[level-membership-for-cardiovascular-category]
CHAPTER 63 Restrictive Cardiomyopathy
Restrictive cardiomyopathy (RCM) is the least common cardiomyopathy, and is characterized by diastolic dysfunction with restrictive ventricular filling with normal or near-normal systolic function and wall thickness.1 RCM may be idiopathic or associated with other infiltrative diseases, such as amyloidosis, endomyocardial disease, sarcoidosis, iron deposition disease, and storage diseases. Numerous other diseases may have a prominent restrictive component. Presentation of RCM is variable, and diagnosis is often difficult. The prognosis for most forms of RCM is poor, and it is important to distinguish RCM from constrictive pericarditis, which may have a similar clinical presentation
CARDIAC AMYLOIDOSIS
Prevalence and Epidemiology
Primary amyloidosis is a rare but devastating disease, with an incidence of 9 per 1 million and mean survival of approximately 13 months after diagnosis.2 Cardiac involvement in primary amyloidosis is common, with 60% of patients exhibiting ECG or echocardiographic abnormalities. Death is attributed to cardiac causes in at least 50% of patients with primary amyloidosis who die either from heart failure or from a malignant arrhythmia.2
Senile amyloidosis predominantly affects men older than 70 years and involves the heart in 25% of individuals older than 80 years.3 Senile cardiac amyloidosis is often clinically silent; however, extensive amyloid deposition can lead to significant clinical symptoms.
Etiology and Pathophysiology
Amyloid deposits, regardless of their protein composition, all have a characteristic appearance on light microscopy, staining pink with Congo red dye and exhibiting apple-green birefringence under polarized light microscopy. Nearly all organ systems can be involved, including the kidneys, heart, blood vessels, central and peripheral nervous system, liver, bowel, lungs, eyes, skin, and bones. Cardiac amyloidosis is a devastating progressive process that leads to congestive heart failure, angina, and arrhythmias.1
In addition to mechanical effects on myocardial stiffness, amyloid deposition induces oxidative stress that depresses myocyte contractile function. Myocardial ischemia may also result from microvascular disease. Amyloid deposits typically spare the epicardial vessels, whereas involvement of intramyocardial vasculature is seen in more than 90% of patients with AL amyloidosis.4
Manifestations of Disease
Imaging Techniques and Findings
Ultrasonography
Increased wall thickness without dilation of the ventricular cavity and preserved systolic function until relatively advanced stages of the disease are hallmarks of cardiac amyloidosis on echocardiography (Fig. 63-1). Amyloid deposits may involve nearly all regions of the heart, including valves, myocardium, interatrial septum, and pericardium, and manifestations of this involvement can be seen as multivalvular regurgitation, thickening of the interatrial septum, atrial dilation, pericardial effusions, and diffuse thickening of the right ventricular and left ventricular (LV) myocardium.5 A classic finding of myocardial amyloidosis is a granular sparkling pattern on two-dimensional echocardiography. This pattern is not specific for amyloidosis, however, and can be seen in patients with hypertensive cardiomyopathy, glycogen storage disorders, and hypertrophic cardiomyopathy. Atrial and ventricular thrombi are common findings, particularly in advanced disease.
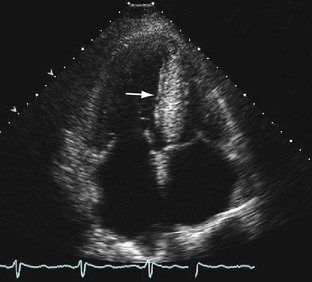
 FIGURE 63-1
FIGURE 63-1Pulsed wave Doppler echocardiography is helpful in assessing diastolic dysfunction in cardiac amyloidosis. The initial diastolic abnormality is abnormal relaxation (grade 1 diastolic dysfunction) resulting from increased ventricular wall thickness; the pattern becomes restrictive (grades 3 to 4) when progressive amyloid infiltration decreases LV compliance and increases left atrial pressure. The filling pattern may normalize temporarily (pseudonormalization—grade 2 diastolic dysfunction) as a result of combined relaxation abnormality and moderate increase in left atrial filling pressure before becoming frankly restrictive. Deceleration time is an important prognostic variable in cardiac amyloidosis; the average survival for patients with a deceleration time less than 150 ms is less than 1 year versus 3 years for patients with a deceleration time greater than 150 ms.6 A combination of LV wall thickness greater than 15 mm and fractional shortening of less than 20% (thought to reflect combined systolic and diastolic dysfunction) is associated with a median survival of 4 months.7 Right ventricular function has also been correlated with poor prognosis in patients with cardiac amyloidosis.8
Tissue Doppler imaging (TDI) has emerged more recently as a useful technique in assessment of LV regional wall motion and diastolic dysfunction in patients with cardiac amyloidosis. Koyama and colleagues9 showed that TDI measurements differentiated patients without from patients with cardiac amyloidosis, and amyloidosis patients with and without heart failure. TDI more clearly documented diastolic function than conventional Doppler-derived indices. Myocardial strain and strain rate imaging have also been investigated in cardiac amyloidosis, and these techniques have documented early impairment in systolic function before the onset of clinical heart failure.10
Computed Tomography
ECG gated contrast-enhanced CT reveals many of the findings discussed previously with regard to echocardiography: ventricular thickening with preserved systolic function, enlarged atria, and pericardial and pleural effusions (Fig. 63-2). Little evidence currently available suggests that CT is useful in the initial diagnosis of cardiac amyloidosis or in distinguishing amyloidosis from other causes of RCM.
