[level-membership-for-hematology-oncology-and-palliative-medicine-category]
65 Retinoblastoma
Retinoblastoma was first described by Petras Pawius of Amsterdam as early as 1657. However, James Wardrop, an Edinburgh ophthalmologist, was officially credited for recognizing retinoblastoma in 1809, when he documented the first enucleation successfully performed to treat retinoblastoma.1 Two centuries later, enucleation is still used to treat retinoblastoma, along with other methods such as external beam radiation therapy, brachytherapy, cryotherapy, and laser therapy. Newer modalities that have recently gained popularity include chemoreduction and transpupillary thermotherapy. In many cases, treatment can help preserve both patients’ eyes and vision. In the United States, the current 5-year survival rate for children with retinoblastoma is 98%.2
Incidence
Surveys suggest a relatively constant occurrence of retinoblastoma during the last century throughout the world. The frequency of retinoblastoma has been documented as 1 in 14,000 to 1 in 34,000 live births.3 The incidence in the United States is relatively low, at 3.58 cases per million children under the age of 15 years, and is closely correlated with age. For ages 1 to 4 years, the incidence is 10.6 per million; for 5 to 9 years, 1.53 per million; and for 10 to 14 years, 0.27 per million.4 Approximately 350 new cases are diagnosed in the United States each year, and retinoblastoma is the seventh most common cancer in the pediatric population in this country.
Retinoblastoma appears to have no predilection for any particular racial group or gender. Retinoblastoma can affect one or both eyes. The right eye is affected as frequently as the left eye. Unilateral retinoblastoma occurs in 75% of all cases diagnosed, whereas bilateral disease occurs in 25% of cases. All patients with bilateral retinoblastoma have multifocal disease (more than one independent tumor focus in each eye) with 5 as the mean number of tumors distributed between the two eyes, and a range of 2 to 20 tumors. Of the patients with unilateral disease, only 15% have multifocal disease.5
Genetics
Retinoblastoma was one of the disease models that has been used to demonstrate the genetic nature of cancer. Knudson proposed the now classic two-hit model in 1971 after noting that the timing of tumor development (younger age at diagnosis of bilateral tumors than of unilateral tumors) suggested a mechanism in which at least two events would be responsible for the development of the tumor.6 He hypothesized that patients with multifocal bilateral disease were germline carriers for the first hit, with only one second hit necessary for the development of retinoblastoma. Unilateral unifocal patients, he determined, usually had a normal germline genome, but developed both hits in the progenitor tumor cell.
Since the Rb1 gene was isolated in 1986 by Friend et al. many studies have explored its location and function. The gene is located on the long arm of chromosome 13, band 14.2.7 Further characterization of the gene has revealed that it spans 200 kb and is composed of 27 exons. The gene encodes a 4.7 kb mRNA transcript, which is expressed in all adult tissues. The 110 kD nuclear phosphoprotein consists of 928 amino acids.
The protein encoded by the gene is a regulator at the cell cycle checkpoint between G1 and entry into the S-phase.8 The phosphorylation pattern of p110 RB varies during the cell cycle and the current model suggests that the unphosphorylated normal RB1 protein binds transcriptional regulators that promote entry into the S-phase. When the normal RB1 protein is phosphorylated, it dissociates from E2F, freeing it to bind to DNA and stimulate transcription of downstream genes that promote progression through the cell cycle. Loss of normal RB1 function, as in the case of the tumors, presumably allows for uncontrolled entry into the S-phase, more rapid cell cycling, and rapid cell division. The retinoblastoma protein is thus a dominant suppressor of tumor formation, making the Rb1 gene a member of the tumor suppressor class of genes.
Mutations in the Rb1 gene seem to occur throughout the gene. The majority of the germline mutations are nonsense or frameshift, introducing a premature stop codon and therefore producing a truncated and presumably nonfunctional protein. Other types of mutations such as missense, aberrant splice mutations, and mutations in promotor sequences, can result in a reduced penetrance or decreased expressivity form of the disease. Only about 2% to 3% of tumors demonstrate karyotypically visible larger deletions of material in the 13q14 band. Some of the children with this cytogenetic abnormality in their germline have a constitutional disease with severe developmental delay and dysmorphic features, named the 13q syndrome, whereas others are phenotypically normal other than their retinoblastoma. Evidence suggests that retinoblastoma, like other cancers such as colon carcinomas and glial brain tumors, requires other genetic abnormalities to occur before transformation into malignancy. This finding has stimulated many studies focused on subsequent mutations that occur after the initial mutation in the Rb1 gene. Candidate oncogenes and tumor suppressor genes include KIF14, MDM4, MYCN, E2F3, DEK, and CDH11.9
Genetic counseling for the families of retinoblastoma patients is complex and challenging (Table 65-1). Epidemiologic observation reveals that a parent with bilateral retinoblastoma has roughly a 45% chance of having each child with retinoblastoma, although the retinoblastoma may not be present at the child’s birth in either eye. Of those parents with bilateral disease whose children develop unilateral disease, 96% of the children will have multifocal tumors and most of them will be diagnosed within the first 6 months of life. Since all of these children (of bilateral parents) have a germinal mutation, 45% of their children will also develop retinoblastoma.
Table 65-1 Genetic Counseling for Retinoblastoma
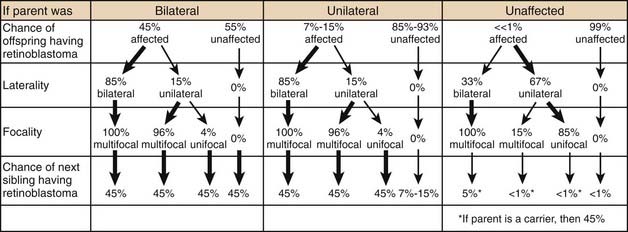
Two of the most common and challenging types of families who require genetic counseling are: (1) families in which neither parent has a history of retinoblastoma but the couple already has one child with the disease and wants to know the chances that the next will also have retinoblastoma, and (2) families in which one parent has a history of unilateral retinoblastoma and the couple wants to know the risk to future children of developing the disease. In the first scenario, an ophthalmoscopic examination of both parents’ eyes should be performed. The reason for this examination is that 1% to 2% of such parents will demonstrate on fundoscopic examination a rare benign clinical entity called a retinoma. There is debate as to the exact characterization of these lesions, but they likely represent either retinoblastoma precursor lesions as a result of the loss of the RB1 gene without the subsequent acquisition of other mutations that are required for progression to the malignant state or spontaneously regressed or arrested retinoblastoma.10 These parents should be counseled that although they have never been treated for retinoblastoma, they may harbor a germinal mutation, and if they have multifocal retinomas, each of their children will have a 45% chance of inheriting the mutation. Parents with no ophthalmoscopic evidence of retinoblastoma can be told that the chance that their next child will have retinoblastoma is between 1% and 5%. In the small percentage of cases in which the second child does develop the disease, it is presumed that one of the parents carries a germinal mutation although he or she is phenotypically normal. Several have been theories fashioned to explain these rare cases, including epigenetic modulation and somatic mosaicism in the parent. Additionally, some large kindreds have demonstrated the presence of two independent, distinct somatic mutations in different family members.
In these scenarios as well as others, parents of children with retinoblastoma are often eager to undergo genetic analysis to determine the risk that siblings or future children will develop the disease. Some centers have devised economical ways to routinely offer genetic screening to all retinoblastoma patients with the ability to detect sequence alterations in the Rb1 gene in about 80% to 90% of patients with a germinal mutation in peripheral blood or tumor tissue available.11 These programs utilize multiplex ligation–dependent probe amplification (MLPA) to detect large deletions or duplications, microsatellite analysis to detect loss of heterozygosity (LOH), and denaturing high-performance liquid chromatography (D-HPLC) analysis to detect point mutations and small insertions or deletions, and quantitative multiplex polymerase chain reaction (PCR) of short fluorescent fragments (QMPSF).
Finally, because the gene is expressed in all adult tissues and the RB1 protein plays such a vital role in controlling cell proliferation, all patients who carry a germinal Rb1 mutation are at risk for the development of additional nonocular cancers. These cancers can occur as late as 40 years or more after the initial development of retinoblastoma. The incidence of these cancers is significantly increased in patients who receive external beam radiation, especially when given before the age of 1 year. Patients should be educated about their risk factors for developing these cancers, as it is these cancers, not retinoblastoma, that are the leading cause of death in patients with the germinal RB1 mutation.12 See “Select Results” later in this chapter for more information on additional nonocular cancers.
Anatomy
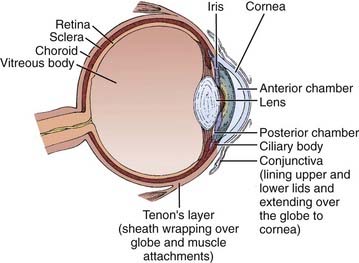
Figure 65-1 illustrates the important anatomic structures of the eye. The inner layer is the retina, where the photoreceptors are located. Retinoblastoma is thought to originate from an unidentified progenitor cell in this layer. The normal retina extends from the posterior “pole” forward to a region just behind the lens, in cross-section called the ora serrata. The middle layer of the eye is the choroid; this pigmented structure is continuous anteriorly with the ciliary body and iris, and together these are considered to be the uveal tract. The outermost layer of the eye is the sclera, a tough and radioresistant structure that fuses with the cornea anteriorly. The anterior chamber of the eye is between the cornea and the iris; the posterior chamber is just behind it, between the iris and the lens. Both chambers are filled with aqueous humor, produced by the ciliary body. Behind the lens is the vitreous chamber, with vitreous humor, a thick clear gel-like substance. In advanced retinoblastoma, “seeding” of the tumor is noted in this chamber.
Pathology
The diagnosis of retinoblastoma is routinely made without pathologic confirmation. Needle biopsies are rarely, if ever, indicated, as puncturing the eye can lead to extraocular extension and orbital invasion. Aqueous taps, previously explored as a possible diagnostic tool, are inappropriate for the same reason.13
When enucleation is the treatment of choice, histopathologic examination of the globe can be useful in providing information about potential extraocular extension (Fig. 65-2). For optimal prognostic information, the globe should be fixed for at least 24 hours, with the optic nerve preserved for separate evaluation.14
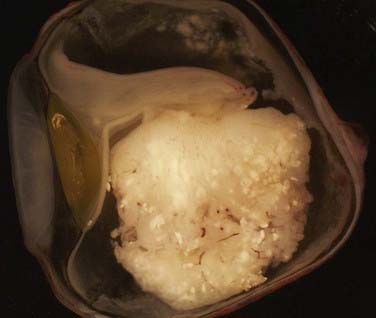
FIGURE 65-2 • Gross section of a globe, showing extensive disease in the retina, vitreous body, choroid, and optic nerve.
