cm are recommended for cutaneous melanomas greater than 4 mm in thickness to prevent potential local recurrence in or around the scar site and clinically involved nodal basin.53 Surgery for metastatic lesions is primarily palliative.
At present, there is no evidence that elective regional lymph node dissection is of benefit for patients with stage I melanoma. Its role is less clear for patients with melanomas 1 to 4 mm thick, although three randomized, prospective trials failed to show increased survival in patients with melanoma of the extremities.54–57 Stage IV patients appear to receive no benefit from lymph node dissection, except for palliation. Because stage I patients generally do well with surgical excision, there is little role for adjunctive therapy in this group. For later stage patients, higher rates of treatment failure exist, thus making adjuvant treatment frequently necessary. Additional treatment modalities include chemotherapy (e.g., dacarbazine, the nitrosoureas), immunotherapy (interferon alfa, interleukin-2, tumor-infiltrating lymphocytes), monoclonal antibodies, and gene therapy. All are associated with, at best, modest improvements in survival, although complete responses have been reported in some patients. Three prospective, multicenter, randomized controlled trials have been conducted to assess the effect of adjuvant high-dose interferon alfa-2b on relapse-free survival (RFS) and overall survival (OS) rates in patients with high-risk melanoma (primary tumors ≥4 mm depth and regional nodal disease). One clinical trial showed an 11% increase (26% to 37%) in RFS rates at 5 years in the interferon alfa-2b treatment group versus the observation arm. Similarly, this trial showed an increase in 5-year OS rates from 37% to 46% (median OS times, 2.78 to 3.82 years) in the treatment arm versus the observation arm.58 A second trial comparing the administration of high- and low-dose interferon alfa-2b showed an increase in estimated 5-year RFS rates from 35% in the observation arm to 44% in the high-dose arm. No significant increase in the RFS rate was associated with low-dose interferon administration. Importantly, no difference in OS rate was seen in the interferon-treated groups (high- or low-dose) versus the observation arm.59 The most recent trial compared the use of standard high-dose interferon alfa-2b with GM2 ganglioside vaccine (GMK). The study was closed prematurely because of the observation of a significant benefit for interferon alfa-2b treatment relative to GMK treatment with regard to both RFS and OS rates.60 Yet the potential benefits of high-dose interferon must be weighed against its substantial tolerability and toxicity issues, including the yearlong duration of therapy, commonly associated flu-like symptoms, and potential for significant adverse reactions. Pegylation of interferon alfa-2b, which allows prolonged weekly self-administered therapy, has recently been shown to have a sustained effect on recurrence-free survival in stage III melanoma patients versus observation alone (hazard ratio, 0.82; P = .01).61
The prognosis for patients with melanoma worsens with increasing stage. With stage Ia disease (≤1-mm lesions with no ulceration), the 10-year survival rate is 87.9% as opposed to 6% to 15.7% in patients with stage IV disease (distant metastases).62 The relative resistance of melanoma to current adjuvant therapy points to the importance of prevention, especially given the evidence that reduction of sun exposure, use of sunscreen, and skin self-examination may significantly reduce the risk for disease.63 However, many treatments that target specific molecular mechanisms are currently under investigation. For example, mutations in oncogenes and loss of heterozygosity of tumor suppressor genes have been a major focus of targeted molecular therapies. The RAS–mitogen-activated protein kinase (MAPK) pathway is targeted by sorafenib (a tyrosine kinase inhibitor). It inhibits BRAF, vascular endothelial growth factor receptor 2 (VEGFR-2), VEGFR-3, c-KIT, and platelet-derived growth factor receptor-β (PDGFR-β). Sorafenib has not been shown to be effective as a single agent, but in combination with chemotherapy, it is associated with an improvement in response rate and progression-free survival.64 Consequently, an Eastern Cooperative Oncology Group (ECOG) phase III trial was initiated to assess the use of carboplatin and paclitaxel with or without sorafenib. Another pathway that has been targeted is the phosphatidylinositol-3′-kinase (PI3K)/Akt pathway. The target is mTOR (mammalian target of rapamycin), a serine-threonine kinase that functions downstream of Akt (protein kinase B); once activated, mTOR will phosphorylate several regions that control cell survival, proliferation, and invasiveness. Rapamycin is a macrolide antibiotic that inhibits mTOR and may block the translation of proteins required for progression through the cell cycle.64 Derivatives of rapamycin (temsirolimus, everolimus) are currently in clinical trials to evaluate their efficacy as combined agents.
