[level-membership-for-surgery-category]
Chapter 5 Reproductive Genetics
INTRODUCTION
We learn quickly in life that variability exists between individuals. For many of us the differences are as simple as hair or eye color. For others, the differences are profound and can take the form of a severe birth defect or syndrome. As a whole, these differences add up and cause significant morbidity and mortality. A Canadian study has found that approximately 12% of individuals suffer health problems related to or caused by genetic disease from birth to early adulthood.1 Genetics is the study of traits and inherited differences between individuals; as Gregor Mendel demonstrated, we were able to learn the principles of inheritance without knowing about or understanding DNA and its organization in the genome. The traditional study of medicine and medical genetics is, however, about to be revolutionized. In this technological revolution of genomic medicine, we see that a fundamental understanding of genetics and the principles of inheritance are still required.
The practice of clinical medical genetics has been significantly transformed over the past 15 years, from a somewhat arcane specialty dealing with prenatal diagnosis, chromosome abnormalities, and dysmorphology to a challenging and cutting edge specialty taking the lead in introducing genomic medicine to the broader field of medicine. In 2001, the Human Genome Project announced that a rough sequence of the human genome had been completed.2,3 In its original iteration the Human Genome Project was an attempt to sequence and determine the linear sequence of 3 billion base pairs and map the individual genes encoded by this sequence. Genomics is the study of our complete set of genes, their functions and interactions with themselves and their environment, and how genetic variation contributes to disease risk and response to treatment (i.e., pharmacogenomics). This sequencing and mapping effort has now spawned functional genomics, which seeks to identify gene function, regulation, and gene interaction.
The knowledge and technologies learned from this effort have, and will have, as profound an impact on the field of reproductive medicine as any other. The physician in obstetrics and gynecology will not only have to have an understanding of genetics and the principles of inheritance, but he or she will also have to understand genomics and its derivative technologies and how these will affect the risk, diagnosis, and treatment of medical disorders. Physicians will not only be practicing the traditional paradigm of diagnosis and treatment of disease, but will also be recognizing and treating genomically derived disease risk prior to disease manifestation. They must be ready to deal with all the ethical, legal, and social implications of this revolution. Although the era of genomic medicine will bring new tools to individualize disease risk, emphasize prevention, and treat disease, the simple basic recording and understanding of the family history will still be a fundamental component of this new era.4
THE HUMAN GENOME
The Human Genome Project has resulted in some surprising revelations.2,3,5 Less than 2% of the human genome has genes that code for proteins. This was a surprising finding because it was thought that the human genome would reflect the complexity of human development and it was estimated that the human genome would contain up to 120,000 genes. The first report of the Human Genome Project estimated that the human genome contained only 30,000 to 35,000 genes. Drosophila melanogaster had been found to have 14,000 genes and the mustard plant Arabidopsis thaliana 26,000 genes.6,7 It is likely now that the complexity of human growth and development and its relation to disease is not going to be explained by just new gene discovery. The previous dogma that one gene produces one protein has now been replaced by the theory of alternative splicing, in which a gene’s exons or coding regions are shuffled to produce alternative forms of a related protein (Fig. 5-1).
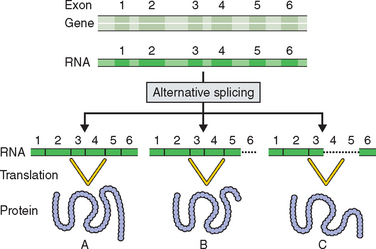
Exons are sequences of a gene that are transcribed into a protein. Introns are regions of a gene that do not code for a protein. When the mRNA of a gene is translated, exons are spliced together. Exons have been found to comprise only 1% of the human genome and introns about 25%. Typically more than 25 times the amount of DNA in a gene is not associated with protein structure and function. This has significant implications for DNA testing and interpretation of a test’s sensitivity and specificity. Regions of genes on chromosomes have been described as existing in gene-rich oases separated by gene-sparse deserts.
Genetic Variation
The human genome contains just over 3 billion base pairs and the sequence is about 99.9% identical in all individuals. It is a surprising finding that human genetic variability may be due to just 0.1% difference in the human genome sequence. A single-nucleotide polymorphism, or SNP, is the most common DNA variant in the genome, with about 10 million occurrences. SNPs are single-base substitutions and occur on average in 1 in every 1250 nucleotides. SNPs are polymorphisms in that a base substitution does not cause a change in the phenotype; they are found in more than 1% of the population. Because of their ubiquity in the human genome SNPs have become invaluable in identifying sequence changes associated with disease risk through traditional linkage studies and population-based association studies. Although not thought to alter protein coding, SNPs have been associated with diseases by their presence in regulatory control regions or introns. SNPs may also determine a disease’s response to treatment, as with the angiotensin-II type 1 receptor polymorphism and its association with congestive heart failure.8
CHROMOSOMAL BASIS OF INHERITANCE
Since Gregor Mendel’s work with the garden pea elucidating the mechanisms of inheritance, genetics has focused on single-gene defects. Modes of inheritance have now been identified for thousands of conditions and catalogued in the online compendium Online Mendelian Inheritance in Man, OMIM at http://www.ncbi.nlm.gov/omim/.
CHROMOSOME ABNORMALITIES
Chromosome Structure
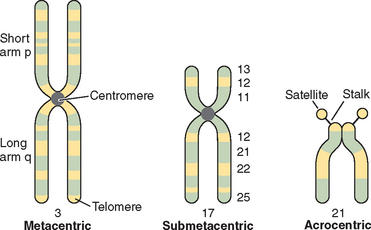
Chromosomes are composed of short arms, p, and long arms, q. The chromosome has a primary constriction, or centromere, where the microtubules attach for cell division. The telomeres, or tips of the chromosomes, are capped with a repeating sequence TTAGGG that is critical for the maintenance of chromosome integrity. The relative position of the centromere further delineates the structure of the chromosome. In humans the acrocentric chromosomes 13, 14, 15, 21, and 22 are characterized by stalks and satellites where genes for ribosomal RNA are located (Fig. 5-2).
Numerical Abnormalities
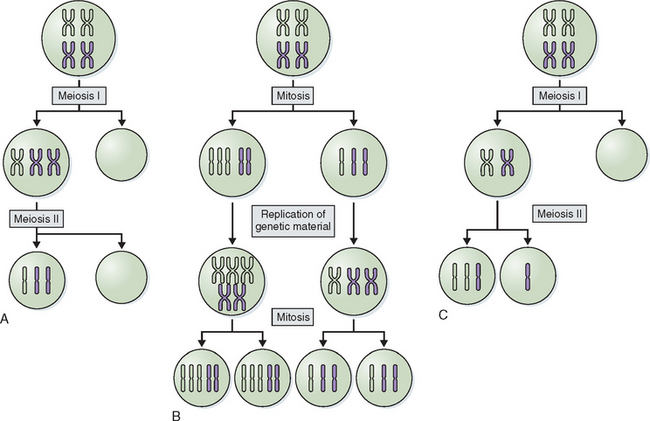
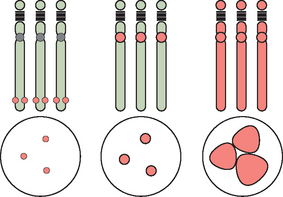
Abnormalities of chromosome number are the most commonly recognized clinical chromosome abnormalities. Structural chromosome anomalies contribute significantly to birth defects, infertility, and recurrent pregnancy loss. Abnormalities in chromosome number generally arise from mistakes at cell division where there is either gain or loss, or both, of chromosomes in daughter cells, or nondisjunction. Nondisjunction can occur either during meiosis or mitosis. Cells resulting from this occurrence are aneuploid because their chromosome number is not a multiple of the haploid number 23. Nondisjunction can occur during either meiosis I or meiosis II. Either can produce an aneuploid conception (Fig. 5-3). Nondisjunction or anaphase lag can occur at mitosis, causing mosaicism, the presence of two or more cell lines in an individual. Mosaicism is often seen with the sex chromosome abnormality Turner’s syndrome, where up to 50% of cases have some form of mosaicism.9 The common numerical chromosome abnormalities are listed in Table 5-1. Numerical chromosome abnormalities can also involve multiples of the haploid number of 23 chromosomes. Triploidy, with 69 chromosomes, usually arises from the fertilization of a single egg by two sperm. Triploid conceptions are seen in about 15% of chromosomally abnormal miscarriages and occasionally survive to term. Tetraploidy has a modal number of 92 chromosomes and occurs in a small percentage of spontaneous losses.
| Chromosome | Clinical Findings |
|---|---|
| Trisomy 13 | Severe CNS abnormalities, holoprosencephaly, microphthalmia, coloboma, cleft lip and palate, abnormal auricles, polydactyly, cardiac defects |
| Trisomy 18 | CNS malformations, prominent occiput, micrognathia, small mouth, low-set ears, hypoplastic nails, overlapping fingers, cardiac defects |
| Trisomy 21 | Microcephaly, flat occiput, Brushfield spots, epicanthal folds, simian crease, conotruncal cardiac defects, redundant skin on nape of neck, hypotonia |
| XXX | Normal female phenotype, tall stature, may have some learning and developmental disabilities, normal fertility |
| 45,X Turner’s | Short stature, gonadal failure, absent secondary sex characteristics, webbed neck, low-set hairline, coarctation of the aorta, horseshoe kidney, may have some spatial learning disabilities |
| XXY Klinefelter’s | May be tall, infertile, small testis, learning disabilities, gynecomastia |
Structural Chromosome Abnormalities
Structural chromosomal rearrangements occur when chromosomes break and the original architecture is not restored. Chromosome rearrangements are balanced when the diploid genetic state is maintained. When rearrangements are unbalanced, they result in aneuploidy for one or more chromosome segments. These structural rearrangements may segregate and be termed familial, or they may occur as a first or new event, de novo. Balanced familial chromosome rearrangements are in most cases truly balanced and represent little risk of birth defects or mental retardation. De novo chromosome rearrangements that appear balanced carry a small risk of aneuploidy at the molecular level of about 5% for birth defects and developmental delay.