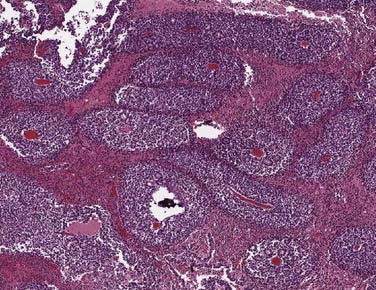
Under the microscope, the appearance of the retinoblastoma cells can range from small, round, undifferentiated cells with frequent mitoses, as seen in Fig. 65-3, to differentiated cells that form rosettes called Flexner-Wintersteiner rosettes.14 Important observations in assessing the risk of the child for distant or local relapse include careful assessment of the degree of invasion of tumor cells beyond the retina and into the choroid and sclera, and assessment of the degree of tumor invasion into the optic nerve beyond the region of the lamina cribrosa.15
Clinical Presentation
The presenting signs and symptoms of retinoblastoma vary depending on the geographic location in which the child presents. In developing countries, children can develop extraocular disease by the time they are diagnosed, with proptosis and an orbital mass. In these cases, the retinoblastoma has often extended directly into the orbit, causing rupture of the globe (Fig. 65-4). Regional nodal metastasis may be found in the preauricular or submandibular regions or the children may have widespread metastatic disease. These children are older at diagnosis (age 4 to 6 years) than patients in the United States, and few survive. In the United States, when there is no family history of disease, the median age at presentation is 1 year for bilateral retinoblastoma and 2 years for unilateral disease. Most children in the United States present with only intraocular disease and are diagnosed with signs rather than symptoms.
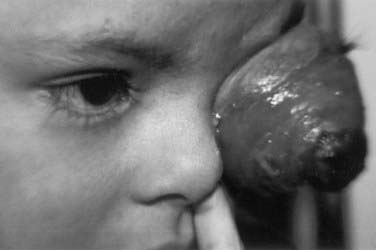
FIGURE 65-4 • A child with extensive orbital disease: the most common presentation in the developing world.
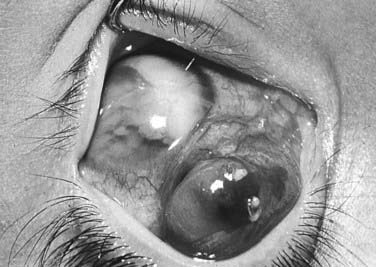
In the United States, the most common presenting sign of retinoblastoma (60% of cases) is leukocoria or a white pupillary reflex that is sometimes referred to as a cat’s eye reflex. Leukocoria can be the result of the tumor itself, a secondary retinal detachment associated with the tumor, or a reflection of light from the white mass at the posterior portion of the eye (Fig. 65-5).
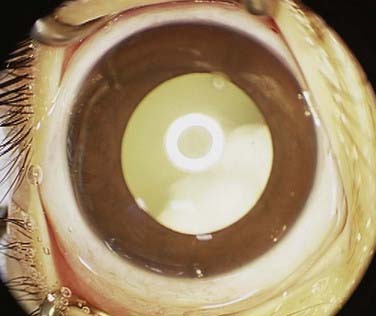
FIGURE 65-5 • A child with the “cat’s eye” reflex, with retinoblastoma visualized as the white material in the pupil.
The second most common sign is strabismus, misalignment of the two eyes. Of the 20% to 25% of patients who present with this sign, 10.5% have eyes crossed in (esotropia) and 5.2% out (exotropia).16 Esotropia in children is generally more common than exotropia, such that an infant with exotropia must be suspected to have retinoblastoma until proved otherwise. The strabismus in retinoblastoma patients is caused by tumor or retinal detachment located in the fovea of the eye, the area of central vision. Although patient survival is independent of whether patients present with strabismus or leukocoria, ocular survival rates are significantly lower for patients who present with leukocoria, as the disease tends to be more advanced at presentation in these cases.17
Presenting signs in the United States that occur in less than 5% of cases include: anisocoria (different-sized pupils); heterochromia (different-colored irides); hyphema (blood in the anterior chamber); tumor hypopyon (tumor in the anterior chamber); nystagmus, a unilateral fixed and dilated pupil; and failure to thrive. In cases in which the patient has gross chromosomal abnormalities, children can present with other findings, such as absence or hyperplasia of the thumbs, mental retardation, prominent nasofrontal bones, large malformed ears, hypospadias, bifid scrotum, and extra digits. Only 7% of patients are detected on routine pediatric screening by a pediatrician, whereas 72% of cases are detected by a family member or friend.17
When there is a family history of retinoblastoma, diagnosis tends to occur earlier, with a median age at diagnosis of 5 months for patients with unilateral disease and 11 months for patients with bilateral disease.18 Sixty percent of all retinoblastoma patients diagnosed under 6 months have no signs or symptoms, but are examined and diagnosed because of a family history of retinoblastoma.19 At our center, children with a family history of retinoblastoma typically undergo frequent serial fundoscopic examinations beginning within 24 to 48 hours of birth and continuing until at least 28 months of age. Twenty-eight months is chosen as the earliest possible time to stop examining these patients because it is the oldest age at which a patient with a positive family history of retinoblastoma has developed his or her first tumor in a previously documented disease-free eye.20 These serial examinations have a significant impact on ocular outcome. Patients who began undergoing screening examinations at our center as newborns because of a family history had a 68% ocular 5-year survival rate, whereas nonscreened patients with a family history had only a 38% ocular survival.17
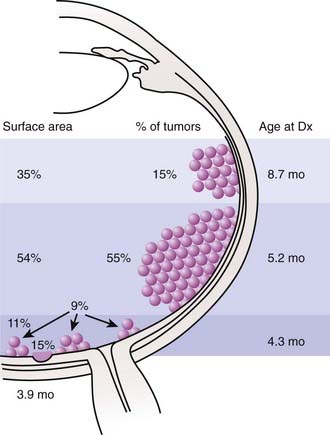
The anatomic location of these tumors within the retina and the age at which the lesions develop have been well studied (Fig. 65-6). Retinoblastoma may be present anywhere in the retina at birth, but as age at tumor development increases, tumors develop progressively farther into the periphery. Thus macular tumors present earliest and peripheral anterior tumors present later. In our center, the average macular tumor is diagnosed at 5.6 months and never presents after 15.5 months. In contrast, peripheral tumors are diagnosed at an average of 16.4 months and can present as late as 8 years of age. This pattern of ocular tumor development has important implications for the radiation oncologist. Since many young children will have no tumors in the anterior retina, there is no need to treat this area, thereby decreasing the incidence of radiation-induced cataracts. However, future development of new peripheral tumors should be expected and the family should be informed of this likelihood. The development of these tumors does not represent a failure of radiation, but rather the inevitable process of tumorigenesis in genetically altered cells. Furthermore, the family of a child with multifocal unilateral retinoblastoma can be assured that new tumors that develop in the fellow eye will rarely originate in the fovea, so the outlook for vision in the fellow eye is typically favorable.
Routes of Spread
The natural course of untreated retinoblastoma follows one of five routes of progression: contiguous spread through the choroid, sclera, and orbit; extension along the optic nerve directly into the brain; invasion of the subarachnoid space and leptomeninges via the cerebrospinal fluid; hematogenous spread to bone marrow, bone, liver, and dissemination via conjunctiva through the lymphatic system.8 In the United States, the few patients with unilateral retinoblastoma who develop metastases usually have them at the time of diagnosis. The majority metastases occur within 5 years of diagnosis.18
Diagnosis of Retinoblastoma
All retinoblastoma tumors are drawn on an expanded concentric circle drawing of the globe called a fundus diagram, used to document the presence, location (in reference to the optic nerve and fovea of the eye), and size of the retinoblastoma tumors. Some ophthalmologists document the presence of tumors in the posterior pole by taking color photographs with a hand-held fundus camera while the patient is under anesthesia. Figure 65-7 demonstrates both techniques in the same child. The original tumor drawing is referred to during follow-up visits, and new drawings are composed to document treatment response. Although the ophthalmoscopic examination should always be considered the primary method for evaluation of retinoblastoma, several ancillary tests can assist in the diagnosis, especially when there is no clear view of the fundus.
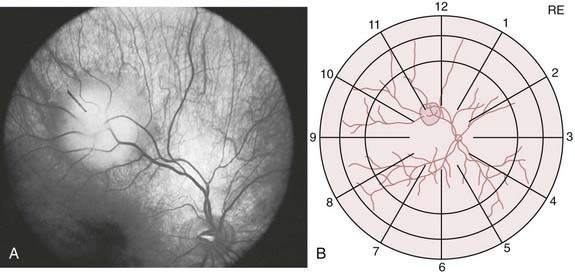
FIGURE 65-7 • A fundus photograph of an eye with retinoblastoma (A) and the corresponding sketch of the disease in the eye diagram (B).
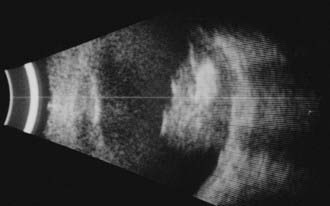
Ophthalmic ultrasonography is one of the most frequently performed ancillary tests for retinoblastoma. It is noninvasive, safe, repeatable, and immediately interpretable. Ultrasonography should be performed on both eyes and can be performed with or without general anesthesia. B-scan ultrasonography reveals a two-dimensional (2D) cross-sectional view of the eye, confirms the presence and the relationship of the solid tumor to other anatomic structures within the eye, and detects the size and shape of the tumors (Fig. 65-8). B-scan can detect orbital involvement, optic nerve invasion (sometimes), extrascleral extension, and calcification. Because there is calcium (which exhibits high reflectivity on ultrasonographic scans) in the majority of retinoblastoma tumors, high amplitude echoes will remain on the screen even if the gain or sensitivity is lowered. Shadowing defects posterior to the tumor may be present and are caused by the absorption of sound and high reflectivity. A-scan ultrasonography evaluates the internal characteristics and vascularity of the tumor and measures the height of the tumor.