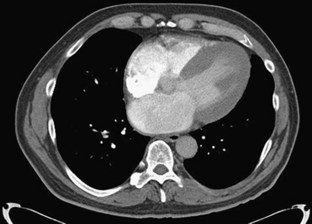
 FIGURE 63-2
FIGURE 63-2Magnetic Resonance Imaging
MRI has shown considerable promise in diagnosis and characterization of cardiac amyloidosis.11–14 Cine steady-state free precession (SSFP) images readily show findings of ventricular thickening with normal chamber size, atrial enlargement, and preserved systolic function (Fig. 63-3). Pleural and pericardial effusions are common and are well depicted on MRI. Impaired diastolic relaxation is often appreciated on cine SSFP images, and mitral inflow measurements can be obtained using cine phase contrast pulse sequences to obtain information analogous to Doppler echocardiography.
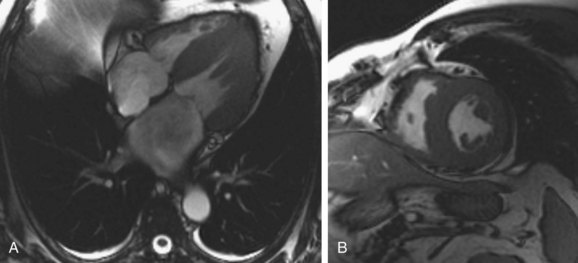
 FIGURE 63-3
FIGURE 63-3After administration of contrast medium, striking abnormalities are often seen on MDE pulse sequences, with patients with cardiac amyloidosis typically showing diffuse irregular hyperenhancement in noncoronary distributions (Fig. 63-4). A circumferential subendocardial hyperenhancement pattern has been described and correlated with predominant amyloid deposition in the subendocardial myocardium; however, in our experience, patterns of hyperenhancement are quite variable. Right ventricular late enhancement is a notable feature of amyloidosis and can help to distinguish this from hypertrophic cardiomyopathy with foci of enhancing fibrotic tissue.
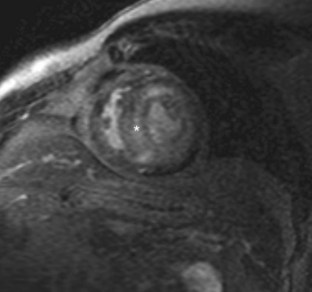
 FIGURE 63-4
FIGURE 63-4Abnormalities of myocardial nulling are also common in amyloidosis and can help to distinguish this disease from other pathologies. A cine multi-TI inversion recovery sequence, in which each image or phase is acquired with a slightly longer inversion time (TI), is often used to select the optimal TI for the delayed enhancement acquisition. As TI increases, blood and myocardium pass through a null point at which signal is minimized. Generally, the blood pool contains a higher concentration of gadolinium, has a shorter T1 relaxation time, and passes through the null point before myocardium. In many amyloid patients, this progression is reversed, with myocardial tissue reaching the null point before the blood pool (Fig. 63-5).
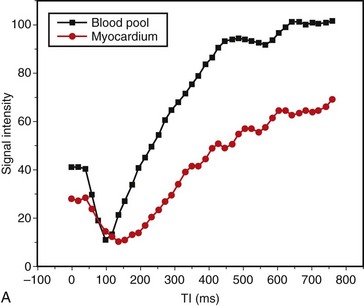
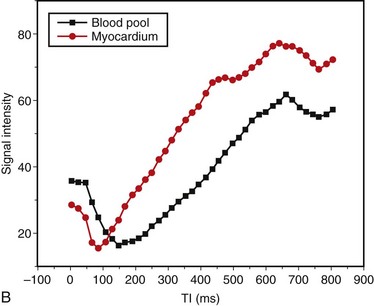
 FIGURE 63-5
FIGURE 63-5Nuclear Medicine
Nuclear medicine has generally played a minor role in the diagnosis and characterization of cardiac amyloidosis. Various tracers, mainly phosphonates, have been used to assess patients with suspected cardiac involvement, with heterogeneous results. More recent developments include the use of Tc 99m DPD scintigraphy to distinguish between AL and senile or transthyretin-related cardiac amyloidosis in patients with known cardiac amyloidosis.15
EOSINOPHILIC ENDOMYOCARDIAL DISEASE
Prevalence and Epidemiology
It is unclear whether these are two distinct diseases or different manifestations of the same underlying pathologic process. IHES occurs in temperate countries, has a more aggressive and rapidly progressive course, and is related to hypereosinophilia, whereas endomyocardial fibrosis occurs most commonly in equatorial Africa and is not definitely related to hypereosinophilia. Endomyocardial fibrosis occurs mainly in children and adolescents belonging to the poorest groups of the population and is an endemic cause of heart disease in certain tropical areas, such as the coastal regions of Mozambique, where 9% of the population is affected.16 The etiology of endomyocardial fibrosis is unclear; however, in most cases eosinophilia is seen in the initial inflammatory process, and histologically the endocardial lesions are similar to those seen in IHES. An alternative hypothesis has focused on an animal protein–deficient cassava diet, which has induced endomyocardial fibrosis in an animal model.
