[level-membership-for-neurosurgery-category]
CHAPTER 151 Sarcoidosis, Tuberculosis, and Xanthogranuloma
Sarcoidosis
Sarcoidosis is a chronic, multisystem granulomatous disease of unknown cause. It can occur in any racial or ethnic population and most often develops in persons between the ages of 20 and 40 years. In the United States, its prevalence ranges from 10 to 40 per 100,000, with an approximately 10 : 1 ratio of black to white patients and a slight female preponderance.1,2 Any organ system can be involved in sarcoidosis, with the lungs, lymph nodes, skin, and eyes being the most common. Five percent of patients have neurological manifestations.3–8 Of these patients with neurosarcoidosis (NS), 50% have neurological problems at the time of initial diagnosis. In a third of these patients, more than one neurological manifestation of their disease is already present or will develop.
Histology and Pathophysiology
Although the precise cause of sarcoidosis is not known, there is evidence that the disease results from dysregulation of the cellular immune responses to foreign antigens or self-antigens, thereby leading to granuloma formation.1 These granulomas are composed of epithelioid cells, histiocytes, T cells, monocytes, and fibroblasts. Granuloma formation can ultimately lead to fibrosis and organ dysfunction.
Clinical Features
Cranial Neuropathy
Cranial neuropathy is the most frequently encountered neurological manifestation of sarcoidosis; it occurs in approximately 75% of patients with NS.8 More than 50% of patients have involvement of multiple cranial nerves.
Dysfunction of the facial nerve is the most common cranial neuropathy seen in NS, and it develops in more than 50% of patients.8 The peripheral palsy can be unilateral or bilateral and is often transient.
NS can occur in the subfrontal region and cause an olfactory neuropathy and anosmia. Involvement of the optic nerve is relatively common and is seen in as many as 5% of patients.8 Funduscopic examination can demonstrate optic atrophy, optic disc swelling or papilledema, or retrobulbar optic neuritis.9 In patients with visual or optic abnormalities, however, it must be remembered that sarcoidosis affects the eye as a uveitis far more often than the optic nerve.
Meningitis
Symptoms of meningeal irritation, including headache, nuchal rigidity, nausea, and rarely, fever, have been reported to occur in 3% to 26% of patients with NS.8 Symptomatic meningeal involvement can take the form of either acute aseptic meningitis or chronic meningitis. Cerebrospinal fluid (CSF) studies generally reveal a mild mononuclear pleocytosis with increased protein. Glucose may be normal or decreased, with no evidence of bacteria. Given the extent of meningeal involvement seen in most patients with NS, symptomatic meningitis is relatively rare. Occasionally, meningeal-based granulomatous mass lesions can develop.
Pituitary and Hypothalamic Sarcoidosis
When sarcoidosis involves the CNS parenchyma, it most commonly affects the hypothalamus and parasellar region.10 This can result in vegetative alterations and neuroendocrinologic dysfunction. Symptoms include disturbances in sleep, appetite, thirst, temperature, and libido. Hypothalamic and pituitary lesions can also cause thyroid, gonadal, and adrenal abnormalities. Altered thirst, antidiuretic hormone deficiency or excess, and hyperprolactinemia have been reported.
Space-Occupying Lesions
Granulomatous masses forming localized space-occupying lesions can occur throughout the CNS but are most frequently found in the cerebral hemispheres.6 They can be manifested as a large, solitary mass mimicking either a primary or metastatic brain tumor.11,12 Alternatively, they can appear as multiple nodules.8 Finally, they can take the form of a dural-based plaque-like mass resembling a meningioma.13–16
Seizures
Generalized or partial seizures have been reported in as many as 22% of patients with NS.17 They are thought to be caused by supratentorial parenchymal involvement in which the granulomatous inflammation is often found in a perivascular distribution.
Hydrocephalus
Hydrocephalus secondary to obstruction of normal CSF flow is a relatively common and potentially lethal clinical feature of NS.18–20 Communicating hydrocephalus can result from extensive meningeal involvement causing decreased CSF resorption through the arachnoid villi. Obstructive hydrocephalus can develop secondary to an intracranial mass or inflammation preventing the flow of CSF through the foramen of Monro, aqueduct of Sylvius, or foramen of Luschka or Magendie.21,22
Spinal Cord Sarcoidosis
Sarcoidosis of the spinal cord is relatively rare. It can occur at any segment of the spinal cord, as well as at the cauda equina, and the granulomatous lesions may be intramedullary or extramedullary.23–30
Peripheral Neuropathy
Peripheral neuropathic manifestations include mononeuropathy, mononeuritis multiplex, and generalized sensory, sensorimotor, and motor neuropathies.31,32 Patients can have a small-fiber sensory or autonomic neuropathy.33 Clinical features resembling Guillain-Barré syndrome have also been described. Nerve biopsy shows granuloma formation in both the epineurial and perineurial spaces, as well as axonal degeneration. There can be associated vascular inflammation, thus suggesting that ischemia may play a role in the nerve damage.
Diagnostic and Imaging Studies
The diagnosis of sarcoidosis is firmly established only when the clinical and paraclinical findings are supported by histologic evidence of widespread noncaseating granulomas.1,2 When neurological findings develop in a patient with sarcoidosis, NS should be considered, although an intercurrent infection or malignancy must be excluded. The following diagnostic criteria for NS are adapted from Zajicek and colleagues36:
When NS is suspected, the patient should be evaluated for evidence of systemic disease and a search for a site suitable for biopsy undertaken. Because corticosteroids can eliminate evidence of systemic inflammation, they should be withheld until the diagnostic evaluation is completed unless severe illness requires their use. The search for systemic sarcoidosis should begin with an examination of the skin and lymph nodes.1,2 Chest radiographs are abnormal in approximately 90% of patients with sarcoidosis, but frequently, additional diagnostic information can be obtained from a computed tomography (CT) scan of the thorax. Serum angiotensin-converting enzyme (ACE) levels can be elevated in patients with sarcoidosis. However, infection or malignancy can cause serum ACE levels to be high, and in patients with isolated NS, serum ACE levels may be normal.37,38 Ophthalmologic examination can reveal uveitis or retinal vasculopathy, as well as conjunctival nodules. Endoscopic nasal and sinus examination can show evidence of mucosal inflammation.
CSF abnormalities occur in NS, but they tend to be nonspecific and thus are not a reliable marker of the disease. CSF opening pressure is elevated in approximately 10% of patients, and protein is increased in two thirds of patients. CSF glucose can be normal or low. Approximately 50% of patients have a predominantly mononuclear pleocytosis. The immunoglobulin G index may be elevated, and oligoclonal bands may be present.6,8,39 The concentration of ACE in CSF may be elevated, but this finding is not specific to NS.40 There are reports of patients deteriorating after lumbar puncture.20
Gallium scanning can be informative; uptake in the lungs and salivary, parotid, and lacrimal glands is suggestive of sarcoidosis and, when a distinctive pattern is found in combination with elevated serum ACE levels, may be highly specific (>95%) for sarcoidosis.41 Whole-body fluorodeoxyglucose positron emission tomography (FDG-PET) is a sensitive technique for highlighting areas of inflammation.42
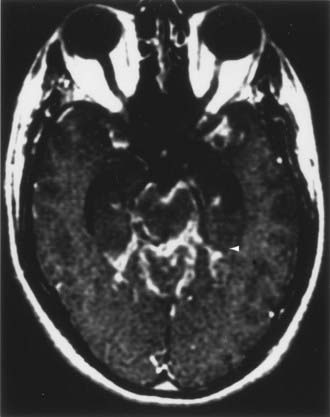
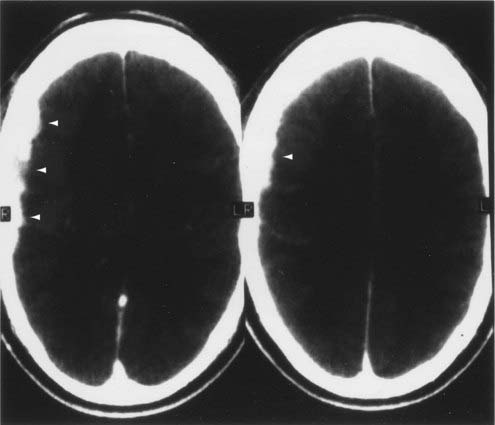
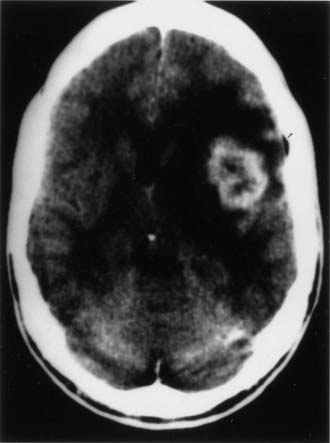
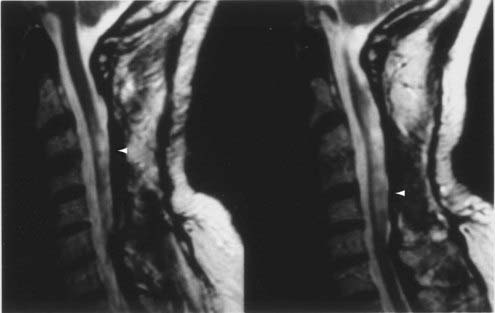
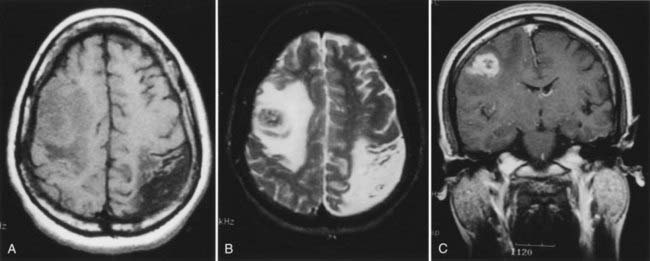
CT and magnetic resonance imaging (MRI) enhance detection of meningeal (Figs. 151-1 and 151-2) and parenchymal disease (Figs. 151-3 and 151-4), even in patients without any neurological findings.
