[level-membership-for-pediatrics-category]Chapter 116
Renal Neoplasms
Nephroblastomatosis Complex
Overview and Pathophysiology: Nephrogenesis is completed by the 36th gestational week. A focus of fetal metanephric blastema or embryonal renal tissue that persists beyond 36 weeks gestation is called a nephrogenic rest. Multiple foci or diffuse nephrogenic rests are termed nephroblastomatosis. Nephrogenic rests are identified in about 1% of neonatal autopsies but usually are no longer found after 4 months of age. Malignant transformation of the fetal metanephric blastema may occur, along with development of a Wilms tumor (nephroblastoma), or nephroblastomatosis may resolve spontaneously.1–3
Nephrogenic rests can occur anywhere in the kidney, depending on when nephrogenesis is interrupted. These rests may be located in the renal lobe (intralobar) or in the cortex that envelops the renal lobe (perilobar). The two types of nephrogenic rests have different appearances, malignant potential, and associated genetic abnormalities. Nephrogenic rests also are classified according to histologic features of development; hyperplastic and neoplastic rests are thought to be active and to have malignant potential, whereas dormant or sclerosing nephrogenic rests are considered inactive.1–4
Intralobar nephrogenic rests are less common than perilobar nephrogenic rests and are more likely to degenerate into a Wilms tumor. Intralobar nephrogenic rests tend to be few and are located randomly in the renal lobe; they are seen in patients with sporadic aniridia, Drash syndrome (i.e., male pseudohermaphrodism and nephritis), and WAGR syndrome (i.e., Wilms tumor, aniridia, genital anomalies, and mental retardation). Patients with sporadic aniridia have a 30% to 40% risk of having a Wilms tumor develop, which represents the greatest likelihood of all of the genetic and syndromic abnormalities associated with nephroblastomatosis.1–4
Perilobar nephrogenic rests are multiple and are located at the corticomedullary junction or in the cortex. Also called diffuse perilobar nephrogenic rests or diffuse perilobar nephroblastomatosis, they are found in persons with hemihypertrophy and in persons with Beckwith-Wiedemann syndrome (i.e., macroglossia, macrosomia, and omphalocele), Perlman syndrome (i.e., fetal gigantism and multiple congenital anomalies), and trisomy 18 syndrome. Patients with hemihypertrophy and Beckwith-Wiedemann syndrome have about a 5% risk of having a Wilms tumor develop.1,3–5
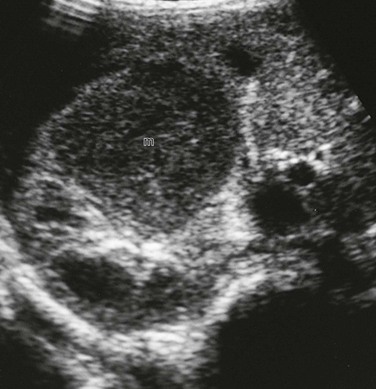
Imaging: Microscopic nephrogenic rests cannot be identified radiologically. Diffuse perilobar nephroblastomatosis and multifocal nephroblastomatosis can be evaluated through ultrasonography, computed tomography (CT), and magnetic resonance imaging (MRI). In diffuse perilobar nephroblastomatosis, the affected kidney may be enlarged. On sonography, corticomedullary differentiation is absent. Regions of nephroblastomatosis may be hypoechoic or isoechoic with respect to normal renal cortex (Fig. 116-1). Multifocal nephroblastomatosis is more difficult to identify on sonography.2,3,6
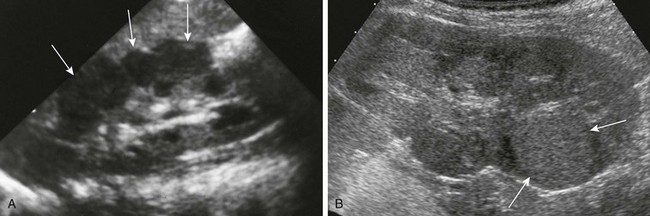
Figure 116-1 Nephroblastomatosis.
A, Diffuse perilobar form. A longitudinal sonogram shows an enlarged right kidney with lobulated contour and hypoechoic cortical masses (arrows). B, Focal intralobar form. A longitudinal sonogram shows a lower pole mass (arrows) that is isoechoic with renal cortex.
CT is more sensitive than ultrasound for the evaluation of nephroblastomatosis. On contrast-enhanced CT, areas of nephroblastomatosis are well defined because they enhance to a lesser extent than does normal renal cortex (Fig. 116-2). Bulky masses of nephroblastomatosis may distort the pelvicalyceal system. Involvement may be symmetric or asymmetric (e-Fig. 116-3). In diffuse perilobar nephroblastomatosis, a thick rind of lower attenuation tissue encases normally enhancing but architecturally distorted parenchyma. In persons with multifocal nephroblastomatosis, multiple round masses of low attenuation are present. Flat or plaquelike areas of involvement may be difficult to identify on CT.2,3,6
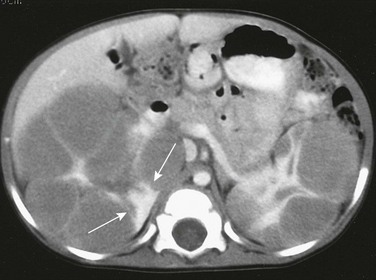
Figure 116-2 Diffuse perilobar nephroblastomatosis with symmetric involvement.