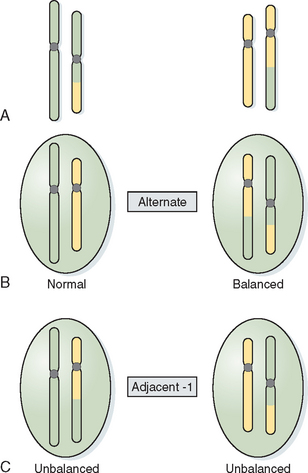
Translocations involve the exchange of chromosome arms between two different chromosomes. Reciprocal translocations occur when there is breakage within two arms and reciprocal exchange of the distal segments, creating a derivative chromosome (Fig. 5-4). In most cases balanced translocation carriers are phenotypically normal but are at risk for producing unbalanced gametes during gametogenesis. In a balanced translocation carrier the types of chromosome segregation can be complex, resulting in a normal segregation pattern, a balanced translocation pattern, and an unbalanced pattern producing a partial trisomy and partial monosomy for the chromosomes involved (see Fig. 5-4).
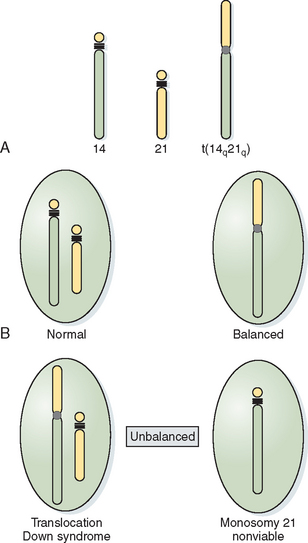
When the short arms of two acrocentric chromosomes are involved in a translocation, the long arms are joined in the centromeric region of one chromosome, with the loss of the short arms of the acrocentric chromosomes producing Robertsonian translocations. Because the short arms of acrocentric chromosomes contain redundant ribosomal genetic material, the loss of this material is of no phenotypic consequence. As with balanced translocations, the products of meiotic segregation can be either balanced or unbalanced (Fig. 5-5).
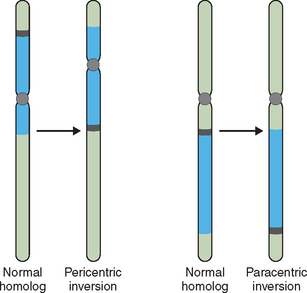
Other structural abnormalities can produce pregnancy loss and birth defects. When two breaks occur in a single chromosome with the interstitial segment flipped 180 degrees at the time of repair, an inversion can occur. If this involves each arm of the chromosome a pericentric inversion is produced. If only a single arm of the chromosome is involved in the inversion, a paracentric inversion is produced (Fig. 5-6). Each has unique and different implications for gamete production and pregnancy loss. With chromosome duplication a chromosome segment of varying size can be duplicated, causing a partial trisomy for this segment. With a deletion, a segment of varying size is missing, causing a genetic imbalance or partial monosomy. This condition, in which a second copy of a gene or segment of chromosome is missing, resulting in an abnormal phenotype or clinical presentation, is known as haploinsufficiency.
Fluorescent In Situ Hybridization
Traditional cytogenetics has always been limited by the band level or resolution of the karyotype. Even with high-resolution banding that could elongate chromosomes and resolve an increasing number of bands per chromosome, one could never be certain that a chromosome was intact at the molecular level. With advances in DNA technology and cytogenetics it is now possible using fluorescent in situ hybridization (FISH) to analyze chromosomes at the molecular level for changes in the DNA. Molecular cytogenetics has now revolutionized cytogenetics by permitting (1) the analysis of DNA structure within a chromosome down to within 10 to 100 kb and (2) the diagnostic analysis of nondividing interphase cells, producing a significant impact on the field of prenatal diagnosis and that of preimplantation genetic diagnosis.10
FISH technology uses DNA probes that can bind or anneal to specific DNA sequences within the chromosome. A denatured probe is incubated with native DNA from a cell that has also been denatured to the single-strand state. The probe substitutes biotin-dUTP or digoxigenin-UTP for thymidine. After the probe has annealed to native DNA, the probe–DNA complex can be detected by adding fluorochrome-tagged avidin that binds to biotin or fluorochrome-labeled antidigoxigenin. This signal can be additionally amplified by adding antiavidin and the complex visualized by fluorescence microscopy. Using several different fluorochromes tagged to different DNA probes, different chromosomes or chromosome segments can be simultaneously visualized within a cell as different colored signals. The ability to detect specific gene segments that are either present or missing has permitted the diagnosis of contiguous gene syndromes at the DNA level as well as translocations in interphase nuclei, often in single cells.
Applications of FISH
FISH technology has been developed in three forms. Centromeric or alpha-satellite probes are relatively chromosome specific and have had probably the broadest application in interphase genetics.11,12 These probes produce somewhat diffuse signals near the centromere with adequate strength, but do not cross-hybridize with chromosomes that have similar centromeric sequences. Single copy probes have now been developed that give discrete signals from a specific band on a chromosome and avoid the issue of cross-hybridization. These can also be used to detect copy number and specific chromosome regions known to be associated with syndromes. Single copy probes and centromeric probes for chromosomes 13, 18, 21, X, and Y have been developed for use in prenatal diagnosis. It is also possible to “paint” whole chromosomes using FISH. Using spectral karyotyping technology that combines mixtures of fluorochromes, it is now possible to produce a unique fluorescent pattern for each individual chromosome in 24 different colors. This technology permits the detection of complex chromosome rearrangements that cannot be seen with traditional cytogenetic techniques (Fig. 5-7).
Prenatal Diagnosis
For an older woman, pregnancy may not be a time of joy but a time of anxiety. Advanced maternal age has a long-known association with an increasing risk of fetal chromosome abnormalities. Amniocentesis done at 16 weeks’ gestation followed by traditional karyotype analysis may take from 10 days to 2 weeks. The use of FISH for preliminary results can expedite a diagnosis and reduce the waiting time for results. Most geneticists and laboratories recommend that FISH not be used alone to make a management decision in pregnancy. FISH studies should always be confirmed with a final karyotype or at minimum correlated with abnormal ultrasound findings or an abnormal maternal serum analyte screen.
Contiguous Gene Syndromes
Contiguous gene syndromes are also known as microdeletion syndromes, or segmental aneusomy.10 These are deletions of a contiguous stretch of chromosome that usually involve multiple genes. The contiguous gene syndromes were first described in 1986 using classical cytogenetic methodologies. Using FISH, submicroscopic deletions can now be identified at the level of DNA; this has permitted the characterization of the smallest deleted region consistently associated with a syndrome, known as the critical region. By identifying the critical region of a syndrome, it is often possible to identify the specific genes that, when missing, are associated with the syndrome (Fig. 5-8). A recent compendium of deletion syndromes has reported 18 deletion and microdeletion syndromes spread over 14 chromosomes.13 Some of the more common deletion and mirodeletion syndromes and their clinical findings are shown in Table 5-2.
