Chapter 18 Retinal and Choroidal Vasculature
Retinal Oxygenation
Introduction
Oxygen is necessary for the existence of mammals because it is required to generate adenosine triphosphate (ATP) oxidatively. Although the partial pressure of oxygen (po2) is 149 mmHg (21% of atmospheric oxygen) at sea level, arterial oxygen content is as low as 75–100 mmHg (10–14%) and tissue po2 is much lower. The oxygen level in inner segments of photoreceptors (mitochondria-rich) after dark adaptation is between 0 and 5 mmHg (0.7%) but up to 20 mmHg in the light. Inner retinal oxygen is normally 20 mmHg, so normoxia depends on the area of retina and dark/light state.1,2 Hypoxia is an oxygen level below normoxia while hyperoxia is achieved by inhaling high levels of oxygen as in the isolette of the neonatal intensive care unit.
The retina is one of the most metabolically active tissues in the body. It has two unique zones of oxygenation.1 The inner retina is supplied with oxygen by the retinal vasculature. The retinal vasculature is autoregulated because it is responsive to changes in systemic oxygen levels, keeping the inner retina at a relatively constant level. If the retinal vasculature is compromised, as in ischemic retinopathies, the retina becomes hypoxic in that area. The outer retina is supplied solely by the choroidal vasculature. Unlike retina, choroidal vessels are not autoregulated, so systemic levels of oxygen control the level of oxygen in choroid. Supply of oxygen to choroid is diminished by stenosis of the ophthalmic artery, which is the branch off the internal carotid that is most likely to be stenosed because it is a right-angle branch.
Comparison of retinal and choroidal vasculatures
Although less than 300 µm apart in distance, the retinal and choroidal vasculatures are vastly different in many attributes in addition to autoregulation. The initial retinal vasculature in human starts forming around 14 weeks’ gestation (WG) by vasculogenesis, development by differentiation and assembly of vascular precursors, angioblasts.3–5 The deep capillary network forms after 20 WG by angiogenesis, development by migration, and proliferation of endothelial cells from existing blood vessels. The driving force for vascular development is physiological hypoxia; metabolic requirements of developing neurons are only met by stimulating the development of a retinal vasculature.6,7 It is mostly a bilayered system, a superficial network, and a deep capillary plexus; however, there are multiple layers of capillaries in the peripapillary region, the radial peripapillary capillaries. There is only one layer of blood vessels in the periphery at the ora serrata where the retina thins to 100 µm. The retinal vasculature is supplied with blood directly by the central retinal artery in humans. The retinal vasculature has a traditional end-arterial hierarchy: arteries branch to arterioles, which supply a capillary network that is drained by venules, and then veins remove the blood from the retina (Fig. 18.1). The retinal vasculature forms the inner blood–retinal barrier (BRB), restricting passage of molecules that do not have receptors or transporters on the luminal surface of the endothelial cells. Capillaries have a lumen diameter of 3.5–6 µm, permitting passage of red blood cells only after deformation of their disc shape. The retinal capillaries and venules have perivascular pericytes and the retina has the highest endothelial cell-to-pericyte ratio in the body, 1 : 1.8
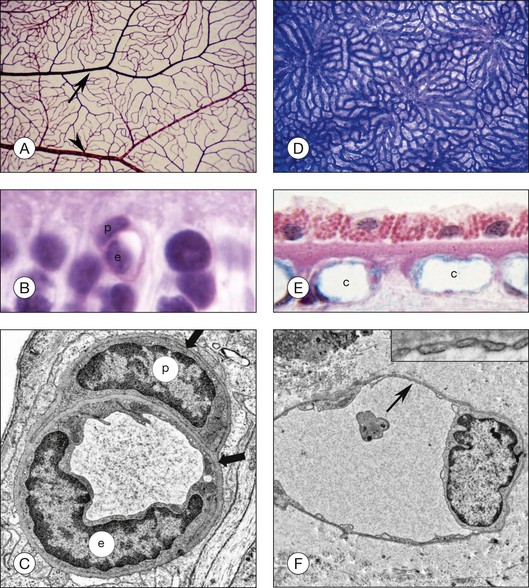
The choroidal vasculature forms well before the retinal vessels (6–9 WG), although its maturation is only completed after 20 WG. It develops by hemovasculogenesis, formation of blood cells and blood vessel cells from a common progenitor, the hemangioblast.9 The choroidal vasculature provides oxygen and nutrients to the photoreceptors. The capillary system, the choriocapillaris, lies directly under the Bruch’s membrane, while intermediate and large blood vessels of the system lie posterior to the capillaries. The short and long ciliary arteries supply blood to the choroidal vasculature while 4–6 vortex veins remove blood from this vast system. Unlike the retina, the hierarchy in choroid is lobular, similar to kidney glomeruli (Fig. 18.1). The lobules change in shape, vascular density, and size depending upon area and the location of feeding arterioles and draining venules also varies by geographic location of the lobule.10 The capillaries are broad and flat, having luminal diameters ranging from 10 to 38 µm in diameter. Another major difference from retina is that the capillaries are fenestrated, allowing the passage of small molecules and solutes through these 60–70-nm pores. The choriocapillaris is sided in that the majority of the fenestrations are on the retinal side as well as all three types of vascular endothelial growth factor (VEGF) receptors.11 Pericytes, however, are mostly on the scleral side of these capillaries. Control of vascular tone in choroid may be accomplished by mast cells, which lie abluminal to arteries and arterioles, or choroidal ganglion cells.12
History of retinal ischemia
Ischemia is the restriction in oxygen supply without considering actual levels of oxygen. Michaelson13,14 and Wise15 hypothesized that areas of vascular loss in retina must be hypoxic because the high metabolic rate requires a continuous supply of oxygen. They observed that neovascularization always formed adjacent to these nonperfused areas and, therefore, an angiogenic factor must be produced by the hypoxic retina. They hypothesized that this factor X must be hypoxia-inducible and diffusible. Subsequently, oxygen was measured directly in retinas of several species and it demonstrated that nonperfused areas were indeed hypoxic.1,2 It was not until 1989 that factor X was discovered, purified, and characterized as vascular endothelial growth factor (VEGF).16 This factor was first shown to be responsible for the increased vascular permeability seen in some retinopathies.17
Normoxia
The studies of Wangsa-Wirawan and Linsenmeier1 and Yu and Cringle2 using oxygen electrodes directly assessed oxygen levels from choroid to vitreous in various species. The oxygen tension is around 70 mmHg in choroid and plummets to zero at the inner segments in the dark (Fig. 18.2). Inner retina is around 10–20 mmHg. There are regional variations in oxygen concentration within the retina. Yu et al.18 showed that oxygen consumption in outer retina is highest in the parafoveal region while inner retinal oxygen in the fovea (approximately 5 mmHg) reflected the lack of a retinal vasculature and the predominantly choroidal source of oxygen.
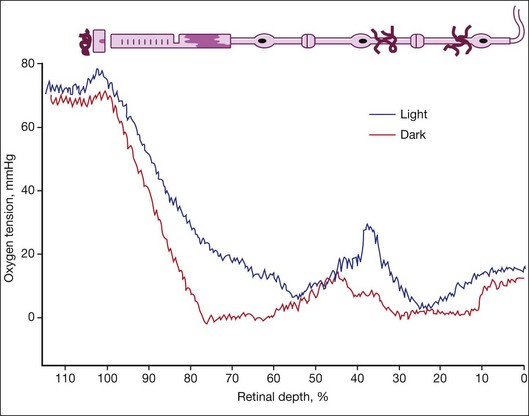
Hyperoxia
Life in utero is hypoxic, so when a child is born prematurely, the normoxic environment is actually hyperoxic. Prematurely born children are placed in 40% oxygen, making their tissue further hyperoxic, which yields vaso-obliteration (endothelial cells die and pericytes and progenitors survive).19 The only direct measurements of oxygen in a model of retinopathy of prematurity (ROP) were performed by Ernest and Goldstick.20 They found in kitten after 80–90% O2 that preretinal po2 over avascular retina was close to zero but was normal over vascularized retina. Vaso-obliteration from hyperoxia does not occur in the choroid of humans and dogs19 but does occur when rats are exposed to hyperoxia.21 Loss of vasculature in vaso-obliteration makes the retina hypoxic when the child is returned to room air. Exposure of the adult vasculature to hyperoxia causes constriction but not vaso-obliteration. During hyperoxia breathing (100% oxygen), the inner retinal po2 remains unchanged due to autoregulation while the choroidal po2 rises to 250 mmHg in cat22 and 220 mmHg in the minipig, due to a lack of metabolic control of the choroidal vasculature.23
Hypoxia
Complex homeostatic mechanisms are designed to maintain O2 concentration in each cell within a narrow range. While O2 consumption increases with the metabolic activity of the organism, exposure to O2 must be limited due to the potentially damaging effects of reactive oxygen species (ROS). Hypoxia, the state of low oxygen concentration, promotes the formation of blood vessels and is important for the formation of a vascular system in embryos.24 Disease occurs when the retina and choroid are deprived of adequate oxygen supply; this can also be described as a mismatch of oxygen supply versus demand at the cellular level within ocular tissues.
The blood O2-carrying capacity is maintained by the O2-regulated production of erythropoietin (EPO), which stimulates the proliferation and survival of red blood cell progenitors. Semenza and coworkers25,26 performed seminal studies to identify hypoxia-inducible factor-1 (HIF-1). HIF-1 orchestrates a pleiotropic adaptive response to hypoxia by inducing the expression of more than 100 genes encoding glycolytic enzymes and glucose transporters (thereby facilitating the glycolytic switch in energy metabolism typically observed under hypoxic conditions), matrix metalloproteinases, and angiogenic, mitogenic, and survival factors, including EPO.27,28 Other molecules upregulated by HIF-1 that have profound effects on vasculature include 5’ nucleotidase, an enzyme that is the major source of the potent vasodilator adenosine in the body, and VEGF. HIFs are vital to development and, in mammals, deletion of the HIF-1 genes results in perinatal death. HIF-1 is expressed in all cell types and functions as a master regulator of oxygen homeostasis by playing critical roles in embryonic development and postnatal physiology.