On CT, the granulomatous lesions of NS are usually isodense or hyperdense on non–contrast-enhanced studies and uniformly enhancing after the administration of contrast medium.8,43 CT can reveal bony erosion of the skull base caused by NS lesions. The orbital contents, including the optic nerve and extraocular muscles, can also be visualized, particularly with sagittal and coronal reconstructions.
The imaging procedure of choice for NS is MRI with gadolinium enhancement.44–47 This test is particularly sensitive for detecting disease of the parasellar region, basal cisterns, and spinal cord. On T1-weighted images, NS lesions are usually hyperintense in relation to normal brain parenchyma, and they enhance after the administration of gadolinium. T2-weighted images often show minimal edema surrounding the lesions, although this finding can be variable and may be related to disease activity. A wide spectrum of NS abnormalities can be seen on MRI and can mimic other neurological diseases. Gadolinium-enhanced MRI can reveal both diffuse (see Fig. 151-1) and local (see Fig. 151-2) leptomeningeal involvement.47,48 The dura often shows marked thickening along with evidence of inflammation (pachymeningitis). Parasellar lesions and intraparenchymal masses (see Fig. 151-3) can also be readily identified, but the latter can easily be mistaken for a neoplasm.49 Involvement of the optic nerves or other cranial nerves can likewise be documented. MRI can identify hydrocephalus, and the presence of transependymal edema on T2-weighted images can be a useful indicator of increased intracranial pressure secondary to obstruction of CSF flow. Finally, MRI can be valuable in the detection of sarcoidosis involving the spinal cord (see Fig. 151-4) and cauda equina.50
In addition to its utility in identifying lesions consistent with NS, gadolinium-enhanced MRI can be used to monitor the efficacy of NS treatment.51 Decreased enhancement and regression of the size of lesions suggest therapeutic efficacy.
Biopsy of the brain, spinal cord, meninges, nerve, or muscle is occasionally performed to confirm a diagnosis of NS in patients with known sarcoidosis who have a neurological problem consistent with NS. Biopsy should also be considered in patients with known systemic sarcoidosis and neurological disease who are progressively deteriorating despite “optimal” treatment to uncover an alternative diagnosis.52 Importantly, biopsy can support a diagnosis of NS in patients in whom the diagnosis is suspected but for which there is no confirmatory systemic documentation. Occasionally, NS is the unexpected consequence of a CNS biopsy, for instance, in a patient with a dural-based mass thought to be a meningioma but on biopsy granulomatous inflammation is found.
Treatment
Because of the relative rarity of NS, there have been no rigorous clinical trials to determine optimal treatment. Given the pathophysiology of sarcoidosis, however, immunosuppressive therapy has been the cornerstone of therapy.6,8
Corticosteroids are the first-line agents for the treatment of NS. The site of neurological involvement, disease severity, and response to treatment generally dictate the dose and duration of corticosteroid therapy. For example, patients with facial nerve palsy or aseptic meningitis are often treated with prednisone, 0.5 mg/kg per day for 2 weeks; patients with myopathy or neuropathy are generally treated with prednisone, 0.5 mg/kg per day for 4 weeks, and then reassessed, with most patients requiring chronic therapy; and patients with a meningeal or parenchymal mass lesion, encephalopathy, or symptomatic hydrocephalus usually require prednisone, 0.5 to 1 mg/kg per day for 4 weeks, before any improvement is appreciated, and most require chronic therapy. Once the acute exacerbation is under control, corticosteroid administration can usually be slowly tapered. Frequent clinical evaluation and, for CNS disease, periodic MRI to assess the extent of enhancement can be helpful in guiding therapy.51,53
Patients who deteriorate despite aggressive corticosteroid therapy, who cannot tolerate corticosteroids, or who have a contraindication to corticosteroids may benefit from alternative therapeutic agents.54–56 Mycophenolate mofetil, cyclosporine,57 azathioprine, methotrexate,58,59 chlorambucil, cyclophosphamide,55 hydroxychloroquine,60 pentoxifylline,61,62 thalidomide,63,64 and infliximab and adalimumab65–68 have all been used to treat NS. There are no studies comparing the efficacy of these alternative treatments in patients with NS. Consequently, selection of agents is often based on ease of use, cost, and desire to avoid the complications of a particular drug. Because a patient’s response to a specific agent cannot be predicted, two or three different agents should be tried before concluding that the patient’s disease is refractory.
Cranial or spinal irradiation has been used for refractory NS and should be considered in patients who fail corticosteroid therapy and trials of alternative agents.69,70 Radiotherapy has also been used in patients with acute, life-threatening disease.
Intracranial and intraspinal mass lesions generally respond well to corticosteroids or alternative agents if the disease has been caught early, before the development of significant ischemia or fibrosis.6,8 Thus, in patients with known systemic sarcoidosis in whom a mass lesion develops, corticosteroid therapy should be initiated promptly as long as there are no significant contraindications and infection and malignancy have been reasonably excluded as diagnoses. In patients without extensive systemic disease, treatment of an intracranial or intraspinal mass can be complicated by the imaging similarities of sarcoidosis, tuberculosis, fungal disease, and neoplasm.43 In patients without known sarcoidosis, it may be necessary to perform a biopsy at the primary site of involvement to determine the nature of a mass lesion. During surgery, effort should be made to minimize disruption of the surrounding parenchyma because NS can be infiltrative and patients can deteriorate after surgical intervention. If the biopsy reveals noncaseating granulomas, further resection should be deferred while appropriate medical management is pursued. Only in highly refractory cases in which a mass lesion persists or enlarges despite optimal immunosuppressive therapy should surgical debulking or resection be considered. The results of these aggressive surgical interventions are often unsatisfactory.71
Patients with chronic, asymptomatic hydrocephalus can be observed clinically, and surgical intervention can be offered if the hydrocephalus worsens or if the patient deteriorates despite medical therapy. These patients must be watched closely, however, because abrupt, life-threatening deterioration can occur. In patients who respond to medical therapy, improvement in CSF flow can occur over time.72
Conclusion
The long-term course of NS has not been clearly defined. Approximately two thirds of patients have a monophasic illness, and the others have either a relapsing-remitting course or progressive disease.72 As many as 10% of NS patients die as a result of the inflammatory disease process or its treatment. These patients typically have CNS parenchymal disease or hydrocephalus. Prompt diagnosis of NS can lead to earlier treatment, and this may improve the outcome.
Tuberculosis
Tuberculosis (TB), a worldwide disease, experienced a resurgence in the United States in the early 1990s, largely related to the human immunodeficiency virus (HIV) epidemic, immigration from countries endemic for TB, transmission of TB in settings such as hospitals and prisons, and the development of multidrug-resistant strains of TB. TB is primarily a disease of the lungs, but extrapulmonary involvement is common. CNS involvement is reported in as many as 10% of immunocompetent patients with TB.73 CNS TB is a defining condition of acquired immunodeficiency syndrome (AIDS), and both TB and CNS involvement are more prevalent in the HIV-infected population.74 CNS involvement can take the form of tuberculous meningitis, intracranial and spinal cord tuberculomas, abscesses, and Pott’s disease. Here, the focus is primarily on neurosurgical management of CNS tuberculomas and abscesses.
Pathogenesis
CNS TB is almost entirely due to infection with Mycobacterium tuberculosis. The bacterium is transmitted by inhalation of aerosolized droplets, and as few as 1 to 10 organisms are sufficient to cause infection. As the bacteria multiply in alveoli and macrophages, a cell-mediated immune response ensues and generates a granulomatous reaction characterized by a tubercle and caseating necrosis. If the infection is not contained, bacteria can spread hematogenously to distant sites such as the CNS. The inciting CNS tubercle is called a Rich focus.75 The location of tuberculous lesions in the brain is related to the pattern of blood flow, and lesions generally involve the corticomedullary junction and periventricular regions. They usually occur in the cerebral hemispheres and basal ganglia in adults and in the cerebellar hemispheres in children; brainstem and spinal lesions are rare.76 Tubercles that rupture into the subarachnoid space cause tuberculous meningitis, whereas deep-seated tubercles cause tuberculomas or abscesses. The immune reaction around a tuberculous focus can also cause vascular inflammation, vasculitis, and edema. Vasculitis can lead to ischemia, as well as poor delivery of drug to affected areas. Furthermore, hydrocephalus is common in CNS TB, especially in children.