| Deletion | Syndrome | Clinical Description |
|---|---|---|
| 4p– | Wolf-Hirschhorn | Microcephaly, hypertelorism, frontal bossing, cleft lip and palate, “Greek helmet” face, mental retardation, hypotonia, cardiac anomalies |
| 5p– | Cri du chat | Microcephaly, characteristic “cat cry,” hypotonia, mental retardation |
| 7q11.23 | Williams | Round face, full lips, stellate iris, supravalvular aortic stenosis, mental delay, “cocktail personality” |
| 11p13 | WAGR | Wilm’s tumor, aniridia, genital abnormalities, mental retardation |
| 15q11-13 | Angelman | Blonde hair, prognathism, seizures, ataxia, laughter, hypotonia, mental retardation |
| 15q11-13 | Prader-Willi | Birth hypotonia, hyperphagia, obesity, short stature, hypogonadism, mental retardation |
| 16p13 | Rubinstein-Taybi | Characteristic facies, beaked nose, microcephaly, mental retardation |
| 17p11 | Smith-Magenis | Brachycephaly, prominent chin, short stature, mental retardation, behavioral phenotype |
| 17p13 | Miller-Dieker | Microcephaly, lissencephaly, growth retardation, seizures, mental retardation |
| 20p12 | Alagille | Cholestasis, heart defects, ocular findings, skeletal defects |
| 22q11 | DiGeorge/CATCH 22 | Thymic and parathyroid hypoplasia, calcium abnormalities, conotruncal heart defects, short stature, behavioral and learning problems |
Telomeres
Telomeres are structures that cap the tips of the long and short arms of chromosomes. They are composed of repeating sequences of TTAGGG and effectively prevent the ends of the chromosomes from fusing together. Telomere probes can be important in sorting out complex translocations that cannot be determined by traditional cytogenetic means. In addition, one of the findings of the Human Genome Project was that the chromosome regions next to telomeres are gene-rich. It has now been shown that submicroscopic subtelomeric deletions are responsible for a significant proportion of genetic morbidity.14
GENERAL PRINCIPLES OF MENDELIAN GENETICS
Single-Gene Disorders
In humans as with all diploid organisms, genes come in pairs on related or homologous chromosomes. The exception to this is the sex chromosomes, the X and the Y in the male. Single-gene disorders are described on the basis of the interaction of these pairs or alleles in the genotype and on how the interaction is expressed in the effect or the phenotype. When the alleles are not identical, the genotypes are heterozygous. When the alleles are identical, the genotype is described as homozygous. Gregor Mendel recognized that single-gene disorders are often recognizable and that these single-gene disorders segregate in families with predictable proportions; they have thus been referred to as mendelian traits.15 If a single-gene disorder manifests itself in the heterozygous state, the condition is inherited in a dominant manner. If the single-gene disorder is only inherited when both alleles are affected or altered, the condition is inherited in a recessive fashion. In addition, single-gene disorders are further characterized according to whether they are transmitted on the autosomes or the sex chromosomes.
Autosomal Dominant Inheritance
The following features characterize autosomal dominant inheritance:
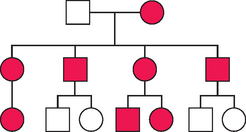
An example of an autosomal dominant pedigree is seen in Figure 5-9. Autosomal dominant inheritance patterns have additional characteristics that the clinician must recognize. Not all individuals in a family who have inherited a dominant condition are affected to the same extent or have the same organ systems affected. This characteristic is known as variable expressivity. This is one reason why it is important in clinical genetics to examine other family members when a proband, or referred patient, is initially evaluated. An autosomal dominant condition is considered fully penetrant if all individuals who inherit the single-gene mutation manifest its characteristics. Many autosomal dominant conditions have reduced penetrance, and not all individuals with the dominant gene manifest its phenotype. For women who inherit a BRCA1 or BRCA2 gene mutation, the lifetime risk of developing a breast cancer is 85%. Therefore, some families that are segregating BRCA1 and BRCA2 mutations will appear to “skip generations” when in fact an individual will carry a mutation but will not develop a breast cancer in her lifetime.
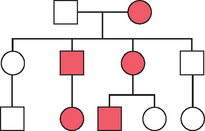
There are in addition other characteristics of autosomal dominant inheritance. Autosomal dominant conditions are rarely found in the homozygous condition and almost always result in embryonic lethality. Finally, autosomal dominant conditions will occur in families with no previous family history. This does not imply nonpaternity, which the clinician must be aware of. For example, new mutations occur in Marfan syndrome in about 25% of cases and about 80% of cases in achondroplasia.16 Parents of such an affected child are unlikely to be at risk for having another affected child; however, for the child, the risk of passing the condition on to an offspring is 50%. Geneticists have all been faced with the clinical scenario where a dominant, sporadic condition occurred in a family with a negative family history. Traditional genetic counseling would consider the recurrence risk negligible. However, most geneticists have seen these conditions recur in families, and empirically the recurrence risk has been as high as 3% to 5%. This phenomenon is usually attributed to germ cell mosaicism caused by a postzygotic nondisjunctional chromosome event or nondisjunctional mutational event. Presumably one of the parents in such a case has a small population of germ cells carrying the mutation. Recurrence risks must therefore be revised upward.
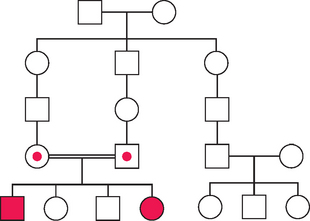
Autosomal Recessive Inheritance
An example of an autosomal recessive pedigree is shown in Figure 5-10. Autosomal recessive inheritance requires an affected individual to be homozygous; therefore, these traits tend to occur less frequently than dominantly inherited disorders. For some conditions, such as cystic fibrosis, in which many of the mutations causing the disorder are known, carrier status can be determined by DNA testing. In other conditions in which the gene product is known, the level of an enzyme, approximately 50% of normal, can determine carrier status.
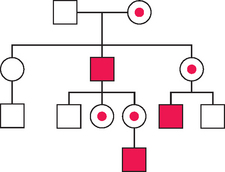
X-linked Inheritance
An example of X-linked recessive inheritance is shown in Figure 5-11. Complete androgen insensitivity (testicular feminization) syndrome is an X-linked recessive disorder that presents as primary amenorrhea. These patients have a 46,XY genotype with a mutation in the androgen receptor gene located on the long arm of the X chromosome (Xq11-12). Mutations of this gene can also cause partial androgen insensitivity with genital androgenization or a milder form with abnormalities of spermatogenesis. These patients have normal-appearing female phenotype but with sparse pubic and axillary hair, short vagina, no müllerian derivatives, and normal testes. The testes can be in the abdomen or inguinal canal. The testes are removed after completion of spontaneous puberty. The serum androgen levels are in the male range.
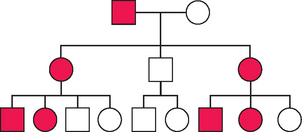
An example of X-linked dominant inheritance is shown in Figure 5-12.
NONTRADITIONAL INHERITANCE
Multifactorial Disorders
There are many birth defects, genetic conditions such as congenital heart disease and spina bifida, and adult onset disorders that cannot be explained by single genes or the traditional patterns of inheritance. Yet, many of these conditions are clearly familial. These conditions are considered to be the result of additive genetic and environmental factors that trigger a threshold for expression in development or adulthood. Multifactorial inheritance attributes disease risk to a combination of multiple genetic factors that interact with an environmental stimulus. Polygenic traits are those diseases attributable to the additive effect of several genes. Often, these two concepts are used interchangeably.
Evidence for multifactorial or familial inheritance has led to a threshold model of genetic liability. This model assumes a continuous normal distribution of genetic risk in the population in the form of a bell-shaped curve. Affected individuals are considered to fall to the far right of the genetic liability curve. Affected first-degree relatives (siblings, parents, and children) would also fall to the right of the curve, but not as far. As the degree of relatedness falls off, the genetic liability falls back to that of the general population.15 Evidence for multifactorial inheritance can be found in the following:
Mitochondrial Inheritance
Unlike nuclear genes, mtDNA follows a maternal line of transmission. Each oocyte contains from 200,000 to 300,000 mtDNA molecules. At fertilization the sperm have few mitochondria, located in the tails, and their contribution of mitochondria to the conception is negligible. An example of mitochondrial or maternal inheritance is shown in Figure 5-13. Both males and females can be affected, but inheritance only occurs from an affected mother. The phenotype of a mitochondrial disorder may be quite variable because of the nature of the mutation itself and its distribution within the population of mitochondria. At fertilization the oocyte contains thousands of mitochondria, both mutant and normal. At cell division, the proportion of mutant and normal mitochondria distributed to each daughter cell is never even. In addition, as the embryo grows new mutations accumulate in the mtDNA. Therefore, any cell or organ system may have proportionately few or many mutant mitochondria. This characteristic of mitochondrial inheritance is known as heteroplasmy. Within a single family there may be different manifestations of the same mitochondrial disease and within an individual the manifestations may change over time. As predicted with disorders of the oxidative phosphorylation system, those organ systems affected are those with the highest energy demands. Examples of mitochondrial disorders include Leber hereditary optic neuropathy, myoclonic epilepsy with red ragged fibers, and mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes.