Antiapoptotic therapies such as dacarbazine (DTIC) and oblimersen (a phosphorothioate antisense oligonucleotide) have shown promise in recent randomized trials. A phase III trial in 771 patients randomized to receive DTIC alone or DTIC plus oblimersen showed no difference in OS, but the overall response rate favored combination treatment (11.7% versus 6.8% for DTIC alone; P = .019).65 Bortezomib, a proteosome inhibitor that controls the nuclear factor NF-κB (a regulator of the expression of genes involved in normal immune responses), has not shown a response in phase II studies but is now being investigated in combination with paclitaxel and carboplatin.
Antiangiogenic therapies optimize the fact that neoangiogenesis is necessary for neoplastic proliferation. Bevacizumab (Avastin) is a recombinant humanized monoclonal antibody to VEGF that has demonstrated efficacy in colorectal cancer.66 A phase II randomized trial of Avastin with or without low-dose interferon in patients with stage IV melanoma demonstrated both tumor regression and stabilization of disease.64 Another agent that targets VEGF is SU5416, and it may have a future role in combination with chemotherapy.67 Thalidomide inhibits basic fibroblast growth factor–induced angiogenesis and has been studied as a single agent or in combination with temozolomide in patients with metastatic melanoma. No objective responses were seen in a phase II clinical trial,68 but disease response (both complete and partial) was observed in stage IV melanoma patients with brain metastases who received combination therapy.69 Immunomodulatory drugs and integrins are other targets of antiangiogenic therapies that are currently being investigated for use against melanoma.
There have been multiple phase I and phase II trials that have been optimistic regarding vaccine therapy for melanoma. However, in a recent review of 13 phase III randomized trials of melanoma vaccines, no significant survival benefit was observed in vaccine recipients.64 Four of these trials have not yet released their final results. A more promising immunotherapeutic target is blockade of cytotoxic T lymphocyte–associated antigen 4 (CTLA-4), which may augment an antitumor immune response. Different antibodies blocking CTLA-4 have been developed, such as MDX-010,70 and they are currently in clinical trials and showing an improvement in disease response rate.
Soft Tissue
These lesions are grouped together in this chapter despite their heterogeneity of origin (bone, vascular, muscle, and so forth) because it is histologic grade rather than cell type that more accurately reflects clinical outcome. In fact, cell type is not part of the prognostic staging system for soft tissue sarcomas as defined by the AJCC, which uses tumor size, nodal status, grade, and metastasis in its staging scheme.71,72
Early-stage (IA, IB, IIA) soft tissue sarcomas are generally treated by surgical excision with a 2-cm margin recommended. Additional therapy may include radiation treatment, but chemotherapy is not generally used given the relatively low risk for metastatic disease in patients with low-grade sarcoma. For lesions on the face and scalp, MMS is also used for small, well-differentiated lesions to preserve normal appearance.72
Higher stage (IIB, IIC, III) sarcomas can be extremely malignant and are associated with lower survival rates. Complete surgical resection is the goal but may be difficult to achieve without affecting function. In such cases, radiation therapy and chemotherapy are often added, either before or after surgery. Chemotherapy trials have studied doxorubicin-based therapy regimens; unfortunately, data that specifically address scalp sarcomas are sparse, and there is little evidence that chemotherapy provides a long-term survival benefit.73,74 Stage IV patients are treated by surgical resection and lymphadenectomy, and radiotherapy is generally administered. Adjuvant therapy is usually given in the context of a clinical trial for eligible patients.