Hypoxia-inducible factor
The activity of HIF depends on the intracellular levels of its inducible alpha subunit. In the presence of oxygen, HIF-1α is hydroxylated on two critical proline residues (Pro402 and Pro564) in the so-called oxygen-dependent degradation domain. Three prolyl hydroxylases have been identified in mammalian cells and use O2 as a substrate to generate 4-hydroxyproline at residues 402 and/or 564 of HIF-1α. The hydroxylation reaction also requires 2-oxoglutarate (α-ketoglutarate) as a substrate and generates succinate as a side product. These prolyl hydroxylases have a high Km for O2 that is slightly above atmospheric concentration; thus O2 is rate-limiting for enzymatic activity under physiological conditions and any change in cellular O2 concentration is directly transduced into changes in the rate of HIF-1α hydroxylation.29
Factor-inhibiting HIF-1 (FIH-1), which was identified in a yeast two-hybrid screen as a protein that interacts with, and inhibits the activity of the HIF-1α transactivation domain,30 functions as asparaginyl hydroxylase.31 As in the case of the prolyl hydroxylases, FIH-1 appears to use O2 and 2-oxoglutarate,32 although it has a Km for O2 that is three times lower than the prolyl hydroxylases.33 Hydroxylation provides a mechanism for regulating protein–protein interactions, similar to the effect of phosphorylation and other posttranslational modifications. However, this hydroxylation occurs in an O2-dependent manner, thus establishing a direct link between cellular oxygenation and HIF-1 activity. Following HIF-1α hydroxylation, the protein becomes targeted for ubiquitination by an E3 ligase complex (including the von Hippel–Lindau (VHL) tumor suppressor protein) and subsequent proteasomal degradation.
Under hypoxic conditions, the HIF prolyl-hydroxylases are inhibited, because these HIF prolyl-hydroxylases utilize oxygen as a cosubstrate. Hypoxia results in an increase in succinate, due to inhibition of the electron transport chain in the mitochondria, which serves to inhibit further HIF prolyl-hydroxylase activity. When stabilized by hypoxic conditions, HIF increases the expression of critical genes that promote survival in low-oxygen conditions, including glycolytic enzymes, which allow ATP synthesis in an oxygen-independent manner. HIF activates the transcription of genes encoding secreted signaling molecules, including angiogenic growth factors and survival factors, cell surface receptors, extracellular matrix proteins and modifying enzymes, transcription factors, cytoskeletal proteins, proapoptotic proteins, and glucose transporters and glycolytic enzymes (Fig. 18.3).29
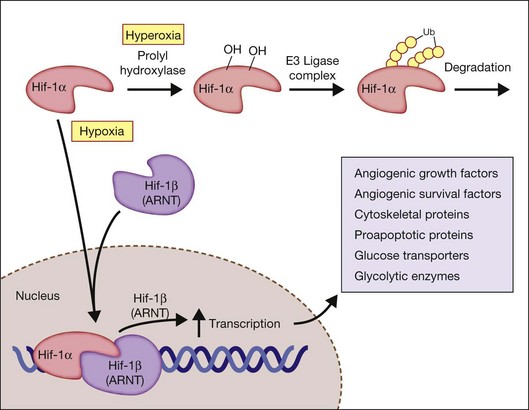
HIF-induced VEGF, stromal-derived factor-1 (SDF-1), and EPO promote neovascularization. HIF-1 acts by binding to HIF-responsive elements in promoters that contain the sequence NCGTG, which is present in the promoters for VEGF, SDF-1, EPO, and many other genes. In addition to hypoxia, other factors such as nuclear factor κB (NF-κB) modulate HIF-1α expression in the presence of normal oxygen pressure. Thus, conditions such as tissue inflammation can lead to local HIF-1α expression.34 HIF-1 DNA-binding activity and target gene expression are induced in cells exposed not only to hypoxia but also to the iron chelator desferrioxamine or to cobalt chloride.35
A structurally and functionally related protein to HIF-1α, designated HIF-2α, is the product of the EPAS1 gene. HIF-2α can also heterodimerize with HIF-1ß.36 HIF-1α:HIF-1ß and HIF-2α:HIF-1ß heterodimers have overlapping yet distinct target gene specificities.37 HIF-2α, unlike HIF-1α, is not expressed in all cell types and HIF-2α can be inactivated by cytoplasmic sequestration. This “compartmentalization” of oxygen-sensitive signaling components also influences the hypoxic response.38,39
HIF deficiency and its resultant pathology
O2 delivery to cells of the developing embryo becomes limited by diffusion such that establishment of a functioning circulatory system is required for embryonic survival by embryonic day 9 (E9) in the mouse. In wild-type mouse embryos, HIF-1α expression increases dramatically between E8.5 and E9.5, whereas embryos that lack HIF-1α expression die between E9.5 and E10.5 and show cardiac malformations, vascular regression, and massive cell death.40 Complete HIF-2α deficiency is also associated with embryonic lethality41 and because the embryos survive longer than HIF-1a–/– mice, effects on multiple organ systems can be demonstrated.42
Complete HIF-1α deficiency results in developmental defects; however, partial HIF-1α deficiency is sufficient to result in impaired responses to physiological stimuli. A particularly dramatic example is the loss of O2 sensing in the carotid body of HIF-1a+/– mice.43 Although the carotid bodies are anatomically and histologically normal and depolarize normally in response to cyanide application, they show essentially no response to hypoxia. Thus partial HIF-1α deficiency in the carotid body results in a complete loss of the ability to sense and/or respond to changes in the arterial Po2 by stimulation of the central nervous system cardiorespiratory centers. The HIF-1 target genes that are critical for O2 sensing and/or efferent responses by the carotid body have not been identified.
Mice with HIF-1α conditionally knocked out using PAX6-Cre have delayed development of the outer retinal plexus but not the superficial or deep plexus.44 However, when HIF-1α was knocked down only in Müller cells using a Cre-LOX system, and the animals were made diabetic with streptozotocin, vascular permeability in retina was reduced and leukostasis and overproduction of VEGF and intercellular adhesion molecule (ICAM)-1 were attenuated in adult mice.45
Another dramatic phenotype is the complete inability of HIF-1α–/– myeloid cells (granulocytes and macrophages) to respond to inflammatory stimuli.46 Myeloid cells are dependent on glycolysis for ATP generation, perhaps reflecting the hypoxic microenvironment that is often associated with inflammation and infection. HIF-1α deficiency results in ATP deficiency, which impairs critical myeloid cell functions such as aggregation, motility, invasion, and bacterial killing. HIF-1 also plays critical roles in B-lymphocyte development47 and T-lymphocyte activation.48
HIF-activated genes relevant to physiological and pathological ocular angiogenesis
The paragraphs above provide a brief summary of the critical role of HIF-1α in oxygen sensing, development, and physiology. HIF-1α plays an equally important role in disease pathophysiology, including retinal diseases. As a result, there is considerable interest in HIF-1 α as a therapeutic target.49 In cardiovascular diseases, increased HIF-1α activity induced as a result of HIF-1α gene therapy,50 small-molecule inhibitors of prolyl hydroxylase activity,51 or inhibitors of HIF-1α–VHL interaction52 may provide a means to stimulate neovascularization in ischemic tissue. In contrast, small-molecule inhibitors of HIF-1 activity may be useful as antiangiogenic agents. However, because HIF-1α functions as a global regulator of oxygen homeostasis, it may not be a useful therapeutic target if the treatment results in unintended and undesirable side-effects.
An alternative therapeutic approach that may be particularly relevant to the treatment of ocular pathology is to focus on modulation of HIF-1α target genes. However, the protein products of these target genes must also be delivered in a precise and perfectly timed manner. EPO is an oxygen-regulated hormone stimulating erythrocyte production and is critical for retinal angiogenesis. Increasing EPO expression in phase 1 of the murine ROP model (postnatal days 7–12) is protective and results in less neovascularization during phase 2 (postnatal days 12–17).53 In contrast, EPO mRNA expression levels in retina are highly elevated during the hypoxia-induced proliferation phase of retinopathy (phase 2) and inhibition of retinal EPO mRNA expression with RNA interference results in suppressed retinal neovascularization.53
The best-known gene activated by HIF-1α is VEGF, first identified as a potent promoter of vascular permeability17 and endothelial cell proliferation.15 VEGF has become known as a master regulator of angiogenesis.54 Tight control of physiologic VEGF levels is required for proper embryological development.55 Although it was initially thought that the postembryonic role of VEGF was restricted to a few processes, it is now quite clear that VEGF acts as a pluripotent growth factor essential for a wide variety of physiological processes,56 including maintenance of the adult microvasculature,57 neuronal survival,58 and trophic maintenance of ocular tissues.
VEGF in health and in ocular disease
VEGF is produced by many cell types in the retina, including retinal pigment epithelium (RPE),59 vascular endothelial cells,60 pericytes,60 retinal neurons,61 Müller cells,61 and astrocytes,62 suggesting that VEGF has important functions in ocular homeostasis. RPE-secreted VEGF plays an important role in maintaining the choriocapillaris.11,63,64 VEGF secretion by retinal cells and the RPE is stimulated in response to hypoxia.60 VEGF administration protects retinal neurons from apoptosis.65 Moreover, chronic VEGF inhibition can lead to a significant loss of retinal ganglion cells in normal adult animals.65
While VEGF is critical to maintaining normal ocular function, overproduction of VEGF is deleterious. Elevated levels of VEGF have been strongly implicated in the pathogenesis of ocular neovascular diseases such as neovascular age-related macular degeneration (NV in AMD)66 and proliferative diabetic retinopathy67 as well as diabetic macular edema.68 Elevated VEGF levels are observed in central and branch retinal vein occlusion (CRVO and BRVO),69 neovascular glaucoma,70 and ROP.71 Blocking VEGF action is now an established strategy for the treatment of NV in AMD, with two agents (the RNA aptamer pegaptanib sodium72 and the humanized murine monoclonal antibody antigen-binding fragment ranibizumab73) having received regulatory approval for the intravitreal treatment of NV in AMD.
While current standard of care for NV in AMD uses intravitreal delivery of an anti-VEGF agent on a repeated basis, this routine clinical practice does not address all the nuances of VEGF biology. The biology of the VEGF proteins is extremely complex. The VEGF family members are part of a superfamily of cysteine knot proteins and include VEGF-B, -C, -D, and placental growth factor (PlGF). Alternative splicing events for VEGFA give rise to at least 14 subtypes of VEGF, namely, VEGF111, VEGF121, VEGF121b, VEGF145, VEGF145b, VEGF148, VEGF162, VEGF165, VEGF165b, VEGF183, VEGF183b, VEGF189, VEGF189b,74 and VEGF206.75,76 Following the discovery of the antiangiogenic isoform of VEGF, VEGF165b, and its associated family of isoforms, a further layer of complexity was added to understanding the regulation of VEGF.
All VEGF isoforms are essential regulators of angiogenesis and vascular permeability. VEGFs elicit their intracellular activities via the activation of two receptor tyrosine kinases (RTKs): VEGFR-1 and -2. VEGFR-1, a high-affinity fms-like tyrosine kinase-1,77 and VEGFR-2, a kinase insert domain-containing receptor,78 are transmembrane glycoproteins consisting of a seven-tandem immunoglobulin-like domains, which serves as the extracellular ligand-binding region, a single-transmembrane domain, and a cytoplasmic domain consisting of two tyrosine kinase catalytic domains. Moreover, it has also been reported that a family of cell surface glycoproteins, particularly neuropilin-1, act as isoform-specific coreceptors for VEGF-A.79 The VEGF family and their respective receptors are outlined in Fig. 18.4.