Manifestations of Disease
Imaging Techniques and Findings
Ultrasonography
Characteristic findings on echocardiography include apical obliteration of the involved ventricle with echogenic material, and thickening and adherence to the ventricular wall of the posterior atrioventricular valvular chordae tendineae cordis and adjacent papillary muscles, with enlargement of the corresponding atrium (Fig. 63-6).17 Doppler echocardiography may reveal typical findings of restriction, including shortened deceleration time and reduced isovolumetric relaxation time. Contrast-enhanced techniques may aid in visualizing the rim of hypoenhancing thrombus and subendocardial fibrotic tissue.
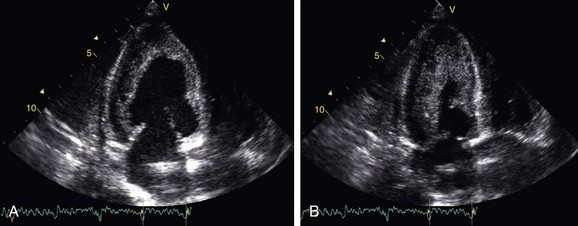
 FIGURE 63-6
FIGURE 63-6Computed Tomography
Contrast-enhanced CT may show hypoenhancing thrombus or fibrotic material along the endocardial surface of the myocardium, which should suggest the diagnosis (Fig. 63-7).18 Atrial enlargement may also be seen.
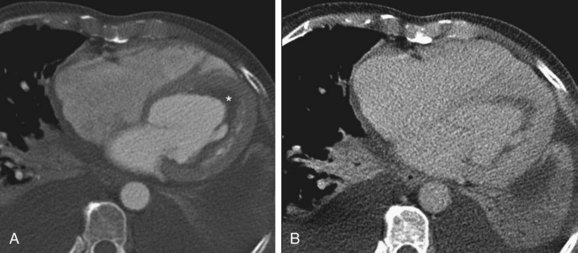
 FIGURE 63-7
FIGURE 63-7Magnetic Resonance Imaging
MRI findings of endomyocardial fibrosis typically include subendocardial thrombus and fibrosis, which can be visualized on SSFP, myocardial perfusion, and MDE pulse sequences. First-pass contrast-enhanced perfusion sequences typically reveal perfusion defects along the endocardial margin of the myocardium in the left ventricle and often right ventricle. Hyperenhancing fibrotic granulation tissue may surround a core of nonenhancing thrombus and can be seen on MDE imaging (Fig. 63-8).19–21
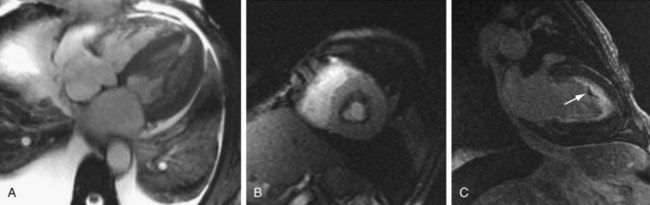
 FIGURE 63-8
FIGURE 63-8SIDEROTIC CARDIOMYOPATHY
Prevalence and Epidemiology
Cardiac failure as a result of transfusional iron overload is the most common cause of death in patients with thalassemia major, with more than 50% of these patients dying before age 35.23 Sickle cell and other hereditary anemias are also common causes of transfusional iron overload. Primary hemochromatosis is an autosomal recessive disorder affecting approximately 1 in 220 individuals and is an important cause of siderotic cardiomyopathy.24
Etiology and Pathophysiology
Iron overload occurs as a result of either excessive absorption or repeated transfusions. In hemochromatosis, iron is deposited in periportal hepatocytes and, in severe disease, the pancreas, heart, and endocrine organs. In thalassemia, iron overload results from overabsorption and transfusional siderosis. Transfusional iron is deposited in reticuloendothelial cells of the spleen, liver, and bone marrow. In severe cases, iron also accumulates in parenchymal cells of the liver, heart, and endocrine organs. At normal body iron levels, plasma iron is bound to transferrin, which prevents catalytic activity and free radical production. When transferrin is fully saturated, surplus iron appears as non–transferrin-bound iron (NTBI), and enters cells where it is stored as ferritin and hemosiderin. NTBI, probably by virtue of its more accessible unpaired electrons, promotes free radical formation and consequent damage to membrane lipids and proteins. As iron accumulates in the heart, there is little effect on function until a threshold is reached where the iron storage capacity is exhausted, and NTBI begins to appear. Excessive iron in myocytes may impair Na+,K+-ATPase function, reduce mitochondrial activity, and increase lysosomal fragility.25
Manifestations of Disease
Clinical Presentation
Initially, myocardial iron overload is asymptomatic, with a mild increase in LV wall thickness and mild LV dilation. Diastolic dysfunction usually precedes systolic abnormalities and is characterized by a restrictive filling pattern. Classic cardiac abnormalities of excess iron deposition include congestive heart failure and dysrhythmias. ECG abnormalities are present in 35% of symptomatic patients, including ventricular ectopies, supraventricular and ventricular tachycardias, ventricular fibrillation, and heart block.26
Imaging Techniques and Findings
Ultrasonography
More recent investigation has shown that assessment of pulmonary venous flow and TDI echocardiographic indices add useful information in assessing patients with hemochromatosis, and that alterations in these parameters may be seen before symptomatic involvement.27 TDI has also been used to assess thalassemic patients with siderotic cardiomyopathy. Wall motion abnormalities were significantly more common in patients with myocardial iron overload than in patients without overload, and may represent an early sign of cardiac disease in patients with preserved systolic function.28
Magnetic Resonance Imaging
Several techniques for measuring T2* are available. Probably the most commonly employed clinical method is an ECG gated multi-echo gradient-echo sequence, which acquires a series of images in the same location with progressively longer echo times (TE). The signal intensity of each image pixel or of a user-drawn region of interest can be plotted versus TE and the resulting curve can be fit to an exponential decay function and solved for T2* or R2*, which can be related to the tissue iron concentration on the basis of calibration curves constructed from animal models or from human biopsy data (Fig. 63-9). Wood and colleagues29 used an animal model to show that MRI measurements of T2 and T2* can quantify cardiac and hepatic iron concentrations.