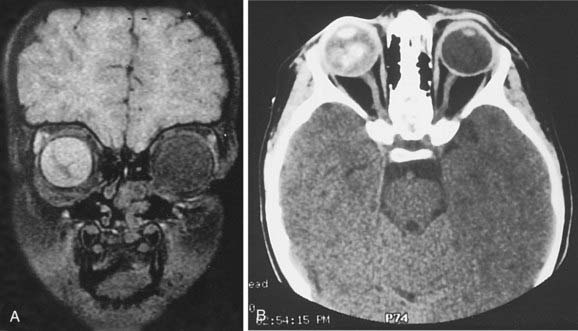
Computed tomography (CT) scans may no longer be routinely appropriate for retinoblastoma patients, as analysis has suggested an increased lifetime risk of other cancers in pediatric patients subjected to this imaging modality.21 Instead, as part of an extent-of-disease work-up, magnetic resonance imaging (MRI) is routinely performed (Fig. 65-9A, B). In addition to its excellent resolution in the diagnosis of extraocular soft tissue disease, MRI can readily distinguish between retinoblastoma and Coats’ disease (see “Differential Diagnosis of Retinoblastoma”), as Coats’ disease appears brighter than retinoblastoma on T2-weighted images due to proteinaceous exudate.8 One disadvantage of MRI is that calcification, a key feature of retinoblastoma, is more easily demonstrated with CT than with MRI.
Differential Diagnosis of Retinoblastoma
Retinoblastoma can be simulated by several other ophthalmic tumors and ophthalmic disorders (Table 65-2). The lesion that most commonly simulates retinoblastoma is Coats’ disease. Coats’ disease is a nonheritable, primarily unilateral anomaly that affects mostly boys, with a later age at diagnosis than retinoblastoma (mean age at diagnosis is 6 years). Ophthalmic examination reveals telangiectatic vessels of the retina with intraretinal and subretinal exudates and an exudative retinal detachment. Presence of a yellow-green sheen on indirect ophthalmoscopic examination aids in differentiating Coats’ from retinoblastoma. When rubeosis iridis, glaucoma, and corneal edema develop, the fundus may be impossible to view. Under these circumstances, ultrasonography can be helpful.
Ocular toxocariasis also commonly mimics retinoblastoma. Nematode endophthalmitis and solitary granulomas are the two forms most often confused with retinoblastoma. Patients with nematode endophthalmitis present at an average age of 6 years with pain, photophobia, inflammatory signs including vascular injection, cataracts with cells and flare on the slit-lamp examination, and sometimes posterior synechiae. Patients with solitary granulomas rarely have external ophthalmic inflammatory signs but present with a white, pearl, or yellow ball-shaped mass. Mild or severe vitreal traction may be seen.6
Staging of Retinoblastoma
The Reese-Ellsworth Classification scheme is the most commonly used classification system for describing intraocular tumors (Table 65-3).22 It is not a true staging scheme, for untreated patients do not progress from group I to higher groups. It was originally developed to predict prognosis in eyes that were treated with lateral photons via external beam irradiation and other vision-preserving techniques. A higher numeric classification signifies that a tumor is located more anteriorly and that there is a decreased success rate treating the eye with lateral port external beam radiation. Despite its limited applicability, it has served as an excellent ocular reference for comparison of different series and treatment schemes. Other classification systems have been proposed to predict prognosis with chemoreduction treatments and have similar limitations as the Reese-Ellsworth Classification scheme, the most recent being the international classification system: we currently utilize both.23 The international classification for retinoblastoma classifies tumors as group A if they are less than 3 mm in size. Group B tumors are larger than 3 mm, or are within 1.5 mm. of the optic nerve, or within 3 mm of the fovea, or have less than 3 mm of surrounding subretinal fluid. Group C contains tumors if they have subretinal or vitreous seeds within 3 mm from the retinoblastoma. Group D contains tumors if they have subretinal or vitreous seeds greater than 3 mm from the retinoblastoma. Group E eyes have tumor in more than 50% of the eye, or neovascular glaucoma, invasion of postlaminar optic nerve, choroid (2 mm or more), sclera, orbit, or anterior chamber.23 No single scheme that includes patients with extraocular retinoblastoma has been widely accepted among ophthalmologists. In our center, we used the Abramson-Grabowski Staging System for extraocular disease (see Table 65-3). The TNM system of the American Joint Committee on Cancer, shown for comparison in Table 65-4, is not commonly used. Currently we use the international staging system.24
| Reese-Ellsworth—Intraocular |
| GROUP I |
CNS, Central nervous system; CSF, cerebrospinal fluid; DD, disc diameter.
Table 65-4 Classification of Retinoblastoma Rights were not granted to include this table in electronic media. Please refer to the printed book.
Treatment of Retinoblastoma
The primary goal of retinoblastoma treatment is to ensure the survival of these children. Secondary but also important goals include retention of eyes and of vision. A final goal is the avoidance treatment side effects such as second malignancies, facial bony deformities, or other physical changes that can affect functional well-being.25 Treatment approaches are guided by the presence of intraocular or extraocular disease. This section will concentrate on the treatment of intraocular disease. Because most of the children survive their retinoblastoma, long-term complications of treatment must be factored into initial treatment decisions.
The treatment modalities currently used for intraocular disease include enucleation, external beam radiation therapy, cryotherapy, photocoagulation and transpupillary thermotherapy, episcleral brachytherapy, and systemic, intra-arterial, and periocular chemotherapy.26–28 Intra-arterial chemotherapy is a recent technique designed to deliver chemotherapy directly to the arterial circulation of the affected eye, minimizing the systemic dose and complications while maximizing the concentration to the tumor. This technique shows encouraging preliminary results.28 Localized excision of the tumor from within the eye is never performed because of the risk of tumor seeding during the procedure (Fig. 65-10).
Enucleation
Enucleation is still widely used for retinoblastoma worldwide. Patients considered for enucleation include those with: unilateral or bilateral Reese-Ellsworth group V eyes, active tumor in a blind eye, and painful glaucoma from tumor invasion. Patients are also considered for enucleation if they have failed all other forms of treatment, or if they have active tumor and cannot be followed.23 In the past, some centers have advocated, in bilateral cases, enucleation of the eye with worse disease and treatment of the fellow eye with external beam radiation or other focal techniques.25 In our center, however, we find that it is often difficult to predict which eye will have disease progression and which one will respond to treatment. An eye that appears worse at diagnosis may in fact result in better vision after treatment than the fellow eye. Therefore, we believe that in a case of bilateral disease in which neither eye meets the criteria for enucleation, but both eyes have disease too advanced for focal techniques, bilateral external beam radiation or chemotherapy may be attempted if the family understands the short- and long-term risks.
Cryotherapy
Cryotherapy was first used for retinoblastoma by Lincoff in 1967 and has since been used extensively.29–31 Cryoprobes, which look like blunted pencils, are applied to the scleral surface corresponding to the location of the intraocular tumor focus. Under direct visualization, the tumor to be treated is identified by pressing the probe on the sclera (“indenting”) and a foot switch is pressed to commence the freezing. The tumor is completely covered by ice within 1 or 2 minutes. It has been shown that the tumors are frozen quickly (faster than −90° C per minute), forming intracellular ice crystals that expand and lead to cell death by causing protein denaturation, pH changes, and finally cell membrane rupture. There may also be a local vascular effect created by immediate slowing of blood flow, subsequent destruction of endothelial cells, and a possible local immunologic effect.
Cryotherapy may be used as the primary treatment for small peripheral retinoblastomas or as secondary treatment for recurrent tumors treated previously. Size and location of the tumor are factors that determine success with cryotherapy. Retinoblastomas that are successfully treated with cryotherapy include small tumors (4.5 to 6 mm) not located at the vitreous base. The average tumor this size requires several treatments, and sessions are usually scheduled 3 to 6 weeks apart. Larger tumors require more treatments and have a poorer chance of success. Tumors with widespread vitreous involvement and seeds that settle at the vitreous base rarely respond to this treatment.30,32,33 However, cryotherapy is uniquely useful for tumors with localized vitreous seeding overlying the tumor apex. In these cases, the freeze is extended in area to include the location of the seeds.30
Tumors treated with cryotherapy will ultimately shrink and completely disappear if treatment is successful. In our center, 90% of tumors that are less than 3 mm in diameter are cured permanently.30 The cryotherapy scars are flat and pigmented from the reaction of the overlying retinal pigment epithelium (RPE).
Photocoagulation
Light coagulation with the xenon arc photocoagulator was first described by Meyer-Schwickerath in 1957, who demonstrated that intense, focused, white light could be used to treat intraocular retinoblastomas.20 Currently, patients are treated while under general anesthesia with their pupils dilated. One to three barriers of photocoagulation burns are aimed through the anterior chamber at the tumor’s feeding vessels. The tumor itself is never treated because it does not absorb very much of the light energy due to its color. When the tumor’s blood supply is destroyed, the tumor begins to involute within 1 week of treatment. Traditionally, xenon arc photocoagulation (wavelength 250 to 1500 nm) was used, but currently tumors can be treated with argon lasers in the visible range (wavelength 488 to 536 nm) or diode/infrared lasers in the invisible range (wavelength 810 nm) with success.9
Photocoagulation is employed as primary treatment for select, small retinoblastomas. The following tumor features correlate with the success of photocoagulation: small size (4.5 mm), anterior location, and low elevation (tumor ≤50% of the base diameter).34 Photocoagulation is not used for tumors that involve the optic disc or fovea, as this technique would result in loss of central vision. In such cases, external beam radiation or chemoreduction are typically used.
More than 70% of tumors with the previously mentioned characteristics are cured with photocoagulation.34 Success is attained when a flat scar develops, and long-term recurrence of photocoagulated tumors is extremely unusual. Potential complications of photocoagulation include cataracts, iris burns, vitreous hemorrhage, retinal detachment, and traction effects.
Transpupillary Thermotherapy
Transpupillary thermotherapy (TTT) was first used as treatment for retinoblastoma by Lagendijk, although other researchers in the Netherlands had previously studied its use as treatment for choroidal melanoma.35 Thermal energy is delivered from an 810-nm infrared ophthalmic laser with modifications to the laser’s hardware and software. The mechanism by which TTT causes tumor cell death is different from the mechanism by which classic laser photocoagulation destroys tumors. With TTT, the temperature is thought to be lower (45° C to 60° C), and the thermal effect leads to apoptosis rather than burning. Because the effects of TTT rely on the direct killing of tumor cells, the laser beam is aimed directly at the tumor rather than at the feeding vessels as in photocoagulation.