Vascular Tissue
Angiosarcoma
Scalp angiosarcomas are rare malignant tumors of blood vessels characterized by anaplastic spindle cells and malformed vascular channels (if any are identified at all) lined with poorly differentiated endothelial cells. Potential risk factors include increasing age, radiotherapy, exposure to arsenic compounds, and sun exposure. Clinically, these tumors appear as flat or raised red-violet lesions. They tend to be multicentric, solid, heavily vascular, and locally aggressive, with microscopic tissue invasion. Treatment may include wide surgical resection, multidrug chemotherapy, radiotherapy, and MMS.75–77 Recent studies have suggested that recombinant interleukin-2, administered systemically and to the tumor directly, may also prolong survival when combined with radiotherapy.78 Unfortunately, the disease has a high rate of recurrence and frequently metastasizes. Because initial tumor size is an important prognostic factor, early detection and treatment are essential.
Kaposi’s Sarcoma
Kaposi’s sarcoma is a neoplasm of unclear cause and multiple variants that is frequently diagnosed in individuals infected with human immunodeficiency virus. As is true for angiosarcoma, from which it is histologically difficult to differentiate, Kaposi’s sarcoma lesions are thought to originate from stem cells that proliferate in varying degrees toward blood vessel endothelium. Lesions are frequently seen on the skin, including the face and scalp, and appear as multifocal red to violet patches. Treatment of these lesions depends on the variant, the patient’s immunologic status and age, and the location. Treatment should be individualized but may include surgical excision, radiotherapy, chemotherapy, and discontinuation of immunosuppressive therapy.79
Scalp Reconstructive Procedures
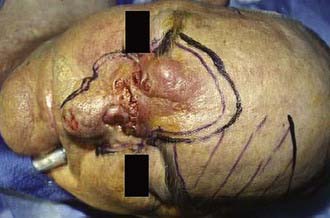
In many cases, the natural mobility of the scalp and surrounding tissues allows simple reapproximation and closure of the surgical site after excision of a scalp tumor. Yet in some instances additional techniques may be needed to close the wound. Simple maneuvers include the use of back cuts, galeal scoring, or scalp stretching. Galeal scoring consists of making small incisions through the galea parallel to the leading edge of the flap. These incisions are made at 0.5-cm intervals while taking care to preserve the blood supply to the flap. A number of local skin flaps are commonly used to repair surgical defects on the scalp, including rotation, advancement, and transposition (e.g., rhomboid) flaps (Fig. 149-5).80 Although rotation and advancement flaps are popular techniques for scalp and forehead defects, in acute wound closure (especially in the anterior direction for bald men), care must be taken to maintain the natural location of the hairline and minimize the loss of hair-bearing regions of the scalp. When wound closure is not urgent (e.g., when there is no likelihood of cerebrospinal fluid leaks and the wound bed is vascularized), scalp expansion with tissue expanders may assist in closing the defects. This is a safe but time-consuming technique with a complication rate that varies from 3% to 17% in the scalp.81,82 It is useful in secondary revisions and when removing giant congenital melanocytic nevi. The surgeon must be vigilant for possible complications of flap management, including hematoma, infection, anesthesia, hypoesthesia, alopecia, and flap necrosis secondary to vascular compromise. In the event of a large scalp defect that cannot be repaired with skin flaps, skin grafting remains a possibility. It is important to note that this option is feasible only if the initial flap has been raised above the pericranium because skin grafts do not regularly “take” on bone without a preparatory step of removing the outer table of the skull and allowing granulation tissue to develop.
, 1997 Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Sarcoma Meta-analysis Collaboration. Lancet. 1997;350:1647-1654.
American Joint Committee on Cancer. Malignant melanoma of the skin. In AJCC Cancer Staging Manual, 5th ed, Philadelphia: Lippincott-Raven; 1997:163-170.
Ashton KJ, Carless MA, Griffiths LR. Cytogenetic alterations in nonmelanoma skin cancer: a review. Genes Chromosomes Cancer. 2005;43:239-248.