Tuberculomas are usually solitary lesions.77 They are composed of a necrotic caseous center surrounded by a capsule consisting of fibroblasts, epithelioid cells, Langhans giant cells, and lymphocytes. This composition gives tuberculomas a firmness that is different from pyogenic abscesses or malignant gliomas. Calcification can occur in a concentric ring at the margins of the caseous core, as well as at the center of the core, and result in a target sign on CT. Supratentorial tuberculomas are commonly deep, but they can be dural based and mimic meningiomas.78–80 Additionally, cerebellar tuberculomas are frequently in contact with the pia, which makes them difficult to remove without contamination of the subarachnoid space.81
When the caseous core of a tuberculoma liquefies, a tuberculous abscess results. Tuberculous abscesses are usually larger and produce more edema than tuberculomas do. In contrast to tuberculomas, which may not consistently produce positive TB cultures, the liquefied core of an abscess usually contains an odorless, green purulence teeming with acid-fast bacilli. Additionally, abscess walls generally have less granulomatous reaction, although this finding is variable.82 These features give tuberculous abscesses an appearance similar to pyogenic abscesses on imaging studies.
Clinical Features
Because tuberculomas usually represent reactivation of a latent tuberculous focus, patients generally do not have active TB symptoms when initially seen. For instance, fever was reported in only 1 of 18 patients with tuberculomas.81 Another report found that just 7 of 12 patients with tuberculomas had signs of extracranial TB at initial evaluation.83 Tuberculous meningitis has been reported to appear simultaneously with tuberculomas in 3% of cases.84 Most commonly, tuberculomas appear as space-occupying lesions in the CNS. Because they are distributed hematogenously, they can occur anywhere and thus have a spectrum of clinical characteristics mimicking brain tumors. Seizures are one of the most common symptoms and occur in up to 85% of patients.81,83,84 Signs and symptoms related to elevated intracranial pressure are common. Symptoms of focal mass lesions occur in about 70% of patients with supratentorial and cerebellar lesions and in 100% of patients with brainstem lesions.84 Tuberculomas have been unexpected findings during surgery for cerebral gliomas, acoustic schwannomas, cavernous sinus meningiomas, and pituitary adenomas.85 Tuberculous abscesses usually have a more accelerated time course and are commonly associated with fever, headache, and focal neurological signs.
Diagnostic and Imaging Studies
CNS tuberculomas account for 0.5% to 2% of all space-occupying intracranial lesions in Western countries.81,86 In developing countries, 10% to 30% of intracranial masses are tuberculomas.76 The diagnosis of CNS tuberculoma should be considered in anyone with a CNS mass lesion who also has risk factors for TB. Such risk factors include emigration from TB-endemic areas, history of pulmonary TB, and exposure to populations with high rates of TB. Absence of these risk factors does not eliminate the possibility of TB, however, because less than half of patients with tuberculomas have a history of TB.84
Laboratory studies may aid in the diagnosis of TB, but they have only intermediate sensitivity and specificity, especially in immunocompromised patients. The tuberculin skin test is positive in up to 85% of immunocompetent patients,83,87,88 but immunocompromised patients may be anergic. Chest radiographs show signs of TB in 30% to 60% of patients.81,83,89 Mature tuberculomas are thought to be isolated from the subarachnoid space because of a thick capsule, although tuberculomas coexist with tuberculous meningitis in 3% to 10% of patients.84,90 Thus, CSF studies may be helpful in making the diagnosis of TB. CSF studies typically show a mild, nonspecific increase in protein content with normal or low glucose. M. tuberculosis is notoriously difficult to culture, and cultures may take days or weeks to become positive. The sensitivity of CSF TB polymerase chain reaction for the diagnosis of CNS infection is approximately 60% to 70% with a specificity of 97% to 100%.91,92 It is imperative to treat empirically when the suspicion of TB is high while the results of diagnostic studies are awaited.
Tuberculomas are multiple in 10% to 25% of patients.84,85,87 The appearance of tuberculomas on CT depends in part on the maturity of the tuberculoma. Immature tuberculomas are initially nonenhancing with low attenuation on CT.93 They are rarely detected at this stage, however, because they are seldom symptomatic. At this early stage, they can be confused with a low-grade glioma, infarction, or cholesteatoma. As the granuloma forms, tuberculomas become isointense or slightly hyperdense on CT, with solid, ring, or mixed contrast enhancement. A circumscribed nodule with a central spot of radiolucency is highly suggestive of caseation and tuberculoma.93 Central calcification in a ring-enhancing lesion, or the “target sign,”94 is suggestive of but not pathognomonic for tuberculoma.95 Healed tuberculomas often leave calcifications. Based on the size of the lesion and the degree of midline shift, it is possible to differentiate tuberculomas from the small granulomas of cysticercosis;96 however, CT alone may not be specific enough to diagnose tuberculomas. CT has high sensitivity but a positive predictive value of only 33% for diagnosing CNS tuberculomas when used with the clinical history.97 Tuberculous abscesses are hypodense and ring enhancing on CT, with findings similar to those of pyogenic abscesses. Because TB abscesses behave more aggressively, edema and mass effect are common.
On MRI, tuberculomas are typically isodense (to gray matter) centrally on T1-weighted images and isointense to hypointense on T2-weighted images.98,99 An example of this appearance is shown in Figure 151-5. This central isointensity (or mixed isointensity and hyperintensity) has been correlated with caseous necrosis.100 A slightly hyperintense rim is commonly seen on T1-weighted images and corresponds to the fibrous collagen capsule of a mature tuberculoma. A second rim of hypointense signal has also been reported and is thought to correspond to a layer of outer inflammatory infiltrates. Tuberculomas typically show ring enhancement with gadolinium. The enhancement pattern can be a single ring or a series of concentric rings. Mature tuberculomas can be differentiated from pyogenic abscesses by the central hyperintensity usually seen in pyogenic abscesses on T2-weighted images. However, noncaseating tuberculous lesions can be hyperintense on T2-weighted images, with nodular enhancement. Tuberculous abscesses similarly show a central area of hyperintensity on T2-weighted images. The variability in appearance of tuberculomas and tuberculous abscesses on MRI and CT sometimes makes them difficult to differentiate from neoplasms, pyogenic abscesses, and other granulomatous diseases such as sarcoidosis, cysticercosis, and toxoplasmosis.
Because of the difficulty in diagnosing and differentiating tuberculomas from other processes, CT-guided stereotactic biopsy is advocated. Typically, if a patient does not respond to empirical TB chemotherapy within 6 to 8 weeks, stereotactic biopsy is indicated.101–103 Although earlier series reported a low yield rate for the diagnosis of TB, more recent series report a diagnostic yield as high as 85%.104 Histopathologic evidence of TB includes epithelioid cell granulomas with or without Langhans giant cells and caseous necrosis. M. tuberculosis is generally difficult to grow from cultures of these biopsy specimens. Mature tuberculomas typically have a tough fibrous capsule that may be difficult to penetrate with the blunt stereotactic equipment. In these cases, the resulting biopsy sample may be representative of the brain tissue surrounding the tuberculoma. With improved stereotactic techniques, CT-guided biopsy may be a safe and effective means of obtaining a tissue diagnosis of TB. Such confirmation of TB would allow a more directed approach to treatment and follow-up.
Treatment
The mainstay of therapy for CNS tuberculoma is medical management.105 Because tuberculomas exert a mass effect with associated edema, corticosteroids are important in their management. First-line anti-TB drugs include isoniazid, rifampin, pyrazinamide, and ethambutol; these drugs are administered for 2 months, followed by 9 to 12 months of isoniazid and rifampin. However, a study that followed the evolution of tuberculomas by CT suggested that a shorter course may be sufficient. When resistance is suspected, at least four drugs—and sometimes five or six—are necessary until sensitivities can be determined. With optimal medical therapy, mortality is generally less than 10%.
If the patient is stable neurologically, medical therapy is usually given a 2-month trial.106 This period is necessary because reports show that medical management of intracranial tuberculomas sometimes leads to initial paradoxical expansion of the lesion.107–109 This atypical response is thought to occur from a heightened immune response, the HIV-infected patient immune reconstitution syndrome,110 around the tuberculoma capsule as immunogenic bacilli proteins are liberated through the destruction of mycobacteria by chemotherapeutic agents. Furthermore, the immune reaction around the tuberculoma can lead to a perilesional granulomatous vasculitis, thus making it more difficult for tuberculostatic drugs to reach the tuberculoma. In these cases, corticosteroids are helpful in managing the immune-mediated progression of tuberculous lesions. Antituberculous therapy should be continued; these patients are not considered “treatment failures.”110
Although conservative therapy is effective, tuberculomas are space-occupying lesions, and accordingly, they can cause a mass effect, midline shift, and hydrocephalus. CSF diversion with ventricular shunts is occasionally necessary in patients with hydrocephalus secondary to basilar meningitis or obstructive tuberculomas. Surgical resection is generally indicated for tuberculomas that do not respond to medical therapy and for large, solitary lesions.85,104 With the use of perioperative antituberculous medication and corticosteroids, surgery for tuberculomas is relatively safe and effective.
Because tuberculous abscesses usually have a more rapid clinical course than tuberculomas do, the mainstay of therapy for abscesses is early surgical excision with a full course of anti-TB therapy.111,112 A second option is stereotactic aspiration with a course of anti-TB therapy.113,114 With advances in stereotactic techniques, stereotactic aspiration successfully treats tuberculous abscesses. Stereotactic aspiration is an important option for patients with deep-seated and surgically inaccessible lesions. After aspiration, patients must be monitored carefully because repeat aspirations are sometimes needed for multiloculated collections or because of reaccumulation of the abscess.
