CHAPTER 24 RETINAL DISEASE
The retina reflects many of the pathophysiological processes that occur in the central nervous system (CNS). Indeed the retinal appearances may be diagnostic but, conversely, can be nonspecific; for example, pigmentary retinal changes are part of the diagnostic criteria for Kearns-Sayre syndrome but are also present in many other diseases. Furthermore, in other diseases, the retina may appear clinically normal, but psychophysical and electrophysiological testing reveal abnormalities.
Uveitis, inflammation of the uveal tract, can occur in association with neurological disease. In this chapter, the classification recommended by the Standardization of Uveitis Nomenclature Working Group is used.1 In this classification, the primary site of inflammation in anterior uveitis is the anterior chamber. For intermediate uveitis, it is the vitreous humor. The primary site of inflammation in posterior uveitis can be the retina or choroid. Panuveitis is used when there is no predominant site of inflammation, but inflammation occurs in the anterior chamber, vitreous humor, and retina and/or choroid.
UVEOMENINGEAL SYNDROMES
Sarcoidosis
Sarcoidosis is a multisystem granulomatous disorder of unknown etiology that can affect the eye and the CNS, as well as the lungs, skin, and other organs. Of all patients with sarcoidosis, 25% to 60% have ocular involvement, which may be the manifesting sign. Sarcoidosis can affect most ocular tissues, but the most common ocular manifestations are uveitis and conjunctival nodules. Uveitis can occur in about 30% of patients before nonocular signs.2
A 10-year follow-up study of patients with uveitis caused by sarcoidosis or presumed sarcoidosis revealed that over this period, the disease spread most often to the CNS.3 Posterior segment involvement is frequently associated with neurological disease.4,5 It may be more common in white patients, especially elderly women,2 although this has not been confirmed by all studies.6 The optic nerve may become swollen because of intraocular inflammation, when visual acuity is usually preserved, or it may be involved either directly by granulomas or as a result of meningeal changes, when loss of visual acuity and field result from compressive optic neuropathy.
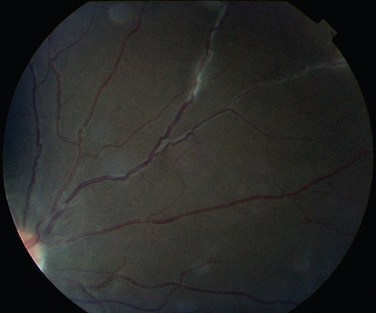
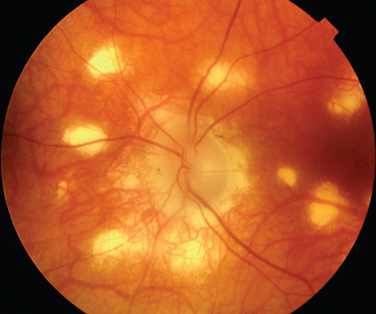
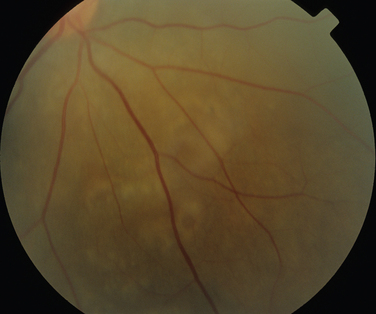
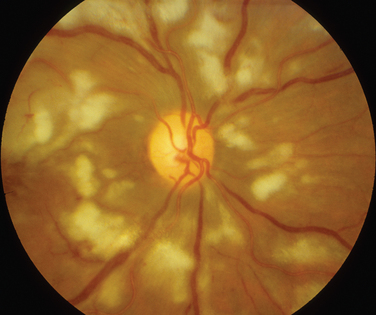
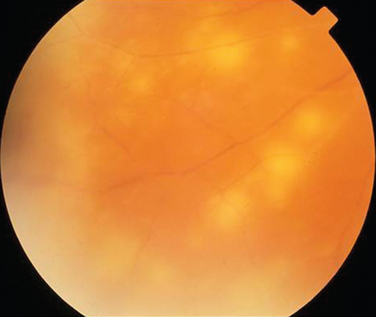
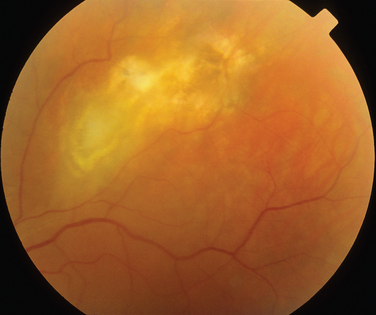
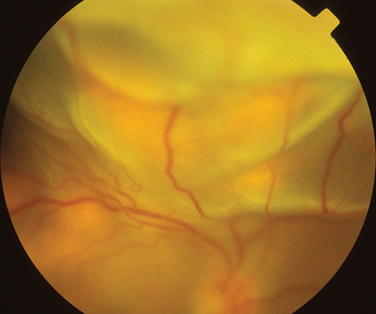
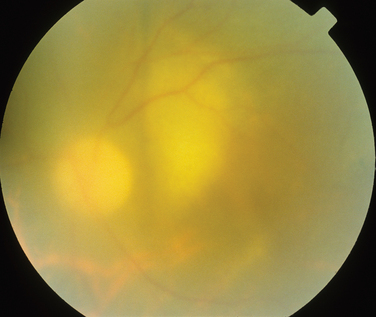
The classic sign of retinal involvement by sarcoidosis is retinal periphlebitis (Fig. 24-1),7–9 which may be described as similar to candle wax dripping. Other vascular changes include retinal hemorrhages, vein occlusions, neovascularization, and possibly retinal arteriolitis with aneurysm or ectasia formation.10 Focal subretinal lesions have been described, but these do not appear to affect vision.9 Confluent choroidal infiltrates have been described.11 Appearances similar to that of a multifocal choroiditis (Fig. 24-2),12,13 and a serpiginous choroidopathy have been described.14 Multiple sclerosis may include a similar ocular inflammatory manifestation, but the choroid is not involved.
Behçet’s Disease
Behçet’s disease is characterized by recurrent episodes of orogenital aphthae and by systemic and retinal venous thrombosis. It remains a clinical diagnosis, and diagnostic criteria have been published.15 The CNS manifestations of Behçet’s disease can be categorized into parenchymal and nonparenchymal involvement (neurovascular Behçet’s disease). Parenchymal CNS manifestations include brainstem and hemisphere involvement, spinal cord lesions, and meningoencephalitis. Nonparenchymal involvement includes dural sinus thrombosis, arterial occlusion, and arterial aneurysms. These categories have different clinical and prognostic properties.16 The ocular manifestations in the parenchymal category include optic neuritis and ischemic optic neuropathy.17–19 Papilledema caused by intracranial hypertension secondary to dural sinus thrombosis occurs in neurovascular Behçet’s disease20,21 and can result in significant visual loss. Intracranial hypertension can, however, occur in the absence of venous sinus thrombosis or meningitis.18
Inflammatory eye disease occurs in approximately 70% of all patients who may have additional neurological or neuro-ophthalmological disease. The inflammation usually occurs after the onset of oral aphthosis, but the delay between the two may be as long as 14 years.22 In approximately 10% of patients, intraocular inflammation is the manifesting feature,23 and in rare cases, neither oral nor genital ulcers may occur at all.24 Usually the involvement is bilateral.
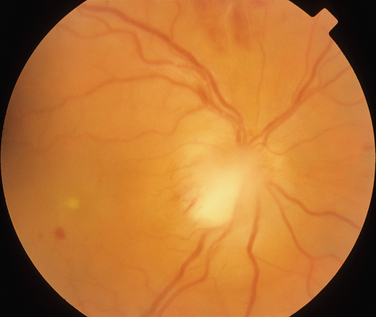
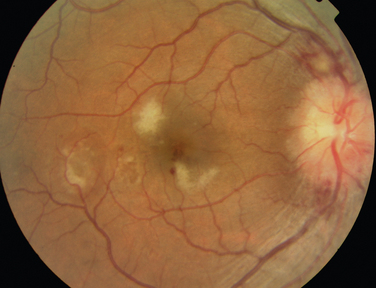
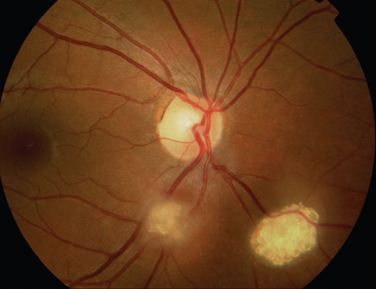
The main ocular finding is panuveitis, although there may be differing degrees of anterior and posterior segment involvement. The anterior uveitis may be so severe that the inflammatory cells precipitate in the inferior portion of the anterior chamber, forming a hypopyon. Retinal vein occlusion is the most characteristic fundal sign, but others include retinal perivasculitis and retinal infiltrates. The perivasculitis involves mainly the veins and less frequently the arteries. The retinal infiltrates are collections of lymphocytes in the superficial retina and are pathognomonic of Behçet’s disease (Fig. 24-3). They resolve spontaneously and carry no visual morbidity. However, recurrent retinal vein occlusions can result in total attenuation of all vessels and consequent optic atrophy. Macular edema may also cause visual loss. Unfortunately, the visual prognosis is poor in spite of the development of new immunosuppressive therapies. Up to 15% of patients are unresponsive to these therapies,25 and the disease may remain active for many years.
Vogt-Koyanagi-Harada Disease
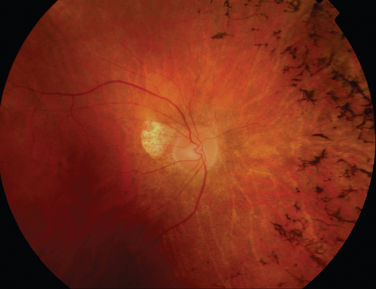
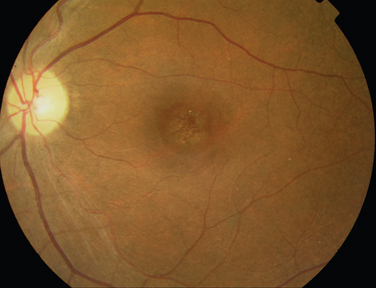
Like Behçet’s disease, it is diagnosed on clinical findings. Diagnosis can be difficult because there are no specific tests and the clinical features are dependent on the stage of the disease at which the patient is examined. Diagnostic criteria have been published.26 One of the following neurological features has to be present for the diagnosis of VKH: meningismus, tinnitus or cerebrospinal pleocytosis. The ocular features have been divided into early and late manifestations of the disease. In the early manifestation, the retinal and choroidal features are mainly in the form of serous retinal detachments. In the late manifestations, choroidal depigmentation that results in a mottled “sunset glow” appearance, nummular chorioretinal depigmented scars, and retinal pigment epithelium clumping and/or migration26 may be present (Fig. 24-4). Recurrent or chronic uveitis also occurs in the late stage of VKH. The criteria include the presence of bilateral ocular involvement. The integumentary findings are alopecia, poliosis, or vitiligo. A further neurological association with VKH is Guillain-Barré syndrome. Three patients who developed VKH within 3 months of having Guillain-Barré syndrome have been reported.27
RHEUMATOLOGICAL DISEASES AND THE SYSTEMIC VASCULITIDES
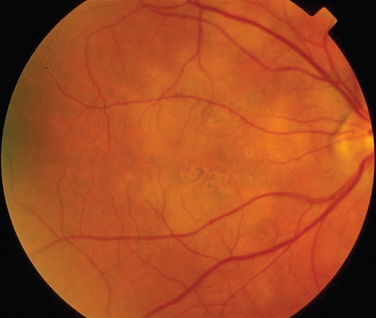
Rheumatoid arthritis is associated with corneal and sclera changes but not usually retinal ones. Retinal involvement is, however, a common ocular feature of systemic lupus erythematosus. Indeed, the presence of ocular features may mirror disease activity elsewhere.28,29 Retinal arteriolar occlusion may occur, and multiple cotton-wool spots may be seen (Fig. 24-5). In rare cases, venulitis develops. Choroidal involvement can also occur. A postmortem study of patients with systemic lupus erythematosus affecting the brain and eye demonstrated vasculitis in the brain and the choroidal vessels.30 A number of retinal arterioles were occluded with thrombus, but there was no evidence of inflammation of the retinal vessels. Emboli from a diseased heart valve or deposition of immune complexes may have caused the occlusion.
Large Artery
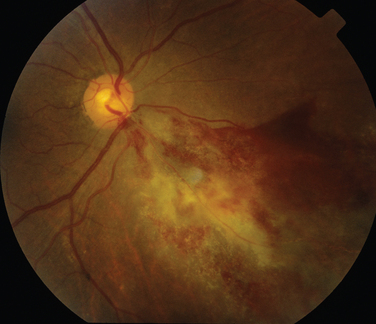
Giant cell arteritis is a necrotizing vasculitis targeting large and medium-sized arteries and affects mainly white patients older than 50 years. Anterior ischemic neuropathy is the most common cause of visual loss in giant cell arteritis, but patients can suffer from either central retinal artery occlusions or, less commonly, branch retinal artery occlusions. Compromised blood flow to the retina may result in signs of ischemia such as cotton-wool spots and hemorrhages. Posture can also affect the ischemia in that vision may be reduced in the upright position, in relation to lying down.31 Poor perfusion of the choroid may result in reduced vision, which can be reversible, but permanent infarction can occur (Fig. 24-6). Treatment is aimed primarily at preventing both visual loss in the second eye and the development of stroke.