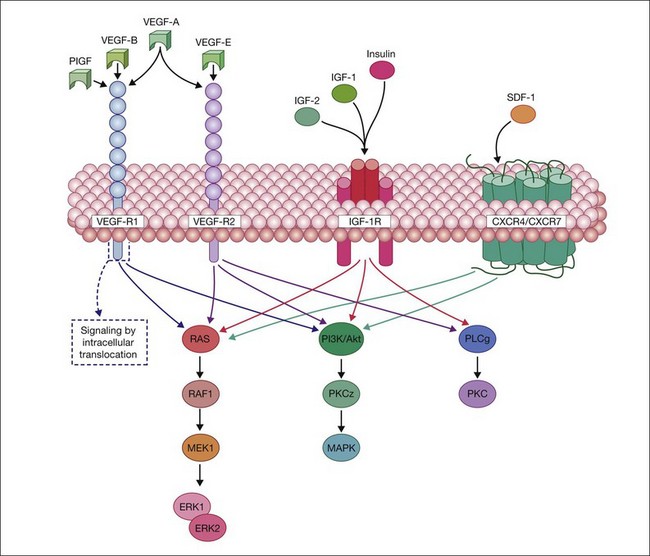
Ligand binding to the extracellular domain of VEGFR-2 results in a maximal increase of kinase activity following the induction of receptor dimerization and subsequent phosphorylation of tyrosine residues on the intracellular domain of the receptor.80 This event is crucial for the recruitment of additional signaling molecules that contain Src homology 2 or phosphotyrosine binding domains, which mediate further downstream signaling cascades.81 The association of RTKs with coreceptors, such as NP-1, in the case of VEGFR-2:VEGF165 signaling/interaction, can enhance the functional signal transduction and facilitate diverse cellular responses.80 VEGFR1/R2 signaling activates RAS, raf1, MEK1, and ERK1/ERK2 and stimulates PI3K/AKT/PKCz/MAPK pathways to mediate proliferation, migration, and cell survival (Fig. 18.4).
VEGFR-2 is the major mediator of angiogenic signaling in endothelial cells and is required for de novo vessel formation, vasculogenesis, and for angiogenesis, the formation of vessels from pre-existing vasculature.82 The pathways leading to VEGFR internalization and the role of receptor degradation in VEGF signaling remain controversial and differ for VEGFR-1 and -2. VEGFRs generate signal output at the plasma membrane and on their way to degradation through endocytic vesicles,83 whereas, in unstimulated cells, VEGFR-2 is predominantly located in recycling endosomes identified by Rab4 and/or Rab5.84 VEGFR internalization is clathrin-mediated and transport is further directed by the endosomal sorting complex required for transport proteins.85 VEGFR signaling is also regulated by ubiquitination, not only of the receptor itself, but also of receptor-associated signaling molecules.86 Specific VEGFR trafficking regulates biological output, as shown for arterial morphogenesis, for example.87
The molecular basis for ligand specificity of VEGFR signaling is poorly understood. It is well accepted however that VEGF receptors can associate with distinct coreceptors such as neuropilins, integrins, semaphorins, or heparan sulfate glycosaminoglycans, and engage distinct signaling molecules giving rise to specific signal output. Ligand-specific signaling may also result from receptor trafficking to specific cellular compartments, including the nucleus,88 where receptors encounter distinct signaling molecules.89
In addition to EPO and VEGF, SDF-1 is hypoxia-regulated and ischemic tissues express high levels of SDF-1 to recruit reparative cells to the injured region. SDF-1 activation of either CXCR-4 or CXCR-7 results in stimulation of the Ras/Raf/Mek/ERK pathway and the PI3K/Akt pathway to promote endothelial cell proliferation and neovascularization. Thus ligand–receptor interaction of VEGF to VEGF-R1 and VEGF-R2 and SDF-1 to CXCR4/CXCR-7 and their subsequent internalization sets in motion the cascade of cellular effects of VEGF and SDF-1 (Fig. 18.4). The VEGF and SDF-1 signaling pathways appear to be intimately connected with HIF-1 activation (Fig. 18.3), as the promoters of each of these factors contain a HIF response element. Because hypoxic tissue releases SDF-1 and VEGF, varying O2 concentrations would have an effect on expression of the receptors for these factors.
Bone marrow-derived progenitor cells (BMPC) and vascular repair
In conditions like diabetic retinopathy and ROP, areas of retinal vasodegeneration occur and lead to retinal ischemia which in turn induces the expression of hypoxia-regulated angiogenic factors. Typically, BMPCs robustly respond to these factors, including VEGF and SDF-1.90
Importantly, all hypoxia-regulated angiogenic factors are modulators of bone marrow-derived stem cells. Specifically, hematopoietic stem cells and other BMPCs reside in the bone marrow and are mobilized into the peripheral circulation by increased levels of EPO, SDF-1, and VEGF released by the ischemic tissue. Hematopoietic stem cell/BMPC mobilization occurs in response to vascular injury throughout the body and, when functioning properly, leads to revascularization of injured areas.91 In healthy individuals, bone marrow-derived CD34+ endothelial progenitor cells as well as other BMPC successfully orchestrate the reparative process. These BMPC demonstrate a limited ability to differentiate directly into endothelial cells and form components of new blood vessels by vasculogenesis, but show marked ability to provide paracrine support for the resident vasculature. This paracrine support facilitates resident endothelial cell recovery.
Disease-associated BMPC dysfunction
In diabetes, for example, BMPC are dramatically altered and cannot facilitate the repair process. Diabetic individuals have fewer circulating CD34+ cells and an increased number of inflammatory BMPC such as CD14+ cells than nondiabetics.92–94 This diabetes-related bone marrow dysfunction is closely linked to the impaired healing response experienced by many diabetic patients and to the vasodegenerative aspect of diabetic macro- and microvascular complications.94–96 Diabetes-induced BMPC defects occur in part due to uncoupling of nitric oxide synthases, enhanced NADPH oxidase activity, and increased generation of ROS such as superoxide and peroxynitrite (ONOO-)97 within BMPC. While stem and progenitor cells are deemed more resistant to oxidative stress,98 the highly oxidative diabetic milieu has a clearly detrimental effect on the function of these cells.99 Prolonged oxidant exposure reduces reparative function100 by impairing antioxidant defense enzymes. Previously, we and others have shown that diabetic CD34+ cells exhibit decreased migration and adhesion activities in vitro, and consequently reduce recruitment to areas of injury.96 In addition to oxidative stress, other key mechanisms implicated in diabetes-induced BMPC dysfunction include a reduction of cathepsin L activity101 and an upregulation of thrombospondin-1.100,102 While these functional defects are profound, strategies that successfully reverse BMPC defects in diabetics have included: (1) enhancement of angiogenic stimulus by increasing BMPC mobilization using granulocyte colony-stimulating factor and targeting SDF-1103; (2) use of nitric oxide donors to correct migration and promote cell deformability104; (3) enhancing cell interactions with substrate proteins to increase attachment to basement membranes105; (4) reducing high levels of endogenous transforming growth factor-β to normal levels106; and (5) treatment with rosiglitazone107 or atorvastatin.108
In addition to CD34+ cells, other populations of bone marrow cells may attempt vascular repair, such as CD14+ cells, discussed above, which can, under select circumstances, form endothelial-like cells109; however, the blood vessels formed by the endothelial cells of CD14+ origin eventually generate pathological blood vessels with increased permeability and contribute to the pathology of diabetic retinopathy.
We and others have been particularly interested in a novel factor expressed in increased concentrations in hypoxic tissue, insulin-like growth factor-binding protein 3 (IGFBP-3). While the standard IGF-dependent actions of the family of IGF-binding proteins have been well described, recently several IGF-independent actions have been discovered for IGFBPs, including IGFBP-3. These IGF-1 independent actions have been characterized as regulating cell fate and apoptosis.110–113 The role of IGFBP-3 in the control of cell growth remains an area under intense study, since IGFBP-3 may enhance or suppress cell growth, depending on specific conditions.112,114 In the retina, multiple forms of IGFBP (2–5) are secreted by retinal endothelial cells.113 IGFBP-3 has been shown to enhance cell proliferation in retinal endothelial cells and to decrease the formation of neovascular tufts in a murine model of oxygen-induced retinopathy.115,116 These studies suggest that IGFBP-3 may be vascular-protective in the retina. Recently, we also have demonstrated that IGFBP-3 is neuroprotective in the retina and reduces injury-induced retinal inflammation.117
Key factors that modulate VEGF function in the retina
The retina is one of the last organs to become vascularized in the fetus. The vasculogenic part of the process of human retinal vascularization begins in about gestational week 14 and appears to depend on a physiological hypoxia7,118 brought about by an increase in metabolic demand within the developing retina.3–5 This physiological hypoxia induces the local release of VEGF which, together with IGF-1, regulates angiogenesis and therefore normal vascularization of the retina.118,119 Retinal vascularization is complete by 36–40 WG.
ROP is a two-phase disease in which the phases are mirror images; the controlling growth factors are deficient in phase 1 and in excess in phase 2. Phase 2 involves uncontrolled proliferative growth of retinal blood vessels in response to hypoxia. The therapeutic intention would be to prevent the first phase of cessation of vessel and neural retinal development and then the second destructive phase would be prevented. This has been successfully performed using exogenous EPO, VEGF, and IGFBP-3 during phase 1 to prevent phase 2. These two phases, seen in premature infants, can be duplicated in animal models of ROP. Hyperoxia causes vaso-obliteration and cessation of normal retinal blood vessel development, which mimics phase 1 of ROP. IGF-1 levels rise in the third trimester of pregnancy but not in the preterm infant. The low IGF-1 in the preterm infant is due to the infant no longer being exposed to the support of the maternal environment, including maternal sources of IGF-1. IGF-1 levels rise slowly after preterm birth, as these babies are unable to produce adequate IGF-1 compared to term infants.120 In these premature infants, IGF-1 is further reduced by poor nutrition,121 acidosis, hypothyroxinemia, and sepsis.122 IGF-1 appears important for retinal and brain growth.119 Low levels of IGF-1 appear to play an important role in the early cessation of retinal growth that precipitates ROP.
IGF-1 mediates its effects through activation of the IGF-1 receptor (IGF-1R) (Fig. 18.3). IGF-1R is a receptor tyrosine kinase and is well established as a key regulator of cell growth and survival with activation of Ras-ERK pathway, the PI3K/Akt pathway and PKC (Fig. 18.4). Insulin and IGF-2 can also signal using the IGF-R. There is also a growing body of data to support a role for the structurally and functionally related insulin receptor (IR) in cell survival, even though its major function has been to modulate metabolism. Bidirectional cross-talk between IGF-1R and IR is observed, where specific inhibition of either receptor confers a compensatory increase in activity for the reciprocal receptor.