Xanthogranuloma
Xanthogranulomas of the CNS represent a group of uncommon and poorly understood tumefactions. Xanthogranulomas are generally associated with the choroid plexus of the lateral ventricles but can also occur in the third ventricle.115–118 Previous descriptions applied the terms xanthoma and cholesterol granuloma to these lesions.119 Cholesterol granulomas of the petrous apex are histologically similar lesions that may be related to xanthogranulomas. Although xanthogranulomas may represent a heterogeneous group of lesions, they are uniformly benign. Most xanthogranulomas of the choroid plexus are asymptomatic and are found incidentally on imaging. Symptomatic lesions are rare, and symptoms arise from mass effect. Xanthogranulomas have been reported in all ages, with a mean age of 50 years.119 The incidence of xanthogranulomas of the choroid plexus at autopsy is estimated to be 1.6% to 7%.115,118
Histopathology and Pathogenesis
Histologically, xanthogranulomas of the lateral ventricles and the third ventricle, as well as cholesterol granulomas of the petrous apex, have several features in common. All three lesions are composed of granulomatous reactions containing foam cells or cholesterol deposits in the form of cholesterol clefts.117,120,121 Foam cells resemble desquamated epithelial cells and are morphologically indistinguishable from lipid-laden macrophages.116 Often intermixed with the lipid-containing cells are hemosiderin deposits and other blood breakdown products. Surrounding these elements is an inflammatory reaction composed of macrophages, multinucleated giant cells, lymphocytic infiltrates, plasma cells, eosinophilic granulocytes, calcifications with psammoma bodies, and a fibrous stroma. In the past, the term xanthoma was used interchangeably with xanthogranuloma, but more recent investigations use xanthoma to indicate aggregates of foam cells or xanthoma cells in the stroma of the choroid plexus without the granulomatous reaction, hemorrhage by-products, cholesterol, calcifications, or psammoma bodies.119
The typical xanthogranuloma in the glomus of the choroid plexus of the lateral ventricle is often bilateral and does not generally contain epithelial elements or cystic components.115,116 By comparison, xanthogranulomas of the third ventricle are frequently associated with colloid cysts or contain cystic components.117 The cystic component consists of epithelial cells surrounding cyst fluid that is often positive for mucicarmine and periodic acid–Schiff (a pattern typical for a colloid cyst). The contents of the cyst can be quite variable, ranging from hemorrhagic to cholesterol-laden yellow fluid.122–125
Xanthogranulomas and cholesterol granulomas histologically similar to those found in the ventricles have been reported in the sellar region, as well as in the petrous apex. Xanthogranuloma of the sellar region distinct from adamantinomatous or papillary craniopharyngioma with xanthogranulomatous change has been reported.126 These lesions exhibit cholesterol clefts, lymphoplasma cellular infiltrates, hemosiderin deposits, fibrosis, and foreign body giant cells. These lesions were also found to be histologically distinct from ameloblastomas.
Cholesterol granulomas of the petrous apex similarly exhibit histologic features of xanthogranuloma.120,127 These lesions, which can be manifested as a mass in the cerebellopontine angle, are composed histologically of an inflammatory granulation reaction with multinucleated giant cells around cholesterol crystals and clefts. Hemorrhagic by-products are thought to be associated with nearly all cases, and bone destruction is common as a result of expansile growth.
The cause and pathogenesis of xanthogranulomas remain obscure. Shuangshoti and Netsky116 proposed that xanthogranulomas arise from a macrophage and foreign body giant cell inflammatory response to lipid and perhaps hemorrhage by-products. The lipid is proposed to arise from desquamated epithelial cells that enter the stroma of the choroid plexus and degenerate. Wolf and colleagues,118 as well as Ayres and Haymaker,115 proposed that the desquamated epithelial cells are not of neuroepithelial origin but originate from a leptomeningeal source. Additionally, Hadfield and coworkers124 and Shuangshoti and associates117 suggested that hemorrhage may be an important factor in the pathogenesis of the granulomatous reaction. In addition, for third ventricular lesions associated with colloid cysts, Hadfield and coworkers124 proposed that the colloid material itself might incite a granulomatous reaction.
Although the origin of foam cells, cholesterol, and hemorrhage in choroid plexus xanthogranulomas remains in dispute, a proposed pathogenic mechanism for cholesterol granulomas of the petrous apex may shed light on the debate.120 Cholesterol granulomas are thought to develop in pneumatized bone, such as the petrous bone, when air exchange and drainage of the bone are disrupted. The interference with air exchange results in decreased oxygen tension and hypoxia, which is thought to cause mucosal edema, rupture of blood vessels, and hemorrhage. The stagnant hemorrhage incites an inflammatory reaction that results in the accumulation of cholesterol crystals and hemorrhage by-products. Xanthogranulomas and cholesterol granulomas may therefore be a generalized reaction to blood and other irritating substances such as colloid and may be a common inflammatory end point for a variety of inciting causes.
Clinical Features
Most choroid plexus xanthogranulomas are asymptomatic. Symptomatic patients have a diverse range of initial symptoms generally related to a mass effect.117,121,122,128–133 For lesions associated with the choroid plexus in the lateral or third ventricle, the most serious clinical finding is hydrocephalus secondary to ventricular obstruction. This situation occurs more commonly with xanthogranulomas of the third ventricle.125,134,135 Patients with benign tumors in the third and lateral ventricles may be at risk for sudden death; xanthogranuloma in the third ventricle has been reported to cause sudden death.124
Imaging Findings
Because xanthogranulomas are commonly asymptomatic, they are often found incidentally on head CT or MRI studies. Xanthogranulomas are heterogeneous in content, so they do not have uniform findings on CT or MRI. CT typically shows a round, discrete mass in the lateral ventricles associated with the glomus. They can be isodense,132 slightly hyperdense,134 or of mixed intensity.136,137 Punctate calcification is common. Lesions can be homogeneously enhancing,134 heterogeneously enhancing,136 or nonenhancing.130 Central hypodensity on non–contrast-enhanced CT and low central attenuation on contrast-enhanced CT have also been reported.136,137 These findings on CT make xanthogranulomas difficult to differentiate from colloid cysts.132
Findings on MRI are equally inconsistent. Because xanthogranulomas are associated with lipid, cholesterol, and hemorrhage by-products at different stages of degradation and in different amounts, MRI findings can vary greatly. Kadota and associates134 and Brück and colleagues121 suggested that xanthogranulomas should exhibit hypointensity on T2-weighted images, isointensity or hyperintensity on T1-weighted images, and homogeneous contrast enhancement on T1-weighted images. Hyperintense lesions on T1- and T2-weighted images without enhancement, as well as isointense T1-weighted images and hyperintense T2-weighted images with rim enhancement, have been seen as well.125,132 The diversity of findings on imaging studies makes accurate differentiation of xanthogranulomas from other ventricular tumors or colloid cysts difficult.
Treatment
Although xanthogranulomas are generally benign lesions, they are a risk for causing sudden death from obstructive hydrocephalus and herniation.124 Furthermore, massive hematomas can develop in xanthogranulomas and cause hypothalamic dysfunction and death.125 For these reasons, symptomatic lateral ventricular lesions and third ventricular lesions should be removed surgically.130 Although colloid cysts have been treated successfully with stereotactic aspiration, they have a high recurrence rate.138 Cystic xanthogranulomas are more difficult to aspirate because they have a thicker wall, more viscous fluid, and a tendency to bleed.125,132 Thus, radical extirpation should be the treatment of choice. As a cautionary note, xanthogranulomas may be more difficult to remove than colloid cysts because of adhesion to the choroid plexus. Both transcortical and transcallosal approaches have been used successfully for lesions in the third ventricle.
American Thoracic Society, Centers for Disease Control and Prevention, and Infectious Diseases Society of America. Treatment of tuberculosis. Available at http://www.cdc.gov/mmwR/preview/mmwrhtml/rr5211a1.htm Accessed May 14, 2008
Burger PC, Scheithauer BW, Vogel FS. Surgical Pathology of the Nervous System and Its Coverings. New York: Churchill Livingstone; 1991.
Christoforidis GA, Spickler EM, Recio MV, et al. MR of CNS sarcoidosis: correlation of imaging features to clinical symptoms and response to treatment. AJNR Am J Neuroradiol. 1999;20:655-669.
Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis N Engl J Med. 2007;357:2153-2165.
Junger SS, Stern BJ, Levine SR, et al. Intramedullary spinal sarcoidosis: clinical and magnetic resonance imaging characteristics. Neurology. 1993;43:333-337.
Luke RA, Stern BJ, Krumholz A, et al. Neurosarcoidosis: the long term clinical course. Neurology. 1987;37:461-463.
Scott TF. Neurosarcoidosis: progress and clinical aspects. Neurology. 1993;43:8-12.
Scott TF, Yandora K, Valeri A, et al. Aggressive therapy for neurosarcoidosis: long-term follow-up of 48 treated patients. Arch Neurol. 2007;64:691-696.
Stern BJ, Krumholz A, Johns C, et al. Sarcoidosis and its neurological manifestations. Arch Neurol. 1985;42:909-917.
Zajicek JP, Scolding NJ, Foster O, et al. Central nervous system sarcoidosis—diagnosis and management. Q.J.M. 1999;92:103-117.
Zouaoui A, Maillard JC, Dormont D, et al. MRI in neurosarcoidosis. J Neuroradiol. 1992;19:271-284.
1 Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. N Engl J Med. 2007;357:2153-2165.
2 Johns CJ, Michele TM. The clinical management of sarcoidosis: a 50-year experience at the Johns Hopkins Hospital. Medicine (Baltimore). 1999;78:65-111.