Takayasu’s disease is another disease of the large vessels. Retinal disease is present in fewer than 50% of patients with Takayasu’s disease.32 The retinal changes include vessel dilation, tortuosity of vessels, arteriovenous anastomoses, and hemorrhages. In many patients, hypertension is probably the major cause of retinal changes; in some patients, however, the changes may result from reduced flow to the retinal circulation (i.e., a hypotensive retinopathy).32
Medium-Sized Artery
Ocular involvement occurs in 10% to 20% of patients with polyarteritis nodosa. Retinal artery occlusion, ischemic retinopathy, exudative retinal detachment,33 and choroidal infarcts have been described.34 Kawasaki’s disease tends not to affect the retina, although one postmortem study did yield evidence of retinal ischemia.35
Medium-Sized and Small Vessels
Wegener’s granulomatosis is characterized by a systemic vasculitis and necrotizing granulomatous lesions that may be found in the respiratory tract, kidney, orbit, and brain. Between 30% and 50% of patients with Wegener’s granulomatosis have neurological involvement.36,37 The patterns of neurological involvement have been described as granulomatosis invasion of the orbit, granulomas of the brain and meninges, and vasculitis of the nervous system.36 The orbital involvement usually is from the extension of disease in the paranasal sinuses. Orbital involvement may be the initial sign of the disease, manifesting with orbital pain, rapidly progressing proptosis, and limited eye movements with associated granulomatosis infiltration of adjacent structures. Cranial nerves may also be involved; the optic nerve, the abducens nerve, and the facial nerve are the most commonly affected.37 Retinal and choroidal involvement is rare but may manifest as retinitis, vein occlusion, or an intermediate uveitis.38 Some of these changes, however, may have been secondary to hypertension or cytomegalovirus–associated retinitis. A number of patients taking cyclophosphamide for Wegener’s granulomatosis have developed cytomegalovirus-associated retinitis (Fig. 24-7).39–41 In patients with the limited form of Wegener’s granulomatosis, the neuro-opthalmological features may, again, be the earliest feature of the disease.42 A similar pattern of neuro-ophthalmological involvement is seen, the orbit being affected more frequently than the retina.43
Ocular involvement in the Churg-Strauss syndrome can be in the form of either chronic orbital pseudotumor or ischemic vasculitis. The retinal manifestations of the latter group include retinal artery occlusions.44 Cotton-wool spots in the retina have been described in a patient with microscopic polyangiitis. The patient also had a very severe uveitis that resulted in a hypopyon.45
MULTIPLE SCLEROSIS
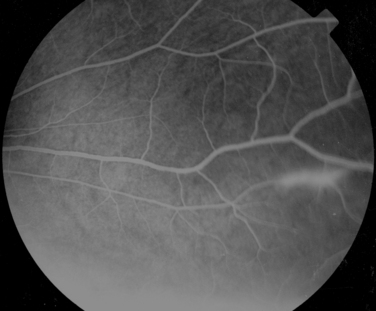
Manifestations of multiple sclerosis in the eye other than optic neuritis include uveitis and retinal changes, such as periphlebitis or sheathing or cuffing of the retinal veins by lymphocytes and plasma cells.46–49 This may occur in isolation or as part of an intermediate uveitis. Periphlebitis may occur in up to 36% of patients with multiple sclerosis50,51 and is usually asymptomatic. These changes are often in the peripheral retina and so cannot be viewed through a direct ophthalmoscope trained on an undilated pupil. Indeed, the changes may be visible only on fluorescein angiography (Fig. 24-8).52 Focal perivenous hemorrhage can also occur. The presence of vascular changes and/or ocular inflammation in patients with optic neuritis is associated with a greater risk of developing multiple sclerosis.52,53 It has been suggested that periphlebitis is a marker for disease activity,50,54 but this has not been confirmed.55 Vascular sheathing can occur in other diseases such as sarcoidosis, systemic lupus erythematosus, Wegener’s granulomatosis, and Behçet’s disease.56 Multiple sclerosis can be differentiated from sarcoidosis inasmuch as it is not associated with choroidal lesions.
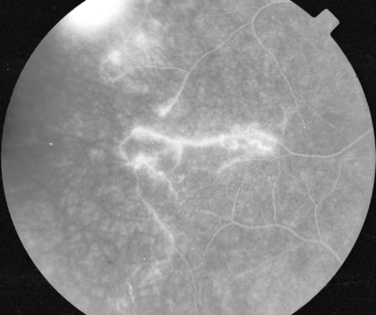
Uveitis
Studies of records of patients at multiple sclerosis clinics have revealed that 1% to 2% have uveitis, which is a higher frequency than in the general population.57,58 Asymptomatic uveitis has been found in a range from none59 to 18%60 of patients with multiple sclerosis. This range probably reflects the difference in disease activity of the patients in the studies. All types of uveitis are observed; one form of intermediate uveitis, pars planitis, is probably the most common,57,61,62 but this has not been found in all studies.59
There does not seem to be any correlation between the type of multiple sclerosis and uveitis or between the degree of neurological disability and the type of uveitis,57,59 although the presence of optic atrophy may be protective against uveitis.60
Visual Loss
In patients with multiple sclerosis and uveitis, visual loss cannot be assumed to result from optic neuritis, because the complications of uveitis such as macular edema63,64 and retinal vasculitis with associated neovascularization63,65 can also result in visual loss. The features that suggest a diagnosis of uveitis rather than optic neuritis include photophobia, floaters, redness of the eye, and the absence of a relative afferent pupillary deficit. Nevertheless, many patients with multiple sclerosis–associated uveitis retain useful vision if it is treated appropriately.62
Neuroretinitis
Neuroretinitis is a descriptive term for optic disc swelling and macular exudates (Fig. 24-9) that classically results from infective causes (discussed in the next section). One study has suggested that the presence of neuroretinitis indicated that the patient did not have demyelinating disease.66 Thus, neuroretinitis should be differentiated from cases of optic neuritis that manifest with papillitis. However, in a retrospective review of 35 patients with neuroretinitis, 3 patients were found to have multiple sclerosis, although they also had received interferon β therapy.67
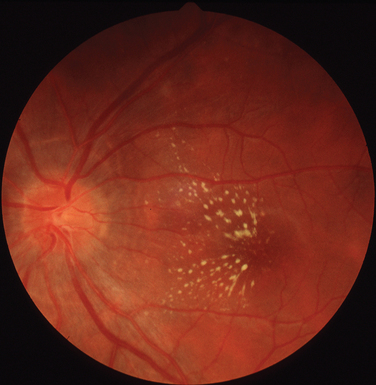
INFECTIONS
Infectious Causes of Neuroretinitis
As discussed, the presence of a swollen disc and macular exudates in a starlike pattern is known as neuroretinitis and is classically caused by infection. However, other causes such as hypertension can result in the same clinical picture. A number of infections are classically associated with neuroretinitis and can have neurological associations. These include cat-scratch disease, Lyme disease, and syphilis. In very rare cases, toxoplasmosis manifests this appearance.68 The ocular features of these other conditions are discussed as follows, except for toxoplasmosis, which is discussed elsewhere.
Cat scratch fever is caused by Bartonella henselae. There may be an associated uveitis, and the other fundal appearances include discrete retinal or choroidal lesions, which are more common than in classic neuroretinitis.69
Lyme disease is a spirochetal infection that results from tickborne transmission of Borrelia burgdorferi. Conjunctivitis is the most common ocular manifestation of early Lyme disease and occurs in approximately 10% of cases. Uveitis is a relatively rare manifestation of Lyme disease, but it may occur in the later stages. Vitritis, choroiditis optic neuritis, and motility problems have also been described.70–74 Exudative detachment associated with the choroiditis74 has been reported, as has a case of retinal vasculitis.75
Syphilis is caused by Treponema pallidum. Between 1995 and 2003, the increase in the incidence of syphilis in the United Kingdom has been more than 10-fold.76 Ocular disease typically occurs in secondary syphilis but is also observed in tertiary syphilis. Uveitis is the most common ocular manifestation in both these stages of the disease.77,78 The retinal changes are essentially similar in both secondary and tertiary syphilis. In keeping with its moniker, the “great imitator,” the disease can affect all structures of the eye. The combination of good visual acuity, reasonable visual fields, mild panuveitis, swollen disc, and afferent papillary defect is highly suggestive of syphilitic infection.
In addition to neuroretinitis, the retinal findings include chorioretinitis, retinal vasculitis, serous retinal detachment, and necrotizing retinitis (Fig. 24-10). The necrotizing retinitis may be such that it is difficult to clinically distinguish from acute retinal necrosis (ARN) (discussed later in this chapter). The diagnosis is established by the presence of other systemic features.79 The association with HIV is discussed elsewhere in this chapter.
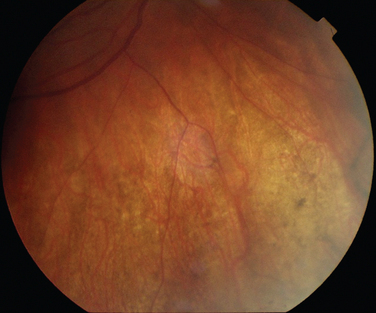
Tuberculosis
Choroidal involvement is probably the most common form of ocular involvement in tuberculosis. Usually it is in the form of choroidal tubercles that are white, gray, or yellow lesions (Fig. 24-11). The patient neither has to be seriously unwell nor has to have miliary tuberculosis for ocular disease to be present.80,81 The ocular disease may manifest many years after the tuberculosis has been found and treated elsewhere in the body.82 Other manifestations include multifocal choroiditis or serpiginous-like choroiditis.
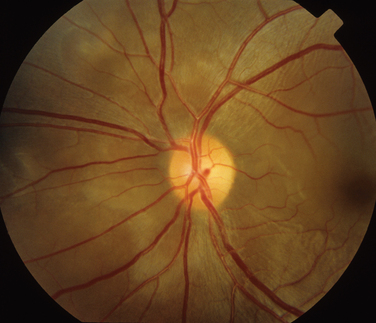
Retinal involvement can result from either choroidal extension or bloodborne spread. Retinal involvement can be either as tubercles or as a diffuse retinitis. Neovascularization and retinal vasculitis can also occur.83,84
Whipple’s Disease
Whipple’s disease is characterized by arthralgia, abdominal pain, and weight loss. The causative organism is Tropheryma whippelii. Oculomasticatory myorrhythmia is believed to be pathognomonic, but fundal changes also occur. These include vitreous opacities, diffuse chorioretinal inflammation with capillary involvement, and retinitis.85–89
Herpetic Infections
Herpesviruses cause ARN. The clinical characteristics for the diagnosis are focal, well-demarcated areas of retinal necrosis, which progresses in a rapid and circumferential manner; occlusive vasculopathy; and a prominent inflammatory reaction (Fig. 24-12).90 ARN may occur in isolation or in association with other forms of herpes infection, such as herpes zoster ophthalmicus. ARN may also be present in association with either meningitis or encephalitis. It appears that encephalitis, either concurrent or past, is more likely to be associated with herpes simplex virus type 1 infection, whereas herpes simplex virus type 2 is more likely to be involved in the presence of meningitis, whether past or concurrent.91 Genotypical analysis in two patients who had ARN after encephalitis revealed identical strains of herpes simplex virus type 1 in the brain and eye, suggestive of brain-to-eye transmission of the infection in these patients.92 ARN may occur concurrently with encephalitis or up to 20 years later.93 A case of cerebral vasculitis in which ARN was the only manifestation of any herpetic infection has been reported.94
Subacute Sclerosing Panencephalitis
Subacute sclerosing panencephalitis can occur months to years after measles infection. It usually affects children or young adults, but a case in a 49-year-old man has been described.95 Ocular involvement occurs in about 50% of cases and most often develops at the same time as neurological symptoms. Chorioretinitis with subsequent scarring is the most common retinal appearance. In rare cases, the ocular signs precede the neurological ones.96
Human Immunodeficiency Virus
The retinal manifestations of HIV can be subdivided into those resulting primarily from the HIV infection, from associated infections, and from malignancies. The incidence of many of these conditions appears to have decreased since the introduction of highly active antiretroviral therapy.97 Diagnosis can be difficult because the ocular signs may differ from those in an immunocompetent individual, and there may be two concurrent infections. Also interesting is that although HIV has contributed to the increase in incidence of tuberculosis, this may not have been mirrored by an increase of ocular tuberculosis. An autopsy study of eyes from 235 patients with HIV infection revealed intraocular tuberculosis in only two eyes.98
Retinal Manifestations of HIV Infection
Microvasculopathy is the most common ocular manifestation of HIV infection, being present in 40% to 60% of patients,99 and appears to be inversely related to the CD4 count.100 In the retina, it is most frequently represented by cotton-wool spots, which are usually asymptomatic and resolve after about 6 weeks. Hemorrhages may occur, particularly in patients with low platelet counts or diabetes. They are a response to the HIV infection. Less commonly, patients may develop macular ischemia,101 which is also probably caused by the primary infection. Large retinal vessel disease has been described,102 but this is rare. Progressive visual loss has been described in a patient with HIV infection who had a clinically normal retina but abnormal photoreceptor function on electrophysiological testing.103 This process continued even when the viral load was undetectable.
Cytomegalovirus-Associated Retinitis
Cytomegalovirus-associated retinitis is the commonest ocular infection in patients with acquired immunodeficiency syndrome (Fig. 24-7). However, its incidence has dropped since the introduction of highly active antiretroviral therapy.104 It rarely occurs in patients with a CD4 count lower than 50.100 Cytomegalovirus-associated retinitis occurs mainly in patients with HIV infection but can occur in patients who are immunocompromised for other reasons.41,105 There are two main forms of clinical presentation: a fulminant form and an indolent form. The fulminant form is characterized by confluent retinal necrosis with hemorrhage that in most cases develops in the posterior retina, whereas the indolent form has a more granular form. Up to 15% of patients with cytomegalovirus-associated retinitis are asymptomatic; therefore, screening is required for patients with HIV infection who have low CD4 counts and positive cytomegalovirus serological profiles.