Although fluctuating oxygen has long been associated with the development of ROP, oxygen-regulated factors like VEGF appear to be directly modulated by IGF-1. IGF-1 is a key growth factor in early retinal development. IGF-1 controls maximum VEGF activation of the Akt endothelial cell survival pathway (Fig. 18.3). Thus loss of IGF-1 leads to loss of VEGF signaling and retinal vaso-obliteration. This vaso-obliteration leads to phase 2 of ROP which is the proliferative phase. At this point, suppression of IGF-1 and VEGF can reduce neovascularization. Thus IGF-1 is critical to normal retinal vascular development and a lack of IGF-1 in the early neonatal period is associated with lack of vascular growth and with subsequent proliferative ROP. In IGF-1-null mice, the retinal blood vessels grow more slowly than in those of normal mice, a pattern very similar to that seen in premature babies with ROP.
Adult retinal hypoxia and etiology
Diabetic retinopathy
Much like ROP, diabetic retinopathy has a vaso-obliteration phase that leads to a proliferative phase. The vaso-obliteration is not due to hyperoxia but rather to vaso-occlusion and vasodegeneration of the microvasculature, setting up the ischemic environment that leads to the vasoproliferative end-stage condition. Clinically the early pathology has been classified as nonproliferative (microaneurysms, exudates, leakage, capillary nonperfusion) resulting in hypoxia and the end-stage pathology as proliferative (preretinal neovascularization). The purely vasodegenerative, nonproliferative form of the disease is by far the most common and represents a disease of the neurovascular unit, resulting in dysfunction and eventual death of several of the key cells that maintain the BRB: pericytes, vascular endothelial cells, Müller glia and neurons. Kohner and Henkind elegantly demonstrated that, in diabetic individuals, areas of nonperfusion on fluorescein angiography are associated with acellular capillaries in trypsin digests.123 Direct measurement of oxygen in diabetic cat retinas demonstrated that even small aneurysms can result in a decrease in retinal interstitial oxygen.124
There are many mechanisms implicated in the pathogenesis of diabetic retinopathy but one that has gained considerable attention in the last decade is inflammation. The environment of hyperglycemia, abnormal lipids, increased oxidative stress, elevated serum and tissue advanced glycation endproducts (AGE)/receptor for AGE, increased serum/tissue cytokines, elevated blood pressure, and endoplasmic reticulum stress are the likely initiators of inflammation.125 Pathways of inflammation converge with pathways of endothelial dysfunction and coagulation to accelerate the pathogenesis of this disease.126 Initially nonspecific indicators of inflammation such as white-cell count and fibrinogen were found to be predictive of incident diabetes.127 Subsequently plasminogen activator inhibitor-1 (PAI-1), C-reactive protein, and fibrinogen were shown to be independent predictors.128 These observations are supported by several other prospective studies, in which tissue plasminogen activator, another marker of reduced fibrinolysis,129 and von Willebrand factor, a marker of endothelial injury, were predictive.130 In one clinical study of type 2 diabetics, adhesion molecules were higher in subjects with retinopathy than those without,131 and in another population-based cohort, composite scores of both inflammatory and endothelial function markers were strongly associated with the presence of diabetic retinopathy.132 Similarly, E-selectin values were found to be increased in a group with type 1 diabetes and retinopathy.133 Adiponectin was increased in the advanced stages of retinopathy.134 These results should be interpreted cautiously, however, as serum markers are not necessarily indicators of tissue events. However, P-selectin, ICAM-1, and polymorphonuclear leukocyte numbers are all elevated in human diabetic retina.135 Experimental work in diabetic rat retinas later demonstrated that inflammatory cytokine-mediated leukostasis occurs early in diabetic retina and neutralizing ICAM-1 and CD-18 prevents it.136–138 From these studies, it appears that inflammation-mediated EC injury and vaso-occlusion may cause nonperfusion and subsequrent hypoxia in diabetic retina.139,140
The hypothesis that inflammation is critical to the development of diabetic retinopathy arose from initial reports that diabetic patients taking salicylates to treat rheumatoid arthritis had a lower-than-expected incidence of diabetic retinopathy.141 The subsequent decades demonstrated an increase in inflammatory markers and growth factors in the diabetic vitreous and retina. Recently microarray analyses substantiated a marked inflammatory response in the retinas of diabetic rodents.142 Confirmation of the importance of inflammatory factors and growth factors is supported by additional rodent studies that show that blocking these factors prevents the development of lesions characteristic of the retinopathy in animals. Specific inflammatory molecules that have been shown to contribute to structural or functional alterations that are characteristic of the retinopathy include NF-κβ143; inducible nitric oxide synthase143; cytochrome c oxidase143; ICAM140; 5-lipoxygenase144; interleukin-1β145; tumor necrosis factor (TNF)-α146; and VEGF.67,147 Inflammation can intensify the generation of AGEs that are produced in response to hyperglycemia and increased oxidative stress.
Retinal vein occlusion (RVO)
Occlusion of large retinal blood vessels is a common occurrence, which results in retinal hypoxia. RVO is the second most common sight-threatening retinal vascular disorder after diabetic retinopathy.148 RVO represents an obstruction of the retinal venous system that involves either the central retinal vein or a branch retinal vein. RVO is typically due to external compression or disease of the vein wall, such as is seen in vasculitis.149 Central retinal artery occlusion (CRAO) results in sudden, catastrophic visual loss and branch retinal arteriolar occlusion (BRAO) causes sudden segmental visual loss and may recur to involve other branch retinal arterioles. CRAO studies have shown that the ischemic retinal whitish opacity and swelling of CRAO are essentially located in the perifoveolar region of the macula. Oxygen supply and nutrition from the choroidal vascular bed to the thinner peripheral retina help in its much longer survival and the maintenance of peripheral visual fields. The diagnosis is clinical and based on the observation of the ocular fundus: venous dilatation and tortuosity, flame-shaped retinal hemorrhages, retinal edema and cotton-wool exudates affecting all the retinal sectors (in CRVO) or the sector of the retina drained by the affected vein in BRVO. Open-angle glaucoma is the most frequent local alteration predisposing to RVO as it compromises venous outflow by increasing intraocular pressure. Raised intraocular pressure causes external compression of the central retinal vein as it passes through the lamina cribrosa, resulting in turbulent blood flow distal to the compression leading to thrombus formation.
The natural history of RVO is highly variable; in some cases the retinal findings progressively disappear and there is a good visual outcome, while in other cases severe complications like ocular neovascularization (proliferative retinopathy), vitreous hemorrhage, neovascular glaucoma, and macular edema develop. Using retinal oximetry in BRVO, venular saturation was found to be highly variable between patients (12–93%)150; this was attributed to variable severity of the disease, recanalization, degree of occlusion, collateral vasculature, or tissue atrophy. The majority of patients with CRVO have signs of macular edema at presentation whereas only 5–15% of eyes with BRVO develop macular edema over the first year. Venous collateral channels represent tortuous vessels that develop locally, mainly around the optic disc, and are usually associated with a long-standing vascular obstruction. Vitreous hemorrhage develops in 10% of eyes with CRVO within 9 months of presentation and in about 40% of eyes with BRVO.148 If there is restoration of circulation in the central retinal artery, the retinal capillaries in the central, thickest part of the macular region do not refill because of compression by the surrounding swollen superficial retinal tissue, resulting in the “no-reflow phenomenon,”151and consequently in permanent ganglion cell death in the nonperfused retina; the area of central retinal capillary nonfilling may vary from eye to eye depending upon the severity of retinal swelling in the macular region. This results in the variable size of the permanent central scotoma. In considering the outcome of CRAO, it is important to consider retinal tolerance time to acute retinal ischemia. The chance of recovery of vision only exists as long as the retina has reversible ischemic damage. The retina suffers no detectable damage with CRAO of up to 97 minutes, but after that, the longer the CRAO, the more extensive the irreversible ischemic retinal damage.
It is generally believed that the CRAO is always either embolic or thrombotic in origin. Embolism is far more common than thrombosis,152 as was pointed out almost a century ago by Coats.153,154 An inflammatory etiology is also postulated in RVO as RVO is associated with immunological diseases in young patients. To support this contention, patients can experience prompt resolution of symptoms with the use of periocular steroids.155 Giant cell arteritis is an important and well-known cause of CRAO, and is an ophthalmic emergency because of the high risk of bilateral visual loss, which is preventable.
Elevated levels of PAI-1, lipoprotein(a), and hyperhomocysteinemia, and low circulating levels of folic acid, vitamin B12 and vitamin B6 have been implicated in the pathogenesis of this disease.156 While the pathogenesis is complex and largely unknown, medical treatment includes identification and correction of vascular risk factors. The use of fluorescein fundus angiography before (showing occlusion of the central retinal artery) and immediately after thrombolysis may show improvement in and/or restoration of retinal circulation and retinal function. It is also important to consider that fibrinolytic agents can dissolve only platelet fibrin emboli.157 Retinal emboli are made of 74% cholesterol, 10.5% calcific material, and only 15.5% of platelet fibrin. Fibrinolytic agents cannot dissolve cholesterol or calcified material. Therefore, there is no scientific rationale for the use of fibrinolytic agents in at least 85% of CRAO cases. For the remaining 15%, if the diagnosis is made within 15 days from onset of clinical manifestations, low-molecular-weight heparins at anticoagulant doses are typically used 10–15 days followed by half dose for a total of 90 days.158 No data are available on the possible role of antithrombotic/antiplatelet strategies in the long-term prevention of recurrent RVO159; however, they are routinely prescribed to most patients with diabetes, hypertension, or arterial disease. Of Virchow’s three classical factors that play a role in thrombogenesis – stasis, vessel wall damage, and hypercoagulability – the first two have long been reported in patients with RVO, whereas the third has not been sufficiently investigated until recently.
Sickle-cell disease (SCD)
The pathological processes involved in SCD can affect virtually every vascular bed including the retina and in its advanced stages has the potential to cause blindness.160 Classification of ophthalmic manifestations of SCD in the retina is based on the presence or absence of vascular proliferation, which is the most important precursor of blinding complications and precedes development of a vitreous hemorrhage or retinal detachment. Ischemia occurs due to obstruction of capillaries with sickle red blood cells and subsequent thrombus formation. The subsequent scenario for disease pathogenesis is similar to that for ROP, diabetic retinopathy, and RVO, described above, except that there is no retinal leakage of retinal blood vessels only from preretinal neovascularization, even though there is loss of peripheral vasculature and elevation of VEGF.161 Neovascularization that occurs can lead to fibrosis that can then result in retinal detachment.