3 Oksanen V. Neurosarcoidosis. Sarcoidosis. 1994;11:76-79.
4 Delaney P. Neurological manifestations of sarcoid. Ann Intern Med. 1977;87:336-345.
5 Heck AW, Phillips LH. Sarcoidosis and the nervous system. Neurol Clin. 1989;7:641-654.
6 Scott TF. Neurosarcoidosis: progress and clinical aspects. Neurology. 1993;43:8-12.
7 Sharma OP, Sharma AM. Sarcoidosis of the nervous system: a clinical approach. Arch Intern Med. 1991;151:1317-1321.
8 Stern BJ, Krumholz A, Johns C, et al. Sarcoidosis and its neurological manifestations. Arch Neurol. 1985;42:909-917.
9 Stern BJ, Corbett J. Neuro-ophthalmologic manifestations of sarcoidosis. Currt Treat Options Neurol. 2007;9:63-71.
10 Bihan H, Christozova V, Dumas JL, et al. Sarcoidosis: clinical, hormonal, and magnetic resonance imaging (MRI) manifestations of hypothalamic-pituitary disease in 9 patients and review of the literature. Medicine (Baltimore). 2007;86:259-268.
11 Clark WC, Acker JD, Dohan FC, et al. Presentation of central nervous system sarcoidosis as intracranial tumors. J Neurosurg. 1985;63:851-856.
12 Powers WJ, Miller FM. Sarcoidosis mimicking glioma: case report and review of intracranial sarcoidosis-like mass lesions. Neurology. 1981;31:907-910.
13 Healton EB, Zito G, Chauhan P, et al. Intracranial subdural sarcoid granuloma. J Neurosurg. 1982;56:728-731.
14 Osenbach RK, Blumenkopf B, Ramirez H, et al. Meningeal neurosarcoidosis mimicking convexity en-plaque meningioma. Surg Neurol. 1986;26:387-390.
15 Sethi KD, Taher EG, Patel BR, et al. Dural sarcoidosis presenting with transient neurologic symptoms. Arch Neurol. 1986;43:595-597.
16 Stubgen JP. Neurosarcoidosis presenting as a retroclival mass. Surg Neurol. 1995;43:85-87.
17 Krumholz A, Stern BJ, Stern EG. Clinical implications of seizures in neurosarcoidosis. Arch Neurol. 1991;48:842-844.
18 Maisel JA, Lynam T. Unexpected sudden death in a young pregnant woman: unusual presentation of neurosarcoidosis. Ann Emerg Med. 1996;28:94-97.
19 Schlitt MS, Duvall ER, Bonnin J, et al. Neurosarcoidosis causing ventricular loculation, hydrocephalus, and death. Surg Neurol. 1986;26:67-71.
20 Scott TF. Cerebral herniation after lumbar puncture in sarcoid meningitis. Clin Neurol Neurosurg. 2000;102:26-28.
21 Lukin RR, Chambers AA, Soleimanpour M. Outlet obstruction of the fourth ventricle in sarcoidosis. Neuroradiology. 1996;10:65-68.
22 Spencer N, Ross G, Helm G, et al. Aqueductal obstruction in sarcoidosis. Clin Neuropathol. 1989;8:158-161.
23 Banerjee T, Hunt WE. Spinal cord sarcoidosis. J Neurosurg. 1993;36:490-493.
24 Campbell JN, Black P, Ostrow PT. Sarcoid of the cauda equina: case report. J Neurosurg. 1977;47:109-112.
25 Fried ED, Landau AJ, Sher JH, et al. Spinal cord sarcoidosis: a case report and review of the literature. J Assoc Acad Minor Phys. 1993;4:132-137.
26 Hitchon PW, Haque AW, Olson JJ, et al. Sarcoidosis presenting as an intramedullary spinal cord mass. Neurosurgery. 1985;15:86-90.
27 Junger SS, Stern BJ, Levine SR, et al. Intramedullary spinal sarcoidosis: clinical and magnetic resonance imaging characteristics. Neurology. 1993;43:333-337.
28 Kelly RB, Mahoney PD, Cawley KM. MR demonstration of spinal cord sarcoidosis: report of a case. AJNR Am J Neuroradiol. 1988;9:197-199.
29 Levivier M, Brotchi J, Baleriaux D, et al. Sarcoidosis presenting as an isolated intramedullary tumor. Neurosurgery. 1991;29:271-276.
30 Zajicek J. Sarcoidosis of the cauda equina: a report of three cases. J Neurol. 1990;237:424-426.
31 Oh SJ. Sarcoid polyneuropathy: a histologically proved case. Arch Neurol. 1980;7:178-181.
32 Zuniga G, Ropper AH, Frank J. Sarcoid peripheral neuropathy. Neurology. 1984;41:1558-1561.
33 Hoitsma E, Marziniak M, Faber CG, et al. Small fibre neuropathy in sarcoidosis. Lancet. 2002;359:2085.
34 Ando DG, Lynch JP, Fantone JC. Sarcoid myopathy with elevated creatine phosphokinase. Am Rev Respir Dis. 1985;131:298-300.
35 Silverstein A, Siltzbach LE. Muscle involvement in sarcoidosis. Arch Neurol. 1969;21:235-241.
36 Zajicek JP, Scolding NJ, Foster O, et al. Central nervous system sarcoidosis—diagnosis and management. Q.J.M. 1999;92:103-117.
37 DeRemee R, Rohrbach M. Serum angiotensin converting enzyme activity in evaluating the clinical course of sarcoidosis. Ann Intern Med. 1980;92:361-365.
38 Shultz T, Miller WC, Bedrossian CW. Clinical application of measurement of angiotensin-converting-enzyme level. JAMA. 1979;242:439-441.
39 Mitchell JD, Yap PL, Milne LA, et al. Immunological studies on the cerebrospinal fluid in neurological sarcoidosis. J Neuroimmunol. 1985;7:249-253.
40 Dale JC, O’Brien JF. Determination of angiotensin-converting enzyme levels in cerebrospinal fluid is not a useful test for the diagnosis of neurosarcoidosis. Mayo Clinic Proc. 1999;74:535.
41 Israel H, Gushue G, Park C. Assessment of gallium-67 scanning in pulmonary and extrapulmonary sarcoidosis. Ann N Y Acad Sci. 1988;465:455-462.
42 Dubey N, Miletich R, Wasay M, et al. Role of fluorodeoxyglucose positron emission tomography in the diagnosis of neurosarcoidosis. J Neurol Sci. 2002;205:77-81.
43 Ellis PK, Bell KE. The radiological investigation of neurosarcoidosis. Ulster Med J. 1995;64:101-104.
44 Kadakia JK, Collette PM, Sharma OP. Role of magnetic resonance imaging in neurosarcoidosis. Sarcoidosis. 1993;10:98-99.
45 Sherman JL, Stern BJ. Sarcoidosis of the CNS: comparison of unenhanced and enhanced MR images. AJNR Am J Neuroradiol. 1990;11:915-923.
46 Zouaoui A, Maillard JC, Dormont D, et al. MRI in neurosarcoidosis. J Neuroradiol. 1992;19:271-284.
47 Ahn HS, Stern BJ, Fearnow EC. Imaging case of the month. Md Med J. 1992;41:921-922.
48 Khaw KT, Manji H, Britton J, et al. Neurosarcoidosis—demonstration of meningeal disease by gadolinium enhanced magnetic resonance imaging. J Neurol Neurosurg Psychiatry. 1991;54:499-502.
49 Handler MS, Johnson LM, Dick AR, et al. Neurosarcoidosis with unusual MRI findings. Neuroradiology. 1993;35:146-148.
50 Sauter MK, Panitch HS, Kristt DA. Myelopathic neurosarcoidosis: diagnostic value of enhanced MRI. Neurology. 1991;41:150-151.
51 Lexa FJ, Grossman RI. MR of sarcoidosis in the head and spine: spectrum of manifestations and radiographic response to steroid therapy. AJNR Am J Neuroradiol. 1994;15:973-982.
52 Peeples DM, Stern BJ, Jiji V, et al. Germ cell tumors masquerading as central nervous system sarcoidosis. Arch Neurol. 1991;48:554-556.
53 Christoforidis GA, Spickler EM, Recio MV, et al. MR of CNS sarcoidosis: correlation of imaging features to clinical symptoms and response to treatment. AJNR Am J Neuroradiol. 1999;20:655-669.
54 Agbogu BN, Stern BJ, Sewell C, et al. Therapeutic considerations in patients with refractory neurosarcoidosis. Arch Neurol. 1995;52:875-879.
55 Baughman RP, Lower EE. Steroid-sparing alternative treatments for sarcoidosis. Clin Chest Med. 1997;18:853-864.
56 Scott TF, Yandora K, Valeri A, et al. Aggressive therapy for neurosarcoidosis: long-term follow-up of 48 treated patients. Arch Neurol. 2007;64:691-696.
57 Stern BJ, Schonfeld SA, Sewell C, et al. The treatment of neurosarcoidosis with cyclosporine. Arch Neurol. 1992;49:1065-1072.
58 Lower EE, Broderick JP, Brott TG, et al. Diagnosis and management of neurological sarcoidosis. Arch Intern Med. 1997;157:1864-1868.
59 Soriano FG, Caramelli P, Nitrini R, et al. Neurosarcoidosis: therapeutic success with methotrexate. Postgrad Med J. 1990;66:142-143.