Brennan MF, Casper ES, Harrison LB. Soft tissue sarcoma. In: DeVita VTJr, Hellman S, Rosenberg SA, editors. Cancer: Principles and Practice of Oncology. 5th ed. Philadelphia: Lippincott-Raven; 1997:1738-1788.
Brown KM, Macgregor S, Montgomery GW, et al. Common sequence variants on 20q11.22 confer melanoma susceptibility. Nat Genet. 2008;40:838-840.
Butler GJ, Neale R, Green AC, et al. Nonsteroidal anti-inflammatory drugs and the risk of actinic keratoses and squamous cell cancers of the skin. J Am Acad Dermatol. 2005;53:966-972.
Byrd KM, Wilson DC, Hoyler SS, et al. Advanced presentation of melanoma in African Americans. J Am Acad Dermatol. 2004;50:21-24.
Cress RD, Holly EA. Incidence of cutaneous melanoma among non-Hispanic whites, Hispanics, Asians, and blacks: an analysis of California Cancer Registry data, 1988-93. Cancer Causes Control. 1997;8:246-252.
Demierre MF, Nathanson L. Chemoprevention of melanoma: an unexplored strategy. J Clin Oncol. 2003;21:158-165.
Dummer R, Hauschild A, Jost L. Cutaneous malignant melanoma: ESMO clinical recommendations for diagnosis, treatment and follow-up. Ann Oncol. 2008;19(suppl 2):ii86-88.
Fecher LA, Cummings SD, Keefe MJ, et al. Toward a molecular classification of melanoma. J Clin Oncol. 2007;25:1606-1620.
Geller AC, Miller DR, Annas GD, et al. Melanoma incidence and mortality among US whites, 1969-1999. JAMA. 2002;288:1719-1720.
Gurlek A, Alaybeyoglu N, Demir CY, et al. Aesthetic reconstruction of large scalp defects by sequential tissue expansion without interval. Aesthetic Plast Surg. 2004;28:245-250.
Haluska FG, Hodi FS. Molecular genetics of familial cutaneous melanoma. J Clin Oncol. 1998;16:670-682.
Kacker A, Antonescu CR, Shaha AR. Multifocal angiosarcoma of the scalp: a case report and review of the literature. Ear Nose Throat J. 1999;78:302-305.
PDQ—Kaposi’s sarcoma . PDQ—Kaposi’s sarcoma. National Cancer Institute Resource Page. Available at http://cancernet.nci.nih.gov/clinpdq/soa/Kaposi’s_sarcoma_Physician.html
Quinn AG, Sikkink S, Rees JL. Basal cell carcinomas and squamous cell carcinomas of human skin show distinct patterns of chromosome loss. Cancer Res. 1994;54:4756-4759.
Roewert-Huber J, Lange-Asschenfeldt B, Stockfleth E, et al. Epidemiology and aetiology of basal cell carcinoma. Br J Dermatol. 2007;157(suppl 2):47-51.
Rowe DE, Carroll RJ, Day CLJr. Prognostic factors for local recurrence, metastasis, and survival rates in squamous cell carcinoma of the skin, ear, and lip. Implications for treatment modality selection. J Am Acad Dermatol. 1992;26:976-990.
Salasche SJ. Epidemiology of actinic keratoses and squamous cell carcinoma. J Am Acad Dermatol. 2000;42:4-7.
Shumate CR, Carlson GW, Giacco GG, et al. The prognostic implications of location for scalp melanoma. Am J Surg. 1991;162:315-319.
Stockfleth E, Kerl H. Guidelines for the management of actinic keratoses. Eur J Dermatol. 2006;16:599-606.
Thomas RM, Amonette RA. Mohs micrographic surgery. Am Fam Physician. 1998;37:135-142.
Tsai KY, Tsao H. The genetics of skin cancer. Am J Med Genet C Semin Med Genet. 2004;131C:82-92.
Tsai S, Sabel MS. Translational research in melanoma. Surg Oncol Clin N Am. 2008;17:391-419. ix-x
Valverde P, Healy E, Jackson I, et al. Variants of the melanocyte-stimulating hormone receptor gene are associated with red hair and fair skin in humans. Nat Genet. 1995;11:328-330.