Imprinting
Pronuclear transplantation studies in mice have shown a reversible effect of the sex of the parent on the embryo. Mice can be conceived with both haploid chromosome complements coming from either the father or the mother. Mouse zygotes with paternally derived genes have arrested embryonic development but near-normal placental development. Zygotes with maternally derived genes have embryonic development but arrested placental development. Thus it became apparent that specific genes are differentially marked, or imprinted, at gametogenesis depending on whether they are inherited from the mother or father. This process is known as genomic imprinting. An example of this has long been familiar to the obstetrician/gynecologist. Hydatidiform moles are placental tumors without embryonic tissue with a genome derived from two paternal sets of haploid chromosomes. Ovarian teratomas are embryonic tumors without placental tissue derived from two maternal sets of haploid chromosomes. Triploidy also demonstrates examples of genomic imprinting. Android embryos with two sets of paternal chromosomes and a single set of maternal chromosomes occur as partial moles. Gynoid embryos with two sets of maternal chromosomes and a single set of paternal chromosomes occur as growth-restricted fetuses with abnormal placentas. Imprinting is accomplished by the reversible process of methylation.
The role of imprinting in human genetic syndromes was revealed by a cytogenetic paradox. Prader-Willi syndrome is a syndrome of hypotonia at birth, moderate developmental delay, hypogonadotrophic hypogonadism, small hands and feet, uncontrolled appetite, and obesity. In most cases it is associated with a chromosome 15 deletion at 15q11-13. The phenotype of Angelman syndrome, quite different from that of Prader-Willi, is associated with severe mental retardation, inappropriate laughter, ataxic movements, and seizures. It is also associated with a deletion of 15q11-13. The dilemma of how two very distinct syndromes could be caused by the same cytogenetic deletion was solved when molecular studies showed that the deletion of Prader-Willi syndrome was always paternally derived and the deletion associated with Angelman syndrome was maternally derived.17 Individuals who inherit the 15q11-13 deletion paternally have Prader-Willi syndrome because a critical region that is needed for normal development is inactivated or imprinted during maternal meiosis and the corresponding region on the paternal chromosome, which must remain active for normal development, is lost in the deletion. Infants with Angelman syndrome have a maternally derived deletion because the critical region needed for normal development is inactivated or imprinted in the paternally derived chromosome and lost in the maternal deletion.
Uniparental Disomy
For a subset of patients with Prader-Willi syndrome and Angelman syndrome no cytogenetic deletion can be detected, nor were submicroscopic deletions detected by FISH. Molecular studies did however reveal that both copies of the chromosome were inherited from the same parent. For Prader-Willi syndrome without a deletion both copies of chromosome 15 were maternal in origin.18 For Prader-Willi syndrome the critical region of chromosome 15 is maternally imprinted, and if no paternally derived copy is present, there are no functioning paternal genes for this critical region. Uniparental inheritance of two copies of the same chromosome from one parent is termed uniparental disomy and has been shown to cause genetic morbidity and specific syndromes.
Genomic imprinting and uniparental disomy have been reported for a number of chromosomes, including 6, 7, 14, 15, and 16. The most common mechanism causing uniparental disomy is believed to be trisomic rescue (i.e., the loss of a chromosome after a trisomic conception that reestablished the diploid number of 46 chromosomes). On average, two thirds of the time the chromosome lost will be that from the parent who contributed the extra chromosome. However, one third of the time the chromosome lost will be from the other parent, resulting in uniparental genetic syndromes; it can also be a cause of more common genetic disease. If a parent is a carrier for a recessive genetic disorder and conceives an infant with uniparental disomy, the infant can be affected with the recessive condition if two identical copies of the same chromosome are inherited from the same parent. Several cases of cystic fibrosis have been reported in individuals with uniparental disomy for chromosome 7.19
Trinucleotide Repeat Disorders
Fragile X syndrome is one of the most common inherited mental retardation syndromes, occurring in about 1 in 12,000 males and 1 in 2500 females. Fragile X males also have behavioral problems and characteristic features, including a long face with prognathia, large ears, and macroorchidism. Individuals are often diagnosed or labeled with autism. Affected females have a similar but milder phenotype and are developmentally less delayed.20 Fragile X syndrome was originally identified as a fragile site on the X chromosome at Xq27.3 under cell culture conditions deficient in folate.
Fragile X syndrome is caused by expanded number of CGG repeats in the promoter region of the FMR-1 gene. The inheritance and its consequences can be complex, as shown in Table 5-3. Normal X chromosomes have between 6 and 54 CGG repeats. Individuals with 40 to 54 repeats are in a gray zone with the possibility of expansion. Males and females with between 55 and 230 repeats carry premutations; males are called transmitting males because they can pass the premutation on to daughters, who are then at risk of expanding the allele to their offspring. In general males only transmit premutations to their daughters. This change of normal CGG repeat number to full mutation is likely a multistep process occurring over several generations. There is now evidence in the literature to suggest that the premutation state in a male carrier may not be harmless. Reports have suggested that a neurologic syndrome can develop with cerebellar and parkinsonian features and decline of cognitive function.21 Although women who are carriers for the fragile X premutation are not cognitively affected, new concerns have also arisen over their long-term functioning. Premutation carriers have shown subtle changes in psychological and cognitive functioning. For women premutation carriers there is also a concern for premature menopause, with a 4% risk by age 30 and a 25% risk for ovarian failure by age 40.22 These authors also reported an imprinting effect for premature ovarian failure with paternally inherited premutations responsible for ovarian failure.

Two mechanisms have been proposed to cause the fragile X phenotype when the CGG repeats are expanded. The repeat sequence is located in the 5′ untranslated region of the gene. When this region expands beyond 230 copies of CGG it is hypermethylated and the FMR-1 allele with the expansion is inactivated.23 Not all fragile X chromosomes are methylated. It has also been proposed that the expanded CGG transcript is unstable, such that the mRNA is not transcribed, leading to an absence of the gene product.
GENETICS OF SEX DETERMINATION AND SEXUAL DIFFERENTIATION
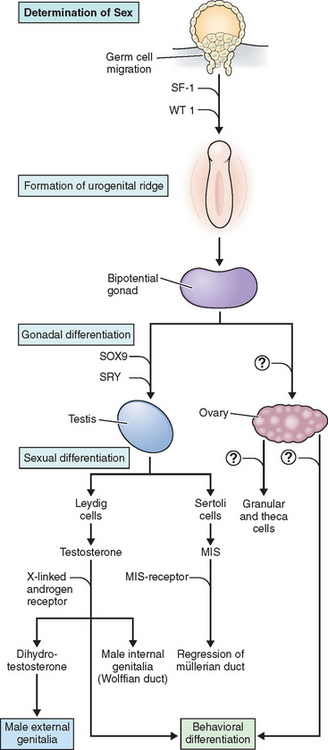
This development takes place on the background of a common, undifferentiated genital blueprint. The blueprint of the undifferentiated primordial genitalia includes paired gonads, paired internal ducts, a genital sinus, labial scrotal folds, and a genital tubercle. The default blueprint for gonadal and genital development is female. In the absence of a Y chromosome and SRY expression the primordial gonad develops into an ovary. The underlying molecular mechanisms for ovarian development are not as well understood as the molecular biology of testicular development. Abnormalities of sex determination and sexual differentiation appear to be determined mostly by mutations in male sexual determination in the background of a female blueprint.
The testis, differentiated under the influence of the SRY gene, secrete testosterone, müllerian inhibiting substance (MIS), and an insulin-like growth factor.24 Testosterone induces Wolffian duct differentiation into the vas deferens, epididymis, and seminal vesicles of the male reproductive tract. The MIS inhibits the differentiation of the müllerian ducts. In the absence of the SRY gene and the production of male hormones and in the presence of a female gonad, the müllerian ducts persist and differentiate into the fallopian tube, uterus, cervix, and the proximal portion of the vagina. In the presence of an SRY-determined testis secreting male hormones, the undifferentiated external genitalia develop into a penis and scrotum and male secondary sexual characteristics. In the absence of the SRY gene and the presence of female hormones secreted by the ovary, the undifferentiated external gonad develops into the distal vagina, vulva, clitoris, and female secondary sexual characteristics. This process is depicted in Figure 5-14.