60 Sharma OP. Effectiveness of chloroquine and hydroxychloroquine in treating selected patients with sarcoidosis with neurological involvement. Arch Neurol. 1998;55:1248-1254.
61 Marques LJ, Zheng L, Poulakis N, et al. Pentoxifylline inhibits TNF-alpha production from human alveolar macrophages. Am J Respir Crit Care Med. 1999;159:508-511.
62 Zabel P, Entzian P, Dalhoff K, et al. Pentoxifylline in treatment of sarcoidosis. Am J Respir Crit Care Med. 1997;155:1665-1669.
63 Lee JB, Koblenzer PS. Disfiguring cutaneous manifestation of sarcoidosis treated with thalidomide: a case report. J Am Acad Dermatol. 1998;39:835-838.
64 Rousseau L, Beylot-Barry M, Doutre MS, et al. Cutaneous sarcoidosis successfully treated with low doses of thalidomide. Arch Dermatol. 1998;134:1045-1046.
65 Pettersen JA, Zochodne DW, Bell RB, et al. Refractory neurosarcoidosis responding to infliximab. Neurology. 2002;59:1660.
66 Katz JM, Bruno MK, Winterkorn JMS, et al. The pathogenesis and treatment of optic disc swelling in neurosarcoidosis: a unique therapeutic response to infliximab. Neurology. 2003;60:426-430.
67 Pritchard C, Nadarajah K. Tumour necrosis factor [alpha] inhibitor treatment for sarcoidosis refractory to conventional treatments: a report of five patients. Ann Rheum Dis. 2004;63:318-320.
68 Sweiss NJ, Baughman RP. Tumor necrosis factor inhibition in the treatment of refractory sarcoidosis: slaying the Dragon? J Rheumatol. 2007;34:2129-2131.
69 Bejar JM, Kerby GR, Zeigler DK, et al. Treatment of central nervous system sarcoidosis with radiotherapy. Am J Med. 1982;73:605-608.
70 Motta M, Alongi F, Bolognesi A, et al. Remission of refractory neurosarcoidosis treated with brain radiotherapy: a case report and a literature review. Neurologist. 2008;14:120-124.
71 Cahill DW, Salcman MD. Neurosarcoidosis: a review of the rarer manifestations. Surg Neurol. 1980;15:86-90.
72 Luke RA, Stern BJ, Krumholz A, et al. Neurosarcoidosis: the long term clinical course. Neurology. 1987;37:461-463.
73 Udani PM, Parekh UC, Dastur DK. Neurological and related syndromes in CNS tuberculosis: clinical features and pathogenesis. J Neurol Sci. 1971;14:341-357.
74 Whiteman M, Espinoza L, Post JD, et al. Central nervous system tuberculosis in HIV-infected patients: clinical and radiographic findings. AJNR Am J Neuroradiol. 1995;16:1319-1327.
75 Rich AR, MacCordick HA. The pathogenesis of tuberculosis. Bull Johns Hopkins Hosp. 1933;52:5-10.
76 Dastur DK, Lalitha VS, Prabhakar V. Pathological analysis of intracranial space-occupying lesions in 1000 cases including children. Part 1. Age, sex and pattern; and the tuberculomas. J Neurol Sci. 1968;6:575-592.
77 Burger PC, Scheithauer BW, Vogel FS. Surgical Pathology of the Nervous System and Its Coverings. New York: Churchill Livingstone; 1991.
78 Isenmann S, Zimmermann DR, Wichmann W, et al. Tuberculoma mimicking meningioma of the falx cerebri: PCR diagnosis of mycobacterial DNA from formalin-fixed tissue. Clin Neuropathol. 1996;15:155-158.
79 Lindner A, Schneider C, Hofmann E, et al. Isolated meningeal tuberculoma mimicking meningioma: case report. Surg Neurol. 1995;43:81-84.
80 Ng SH, Tang LM, Lui TN, et al. Tuberculoma en plaque: CT. Neuroradiology. 1996;38:453-455.
81 Sibley WA, O’Brien JL. Intracranial tuberculomas: a review of clinical features and treatment. Neurology. 1956;6:157-165.
82 Tyson G, Newman P, Strachan WE. Tuberculous brain abscess. Surg Neurol. 1978;10:323-325.
83 Mayers MM, Kaufman DM, Miller MH. Recent cases of intracranial tuberculomas. Neurology. 1978;28:256-260.
84 Arseni C. Two hundred and one cases of intracranial tuberculoma treated surgically. J Neurol Neurosurg Psychiatry. 1958;21:308-311.
85 Artco M, De Caro GMF, Carloia S, et al. Advances in diagnosis, treatment and prognosis of intracerebral tuberculomas in the last 50 years: report of 21 cases. Neurochirurgie. 1999;45:129-133.
86 Maurice-Williams RS. Tuberculomas of the brain in Britain. J Postgrad Med. 1972;48:678-681.
87 Bagga A, Kalra V, Ghai OP. Intracranial tuberculoma. Clin Pediatr (Phila). 1988;27:487-490.
88 Harder E, Al-Kawi MZ, Carney P. Intracranial tuberculoma: conservative management. Am J Med. 1983;74:570-576.
89 Loizou LA, Anderson M. Intracranial tuberculomas: correlation of computerized tomography with clinico-pathological findings. Q.J.M. 1982;51:104-114.
90 García-Moncó JC. Central nervous system tuberculosis. Neurol Clin. 1999;17:737-759.
91 Bonington A, Strang JI, Klapper PE, et al. Use of Roche AMPLICOR Mycobacterium tuberculosis PCR in early diagnosis of tuberculous meningitis. J Clin Microbiol. 1998;36:1251-1254.
92 Michael JS, Lalitha MK, Cherian T, et al. Evaluation of polymerase chain reaction for rapid diagnosis of tuberculous meningitis. Indian J Tuberc. 2002;49:133-137.
93 Vengsarkar US, Pisipaty RP, Parekh B, et al. Intracranial tuberculoma and the CT scan. J Neurosurg. 1986;64:568-574.
94 Welchman JM. Computerized tomography of intracranial tuberculomata. Clin Radiol. 1979;30:567-573.
95 Bargalló J, Berenguer J, García-Barrionuevo J, et al. The “target sign”: is it a specific sign of CNS tuberculoma? Neuroradiology. 1996;38:547-550.
96 Rajshekhar V, Haran RP, Prakash S, et al. Differentiating solitary small cysticercous granulomas and tuberculomas in patients with epilepsy. J Neurosurg. 1993;78:402-407.
97 Selvapandian S, Rajshekhar V, Chandy MJ, et al. Predictive value of computed tomography–based diagnosis of intracranial tuberculomas. Neurosurgery. 1994;35:845-850.
98 Gupta RK, Jena A, Singh AK, et al. Role of magnetic resonance (MR) in the diagnosis and management of intracranial tuberculomas. Clin Radiol. 1990;41:120-127.
99 Wasay M, Kheleani BA, Moolani MK, et al. Brain CT and MRI findings in 100 consecutive patients with intracranial tuberculoma. J Neuroimaging. 2003;13:240-247.
100 Kim TK, Chang KH, Kim CJ, et al. Intracranial tuberculoma: comparison of MR with pathologic findings. AJNR Am J Neuroradiol. 1995;16:1903-1908.
101 Bouchama A, Zuheir Al-Kawi M, Kanaan I, et al. Brain biopsy in tuberculoma: the risks and benefits. Neurosurgery. 1991;28:405-409.
102 Tandon PN. Brain biopsy in tuberculoma: the risks and benefits. Neurosurgery. 1992;30:301.
103 Al-Mefty O. Intracranial tuberculoma. J Neurosurg. 1986;65:572-573.
104 Mohanty A, Santosh V, Anandh B, et al. Diagnostic efficacy of stereotactic biopsies in intracranial tuberculomas. Surg Neurol. 1999;52:252-258.
105 American Thoracic Society, Centers for Disease Control and Prevention, and Infectious Diseases Society of America. Treatment of tuberculosis. Available at http://www.cdc.gov/mmwR/preview/mmwrhtml/rr5211a1.htm, May 14, 2008. Accessed
106 Awada A, Daif AK, Pirani M, et al. Evolution of brain tuberculomas under standard antituberculous treatment. J Neurol Sci. 1998;156:47-52.
107 Chambers ST, Record C, Hendrickse WA, et al. Paradoxical expansion of intracranial tuberculoma during chemotherapy. Lancet. 1984;2:181-184.
108 Hejazi N, Hassler W. Multiple intracranial tuberculomas with atypical response to tuberculostatic chemotherapy: review of the literature and own experience. Acta Neurochir (Wien). 1997;139:194-202.
109 Rao GP, Nadh BR, Hemaratnan A, et al. Paradoxical progression of tuberculous lesions during chemotherapy of central nervous system tuberculosis. J Neurosurg. 1995;83:359-362.
110 Wasay M. Central nervous system tuberculosis and paradoxical response. S Med J. 2006;99:331-332.
111 Bannister CM. A tuberculous abscess of the brain: case report. J Neurosurg. 1970;33:203-206.
112 Whitener DR. Tuberculous brain abscess. Arch Neurol. 1978;35:148-155.
113 Mohanty A, Venkatarama SK, Vasudev MK, et al. Role of stereotactic aspiration in the management of tuberculous brain abscess. Surg Neurol. 1999;51:443-447.
114 Rajshekhar V, Chandy MJ. CT-guided stereotactic surgery in the management of intracranial tuberculomas. Br J Neurosurg. 1993;7:665-671.