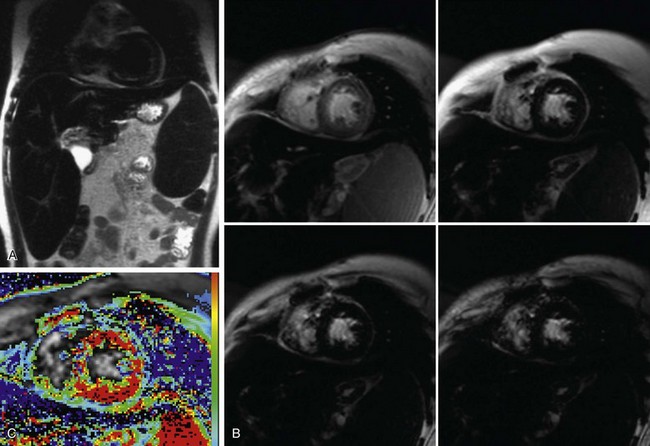
 FIGURE 63-9
FIGURE 63-9Measurement of myocardial T2* has been shown to have clinical utility. Anderson and associates30 showed a progressive decline in myocardial ejection fraction as T2* decreased in thalassemia patients, and found that all patients with ventricular dysfunction had a myocardial T2* less than 20 ms. Myocardial T2* measurements have also been used to follow reversal of siderotic cardiomyopathy with intravenous desferrioxamine.31
CARDIAC SARCOIDOSIS
Prevalence and Epidemiology
Sarcoidosis is common and affects individuals of both sexes, and almost all ages, races, and geographic locations. There is remarkable diversity in the prevalence of sarcoid among ethnic and racial groups, with a prevalence of 1 to 64 per 100,000 worldwide.32 The main organ systems targeted are the lungs and lymph nodes of the thorax. The estimated incidence of cardiac involvement is 4% to 5%, although autopsy series have found higher rates of 20% to 25%.33,34 In Japan, cardiac involvement is present in 58% of patients and is responsible for 85% of deaths from sarcoidosis.35
Manifestations of Disease
Clinical Presentation
Prognosis and disease course in sarcoidosis vary greatly. In many cases, granulomas resolve spontaneously. This is particularly true in patients presenting asymptomatically with hilar and mediastinal adenopathy, who are likely to experience spontaneous resolution within 2 years. Patients who are symptomatic at presentation are less likely, however, to experience spontaneous recovery. The prognosis with cardiac involvement is significantly worse, and it is estimated that 5% to 8% of these patients eventually die of their disease.36
Imaging Techniques and Findings
Radiography
Plain radiographs provide no information regarding cardiac sarcoidosis. The presence of typical signs of mediastinal and pulmonary sarcoidosis (bilateral hilar and right paratracheal adenopathy with or without interstitial pulmonary infiltrates) in the setting of cardiac symptoms consistent with sarcoidosis should suggest the diagnosis (Fig. 63-10A).
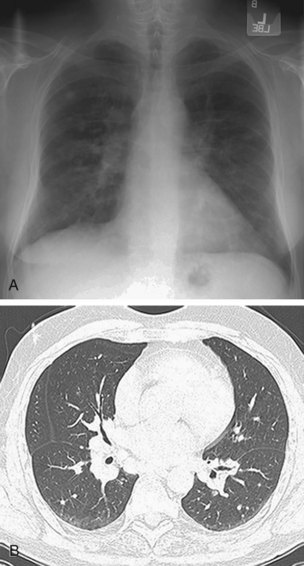
 FIGURE 63-10
FIGURE 63-10Ultrasonography
Abnormalities on echocardiography, including increased or decreased wall thickness, ventricular dilation, functional impairment, mitral regurgitation, impaired diastolic relaxation, and pericardial effusion, have been reported in 14% to 40% of patients with sarcoidosis. Echocardiography is usually the first examination performed when the diagnosis is suspected, and may show regional wall motion abnormalities and thickening of the interventricular septum, with bright echoes suggesting infiltration. Alternatively, the ventricles might appear thinned with global dysfunction and aneurysm formation. Diastolic dysfunction may be seen during the initial interstitial inflammatory stage when systolic function is still normal. RCM with dominant diastolic dysfunction may occur with extensive infiltration.37,38
Computed Tomography
CT plays a similar role to radiography; however, it is considerably more sensitive than plain radiographs for detecting mediastinal and hilar adenopathy (see Fig. 63-10B). CT is the test of choice for detecting pulmonary involvement in sarcoidosis, including nodules in a subpleural and bronchovascular distribution, pulmonary fibrosis, ground-glass opacities, bronchiectasis, and cystic changes. There is little evidence in the literature to suggest that CT provides information leading to a specific diagnosis of cardiac sarcoidosis.