Gene Disorders Causing Abnormal Gonadal Development
SRY
With the discovery of Klinefelter’s males having an 47,XXY karyotype and the description of Turner’s females as having a 45,X karyotype, it appeared that the presence of a Y chromosome and not the number of X chromosomes resulted in male differentiation in mammalian embryos. The gene on the Y chromosome responsible for male sex determination was named TDF, or testis-determining factor but was not easily identified. Because the X and the Y chromosome pair and segregate during meiosis just like the autosomes, it had been proposed that the X and Y chromosomes have autosomal-like regions in their distal chromosome arms, or pseudoautosomal regions allowing for pairing and segregation during meiosis. Magenis and coworkers in 1982 reported that 46,XX males have translocations of the Yp region to one of their Xp regions, demonstrating one mechanism for primary sex reversal.25 The occurrence of synapsis and crossovers between the Yp pseudoautosomal regions more proximal to the centromere resulted in the transfer of TDF to the X chromosome and resulted in the construction of deletion maps of the Y chromosome that led to the identification of SRY as the TDF.26,27 The discovery of mutations in the SRY gene in XY pure gonadal dysgenesis confirmed SRY as the candidate gene for TDF.28,29
WT1, Frasier, and Denys-Drash Syndromes
The genes that control the development of the early indifferent gonad have been more difficult to define. These genes may also be involved in renal development, and many potential regulatory pathways may be involved. WT1, or Wilms’ tumor gene, is a complex gene expressed in early intermediate mesoderm formation prior to urogenital development and when mutated can lead to malformation in renal and gonadal development. Streak gonads, renal abnormalities, and nephrotic syndrome characterize Frasier syndrome. It is caused by mutations in the WT1 gene. This gene codes for a DNA-binding transcription factor with 24 different isoforms due to alternative splicing. Alternative splice donor sites at the end of exon 9 lead to the insertion (+) or deletion (–) of three amino acids—lysine, threonine, and serine (KTS)—between the third and fourth zinc fingers of the transcription factor. XY patients with Frasier syndrome have loss of the +KTS isoform of WT1, causing decreased production of the SRY protein in the urogenital ridge and XY sex reversal. The phenotype is that of a female with gonadal dysgenesis with streak gonads, müllerian duct structures, and nephropathy.
Individuals with Denys-Drash syndrome are 46,XY with some testosterone production and MIS production. Müllerian duct regression occurs, as does partial testicular development. They have a male phenotype but may have hypospadias and undescended testis and male pseudohermaphroditism. These individuals are +KTS and have mutations outside the KTS region that are believed to affect gonads later in development, causing a less severe phenotype.30 These individuals are at risk for Wilms’ tumors and nephropathy that can lead to end-stage renal disease.
SF1
SF1, a steroidogenic factor, is a zinc-finger DNA-binding transcription factor involved with steroid biogenesis. It is expressed during development in the adrenals, gonads, and the pituitary. It appears to have a role in growth and maintenance of the indifferent gonad. SF1 mutations have now been reported with 46,XX sex reversal and adrenal insufficiency shortly after birth.31
DAX1 Gene Mutations and Duplications
DAX1 is an X-linked gene that is expressed in part of the developing embryo, specifically the hypothalamus, pituitary, adrenal, and the gonads.32 Alterations in DAX1 expression are associated with sex reversal and adrenal hypoplasia congenita. DAX1 is a nuclear receptor protein named for dosage-sensitive sex reversal, adrenal hypoplasia congenita, critical region on the X chromosome, and gene 1. DAX1 appears to be part of a class of genes that antagonize male development. Mutations or deletions of the DAX1 gene cause adrenal hypoplasia congenita. Duplications of this gene cause sex-reversed XY females. Mutations in DAX1 cause adrenal hypoplasia congenita with adrenal insufficiency; abnormal pituitary development causes hypogonadotropic hypogonadism.
Campomelic Dysplasia and SOX9
SOX9 was identified in 1994 as a downstream regulator of male sexual differentiation and was also implicated in skeletal development.33,34 The SOX9 gene is located on chromosome 17q and shares some homology with the SRY gene; hence its name SOX for Sex-related-box. It functions as a transcription factor and in humans is expressed in the testis, kidney, chrondrocytes, liver, and brain. SOX9 plays a significant dose-dependent role in male sex determination, as shown by haploinsufficiency for SOX9 causing either sex reversal or ambiguous sexual differentiation. Increased dosages of SOX9 from duplication have been shown to cause autosomal sex reversal in XX individuals.35 SOX9 also plays a role in MIS activation. In addition SOX9 plays a role in bone and cartilage development and may regulate COL2A1 expression, as seen in the skeletal abnormalities of campomelic dysplasia.
ABNORMALITIES OF THE FEMALE REPRODUCTIVE TRACT
Abnormalities of the female reproductive tract are thought to occur in up to 3% of live births.36 Abnormalities of the female reproductive tract affect the fallopian tubes, uterus, cervix, or vagina. The spectrum of abnormalities includes agenesis, atresia, and septations. With a female conception and the differentiation of the bipotential gonad into the ovary, the Wolffian ducts degenerate and the müllerian ducts differentiate into the female reproductive system. The molecular basis for female reproductive tract development in humans is not well described. More is known about the development of the female reproductive tract in mice through knockout studies. The process of development of the müllerian ducts involves apoptosis, or programmed cell death. This process is highly regulated and involves many genes, including the bcl-2 gene, which protects cells from apoptosis. If expression of this gene is absent at a critical time when cells of the fused ducts were to undergo resorption, the septum may persist. A number of human genetic syndromes have been described that affect female reproductive tract formation, differentiation, and regression.
Human genetic syndromes involving abnormalities of female reproductive tract formation include Kaufman-McKusick syndrome, Mayer-Rokitansky-Küster-Hauser syndrome, and maturity-onset diabetes of the young type V. Kaufman-McKusick syndrome is an autosomal recessive syndrome originally described in the Amish population with polydactyly, congenital heart disease, and hydrometrocolpos.37 Males can be affected but have only the hand and cardiac findings. The failure to cannalize the uterovaginal plate during embryogenesis produces a transverse vaginal septum, causing uterine distension and organ compression. These septa tend to occur in the upper one third of the vagina. This condition is related to Bardet-Biedl syndrome, which is made up of retinopathy, learning disabilities, obesity, polydactyly, renal anomalies, and male hypogonadism.38 The MKKS gene codes for a protein that functions as a chaperonin, a group of proteins that assist in protein folding as the protein is produced from the ribosomes.39 How mutations in the MKKS gene cause Kaufman-McKusick syndrome or the more variable Bardet-Biedl syndrome is not clearly understood.
Hand-foot-genital syndrome is an autosomal dominant disorder characterized by shortening of the thumbs and great toes, fifth finger clinodactyly, incomplete müllerian fusion with or without a double cervix and longitudinal vaginal septum, and renal anomalies. Males may have hypospadias. Hand-foot-genital syndrome is caused by mutations in the HOXA13 gene and is inherited as an autosomal dominant condition.40 Hand-foot-genital syndrome is caused by any mutation producing haploinsufficiency in the HOXA13 gene. HOXA13 is a member of the HOX group of transcription factors expressed in distal structures of the limb and the müllerian ducts. Its exact role in müllerian duct fusion and differentiation is unclear. Fryns’ syndrome is a multiple malformation syndrome consisting of cleft lip and palate, CNS abnormalities, renal tract malformations, cardiac defects, and müllerian fusion abnormalities. It is usually lethal and is inherited in an autosomal recessive manner.
GENETICS OF MALE INFERTILITY
Oligospermia (concentrations <5 million/mL) or azoospermia can be attributed to chromosome abnormalities in approximately 3% to13% of cases.41 The chromosome abnormalities include sex chromosome abnormalities such as 47,XXY, 47,XYY, sex chromosome mosaics, and structural Y chromosome abnormalities. Autosomal chromosome abnormalities include reciprocal translocations, Robertsonian translocations, inversions, and other structural abnormalities. The most common chromosomal abnormality associated with nonobstructive azoospermia is 47,XXY, Klinefelter’s syndrome. The most common gene disorders associated with congenital obstructive azoospermia are mutations of the cystic fibrosis transmembrane conductance regulator gene.
PREIMPLANTATION GENETIC DIAGNOSIS
For couples who have had a child die from a congenital genetic disorder or who have conceived and then terminated a wanted but affected pregnancy, the option of prenatal diagnosis and pregnancy termination is not attractive. These couples have welcomed an alternative to traditional prenatal diagnosis and pregnancy termination. Preimplantation genetic diagnosis (PGD) is a prepregnancy form of prenatal diagnosis that combines routine IVF with embryo biopsy to identify affected pregnancies before embryo transfer. PGD has been used in the clinical setting now for almost 15 years. It is now used in the prevention and treatment of three groups of genetic disorders.
First, it has been used in the prevention of single-gene recessive disorders when the parents are carriers and the recurrence risk is 25%. Cystic fibrosis was the first such condition to be identified by PGD.42 Individuals affected with autosomal dominant disorders have a recurrence risk of 50%. Examples of autosomal dominant disorders identified by PGD include Marfan syndrome and Huntington disease. Because PGD requires the analysis of DNA from single cells, polymerase chain reaction (PCR) has been the primary method used for the analysis. Most analyses have been done with nested multiplex and fluorescent multiplex PCR.