115 Ayres WW, Haymaker W. Xanthoma and cholesterol granuloma of the choroid plexus. J Neuropathol Exp Neurol. 1960;19:280-295.
116 Shuangshoti S, Netsky MG. Xanthogranuloma (xanthoma) of choroid plexus: the origin of foamy (xanthoma) cells. Am J Pathol. 1966;48:503-533.
117 Shuangshoti S, Phonprasert C, Suwanwela N, et al. Combined neuroepithelial (colloid) cyst and xanthogranuloma (xanthoma) in the third ventricle. Neurology. 1975;25:547-552.
118 Wolf A, Cowen D, Graham S. Xanthomas of the choroid plexus in man. J Neuropathol Exp Neurol. 1950;9:286-297.
119 Muenchau A, Laas R. Xanthogranuloma and xanthoma of the choroid plexus: evidence for different etiology and pathogenesis. Clin Neuropathol. 1997;16:72-76.
120 Brodkey JA, Robertson JH, Shea JJ, et al. Cholesterol granulomas of the petrous apex: combined neurosurgical and otological management. J Neurosurg. 1996;85:625-633.
121 Brück W, Sander U, Blanckenberg P, et al. Symptomatic xanthogranuloma of choroid plexus with unilateral hydrocephalus: case report. J Neurosurg. 1991;75:324-327.
122 Antunes JL, Kvam D, Ganti SR, et al. Mixed colloid cysts–xanthogranulomas of the third ventricle. Surg Neurol. 1981;16:256-261.
123 Gherardi R, Nguyen JP, Gaston A, et al. Symptomatic xanthogranuloma of the third ventricle: a clinicopathological report. Eur Neurol. 1984;23:156-162.
124 Hadfield MG, Ghatak NR, Wanger GP. Xanthogranulomatous colloid cyst of the third ventricle. Acta Neuropathol (Berl). 1985;66:343-346.
125 Tomita H, Tamaki N, Korosue K, et al. Xanthogranuloma with massive hematoma in the third ventricle: case report. Neurosurgery. 1996;39:591-594.
126 Paulus W, Honegger J, Keyvani K, et al. Xanthogranuloma of the sellar region: a clinicopathological entity different from adamantinomatous craniopharyngioma. Acta Neuropathol (Berl). 1999;97:377-382.
127 Eisenberg MB, Haddad G, Al-Mefty O. Petrous apex cholesterol granulomas: evolution and management. J Neurosurg. 1997;86:822-829.
128 Godersky JC, Rockswold G, Larson DA. Xanthogranuloma of the third ventricle producing hydrocephalus. Neurosurgery. 1980;7:68-70.
129 Jaer Ø, Løken AC, Nesbakken R. Hydrocephalus due to xanthogranuloma: case report. J Neurosurg. 1973;39:659-661.
130 Montaldi S, Deruaz JP, Cai ZT, et al. Symptomatic xanthogranuloma of the third ventricle: report of two cases and review of the literature. Surg Neurol. 1989;32:200-205.
131 Rush JL, Kusske JA, Porter RW, et al. Xanthogranulomas of the third ventricle. Neurosurgery. 1979;4:329-333.
132 Tatter SB, Ogilvy CS, Golden JA, et al. Third ventricular xanthogranulomas clinically and radiologically mimicking colloid cysts. J Neurosurg. 1994;81:605-609.
133 Wiot JG, Lukin RR, Tomsick TA. Xanthogranuloma of the third ventricle. AJNR Am J Neuroradiol. 1989;10(5 suppl):S57.
134 Kadota T, Mihara N, Tsuji N, et al. MR of xanthogranuloma of the choroid plexus. AJNR Am J Neuroradiol. 1995;17:1595-1597.
135 Razavi-Encha F, Gray F, Gaston A, et al. Symptomatic xanthogranuloma of the choroid plexus of the third ventricle: a new case with ultrastructural study. Surg Neurol. 1987;27:569-574.
136 Handagoon P, Pitakdamrongwong N, Shuangshoti S. Xanthogranulomas of choroid plexus. Neuroradiology. 1987;29:172-173.
137 Terao H, Kobayashi S, Teraoka A, et al. Xanthogranulomas of the choroid plexus in a neuro-epileptic child: case report. J Neurosurg. 1978;48:649-653.
138 Mathiesen T, Grane P, Lindquist C, et al. High recurrence rate following aspiration of colloid cysts in the third ventricle. J Neurosurg. 1993;78:748-752.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
CHAPTER 151 Sarcoidosis, Tuberculosis, and Xanthogranuloma
Sarcoidosis
Sarcoidosis is a chronic, multisystem granulomatous disease of unknown cause. It can occur in any racial or ethnic population and most often develops in persons between the ages of 20 and 40 years. In the United States, its prevalence ranges from 10 to 40 per 100,000, with an approximately 10 : 1 ratio of black to white patients and a slight female preponderance.1,2 Any organ system can be involved in sarcoidosis, with the lungs, lymph nodes, skin, and eyes being the most common. Five percent of patients have neurological manifestations.3–8 Of these patients with neurosarcoidosis (NS), 50% have neurological problems at the time of initial diagnosis. In a third of these patients, more than one neurological manifestation of their disease is already present or will develop.
Histology and Pathophysiology
Although the precise cause of sarcoidosis is not known, there is evidence that the disease results from dysregulation of the cellular immune responses to foreign antigens or self-antigens, thereby leading to granuloma formation.1 These granulomas are composed of epithelioid cells, histiocytes, T cells, monocytes, and fibroblasts. Granuloma formation can ultimately lead to fibrosis and organ dysfunction.
Clinical Features
Cranial Neuropathy
Cranial neuropathy is the most frequently encountered neurological manifestation of sarcoidosis; it occurs in approximately 75% of patients with NS.8 More than 50% of patients have involvement of multiple cranial nerves.
Dysfunction of the facial nerve is the most common cranial neuropathy seen in NS, and it develops in more than 50% of patients.8 The peripheral palsy can be unilateral or bilateral and is often transient.
NS can occur in the subfrontal region and cause an olfactory neuropathy and anosmia. Involvement of the optic nerve is relatively common and is seen in as many as 5% of patients.8 Funduscopic examination can demonstrate optic atrophy, optic disc swelling or papilledema, or retrobulbar optic neuritis.9 In patients with visual or optic abnormalities, however, it must be remembered that sarcoidosis affects the eye as a uveitis far more often than the optic nerve.
Meningitis
Symptoms of meningeal irritation, including headache, nuchal rigidity, nausea, and rarely, fever, have been reported to occur in 3% to 26% of patients with NS.8 Symptomatic meningeal involvement can take the form of either acute aseptic meningitis or chronic meningitis. Cerebrospinal fluid (CSF) studies generally reveal a mild mononuclear pleocytosis with increased protein. Glucose may be normal or decreased, with no evidence of bacteria. Given the extent of meningeal involvement seen in most patients with NS, symptomatic meningitis is relatively rare. Occasionally, meningeal-based granulomatous mass lesions can develop.
Pituitary and Hypothalamic Sarcoidosis
When sarcoidosis involves the CNS parenchyma, it most commonly affects the hypothalamus and parasellar region.10 This can result in vegetative alterations and neuroendocrinologic dysfunction. Symptoms include disturbances in sleep, appetite, thirst, temperature, and libido. Hypothalamic and pituitary lesions can also cause thyroid, gonadal, and adrenal abnormalities. Altered thirst, antidiuretic hormone deficiency or excess, and hyperprolactinemia have been reported.
Space-Occupying Lesions
Granulomatous masses forming localized space-occupying lesions can occur throughout the CNS but are most frequently found in the cerebral hemispheres.6 They can be manifested as a large, solitary mass mimicking either a primary or metastatic brain tumor.11,12 Alternatively, they can appear as multiple nodules.8 Finally, they can take the form of a dural-based plaque-like mass resembling a meningioma.13–16
Seizures
Generalized or partial seizures have been reported in as many as 22% of patients with NS.17 They are thought to be caused by supratentorial parenchymal involvement in which the granulomatous inflammation is often found in a perivascular distribution.
Hydrocephalus
Hydrocephalus secondary to obstruction of normal CSF flow is a relatively common and potentially lethal clinical feature of NS.18–20 Communicating hydrocephalus can result from extensive meningeal involvement causing decreased CSF resorption through the arachnoid villi. Obstructive hydrocephalus can develop secondary to an intracranial mass or inflammation preventing the flow of CSF through the foramen of Monro, aqueduct of Sylvius, or foramen of Luschka or Magendie.21,22
Spinal Cord Sarcoidosis
Sarcoidosis of the spinal cord is relatively rare. It can occur at any segment of the spinal cord, as well as at the cauda equina, and the granulomatous lesions may be intramedullary or extramedullary.23–30
Peripheral Neuropathy
Peripheral neuropathic manifestations include mononeuropathy, mononeuritis multiplex, and generalized sensory, sensorimotor, and motor neuropathies.31,32 Patients can have a small-fiber sensory or autonomic neuropathy.33 Clinical features resembling Guillain-Barré syndrome have also been described. Nerve biopsy shows granuloma formation in both the epineurial and perineurial spaces, as well as axonal degeneration. There can be associated vascular inflammation, thus suggesting that ischemia may play a role in the nerve damage.