Indications for TTT have not yet been established. In the largest series to date, patients with viable retinoblastoma within the retina or subretinal space with less than 1 mm of overlying subretinal fluid were included.36 Larger tumors and tumors with vitreous seeding were excluded. Our group treats primarily tumors that are 3 mm or less in base diameter and are located in the posterior pole.
Limited studies demonstrating the effectiveness of TTT have been completed. Shields et al. have published the largest series of retinoblastomas treated with chemotherapy and TTT to date, in which 188 retinoblastomas in 80 eyes of 58 patients were treated.36 One hundred eight of the tumors were treated simultaneously with systemic chemotherapy and TTT, and mean follow-up was 12 months. Although 86% of the tumors demonstrated regression, complications were significant and included focal iris atrophy, focal paraxial lens opacity, and sector optic disc atrophy. Macular tumors specifically were found to have high control rate (100%) for Reese-Ellsworth Classification groups I to IV and 83% for group V eyes with a similar side-effect profile.37
Chemotherapy (Chemoreduction)
Systemic chemotherapy for intraocular retinoblastoma has been used since 1953, when Kupfer used intravenous nitrogen mustard as a primary treatment for retinoblastoma.38 In 1955, Reese et al. used intracarotid triethylenemelamine (TEM) in combination with external beam radiation in order to decrease the required dose of radiation.39 Currently, “chemoreduction” (using chemotherapy to reduce the size of tumors) is an area of active clinical and basic science research, motivated by the desire to avoid enucleation and external beam radiation in children at risk for developing nonocular cancers.9
Most studies of chemoreduction for retinoblastoma have used vincristine, carboplatin, and an epipodophyllotoxin, either etoposide or teniposide. Currently, six cycles of vincristine, carboplatin and etoposide are employed as the standard starting regimen. The addition of cyclosporine as a P-glycoprotein inhibitor has been suggested to decrease the ability of tumor cells to transport antineoplastic drugs from the intracellular space, avoiding multidrug resistance.40,41 Currently, choice of agents as well as number and frequency of cycles varies at different institutions.
The results of studies examining chemoreduction followed by focal therapies have been most promising for patients with Reese-Ellsworth group I to III eyes. For these patients, several authors have demonstrated that enucleation can be successfully avoided almost 100% of the time.40–50 Results for patients with Reese-Ellsworth groups IV and V eyes have been more discouraging. A meta-analysis of studies demonstrated that only 30% of group IV and V eyes avoid both external beam radiation and enucleation. Forty-seven percent of eyes require external beam radiation but avoid enucleation, and 35% of eyes require enucleation (with or without previous external beam radiation).51 More recent publications have reported successful avoidance of both external beam radiation and enucleation in 17% to 83% of 132 group V eyes.51
A potential complication of chemoreduction is the development of secondary nonocular cancers. Several agents used in current chemoreduction studies for retinoblastoma have previously been demonstrated to increase the risk for secondary cancers either in retinoblastoma patients or in patients treated with these drugs for other primary malignancies. Cases of acute myelogenous leukemia have been reported in children who have received chemotherapy for their initial disease.52 The follow-up periods of chemoreduction studies with retinoblastoma patients to date are inadequate to assess the development of second cancers in these patients. Studies with long-term follow-up are needed.
The Role of Radiation Therapy
External beam radiation therapy has been successfully employed for retinoblastoma since Hilgartner experimented with x-ray treatments in Austin, Texas in 1903.53 Early experience demonstrated the efficacy of tumor regression with radiation therapy. However, significant complications were frequently observed due to the techniques employed.54,55 Complications included keratitis sicca, keratinization of conjunctiva and sclera, lachrymal gland atrophy/fibrosis, loss of lashes, corneal ulcers/perforation, hyphema, rubeosis iridis, glaucoma, iritis/uveitis, cataract, vitreous hemorrhage, retinal vascular damage, optic nerve infarction, fat atrophy in the orbit, and arrest of orbital growth.23 Currently most of these complications have been eliminated or minimized by reducing the total radiation dose, by changing the source of radiation to high energy photons and electrons and positioning of the portals, and by using fractionated doses.
Simulation
A decade ago, we first analyzed our experience with a combination of lateral photon and anterior fields, employing a lens block and general anesthesia when the anterior electron field was treated. We found that because of the small size of the lens and the relationship of the posteriorly located tumor to the shadow cast by the lens block, local recurrences were higher in this group of children, and development of cataracts was similar to the lateral technique. Thus, we favor the lateral technique.56
The simulation procedure begins by placing the child straight in the mold, employing lateral lasers and lateral landmarks of the eye and orbit. A third laser is placed midline down the child’s nose if both eyes are to be treated, or through the pupil of the treated eye if only one eye is to be treated. The gantry is then rotated 90 degrees and, with the use of a CT or MRI scan of the globe to reference lens position, and a split beam or independent jaws, the central ray of the beam is placed 2 to 3 mm posterior to the surgical limbus, corresponding to the posterior pole of the lens (Fig. 65-11). This structure anatomically correlates with the anterior extent of the retina at the ora serrata. In a child under the age of 1 year, a field size measuring 3 cm wide and 4 cm high is an adequate field size, around which blocks are designed, shaping the field to the bony orbit and including 1 cm of optic nerve as it exits the globe. Care must be taken to visualize the pituitary fossa and shield it from the primary beam. We use Cerrobend to define the entire field in all directions, minimizing the penumbra when compared to using the jaws (Fig. 65-12). Similar results can also be achieved using a CT-simulator.


Radiation Treatment Plan
Because of the marked variation in the relationship of the globe to the bony orbit, all treatment planning is done from CT or MRI images. A transaxial image that demonstrates the lens as well as soft tissue and bone landmarks is selected. Fig. 65-13 illustrates a plan for patients with bilateral disease. The prescription isodoses cover the retina from the equator posteriorly. A dose gradient exists anteriorly toward the lens. As noted earlier, because of the philosophy of our group, every effort is made to spare the lens, and this sometimes results in less than the prescription dose to the most anterior portion of the retina. We find these areas are easy to monitor with post-treatment examinations under anesthesia and are amenable to cryotherapy should the need arise to treat new tumors in the region.
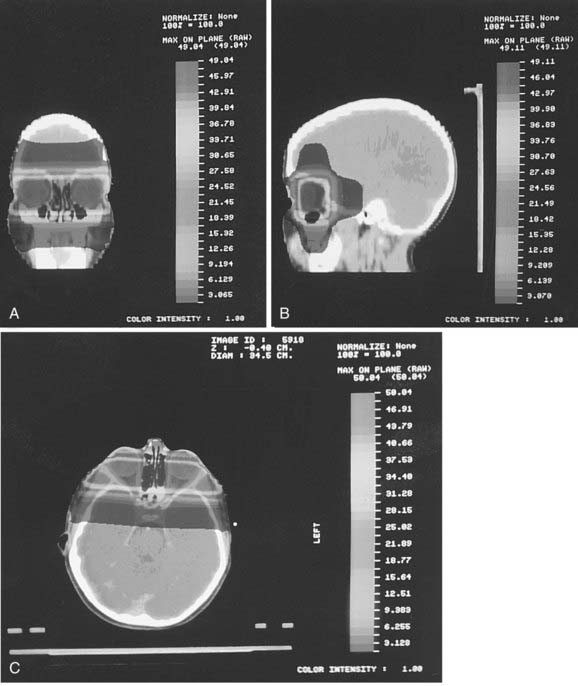
Proton Therapy
Proton therapy for the pediatric age group has a dosimetric advantage over photon therapy for many anatomic sites, and retinoblastoma has been included in several dosimetric demonstration studies. The group at the M.D. Anderson Cancer Center chose three patients who had completed radiation treatment for unilateral intraocular retinoblastoma with conventional photon therapy. The physics planning group then replanned each child with protons as well as several other radiation techniques described in the literature. Target coverage was 100% with protons, but also excellent with both 3D and IMRT planning (95% and 96%). Dose to the lens was significant with all three techniques. Dose to the bony orbit and the optic nerve were markedly reduced with the proton plan.56a
Retinoblastoma patients have been treated at the Harvard Cyclotron and then at the newer Northeast Proton Center at the Massachusetts General Hospital since 1986. A recent publication from that group describes the clinical data as “currently under review.” In the same paper, treatment plans for hypothetical retinoblastoma lesions in the nasal, central and temporal locations of the posterior retina are depicted. The study concludes that that proton treatment has the “potential to precisely irradiate retinoblastoma while staying below threshold doses for the bony growth centers.” Low doses to the lens are also achieved in this treatment planning exercise.56b
Episcleral Brachytherapy
Episcleral brachytherapy was pioneered by the British eye surgeon Henry Stallard. He designed cobalt applicators that were curved to fit a child’s eye with suture holes built in to attach the plaque to the sclerae. The plaques were left in place for 3 to 7 days and radiation was delivered at 40 Gy to the tumor apex; in recent years, this technique has been refined.57
Following plaque treatment, tumors show a distinct response known as type IV radiation regression pattern.58 A sharp demarcation distinguishes the profound radiation effect from an area where no effect is seen (this occurs in <1 mm of retina). The tumor, overlying retina, and choroid disappear (Fig. 65-14). Curiously, some overlying vessels may persist. Local tumor recurrence is rare.
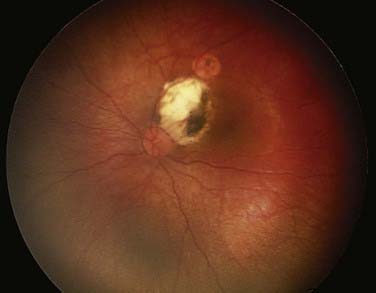
FIGURE 65-14 • A type IV regression pattern after plaque therapy, illustrating the white sclera and calcium deposits.
Side effects from plaque therapy for retinoblastoma are far less common than from plaques for melanoma. The apical dose of a melanoma is usually 85 Gy, whereas for retinoblastoma it is 40 Gy. Melanomas range from 2.5 to 10 mm in height whereas retinoblastomas usually range from 2 to 4 mm in height. The difference in the doses delivered to the surrounding retina and underlying choroid are significant. Thus optic neuropathy and radiation retinopathy from treated retinoblastoma tumors is unusual however can be seen in up to 24% children that have been treated with chemotherapy.59 Cataracts will occur when both iodine and cobalt plaques are placed over anterior retinoblastoma tumors. The cataracts may not be evident for years and usually do not require surgery.