Ocular ischemic syndrome (OIS)
OIS occurs at a mean age of 65 years and is rare before the age of 50. Men are affected twice as often as women,162 reflecting their higher incidence of atherosclerotic disease; however, no racial predilection exists. Bilateral involvement may occur in up to 22% of cases.163,164 Sturrock and Mueller estimated 7.5 cases per million persons every year,165 but this is likely an underestimation as OIS can be easily misdiagnosed. Kearns166 reported that, of patients with occlusion of the internal carotid artery undergoing surgical anastomosis between the superficial temporal artery and the middle cerebral artery, 18% presented with OIS. Up to 29% of patients with a symptomatic carotid artery occlusion manifest retinal vascular changes that are usually asymptomatic; however, 1.5% of them progress per year to symptomatic OIS.167 OIS develops especially in patients with poor collateral circulation between the internal and external carotid arterial systems. Insufficient collateral vascular flow in OIS patients explains the frequent association with cerebral infarctions and the poor neurologic outcomes.168 Degree of internal carotid artery stenosis, presence of collateral vessels, and compensation by collaterals are important in assessing OIS disease severity. Also if the OIS is bilateral or there are associated systemic vascular diseases, this tends to worsen the prognosis.
Retinal detachment
Retinal detachment causes the sensory retina to be distant from choriocapillaris, thus reducing its oxygen supply and resulting in photoreceptor degeneration. Linsenmeier and Padnick-Silver169 demonstrated in cat that retinal detachment resulted in a significant decrease in outer retinal oxygen, having a serious metabolic effect on photoreceptors. In subsequent work, Linsenmeier demonstrated that hyperoxia may have clinical benefit after retinal detachment because it normalized photoreceptor oxygen consumption and prevented photoreceptor dysfunction.170 Furthermore, hyperoxia prevents proliferation and reactivity of retinal Müller cells in the detached feline retina, limiting retinal injury.171
Consequences of retinal ischemia
In 1971, Judah Folkman reported in the New England Journal of Medicine that all cancer tumors are angiogenesis-dependent.172 If a tumor could be stopped from growing its own blood supply, he surmised, it would wither and die. Though his hypothesis was initially disregarded by most experts in the field, Folkman persisted with his research. After more than a decade, his theory became widely accepted and is now at the center of our understanding of ocular angiogenesis. Retinal neovascularization is defined as a state where new pathologic vessels originate from the existing retinal veins and extend along the inner surface of the retina.
Vascular permeability
Growth factors such as VEGF have been implicated in both retinal neovascularization and vascular hyperpermeability.173 Antibodies to VEGF improve visual function in patients with diabetic macular edema.174 In experimental diabetes and in VEGF-induced permeability, alterations of the tight junction (TJ) complex of microvascular endothelial cells alters the BRB.175 A role of classical PKC isoforms (cPKCs) but especially PKCβ in regulating VEGF-induced vascular permeability is well accepted.176 VEGF activation of PKCβ leads to phosphorylation and reorganization of the TJ complex, increasing vessel wall permeability.177 VEGF increases the phosphorylation of the TJ protein, occludin, at multiple sites,178 including Ser490.179 Phosphorylation at Ser490 allows subsequent ubiquitination and endocytosis of occludin and fosters breaks in the TJ.180 Despite this well-supported mechanism, the PKCβ inhibitor ruboxistaurin failed to achieve Food and Drug Administration approval for diabetic retinopathy. cPKC inhibition also failed to prevent TNF-α-induced permeability,181 a proinflammatory cytokine also implicated in diabetic retinopathy. Thus targeting permeability in the retina continues to be a difficult clinical problem.
Adult choroidal ischemia
Diabetic choroidopathy was first described by Hidiyat and Fine.182 The use of alkaline phosphatase enzyme histochemistry on human choroid permitted quantification of vascular loss in choroid. Viable vessels were positive for alkaline phosphatase while acellular, dysfunctional capillaries lacked it, and choroidal neovascularization had the greatest activity.10 This technique demonstrated that there was four times greater loss in choriocapillaris in diabetic subjects than in aged control subjects.183 Loss of alkaline phosphatase activity was associated with the presence of polymorphonuclear leukocytes.184 Areas of choriocapillaris loss were also associated with choroidal neovascularization and Bruch’s membrane deposits. A consequence of choriocapillaris dropout is loss of outer retinal oxygenation. This may be the reason for loss of blue cones and other cones in diabetic retina when there is no retinopathy present.185,186
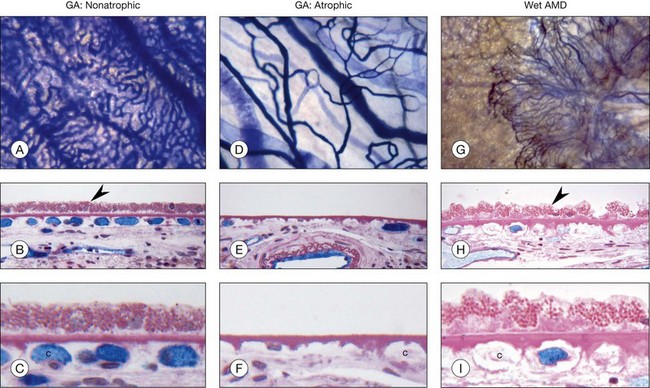
Decreased choroidal blood flow has been measured in AMD.187–190 Using alkaline phosphatase activity and quantification of RPE in AMD, the loss of choriocapillaris has been demonstrated in exudative or wet AMD as well as geographic atrophy (Fig. 18.5). RPE loss occurs first in geographic atrophy and then choriocapillaris degeneration occurs. Although some capillaries survive in the area of RPE atrophy, a 50% loss of vasculature occurs and the surviving capillaries are highly constricted.191,192 This undoubtedly contributes to photoreceptor loss in the area of degeneration. Capillary loss occurs in exudative AMD as well but in the presence of a complete RPE monolayer (Fig. 18.5). This suggests that the RPE in this area are hypoxic and VEGF production may be increased, causing choroidal neovascularization. Large-vessel stenosis is also often observed in AMD choroid, suggesting that the choriocapillaris blood supply may be limited as well. Unfortunately, we have no way of measuring oxygen directly in the choroid so we can only assume ischemia occurs when there is loss of viable choriocapillaris and stenosis of large and intermediate choroidal blood vessel.
Conclusions
Both too little and too much oxygen can be damaging to the retina. Retina cannot function properly nor survive without the support of two vasculatures: retinal and choroidal. If either vasculature is dysfunctional, retinal hypoxia occurs and HIF-1α becomes stable and induces expression of key factors to assist in retinal cell survival. Reduced retinal oxygenation causes impaired neuroretinal activity in young healthy persons.193 One key factor upregulated by HIF-1α is VEGF, which stimulates new angiogenesis and increased vascular permeability. These pathological events can be controlled by destroying ischemic retina (photocoagulation), inhibiting VEGF, or reducing levels of HIF-1α.
1 Wangsa-Wirawan ND, Linsenmeier RA, Retinal oxygen. Fundamental and clinical aspects. Arch Ophthalmol. 2003;121:547–557.
2 Yu DY, Cringle SJ. Oxygen distribution and consumption within the retina in vascularised and avascular retinas and in animal models of retinal disease. Prog Retin Eye Res. 2001;20:175–208.
3 Chan-Ling T, McLeod DS, Hughes S, et al. Astrocyte–endothelial cell relationships during human retinal vascular development. Invest Ophthalmol Vis Sci. 2004;45:2020–2032.
4 Hasegawa T, McLeod DS, Prow T, et al. Vascular precursors in developing human retina. Invest Ophthalmol Vis Sci. 2008;49:2178–2192.
5 McLeod DS, Hasegawa T, Prow T, et al. The initial fetal human retinal vasculature develops by vasculogenesis. Dev Dyn. 2006;235:3336–3347.
6 Stone J, Itin A, Alon T, et al. Development of retinal vasculature is mediated by hypoxia-induced vascular endothelial growth factor (VEGF) expression by neuroglia. J Neurosci. 1995;15:4738–4747.
7 Chan-Ling T, Gock B, Stone J. The effect of oxygen on vasoformative cell division: Evidence that ‘physiological hypoxia’ is the stimulus for normal retinal vasculogenesis. Invest Ophthalmol Vis Sci. 1995;36:1201–1214.
8 Cogan DG, Kuwabara T. The mural cell in perspective. Arch Ophthalmol. 1967;78:133–139.
9 Hasegawa T, McLeod DS, Bhutto IA, et al. The embryonic human choriocapillaris develops by hemo-vasculogenesis. Dev Dyn. 2007;236:2089–2100.
10 McLeod DS, Lutty GA. High resolution histologic analysis of the human choroidal vasculature. Invest Ophthalmol Vis Sci. 1994;35:3799–3811.
11 Blaauwgeers HG, Holtkamp GM, Rutten H, et al. Polarized vascular endothelial growth factor secretion by human retinal pigment epithelium and localization of vascular endothelial growth factor receptors on the inner choriocapillaris. Evidence for a trophic paracrine relation. Am J Pathol. 1999;155:421–428.
12 Lutjen-Drecoll E. Choroidal innervation in primate eyes. Exp Eye Res. 2006;82:357–361.
13 Michaelson IC. The mode of development of the vascular system of the retina, with some observations on its significance for certain retinal diseases. Trans Ophthalmol Soc UK. 1948;68:137–180.
14 Michaelson IC. Retinal circulation in man and animals. Springfield: CC Thomas; 1954.
15 Wise GN. Retinal neovascularization. Trans Am Ophthalmol Soc. 1956;96:729–826.
16 Leung DW, Cachianes G, Kuang WJ, et al. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246:1306–1309.
17 Senger DR, Galli SJ, Dvorak AM, et al. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 1983;219:983–985.
18 Yu DY, Cringle SJ, Su EN. Intraretinal oxygen distribution in the monkey retina and the response to systemic hyperoxia. Invest Ophthalmol Vis Sci. 2005;46:4728–4733.
19 McLeod DS, Brownstein R, Lutty GA. Vaso-obliteration in the canine model of oxygen-induced retinopathy. Invest Ophthalmol Vis Sci. 1996;37:300–311.
20 Ernest JT, Goldstick TK. Retinal oxygen tension and oxygen reactivity in retinopathy of prematurity in kittens. Invest Ophthalmol Vis Sci. 1984;25:1129–1134.
21 Hardy P, Peri KG, Lahaie I, et al. Increased nitric oxide synthesis and action preclude choroidal vasoconstriction to hyperoxia in newborn pigs. Circ Res. 1996;79:504–511.
22 Linsenmeier RA, Yancy CM. Effects of hyperoxia on the oxygen distribution in the intact cat retina. Invest Ophthalmol Vis Sci. 1989;30:612–618.
23 Pournaras CJ, Riva CE, Tsacopoulos M, et al. Diffusion of O2 in the retina of anesthetized miniature pigs in normoxia and hyperoxia. Exp Eye Res. 1989;49:347–360.
24 Benizri E, Ginouves A, Berra E. The magic of the hypoxia-signaling cascade. Cell Mol Life Sci. 2008;65:1133–1149.
25 Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992;12:5447–5454.
26 Wang GL, Jiang BH, Rue EA, et al. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92:5510–5514.
27 Gariboldi MB, Ravizza R, Monti E. The IGFR1 inhibitor NVP-AEW541 disrupts a pro-survival and pro-angiogenic IGF-STAT3-HIF1 pathway in human glioblastoma cells. Biochem Pharmacol. 2010;80:455–462.