1 Salasche SJ. Epidemiology of actinic keratoses and squamous cell carcinoma. J Am Acad Dermatol. 2000;42:4-7.
2 Glogau RG. The risk of progression to invasive disease. J Am Acad Dermatol. 2000;42:23-24.
3 Tutrone WD, Saini R, Caglar S, et al. Topical therapy for actinic keratoses, II: diclofenac, colchicine, and retinoids. Cutis. 2003;71:373-379.
4 Tutrone WD, Saini R, Caglar S, et al. Topical therapy for actinic keratoses, I: 5-fluorouracil and imiquimod. Cutis. 2003;71:365-370.
5 Stockfleth E, Kerl H. Guidelines for the management of actinic keratoses. Eur J Dermatol. 2006;16:599-606.
6 Hsi ED, Svoboda-Newman SM, Stern RA, et al. Detection of human papillomavirus DNA in keratoacanthomas by polymerase chain reaction. Am J Dermatopathol. 1997;19:10-15.
7 Roewert-Huber J, Lange-Asschenfeldt B, Stockfleth E, et al. Epidemiology and aetiology of basal cell carcinoma. Br J Dermatol. 2007;157(suppl 2):47-51.
8 Binstock JH, Stegman SJ, Tromovitch TA. Large, aggressive basal-cell carcinomas of the scalp. J Dermatol Surg Oncol. 1981;7:565-569.
9 Gallagher RP, Hill GB, Bajdik CD, et al. Sunlight exposure, pigmentary factors, and risk of nonmelanocytic skin cancer. I. Basal cell carcinoma. Arch Dermatol. 1995;131:157-163.
10 Tsai KY, Tsao H. The genetics of skin cancer. Am J Med Genet C Semin Med Genet. 2004;131C:82-92.
11 Miller DL, Weinstock MA. Nonmelanoma skin cancer in the United States: incidence. J Am Acad Dermatol. 1994;30:774-778.
12 Diepgen TL, Mahler V. The epidemiology of skin cancer. Br J Dermatol. 2002;146(suppl 61):1-6.
13 Quinn AG, Sikkink S, Rees JL. Basal cell carcinomas and squamous cell carcinomas of human skin show distinct patterns of chromosome loss. Cancer Res. 1994;54:4756-4759.
14 Ziegler A, Jonason AS, Leffell DJ, et al. Sunburn and p53 in the onset of skin cancer. Nature. 1994;372:773-776.
15 Ashton KJ, Carless MA, Griffiths LR. Cytogenetic alterations in nonmelanoma skin cancer: a review. Genes Chromosomes Cancer. 2005;43:239-248.
16 Beahrs OH, Henson DE, Hutter RVP, et al, editors. American Joint Committee on Cancer Manual for Staging of Cancer, 4th ed, Philadelphia: J. B. Lippincott, 1992.
17 Carneiro RV, Sotto MN, Azevedo LS, et al. Acitretin and skin cancer in kidney transplanted patients. Clinical and histological evaluation and immunohistochemical analysis of lymphocytes, natural killer cells and Langerhans’ cells in sun exposed and sun protected skin. Clin Transplant. 2005;19:115-121.
18 Butler GJ, Neale R, Green AC, et al. Nonsteroidal anti-inflammatory drugs and the risk of actinic keratoses and squamous cell cancers of the skin. J Am Acad Dermatol. 2005;53:966-972.
19 Wollina U, Hansel G, Koch A, et al. Oral capecitabine plus subcutaneous interferon alpha in advanced squamous cell carcinoma of the skin. J Cancer Res Clin Oncol. 2005;131:300-304.
20 Rowe DE, Carroll RJ, Day CLJr. Prognostic factors for local recurrence, metastasis, and survival rates in squamous cell carcinoma of the skin, ear, and lip. Implications for treatment modality selection. J Am Acad Dermatol. 1992;26:976-990.