Herpetic Infections Associated with HIV Infection
Necrotizing herpetic retinopathy represents a continuum of posterior segment inflammation caused by herpesviruses. The best recognized are ARN and progressive outer retinal necrosis. Usually ARN occurs in immunocompetent patients or in patients with HIV infection but minimal immune dysfunction; progressive outer retinal necrosis occurs in patients with HIV and significant immune compromise. Progressive outer retinal necrosis consists of a retinitis, often in the posterior pole, that is not usually associated with an inflammatory reaction. However, the two diseases can occur simultaneously, one eye having ARN and the other progressive outer retinal necrosis.106 Both necessitate intraocular and systemic antiviral treatment.
Syphilis in HIV Infection
Ocular syphilis has been discussed elsewhere, but it has a number of significant features in relation to HIV infection. It can develop when CD4 counts are higher than 200 and can be the manifesting sign of HIV infection. The ocular findings are similar to those in patients without HIV infection, but a rare appearance, believed to be more common in patients infected with both HIV and T. pallidum, has been described. This is a placoid chorioretinopathy, consisting of large, yellowish placoid lesions with faded centers in the region of the macula and optic disc.107 There are two important features about syphilis infections in HIV-seropositive individuals. First, it can mimic the appearance of other ocular conditions such as cytomegalovirus-associated retinitis, making diagnosis and treatment difficult. Second, ocular syphilis is associated with CNS involvement in 85% to 100% of patients with HIV infection, as opposed to 35% to 40% of HIV-seronegative patients.108,109
Toxoplasmosis in HIV Infection
Toxoplasmosis retinitis can be similar to cytomegalovirus-associated retinitis, but there is usually more inflammation, and it is less likely to be associated with hemorrhage. The classic appearance in an immunocompetent patient is that of a fluffy white chorioretinal lesion in association with an area of scarring in one eye. However, in immunosuppressed patients, it can be bilateral, multifocal, and not associated with an old scar (Fig. 24-13). All of these features are suggestive of a primary infection rather than a reactivation of the condition. More than 50% of patients with ocular toxoplasmosis may have simultaneous toxoplasmosis cerebritis.99 Other fundal appearances include choroiditis, numerous scattered white lesions (a “miliary” pattern),110 a diffuse necrotizing retinitis,111,112 and punctate lesions in the outer retina.113
Malignancies Associated with HIV Infection
In patients with HIV infection, ocular non-Hodgkin lymphoma is usually associated with CNS and systemic involvement. The fundal features include necrotizing retinitis, choroidal infiltrates, and vitritis, but these appearances can also be as a result of infection by syphilis, toxoplasmosis, or viruses.114–118 Therefore, the diagnosis must be considered if the retinitis is unresponsive to antiviral, antisyphylis, and antitoxoplasmosis treatment.
Fungal Infections
A number of fungi can affect the brain and the retina. These are most often present in patients who are immunocompromised. The incidence of ocular fungal infections, such as Pneumocystis and Cryptococcus organisms, in HIV-seropositive patients appears to have decreased since the introduction of highly active antiretroviral therapy.97
Mucormycosis
Mucormycosis is an acute fungal infection that is rapidly invasive and carries a high mortality rate. Diabetes is a common predisposing factor. There are a number of clinical forms of mucormycosis, the most common being rhino-orbito-cerebral. This form affects the eye, and a necrotic eschar of the nose or hard palate is a characteristic sign. Although orbital cellulitis is the most common form of ocular involvement, serous retinal detachment and retinitis119 and choroidal ischemia have been described.120,121
Cryptococcus
Cryptococcal infection occurs mainly in patients who are HIV-seropositive. Up to 25% of patients with cryptococcal meningitis have neuro-ophthalmological lesions, which makes it the most common cause of acquired immunodeficiency syndrome–related neuro-ophthalmological lesions.99 Cryptococcal choroiditis is, however, rare. It may be may be multifocal, solitary, or confluent (Fig. 24-14).122 The lesions may initially be asymptomatic. Progressive visual loss may occur as a result of papilledema and also fungal optic nerve sheath invasion.123
Pneumocystis Carinii
One of the ocular features of P. carinii is choroiditis.124 It is classically bilateral and multifocal. The lesions are distinctive, being yellowish and well demarcated. They are slowly progressive, and usually vision is unaffected. There is not usually any associated ocular inflammation.124,125 In HIV-seropositive patients with presumed P. carinii choroidopathy, the majority had used inhaled pentamidine as prophylaxis against recurrent Pneumocystis-related pneumonia.125
MITOCHONDRIAL DISEASES
Pigmentary changes in the retina may occur in patients with mitochondrial disease. These can take a variety of forms and include appearances that have been described as pigment clumping, atrophy, salt-and-pepper retinopathy, and appearances of classic retinitis pigmentosa (Fig. 24-15). Involvement of the macula can occur, as can vascular attenuation. The most common appearance is that of a salt-and-pepper retinopathy. There does not, however, appear to be any link between the presence or type of retinal pigmentary change, the type of genetic defect, biochemical abnormality, and any clinical features.
A pigmentary retinopathy is a major diagnostic criterion for the diagnosis of Kearns-Sayre syndrome (the other features being a chronic progressive external ophthalmoplegia; onset before the age of 20; and cardiac conduction abnormalities, elevated cerebrospinal fluid protein levels, or cerebellar dysfunction). Retinal pigmentary degeneration can also be present in patients with chronic progressive external ophthalmoplegia and no other neurological or systemic abnormalities, as well as in otherwise unaffected relatives.126
Pigmentary retinopathy is also a major diagnostic feature of the syndrome of neurogenic muscle weakness, ataxia, and retinitis pigmentosa (NARP). However, patients who have no retinopathy, no subtle pigmentary retinopathy, and no severe bone spicule pattern have been identified with the same mutation.126 Furthermore, a patient in whom the retinal changes progressed from an initial salt-and-pepper retinopathy to typical retinitis pigmentosa over an 8-year period has been described.127
Other mitochondrial diseases in which retinal changes have been reported include the syndrome of myoclonic epilepsy and ragged red fibers and the syndrome of mitochondrial encephalopathy, lactic acidosis, and strokelike episodes (MELAS). The retinal changes in MELAS are in the form of pigmentary changes in the macula.128,129 MELAS is associated with the 3243 mutation, which can also manifest as maternally inherited diabetes and deafness. Macular changes similar to those in MELAS have also been reported in these patients,130,131 which demonstrates the marked overlap between clinical syndromes.
Funduscopic abnormalities may be present in patients with Leber’s hereditary optic neuropathy and in their asymptomatic maternal relatives. Especially during the acute phase of visual loss, there may be hyperemia of the optic nerve head, dilation and tortuosity of vessels, hemorrhages, circumpapillary telangiectatic microangiopathy, or circumpapillary nerve fiber layer swelling (pseudoedema). There may be cupping of the optic disc and arterial attenuation.126 In 2 of 20 patients with Leber’s hereditary optic neuropathy, pigmentary changes were noted at the retina.128
NEOPLASIA
Metastatic Disease
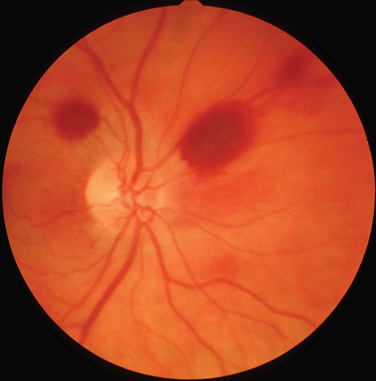
Metastases are probably the most common form of intraocular malignancy and can be associated with concurrent neurological involvement. They tend to involve the choroid but can affect the retina or vitreous humor (Fig. 24-16).132,133 Lung and breast carcinoma are the most common sources of ocular metastases and may be the manifesting sign of the disease.
Lymphoma
Primary intraocular lymphoma may occur in isolation, without involvement of the CNS. However, because the ocular and the CNS components show identical cytological features and phenotypical expression, the two entities are often considered as one entity: primary CNS lymphoma. Involvement of the eyes in this condition has been reported to occur in 12% to 25% of cases,134–137 and 56% to 80% of patients with intraocular lymphoma eventually develop intracranial involvement.138–142 As yet, it is unknown whether lymphoma cells from the eye can invade the CNS or whether the disease arises multifocally, in the eye and CNS. Experimental data from an animal model suggest that invasion of the CNS from the eye does not occur.143
The clinical features are dependent on the site of lymphoma involvement. The only sign of the disease may be vitreous cellular infiltration with resulting floaters and reduction of acuity. Because this is typically the appearance of a posterior uveitis, ocular lymphomas are described as being one of the masquerade syndromes. Diagnosis at this stage can therefore be difficult. The classic lesions are creamy yellow subretinal infiltrates (Fig. 24-17), with overlying retinal pigment epithelial detachments,142 but they may take on many forms, such as discrete white lesions, suggestive of ARN or toxoplasmosis144; branch retinal artery obstruction with coexistent multifocal chorioretinal scars145; and retinal vasculitis.
Paraneoplastic Syndromes
Carcinoma-Associated Retinopathy
Carcinoma-associated retinopathy (CAR) was first described in 1976.146 It is associated most commonly with small cell lung cancer and next most commonly with gynecological and breast cancers. Cases associated with other cancers such as non–small cell lung, colon, pancreatic, prostate, larynx, and bladder cancers and lymphoma have also been reported. Overall, the incidences among men and women are equal. Visual loss is usually subacute and bilateral, preceding tumor diagnosis in about 50% of cases. Patients may complain of positive visual phenomena, visual field loss, and night blindness. Initially there may be no retinal signs, but arteriolar narrowing and pigmentary retinal changes develop. Sheathing of the retinal vessels may develop. Electrophysiological testing demonstrates that both rods and cones are affected.
The first antigen shown to represent the source of autoimmunity in patients with CAR was the 23-kD protein recoverin.147,148 This is a calcium-binding protein that regulates phosphorylation of the visual pigment rhodopsin during visual transduction. A number of other antigens have since been reported to be associated with CAR. The next most commonly found antigen is the 46-kD protein enolase,149 although a number have now been identified (see review by Chan150).
However, the presence of neither antirecoverin nor antienolase antibodies is diagnostic for CAR. A few patients with antirecoverin antibodies but with no evidence of malignancy have been described.151,152 Up to two thirds of patients with antienolase antibody–associated retinopathy have no evidence of malignancy.153,154 Furthermore, in patients with antienolase antibodies, the retinopathy often develops after the detection of cancer, and the disease course is usually less severe than in patients with antirecoverin antibodies.154
Melanoma-Associated Retinopathy
Because of the relatively greater decrease in incidence of lung cancers in comparison with melanomas, it has been suggested that melanoma-associated retinopathy (MAR) is becoming more common than CAR.150 There are a number of differences between MAR and CAR. In MAR, the diagnosis of melanoma has often been made before the development of visual problems, and the incidence of MAR is higher in men. Patients tend to complain of shimmering vision and night blindness. The degree of visual loss is less severe than in CAR. As in CAR, the initial retinal appearances may be normal, but retinal pigment epithelial irregularity, retinal arteriolar attenuation, and optic disc pallor are present in cases in which the symptoms have been present for some time. Vitritis and retinal periphlebitis have been reported.155
Autoantibodies from MAR sera were shown to stain rod bipolar cells in the human retina.156 The specific antigen responsible has not been identified. Retinal cells other than bipolar cells have implicated, and antibodies against a variety of antigens have been identified (see Chan150).
Paraneoplastic Optic Neuritis with Retinitis
Paraneoplastic ophthalmological syndromes are usually retinopathies, but in rare cases, the optic nerve is affected. In some patients, both paraneoplastic optic neuritis and a retinopathy coexist. This has been described in patients with small cell lung carcinoma. None had antirecoverin antibodies, but all had a distinct immunoglobulin G marker antibody to collapsing response-mediator protein-5.157
Bilateral Diffuse Uveal Melanocytic Proliferation
This rare condition often precedes the diagnosis of cancer. Patients present with visual loss and are found to have many round or oval red patches at the level of retinal pigment epithelium and pigmented and nonpigmented melanocytic lesions of the uveal tract. An ultrasonogram reveals extremely thickened choroid. Retinal detachment and cataracts may develop.158 Approximately 25% of patients develop pigmentation of their skin or mucous membranes.159 The most commonly associated neoplasms are ovarian cancers in women and lung and pancreas cancers in men, although it has also been reported in kidney, colon, breast, and esophageal cancers.159,160
MOVEMENT DISORDERS AND ATAXIA
Most often the neuro-ophthalmological findings in patients with movement disorders are those of the oculomotor system. Patients with Parkinson’s disease often complain of blurred vision. Retinal appearances are normal, however, but functional abnormalities are present on psychophysical and electrophysiological testing. Some of these are reversible with levodopa (see Jackson and Owsley161 for a review). Patients with Huntington’s disease also demonstrate abnormalities of retinal function on psychophysical testing.162 However, about 50% of patients with Guam amyotrophic lateral sclerosis–parkinsonism–dementia complex (Lytico-Bodig disease) do have retinal pigmentary changes.163
A number of rare movement disorders are associated with retinal changes. Pantothenate kinase–associated neurodegeneration (formerly known as Hallervorden-Spatz syndrome),164 which is caused by a mutation in the PANK2 gene, is characterized by dystonia, parkinsonism, iron accumulation in the brain, and occasionally retinopathy. Electrophysiological abnormalities have been observed in affected patients who have clinically normal eyes.165 The syndrome of hypoprebetalipoproteinemia, acanthocytosis, retinitis pigmentosa, and pallidal degeneration, which is also caused by a PANK2 mutation,166 also has a pigmentary retinopathy.