Magnetic Resonance Imaging
MRI offers many advantages in imaging patients with suspected cardiac sarcoidosis. Acute myocardial inflammation resulting from sarcoid infiltration may be seen as regions of focal thickening with increased signal intensity on T2-weighted black blood images. Perfusion images or early T1-weighted postcontrast images may show increased contrast enhancement of affected myocardium, and MDE images may show epicardial patchy hyperenhancement, reflecting edema and myocardial injury. Focal wall motion abnormalities can be identified on cine SSFP images. Late changes include wall thinning and delayed hyperenhancement thought to reflect chronic scarring (Fig. 63-11). These changes may be difficult to distinguish from chronic infarction, although they tend to be in a noncoronary distribution and may spare the subendocardium. The appearance of cardiac sarcoidosis is very similar to that of myocarditis, and distinguishing between these two entities on the basis of MRI findings alone may be quite difficult.
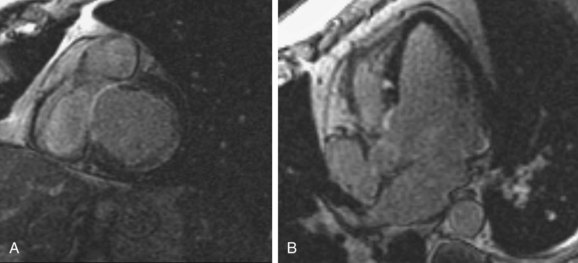
 FIGURE 63-11
FIGURE 63-11Several more recent studies have evaluated the efficacy of MRI in detecting cardiac sarcoidosis. Smedema and colleagues39 assessed 55 patients with pulmonary sarcoidosis who had evaluation for cardiac involvement consisting of ECG, echocardiography, thallium 201 scintigraphy, and MRI. MRI detected cardiac involvement in an additional six patients compared with the other techniques. The extent of delayed hyperenhancement correlated with disease duration, ventricular function, mitral regurgitation, and presence of ventricular tachycardia. Patel and coworkers40 assessed 58 sarcoidosis patients without cardiac symptoms and reported a twofold higher rate of cardiac involvement with gadolinium-enhanced MRI compared with evaluation with ECG and echocardiography.
Nuclear Medicine
Ohira and associates41 compared 18FDG-PET and MRI in assessing 21 patients with suspected cardiac sarcoidosis. According to the Japanese guidelines, 8 of 21 patients were diagnosed with cardiac sarcoidosis. PET had sensitivity and specificity of 88% and 38% versus 75% and 77% for MRI. The specificity of 18FDG-PET was lower in this study than in previous trials, and the authors speculated that some of the false-positive results might represent cases in which early subclinical involvement had been detected. 18FDG accumulates in cells with augmented glucose uptake, such as inflammatory cells or ischemic myocardial cells.
ADDITIONAL CAUSES OF RESTRICTIVE CARDIOMYOPATHY
Additional causes of RCM are as follows:
DIFFERENTIAL DIAGNOSIS
The absence of pericardial disease on imaging suggests RCM. The imaging findings in RCM may be subtle, and even when a diagnosis of RCM is made, it is often difficult to reach an exact diagnosis. Patients with amyloidosis are occasionally mistakenly thought to have hypertrophic cardiomyopathy, particularly when ventricular thickening is asymmetric, and there is systolic anterior motion of the mitral valve. Common imaging features discussed previously are listed in Table 63-1.
| Disease | Imaging Features |
|---|---|
| Cardiac amyloidosis | General: Ventricular thickening without dilation, dilated atria, pleural and pericardial effusions, diastolic dysfunction with preserved systolic function |
| Echocardiography: Granular sparkling myocardium | |
| MRI: Diffuse circumferential subendocardial enhancement with difficulties achieving suitable myocardial nulling on MDE imaging | |
| Eosinophilic endomyocardial disease | General: Apical subendocardial fibrosis and thrombus, atrial enlargement |
| Echocardiography and angiography: Apical obliteration | |
| MRI: Apical subendocardial hyperenhancement with nonenhancing thrombus on MDE | |
| Siderotic cardiomyopathy | General: Diffuse diastolic and systolic LV dysfunction |
| MRI: Decreased signal intensity on all MRI sequences, but most notably gradient-echo sequences; myocardial iron deposition can be measured using T2 and T2* imaging techniques | |
| Cardiac sarcoidosis | General: Mediastinal and hilar adenopathy, regional wall motion abnormalities with involvement of the basal septum |
| MRI: Subepicardial hyperenhancement on MDE | |
| Nuclear medicine: Reverse distribution (resting defects that disappear on stress images) on thallium and sestamibi scintigraphy, focal uptake in cardiac and extracardiac sites with gallium and PET |
TREATMENT OPTIONS
Medical
Sarcoidosis is unique among potential life-threatening disease because many patients do not require treatment. Approximately two thirds of patients experience spontaneous regression.42 Treatment of cardiac sarcoidosis usually involves corticosteroids, immunosuppressive therapy, or both, although no randomized controlled trials have substantiated their efficacy.
INFORMATION FOR THE REFERRING PHYSICIAN
Reports for the referring physician should emphasize the presence or absence of pericardial disease and features suggestive of RCM (see Table 63-1), highlighting the possible etiologies.





Dubrey SW, Bell A, Mittal TK. Sarcoid heart disease. Postgrad Med J. 2007;83:618-623.
Hassan W, Al-Sergani H, Mourad W, et al. Amyloid heart disease: new frontiers and insights in pathophysiology, diagnosis, and management. Tex Heart Inst J. 2005;32:178-184.