21 Friedman RJ, Rigel DS, Nossa R. Basal cell and squamous cell carcinoma of the skin. In: Murphy GP, Lawrence W, Lenhard RE, editors. American Cancer Society Textbook of Clinical Oncology. 2nd ed. Atlanta: American Cancer Society; 1995:330-341.
22 Thomas RM, Amonette RA. Mohs micrographic surgery. Am Fam Physician. 1998;37:135-142.
23 Luce EA. Oncologic considerations in nonmelanotic skin cancer. Clin Plast Surg. 1995;22:39-50.
24 Green A, Swerdlow AJ. Epidemiology of melanocytic nevi. Epidemiol Rev. 1989;11:204-221.
25 Greene MH, Clark WHJr, Tucker MA, et al. Acquired precursors of cutaneous malignant melanoma. The familial dysplastic nevus syndrome. N Engl J Med. 1985;312:91-97.
26 Fraser MC, Goldstein AM, Tucker MA. The genetics of melanoma. Semin Oncol Nurs. 1997;13:108-114.
27 Geller AC, Miller DR, Annas GD, et al. Melanoma incidence and mortality among US whites, 1969-1999. JAMA. 2002;288:1719-1720.
28 Fecher LA, Cummings SD, Keefe MJ, et al. Toward a molecular classification of melanoma. J Clin Oncol. 2007;25:1606-1620.
29 Demierre MF, Nathanson L. Chemoprevention of melanoma: an unexplored strategy. J Clin Oncol. 2003;21:158-165.
30 Rhodes AR, Weinstock MA, Fitzpatrick TB, et al. Risk factors for cutaneous melanoma. A practical method of recognizing predisposed individuals. JAMA. 1987;258:3146-3154.
31 Cress RD, Holly EA. Incidence of cutaneous melanoma among non-Hispanic whites, Hispanics, Asians, and blacks: an analysis of California Cancer Registry data, 1988-93. Cancer Causes Control. 1997;8:246-252.
32 Tucker MA, Halpern A, Holly EA, et al. Clinically recognized dysplastic nevi. A central risk factor for cutaneous melanoma. JAMA. 1997;277:1439-1444.
33 Rodenas JM, Delgado-Rodriguez M, Farinas-Alvarez C, et al. Melanocytic nevi and risk of cutaneous malignant melanoma in southern Spain. Am J Epidemiol. 1997;145:1020-1029.
34 Haluska FG, Hodi FS. Molecular genetics of familial cutaneous melanoma. J Clin Oncol. 1998;16:670-682.
35 Lowe SW, Sherr CJ. Tumor suppression by Ink4a-Arf: progress and puzzles. Curr Opin Genet Dev. 2003;13:77-83.
36 Bishop DT, Demenais F, Goldstein AM, et al. Geographical variation in the penetrance of CDKN2A mutations for melanoma. J Natl Cancer Inst. 2002;94:894-903.
37 Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949-954.
38 James MR, Roth RB, Shi MM, et al. BRAF polymorphisms and risk of melanocytic neoplasia. J Invest Dermatol. 2005;125:1252-1258.
39 Valverde P, Healy E, Jackson I, et al. Variants of the melanocyte-stimulating hormone receptor gene are associated with red hair and fair skin in humans. Nat Genet. 1995;11:328-330.
40 Kennedy C, ter Huurne J, Berkhout M, et al. Melanocortin 1 receptor (MC1R) gene variants are associated with an increased risk for cutaneous melanoma which is largely independent of skin type and hair color. J Invest Dermatol. 2001;117:294-300.
41 Easton DF. Consortium TBCL: cancer risks in BRCA2 mutation carriers. J Natl Cancer Inst. 1999;91:1310-1316.
42 Brown KM, Macgregor S, Montgomery GW, et al. Common sequence variants on 20q11.22 confer melanoma susceptibility. Nat Genet. 2008;40:838-840.
43 Shumate CR, Carlson GW, Giacco GG, et al. The prognostic implications of location for scalp melanoma. Am J Surg. 1991;162:315-319.