Aceruloplasminemia is a rare condition that was first described in 1987.167 It also results in iron overload in the brain, retina, and pancreas. The first patient described was Asian, and the retinal changes were noted in the midperipheral region.167 Subsequently a case was described in a white patient, but the changes were at the macula.168
In Wilson’s disease, there is also a deficiency of ceruloplasmin, but the biochemical problem relates to copper metabolism. Retinal changes have been reported in addition to the familiar Kayser-Fleischer rings in the cornea. However, it is unclear whether these were caused by long-term therapy with pencillamine.169
A number of the ataxias are also associated with retinal pigmentary changes. These include Bassen-Kornzweig disease,170 Refsum’s disease,171 and adult-onset spinocerebellar syndrome with idiopathic vitamin E deficiency.172 A deficiency of vitamin E may cause the initial retinopathy; the treatment of these conditions includes vitamin E supplementation.173
Although there are many forms of spinocerebellar ataxia with oculomotor abnormalities, spinocerebellar ataxia type 7 can be differentiated from the other forms by the associated retinal changes. These take the form of progressive macular changes with visual loss (Fig. 24-18).174 Electrophysiological studies have demonstrated that the functional defects are greater than expected from the clinical appearance.175
MUSCLE DISEASES
Patients with facioscapulohumeral muscular dystrophy often have retinal telangiectasia. In rare cases, this can have significant visual consequences, such as exudative retinal detachment.176 Myotonic dystrophy is most frequently associated with cataract, but retinal pigmentary changes are also present. They can be peripheral or affect the macula and may slowly progress.177 The classic neuro-ophthalmological feature of dermatomyositis is the heliotrope rash of the eyelids, but a vasculitic retinopathy with retinal hemorrhages and cotton-wool spots may occur. With immunosuppression, visual recovery is usually but not always complete.178
PHACOMATOSES
Neurofibromatosis
Neurofibromatosis Type 1
The diagnostic criteria for neurofibromatosis type 1 include optic gliomas and Lisch nodules, which are hamartomas of the pigment epithelium. Nevertheless, retinal involvement is rare and generally nonspecific. The retinal lesions include a pigmentary retinopathy,179 retinal hamartomas,180,181 and capillary hemangiomas and combined hamartomas of the retina and pigment epithelium.181 Mild pigmentary changes that resemble cutaneous café au lait spots have been described.180 Myelinated fibers may be present more frequently than in the normal population,182 and vascular occlusions have also been reported,183 but it is unclear whether these are merely coincidental. Neurofibromas of the uveal tract can occur in up to 50% of patients.182,184–189 These are usually pale yellow nodules, but the entire uvea may be thickened by a diffuse neurofibroma. Choroidal nevi may occur more frequently in patients with neurofibromatosis type 1 than in the normal population.190
Neurofibromatosis Type 2
Cataracts and retinal changes are common in patients with neurofibromatosis type 2, whereas Lisch nodules are not as common as in neurofibromatosis type 1.191–196 Indeed, the ocular findings may be the first manifestation of the disease.194 The retinal changes can take the forms of epiretinal membranes,196–198 hamartomas of the retina,194,195 and combined pigment epithelial and retinal hamartomas.191,193,198 Retinal hamartomas are not exclusively associated with severe neurofibromatosis type 2, and neither the type nor the location of the germline neurofibromatosis type 2 mutation is the sole determinant of retinal abnormalities, which can be variably expressed in families with neurofibromatosis type 2.199
Tuberous Sclerosis
Hamartomas of the retina are the most prominent ocular manifestation of tuberous sclerosis, present in about 50% of patients and bilateral in 25% of cases. They arise from the ganglion cell layers and infiltrate all layers of the retina. They do not tend to grow or interfere with vision. The appearances are varying, ranging form semitransparent structures to opaque multinodular lesions (Fig. 24-19). Although most lesions remain stable, becoming calcified over time, they can develop in areas of previously normal-appearing retina.200 A case of a patient with a giant cell astrocytoma of the retina with atypical histopathological features and local aggressive behavior has been reported.201 Depigmented pigment epithelial lesions may be present. These can have a shape similar to that of cutaneous mountain ash leaf spots.202–204
Von Hippel-Lindau Disease
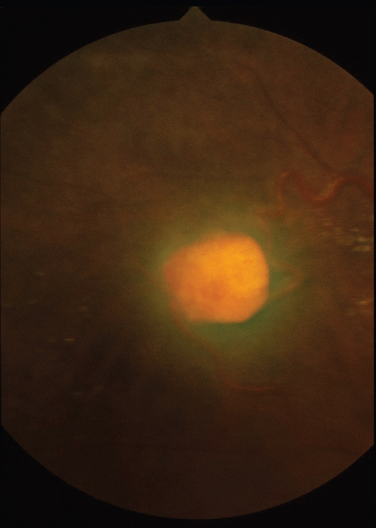
The retinal capillary hemangioma is the most frequent and the earliest manifestation of von Hippel-Lindau disease (Fig. 24-20).205–209 The other systemic features include hemangiomas of the CNS; renal cell carcinoma; pheochromocytoma; and renal, pancreatic, and epididymal cysts. The retinal capillary hemangioma is usually an orange-red, circumscribed round lesion. It can enlarge and develop a feeder vessel, and fluid may extravasate, leading to a retinal detachment. Thus, patients should have an annual ophthalmological assessment with dilated funduscopy, because the lesions may be small and situated in the retinal periphery. Retinal capillary hemangiomas can also develop around the optic disc. A rare retinal feature of von Hippel-Lindau disease are retinal “twin vessels,”210 defined as a paired retinal arteriole and venule that are separated by less than the diameter of one venule. Twin vessels are of normal caliber and look like normal retinal vessels except for their course. Retinal capillary hemangiomas not associated with von Hippel-Lindau disease do occur but at a later age (48 years) than in patients with von Hippel-Lindau disease (25 years).211 Nevertheless, the systemic features of von Hippel-Lindau disease must be ruled out in patients presenting with a solitary capillary hemangioma. The presence of multiple retinal capillary hemangiomas (two or more) indicates the presence of underlying von Hippel-Lindau disease.212
Sturge-Weber Syndrome
The chief components of the Sturge-Weber syndrome are a cutaneous hemifacial angioma and an ipsilateral angioma of the leptomeninges and brain. Glaucoma is the most common ocular association. Choroidal angiomas are also present; these can affect one or both eyes. Most commonly the angiomas are diffuse, obscuring the normal choroidal markings.213 More rarely, they are localized and may be associated with a serous retinal detachment.214 The retinal vasculature may be abnormal.215
Wyburn-Mason Syndrome
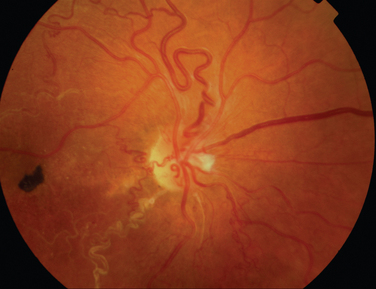
The classic Wyburn-Mason syndrome consists of an intracranial arteriovenous malformation and a separate retinal one. The retinal arteriovenous malformation has also been described as an arteriovenous aneurysm or racemose angioma. It is usually unilateral and involves the posterior pole, thus affecting the vision. Often it is stable, but it may enlarge (Fig. 24-21).216,217
CEREBROVASCULAR DISEASE
Retinopathy
The retinal and cerebral arterioles share common anatomy and physiology. Because the retina is readily visualized, it provides an opportunity to assess the retinal and, by inference, the cerebral circulation. Studies in people with hypertension have shown that signs of retinopathy are associated with both subclinical and clinical stroke.219–222 Retinal digital photography has been used in large population-based studies to investigate the relationship between retinal findings and systemic disease, including stroke. The images are analyzed in a standardized manner and are used to look for features of retinopathy such as microaneurysms, retinal hemorrhages, cotton-wool spots, arteriovenous nicking, and arteriolar narrowing. The studies include the Atherosclerosis Risk in Communities study, the Beaver Dam Eye Study, and the Blue Mountains Eye Study.
The Atherosclerosis Risk in Communities study has demonstrated that after controlling of stroke risk factors such as diabetes and hypertension, the presence of retinopathy was predictive of an incident stroke.223 Retinopathy was also associated with cognitive decline224 and with magnetic resonance imaging findings such as white matter lesions225 and cerebral atrophy.226 The risk of stroke was higher in patients who had both retinopathy and changes noted on cerebral magnetic resonance imaging.225
Retinal Emboli and Arteriolar Occlusion
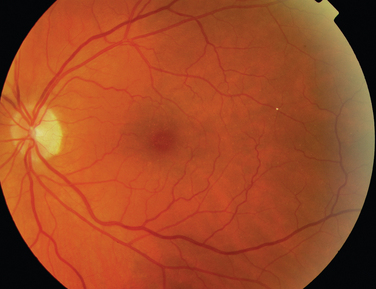
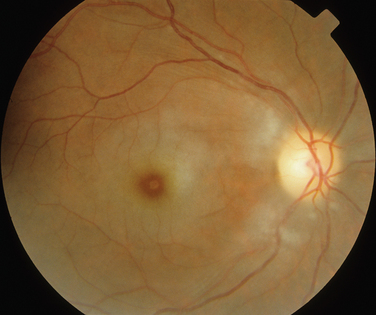
Retinal emboli are usually present at the bifurcation of retinal arterioles and may be reflective or nonreflective. A variety of emboli have been described, but clinically a reliable distinction cannot always be made (Fig. 24-22).227 Emboli are also present in up to about 40% of central retinal artery occlusions (Fig. 24-23) and 70% of branch retinal artery occlusions.228,229 The emboli usually originate from the carotid arteries or a cardiac source. The carotid vessels are the commonest source of emboli in patients with amaurosis fugax or retinal artery occlusion and of asymptomatic emboli,230 but often no embolic source is found.230,231 In elderly patients, the absence of an embolus in a retinal artery occlusion necessitates the exclusion of giant cell arteritis as a diagnosis. Furthermore, the presence of an embolus that is asymptomatic232 or in association with a retinal artery occlusion233 is not a good predictor of carotid artery stenosis. This may be because the emboli result from plaques and are not associated with stenosis per se.232
Emboli may be asymptomatic. It has been shown that for men with asymptomatic cholesterol emboli, there is an increased risk of stroke, but not myocardial infarction,234 and that patients with an embolus, symptomatic or not, have a reduced survival rate.228 Again, the analysis of digital photographs in large populations has added further information about emboli. The Beaver Dam Eye study found that for asymptomatic retinal emboli, the 10-year cumulative incidence was 1.5%.235 The incidence rose with age, from 1.0% in subjects aged 43 to 54 years to 2.2% in those aged 65 years or older at baseline. They are more common in men than in women. They appear to be transitory, and in approximately 30% of affected eyes, they are multiple. Bilateral ocular involvement is rare. The presence of asymptomatic retinal emboli is associated with carotid artery disease236 and an increased risk of death from stroke, independent of any other risk factors.235
Ocular Ischemic Syndrome and Other Ocular Associations with Carotid Artery Disease
Severe carotid stenosis is associated with venous stasis retinopathy. In this condition, there is an insidious onset of blurred vision in the affected eye. Patients may complain of episodes of transient monocular visual loss precipitated by exercise, eating, or bright lights. Examination reveals dilated and tortuous retinal veins, peripheral hemorrhages, and an easily induced pulsation of the central retinal artery with digital pressure.237,238
Venous stasis retinopathy may progress to ocular ischemic syndrome. This is a severe form of chronic ischemia caused by hypoperfusion of the eye, which affects both its anterior and posterior segments. Neovascularization of the iris, disc, and retina may occur. In patients with minimal or no neovascular changes, carotid endarterectomy appears to improve blood flow through the ophthalmic and central retinal arteries.239 It also appears to prevent progression of ocular ischemic syndrome, if not improve vision in all patients.239 Percutaneous angioplasty and stenting have also been used effectively in a number of patients who had lesions that were not amenable to endarterectomy.240
Patients with carotid occlusive disease may develop neurological symptoms at the same time as a central retinal artery occlusion.240 Carotid artery dissection is classically associated with a painful Horner’s syndrome, but both central and branch retinal artery occlusions can be present in this condition.241–243 Carotid artery disease is also associated with retinal vein occlusions.236
Retinal Vasculitis and Stroke
One form of retinal vasculitis is localized to the eye, and fluorescin angiography demonstrates ischemic retinal changes. This condition, known as idiopathic ischemic retinal vasculitis, has a worse visual prognosis than does retinal vasculitis with no associated ischemia.244 Furthermore, after a minimum follow up of 5 years, almost one third of patients in one study had suffered a stroke and/or myocardial infarction.245
Secondary Causes of Stroke
Coagulopathies that are associated with stroke, such as hyperhomocysteinemia and antiphospholipid syndrome, also have retinal manifestations such as retinal artery and vein occlusions.246,247
Cerebral Small-Vessel Disease
Susac’s Syndrome
Susac’s syndrome consists of the triad of encephalopathy, branch retinal artery occlusions, and hearing loss, which occurs most commonly in affected young women.248,249 The branch retinal artery occlusions are often bilateral and may be the manifesting features of the illness, or they may occur later in the clinical course. Fluorescein angiography may reveal focal leakage from retinal arterioles even in the absence of frank occlusion (Fig. 24-24). Such leakage can be used to measure disease activity. The encephalopathy manifests with headache, confusion, memory loss, behavioral changes, dysarthria, and occasional mutism. The hearing loss is usually bilateral and frequently associated with tinnitus and vertigo. Although the presence of encephalopathy, branch retinal artery occlusions, and hearing loss is pathognomonic for Susac’s syndrome, not all elements may be present initially. The condition usually stabilizes after a period of 2 to 4 years. On magnetic resonance images, lesions of the corpus callosum are often present.250
Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy
CADASIL is caused by mutations of the Notch3 gene, which is located on chromosome 19. In patients with CADASIL vision is usually preserved, although retinal findings such as peripapillary arteriolar sheathing, arteriolar narrowing, ateriovenous nicking,251 and cotton wool spots252 may be present. Visual loss may take the form of a hemianopic defect caused by stroke252 or anterior ischemic optic neuropathy.253 In asymptomatic patients, there may be electrophysiological evidence of retinal dysfunction.254 A histopathological study of four eyes from two patients with CADASIL demonstrated loss of vascular smooth muscle cells in the central retinal artery and its branches, as well as the optic disc. The choroidal circulation was spared, which demonstrated a differential involvement of small blood vessels.255
Other Hereditary Diseases of the Small Vessels
There are other rare hereditary conditions that affect the retinal and cerebral circulation. Patients with autosomal dominant vascular retinopathy, migraine, and Raynaud’s phenomenon may have a retinopathy that appears very similar to that of diabetic retinopathy. Indeed, a few patients may develop retinal neovascularization, which results in poor vision.256,257 Three other diseases with retinopathy have been linked to the same locus on chromosome 3p21. These are cerebroretinal vasculopathy; the syndrome of hereditary endotheliopathy, retinopathy, nephropathy, and stroke (HERNS); and hereditary vascular retinopathy.258 The retinal vascular abnormality constitutes part of the disease title in the syndrome of hereditary infantile hemiparesis, retinal arteriolar tortuosity, and leukoencephalopathy.259 Fabry’s disease is also accompanied by retinal vascular changes.260 The practical implication of these findings is that all patients with small-vessel disease probably require ophthalmoscopy.261 Finally, of interest is that a high prevalence of migraine has been in reported in CADASIL; HERNS; hereditary vascular retinopathy; and the syndrome of hereditary infantile hemiparesis, retinal arteriolar tortuosity, and leukoencephalopathy.