Diagnostic and Imaging Studies
The diagnosis of sarcoidosis is firmly established only when the clinical and paraclinical findings are supported by histologic evidence of widespread noncaseating granulomas.1,2 When neurological findings develop in a patient with sarcoidosis, NS should be considered, although an intercurrent infection or malignancy must be excluded. The following diagnostic criteria for NS are adapted from Zajicek and colleagues36:
When NS is suspected, the patient should be evaluated for evidence of systemic disease and a search for a site suitable for biopsy undertaken. Because corticosteroids can eliminate evidence of systemic inflammation, they should be withheld until the diagnostic evaluation is completed unless severe illness requires their use. The search for systemic sarcoidosis should begin with an examination of the skin and lymph nodes.1,2 Chest radiographs are abnormal in approximately 90% of patients with sarcoidosis, but frequently, additional diagnostic information can be obtained from a computed tomography (CT) scan of the thorax. Serum angiotensin-converting enzyme (ACE) levels can be elevated in patients with sarcoidosis. However, infection or malignancy can cause serum ACE levels to be high, and in patients with isolated NS, serum ACE levels may be normal.37,38 Ophthalmologic examination can reveal uveitis or retinal vasculopathy, as well as conjunctival nodules. Endoscopic nasal and sinus examination can show evidence of mucosal inflammation.
CSF abnormalities occur in NS, but they tend to be nonspecific and thus are not a reliable marker of the disease. CSF opening pressure is elevated in approximately 10% of patients, and protein is increased in two thirds of patients. CSF glucose can be normal or low. Approximately 50% of patients have a predominantly mononuclear pleocytosis. The immunoglobulin G index may be elevated, and oligoclonal bands may be present.6,8,39 The concentration of ACE in CSF may be elevated, but this finding is not specific to NS.40 There are reports of patients deteriorating after lumbar puncture.20
Gallium scanning can be informative; uptake in the lungs and salivary, parotid, and lacrimal glands is suggestive of sarcoidosis and, when a distinctive pattern is found in combination with elevated serum ACE levels, may be highly specific (>95%) for sarcoidosis.41 Whole-body fluorodeoxyglucose positron emission tomography (FDG-PET) is a sensitive technique for highlighting areas of inflammation.42
CT and magnetic resonance imaging (MRI) enhance detection of meningeal (Figs. 151-1 and 151-2) and parenchymal disease (Figs. 151-3 and 151-4), even in patients without any neurological findings.
On CT, the granulomatous lesions of NS are usually isodense or hyperdense on non–contrast-enhanced studies and uniformly enhancing after the administration of contrast medium.8,43 CT can reveal bony erosion of the skull base caused by NS lesions. The orbital contents, including the optic nerve and extraocular muscles, can also be visualized, particularly with sagittal and coronal reconstructions.
The imaging procedure of choice for NS is MRI with gadolinium enhancement.44–47 This test is particularly sensitive for detecting disease of the parasellar region, basal cisterns, and spinal cord. On T1-weighted images, NS lesions are usually hyperintense in relation to normal brain parenchyma, and they enhance after the administration of gadolinium. T2-weighted images often show minimal edema surrounding the lesions, although this finding can be variable and may be related to disease activity. A wide spectrum of NS abnormalities can be seen on MRI and can mimic other neurological diseases. Gadolinium-enhanced MRI can reveal both diffuse (see Fig. 151-1) and local (see Fig. 151-2) leptomeningeal involvement.47,48 The dura often shows marked thickening along with evidence of inflammation (pachymeningitis). Parasellar lesions and intraparenchymal masses (see Fig. 151-3) can also be readily identified, but the latter can easily be mistaken for a neoplasm.49 Involvement of the optic nerves or other cranial nerves can likewise be documented. MRI can identify hydrocephalus, and the presence of transependymal edema on T2-weighted images can be a useful indicator of increased intracranial pressure secondary to obstruction of CSF flow. Finally, MRI can be valuable in the detection of sarcoidosis involving the spinal cord (see Fig. 151-4) and cauda equina.50
In addition to its utility in identifying lesions consistent with NS, gadolinium-enhanced MRI can be used to monitor the efficacy of NS treatment.51 Decreased enhancement and regression of the size of lesions suggest therapeutic efficacy.
Biopsy of the brain, spinal cord, meninges, nerve, or muscle is occasionally performed to confirm a diagnosis of NS in patients with known sarcoidosis who have a neurological problem consistent with NS. Biopsy should also be considered in patients with known systemic sarcoidosis and neurological disease who are progressively deteriorating despite “optimal” treatment to uncover an alternative diagnosis.52 Importantly, biopsy can support a diagnosis of NS in patients in whom the diagnosis is suspected but for which there is no confirmatory systemic documentation. Occasionally, NS is the unexpected consequence of a CNS biopsy, for instance, in a patient with a dural-based mass thought to be a meningioma but on biopsy granulomatous inflammation is found.
Treatment
Because of the relative rarity of NS, there have been no rigorous clinical trials to determine optimal treatment. Given the pathophysiology of sarcoidosis, however, immunosuppressive therapy has been the cornerstone of therapy.6,8
Corticosteroids are the first-line agents for the treatment of NS. The site of neurological involvement, disease severity, and response to treatment generally dictate the dose and duration of corticosteroid therapy. For example, patients with facial nerve palsy or aseptic meningitis are often treated with prednisone, 0.5 mg/kg per day for 2 weeks; patients with myopathy or neuropathy are generally treated with prednisone, 0.5 mg/kg per day for 4 weeks, and then reassessed, with most patients requiring chronic therapy; and patients with a meningeal or parenchymal mass lesion, encephalopathy, or symptomatic hydrocephalus usually require prednisone, 0.5 to 1 mg/kg per day for 4 weeks, before any improvement is appreciated, and most require chronic therapy. Once the acute exacerbation is under control, corticosteroid administration can usually be slowly tapered. Frequent clinical evaluation and, for CNS disease, periodic MRI to assess the extent of enhancement can be helpful in guiding therapy.51,53
Patients who deteriorate despite aggressive corticosteroid therapy, who cannot tolerate corticosteroids, or who have a contraindication to corticosteroids may benefit from alternative therapeutic agents.54–56 Mycophenolate mofetil, cyclosporine,57 azathioprine, methotrexate,58,59 chlorambucil, cyclophosphamide,55 hydroxychloroquine,60 pentoxifylline,61,62 thalidomide,63,64 and infliximab and adalimumab65–68 have all been used to treat NS. There are no studies comparing the efficacy of these alternative treatments in patients with NS. Consequently, selection of agents is often based on ease of use, cost, and desire to avoid the complications of a particular drug. Because a patient’s response to a specific agent cannot be predicted, two or three different agents should be tried before concluding that the patient’s disease is refractory.
Cranial or spinal irradiation has been used for refractory NS and should be considered in patients who fail corticosteroid therapy and trials of alternative agents.69,70 Radiotherapy has also been used in patients with acute, life-threatening disease.
Intracranial and intraspinal mass lesions generally respond well to corticosteroids or alternative agents if the disease has been caught early, before the development of significant ischemia or fibrosis.6,8 Thus, in patients with known systemic sarcoidosis in whom a mass lesion develops, corticosteroid therapy should be initiated promptly as long as there are no significant contraindications and infection and malignancy have been reasonably excluded as diagnoses. In patients without extensive systemic disease, treatment of an intracranial or intraspinal mass can be complicated by the imaging similarities of sarcoidosis, tuberculosis, fungal disease, and neoplasm.43 In patients without known sarcoidosis, it may be necessary to perform a biopsy at the primary site of involvement to determine the nature of a mass lesion. During surgery, effort should be made to minimize disruption of the surrounding parenchyma because NS can be infiltrative and patients can deteriorate after surgical intervention. If the biopsy reveals noncaseating granulomas, further resection should be deferred while appropriate medical management is pursued. Only in highly refractory cases in which a mass lesion persists or enlarges despite optimal immunosuppressive therapy should surgical debulking or resection be considered. The results of these aggressive surgical interventions are often unsatisfactory.71
Patients with chronic, asymptomatic hydrocephalus can be observed clinically, and surgical intervention can be offered if the hydrocephalus worsens or if the patient deteriorates despite medical therapy. These patients must be watched closely, however, because abrupt, life-threatening deterioration can occur. In patients who respond to medical therapy, improvement in CSF flow can occur over time.72
Conclusion
The long-term course of NS has not been clearly defined. Approximately two thirds of patients have a monophasic illness, and the others have either a relapsing-remitting course or progressive disease.72 As many as 10% of NS patients die as a result of the inflammatory disease process or its treatment. These patients typically have CNS parenchymal disease or hydrocephalus. Prompt diagnosis of NS can lead to earlier treatment, and this may improve the outcome.
Tuberculosis
Tuberculosis (TB), a worldwide disease, experienced a resurgence in the United States in the early 1990s, largely related to the human immunodeficiency virus (HIV) epidemic, immigration from countries endemic for TB, transmission of TB in settings such as hospitals and prisons, and the development of multidrug-resistant strains of TB. TB is primarily a disease of the lungs, but extrapulmonary involvement is common. CNS involvement is reported in as many as 10% of immunocompetent patients with TB.73 CNS TB is a defining condition of acquired immunodeficiency syndrome (AIDS), and both TB and CNS involvement are more prevalent in the HIV-infected population.74 CNS involvement can take the form of tuberculous meningitis, intracranial and spinal cord tuberculomas, abscesses, and Pott’s disease. Here, the focus is primarily on neurosurgical management of CNS tuberculomas and abscesses.