Shah KB, Inoue Y, Mehra MR. Amyloidosis and the heart. Arch Intern Med. 2006;166:1805-1813.
1 Richardson P, McKenna W, Bristow M, et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation. 1996;93:841-842.
2 Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol. 1995;32:45-59.
3 Cornwell GGIII, Murdoch WL, Kyle RA, et al. Frequency and distribution of senile cardiovascular amyloid: a clinicopathologic correlation. Am J Med. 1983;75:618-623.
4 Hassan W, Al-Sergani H, Mourad W, et al. Amyloid heart disease: new frontiers and insights in pathophysiology, diagnosis, and management. Tex Heart Inst J. 2005;32:178-184.
5 Roberts WC, Waller BF. Cardiac amyloidosis causing cardiac dysfunction: analysis of 54 necropsy patients. Am J Cardiol. 1983;52:137-146.
6 Klein AL, Hatle LK, Taliercio CP, et al. Prognostic significance of Doppler measures of diastolic function in cardiac amyloidosis: a Doppler echocardiography study. Circulation. 1991;83:808-816.
7 Cueto-Garcia L, Roeder GS, Kyle RA, et al. Echocardiographic findings in systemic amyloidosis: spectrum of cardiac involvement and relation to survival. J Am Coll Cardiol. 1985;6:737-743.
8 Ghio S, Perlini S, Palladini G, et al. Importance of the echocardiographic evaluation of right ventricular function in patients with AL amyloidosis. Eur J Heart Fail. 2007;9:808-813.
9 Koyama J, Ray-Sequin PA, Falk RH. Prognostic significance of ultrasound myocardial tissue characterization in patients with cardiac amyloidosis. Circulation. 2002;106:556-561.
10 Bellavia D, Abraham TP, Pellikka PA, et al. Detection of left ventricular systolic dysfunction in cardiac amyloidosis with strain rate echocardiography. J Am Soc Echocardiogr. 2007;20:1194-1202.
11 Celletti F, Fattori R, Napoli G, et al. Assessment of restrictive cardiomyopathy of amyloid or idiopathic etiology by magnetic resonance imaging. Am J Cardiol. 1999;83:798-801.
12 Maceira AM, Joshi J, Prasad SK, et al. Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation. 2005;111:186-193.
13 Krombach GA, Hahn C, Tomars M, et al. Cardiac amyloidosis: MR imaging findings and T1 quantification, comparison with control subjects. J Magn Reson Imaging. 2007;25:1283-1287.
14 Cheng ASH, Banning BP, Mitchell ARJ, et al. Cardiac changes in systemic amyloidosis: visualization by magnetic resonance imaging. Int J Cardiol. 2006;113:e21-e23.
15 Perugini E, Guidalotti PL, Salvi F, et al. Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. J Am Coll Cardiol. 2005;46:1076-1084.
16 Marijon E, Ou P. What do we know about endomyocardial fibrosis in children of Africa? Pediatr Cardiol. 2006;27:523-524.
17 Hassan WM, Fawzy ME, Al Helaly S, et al. Pitfalls in diagnosis and clinical, echocardiographic, and hemodynamic findings in endomyocardial fibrosis: a 25-year experience. Chest. 2005;128:3985-3992.
18 Salanitri GC. Endomyocardial fibrosis and intracardiac thrombus occurring in idiopathic hypereosinophilic syndrome. AJR Am J Roentgenol. 2005;184:1432-1433.
19 Syed IS, Martinez MW, Feng DL, et al. Cardiac magnetic resonance imaging of eosinophilic endomyocardial disease. Int J Cardiol. 2008;126:e50-e52.
20 Puvaneswary M, Joshua F, Ratnarajah S. Idiopathic hypereosinophilic syndrome: magnetic resonance imaging findings in endomyocardial fibrosis. Austr Radiol. 2001;45:524-527.
21 Alter P, Maisch B. Endomyocardial fibrosis in Churg-Strauss syndrome assessed by cardiac magnetic resonance imaging. Int J Cardiol. 2006;108:112-113.
22 Namboordiri KK, Bohora S. Images in cardiology: clenched fist appearance in endomyocardial fibrosis. Heart. 2006;92:720.
23 Modell B, Khan M, Darlison M. Survival in beta thalassaemia major in the UK: data from the UK thalassaemia register. Lancet. 2000;355:2051-2052.
24 Hanson EH, Imperatore G, Burke W. HFE gene and hereditary hemochromatosis: a HuGE review. Human Genome Epidemiology. Am J Epidemiol. 2001;154:193-206.
25 Hershko C, Link G, Cabantchik I. Pathophysiology of iron overload. Ann N Y Acad Sci. 1998;850:191-201.
26 Niederau C, Fischer R, Purschel A, et al. Long term survival in patients with hereditary haemochromatosis. Gastroenterology. 1996;110:1107.
27 Palka P, Macdonald G, Lange A, et al. The role of Doppler left ventricular filling indexes and Doppler tissue echocardiography in the assessment of cardiac involvement in hereditary hemochromatosis. J Am Soc Echocardiogr. 2002;15:884-890.
28 Vogel M, Anderson LJ, Holden S, et al. Tissue Doppler echocardiography in patients with thalassaemia detects early myocardial dysfunction related to myocardial iron overload. Eur Heart J. 2003;24:113-119.