28 Jiang BH, Zheng JZ, Leung SW, et al. Transactivation and inhibitory domains of hypoxia-inducible factor 1alpha. Modulation of transcriptional activity by oxygen tension. J Biol Chem. 1997;272:19253–19260.
29 Semenza GL. Hydroxylation of HIF-1: oxygen sensing at the molecular level. Physiol (Bethesda). 2004;19:176–182.
30 Mahon PC, Hirota K, Semenza GL. FIH-1: a novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001;15:2675–2686.
31 Lando D, Peet DJ, Whelan DA, et al. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science. 2002;295:858–861.
32 Lee C, Kim SJ, Jeong DG, et al. Structure of human FIH-1 reveals a unique active site pocket and interaction sites for HIF-1 and von Hippel–Lindau. J Biol Chem. 2003;278:7558–7563.
33 Kukkola L, Koivunen P, Pakkanen O, et al. Collagen prolyl 4-hydroxylase tetramers and dimers show identical decreases in Km values for peptide substrates with increasing chain length: mutation of one of the two catalytic sites in the tetramer inactivates the enzyme by more than half. J Biol Chem. 2004;279:18656–18661.
34 van Uden P, Kenneth NS, Rocha S. Regulation of hypoxia-inducible factor-1alpha by NF-kappaB. Biochem J. 2008;412:477–484.
35 Wang GL, Semenza GL. Desferrioxamine induces erythropoietin gene expression and hypoxia-inducible factor 1 DNA-binding activity: implications for models of hypoxia signal transduction. Blood. 1993;82:3610–3615.
36 Tian H, McKnight SL, Russell DW. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997;11:72–82.
37 Sowter HM, Raval RR, Moore JW, et al. Predominant role of hypoxia-inducible transcription factor (Hif)-1alpha versus Hif-2alpha in regulation of the transcriptional response to hypoxia. Cancer Res. 2003;63:6130–6134.
38 Park SK, Dadak AM, Haase VH, et al. Hypoxia-induced gene expression occurs solely through the action of hypoxia-inducible factor 1alpha (HIF-1alpha): role of cytoplasmic trapping of HIF-2alpha. Mol Cell Biol. 2003;23:4959–4971.
39 Makino Y, Cao R, Svensson K, et al. Inhibitory PAS domain protein is a negative regulator of hypoxia-inducible gene expression. Nature. 2001;414:550–554.
40 Compernolle V, Brusselmans K, Franco D, et al. Cardia bifida, defective heart development and abnormal neural crest migration in embryos lacking hypoxia-inducible factor-1alpha. Cardiovasc Res. 2003;60:569–579.
41 Peng J, Zhang L, Drysdale L, et al. The transcription factor EPAS-1/hypoxia-inducible factor 2alpha plays an important role in vascular remodeling. Proc Natl Acad Sci U S A. 2000;97:8386–8391.
42 Scortegagna M, Morris MA, Oktay Y, et al. The HIF family member EPAS1/HIF-2alpha is required for normal hematopoiesis in mice. Blood. 2003;102:1634–1640.
43 Kline DD, Peng YJ, Manalo DJ, et al. Defective carotid body function and impaired ventilatory responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1 alpha. Proc Natl Acad Sci U S A. 2002;99:821–826.
44 Caprara C, Thiersch M, Lange C, et al. HIF1A is essential for the development of the intermediate plexus of the retinal vasculature. Invest Ophthalmol Vis Sci. 2011;52:2109–2117.
45 Lin M, Chen Y, Jin J, et al. Ischaemia-induced retinal neovascularisation and diabetic retinopathy in mice with conditional knockout of hypoxia-inducible factor-1 in retinal Muller cells. Diabetologia. 2011;54:1554–1566.
46 Cramer T, Yamanishi Y, Clausen BE, et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–657.
47 Kojima H, Gu H, Nomura S, et al. Abnormal B lymphocyte development and autoimmunity in hypoxia-inducible factor 1alpha -deficient chimeric mice. Proc Natl Acad Sci U S A. 2002;99:2170–2174.
48 Makino Y, Nakamura H, Ikeda E, et al. Hypoxia-inducible factor regulates survival of antigen receptor-driven T cells. J Immunol. 2003;171:6534–6540.
49 Giaccia A, Siim BG, Johnson RS. HIF-1 as a target for drug development. Nat Rev Drug Discov. 2003;2:803–811.
50 Kelly BD, Hackett SF, Hirota K, et al. Cell type-specific regulation of angiogenic growth factor gene expression and induction of angiogenesis in nonischemic tissue by a constitutively active form of hypoxia-inducible factor 1. Circ Res. 2003;93:1074–1081.
51 Hirsila M, Koivunen P, Gunzler V, et al. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J Biol Chem. 2003;278:30772–30780.
52 Willam C, Masson N, Tian YM, et al. Peptide blockade of HIFalpha degradation modulates cellular metabolism and angiogenesis. Proc Natl Acad Sci U S A. 2002;99:10423–10428.
53 Chen J, Smith LE. A double-edged sword: erythropoietin eyed in retinopathy of prematurity. J AAPOS. 2008;12:221–222.
54 Ferrara N. Vascular endothelial growth factor as a target for anticancer therapy. Oncologist. 2004;9(Suppl 1):2–10.
55 Carmeliet P, Ferreira V, Breier G, et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–439.
56 Alon T, Hemo I, Itin A, Pe’er J, et al. Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nat Med. 1995;1:1024–1028.
57 Baffert F, Le T, Thurston G, et al. Angiopoietin-1 decreases plasma leakage by reducing number and size of endothelial gaps in venules. Am J Physiol Heart Circ Physiol. 2006;290:H107–H118.
58 Saint-Geniez M, Maharaj AS, Walshe TE, et al. Endogenous VEGF is required for visual function: evidence for a survival role on muller cells and photoreceptors. PLoS ONE. 2008;3:e3554.
59 Adamis AP, Shima DT, Yeo KT, et al. Synthesis and secretion of vascular permeability factor/vascular endothelial growth factor by human retinal pigment epithelial cells. Biochem Biophys Res Commun. 1993;193:631–638.
60 Aiello LP, Northrup JM, Keyt BA, et al. Hypoxic regulation of vascular endothelial growth factor in retinal cells. Arch Ophthalmol. 1995;113:1538–1544.
61 Famiglietti EV, Stopa EG, McGookin ED, et al. Immunocytochemical localization of vascular endothelial growth factor in neurons and glial cells of human retina. Brain Res. 2003;969:195–204.
62 Sandercoe TM, Geller SF, Hendrickson AE, et al. VEGF expression by ganglion cells in central retina before formation of the foveal depression in monkey retina: evidence of developmental hypoxia. J Comp Neurol. 2003;462:42–54.
63 Marneros AG, Fan J, Yokoyama Y, et al. Vascular endothelial growth factor expression in the retinal pigment epithelium is essential for choriocapillaris development and visual function. Am J Pathol. 2005;167:1451–1459.
64 Saint-Geniez M, Kurihara T, Sekiyama E, et al. An essential role for RPE-derived soluble VEGF in the maintenance of the choriocapillaris. Proc Natl Acad Sci U S A. 2009;106:18751–18756.
65 Nishijima K, Ng YS, Zhong L, et al. Vascular endothelial growth factor-A is a survival factor for retinal neurons and a critical neuroprotectant during the adaptive response to ischemic injury. Am J Pathol. 2007;171:53–67.
66 Ng EW, Adamis AP. Targeting angiogenesis, the underlying disorder in neovascular age-related macular degeneration. Can J Ophthalmol. 2005;40:352–368.
67 Kunz Mathews M, Merges C, McLeod DS, et al. Vascular endothelial growth factor (VEGF) and vascular permeability changes in human diabetic retinopathy. Invest Ophthalmol Vis Sci. 1997;38:2729–2741.
68 Starita C, Patel M, Katz B, et al. Vascular endothelial growth factor and the potential therapeutic use of pegaptanib (Macugen) in diabetic retinopathy. Dev Ophthalmol. 2007;39:122–148.
69 Aiello LP, Cahill MT, Cavallerano JD. Growth factors and protein kinase C inhibitors as novel therapies for the medical management diabetic retinopathy. Eye (Lond). 2004;18:117–125.
70 Tripathi RC, Li J, Tripathi BJ, et al. Increased level of vascular endothelial growth factor in aqueous humor of patients with neovascular glaucoma. Ophthalmology. 1998;105:232–237.
71 Lashkari K, Hirose T, Yazdany J, et al. Vascular endothelial growth factor and hepatocyte growth factor levels are differentially elevated in patients with advanced retinopathy of prematurity. Am J Pathol. 2000;156:1337–1344.
72 Chakravarthy U. Age related macular degeneration. Br Med J. 2006;333:869–870.
73 Rosenfeld PJ, Brown DM, Heier JS, et al. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med. 2006;355:1419–1431.
74 Miller-Kasprzak E, Jagodzinski PP. 5-Aza-2’-deoxycytidine increases the expression of anti-angiogenic vascular endothelial growth factor 189b variant in human lung microvascular endothelial cells. Biomed Pharmacother. 2008;62:158–163.
75 Anthony FW, Wheeler T, Elcock CL, et al. Short report: identification of a specific pattern of vascular endothelial growth factor mRNA expression in human placenta and cultured placental fibroblasts. Placenta. 1994;15:557–561.
76 Woolard J, Bevan HS, Harper SJ, et al. Molecular diversity of VEGF-A as a regulator of its biological activity. Microcirculation. 2009;16:572–592.
77 de Vries C, Escobedo JA, Ueno H, et al. The fms-like tyrosine kinase, a receptor for vascular endothelial growth factor. Science. 1992;255:989–991.
78 Quinn TP, Peters KG, De Vries C, et al. Fetal liver kinase 1 is a receptor for vascular endothelial growth factor and is selectively expressed in vascular endothelium. Proc Natl Acad Sci U S A. 1993;90:7533–7537.
79 Soker S, Takashima S, Miao HQ, et al. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell. 1998;92:735–745.
80 Schlessinger J. New roles for Src kinases in control of cell survival and angiogenesis. Cell. 2000;100:293–296.
81 Hubbard SR. Structural analysis of receptor tyrosine kinases. Prog Biophys Mol Biol. 1999;71:343–358.
82 Shalaby F, Rossant J, Yamaguchi TP, et al. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376:62–66.
83 Scott A, Mellor H. VEGF receptor trafficking in angiogenesis. Biochem Soc Trans. 2009;37:1184–1188.
84 Jopling HM, Odell AF, Hooper NM, et al. Rab GTPase regulation of VEGFR2 trafficking and signaling in endothelial cells. Arterioscler Thromb Vasc Biol. 2009;29:1119–1124.