When plaque therapy is used as a salvage technique in eyes that have failed other combinations of therapy, including external beam radiation, 50% to 89% of such eyes can be salvaged.60,61 Plaques have not been shown to increase the incidence of second tumors in patients who have also received external beam radiation.
In an attempt to treat the entire eye while avoiding dose to the bony orbit and sinuses, a group in South Africa reported on using a custom-designed iodine-125 applicator with four “claws” that attach to a periorbital anchoring ring. Each “claw” is designed to fit between an extraocular muscle group and is loaded with three iodine-125 seeds. Promising early results were seen in patients with Reese-Ellsworth groups I to III disease, although considerable dose inhomogeneity was noted across the target volume.62
Normal Tissue Tolerance
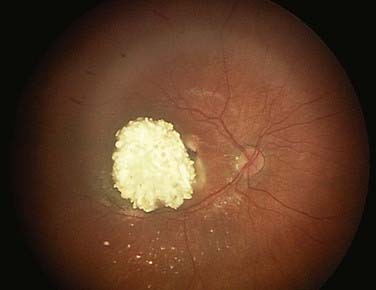
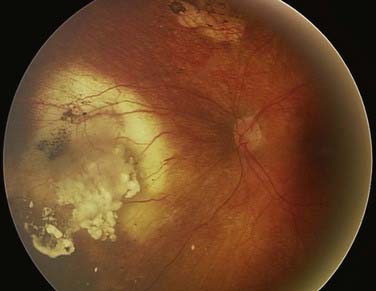
The late response of the tumor and the appearance of the retina itself have been well described by our group. With the use of indirect ophthalmoscopy when the treated tumor shrinks, the tumor loses its vascularity and picks up calcium; this is classified as a type I radiation regression pattern (Fig. 65-15). In contrast, a type II regression pattern is defined as less dramatic shrinkage of the tumor, ranging from 25% to 50%, with the lesion appearing a lucent gray color with no calcium. A type III regression pattern is a combination of type I and type II and is the type we see most frequently (Fig. 65-16). When plaque radiotherapy is used in the treatment, because of the increased sclera-to-tumor dose ratio, the regression pattern seen is described as a type IV regression pattern, notable for destruction of the overlying retina with a white appearance signifying visible sclera (see Fig. 65-14). In recent years, a fifth regression pattern has been observed, labeled type 0. This results when the posttreatment examination of the retina reveals no evidence at all of the previously existing retinoblastoma. It is uncommon to observe except in the smallest-sized presenting tumors.63
Facial and temporal bone hypoplasia can occur following external beam radiation in very young children. It is most marked when both eyes are treated with parallel opposing fields and in children who are under the age of 6 months at diagnosis.64 Because of the rapid head growth in the infant, this hypoplasia is noted to a lesser degree in older children. With the use of the three-field–modified lateral beam technique, the hypoplasia is minimized because of the smaller entrance dose at each of the three fields.
Select Results
In the United States, retinoblastoma is a highly curable childhood cancer. Cancer registry reports in Europe and the United States have demonstrated 5-year survival rates of 90% and 98%, respectively.2,65 Survival of children treated with external beam radiation, regardless of laterality, Reese-Ellsworth staging, or technique, ranges from 85% to 100%, excluding old orthovoltage series results.66–72
Local control in the irradiated eye, defined as preservation of the eye and based on seven large series, is shown in Table 65-5.66–72 In many of these series, postradiation therapy consisting of cryotherapy, photocoagulation, or further radiation was given; however, because of the difficulties in separating recurrence of previously treated tumors from new tumors located elsewhere in the retina, preservation of the eye is the definition of control that is used. Of note in the Stanford series, most eyes were enucleated because of complications from the higher doses of radiation, not because of tumor recurrence.66
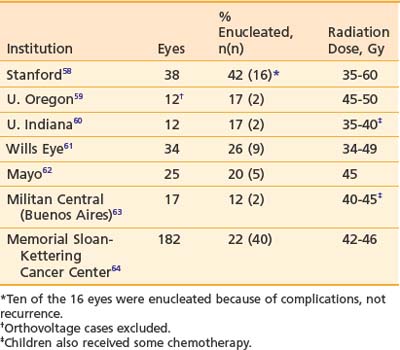
Despite these excellent results, the potential late sequelae of external beam radiation for retinoblastoma patients are serious. As noted previously, patients carrying the germinal Rb1 mutation are in danger of developing additional nonocular cancers, and the use of external beam radiation increases the risk for some of these cancers significantly (Table 65-6).73–76 Cumulative incidence reports of second nonocular cancers among retinoblastoma survivors vary, but it is estimated that a rate of 1% per year of life approximates the risk.77 Patients who develop a second cancer and then survive that cancer have an increased risk for the development of additional nonocular tumors of approximately 2% per year from the time of second tumor diagnosis.78 The average latency period between subsequent tumor diagnoses becomes progressively shorter with each additional cancer that develops.
Table 65-6 Risk Factors for Second Nonocular Cancer Development in Retinoblastoma Survivors
| Increase in Incidence Factor of Second Cancers | |
|---|---|
| Presence of germinal mutation in RB1 gene | 1000× increase65,66 |
| External beam radiation | 400-600× increase66 |
| External beam radiation given at younger than 1 year of age | 2-8× increase67,68 |
| Dose of external beam radiation | 12× increase at 60 Gy66 |
| Presence of lipomas | 8× increase69 |
Second nonocular neoplasms observed in survivors of germinal retinoblastoma include, in order of most common to least: osteosarcomas (bone, nasal cavities, and nasopharynx), soft tissue sarcomas (connective and soft tissues, nasal cavities, orbit, and other sites), pineoblastomas, cutaneous melanomas, brain tumors, Hodgkin’s disease, lung cancer, and breast cancer.74–80 Survivors of hereditary retinoblastoma are also at increased risk for the development of lipomas throughout the body. For unclear reasons, lipomas seem to be a clinical marker for a risk of second cancer development.77
In recent years, the effects of external beam radiation on the development of these cancers have been studied extensively. Children irradiated during the first year of life are between two and eight times as likely to develop osteogenic sarcomas of the skull and face bones and soft tissue sarcomas of the head as those children radiated after the age of 1 year.75,76 Patients treated with orthovoltage, higher doses, and older methods of radiation delivery that lead to increased superficial skin and bone exposure are at higher risk for subsequent tumor development in the radiation field.79 The dose–response curve for the development of soft tissue sarcomas in bilateral retinoblastoma patients has been described. The odds ratio at 0 to 4.9 Gy was 1.0; at 5 to 9.9 Gy, 1.91; at 10 to 29.9 Gy, 4.6; at 30 to 59.9 Gy, 6.4, and at greater than 60 Gy, 11.7.74 Additional nonocular cancers develop both within and outside the field of radiation. Our group has reported that in irradiated patients who develop second malignancies, the tumors are within the radiation field two thirds of the time and outside the field one third of the time. In nonirradiated patients who develop second tumors, the tumors are outside the hypothetic field two thirds of the time and within the hypothetic field one third of the time.73,74
Fig. 65-17 demonstrates the timing of the development of each type of second cancer in retinoblastoma survivors. In general, external beam radiation therapy given before the age of 1 year both increases the risk that patients will develop certain nonocular cancers and causes the cancers to develop at an earlier age. For example, soft tissue sarcomas of the head, which occur at approximately age 30 in nonirradiated patients, can occur as early as the teenage years in patients irradiated at less than 1 year of age. Osteogenic sarcomas of the skull, for which nonirradiated patients carry a 1 : 1000 risk per year of developing, occur four times more commonly during the late teenage years in patients radiated under 1 year of age.
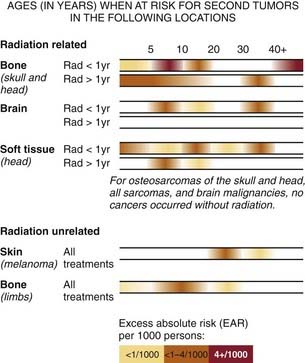
FIGURE 65-17 • Risk of developing different types of second cancers for germinal retinoblastoma survivors from birth to age 40.
(From David H. Abramson, MD; Christopher M. Frank, 1997, unpublished handout.)
Our group published a report on the largest series of retinoblastoma survivors who developed a second cancer, survived, and went on to develop third, fourth, or fifth nonocular tumors.79 Survivors of retinoblastoma who develop second malignancies and survive are at an even higher risk for the development of additional cancers than they were for the development of a second tumor. The distribution of tumor sites in the second-tumor group suggests a nonrandom pattern of third, fourth, and fifth tumor development. Of patients with skin cancers as their second tumors, skin cancers also represent most of the third, fourth, and fifth tumors that develop in this group. Of patients treated for a second tumor in the skull in whom a third tumor develops, most are diagnosed with a soft tissue sarcoma in the head as the third tumor. The locations and expected ages at which soft tissue sarcomas of the head and osteogenic sarcomas of the long bones develop are consistent with the patterns observed in second tumors (see Fig. 65-17).
1 Spencer WH Ophthalmic Pathology; Vol 1. 1986; WB Saunders, Philadelphia.
2 Novakovic B. U.S. childhood cancer survival, 1973–1987. Med Pediatr Oncol. 1994;23:480.
3 Abramson DH. Retinoblastoma: diagnosis and management. CA Cancer J Clin. 1982;32:130.
4 Abramson DH. Retinoblastoma. Pediatr Emerg Casebook. 1985;3:3.
5 Abramson DH. The diagnosis of retinoblastoma. Bull N Y Acad Med. 1988;64:283.
6 Knudson AGJr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68:820.
7 Friend SH, Bernards R, Rogelj S, et al. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature. 1986;323:643.
8 Abramson DH, Dunkel IJ, McCormick B. Neoplasms of the eye. Cancer Med. 2000:1083.
9 Corson TW, Gallie BL. One hit, two hits, three hits, more? Genomic changes in the development of retinoblastoma. Genes Chromosomes Cancer. 2007;46:617-634.
10 Dimaras H, Khetan V, Halliday W, et al. Loss of RB1induces non-proliferative retinoma: increasing genomic instability correlates with progression to retinoblastoma. Hum Mol Genet. 2008;17:1363-1372.
11 Houdayer C, Gauthier-Villars M, Laugé A, et al. Comprehensive screening for constitutional RB1mutations by DHPLC and QMPSF. Hum Mutat. 2004;23:193-202.