Preimplantation genetic diagnosis has also been used in the diagnosis and prevention of recurrent miscarriages when one partner is a reciprocal translocation or Robertsonian translocation carrier. Two techniques have been used for treating couples who are carriers. For women who are translocation carriers, the first polar body can be biopsied shortly after retrieval, prior to fertilization, and can be evaluated by whole chromosome FISH. Because this technique can only be used with female carriers, the more commonly used technique is that of embryo biopsy. For Robertsonian translocations two FISH probes are required, each located at the telomeric end of the chromosomes involved. For reciprocal translocations three probes with different reporter fluorochromes are required. Two probes must be for the centromeric region of each chromosome, and one probe must be at the telomeric end of one chromosome. Embryos with an unbalanced chromosome complement can be distinguished from those with a balanced complement, either normal or balanced carriers. Unfortunately studies have shown that embryos from translocation carriers have a high incidence of chromosome abnormalities and that pregnancy rates from these couples are low.43,44
There is a long-known relation between maternal age and chromosome abnormalities. It is also recognized that about 60% of pregnancies that miscarry have chromosome abnormalities. Therefore, it is not surprising that PGD has been used in combination with IVF when a couple is at risk of aneuploidy by virtue of advanced maternal age. IVF success rates may be improved when PGD and aneuploidy screening are used in women age 35 to 40.45 Aneuploidy screening has now become the most common indication for PGD.
Routine IVF protocols are used for ovarian stimulation and oocyte retrieval. Oocytes are fertilized in vitro or by ICSI and the embryo grown to the six-cell or eight-cell stage. The embryo is then held in culture by a pipette and the zona pellucida opened by either an acid solution or laser beam; a biopsy pipette is inserted to aspirate one or two blastomeres for genetic analysis. An advantage of embryo biopsy is that more than one cell can be biopsied and analyzed. An alternative to embryo biopsy is polar-body biopsy of an oocyte for carrier status. The group from Chicago has pioneered this technique and shown it to be effective in the identification of single-gene disorders.46
With an estimated 1 in 10 individuals infertile, assisted reproductive technologies (ART) are now responsible for up to 3% of annual births in some Western countries.47 Although ART has been considered safe, registries have been monitoring the children conceived through these technologies. A possible association between ART and Beckwith-Wiedemann syndrome has now been reported by three registries around the world.48 Caused by a defect located at chromosome 11p15, Beckwith-Wiedemann syndrome is characterized by organ overgrowth, macroglossia, and abdominal wall defects. These studies reported about a fourfold increased risk of Beckwith-Wiedemann syndrome with ART. An association between ART and Angelman syndrome has also been reported. Both of these syndromes are associated with imprinted contiguous gene clusters. About 10% of sporadic cases of Angelman syndrome and about 50% of sporadic Beckwith-Weidemann syndrome are due to epigenetic defects in imprinting. In all cases of Angelman syndrome reported and in 13 of 19 cases of Beckwith-Weidemann syndrome reported, there was loss of methylation of the maternal allele for these genes. This has raised speculation that imprinted genes may be at risk for loss of methylation during preimplantation and that ART may be responsible, whether due to ICSI or the culture conditions in vitro. These reports suggest the need for longitudinal studies of children conceived by ART and long-term evaluation of their development.
1 Baird P. Genetic disorders in children and young adults: A population study. Am J Hum Genet. 1988;42:677.
2 Lander E, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860-921.
3 Venter J, et al. The sequence of the human genome. Science. 2001;291:1304-1351.
4 Guttmacher AE, Collins FS, Carmona R. The family history—more important than ever. NEJM. 2004;351:2333-2336.
5 Subramanian G, et al. Implications of the human genome for understanding human biology and medicine. JAMA. 2001;286:2296-2307.
6 Liang F, et al. Gene index analysis of the human genome estimates approximately 120,000 genes. Nat Genet. 2000;25:239-240.
7 Dickson D. Gene estimate rises as US and UK discuss freedom of access. Nature. 1999;401:311.
8 Andersson B, Blange I, Sylven C. Angiotensin-II type 1 receptor gene polymorphism and long-term survival in patients with idiopathic congestive heart failure. Eur J Heart Fail. 1999;1:363-369.
9 Sybert VP, McCauley E. Turner’s syndrome. NEJM. 2004;351:1227-1238.
10 Clark BA, Schwartz S. Molecular cytogenetics. Reed G, Claireaus AE, Cockburn F, editors. Diseases of the Fetus and Newborn, 2nd ed., Vol. II. London: Chapman and Hall, 1995;1121-1129.
11 Maluelidis L. Individual interphase chromosome domains revealed by in situ hybridization. Hum Genet. 1985;71:288-293.
12 Pinkel D, Straume T, Gray J. Cytogenetic analysis using quantitative, high-sensitivity, fluorescence hybridization. Proc Natl Acad Sci USA. 1986;85:2934-2938.
13 Gardner R, Sutherland G. Chromosome abnormalities and genetic counseling. In: Motulsky A., et al, editors. Oxford Monographs on Medical Genetics. 3rd ed. Oxford: Oxford University Press; 2004:577.
14 Knight S, Flint J. Perfect endings: A review of subtelomeric probes and their use in clinical diagnosis. J Med Genet. 2000;37:401-409.
15 Dickerman L, Park V, Clark BA. Genetic aspects of perinatal disease and prenatal diagnosis. In: Fanaroff A, Martin R, editors. Neonatal-Perinatal Medicine: Diseases of the Fetus and Infant. 6th ed. New York: Mosby Year Book; 1997:26.
16 Harper P. Practical Genetic Counseling, 5th ed. Oxford: Butterworth Heinemann, 1988.
17 Knoll J, et al. Angelman and Prader-Willi syndromes share a common chromosome 15 deletion but differ in parental origin of deletion. Am J Med Genet. 1989;32:285.
18 Nicholls R, et al. Genetic imprinting suggested by maternal heterodisomy in nondeletion Prader-Willi syndrome. Nature. 1989;342:281-285.
19 Spence J, et al. Uniparental disomy as a mechasnism for human genetic disease. Am J Hum Genet. 1988;42:217-226.
20 Hagerman R, et al. Fragile X Syndrome: Diagnosis, Treatment, and Research. Baltimore: Johns Hopkins University Press, 1991.
21 Leehey M, et al. The fragile X premutation presenting as essential tremor. Arch Neurol. 2003;60:117-121.
22 Hundscheid R, et al. Imprinting effect in premature ovarian failure confined to paternally inherited fragile X premutations. Am J Hum Genet. 2000;66:413-418.
23 Verkerk A, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905-914.
24 Nef S, Parada L. Hormones in male sexual development. Genes Dev. 2000;14:3075-3086.
25 Magenis R, et al. Translocation (X;Y)(p22.33;p11.2) in XX males: Etiology of male phenotype. Hum Genet. 1982;62:271-276.
26 Vergnaud G, et al. A deletion map of the Y chromosome based on DNA hybridization. Am J Hum Genet. 1986;38:109-124.
27 Sinclair A, et al. A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature. 1990;346:240-244.
28 Berta P, et al. Genetic evidence equating SRY and the testis-determining factor. Nature. 1990;348:448-450.
29 Jager R, et al. A human XY female with a frame shift mutation in the candidate testis-determining gene SRY.. Nature. 1990;348:452-454.
30 Little M, et al. DNA binding capacity of the WT1 protein is abolished by Denys-Drash syndrome WT1 point mutations. Hum Mol Genet. 1995;4:351-358.
31 Achermann J, et al. A mutation in the gene encoding steroidogenic factor-1 causes XY sex reversal and adrenal failure in humans. Nat Genet. 1999;22:125-126.
32 McCabe E. Adrenal hypoplasias and aplasias. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Diseases. 8th ed. New York: McGraw-Hill; 2001:4263-4274.
33 Foster J, et al. Campomelic dysplasia and autosomal sex reversal caused by mutations in an SRY-related gene. Nature. 1994;372:525-530.
34 Wagner T, et al. Autosomal sex reversal and campomelic dysplasia are caused by mutations in and around the SRY-related gene SOX9.. Cell. 1994;79:1111-1120.
35 Huang B, et al. Autosomal XX sex reversal caused by duplication of SOX9.. Am J Med Genet. 1999;87:349-353.
36 Gidwani G, Falcone T. Congenital Malformations of the Female Genital Tract: Diagnosis and Management. Philadelphia: Lippincott Williams & Wilkins, 1999.
37 Kaufman R, Hartmann A, McAlister W. Family studies in congenital heart disease, II: A syndrome of hydrometrocolpos, postaxial polydactyly, and congenital heart disease. Birth Defects Orig Artic Ser. 1972;8:85-87.
38 Beals P, et al. New criteria for improved diagnosis of Bardet-Biedl syndrome: Results of a population survey. J Med Genet. 1999;36:4237-4246.
39 Stone D, et al. Genetic and physical mapping of the McKusick-Kaufman syndrome. Hum Mol Genet. 1998;7:475-481.
40 Mortlock D, Innis J. Mutation of HOXA13 in hand-foot-genital syndrome. Nat Genet. 1997;15:179-180.
41 Dohle G, et al. Genetic risk factors in infertile men with severe oligospermia and azoospermia. Hum Reprod. 2002;17:13-16.
42 Handyside A, et al. Birth of a normal girl after in vitro fertilization and preimplantation diagnositic testing for cystic fibrosis. NEJM. 1992;327:905-909.