44 Swetter SM, Boldrick JC, Jung SY, et al. Increasing incidence of lentigo maligna melanoma subtypes: northern California and national trends 1990-2000. J Invest Dermatol. 2005;125:685-691.
45 Byrd KM, Wilson DC, Hoyler SS, et al. Advanced presentation of melanoma in African Americans. J Am Acad Dermatol. 2004;50:21-24.
46 Breslow A. Thickness, cross-sectional areas and depth of invasion in the prognosis of cutaneous melanoma. Ann Surg. 1970;172:902-908.
47 Clark WHJr, From L, Bernardino EA, et al. The histogenesis and biologic behavior of primary human malignant melanomas of the skin. Cancer Res. 1969;29:705-727.
48 American Joint Committee on Cancer. Malignant melanoma of the skin. In AJCC Cancer Staging Manual, 5th ed, Philadelphia: Lippincott-Raven; 1997:163-170.
49 Agarwala SS, Glaspy J, O’Day SJ, et al. Results from a randomized phase III study comparing combined treatment with histamine dihydrochloride plus interleukin-2 versus interleukin-2 alone in patients with metastatic melanoma. J Clin Oncol. 2002;20:125-133.
50 NIH Consensus Development Panel on Early Melanoma. Diagnosis and treatment of early melanoma. JAMA. 1992;268:1314-1319.
51 Balch CM, Urist MM, Karakousis CP, et al. Efficacy of 2-cm surgical margins for intermediate-thickness melanomas (1 to 4 mm). Results of a multi-institutional randomized surgical trial. Ann Surg. 1993;218:262-267.
52 Veronesi U, Cascinelli N, Adamus J, et al. Thin stage I primary cutaneous malignant melanoma. Comparison of excision with margins of 1 or 3 cm. N Engl J Med. 1988;318:1159-1162.
53 Ringborg U, Andersson R, Eldh J, et al. Resection margins of 2 versus 5 cm for cutaneous malignant melanoma with a tumor thickness of 0.8 to 2.0 mm: randomized study by the Swedish Melanoma Study Group. Cancer. 1996;77:1809-1814.
54 Veronesi U, Adamus J, Bandiera DC, et al. Inefficacy of immediate node dissection in stage 1 melanoma of the limbs. N Engl J Med. 1977;297:627-630.
55 Veronesi U, Adamus J, Bandiera DC, et al. Delayed regional lymph node dissection in stage I melanoma of the skin of the lower extremities. Cancer. 1982;49:2420-2430.
56 Sim FH, Taylor WF, Ivins JC, et al. A prospective randomized study of the efficacy of routine elective lymphadenectomy in management of malignant melanoma. Preliminary results. Cancer. 1978;41:948-956.
57 Balch CM, Soong SJ, Bartolucci AA, et al. Efficacy of an elective regional lymph node dissection of 1 to 4 mm thick melanomas for patients 60 years of age and younger. Ann Surg. 1996;224:255-263.
58 Kirkwood JM, Strawderman MH, Ernstoff MS, et al. Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group Trial EST 1684. J Clin Oncol. 1996;14:7-17.
59 Kirkwood JM, Ibrahim JG, Sondak VK, et al. High- and low-dose interferon alfa-2b in high-risk melanoma: first analysis of intergroup trial E1690/S9111/C9190. J Clin Oncol. 2000;18:2444-2458.
60 Kirkwood JM, Ibrahim JG, Sosman JA, et al. High-dose interferon alfa-2b significantly prolongs relapse-free and overall survival compared with the GM2-KLH/QS-21 vaccine in patients with resected stage IIB-III melanoma: results of intergroup trial E1694/S9512/C509801. J Clin Oncol. 2001;19:2370-2380.
61 Eggermont AM, Suciu S, Santinami M, et al. Adjuvant therapy with pegylated interferon alfa-2b versus observation alone in resected stage III melanoma: final results of EORTC 18991, a randomised phase III trial. Lancet. 2008;372:117-126.