12 Eng C, Li FP, Abramson DH, et al. Mortality from second tumors among long-term survivors of retinoblastoma. J Natl Cancer Inst. 1993;85:1121.
13 Abramson DH. Lactate dehydrogenase and retinoblastoma. In: Jakobiec FA, editor. Ocular and adnexal tumors. Birmingham, Ala: Aesculaoius; 1978:454.
14 Messner EP. Histopathology of retinoblastoma. In: Alberti WE, Sagerman RH, editors. Radiotherapy of intraocular and orbital tumors. Berlin: Springer-Verlag; 1993:131.
15 Ellsworth RM. Retinoblastoma. In: Duane TD, editor. Clinical Ophthalmology. Hagerstown, MD: Harper & Row; 1976:1.
16 Abramson DH, Frank CM, Susman M, et al. Presenting signs of retinoblastoma. J Pediatr. 1998;132:505.
17 Abramson DH, Beaverson K, Sangani P, et al. Retinoblastoma: do the presenting signs have prognostic value for outcome? J Pediatrics. 2003;112:1248-1255.
18 Abramson DH, Ellsworth RM, Grumbach N, et al. Retinoblastoma: survival, age at detection and comparison 1914–1983. J Pediatr Ophthalmol Strabismus. 1985;22:246.
19 Abramson DH, Servodidio CA. Retinoblastoma in the first year of life. Ophthalmic Paediatr Genet. 1992;13:191.
20 Meyer-Schwickerath G. Prophylactic treatment of retinal detachment by light-coagulation. Trans Opthal Soc UK. 1956;76:739-750.
21 Brenner D, Elliston C, Hall E, et al. Estimated risks of radiation-induced fatal cancer from pediatric CT. Am J Roentgenol. 2001;176:289.
22 Reese AB. Tumors of the Eye. New York: Harper & Row; 1963.
23 Shields CL, Shields JA. Basic understanding of current classification and management of retinoblastoma. Curr Opin Ophthalmol. 2006;17:228-234.
24 Chantada G, Doz F, Antoneli CB, et al. A proposal for an international retinoblastoma staging system. Pediatr Blood Cancer. 2006;47:801-805.
25 Abramson DH. Treatment of retinoblastoma. In: Blodi FC, editor. Retinoblastoma. New York: Churchill Livingstone; 1985:3.
26 Seregard S, Kock E, af Trampe E. Intravitreal chemotherapy for recurrent retinoblastoma in an only eye. Br J Ophthalmol. 1995;79:194-195.
27 Abramson DH, Dunkel IJ, Brodie SE, Kim JW, Gobin YP. A Phase I/II Study of Direct Intraarterial (Ophthalmic Artery) Chemotherapy with Melphalan for Intraocular Retinoblastoma Initial Results. Ophthalmology. 2008 Mar 13.
28 Kim JW, Abramson DH, Dunkel IJ. Current management strategies for intraocular retinoblastoma. Drugs. 2007;67:2173-2185.
29 Lincoff H, McLean J, Long R. The cryosurgical treatment of intraocular tumors. Am J Ophthalmol. 1967;63:389.
30 Abramson DH, Ellsworth RM, Rozakis GW. Cryotherapy for retinoblastoma. Arch Ophthalmol. 1982;100:1253.
31 Abramson DH. Cryotherapy in retinoblastoma. In: Jakobiec F, editor. Advanced techniques in ocular surgery. Philadelphia: WB Saunders; 1984:433.
32 Hopping W, Bunke-Schmidt A. Light coagulation and cryotherapy in retinoblastoma. In: Blodi FC, editor. Retinoblastoma. New York: Churchill Livingstone; 1985:95.
33 Shields JA, Parsons H, Shields CL, et al. The role of cryotherapy in the management of retinoblastoma. Am J Ophthalmol. 1989;108:260.
34 Abramson DH. The focal treatment of retinoblastoma with emphasis on xenon arc photocoagulation. Acta Ophthalmol Suppl. 1989;194:3.
35 Lagendijk JJ. A microwave heating technique for the hyperthermic treatment of tumours in the eye, especially retinoblastoma. Phys Med Biol. 1982;27:1313.
36 Shields CL, Santos MC, Diniz W, et al. Thermotherapy for retinoblastoma. Arch Ophthalmol. 1999;117:885.
37 Schefler AC, Cicciarelli N, Feuer W, Toledano S, Murray TG. Macular retinoblastoma: evaluation of tumor control, local complications, and visual outcomes for eyes treated with chemotherapy and repetitive foveal laser ablation. Ophthalmology. 2007;114:162-169.
38 Kupfer C. Retinoblastoma treated with intravenous nitrogen mustard. Am J Ophthalmol. 1953;36:1721.
39 Reese AB, Hyman GA, Merriam GR, et al. Treatment of retinoblastoma by radiation and triethylenemelamine. Arch Ophthalmol. 1955;53:505.
40 Gallie BL, Budning A, DeBoer G, et al. Chemotherapy with focal therapy can cure intraocular retinoblastoma without radiotherapy. Arch Ophthalmol. 1996;114:1321.
41 Chan HS, DeBoer G, Thiessen JJ, et al. Combining cyclosporin with chemotherapy controls intraocular retinoblastoma without requiring radiation. Clin Cancer Res. 1996;2:1499.
42 Murphree AL, Villablanca JG, Deegan WFIII, et al. Chemotherapy plus local treatment in the management of intraocular retinoblastoma. Arch Ophthalmol. 1996;114:1348.
43 Shields CL, Shields JA, Needle M, et al. Combined chemoreduction and adjuvant treatment for intraocular retinoblastoma. Ophthalmology. 1997;104:2101.
44 Kingston JE, Hungerford JL, Madreperla SA, et al. Results of combined chemotherapy and radiotherapy for advanced intraocular retinoblastoma. Arch Ophthalmol. 1996;114:1339.
45 Greenwald MJ, Strauss LC. Treatment of intraocular retinoblastoma with carboplatin and etoposide chemotherapy. Ophthalmology. 1996;103:1989.
46 Shields CL, De Potter P, Himelstein BP, et al. Chemoreduction in the initial management of intraocular retinoblastoma. Arch Ophthalmol. 1996;114:1330.
47 Gunduz K, Shields CL, Shields JA, et al. The outcome of chemoreduction treatment in patients with Reese-Ellsworth group V retinoblastoma. Arch Ophthalmol. 1998;116:1613.
48 Friedman DL, Himelstein B, Shields CL, et al. Chemoreduction and local ophthalmic therapy for intraocular retinoblastoma. J Clin Oncol. 2000;18:12.
49 Wilson MW, Rodriguez-Galindo C, Haik BG, et al. Multiagent chemotherapy as neoadjuvant treatment for multifocal intraocular retinoblastoma. Ophthalmology. 2001;108:2106. discussion 2114–2115
50 Beck MN, Balmer A, Dessing C, et al. First-line chemotherapy with local treatment can prevent external-beam irradiation and enucleation in low-stage intraocular retinoblastoma. J Clin Oncol. 2000;18:2881.
51 Schefler AC, Jockvich ME, Toledano S, Murray TG. Historical and modern approaches to chemotherapy for retinoblastoma. Expert rev Ophthalmol. 2006;1:83-95.
52 Gombos DS, Hungerford J, Abramson DH, et al. Secondary acute myelogenous leukemia in patients with retinoblastoma: is chemotherapy a factor? Ophthalmology. 2007;114:1378-1383.
53 Hilgartner HL. Report of a case of a double glioma treated by x-ray. Tex Med. 1903;18:322.
54 Reese AB, Merriam GR, Martin HE. Treatment of bilateral retinoblastoma by irradiation and surgery. Report on 15-year results. Am J Ophthalmol. 1949;32:175.
55 Martin HE, Reese AB. Treatment of retinal gliomas by the fractionated or divided dose principal of roentgen radiation: preliminary report. Arch Ophthalmol. 1936;16:733.
56 McCormick B, Ellsworth R, Abramson D, et al. Radiation therapy for retinoblastoma: comparison of results with lens-sparing versus lateral beam techniques. Int J Radiat Oncol Biol Phys. 1988;15:567.
56a Lee C, Bilton S, Famiglietti R, et al. Treatment planning with protons for pediatric retinoblastma, medulloblastoma and pelvic sarcoma: how do protons compare with other conformal techniques? Int J Radiat Oncol Biol Phys. 2005;63:362-372.
56b Krengli M, Hug E, Adams J, et al. Proton radiation therapy for retinoblastoma: Comparison of various intraocular tumor locations and beam arrangements. Int J Radiat Oncol Biiol Phys. 2005;61:583-593.
57 Stallard HB. Radiotherapy for malignant intraocular neoplasms. Br J Ophthalmol. 1948;32:681.
58 Buys RJ, Abramson DH, Ellsworth RM, et al. Radiation regression patterns after cobalt plaque insertion for retinoblastoma. Arch Ophthalmol. 1983;101:1206.
59 Shields CL, Mashayekhi A, Sun H, et al. Iodine 125 plaque radiotherapy as salvage treatment for retinoblastoma recurrence after chemoreduction in 84 tumors. Ophthalmology. 2006;113:2087-2092.
60 Abramson DH, Ellsworth RM, Haik BG. Cobalt plaques in advanced retinoblastoma. Retina. 1983;3:12.
61 Shields JA, Shields CL, De Potter P, et al. Plaque radiotherapy for residual or recurrent retinoblastoma in 91 cases. J Pediatr Ophthalmol Strabismus. 1994;31:242.
62 Stannard C, Sealy R, Hering E, et al. Localized whole eye radiotherapy for retinoblastoma using a 125I applicator, “claws.”. Int J Radiat Oncol Biol Phys. 2001;51:399.
63 Abramson DH, McCormick B, Fass D, et al. Retinoblastoma, the long-term appearance of radiated intraocular tumors. Cancer. 1991;67:2753.
64 Imhof SM, Mourits MP, Hofman P, et al. Quantification of orbital and mid-facial growth retardation after megavoltage external beam irradiation in children with retinoblastoma. Ophthalmology. 1996;103:263.
65 Sant M, Capocaccia R, Badioni V. Survival for retinoblastoma in Europe. Eur J Cancer. 2001;37:730.
66 Egbert PR, Donaldson SS, Moazed K, et al. Visual results and ocular complications following radiotherapy for retinoblastoma. Arch Ophthalmol. 1978;96:1826.