Terson’s Syndrome
Vitreous hemorrhage occurring in association with subarachnoid hemorrhage is known as Terson’s syndrome.262 However, intraocular hemorrhages of any type (retinal, subhyaloid, or vitreous) have been documented in 10% to 40% of individuals with subarachnoid hemorrhage,263 and reports have not always made a distinction between such types of hemorrhage (Fig. 24-25).264 Furthermore, preretinal hemorrhage may precede vitreous hemorrhage.263,265 Terson’s syndrome is present in between 3% and 13% of patients with a subarachnoid hemorrhage266 and carries an increased mortality rate in relation to patients who have had a subarachnoid hemorrhage but not vitreous hemorrhage.266 It has also been suggested that mild retinal hemorrhages are associated with a better prognosis than are large preretinal hemorrhages or vitreous hemorrhages.267 An urgent vitrectomy may be required for visual rehabilitation in patients with bilateral vitreous hemorrhage.
METABOLIC DISEASES IN CHILDREN
The most common retinal findings are the presence of a macular cherry-red spot (an appearance similar to that of a central retinal artery occlusion; see Fig. 24-23) and pigmentary retinopathy. The cherry-red spot is classically associated with Tay-Sachs disease. However, a form of this finding is present in many of the metabolic diseases, although not necessarily in all patients affected by them. Also, in other conditions it may have prognostic significance; for example, in Farber’s disease, its presence may be correlated with disease severity.268 Pigmentary retinopathy is also present in a number of conditions, although, again, not in all affected individuals. The clinical pattern of the retinopathy is variable. There are a few striking changes associated with the metabolic diseases, such as the absence of retinal pigmentation observed in phenylketonuria and the presence of bilateral parafoveal ringlets observed in hyperoxaluria type 1.269
Retinal pigmentary changes can be present in a number of the following conditions:
Albert DM, Jakobiec FA, editors. Principles and Practice of Ophthalmology. Philadelphia: WB Saunders, 2000.
Gold DH, Weingeist TA, editors. Color Atlas of the Eye in Systemic Disease. Philadelphia: Lippincott Williams & Wilkins, 2001.
Miller NR, Newman NJ, editors. Walsh and Hoyt’s Clinical Neuro-Ophthalmology. Baltimore: William & Wilkins, 1998.
Riordan-Eva P, Whitcher JP, editors. Vaughan & Asbury’s General Ophthalmology. New York: McGraw-Hill, 2004.
Spalton DJ, Hitchings RA, Hunter P, editors. Atlas of Clinical Ophthalmology, 3rd ed., London: Elsevier, 2004.
1 Jabs DA, Nussenblatt RB, Rosenbaum JT. Standardization of uveitis nomenclature for reporting clinical data. Results of the First International Workshop. Am J Ophthalmol. 2005;140:509-516.
2 Rothova A, Alberts C, Glasius E, et al. Risk factors for ocular sarcoidosis. Doc Ophthalmol. 1989;72:287-296.
3 Edelsten C, Pearson A, Joynes E, et al. The ocular and systemic prognosis of patients presenting with sarcoid uveitis. Eye. 1999;13(Pt 6):748-753.
4 Gould H, Kaufman HE. Sarcoid of the fundus. Arch Ophthalmol. 1961;65:453-456.
5 James DG, Neville E, Langley DA. Ocular sarcoidosis. Trans Ophthalmol Soc U K. 1976;96:133-139.
6 Stavrou P, Linton S, Young DW, et al. Clinical diagnosis of ocular sarcoidosis. Eye. 1997;11(Pt 3):365-370.
7 Walsh F. Ocular importance of sarcoid. Arch Ophthalmol. 1939;21:421-438.
8 Sanders MD, Shilling JS. Retinal, choroidal, and optic disc involvement in sarcoidosis. Trans Ophthalmol Soc U K. 1976;96:140-144.
9 Spalton DJ, Sanders MD. Fundus changes in histologically confirmed sarcoidosis. Br J Ophthalmol. 1981;65:348-358.
10 Rothova A, Lardenoye C. Arterial macroaneurysms in peripheral multifocal chorioretinitis associated with sarcoidosis. Ophthalmology. 1998;105:1393-1397.
11 Cook BEJr., Robertson DM. Confluent choroidal infiltrates with sarcoidosis. Retina. 2000;20:1-7.
12 Lardenoye CW, Van der Lelij A, de Loos WS, et al. Peripheral multifocal chorioretinitis: a distinct clinical entity? Ophthalmology. 1997;104:1820-1826.
13 Thorne JE, Brucker AJ. Choroidal white lesions as an early manifestation of sarcoidosis. Retina. 2000;20:8-15.
14 Edelsten C, Stanford MR, Graham EM. Serpiginous choroiditis: an unusual presentation of ocular sarcoidosis. Br J Ophthalmol. 1994;78:70-71.
15 Criteria for diagnosis of Behçet’s disease. International Study Group for Behçet’s Disease. Lancet. 1990;335:1078-1080.
16 Akman-Demir G, Serdaroglu P, Tasci B. Clinical patterns of neurological involvement in Behçet’s disease: evaluation of 200 patients. The Neuro-Behçet Study Group. Brain. 1999;122(Pt 11):2171-2182.
17 Kansu T, Kirkali P, Kansu E, et al. Optic neuropathy in Behçet’s disease. J Clin Neuroophthalmol. 1989;9:277-280.
18 Kidd D, Steuer A, Denman AM, et al. Neurological complications in Behçet’s syndrome. Brain. 1999;122(Pt 11):2183-2194.
19 Mitra S, Koul RL. Paediatric neuro-Behçet’s disease presenting with optic nerve head swelling. Br J Ophthalmol. 1999;83:1096.
20 Swerdlow RH, Hanna GR. Behçet’s disease: presentation with sagittal sinus thrombosis diagnosed noninvasively. Headache. 1996;36:115-118.
21 Wechsler B, Vidailhet M, Piette JC, et al. Cerebral venous thrombosis in Behçet’s disease: clinical study and long-term follow-up of 25 cases. Neurology. 1992;42:614-618.
22 Lee E-S, Hur W, Bang D, et al. Prognosis of recurrent oral ulceration in Behçet’s disease. In: Godeau P, Wechsler B, editors. Behçet’s Disease. Paris: Elsevier; 1993:291-294.
23 Dilsen N, Konice M, Aral O, et al. Risk factors for vital organ involvement in Behçet’s disease. In: Godeau P, Wechsler B, editors. Behçet’s Disease. Paris: Elsevier; 1993:165-169.
24 Lueck CJ, Pires M, McCartney AC, et al. Ocular and neurological Behçet’s disease without orogenital ulceration? J Neurol Neurosurg Psychiatry. 1993;56:505-508.
25 Muhaya M, Lightman S, Ikeda E, et al. Behçet’s disease in Japan and in Great Britain: a comparative study. Ocul Immunol Inflamm. 2000;8:141-148.
26 Read RW, Holland GN, Rao NA, et al. Revised diagnostic criteria for Vogt-Koyanagi-Harada disease: report of an international committee on nomenclature. Am J Ophthalmol. 2001;131:647-652.
27 Najman-Vainer J, Levinson RD, Graves MC, et al. An association between Vogt-Koyanagi-Harada disease and Guillain-Barré syndrome. Am J Ophthalmol. 2001;131:615-619.
28 Klinkhoff AV, Beattie CW, Chalmers A. Retinopathy in systemic lupus erythematosus: relationship to disease activity. Arthritis Rheum. 1986;29:1152-1156.
29 Gold DH, Morris DA, Henkind P. Ocular findings in systemic lupus erythematosus. Br J Ophthalmol. 1972;56:800-804.
30 Graham EM, Spalton DJ, Barnard RO, et al. Cerebral and retinal vascular changes in systemic lupus erythematosus. Ophthalmology. 1985;92:444-448.
31 Hollenhorst RW. Effect of posture on retinal ischemia from temporal arteritis. Arch Ophthalmol. 1967;78:569-577.
32 Chun YS, Park SJ, Park IK, et al. The clinical and ocular manifestations of Takayasu arteritis. Retina. 2001;21:132-140.
33 Galetta SL. Vasculitis. In: Miller NR, Newman NJ, editors. Walsh and Hoyt’s Clinical Neuro-ophthalmology. Baltimore: Williams & Wilkins; 1998:3725-3886.
34 Hsu CT, Kerrison JB, Miller NR, et al. Choroidal infarction, anterior ischemic optic neuropathy, and central retinal artery occlusion from polyarteritis nodosa. Retina. 2001;21:348-351.
35 Font RL, Mehta RS, Streusand SB, et al. Bilateral retinal ischemia in Kawasaki disease: postmortem findings and electron microscopic observations. Ophthalmology. 1983;90:569-577.
36 Drachman DA. Neurological complications of Wegener’s granulomatosis. Arch Neurol. 1963;8:145-155.
37 Nishino H, Rubino FA, DeRemee RA, et al. Neurological involvement in Wegener’s granulomatosis: an analysis of 324 consecutive patients at the Mayo Clinic. Ann Neurol. 1993;33:4-9.
38 Bullen CL, Liesegang TJ, McDonald TJ, et al. Ocular complications of Wegener’s granulomatosis. Ophthalmology. 1983;90:279-290.
39 Riordan-Eva P, Williams CE, Wing AJ, et al. Retinal neovascularization secondary to cytomegalovirus retinitis in Wegener’s granulomatosis. J R Soc Med. 1993;86:301-302.
40 Tugal-Tutkun I, Kir N, Gul A, et al. Cytomegalovirus retinitis in a patient with Wegener’s granulomatosis. Ophthalmologica. 2000;214:149-152.
41 Agrawal A, Dick AD, Olson JA. Visual symptoms in patients on cyclophosphamide may herald sight threatening disease. Br J Ophthalmol. 2003;87:122-123.
42 Newman NJ, Slamovits TL, Friedland S, et al. Neuroophthalmic manifestations of meningocerebral inflammation from the limited form of Wegener’s granulomatosis. Am J Ophthalmol. 1995;120:613-621.
43 Spalton DJ, Graham EM, Page NG, et al. Ocular changes in limited forms of Wegener’s granulomatosis. Br J Ophthalmol. 1981;65:553-563.
44 Takanashi T, Uchida S, Arita M, et al. Orbital inflammatory pseudotumor and ischemic vasculitis in Churg-Strauss syndrome: report of two cases and review of the literature. Ophthalmology. 2001;108:1129-1133.
45 Mihara M, Hayasaka S, Watanabe K, et al. Ocular manifestations in patients with microscopic polyangiitis. Eur J Ophthalmol. 2005;15:138-142.
46 Arnold AC, Pepose JS, Hepler RS, et al. Retinal periphlebitis and retinitis in multiple sclerosis. I. Pathologic characteristics. Ophthalmology. 1984;91:255-262.
47 Hornstein G. The relation of retinal periphlebitis to multiple sclerosis and other neurological disorders. Acta Neurol Scand. 1971;47:413-425.
48 Rucker CW. Sheathing of the retinal veins in multiple sclerosis. Mayo Clin Proc. 1944;19:176-178.
49 Shaw PJ, Smith NM, Ince PG, et al. Chronic periphlebitis retinae in multiple sclerosis: a histopathological study. J Neurol Sci. 1987;77:147-152.