29 Wood JC, Otto-Duessel M, Aguilar M, et al. Cardiac iron determines cardiac T2*, T2, and T1 in the gerbil model of iron cardiomyopathy. Circulation. 2005;112:535-543.
30 Anderson LJ, Holden S, Davis B, et al. Cardiovascular T2-star magnetic resonance for the early diagnosis of myocardial iron overload. Eur Heart J. 2001;22:2171-2179.
31 Anderson LJ, Westwood MA, Holden S, et al. Myocardial iron clearance during reversal of siderotic cardiomyopathy with intravenous desferrioxamine: a prospective study using T2* cardiovascular magnetic resonance. Br J Haematol. 2004;127:348-355.
32 Crystal RG. Sarcoidosis. In: Kasper DL, Braunwald E, Fauci AS, et al, editors. Harrison’s Principles of Internal Medicine. 16th ed. New York: McGraw-Hill; 2005:2017-2023.
33 Sharma O, Maheshawari A, Thaler K. Myocardial sarcoidosis. Chest. 1993;103:253-258.
34 Ratner SJ, Fenoglio JJJr, Ursell PC. Utility of endomyocardial biopsy in the diagnosis of cardiac sarcoidosis. Chest. 1986;90:528-533.
35 Iwai K, Sekiguti M, Hosoda Y, et al. Racial difference in cardiac sarcoidosis incidence observed at autopsy. Sarcoidosis. 1994;11:26-31.
36 Chestnutt AN. Enigmas in sarcoidosis. West J Med. 1995;162:519-526.
37 Lewin RF, Mor R, Spitzer S, et al. Echocardiographic evaluation of patients with systemic sarcoidosis. Am J Cardiol. 1989;63:478-482.
38 Burstow DJ, Tajik J, Baily KR, et al. Two-dimensional echocardiographic findings in systemic sarcoidosis. Am Heart J. 1985;110:116-122.
39 Smedema JP, Snoep G, van Kroonenburgh MPG, et al. Evaluation of the accuracy of gadolinium-enhanced cardiovascular magnetic resonance in the diagnosis of cardiac sarcoidosis. J Am Coll Cardiol. 2005;45:1683-1690.
40 Patel MR, Cawley PJ, Heitner JF, et al. Delayed enhanced MRI improves the ability to detect cardiac involvement in patients with sarcoidosis (abstract). Circulation. 2004;110(Suppl):2995.
41 Ohira H, Tsujino I, Ishimaru S, et al. Myocardial imaging with 18F FDG positron emission tomography and magnetic resonance imaging in sarcoidosis. Eur J Nucl Med Mol Imaging. 2008;35:933-941.
42 Dubrey SW, Bell A, Mittal TK. Sarcoid heart disease. Postgrad Med J. 2007;83:618-623.
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]
CHAPTER 63 Restrictive Cardiomyopathy
Restrictive cardiomyopathy (RCM) is the least common cardiomyopathy, and is characterized by diastolic dysfunction with restrictive ventricular filling with normal or near-normal systolic function and wall thickness.1 RCM may be idiopathic or associated with other infiltrative diseases, such as amyloidosis, endomyocardial disease, sarcoidosis, iron deposition disease, and storage diseases. Numerous other diseases may have a prominent restrictive component. Presentation of RCM is variable, and diagnosis is often difficult. The prognosis for most forms of RCM is poor, and it is important to distinguish RCM from constrictive pericarditis, which may have a similar clinical presentation
CARDIAC AMYLOIDOSIS
Prevalence and Epidemiology
Primary amyloidosis is a rare but devastating disease, with an incidence of 9 per 1 million and mean survival of approximately 13 months after diagnosis.2 Cardiac involvement in primary amyloidosis is common, with 60% of patients exhibiting ECG or echocardiographic abnormalities. Death is attributed to cardiac causes in at least 50% of patients with primary amyloidosis who die either from heart failure or from a malignant arrhythmia.2
Senile amyloidosis predominantly affects men older than 70 years and involves the heart in 25% of individuals older than 80 years.3 Senile cardiac amyloidosis is often clinically silent; however, extensive amyloid deposition can lead to significant clinical symptoms.
Etiology and Pathophysiology
Amyloid deposits, regardless of their protein composition, all have a characteristic appearance on light microscopy, staining pink with Congo red dye and exhibiting apple-green birefringence under polarized light microscopy. Nearly all organ systems can be involved, including the kidneys, heart, blood vessels, central and peripheral nervous system, liver, bowel, lungs, eyes, skin, and bones. Cardiac amyloidosis is a devastating progressive process that leads to congestive heart failure, angina, and arrhythmias.1
In addition to mechanical effects on myocardial stiffness, amyloid deposition induces oxidative stress that depresses myocyte contractile function. Myocardial ischemia may also result from microvascular disease. Amyloid deposits typically spare the epicardial vessels, whereas involvement of intramyocardial vasculature is seen in more than 90% of patients with AL amyloidosis.4
Manifestations of Disease
Imaging Techniques and Findings
Ultrasonography
Increased wall thickness without dilation of the ventricular cavity and preserved systolic function until relatively advanced stages of the disease are hallmarks of cardiac amyloidosis on echocardiography (Fig. 63-1). Amyloid deposits may involve nearly all regions of the heart, including valves, myocardium, interatrial septum, and pericardium, and manifestations of this involvement can be seen as multivalvular regurgitation, thickening of the interatrial septum, atrial dilation, pericardial effusions, and diffuse thickening of the right ventricular and left ventricular (LV) myocardium.5 A classic finding of myocardial amyloidosis is a granular sparkling pattern on two-dimensional echocardiography. This pattern is not specific for amyloidosis, however, and can be seen in patients with hypertensive cardiomyopathy, glycogen storage disorders, and hypertrophic cardiomyopathy. Atrial and ventricular thrombi are common findings, particularly in advanced disease.