43 Committee of the E.P.C.S. ESHRE Preimplantation Genetic Diagnosis Consortium: Data collection III. Hum Reprod. 2002;17:233-246.
44 Iwarsson E, et al. Highly abnormal cleavage divisions in preimplantation embryos from translocation carriers. Prenat Diagn. 2000;20:1038-1047.
45 Wilton L. Preimplantation genetic diagnosis for aneuploidy screening in early human embryos: A review. Prenat Diagn. 2002;22:312-318.
46 Rechitsky S, et al. Accuracy of preimplantation diagnosis of single-gene disorders by polar body analysis of oocytes. J Assist Reprod Genet. 1999;16:192-198.
47 Evers J. Female subfertility. Lancet. 2002;360:151-159.
48 Gosden R, et al. Rare congenital disorders, imprinted genes, and assisted reproductive technology. Lancet. 2003;361:1975-1977.
[/level-membership-for-surgery-category][not-level-membership-for-surgery-category]
Chapter 5 Reproductive Genetics
INTRODUCTION
We learn quickly in life that variability exists between individuals. For many of us the differences are as simple as hair or eye color. For others, the differences are profound and can take the form of a severe birth defect or syndrome. As a whole, these differences add up and cause significant morbidity and mortality. A Canadian study has found that approximately 12% of individuals suffer health problems related to or caused by genetic disease from birth to early adulthood.1 Genetics is the study of traits and inherited differences between individuals; as Gregor Mendel demonstrated, we were able to learn the principles of inheritance without knowing about or understanding DNA and its organization in the genome. The traditional study of medicine and medical genetics is, however, about to be revolutionized. In this technological revolution of genomic medicine, we see that a fundamental understanding of genetics and the principles of inheritance are still required.
The practice of clinical medical genetics has been significantly transformed over the past 15 years, from a somewhat arcane specialty dealing with prenatal diagnosis, chromosome abnormalities, and dysmorphology to a challenging and cutting edge specialty taking the lead in introducing genomic medicine to the broader field of medicine. In 2001, the Human Genome Project announced that a rough sequence of the human genome had been completed.2,3 In its original iteration the Human Genome Project was an attempt to sequence and determine the linear sequence of 3 billion base pairs and map the individual genes encoded by this sequence. Genomics is the study of our complete set of genes, their functions and interactions with themselves and their environment, and how genetic variation contributes to disease risk and response to treatment (i.e., pharmacogenomics). This sequencing and mapping effort has now spawned functional genomics, which seeks to identify gene function, regulation, and gene interaction.
The knowledge and technologies learned from this effort have, and will have, as profound an impact on the field of reproductive medicine as any other. The physician in obstetrics and gynecology will not only have to have an understanding of genetics and the principles of inheritance, but he or she will also have to understand genomics and its derivative technologies and how these will affect the risk, diagnosis, and treatment of medical disorders. Physicians will not only be practicing the traditional paradigm of diagnosis and treatment of disease, but will also be recognizing and treating genomically derived disease risk prior to disease manifestation. They must be ready to deal with all the ethical, legal, and social implications of this revolution. Although the era of genomic medicine will bring new tools to individualize disease risk, emphasize prevention, and treat disease, the simple basic recording and understanding of the family history will still be a fundamental component of this new era.4
THE HUMAN GENOME
The Human Genome Project has resulted in some surprising revelations.2,3,5 Less than 2% of the human genome has genes that code for proteins. This was a surprising finding because it was thought that the human genome would reflect the complexity of human development and it was estimated that the human genome would contain up to 120,000 genes. The first report of the Human Genome Project estimated that the human genome contained only 30,000 to 35,000 genes. Drosophila melanogaster had been found to have 14,000 genes and the mustard plant Arabidopsis thaliana 26,000 genes.6,7 It is likely now that the complexity of human growth and development and its relation to disease is not going to be explained by just new gene discovery. The previous dogma that one gene produces one protein has now been replaced by the theory of alternative splicing, in which a gene’s exons or coding regions are shuffled to produce alternative forms of a related protein (Fig. 5-1).
Exons are sequences of a gene that are transcribed into a protein. Introns are regions of a gene that do not code for a protein. When the mRNA of a gene is translated, exons are spliced together. Exons have been found to comprise only 1% of the human genome and introns about 25%. Typically more than 25 times the amount of DNA in a gene is not associated with protein structure and function. This has significant implications for DNA testing and interpretation of a test’s sensitivity and specificity. Regions of genes on chromosomes have been described as existing in gene-rich oases separated by gene-sparse deserts.
Genetic Variation
The human genome contains just over 3 billion base pairs and the sequence is about 99.9% identical in all individuals. It is a surprising finding that human genetic variability may be due to just 0.1% difference in the human genome sequence. A single-nucleotide polymorphism, or SNP, is the most common DNA variant in the genome, with about 10 million occurrences. SNPs are single-base substitutions and occur on average in 1 in every 1250 nucleotides. SNPs are polymorphisms in that a base substitution does not cause a change in the phenotype; they are found in more than 1% of the population. Because of their ubiquity in the human genome SNPs have become invaluable in identifying sequence changes associated with disease risk through traditional linkage studies and population-based association studies. Although not thought to alter protein coding, SNPs have been associated with diseases by their presence in regulatory control regions or introns. SNPs may also determine a disease’s response to treatment, as with the angiotensin-II type 1 receptor polymorphism and its association with congestive heart failure.8
CHROMOSOMAL BASIS OF INHERITANCE
Since Gregor Mendel’s work with the garden pea elucidating the mechanisms of inheritance, genetics has focused on single-gene defects. Modes of inheritance have now been identified for thousands of conditions and catalogued in the online compendium Online Mendelian Inheritance in Man, OMIM at http://www.ncbi.nlm.gov/omim/.
CHROMOSOME ABNORMALITIES
Chromosome Structure
Chromosomes are composed of short arms, p, and long arms, q. The chromosome has a primary constriction, or centromere, where the microtubules attach for cell division. The telomeres, or tips of the chromosomes, are capped with a repeating sequence TTAGGG that is critical for the maintenance of chromosome integrity. The relative position of the centromere further delineates the structure of the chromosome. In humans the acrocentric chromosomes 13, 14, 15, 21, and 22 are characterized by stalks and satellites where genes for ribosomal RNA are located (Fig. 5-2).
Numerical Abnormalities
Abnormalities of chromosome number are the most commonly recognized clinical chromosome abnormalities. Structural chromosome anomalies contribute significantly to birth defects, infertility, and recurrent pregnancy loss. Abnormalities in chromosome number generally arise from mistakes at cell division where there is either gain or loss, or both, of chromosomes in daughter cells, or nondisjunction. Nondisjunction can occur either during meiosis or mitosis. Cells resulting from this occurrence are aneuploid because their chromosome number is not a multiple of the haploid number 23. Nondisjunction can occur during either meiosis I or meiosis II. Either can produce an aneuploid conception (Fig. 5-3). Nondisjunction or anaphase lag can occur at mitosis, causing mosaicism, the presence of two or more cell lines in an individual. Mosaicism is often seen with the sex chromosome abnormality Turner’s syndrome, where up to 50% of cases have some form of mosaicism.9 The common numerical chromosome abnormalities are listed in Table 5-1. Numerical chromosome abnormalities can also involve multiples of the haploid number of 23 chromosomes. Triploidy, with 69 chromosomes, usually arises from the fertilization of a single egg by two sperm. Triploid conceptions are seen in about 15% of chromosomally abnormal miscarriages and occasionally survive to term. Tetraploidy has a modal number of 92 chromosomes and occurs in a small percentage of spontaneous losses.
| Chromosome | Clinical Findings |
|---|---|
| Trisomy 13 | Severe CNS abnormalities, holoprosencephaly, microphthalmia, coloboma, cleft lip and palate, abnormal auricles, polydactyly, cardiac defects |
| Trisomy 18 | CNS malformations, prominent occiput, micrognathia, small mouth, low-set ears, hypoplastic nails, overlapping fingers, cardiac defects |
| Trisomy 21 | Microcephaly, flat occiput, Brushfield spots, epicanthal folds, simian crease, conotruncal cardiac defects, redundant skin on nape of neck, hypotonia |
| XXX | Normal female phenotype, tall stature, may have some learning and developmental disabilities, normal fertility |
| 45,X Turner’s | Short stature, gonadal failure, absent secondary sex characteristics, webbed neck, low-set hairline, coarctation of the aorta, horseshoe kidney, may have some spatial learning disabilities |
| XXY Klinefelter’s | May be tall, infertile, small testis, learning disabilities, gynecomastia |
Structural Chromosome Abnormalities
Structural chromosomal rearrangements occur when chromosomes break and the original architecture is not restored. Chromosome rearrangements are balanced when the diploid genetic state is maintained. When rearrangements are unbalanced, they result in aneuploidy for one or more chromosome segments. These structural rearrangements may segregate and be termed familial, or they may occur as a first or new event, de novo. Balanced familial chromosome rearrangements are in most cases truly balanced and represent little risk of birth defects or mental retardation. De novo chromosome rearrangements that appear balanced carry a small risk of aneuploidy at the molecular level of about 5% for birth defects and developmental delay.