67 Gagnon JD, Ware CM, Moss WT, et al. Radiation management of bilateral retinoblastoma: the need to preserve vision. Int J Radiat Oncol Biol Phys. 1980;6:669.
68 Shidnia H, Hornback NB, Helveston EM, et al. Treatment results of retinoblastoma at Indiana University Hospitals. Cancer. 1977;40:2917.
69 Hernandez JC, Brady LW, Shields JA, et al. External beam radiation for retinoblastoma: results, patterns of failure, and a proposal for treatment guidelines. Int J Radiat Oncol Biol Phys. 1996;35:125.
70 Foote RL, Garretson BR, Schomberg PJ, et al. External beam irradiation for retinoblastoma: Patterns of failure and dose-response analysis. Int J Radiat Oncol Biol Phys. 1989;16:823.
71 Zelter M, Damel A, Gonzalez G, et al. A prospective study on the treatment of retinoblastoma in 72 patients. Cancer. 1991;68:1685.
72 Black L, McCormick B, Abramson DH. External beam radiation therapy and retinoblastoma: long term results in the comparison of two techniques. Int J Radiat Oncol Biol Phys. 1995;35:45.
73 Abramson DH, Ellsworth RM, Kitchin FD, et al. Second nonocular tumors in retinoblastoma survivors. Are they radiation-induced? Ophthalmology. 1984;91:1351.
74 Wong FL, Boice JDJr, Abramson DH, et al. Cancer incidence after retinoblastoma. Radiation dose and sarcoma risk. JAMA. 1997;278:1262.
75 Abramson DH, Frank CM. Second nonocular tumors in survivors of bilateral retinoblastoma: a possible age effect on radiation-related risk. Ophthalmology. 1998;105:573. discussion 579–580
76 Moll AC, Imhof SM, Schouten-Van Meeteren AY, et al. Second primary tumors in hereditary retinoblastoma: a register-based study, 1945–1997: Is there an age effect on radiation-related risk? Ophthalmology. 2001;108:1109.
77 Li FP, Abramson DH, Tarone RE, et al. Hereditary retinoblastoma, lipoma, and second primary cancers. J Natl Cancer Inst. 1997;89:83.
78 Abramson DH. Second nonocular cancers in retinoblastoma: a unified hypothesis. The Franceschetti Lecture. Ophthalmic Genet. 1999;20:193.
79 Abramson DH, Melson MR, Dunkel IJ, et al. Third (fourth and fifth) nonocular tumors in survivors of retinoblastoma. Ophthalmology. 2001;108:1868.
80 Kleinerman RA, Tarone RE, Abramson DH, et al. Hereditary retinoblastoma and risk of lung cancer. J Natl Cancer Inst. 2000;92:2037.
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
65 Retinoblastoma
Retinoblastoma was first described by Petras Pawius of Amsterdam as early as 1657. However, James Wardrop, an Edinburgh ophthalmologist, was officially credited for recognizing retinoblastoma in 1809, when he documented the first enucleation successfully performed to treat retinoblastoma.1 Two centuries later, enucleation is still used to treat retinoblastoma, along with other methods such as external beam radiation therapy, brachytherapy, cryotherapy, and laser therapy. Newer modalities that have recently gained popularity include chemoreduction and transpupillary thermotherapy. In many cases, treatment can help preserve both patients’ eyes and vision. In the United States, the current 5-year survival rate for children with retinoblastoma is 98%.2
Incidence
Surveys suggest a relatively constant occurrence of retinoblastoma during the last century throughout the world. The frequency of retinoblastoma has been documented as 1 in 14,000 to 1 in 34,000 live births.3 The incidence in the United States is relatively low, at 3.58 cases per million children under the age of 15 years, and is closely correlated with age. For ages 1 to 4 years, the incidence is 10.6 per million; for 5 to 9 years, 1.53 per million; and for 10 to 14 years, 0.27 per million.4 Approximately 350 new cases are diagnosed in the United States each year, and retinoblastoma is the seventh most common cancer in the pediatric population in this country.
Retinoblastoma appears to have no predilection for any particular racial group or gender. Retinoblastoma can affect one or both eyes. The right eye is affected as frequently as the left eye. Unilateral retinoblastoma occurs in 75% of all cases diagnosed, whereas bilateral disease occurs in 25% of cases. All patients with bilateral retinoblastoma have multifocal disease (more than one independent tumor focus in each eye) with 5 as the mean number of tumors distributed between the two eyes, and a range of 2 to 20 tumors. Of the patients with unilateral disease, only 15% have multifocal disease.5
Genetics
Retinoblastoma was one of the disease models that has been used to demonstrate the genetic nature of cancer. Knudson proposed the now classic two-hit model in 1971 after noting that the timing of tumor development (younger age at diagnosis of bilateral tumors than of unilateral tumors) suggested a mechanism in which at least two events would be responsible for the development of the tumor.6 He hypothesized that patients with multifocal bilateral disease were germline carriers for the first hit, with only one second hit necessary for the development of retinoblastoma. Unilateral unifocal patients, he determined, usually had a normal germline genome, but developed both hits in the progenitor tumor cell.
Since the Rb1 gene was isolated in 1986 by Friend et al. many studies have explored its location and function. The gene is located on the long arm of chromosome 13, band 14.2.7 Further characterization of the gene has revealed that it spans 200 kb and is composed of 27 exons. The gene encodes a 4.7 kb mRNA transcript, which is expressed in all adult tissues. The 110 kD nuclear phosphoprotein consists of 928 amino acids.
The protein encoded by the gene is a regulator at the cell cycle checkpoint between G1 and entry into the S-phase.8 The phosphorylation pattern of p110 RB varies during the cell cycle and the current model suggests that the unphosphorylated normal RB1 protein binds transcriptional regulators that promote entry into the S-phase. When the normal RB1 protein is phosphorylated, it dissociates from E2F, freeing it to bind to DNA and stimulate transcription of downstream genes that promote progression through the cell cycle. Loss of normal RB1 function, as in the case of the tumors, presumably allows for uncontrolled entry into the S-phase, more rapid cell cycling, and rapid cell division. The retinoblastoma protein is thus a dominant suppressor of tumor formation, making the Rb1 gene a member of the tumor suppressor class of genes.
Mutations in the Rb1 gene seem to occur throughout the gene. The majority of the germline mutations are nonsense or frameshift, introducing a premature stop codon and therefore producing a truncated and presumably nonfunctional protein. Other types of mutations such as missense, aberrant splice mutations, and mutations in promotor sequences, can result in a reduced penetrance or decreased expressivity form of the disease. Only about 2% to 3% of tumors demonstrate karyotypically visible larger deletions of material in the 13q14 band. Some of the children with this cytogenetic abnormality in their germline have a constitutional disease with severe developmental delay and dysmorphic features, named the 13q syndrome, whereas others are phenotypically normal other than their retinoblastoma. Evidence suggests that retinoblastoma, like other cancers such as colon carcinomas and glial brain tumors, requires other genetic abnormalities to occur before transformation into malignancy. This finding has stimulated many studies focused on subsequent mutations that occur after the initial mutation in the Rb1 gene. Candidate oncogenes and tumor suppressor genes include KIF14, MDM4, MYCN, E2F3, DEK, and CDH11.9
Genetic counseling for the families of retinoblastoma patients is complex and challenging (Table 65-1). Epidemiologic observation reveals that a parent with bilateral retinoblastoma has roughly a 45% chance of having each child with retinoblastoma, although the retinoblastoma may not be present at the child’s birth in either eye. Of those parents with bilateral disease whose children develop unilateral disease, 96% of the children will have multifocal tumors and most of them will be diagnosed within the first 6 months of life. Since all of these children (of bilateral parents) have a germinal mutation, 45% of their children will also develop retinoblastoma.
Table 65-1 Genetic Counseling for Retinoblastoma
Two of the most common and challenging types of families who require genetic counseling are: (1) families in which neither parent has a history of retinoblastoma but the couple already has one child with the disease and wants to know the chances that the next will also have retinoblastoma, and (2) families in which one parent has a history of unilateral retinoblastoma and the couple wants to know the risk to future children of developing the disease. In the first scenario, an ophthalmoscopic examination of both parents’ eyes should be performed. The reason for this examination is that 1% to 2% of such parents will demonstrate on fundoscopic examination a rare benign clinical entity called a retinoma. There is debate as to the exact characterization of these lesions, but they likely represent either retinoblastoma precursor lesions as a result of the loss of the RB1 gene without the subsequent acquisition of other mutations that are required for progression to the malignant state or spontaneously regressed or arrested retinoblastoma.10 These parents should be counseled that although they have never been treated for retinoblastoma, they may harbor a germinal mutation, and if they have multifocal retinomas, each of their children will have a 45% chance of inheriting the mutation. Parents with no ophthalmoscopic evidence of retinoblastoma can be told that the chance that their next child will have retinoblastoma is between 1% and 5%. In the small percentage of cases in which the second child does develop the disease, it is presumed that one of the parents carries a germinal mutation although he or she is phenotypically normal. Several have been theories fashioned to explain these rare cases, including epigenetic modulation and somatic mosaicism in the parent. Additionally, some large kindreds have demonstrated the presence of two independent, distinct somatic mutations in different family members.
In these scenarios as well as others, parents of children with retinoblastoma are often eager to undergo genetic analysis to determine the risk that siblings or future children will develop the disease. Some centers have devised economical ways to routinely offer genetic screening to all retinoblastoma patients with the ability to detect sequence alterations in the Rb1 gene in about 80% to 90% of patients with a germinal mutation in peripheral blood or tumor tissue available.11 These programs utilize multiplex ligation–dependent probe amplification (MLPA) to detect large deletions or duplications, microsatellite analysis to detect loss of heterozygosity (LOH), and denaturing high-performance liquid chromatography (D-HPLC) analysis to detect point mutations and small insertions or deletions, and quantitative multiplex polymerase chain reaction (PCR) of short fluorescent fragments (QMPSF).
Finally, because the gene is expressed in all adult tissues and the RB1 protein plays such a vital role in controlling cell proliferation, all patients who carry a germinal Rb1 mutation are at risk for the development of additional nonocular cancers. These cancers can occur as late as 40 years or more after the initial development of retinoblastoma. The incidence of these cancers is significantly increased in patients who receive external beam radiation, especially when given before the age of 1 year. Patients should be educated about their risk factors for developing these cancers, as it is these cancers, not retinoblastoma, that are the leading cause of death in patients with the germinal RB1 mutation.12 See “Select Results” later in this chapter for more information on additional nonocular cancers.