Pulsed wave Doppler echocardiography is helpful in assessing diastolic dysfunction in cardiac amyloidosis. The initial diastolic abnormality is abnormal relaxation (grade 1 diastolic dysfunction) resulting from increased ventricular wall thickness; the pattern becomes restrictive (grades 3 to 4) when progressive amyloid infiltration decreases LV compliance and increases left atrial pressure. The filling pattern may normalize temporarily (pseudonormalization—grade 2 diastolic dysfunction) as a result of combined relaxation abnormality and moderate increase in left atrial filling pressure before becoming frankly restrictive. Deceleration time is an important prognostic variable in cardiac amyloidosis; the average survival for patients with a deceleration time less than 150 ms is less than 1 year versus 3 years for patients with a deceleration time greater than 150 ms.6 A combination of LV wall thickness greater than 15 mm and fractional shortening of less than 20% (thought to reflect combined systolic and diastolic dysfunction) is associated with a median survival of 4 months.7 Right ventricular function has also been correlated with poor prognosis in patients with cardiac amyloidosis.8
Tissue Doppler imaging (TDI) has emerged more recently as a useful technique in assessment of LV regional wall motion and diastolic dysfunction in patients with cardiac amyloidosis. Koyama and colleagues9 showed that TDI measurements differentiated patients without from patients with cardiac amyloidosis, and amyloidosis patients with and without heart failure. TDI more clearly documented diastolic function than conventional Doppler-derived indices. Myocardial strain and strain rate imaging have also been investigated in cardiac amyloidosis, and these techniques have documented early impairment in systolic function before the onset of clinical heart failure.10
Computed Tomography
ECG gated contrast-enhanced CT reveals many of the findings discussed previously with regard to echocardiography: ventricular thickening with preserved systolic function, enlarged atria, and pericardial and pleural effusions (Fig. 63-2). Little evidence currently available suggests that CT is useful in the initial diagnosis of cardiac amyloidosis or in distinguishing amyloidosis from other causes of RCM.
Magnetic Resonance Imaging
MRI has shown considerable promise in diagnosis and characterization of cardiac amyloidosis.11–14 Cine steady-state free precession (SSFP) images readily show findings of ventricular thickening with normal chamber size, atrial enlargement, and preserved systolic function (Fig. 63-3). Pleural and pericardial effusions are common and are well depicted on MRI. Impaired diastolic relaxation is often appreciated on cine SSFP images, and mitral inflow measurements can be obtained using cine phase contrast pulse sequences to obtain information analogous to Doppler echocardiography.
After administration of contrast medium, striking abnormalities are often seen on MDE pulse sequences, with patients with cardiac amyloidosis typically showing diffuse irregular hyperenhancement in noncoronary distributions (Fig. 63-4). A circumferential subendocardial hyperenhancement pattern has been described and correlated with predominant amyloid deposition in the subendocardial myocardium; however, in our experience, patterns of hyperenhancement are quite variable. Right ventricular late enhancement is a notable feature of amyloidosis and can help to distinguish this from hypertrophic cardiomyopathy with foci of enhancing fibrotic tissue.
Abnormalities of myocardial nulling are also common in amyloidosis and can help to distinguish this disease from other pathologies. A cine multi-TI inversion recovery sequence, in which each image or phase is acquired with a slightly longer inversion time (TI), is often used to select the optimal TI for the delayed enhancement acquisition. As TI increases, blood and myocardium pass through a null point at which signal is minimized. Generally, the blood pool contains a higher concentration of gadolinium, has a shorter T1 relaxation time, and passes through the null point before myocardium. In many amyloid patients, this progression is reversed, with myocardial tissue reaching the null point before the blood pool (Fig. 63-5).
Nuclear Medicine
Nuclear medicine has generally played a minor role in the diagnosis and characterization of cardiac amyloidosis. Various tracers, mainly phosphonates, have been used to assess patients with suspected cardiac involvement, with heterogeneous results. More recent developments include the use of Tc 99m DPD scintigraphy to distinguish between AL and senile or transthyretin-related cardiac amyloidosis in patients with known cardiac amyloidosis.15
EOSINOPHILIC ENDOMYOCARDIAL DISEASE
Prevalence and Epidemiology
It is unclear whether these are two distinct diseases or different manifestations of the same underlying pathologic process. IHES occurs in temperate countries, has a more aggressive and rapidly progressive course, and is related to hypereosinophilia, whereas endomyocardial fibrosis occurs most commonly in equatorial Africa and is not definitely related to hypereosinophilia. Endomyocardial fibrosis occurs mainly in children and adolescents belonging to the poorest groups of the population and is an endemic cause of heart disease in certain tropical areas, such as the coastal regions of Mozambique, where 9% of the population is affected.16 The etiology of endomyocardial fibrosis is unclear; however, in most cases eosinophilia is seen in the initial inflammatory process, and histologically the endocardial lesions are similar to those seen in IHES. An alternative hypothesis has focused on an animal protein–deficient cassava diet, which has induced endomyocardial fibrosis in an animal model.