85 Salikhova A, Wang L, Lanahan AA, et al. Vascular endothelial growth factor and semaphorin induce neuropilin-1 endocytosis via separate pathways. Circ Res. 2008;103:e71–e79.
86 Ewan LC, Jopling HM, Jia H, et al. Intrinsic tyrosine kinase activity is required for vascular endothelial growth factor receptor 2 ubiquitination, sorting and degradation in endothelial cells. Traffic. 2006;7:1270–1282.
87 Lanahan AA, Hermans K, Claes F, et al. VEGF receptor 2 endocytic trafficking regulates arterial morphogenesis. Dev Cell. 2010;18:713–724.
88 Cai J, Jiang WG, Ahmed A, et al. Vascular endothelial growth factor-induced endothelial cell proliferation is regulated by interaction between VEGFR-2, SH-PTP1 and eNOS. Microvasc Res. 2006;71:20–31.
89 Chen J, Braet F, Brodsky S, et al. VEGF-induced mobilization of caveolae and increase in permeability of endothelial cells. Am J Physiol Cell Physiol. 2002;282:C1053–C1063.
90 Grant MB, Boulton ME, Ljubimov AV. Erythropoietin: when liability becomes asset in neurovascular repair. J Clin Invest. 2008;118:467–470.
91 Takahashi T, Ueno H, Shibuya M. VEGF activates protein kinase C-dependent, but Ras-independent Raf-MEK-MAP kinase pathway for DNA synthesis in primary endothelial cells. Oncogene. 1999;18:2221–2230.
92 Fadini GP, Miorin M, Facco M, et al. Circulating endothelial progenitor cells are reduced in peripheral vascular complications of type 2 diabetes mellitus. J Am Coll Cardiol. 2005;45:1449–1457.
93 Tepper OM, Galiano RD, Capla JM, et al. Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation. 2002;106:2781–2786.
94 Loomans CJ, de Koning EJ, Staal FJ, et al. Endothelial progenitor cell dysfunction: a novel concept in the pathogenesis of vascular complications of type 1 diabetes. Diabetes. 2004;53:195–199.
95 Schatteman GC, Hanlon HD, Jiao C, et al. Blood-derived angioblasts accelerate blood-flow restoration in diabetic mice. J Clin Invest. 2000;106:571–578.
96 Caballero S, Sengupta N, Afzal A, et al. Ischemic vascular damage can be repaired by healthy, but not diabetic, endothelial progenitor cells. Diabetes. 2007;56:960–967.
97 Jarajapu YP, Caballero S, Verma A, et al. Blockade of NADPH oxidase restores vasoreparative function in diabetic CD34+ cells. Invest Ophthalmol Vis Sci. 2011;52:5093–5104.
98 Case J, Ingram DA, Haneline LS. Oxidative stress impairs endothelial progenitor cell function. Antioxid Redox Signal. 2008;10:1895–1907.
99 Togliatto G, Trombetta A, Dentelli P, et al. Unacylated ghrelin rescues endothelial progenitor cell function in individuals with type 2 diabetes. Diabetes. 2010;59:1016–1025.
100 Ingram DA, Lien IZ, Mead LE, et al. In vitro hyperglycemia or a diabetic intrauterine environment reduces neonatal endothelial colony-forming cell numbers and function. Diabetes. 2008;57:724–731.
101 Urbich C, Dernbach E, Rossig L, et al. High glucose reduces cathepsin L activity and impairs invasion of circulating progenitor cells. J Mol Cell Cardiol. 2008;45:429–436.
102 Li SS, Liu Z, Uzunel M, et al. Endogenous thrombospondin-1 is a cell-surface ligand for regulation of integrin-dependent T-lymphocyte adhesion. Blood. 2006;108:3112–3120.
103 Butler JM, Guthrie SM, Koc M, et al. SDF-1 is both necessary and sufficient to promote proliferative retinopathy. J Clin Invest. 2005;115:86–93.
104 Segal MS, Shah R, Afzal A, et al. Nitric oxide cytoskeletal-induced alterations reverse the endothelial progenitor cell migratory defect associated with diabetes. Diabetes. 2006;55:102–109.
105 Bhatwadekar AD, Glenn JV, Li G, et al. Advanced glycation of fibronectin impairs vascular repair by endothelial progenitor cells: implications for vasodegeneration in diabetic retinopathy. Invest Ophthalmol Vis Sci. 2008;49:1232–1241.
106 Bhatwadekar AD, Guerin EP, Jarajapu YP, et al. Transient inhibition of transforming growth factor-beta1 in human diabetic CD34+ cells enhances vascular reparative functions. Diabetes. 2010;59:2010–2019.
107 Sorrentino SA, Bahlmann FH, Besler C, et al. Oxidant stress impairs in vivo reendothelialization capacity of endothelial progenitor cells from patients with type 2 diabetes mellitus: restoration by the peroxisome proliferator-activated receptor-gamma agonist rosiglitazone. Circulation. 2007;116:163–173.
108 Mohler ER, 3rd., Shi Y, Moore J, et al. Diabetes reduces bone marrow and circulating porcine endothelial progenitor cells, an effect ameliorated by atorvastatin and independent of cholesterol. Cytometry A. 2009;75:75–82.
109 Anghelina M, Krishnan P, Moldovan L, et al. Monocytes/macrophages cooperate with progenitor cells during neovascularization and tissue repair: conversion of cell columns into fibrovascular bundles. Am J Pathol. 2006;168:529–541.
110 Chang KH, Chan-Ling T, McFarland EL, et al. IGF binding protein-3 regulates hematopoietic stem cell and endothelial precursor cell function during vascular development. Proc Natl Acad Sci U S A. 2007;104:10595–10600.
111 Granata R, Trovato L, Garbarino G, et al. Dual effects of IGFBP-3 on endothelial cell apoptosis and survival: involvement of the sphingolipid signaling pathways. FASEB J. 2004;18:1456–1458.
112 Franklin SL, Ferry RJ, Jr., Cohen P. Rapid insulin-like growth factor (IGF)-independent effects of IGF binding protein-3 on endothelial cell survival. J Clin Endocrinol Metab. 2003;88:900–907.
113 Liu LQ, Sposato M, Liu HY, et al. Functional cloning of IGFBP-3 from human microvascular endothelial cells reveals its novel role in promoting proliferation of primitive CD34+CD38– hematopoietic cells in vitro. Oncol Res. 2003;13:359–371.
114 Vasylyeva TL, Chen X, Ferry RJ, Jr. Insulin-like growth factor binding protein-3 mediates cytokine-induced mesangial cell apoptosis. Growth Horm IGF Res. 2005;15:207–214.
115 Lofqvist C, Chen J, Connor KM, et al. IGFBP3 suppresses retinopathy through suppression of oxygen-induced vessel loss and promotion of vascular regrowth. Proc Natl Acad Sci U S A. 2007;104:10589–10594.
116 Giannini S, Cresci B, Pala L, et al. IGFBPs modulate IGF-I- and high glucose-controlled growth of human retinal endothelial cells. J Endocrinol. 2001;171:273–284.
117 Kielczewski JL, Hu P, Shaw LC, et al. Novel protective properties of IGFBP-3 result in enhanced pericyte ensheathment, reduced microglial activation, increased microglial apoptosis, and neuronal protection after ischemic retinal injury. Am J Pathol. 2011;178:1517–1528.
118 Chan-Ling TL, Halasz P, Stone J. Development of retinal vasculature in the cat: processes and mechanisms. Curr Eye Res. 1990;9:459–478.
119 Hellstrom A, Engstrom E, Hard AL, et al. Postnatal serum insulin-like growth factor I deficiency is associated with retinopathy of prematurity and other complications of premature birth. Pediatrics. 2003;112:1016–1020.
120 Giudice LC, de Zegher F, Gargosky SE, et al. Insulin-like growth factors and their binding proteins in the term and preterm human fetus and neonate with normal and extremes of intrauterine growth. J Clin Endocrinol Metab. 1995;80:1548–1555.
121 Smith WJ, Underwood LE, Keyes L, et al. Use of insulin-like growth factor I (IGF-I) and IGF-binding protein measurements to monitor feeding of premature infants. J Clin Endocrinol Metab. 1997;82:3982–3988.
122 Smith LE. IGF-1 and retinopathy of prematurity in the preterm infant. Biol Neonate. 2005;88:237–244.
123 Kohner EM, Henkind P. Correlation of fluorescein angiogram and retinal digest in diabetic retinopathy. Am J Ophthalmol. 1970;69:403–414.
124 Linsenmeier RA, Braun RD, McRipley MA, et al. Retinal hypoxia in long term diabetic cats. Invest Ophthalmol Vis Sci. 1998;39:1647–1657.
125 Tang J, Kern TS. Inflammation in diabetic retinopathy. Prog Retin Eye Res. 2011;30:343–358.
126 Kern TS. Contributions of inflammatory processes to the development of the early stages of diabetic retinopathy. Exp. Diabetes Res. 2007;2007:95103.
127 Schmidt MI, Duncan BB, Sharrett AR, et al. Markers of inflammation and prediction of diabetes mellitus in adults (Atherosclerosis Risk in Communities study): a cohort study. Lancet. 1999;353:1649–1652.
128 Festa A, D’Agostino R, Jr., Tracy RP, et al. Elevated levels of acute-phase proteins and plasminogen activator inhibitor-1 predict the development of type 2 diabetes: the insulin resistance atherosclerosis study. Diabetes. 2002;51:1131–1137.
129 Eliasson MC, Jansson JH, Lindahl B, et al. High levels of tissue plasminogen activator (tPA) antigen precede the development of type 2 diabetes in a longitudinal population study. The Northern Sweden MONICA study. Cardiovasc Diabetol. 2003;2:19.
130 Meigs JB, Dupuis J, Liu C, et al. PAI-1 gene 4G/5G polymorphism and risk of type 2 diabetes in a population-based sample. Obesity (Silver Spring). 2006;14:753–758.
131 Orasanu G, Plutzky J. The pathologic continuum of diabetic vascular disease. J Am Coll Cardiol. 2009;53:S35–S42.
132 van Hecke MV, Dekker JM, Nijpels G, et al. Inflammation and endothelial dysfunction are associated with retinopathy: the Hoorn study. Diabetologia. 2005;48:1300–1306.
133 Spijkerman AM, Gall MA, Tarnow L, et al. Endothelial dysfunction and low-grade inflammation and the progression of retinopathy in type 2 diabetes. Diabet Med. 2007;24:969–976.
134 Hadjadj S, Aubert R, Fumeron F, et al. Increased plasma adiponectin concentrations are associated with microangiopathy in type 1 diabetic subjects. Diabetologia. 2005;48:1088–1092.
135 McLeod DS, Lefer DJ, Merges C, et al. Enhanced expression of intracellular adhesion molecule-1 and P-selectin in the diabetic human retina and choroid. Am J Pathol. 1995;147:642–653.
136 Joussen AM, Murata T, Tsujikawa A, et al. Leukocyte-mediated endothelial cell injury and death in the diabetic retina. Am J Pathol. 2001;158:147–152.