50 Tola MR, Granieri E, Casetta I, et al. Retinal periphlebitis in multiple sclerosis: a marker of disease activity? Eur Neurol. 1993;33:93-96.
51 Adams CW, Poston RN, Buk SJ, et al. Inflammatory vasculitis in multiple sclerosis. J Neurol Sci. 1985;69:269-283.
52 Lightman S, McDonald WI, Bird AC, et al. Retinal venous sheathing in optic neuritis: its significance for the pathogenesis of multiple sclerosis. Brain. 1987;110(Pt 2):405-414.
53 Engell T, Sellebjerg F, Jensen C. Changes in the retinal veins in acute optic neuritis. Acta Neurol Scand. 1999;100:81-83.
54 Engell T. Neurological disease activity in multiple sclerosis patients with periphlebitis retinae. Acta Neurol Scand. 1986;73:168-172.
55 Birch MK, Barbosa S, Blumhardt LD, et al. Retinal venous sheathing and the blood-retinal barrier in multiple sclerosis. Arch Ophthalmol. 1996;114:34-39.
56 Graham EM, Stanford MR, Sanders MD, et al. A point prevalence study of 150 patients with idiopathic retinal vasculitis: 1. Diagnostic value of ophthalmological features. Br J Ophthalmol. 1989;73:714-721.
57 Biousse V, Trichet C, Bloch-Michel E, et al. Multiple sclerosis associated with uveitis in two large clinic-based series. Neurology. 1999;52:179-181.
58 Edwards LJ, Constantinescu CS. A prospective study of conditions associated with multiple sclerosis in a cohort of 658 consecutive outpatients attending a multiple sclerosis clinic. Mult Scler. 2004;10:575-581.
59 Schmidt S, Wessels L, Augustin A, et al. Patients with multiple sclerosis and concomitant uveitis/periphlebitis retinae are not distinct from those without intraocular inflammation. J Neurol Sci. 2001;187:49-53.
60 Graham EM, Francis DA, Sanders MD, et al. Ocular inflammatory changes in established multiple sclerosis. J Neurol Neurosurg Psychiatry. 1989;52:1360-1363.
61 Smith JR, Rosenbaum JT. Neurological concomitants of uveitis. Br J Ophthalmol. 2004;88:1498-1499.
62 Zein G, Berta A, Foster CS. Multiple sclerosis-associated uveitis. Ocul Immunol Inflamm. 2004;12:137-142.
63 Towler HM, Lightman S. Symptomatic intraocular inflammation in multiple sclerosis. Clin Exp Ophthalmol. 2000;28:97-102.
64 Wakefield D, Jennings A, McCluskey PJ. Intravenous pulse methylprednisolone in the treatment of uveitis associated with multiple sclerosis. Clin Exp Ophthalmol. 2000;28:103-106.
65 Vine AK. Severe periphlebitis, peripheral retinal ischemia, and preretinal neovascularization in patients with multiple sclerosis. Am J Ophthalmol. 1992;113:28-32.
66 Parmley VC, Schiffman JS, Maitland CG, et al. Does neuroretinitis rule out multiple sclerosis? Arch Neurol. 1987;44:1045-1048.
67 Williams KE, Johnson LN. Neuroretinitis in patients with multiple sclerosis.Ophthalmology. 2004;111:335-340. discussion, Ophthalmology. 2004;111:340-341.
68 Moreno RJ, Weisman J, Waller S. Neuroretinitis: an unusual presentation of ocular toxoplasmosis. Ann Ophthalmol. 1992;24:68-70.
69 Solley WA, Martin DF, Newman NJ, et al. Cat scratch disease: posterior segment manifestations. Ophthalmology. 1999;106:1546-1553.
70 Aaberg TM. The expanding ophthalmologic spectrum of Lyme disease. Am J Ophthalmol. 1989;107:77-80.
71 Balcer LJ, Winterkorn JM, Galetta SL. Neuroophthalmic manifestations of Lyme disease. J Neuroophthalmol. 1997;17:108-121.
72 Breeveld J, Kuiper H, Spanjaard L, et al. Uveitis and Lyme borreliosis. Br J Ophthalmol. 1993;77:480-481.
73 Kuiper H, Koelman JH, Hager MJ. Vitreous clouding associated with Lyme borreliosis. Am J Ophthalmol. 1989;108:453-454.
74 Bialasiewicz AA, Ruprecht KW, Naumann GO, et al. Bilateral diffuse choroiditis and exudative retinal detachments with evidence of Lyme disease. Am J Ophthalmol. 1988;105:419-420.
75 Smith JL, Winward KE, Nicholson DF, et al. Retinal vasculitis in Lyme borreliosis. J Clin Neuroophthalmol. 1991;11:7-15.
76 Trends in infectious syphilis. update on national data to 2003 and current epidemiological data from the London outbreak. Commun Dis Rep CDR Wkly. 2004;14(31):7-10.
77 Barile GR, Flynn TE. Syphilis exposure in patients with uveitis. Ophthalmology. 1997;104:1605-1609.
78 Deschenes J, Seamone CD, Baines MG. Acquired ocular syphilis: diagnosis and treatment. Ann Ophthalmol. 1992;24:134-138.
79 Cubillan LD, Cubillan EA, Berger TG, et al. Syphilitic uveitis and dermatitis. Arch Ophthalmol. 1998;116:696-697.
80 Jabbour NM, Faris B, Trempe CL. A case of pulmonary tuberculosis presenting with a choroidal tuberculoma. Ophthalmology. 1985;92:834-837.
81 Mansour AM, Haymond R. Choroidal tuberculomas without evidence of extraocular tuberculosis. Graefes Arch Clin Exp Ophthalmol. 1990;228:382-383.
82 Gur S, Silverstone BZ, Zylberman R, et al. Chorioretinitis and extrapulmonary tuberculosis. Ann Ophthalmol. 1987;19:112-115.
83 Fountain JA, Werner RB. Tuberculous retinal vasculitis. Retina. 1984;4:48-50.
84 Shah SM, Howard RS, Sarkies NJ, et al. Tuberculosis presenting as retinal vasculitis. J R Soc Med. 1988;81:232-233.
85 Finelli PF, McEntee WJ, Lessell S, et al. Whipple’s disease with predominantly neuroophthalmic manifestations. Ann Neurol. 1977;1:247-252.
86 Avila MP, Jalkh AE, Feldman E, et al. Manifestations of Whipple’s disease in the posterior segment of the eye. Arch Ophthalmol. 1984;102:384-390.
87 Font RL, Rao NA, Issarescu S, et al. Ocular involvement in Whipple’s disease: light and electron microscopic observations. Arch Ophthalmol. 1978;96:1431-1436.
88 Leland TM, Chambers JK. Ocular findings in Whipple’s disease. South Med J. 1978;71:335-338.
89 Rickman LS, Freeman WR, Green WR, et al. Brief report: uveitis caused by Tropheryma whippelii (Whipple’s bacillus). N Engl J Med. 1995;332:363-366.
90 Holland GN. Standard diagnostic criteria for the acute retinal necrosis syndrome. Executive Committee of the American Uveitis Society. Am J Ophthalmol. 1994;117:663-667.
91 Ganatra JB, Chandler D, Santos C, et al. Viral causes of the acute retinal necrosis syndrome. Am J Ophthalmol. 2000;129:166-172.
92 Maertzdorf J, Van der Lelij A, Baarsma GS, et al. Herpes simplex virus type 1 (HSV-1)-induced retinitis following herpes simplex encephalitis: indications for brain-to-eye transmission of HSV-1. Ann Neurol. 2000;48:936-939.
93 Kamel OR, Galloway GD, Trew DR. Delayed onset acute retinal necrosis 20 years following herpetic encephalitis. Eye. 2000;14(Pt 5):788-789.
94 Chung GW, Thulborn KR, Pulido JS, et al. Magnetic resonance imaging of diffuse cerebral vasculitis associated with acute retinal necrosis. Arch Ophthalmol. 2004;122:1719-1720.
95 Gagnon A, Bouchard RW. Fulminating adult-onset subacute sclerosing panencephalitis in a 49-year-old man. Arch Neurol. 2003;60:1160-1161.
96 Berker N, Batman C, Guven A, et al. Optic atrophy and macular degeneration as initial presentations of subacute sclerosing panencephalitis. Am J Ophthalmol. 2004;138:879-881.
97 Goldberg DE, Smithen LM, Angelilli A, et al. HIV-associated retinopathy in the HAART era. Retina. 2005;25:633-649. quiz, Retina 005; 25:682–683.
98 Morinelli EN, Dugel PU, Riffenburgh R, et al. Infectious multifocal choroiditis in patients with acquired immune deficiency syndrome. Ophthalmology. 1993;100:1014-1021.
99 Jabs DA. Ocular manifestations of HIV infection. Trans Am Ophthalmol Soc. 1995;93:623-683.
100 Kuppermann BD, Petty JG, Richman DD, et al. Correlation between CD4+ counts and prevalence of cytomegalovirus retinitis and human immunodeficiency virus-related noninfectious retinal vasculopathy in patients with acquired immunodeficiency syndrome. Am J Ophthalmol. 1993;115:575-582.
101 Akduman L, Feiner MA, Olk RJ, et al. Macular ischemia as a cause of decreased vision in a patient with acquired immunodeficiency syndrome. Am J Ophthalmol. 1997;124:699-702.
102 Conway MD, Tong P, Olk RJ. Branch retinal artery occlusion (BRAO) combined with branch retinal vein occlusion (BRVO) and optic disc neovascularization associated with HIV and CMV retinitis. Int Ophthalmol. 1995;19:249-252.
103 Donahue SP. Human immunodeficiency virus-associated vision loss: electroretinogram attenuation. Am J Ophthalmol. 1998;126:729-731.
104 Holbrook JT, Jabs DA, Weinberg DV, et al. Visual loss in patients with cytomegalovirus retinitis and acquired immunodeficiency syndrome before widespread availability of highly active antiretroviral therapy. Arch Ophthalmol. 2003;121:99-107.
105 Kuo IC, Kempen JH, Dunn JP, et al. Clinical characteristics and outcomes of cytomegalovirus retinitis in persons without human immunodeficiency virus infection. Am J Ophthalmol. 2004;138:338-346.
106 Gariano RF, Berreen JP, Cooney EL. Progressive outer retinal necrosis and acute retinal necrosis in fellow eyes of a patient with acquired immunodeficiency syndrome. Am J Ophthalmol. 2001;132:421-423.
107 Gass JD, Braunstein RA, Chenoweth RG. Acute syphilitic posterior placoid chorioretinitis. Ophthalmology. 1990;97:1288-1297.
108 Becerra LI, Ksiazek SM, Savino PJ, et al. Syphilitic uveitis in human immunodeficiency virus-infected and noninfected patients. Ophthalmology. 1989;96:1727-1730.
109 Levy JH, Liss RA, Maguire AM. Neurosyphilis and ocular syphilis in patients with concurrent human immunodeficiency virus infection. Retina. 1989;9:175-180.
110 Berger BB, Egwuagu CE, Freeman WR, et al. Miliary toxoplasmic retinitis in acquired immunodeficiency syndrome. Arch Ophthalmol. 1993;111:373-376.
111 Lee YF, Chen SJ, Chung YM, et al. Diffuse toxoplasmic retinochoroiditis as the initial manifestation of acquired immunodeficiency syndrome. J Formos Med Assoc. 2000;99:219-223.
112 Parke DW2nd, Font RL. Diffuse toxoplasmic retinochoroiditis in a patient with AIDS. Arch Ophthalmol. 1986;104:571-575.
113 Moraes HVJr. Punctate outer retinal toxoplasmosis in an HIV-positive child. Ocul Immunol Inflamm. 1999;7:93-95.
114 Matzkin DC, Slamovits TL, Rosenbaum PS. Simultaneous intraocular and orbital non-Hodgkin lymphoma in the acquired immune deficiency syndrome. Ophthalmology. 1994;101:850-855.
115 Ormerod LD, Puklin JE. AIDS-associated intraocular lymphoma causing primary retinal vasculitis. Ocul Immunol Inflamm. 1997;5:271-278.
116 Rivero ME, Kuppermann BD, Wiley CA, et al. Acquired immunodeficiency syndrome-related intraocular B-cell lymphoma. Arch Ophthalmol. 1999;117:616-622.
117 Schanzer MC, Font RL, O’Malley RE. Primary ocular malignant lymphoma associated with the acquired immune deficiency syndrome. Ophthalmology. 1991;98:88-91.
118 Stanton CA, Sloan B3rd, Slusher MM, et al. Acquired immunodeficiency syndrome-related primary intraocular lymphoma. Arch Ophthalmol. 1992;110:1614-1617.
119 Kim IT, Shim JY, Yung BY. Serous retinal detachment in a patient with rhino-orbital mucormycosis. Jpn J Ophthalmol. 2001;45:301-304.
120 Newman RM, Kline LB. Evolution of fundus changes in mucormycosis. J Neuroophthalmol. 1997;17:51-52.
121 Kadayifcilar S, Gedik S, Onerci M, et al. Chorioretinal alterations in mucormycosis. Eye. 2001;15:99-102.
122 Verma S, Graham EM. Cryptococcus presenting as cloudy choroiditis in an AIDS patient. Br J Ophthalmol. 1995;79:618-619.
123 Garrity JA, Herman DC, Imes R, et al. Optic nerve sheath decompression for visual loss in patients with acquired immunodeficiency syndrome and cryptococcal meningitis with papilledema. Am J Ophthalmol. 1993;116:472-478.