Anatomy
Figure 65-1 illustrates the important anatomic structures of the eye. The inner layer is the retina, where the photoreceptors are located. Retinoblastoma is thought to originate from an unidentified progenitor cell in this layer. The normal retina extends from the posterior “pole” forward to a region just behind the lens, in cross-section called the ora serrata. The middle layer of the eye is the choroid; this pigmented structure is continuous anteriorly with the ciliary body and iris, and together these are considered to be the uveal tract. The outermost layer of the eye is the sclera, a tough and radioresistant structure that fuses with the cornea anteriorly. The anterior chamber of the eye is between the cornea and the iris; the posterior chamber is just behind it, between the iris and the lens. Both chambers are filled with aqueous humor, produced by the ciliary body. Behind the lens is the vitreous chamber, with vitreous humor, a thick clear gel-like substance. In advanced retinoblastoma, “seeding” of the tumor is noted in this chamber.
Pathology
The diagnosis of retinoblastoma is routinely made without pathologic confirmation. Needle biopsies are rarely, if ever, indicated, as puncturing the eye can lead to extraocular extension and orbital invasion. Aqueous taps, previously explored as a possible diagnostic tool, are inappropriate for the same reason.13
When enucleation is the treatment of choice, histopathologic examination of the globe can be useful in providing information about potential extraocular extension (Fig. 65-2). For optimal prognostic information, the globe should be fixed for at least 24 hours, with the optic nerve preserved for separate evaluation.14
FIGURE 65-2 • Gross section of a globe, showing extensive disease in the retina, vitreous body, choroid, and optic nerve.
Under the microscope, the appearance of the retinoblastoma cells can range from small, round, undifferentiated cells with frequent mitoses, as seen in Fig. 65-3, to differentiated cells that form rosettes called Flexner-Wintersteiner rosettes.14 Important observations in assessing the risk of the child for distant or local relapse include careful assessment of the degree of invasion of tumor cells beyond the retina and into the choroid and sclera, and assessment of the degree of tumor invasion into the optic nerve beyond the region of the lamina cribrosa.15
Clinical Presentation
The presenting signs and symptoms of retinoblastoma vary depending on the geographic location in which the child presents. In developing countries, children can develop extraocular disease by the time they are diagnosed, with proptosis and an orbital mass. In these cases, the retinoblastoma has often extended directly into the orbit, causing rupture of the globe (Fig. 65-4). Regional nodal metastasis may be found in the preauricular or submandibular regions or the children may have widespread metastatic disease. These children are older at diagnosis (age 4 to 6 years) than patients in the United States, and few survive. In the United States, when there is no family history of disease, the median age at presentation is 1 year for bilateral retinoblastoma and 2 years for unilateral disease. Most children in the United States present with only intraocular disease and are diagnosed with signs rather than symptoms.
FIGURE 65-4 • A child with extensive orbital disease: the most common presentation in the developing world.
In the United States, the most common presenting sign of retinoblastoma (60% of cases) is leukocoria or a white pupillary reflex that is sometimes referred to as a cat’s eye reflex. Leukocoria can be the result of the tumor itself, a secondary retinal detachment associated with the tumor, or a reflection of light from the white mass at the posterior portion of the eye (Fig. 65-5).
FIGURE 65-5 • A child with the “cat’s eye” reflex, with retinoblastoma visualized as the white material in the pupil.
The second most common sign is strabismus, misalignment of the two eyes. Of the 20% to 25% of patients who present with this sign, 10.5% have eyes crossed in (esotropia) and 5.2% out (exotropia).16 Esotropia in children is generally more common than exotropia, such that an infant with exotropia must be suspected to have retinoblastoma until proved otherwise. The strabismus in retinoblastoma patients is caused by tumor or retinal detachment located in the fovea of the eye, the area of central vision. Although patient survival is independent of whether patients present with strabismus or leukocoria, ocular survival rates are significantly lower for patients who present with leukocoria, as the disease tends to be more advanced at presentation in these cases.17
Presenting signs in the United States that occur in less than 5% of cases include: anisocoria (different-sized pupils); heterochromia (different-colored irides); hyphema (blood in the anterior chamber); tumor hypopyon (tumor in the anterior chamber); nystagmus, a unilateral fixed and dilated pupil; and failure to thrive. In cases in which the patient has gross chromosomal abnormalities, children can present with other findings, such as absence or hyperplasia of the thumbs, mental retardation, prominent nasofrontal bones, large malformed ears, hypospadias, bifid scrotum, and extra digits. Only 7% of patients are detected on routine pediatric screening by a pediatrician, whereas 72% of cases are detected by a family member or friend.17
When there is a family history of retinoblastoma, diagnosis tends to occur earlier, with a median age at diagnosis of 5 months for patients with unilateral disease and 11 months for patients with bilateral disease.18 Sixty percent of all retinoblastoma patients diagnosed under 6 months have no signs or symptoms, but are examined and diagnosed because of a family history of retinoblastoma.19 At our center, children with a family history of retinoblastoma typically undergo frequent serial fundoscopic examinations beginning within 24 to 48 hours of birth and continuing until at least 28 months of age. Twenty-eight months is chosen as the earliest possible time to stop examining these patients because it is the oldest age at which a patient with a positive family history of retinoblastoma has developed his or her first tumor in a previously documented disease-free eye.20 These serial examinations have a significant impact on ocular outcome. Patients who began undergoing screening examinations at our center as newborns because of a family history had a 68% ocular 5-year survival rate, whereas nonscreened patients with a family history had only a 38% ocular survival.17
The anatomic location of these tumors within the retina and the age at which the lesions develop have been well studied (Fig. 65-6). Retinoblastoma may be present anywhere in the retina at birth, but as age at tumor development increases, tumors develop progressively farther into the periphery. Thus macular tumors present earliest and peripheral anterior tumors present later. In our center, the average macular tumor is diagnosed at 5.6 months and never presents after 15.5 months. In contrast, peripheral tumors are diagnosed at an average of 16.4 months and can present as late as 8 years of age. This pattern of ocular tumor development has important implications for the radiation oncologist. Since many young children will have no tumors in the anterior retina, there is no need to treat this area, thereby decreasing the incidence of radiation-induced cataracts. However, future development of new peripheral tumors should be expected and the family should be informed of this likelihood. The development of these tumors does not represent a failure of radiation, but rather the inevitable process of tumorigenesis in genetically altered cells. Furthermore, the family of a child with multifocal unilateral retinoblastoma can be assured that new tumors that develop in the fellow eye will rarely originate in the fovea, so the outlook for vision in the fellow eye is typically favorable.
Routes of Spread
The natural course of untreated retinoblastoma follows one of five routes of progression: contiguous spread through the choroid, sclera, and orbit; extension along the optic nerve directly into the brain; invasion of the subarachnoid space and leptomeninges via the cerebrospinal fluid; hematogenous spread to bone marrow, bone, liver, and dissemination via conjunctiva through the lymphatic system.8 In the United States, the few patients with unilateral retinoblastoma who develop metastases usually have them at the time of diagnosis. The majority metastases occur within 5 years of diagnosis.18
Diagnosis of Retinoblastoma
All retinoblastoma tumors are drawn on an expanded concentric circle drawing of the globe called a fundus diagram, used to document the presence, location (in reference to the optic nerve and fovea of the eye), and size of the retinoblastoma tumors. Some ophthalmologists document the presence of tumors in the posterior pole by taking color photographs with a hand-held fundus camera while the patient is under anesthesia. Figure 65-7 demonstrates both techniques in the same child. The original tumor drawing is referred to during follow-up visits, and new drawings are composed to document treatment response. Although the ophthalmoscopic examination should always be considered the primary method for evaluation of retinoblastoma, several ancillary tests can assist in the diagnosis, especially when there is no clear view of the fundus.
FIGURE 65-7 • A fundus photograph of an eye with retinoblastoma (A) and the corresponding sketch of the disease in the eye diagram (B).
Ophthalmic ultrasonography is one of the most frequently performed ancillary tests for retinoblastoma. It is noninvasive, safe, repeatable, and immediately interpretable. Ultrasonography should be performed on both eyes and can be performed with or without general anesthesia. B-scan ultrasonography reveals a two-dimensional (2D) cross-sectional view of the eye, confirms the presence and the relationship of the solid tumor to other anatomic structures within the eye, and detects the size and shape of the tumors (Fig. 65-8). B-scan can detect orbital involvement, optic nerve invasion (sometimes), extrascleral extension, and calcification. Because there is calcium (which exhibits high reflectivity on ultrasonographic scans) in the majority of retinoblastoma tumors, high amplitude echoes will remain on the screen even if the gain or sensitivity is lowered. Shadowing defects posterior to the tumor may be present and are caused by the absorption of sound and high reflectivity. A-scan ultrasonography evaluates the internal characteristics and vascularity of the tumor and measures the height of the tumor.
Computed tomography (CT) scans may no longer be routinely appropriate for retinoblastoma patients, as analysis has suggested an increased lifetime risk of other cancers in pediatric patients subjected to this imaging modality.21 Instead, as part of an extent-of-disease work-up, magnetic resonance imaging (MRI) is routinely performed (Fig. 65-9A, B). In addition to its excellent resolution in the diagnosis of extraocular soft tissue disease, MRI can readily distinguish between retinoblastoma and Coats’ disease (see “Differential Diagnosis of Retinoblastoma”), as Coats’ disease appears brighter than retinoblastoma on T2-weighted images due to proteinaceous exudate.8 One disadvantage of MRI is that calcification, a key feature of retinoblastoma, is more easily demonstrated with CT than with MRI.
Differential Diagnosis of Retinoblastoma
Retinoblastoma can be simulated by several other ophthalmic tumors and ophthalmic disorders (Table 65-2). The lesion that most commonly simulates retinoblastoma is Coats’ disease. Coats’ disease is a nonheritable, primarily unilateral anomaly that affects mostly boys, with a later age at diagnosis than retinoblastoma (mean age at diagnosis is 6 years). Ophthalmic examination reveals telangiectatic vessels of the retina with intraretinal and subretinal exudates and an exudative retinal detachment. Presence of a yellow-green sheen on indirect ophthalmoscopic examination aids in differentiating Coats’ from retinoblastoma. When rubeosis iridis, glaucoma, and corneal edema develop, the fundus may be impossible to view. Under these circumstances, ultrasonography can be helpful.
[/not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