137 Joussen AM, Poulaki V, Qin W, et al. Retinal vascular endothelial growth factor induces intracellular adhesion molecule-1 and endothelial nitric oxude synthase expression and initiates early diabetic retinal leukocyte adhesion in vivo. Am J Pathol. 2002;160:501–509.
138 Joussen AM, Poulaki V, Mitsiades N, et al. Nonsteroidal anti-inflammatory drugs prevent early diabetic retinopathy via TNF-α suppression. FASEB J. 2002;16:438–440.
139 Adamis AP. Is diabetic retinopathy an inflammatory disease? Br J Ophthalmol. 2002;86:363–365.
140 Joussen AM, Poulaki V, Le ML, et al. A central role for inflammation in the pathogenesis of diabetic retinopathy. Faseb J. 2004;18:1450–1452.
141 Powell ED, Field RA. Diabetic retinopathy and rheumatoid arthritis. Lancet. 1964;2:17–18.
142 Brucklacher RM, Patel KM, VanGuilder HD, et al. Whole genome assessment of the retinal response to diabetes reveals a progressive neurovascular inflammatory response. BMC Med Genomics. 2008;1:26.
143 Kern TS, Miller CM, Du Y, et al. Topical administration of nepafenac inhibits diabetes-induced retinal microvascular disease and underlying abnormalities of retinal metabolism and physiology. Diabetes. 2007;56:373–379.
144 Gubitosi-Klug RA, Talahalli R, Du Y, et al. 5-Lipoxygenase, but not 12/15- lipoxygenase, contributes to degeneration of retinal capillaries in a mouse model of diabetic retinopathy. Diabetes. 2008;57:1387–1393.
145 Vincent JA, Mohr S. Inhibition of caspase-1/interleukin-1beta signaling prevents degeneration of retinal capillaries in diabetes and galactosemia. Diabetes. 2007;56:224–230.
146 Behl Y, Krothapalli P, Desta T, et al. Diabetes-enhanced tumor necrosis factor-alpha production promotes apoptosis and the loss of retinal microvascular cells in type 1 and type 2 models of diabetic retinopathy. Am J Pathol. 2008;172:1411–1418.
147 Lutty GA, McLeod DS, Merges C, et al. Localization of VEGF in human retina and choroid. Arch Ophthalmol. 1996;114:971–977.
148 Rogers SL, McIntosh RL, Lim L, et al. Natural history of branch retinal vein occlusion: an evidence-based systematic review. Ophthalmology. 2010;117:1094–1101. e1095
149 Laouri M, Chen E, Looman M, et al. The burden of disease of retinal vein occlusion: review of the literature. Eye (Lond). 2011;25(8):981–988.
150 Hardarson HS, Stefansson E. Oxygen saturation in branch vein occlusion. Acta Ophthalmol. 2011. Epub ahead of printing
151 Hayreh SS, Weingeist TA. Experimental occlusion of the central artery of the retina. IV: Retinal tolerance time to acute ischaemia. Br J Ophthalmol. 1980;64:818–825.
152 Hayreh SS, Podhajsky P. Ocular neovascularization with retinal vascular occlusion. II. Occurrence in central and branch retinal artery occlusion. Arch Ophthalmol. 1982;100:1585–1596.
153 Coats G. Obstruction of the central artery of the retina. R. Lond Ophthalmic Hosp Rep. 1905;16:262–306.
154 Coats G. Pathology of obstruction of the centeral artery of the retina. R. Lond Ophthalmic Hosp Rep. 1913;19:45–70.
155 Fong AC, Schatz H, McDonald HR, et al. Central retinal vein occlusion in young adults (papillophlebitis). Retina. 1992;12:3–11.
156 Sofi F, Marcucci R, Fedi S, et al. High lipoprotein (a) levels are associated with an increased risk of retinal vein occlusion. Atherosclerosis. 2010;210:278–281.
157 Arruga J, Sanders MD. Ophthalmologic findings in 70 patients with evidence of retinal embolism. Ophthalmology. 1982;89:1336–1347.
158 Ageno W, Cattaneo R, Manfredi E, et al. Parnaparin versus aspirin in the treatment of retinal vein occlusion. A randomized, double blind, controlled study. Thromb Res. 2010;125:137–141.
159 Marcucci R, Sofi F, Grifoni E, et al. Retinal vein occlusions: a review for the internist. Intern Emerg Med. 2011;6:307–314.
160 Nagpal KC, Goldberg MF, Rabb MF. Ocular manifestations of sickle hemoglobinopathies. Surv Ophthalmol. 1977;21:391–411.
161 Cao J, Kunz Mathews M, McLeod DS, et al. Angiogenic factors in human proliferative sickle cell retinopathy. Br J Ophthalmol. 1999;83:838–846.
162 Brown GC, Magargal LE. The ocular ischemic syndrome. Clinical, fluorescein angiographic and carotid angiographic features. Int Ophthalmol. 1988;11:239–251.
163 Boto de los Bueis A, Fernandez-Prieto A, Ruiz-Martin MM, et al. [Bilateral carotid occlusion in young woman. Clinical and hemodynamic ocular results.]. Arch Soc Esp Oftalmol. 2003;78:227–230.
164 Baatz H, Lange S, Buchner H, et al. [Pseudoangiitis in bilateral ocular ischemia.]. Ophthalmologe. 2007;104:243–245.
165 Sturrock GD, Mueller HR. Chronic ocular ischaemia. Br J Ophthalmol. 1984;68:716–723.
166 Kearns TP. Differential diagnosis of central retinal vein obstruction. Ophthalmology. 1983;90:475–480.
167 Klijn CJ, Kappelle LJ, van Schooneveld MJ, et al. Venous stasis retinopathy in symptomatic carotid artery occlusion: prevalence, cause, and outcome. Stroke. 2002;33:695–701.
168 Costa VP, Kuzniec S, Molnar LJ, et al. Clinical findings and hemodynamic changes associated with severe occlusive carotid artery disease. Ophthalmology. 1997;104:1994–2002.
169 Linsenmeier RA, Padnick-Silver L. Mtabolic dependence of photoreceptors on the choroid in the normal and detached retina. Invest Ophthalmol Vis Sci. 2000;41:3117–3123.
170 Wang S, Linsenmeier RA. Hyperoxia improves oxygen consumption in the detached feline retina. Invest Ophthalmol Vis Sci. 2007;48:1335–1341.
171 Lewis G, Mervin K, Valter K, et al. Limiting the proliferation and reactivity of retinal Muller cells during experimental retinal detachment: the value of oxygen supplementation. Am J Ophthalmol. 1999;128:165–172.
172 Folkman J. Tumor angiogenesis: therapeutic implications. N. Engl J Med. 1971;285:1182–1186.
173 Penn JS, Madan A, Caldwell RB, et al. Vascular endothelial growth factor in eye disease. Prog Retin Eye Res. 2008;27:331–371.
174 Salam A, Mathew R, Sivaprasad S. Treatment of proliferative diabetic retinopathy with anti-VEGF agents. Acta Ophthalmol. 2011;89:405–411.
175 Antonetti DA, Barber AJ, Khin S, et al. Vascular permeability in experimental diabetes is associated with reduced endothelial occludin content: vascular endothelial growth factor decreases occludin in retinal endothelial cells. Penn State Retina Research Group. Diabetes. 1998;47:1953–1959.
176 Aiello L, Bursell S, Clermont A, et al. Vascular endothelial growth factor-induced retinal permeability is mediated by protein kinase C in vivo and suppressed by an orally effective beta-isoform-selective inhibitor. Diabetes. 1997;46:1473–1480.
177 Barber AJ, Antonetti DA, Gardner TW. Altered expression of retinal occludin and glial fibrillary acidic protein in experimental diabetes. The Penn State Retina Research Group. Invest Ophthalmol Vis Sci. 2000;41:3561–3568.
178 Harhaj NS, Felinski EA, Wolpert EB, et al. VEGF activation of protein kinase C stimulates occludin phosphorylation and contributes to endothelial permeability. Invest Ophthalmol Vis Sci. 2006;47:5106–5115.
179 Sundstrom JM, Sundstrom CJ, Sundstrom SA, et al. Phosphorylation site mapping of endogenous proteins: a combined MS and bioinformatics approach. J Proteome Res. 2009;8:798–807.
180 Murakami T, Felinski EA, Antonetti DA. Occludin phosphorylation and ubiquitination regulate tight junction trafficking and vascular endothelial growth factor-induced permeability. J Biol Chem. 2009;284:21036–21046.
181 Aveleira CA, Lin CM, Abcouwer SF, et al. TNF-alpha signals through PKCzeta/NF-kappaB to alter the tight junction complex and increase retinal endothelial cell permeability. Diabetes. 2010;59:2872–2882.
182 Hidayat A, Fine B. Diabetic choroidopathy: light and electron microscopic observations of seven cases. Ophthalmology. 1985;67:512–522.
183 Cao J, McLeod S, Merges CA, et al. Choriocapillaris degeneration and related pathologic changes in human diabetic eyes. Arch Ophthalmol. 1998;116:589–597.
184 Lutty GA, Cao J, McLeod DS. Relationship of polymorphonuclear leukocytes (PMNs) to capillary dropout in the human diabetic choroid. Am J Pathol. 1997;151:707–714.
185 Lovasik J, Kergoat H. Electroretinographic results and ocular vascular perfusion in type I diabetes. Invest Ophthalmol Vis Sci. 1993;34:1731–1743.
186 Holopigian K, Greenstein VC, Seiple W, et al. Evidence for photoreceptor changes in patients with diabetic retinopathy. Invest Ophthalmol Vis Sci. 1997;38:2355–2365.
187 Grunwald J, Hariprasad S, DuPont J, et al. Foveolar choroidal blood flow in age-related macular degeneration. Invest Ophthalmol Vis Sci. 1998;39:385–390.
188 Grunwald JE, Metelitsina TI, Dupont JC, et al. Reduced foveolar choroidal blood flow in eyes with increasing AMD severity. Invest Ophthalmol Vis Sci. 2005;46:1033–1038.
189 Metelitsina TI, Grunwald JE, DuPont JC, et al. Foveolar choroidal circulation and choroidal neovascularization in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2008;49:358–363.
190 Pournaras CJ, Rungger-Brandle E, Riva CE, et al. Regulation of retinal blood flow in health and disease. Prog Retin Eye Res. 2008;27:284–330.
191 McLeod DS, Taomoto M, Otsuji T, et al. Quantifying changes in RPE and choriocapillaris in eyes with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2002;43:1986–1993.
192 McLeod DS, Grebe R, Bhutto I, et al. Relationship between RPE and choriocapillaris in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2009;50:4982–4991.
193 Feigl B. Age-related maculopathy-linking aetiology and pathophysiological changes to the ischaemia hypothesis. Prog Retin Eye Res. 2009;28:63–86.