62 Dummer R, Hauschild A, Jost L. Cutaneous malignant melanoma: ESMO clinical recommendations for diagnosis, treatment and follow-up. Ann Oncol. 2008;19(suppl 2):ii86-88.
63 Berwick M, Halpern A. Melanoma epidemiology. Curr Opin Oncol. 1997;9:178-182.
64 Tsai S, Sabel MS. Translational research in melanoma. Surg Oncol Clin N Am. 2008;17:391-419. ix-x
65 Milward MJ, Bedikian AY, Conry RM, et al. Randomized multination phase III trial of DTIC with or without oblimersen sodium in patients with advanced malignant melanoma. Proc Am Soc Clin Oncol. 2004.
66 Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335-2342.
67 Carson WE, Biber JE, Shah N, et al. A phase II trial of recombinant humanized monoclonal anti-vascular endothelial growth factor (VEGF) antibody in patients with malignant melanoma. Proc Am Soc Clin Oncol. 2003.
68 Reiriz AB, Richter MF, Fernandes S, et al. Phase II study of thalidomide in patients with metastatic malignant melanoma. Melanoma Res. 2004;14:527-531.
69 Hwu WJ, Lis E, Menell JH, et al. Temozolomide plus thalidomide in patients with brain metastases from melanoma: a phase II study. Cancer. 2005;103:2590-2597.
70 Attia P, Phan GQ, Maker AV, et al. Autoimmunity correlates with tumor regression in patients with metastatic melanoma treated with anti–cytotoxic T-lymphocyte antigen-4. J Clin Oncol. 2005;23:6043-6053.
71 Meisenberg BR, Ross M, Vredenburgh JJ, et al. Randomized trial of high-dose chemotherapy with autologous bone marrow support as adjuvant therapy for high-risk, multi-node–positive malignant melanoma. J Natl Cancer Inst. 1993;85:1080-1085.
72 Brennan MF, Casper ES, Harrison LB. Soft tissue sarcoma. In: DeVita VTJr, Hellman S, Rosenberg SA, editors. Cancer: Principles and Practice of Oncology. 5th ed. Philadelphia: Lippincott-Raven; 1997:1738-1788.
73 O’Byrne K, Steward WP. The role of adjuvant chemotherapy in the treatment of adult soft tissue sarcomas. Crit Rev Oncol Hematol. 1998;27:221-227.
74 Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Sarcoma Meta-analysis Collaboration. Lancet. 1997;350:1647-1654.
75 Holden CA. Histogenesis of Kaposi’s sarcoma and angiosarcoma of the face and the scalp. J Invest Dermatol. 1989;93:119S-124S.
76 Kacker A, Antonescu CR, Shaha AR. Multifocal angiosarcoma of the scalp: a case report and review of the literature. Ear Nose Throat J. 1999;78:302-305.
77 Mark RJ, Tran LM, Sercarz J, et al. Angiosarcoma of the head and neck. The UCLA experience 1955 through 1990. Arch Otolaryngol Head Neck Surg. 1993;119:973-978.
78 Ohguri T, Imada H, Nomoto S, et al. Angiosarcoma of the scalp treated with curative radiotherapy plus recombinant interleukin-2 immunotherapy. Int J Radiat Oncol Biol Phys. 2005;61:1446-1453.
79 . PDQ—Kaposi’s sarcoma. National Cancer Institute Resource Page. Available at http://cancernet.nci.nih.gov/clinpdq/soa/Kaposi’s_sarcoma_Physician.html
80 Jackson IT. Local Flaps in Head and Neck Reconstruction. St. Louis: CV Mosby; 1985.
81 Antonyshyn O, Gruss JS, Zuker R, et al. Tissue expansion in head and neck reconstruction. Plast Reconstr Surg. 1988;82:58-68.
82 Gurlek A, Alaybeyoglu N, Demir CY, et al. Aesthetic reconstruction of large scalp defects by sequential tissue expansion without interval. Aesthetic Plast Surg. 2004;28:245-250.