124 Freeman WR, Gross JG, Labelle J, et al. Pneumocystis carinii choroidopathy: a new clinical entity. Arch Ophthalmol. 1989;107:863-867.
125 Shami MJ, Freeman W, Friedberg D, et al. A multicenter study of Pneumocystis choroidopathy. Am J Ophthalmol. 1991;112:15-22.
126 Biousse V, Newman NJ. Neuro-ophthalmology of mitochondrial diseases. Semin Neurol. 2001;21:275-291.
127 Kerrison JB, Biousse V, Newman NJ. Retinopathy of NARP syndrome. Arch Ophthalmol. 2000;118:298-299.
128 Isashiki Y, Nakagawa M, Ohba N, et al. Retinal manifestations in mitochondrial diseases associated with mitochondrial DNA mutation. Acta Ophthalmol Scand. 1998;76:6-13.
129 Latkany P, Ciulla TA, Cacchillo PF, et al. Mitochondrial maculopathy: geographic atrophy of the macula in the MELAS associated A to G 3243 mitochondrial DNA point mutation. Am J Ophthalmol. 1999;128:112-114.
130 Smith PR, Bain SC, Good PA, et al. Pigmentary retinal dystrophy and the syndrome of maternally inherited diabetes and deafness caused by the mitochondrial DNA 3243 tRNA(Leu) A to G mutation. Ophthalmology. 1999;106:1101-1118.
131 Massin P, Virally-Monod M, Vialettes B, et al. Prevalence of macular pattern dystrophy in maternally inherited diabetes and deafness. GEDIAM Group. Ophthalmology. 1999;106:1821-1827.
132 Leys AM, Van Eyck LM, Nuttin BJ, et al. Metastatic carcinoma to the retina: clinicopathologic findings in two cases. Arch Ophthalmol. 1990;108:1448-1452.
133 Spraul CW, Martin DF, Hagler WS, et al. Cytology of metastatic cutaneous melanoma to the vitreous and retina. Retina. 1996;16:328-332.
134 DeAngelis LM, Yahalom J, Heinemann MH, et al. Primary CNS lymphoma: combined treatment with chemotherapy and radiotherapy. Neurology. 1990;40:80-86.
135 Hochberg FH, Miller DC. Primary central nervous system lymphoma. J Neurosurg. 1988;68:835-853.
136 Peterson K, Gordon KB, Heinemann MH, et al. The clinical spectrum of ocular lymphoma. Cancer. 1993;72:843-849.
137 Verbraeken HE, Hanssens M, Priem H, et al. Ocular nonHodgkin’s lymphoma: a clinical study of nine cases. Br J Ophthalmol. 1997;81:31-36.
138 Merchant A, Foster CS. Primary intraocular lymphoma. Int Ophthalmol Clin. 1997;37:101-115.
139 Freeman LN, Schachat AP, Knox DL, et al. Clinical features, laboratory investigations, and survival in ocular reticulum cell sarcoma. Ophthalmology. 1987;94:1631-1639.
140 Chang TS, Byrne SF, Gass JD, et al. Echographic findings in benign reactive lymphoid hyperplasia of the choroid. Arch Ophthalmol. 1996;114:669-675.
141 Rockwood EJ, Zakov ZN, Bay JW. Combined malignant lymphoma of the eye and CNS (reticulum-cell sarcoma). Report of three cases. J Neurosurg. 1984;61:369-374.
142 Corriveau C, Easterbrook M, Payne D. Lymphoma simulating uveitis (masquerade syndrome). Can J Ophthalmol. 1986;21:144-149.
143 Assaf N, Hasson T, Hoch-Marchaim H, et al. An experimental model for infiltration of malignant lymphoma to the eye and brain. Virchows Arch. 1997;431:459-467.
144 Ridley ME, McDonald HR, Sternberg PJr, et al. Retinal manifestations of ocular lymphoma (reticulum cell sarcoma).Ophthalmology. 1992;99:1153-1160. discussion, Ophthalmology. 1992;99:1160-1161.
145 Gass JD, Trattler HL. Retinal artery obstruction and atheromas associated with non-Hodgkin’s large cell lymphoma (reticulum cell sarcoma). Arch Ophthalmol. 1991;109:1134-1139.
146 Sawyer RA, Selhorst JB, Zimmerman LE, et al. Blindness caused by photoreceptor degeneration as a remote effect of cancer. Am J Ophthalmol. 1976;81:606-613.
147 Thirkill CE, Tait RC, Tyler NK, et al. The cancer-associated retinopathy antigen is a recoverin-like protein. Invest Ophthalmol Vis Sci. 1992;33:2768-2772.
148 Polans AS, Burton MD, Haley TL, et al. Recoverin, but not visinin, is an autoantigen in the human retina identified with a cancer-associated retinopathy. Invest Ophthalmol Vis Sci. 1993;34:81-90.
149 Adamus G, Aptsiauri N, Guy J, et al. The occurrence of serum autoantibodies against enolase in cancer-associated retinopathy. Clin Immunol Immunopathol. 1996;78:120-129.
150 Chan JW. Paraneoplastic retinopathies and optic neuropathies. Surv Ophthalmol. 2003;48:12-38.
151 Heckenlively JR, Fawzi AA, Oversier J, et al. Autoimmune retinopathy: patients with antirecoverin immunoreactivity and panretinal degeneration. Arch Ophthalmol. 2000;118:1525-1533.
152 Whitcup SM, Vistica BP, Milam AH, et al. Recoverinassociated retinopathy: a clinically and immunologically distinctive disease. Am J Ophthalmol. 1998;126:230-237.
153 Adamus G, Ren G, Weleber RG. Autoantibodies against retinal proteins in paraneoplastic and autoimmune retinopathy. BMC Ophthalmol. 2004;4:5.
154 Weleber RG, Watzke RC, Shults WT, et al. Clinical and electrophysiologic characterization of paraneoplastic and autoimmune retinopathies associated with antienolase antibodies. Am J Ophthalmol. 2005;139:780-794.
155 Keltner JL, Thirkill CE, Yip PT. Clinical and immunologic characteristics of melanoma-associated retinopathy syndrome: eleven new cases and a review of 51 previously published cases. J Neuroophthalmol. 2001;21:173-187.
156 Milam AH, Saari JC, Jacobson SG, et al. Autoantibodies against retinal bipolar cells in cutaneous melanoma-associated retinopathy. Invest Ophthalmol Vis Sci. 1993;34:91-100.
157 Cross SA, Salomao DR, Parisi JE, et al. Paraneoplastic autoimmune optic neuritis with retinitis defined by CRMP-5-IgG. Ann Neurol. 2003;54:38-50.
158 Gass JD, Gieser RG, Wilkinson CP, et al. Bilateral diffuse uveal melanocytic proliferation in patients with occult carcinoma. Arch Ophthalmol. 1990;108:527-533.
159 O’Neal KD, Butnor KJ, Perkinson KR, et al. Bilateral diffuse uveal melanocytic proliferation associated with pancreatic carcinoma: a case report and literature review of this paraneoplastic syndrome. Surv Ophthalmol. 2003;48:613-625.
160 Chahud F, Young RH, Remulla JF, et al. Bilateral diffuse uveal melanocytic proliferation associated with extraocular cancers: review of a process particularly associated with gynecologic cancers. Am J Surg Pathol. 2001;25:212-218.
161 Jackson GR, Owsley C. Visual dysfunction, neurodegenerative diseases, and aging. Neurol Clin. 2003;21:709-728.
162 Paulus W, Schwarz G, Werner A, et al. Impairment of retinal increment thresholds in Huntington’s disease. Ann Neurol. 1993;34:574-578.
163 Cox TA, McDarby JV, Lavine L, et al. A retinopathy on Guam with high prevalence in Lytico-Bodig. Ophthalmology. 1989;96:1731-1735.
164 Jankovic J, Kirkpatrick JB, Blomquist KA, et al. Late-onset Hallervorden-Spatz disease presenting as familial parkinsonism. Neurology. 1985;35:227-234.
165 Egan RA, Weleber RG, Hogarth P, et al. Neuro-ophthalmologic and electroretinographic findings in pantothenate kinase-associated neurodegeneration (formerly Hallervorden-Spatz syndrome). Am J Ophthalmol. 2005;140:267-274.
166 Houlden H, Lincoln S, Farrer M, et al. Compound heterozygous PANK2 mutations confirm HARP and Hallervorden Spatz syndromes are allelic. Neurology. 2003;61:1423-1426.
167 Miyajima H, Nishimura Y, Mizoguchi K, et al. Familial apoceruloplasmin deficiency associated with blepharospasm and retinal degeneration. Neurology. 1987;37:761-767.
168 Dunaief JL, Richa C, Franks EP, et al. Macular degeneration in a patient with aceruloplasminemia, a disease associated with retinal iron overload. Ophthalmology. 2005;112:1062-1065.
169 Dingle J, Havener WH. Ophthalmoscopic changes in a patient with Wilson’s disease during long-term penicillamine therapy. Ann Ophthalmol. 1978;10:1227-1230.
170 Bassen FA, Kornzweig AL. Malformation of the erythrocytes in a case of atypical retinitis pigmentosa. Blood. 1950;5:381-387.
171 Refsum S. Heredopathia atactica polyneuritiformis: a familial syndrome not hitherto described. A contribution to the clinical study of the hereditary disorders of the nervous system. Acta Psychiatr Scand. 1946;38:1-303.
172 Yokota T, Wada Y, Furukawa T, et al. Adult-onset spinocerebellar syndrome with idiopathic vitamin E deficiency. Ann Neurol. 1987;22:84-87.
173 Berger AS, Tychsen L, Rosenblum JL. Retinopathy in human vitamin E deficiency. Am J Ophthalmol. 1991;111:774-775.
174 Enevoldson TP, Sanders MD, Harding AE. Autosomal dominant cerebellar ataxia with pigmentary macular dystrophy: a clinical and genetic study of eight families. Brain. 1994;117(Pt 3):445-460.
175 Ahn JK, Seo JM, Chung H, et al. Anatomical and functional characteristics in atrophic maculopathy associated with spinocerebellar ataxia type 7. Am J Ophthalmol. 2005;139:923-925.
176 Fitzsimons RB, Gurwin EB, Bird AC. Retinal vascular abnormalities in facioscapulohumeral muscular dystrophy: a general association with genetic and therapeutic implications. Brain. 1987;110(Pt 3):631-648.
177 Kimizuka Y, Kiyosawa M, Tamai M, et al. Retinal changes in myotonic dystrophy: clinical and follow-up evaluation. Retina. 1993;13:129-135.
178 Backhouse O, Griffiths B, Henderson T, et al. Ophthalmic manifestations of dermatomyositis. Ann Rheum Dis. 1998;57:447-449.
179 La Piana FG. Sectoral retinal pigmentation in neurofibromatosis. Ann Ophthalmol. 1977;9:413-422.
180 Cotlier E. Café-au-lait spots of the fundus in neurofibromatosis. Arch Ophthalmol. 1977;95:1990-1992.
181 Destro M, D’Amico DJ, Gragoudas ES, et al. Retinal manifestations of neurofibromatosis: diagnosis and management. Arch Ophthalmol. 1991;109:662-666.
182 Font RL, Ferry AP. The phakomatoses. Int Ophthalmol Clin. 1972;12:1-50.
183 Moadel K, Yannuzzi LA, Ho AC, et al. Retinal vascular occlusive disease in a child with neurofibromatosis. Arch Ophthalmol. 1994;112:1021-1023.
184 Callender GR, Thigpen CA. Two neurofibromas in one eye. Am J Ophthalmol. 1930;13:121-124.
185 Freeman D. Neurofibroma of the choroid. Arch Ophthalmol. 1934;11:641-645.
186 Savino PJ, Glaser JS, Luxenberg MN. Pulsating enophthalmos and choroidal hamartomas: two rare stigmata of neurofibromatosis. Br J Ophthalmol. 1977;61:483-488.
187 Kobrin JL, Blodi FC, Weingeist TA. Ocular and orbital manifestations of neurofibromatosis. Surv Ophthalmol. 1979;24:45-51.
188 Lewis RA, Riccardi VM. Von Recklinghausen neurofibromatosis: incidence of iris hamartomata. Ophthalmology. 1981;88:348-354.
189 Huson S, Jones D, Beck L. Ophthalmic manifestations of neurofibromatosis. Br J Ophthalmol. 1987;71:235-238.
190 Green WR. The uveal tract. In: Spencer WH, editor. Ophthalmic Pathology: An Atlas and Textbook. Philadelphia: WB Saunders; 1996:1864-2099.
191 Landau K, Dossetor FM, Hoyt WF, et al. Retinal hamartoma in neurofibromatosis 2. Arch Ophthalmol. 1990;108:328-329.
192 Mautner VF, Lindenau M, Baser ME, et al. The neuroimaging and clinical spectrum of neurofibromatosis 2.Neurosurgery. 1996;38:880-885. discussion, Neurosurgery. 1996;38:885-886.
193 Rettele GA, Brodsky MC, Merin LM, et al. Blindness, deafness, quadriparesis, and a retinal malformation: the ravages of neurofibromatosis 2. Surv Ophthalmol. 1996;41:135-141.
194 Ragge NK, Baser ME, Riccardi VM, et al. The ocular presentation of neurofibromatosis 2. Eye. 1997;11(Pt 1):12-18.
195 Ragge NK, Baser ME, Klein J, et al. Ocular abnormalities in neurofibromatosis 2. Am J Ophthalmol. 1995;120:634-641.
196 Ragge NK. Clinical and genetic patterns of neurofibromatosis 1 and 2. Br J Ophthalmol. 1993;77:662-672.
197 Kaye LD, Rothner AD, Beauchamp GR, et al. Ocular findings associated with neurofibromatosis type II. Ophthalmology. 1992;99:1424-1429.