Translocations involve the exchange of chromosome arms between two different chromosomes. Reciprocal translocations occur when there is breakage within two arms and reciprocal exchange of the distal segments, creating a derivative chromosome (Fig. 5-4). In most cases balanced translocation carriers are phenotypically normal but are at risk for producing unbalanced gametes during gametogenesis. In a balanced translocation carrier the types of chromosome segregation can be complex, resulting in a normal segregation pattern, a balanced translocation pattern, and an unbalanced pattern producing a partial trisomy and partial monosomy for the chromosomes involved (see Fig. 5-4).
When the short arms of two acrocentric chromosomes are involved in a translocation, the long arms are joined in the centromeric region of one chromosome, with the loss of the short arms of the acrocentric chromosomes producing Robertsonian translocations. Because the short arms of acrocentric chromosomes contain redundant ribosomal genetic material, the loss of this material is of no phenotypic consequence. As with balanced translocations, the products of meiotic segregation can be either balanced or unbalanced (Fig. 5-5).
Other structural abnormalities can produce pregnancy loss and birth defects. When two breaks occur in a single chromosome with the interstitial segment flipped 180 degrees at the time of repair, an inversion can occur. If this involves each arm of the chromosome a pericentric inversion is produced. If only a single arm of the chromosome is involved in the inversion, a paracentric inversion is produced (Fig. 5-6). Each has unique and different implications for gamete production and pregnancy loss. With chromosome duplication a chromosome segment of varying size can be duplicated, causing a partial trisomy for this segment. With a deletion, a segment of varying size is missing, causing a genetic imbalance or partial monosomy. This condition, in which a second copy of a gene or segment of chromosome is missing, resulting in an abnormal phenotype or clinical presentation, is known as haploinsufficiency.
Fluorescent In Situ Hybridization
Traditional cytogenetics has always been limited by the band level or resolution of the karyotype. Even with high-resolution banding that could elongate chromosomes and resolve an increasing number of bands per chromosome, one could never be certain that a chromosome was intact at the molecular level. With advances in DNA technology and cytogenetics it is now possible using fluorescent in situ hybridization (FISH) to analyze chromosomes at the molecular level for changes in the DNA. Molecular cytogenetics has now revolutionized cytogenetics by permitting (1) the analysis of DNA structure within a chromosome down to within 10 to 100 kb and (2) the diagnostic analysis of nondividing interphase cells, producing a significant impact on the field of prenatal diagnosis and that of preimplantation genetic diagnosis.10
FISH technology uses DNA probes that can bind or anneal to specific DNA sequences within the chromosome. A denatured probe is incubated with native DNA from a cell that has also been denatured to the single-strand state. The probe substitutes biotin-dUTP or digoxigenin-UTP for thymidine. After the probe has annealed to native DNA, the probe–DNA complex can be detected by adding fluorochrome-tagged avidin that binds to biotin or fluorochrome-labeled antidigoxigenin. This signal can be additionally amplified by adding antiavidin and the complex visualized by fluorescence microscopy. Using several different fluorochromes tagged to different DNA probes, different chromosomes or chromosome segments can be simultaneously visualized within a cell as different colored signals. The ability to detect specific gene segments that are either present or missing has permitted the diagnosis of contiguous gene syndromes at the DNA level as well as translocations in interphase nuclei, often in single cells.
Applications of FISH
FISH technology has been developed in three forms. Centromeric or alpha-satellite probes are relatively chromosome specific and have had probably the broadest application in interphase genetics.11,12 These probes produce somewhat diffuse signals near the centromere with adequate strength, but do not cross-hybridize with chromosomes that have similar centromeric sequences. Single copy probes have now been developed that give discrete signals from a specific band on a chromosome and avoid the issue of cross-hybridization. These can also be used to detect copy number and specific chromosome regions known to be associated with syndromes. Single copy probes and centromeric probes for chromosomes 13, 18, 21, X, and Y have been developed for use in prenatal diagnosis. It is also possible to “paint” whole chromosomes using FISH. Using spectral karyotyping technology that combines mixtures of fluorochromes, it is now possible to produce a unique fluorescent pattern for each individual chromosome in 24 different colors. This technology permits the detection of complex chromosome rearrangements that cannot be seen with traditional cytogenetic techniques (Fig. 5-7).
Prenatal Diagnosis
For an older woman, pregnancy may not be a time of joy but a time of anxiety. Advanced maternal age has a long-known association with an increasing risk of fetal chromosome abnormalities. Amniocentesis done at 16 weeks’ gestation followed by traditional karyotype analysis may take from 10 days to 2 weeks. The use of FISH for preliminary results can expedite a diagnosis and reduce the waiting time for results. Most geneticists and laboratories recommend that FISH not be used alone to make a management decision in pregnancy. FISH studies should always be confirmed with a final karyotype or at minimum correlated with abnormal ultrasound findings or an abnormal maternal serum analyte screen.
Contiguous Gene Syndromes
Contiguous gene syndromes are also known as microdeletion syndromes, or segmental aneusomy.10 These are deletions of a contiguous stretch of chromosome that usually involve multiple genes. The contiguous gene syndromes were first described in 1986 using classical cytogenetic methodologies. Using FISH, submicroscopic deletions can now be identified at the level of DNA; this has permitted the characterization of the smallest deleted region consistently associated with a syndrome, known as the critical region. By identifying the critical region of a syndrome, it is often possible to identify the specific genes that, when missing, are associated with the syndrome (Fig. 5-8). A recent compendium of deletion syndromes has reported 18 deletion and microdeletion syndromes spread over 14 chromosomes.13 Some of the more common deletion and mirodeletion syndromes and their clinical findings are shown in Table 5-2.
| Deletion | Syndrome | Clinical Description |
|---|---|---|
| 4p– | Wolf-Hirschhorn | Microcephaly, hypertelorism, frontal bossing, cleft lip and palate, “Greek helmet” face, mental retardation, hypotonia, cardiac anomalies |
| 5p– | Cri du chat | Microcephaly, characteristic “cat cry,” hypotonia, mental retardation |
| 7q11.23 | Williams | Round face, full lips, stellate iris, supravalvular aortic stenosis, mental delay, “cocktail personality” |
| 11p13 | WAGR | Wilm’s tumor, aniridia, genital abnormalities, mental retardation |
| 15q11-13 | Angelman | Blonde hair, prognathism, seizures, ataxia, laughter, hypotonia, mental retardation |
| 15q11-13 | Prader-Willi | Birth hypotonia, hyperphagia, obesity, short stature, hypogonadism, mental retardation |
| 16p13 | Rubinstein-Taybi | Characteristic facies, beaked nose, microcephaly, mental retardation |
| 17p11 | Smith-Magenis | Brachycephaly, prominent chin, short stature, mental retardation, behavioral phenotype |
| 17p13 | Miller-Dieker | Microcephaly, lissencephaly, growth retardation, seizures, mental retardation |
| 20p12 | Alagille | Cholestasis, heart defects, ocular findings, skeletal defects |
| 22q11 | DiGeorge/CATCH 22 | Thymic and parathyroid hypoplasia, calcium abnormalities, conotruncal heart defects, short stature, behavioral and learning problems |
Telomeres
Telomeres are structures that cap the tips of the long and short arms of chromosomes. They are composed of repeating sequences of TTAGGG and effectively prevent the ends of the chromosomes from fusing together. Telomere probes can be important in sorting out complex translocations that cannot be determined by traditional cytogenetic means. In addition, one of the findings of the Human Genome Project was that the chromosome regions next to telomeres are gene-rich. It has now been shown that submicroscopic subtelomeric deletions are responsible for a significant proportion of genetic morbidity.14
GENERAL PRINCIPLES OF MENDELIAN GENETICS
Single-Gene Disorders
In humans as with all diploid organisms, genes come in pairs on related or homologous chromosomes. The exception to this is the sex chromosomes, the X and the Y in the male. Single-gene disorders are described on the basis of the interaction of these pairs or alleles in the genotype and on how the interaction is expressed in the effect or the phenotype. When the alleles are not identical, the genotypes are heterozygous. When the alleles are identical, the genotype is described as homozygous. Gregor Mendel recognized that single-gene disorders are often recognizable and that these single-gene disorders segregate in families with predictable proportions; they have thus been referred to as mendelian traits.15 If a single-gene disorder manifests itself in the heterozygous state, the condition is inherited in a dominant manner. If the single-gene disorder is only inherited when both alleles are affected or altered, the condition is inherited in a recessive fashion. In addition, single-gene disorders are further characterized according to whether they are transmitted on the autosomes or the sex chromosomes.
Autosomal Dominant Inheritance
The following features characterize autosomal dominant inheritance:
[/not-level-membership-for-surgery-category]