198 Meyers SM, Gutman FA, Kaye LD, et al. Retinal changes associated with neurofibromatosis 2.Trans Am Ophthalmol Soc. 1995;93:245-252. discussion Trans Am Ophthalmol Soc. 1995;93:252-257.
199 Baser ME, Kluwe L, Mautner VF. Germline NF2 mutations and disease severity in neurofibromatosis type 2 patients with retinal abnormalities. Am J Hum Genet. 1999;64:1230-1233.
200 Zimmer-Galler IE, Robertson DM. Long-term observation of retinal lesions in tuberous sclerosis. Am J Ophthalmol. 1995;119:318-324.
201 Gunduz K, Eagle RCJr, Shields CL, et al. Invasive giant cell astrocytoma of the retina in a patient with tuberous sclerosis. Ophthalmology. 1999;106:639-642.
202 Nyboer JH, Robertson DM, Gomez MR. Retinal lesions in tuberous sclerosis. Arch Ophthalmol. 1976;94:1277-1280.
203 Lucchese NJ, Goldberg MF. Iris and fundus pigmentary changes in tuberous sclerosis. J Pediatr Ophthalmol Strabismus. 1981;18:45-46.
204 Awan KJ. Leaf-shaped lesions of ocular fundus and white eyelashes in tuberous sclerosis. South Med J. 1982;75:227-228.
205 Green JS, Bowmer MI, Johnson GJ. Von Hippel-Lindau disease in a Newfoundland kindred. CMAJ. 1986;134:133-138. 146.
206 Hardwig P, Robertson DM. von Hippel-Lindau disease: a familial, often lethal, multisystem phakomatosis. Ophthalmology. 1984;91:263-270.
207 Lamiell JM, Salazar FG, Hsia YE. von Hippel-Lindau disease affecting 43 members of a single kindred. Medicine (Baltimore). 1989;68:1-29.
208 Maher ER, Yates JR, Harries R, et al. Clinical features and natural history of von Hippel-Lindau disease. Q J Med. 1990;77:1151-1163.
209 Moore AT, Maher ER, Rosen P, et al. Ophthalmological screening for von Hippel-Lindau disease. Eye. 1991;5(Pt 6):723-728.
210 de Jong PT, Verkaart RJ, van de Vooren MJ, et al. Twin vessels in von Hippel-Lindau disease. Am J Ophthalmol. 1988;105:165-169.
211 Chang JH, Spraul CW, Lynn ML, et al. The two-stage mutation model in retinal hemangioblastoma. Ophthalmic Genet. 1998;19:123-130.
212 Neumann HP. Basic criteria for clinical diagnosis and genetic counselling in von Hippel-Lindau syndrome. Vasa. 1987;16:220-226.
213 Susac JO, Smith JL, Scelfo RJ. The “tomato-catsup” fundus in Sturge-Weber syndrome. Arch Ophthalmol. 1974;92:69-70.
214 Bacin F, Bonnet M, Caujolle JP, et al. [Choroidal angioma and Sturge-Weber syndrome.]. J Fr Ophtalmol. 1997;20:405-407.
215 Shin GS, Demer JL. Retinal arteriovenous communications associated with features of the Sturge-Weber syndrome. Am J Ophthalmol. 1994;117:115-117.
216 Hopen G, Smith JL, Hoff JT, et al. The Wyburn-Mason syndrome: concomitant chiasmal and fundus vascular malformations. J Clin Neuroophthalmol. 1983;3:53-62.
217 Effron L, Zakov ZN, Tomsak RL. Neovascular glaucoma as a complication of the Wyburn-Mason syndrome. J Clin Neuroophthalmol. 1985;5:95-98.
218 Brod RD, Shields JA, Shields CL, et al. Unusual retinal and renal vascular lesions in the Klippel-Trénaunay-Weber syndrome. Retina. 1992;12:355-358.
219 Okada H, Horibe H, Yoshiyuki O, et al. A prospective study of cerebrovascular disease in Japanese rural communities, Akabane and Asahi. Part 1: evaluation of risk factors in the occurrence of cerebral hemorrhage and thrombosis. Stroke. 1976;7:599-607.
220 Svardsudd K, Wedel H, Aurell E, et al. Hypertensive eye ground changes: prevalence, relation to blood pressure and prognostic importance. The study of men born in 1913. Acta Med Scand. 1978;204:159-167.
221 Tanaka H, Hayashi M, Date C, et al. Epidemiologic studies of stroke in Shibata, a Japanese provincial city: preliminary report on risk factors for cerebral infarction. Stroke. 1985;16:773-780.
222 Nakayama T, Date C, Yokoyama T, et al. A 15.5-year follow-up study of stroke in a Japanese provincial city. The Shibata Study. Stroke. 1997;28:45-52.
223 Wong TY, Klein R, Couper DJ, et al. Retinal microvascular abnormalities and incident stroke: the Atherosclerosis Risk in Communities Study. Lancet. 2001;358:1134-1140.
224 Wong TY, Klein R, Sharrett AR, et al. Retinal microvascular abnormalities and cognitive impairment in middleaged persons: the Atherosclerosis Risk in Communities Study. Stroke. 2002;33:1487-1492.
225 Wong TY, Klein R, Sharrett AR, et al. Cerebral white matter lesions, retinopathy, and incident clinical stroke. JAMA. 2002;288:67-74.
226 Wong TY, Mosley THJr, Klein R, et al. Retinal microvascular changes and MRI signs of cerebral atrophy in healthy, middleaged people. Neurology. 2003;61:806-811.
227 Sharma S, Pater JL, Lam M, et al. Can different types of retinal emboli be reliably differentiated from one another? An interand intraobserver agreement study. Can J Ophthalmol. 1998;33:144-148.
228 Savino PJ, Glaser JS, Cassady J. Retinal stroke: is the patient at risk? Arch Ophthalmol. 1977;95:1185-1189.
229 Wilson LA, Warlow CP, Russell RW. Cardiovascular disease in patients with retinal arterial occlusion. Lancet. 1979;1:292-294.
230 Babikian V, Wijman CA, Koleini B, et al. Retinal ischemia and embolism: etiologies and outcomes based on a prospective study. Cerebrovasc Dis. 2001;12:108-113.
231 Smit RL, Baarsma GS, Koudstaal PJ. The source of embolism in amaurosis fugax and retinal artery occlusion. Int Ophthalmol. 1994;18:83-86.
232 McCullough HK, Reinert CG, Hynan LS, et al. Ocular findings as predictors of carotid artery occlusive disease: is carotid imaging justified? J Vasc Surg. 2004;40:279-286.
233 Sharma S, Brown GC, Pater JL, et al. Does a visible retinal embolus increase the likelihood of hemodynamically significant carotid artery stenosis in patients with acute retinal arterial occlusion? Arch Ophthalmol. 1998;116:1602-1606.
234 Bruno A, Jones WL, Austin JK, et al. Vascular outcome in men with asymptomatic retinal cholesterol emboli: a cohort study. Ann Intern Med. 1995;122:249-253.
235 Klein R, Klein BE, Moss SE, et al. Retinal emboli and cardiovascular disease: the Beaver Dam Eye Study. Arch Ophthalmol. 2003;121:1446-1451.
236 Wong TY, Larsen EK, Klein R, et al. Cardiovascular risk factors for retinal vein occlusion and arteriolar emboli: the Atherosclerosis Risk in Communities & Cardiovascular Health studies. Ophthalmology. 2005;112:540-547.
237 Hedges TRJr. Ophthalmoscopic findings in internal carotid artery occlusion. Bull Johns Hopkins Hosp. 1962;111:89-97.
238 Kearns TP, Hollenhorst RW. Venous-stasis retinopathy of occlusive disease of the carotid artery. Mayo Clin Proc. 1963;38:304-312.
239 Costa VP, Kuzniec S, Molnar LJ, et al. The effects of carotid endarterectomy on the retrobulbar circulation of patients with severe occlusive carotid artery disease: an investigation by color Doppler imaging. Ophthalmology. 1999;106:306-310.
240 Wilson WB, Leavengood JM, Ringel SP, et al. Transient ocular motor paresis associated with acute internal carotid artery occlusion. Ann Neurol. 1989;25:286-290.
241 McDonough RL, Forteza AM, Flynn HWJ. Internal carotid artery dissection causing a branch retinal artery in a young adult. Am J Ophthalmol. 1998;125:706-708.
242 Newman NJ, Kline LB, Leifer D, et al. Ocular stroke and carotid artery dissection. Neurology. 1989;39:1462-1464.
243 Godfrey DG, Biousse V, Newman NJ. Delayed branch retinal artery occlusion following presumed blunt common carotid dissection. Arch Ophthalmol. 1998;116:1120-1121.
244 Palmer HE, Stanford MR, Sanders MD, et al. Visual outcome of patients with idiopathic ischaemic and non-ischaemic retinal vasculitis. Eye. 1996;10(Pt 3):343-348.
245 Palmer HE, Zaman AG, Edelsten CE, et al. Systemic morbidity in patients with isolated idiopathic retinal vasculitis. Lancet. 1995;346:505-506.
246 Cobo-Soriano R, Sanchez-Ramon S, Aparicio MJ, et al. Antiphospholipid antibodies and retinal thrombosis in patients without risk factors: a prospective case-control study. Am J Ophthalmol. 1999;128:725-732.
247 Cahill MT, Stinnett SS, Fekrat S. Meta-analysis of plasma homocysteine, serum folate, serum vitamin B(12), and thermolabile MTHFR genotype as risk factors for retinal vascular occlusive disease. Am J Ophthalmol. 2003;136:1136-1150.
248 Susac JO, Hardman JM, Selhorst JB. Microangiopathy of the brain and retina. Neurology. 1979;29:313-316.
249 O’Halloran HS, Pearson PA, Lee WB, et al. Microangiopathy of the brain, retina, and cochlea (Susac’s syndrome): a report of five cases and a review of the literature. Ophthalmology. 1998;105:1038-1044.
250 Susac JO, Murtagh FR, Egan RA, et al. MRI findings in Susac’s syndrome. Neurology. 2003;61:1783-1787.
251 Haritoglou C, Rudolph G, Hoops JP, et al. Retinal vascular abnormalities in CADASIL. Neurology. 2004;62:1202-1205.
252 Cumurciuc R, Massin P, Paques M, et al. Retinal abnormalities in CADASIL: a retrospective study of 18 patients. J Neurol Neurosurg Psychiatry. 2004;75:1058-1060.
253 Rufa A, De Stefano N, Dotti MT, et al. Acute unilateral visual loss as the first symptom of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Arch Neurol. 2004;61:577-580.
254 Parisi V, Pierelli F, Fattapposta F, et al. Early visual function impairment in CADASIL. Neurology. 2003;60:2008-2010.
255 Haritoglou C, Hoops JP, Stefani FH, et al. Histopathological abnormalities in ocular blood vessels of CADASIL patients. Am J Ophthalmol. 2004;138:302-305.
256 Storimans CW, Van Schooneveld MJ, Oosterhuis JA, et al. A new autosomal dominant vascular retinopathy syndrome. Eur J Ophthalmol. 1991;1:73-78.
257 Terwindt GM, Haan J, Ophoff RA, et al. Clinical and genetic analysis of a large Dutch family with autosomal dominant vascular retinopathy, migraine and Raynaud’s phenomenon. Brain. 1998;121(Pt 2):303-316.
258 Ophoff RA, DeYoung J, Service SK, et al. Hereditary vascular retinopathy, cerebroretinal vasculopathy, and hereditary endotheliopathy with retinopathy, nephropathy, and stroke map to a single locus on chromosome 3p21.1-p21.3. Am J Hum Genet. 2001;69:447-453.
259 Vahedi K, Massin P, Guichard JP, et al. Hereditary infantile hemiparesis, retinal arteriolar tortuosity, and leukoencephalopathy. Neurology. 2003;60:57-63.
260 Sher NA, Letson RD, Desnick RJ. The ocular manifestations in Fabry’s disease. Arch Ophthalmol. 1979;97:671-676.
261 Dichgans M. A new cause of hereditary small vessel disease: angiopathy of retina and brain. Neurology. 2003;60:8-9.
262 Terson A. De l’hémorrhagie dans le corps vitre au cours de l’hémorrhagie cerebrale. Clin Ophthalmol. 1900;6:309-312.
263 Fahmy JA. Fundal haemorrhages in ruptured intracranial aneurysms. I. Material, frequency and morphology. Acta Ophthalmol (Copenh). 1973;51:289-298.
264 Manschot WA. Subarachnoid hemorrhage: intraocular symptoms and their pathogenesis. Am J Ophthalmol. 1954;38:501-505.
265 Shaw HEJr, Landers MB, Sydnor CF. The significance of intraocular hemorrhages due to subarachnoid hemorrhage. Ann Ophthalmol. 1977;9:1403-1405.
266 McCarron MO, Alberts MJ, McCarron P. A systematic review of Terson’s syndrome: frequency and prognosis after subarachnoid haemorrhage. J Neurol Neurosurg Psychiatry. 2004;75:491-493.
267 Fahmy JA. Fundal haemorrhages in ruptured intracranial aneurysms. II. Correlation with the clinical course. Acta Ophthalmol (Copenh). 1973;51:299-304.
268 Cogan DG. Ocular correlates of inborn metabolic defects. Can Med Assoc J. 1966;95:1055-1065.
269 Small KW, Letson R, Scheinman J. Ocular findings in primary hyperoxaluria. Arch Ophthalmol. 1990;108:89-93.