On a contrast-enhanced computed tomography scan, the kidneys are enlarged and multiple, round, peripheral masses of low attenuation are present. Architectural distortion at this level is pronounced; in the medial portion of the right kidney, only a peripheral area of normally enhancing kidney (arrows) is present.
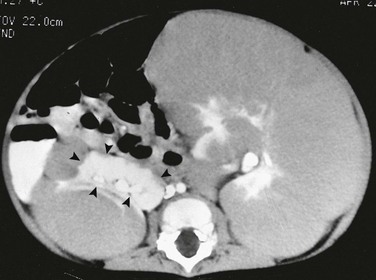
e-Figure 116-3 Diffuse perilobar nephroblastomatosis with asymmetric involvement.
On a contrast-enhanced computed tomography scan, the left kidney is much larger than the right. The architecture of the kidneys is distorted. A portion (arrowheads) of the normally enhancing right kidney consists of parenchyma and calyces.
On MRI, nephroblastomatosis cannot be distinguished from normal renal parenchyma on T1-weighted sequences and is isointense or hyperintense on T2-weighted sequences. Nephroblastomatosis appears hypointense relative to normal renal parenchyma after administration of contrast material on T1-weighted sequences, which are the most sensitive for detection. Active areas of nephroblastomatosis typically are hyperintense on T2-weighted sequences, and inactive areas are hypointense. The signal intensity of nephroblastomatosis is homogeneous, whereas foci of Wilms tumor tend to be heterogeneous.2,3,6–8
Treatment and Follow-up: Although the subject is somewhat controversial, presently no specific treatment is advocated for nephrogenic rests/nephroblastomatosis. Close radiologic follow-up is recommended for children with genetic abnormalities or syndromes associated with nephroblastomatosis. Patients should be monitored to detect Wilms tumor development, because the prognosis for Wilms tumor is best with small lesions. Children with hemihypertrophy or Beckwith-Wiedemann syndrome are at risk for the development of other embryonal tumors, such as hepatoblastoma and adrenal cell carcinoma. No large studies have been performed to establish the optimal screening interval for Wilms tumor surveillance; however, large tumors with metastases that develop within a 6-month period are reported. A baseline CT of nephroblastomatosis-related genetic abnormalities or syndromes at 6 months of age (or at diagnosis if the patient is older than 6 months), followed by ultrasound examinations every 3 to 4 months until the child is 8 years of age, is recommended on the basis of results from the National Wilms Tumor Studies. An area of nephroblastomatosis that grows larger and rounder is suspicious for malignant degeneration.2,3,9–13
Wilms Tumor
Overview, Pathophysiology, and Staging: Wilms tumor is the most common abdominal malignancy of childhood and accounts for 87% of renal masses. Its peak incidence is at 3 to 4 years of age (80% occur in children <5 years), but it has been described in the fetus, neonate, teenager, and adult. Clinical presentation includes a palpable mass, abdominal pain, hematuria, and occasional hypertension (from tumor renin production). As discussed in the Nephroblastomatosis Complex section of this chapter, certain syndromes and genetic abnormalities predispose to development of a Wilms tumor (e-Fig. 116-4). Two loci on chromosome 11 have been implicated in the genesis of Wilms tumors: 11p13 (WT1 gene—WAGR or Drash syndrome) and 11p15 (WT2 gene—Beckwith-Wiedemann syndrome or hemihypertrophy). Bilateral Wilms tumors occur almost exclusively in patients with nephroblastomatosis. Most Wilms tumors arise from the renal parenchyma; however, extrarenal Wilms tumors rarely may develop in the abdomen or at distant sites.1,14–17
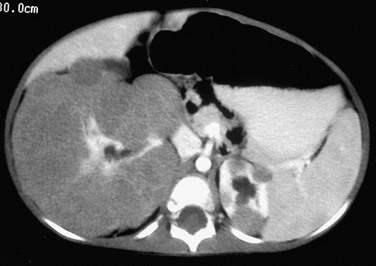
e-Figure 116-4 Diffuse perilobar nephroblastomatosis and a right Wilms tumor.
A contrast-enhanced computed tomography scan shows round, peripheral masses that enhance to a lesser extent than normal renal parenchyma in the upper pole of the left kidney. The right kidney has a thick rind of lobulated, hypoattenuating tissue without a discreet mass. A biopsy showed Wilms tumor on the right, and a nephrectomy was performed.
In the United States, tumor stage (Box 116-1) is determined operatively, and the grade is established on pathologic examination. The classic triphasic Wilms tumor arises from mesodermal precursors of the renal parenchyma (metanephros) and contains blastemal, stromal, and epithelial elements. The “teratoid Wilms tumor” contains tissue not normally found in the kidney (e.g., bone, cartilage, and muscle). Tumors with a favorable histology do not contain any anaplastic changes. The prognosis for tumors with a favorable histology is excellent, even for those at higher stages. In Europe, the staging system is based completely on radiologic findings. Tumors are classified on the basis of their imaging appearance, and chemotherapy is given before definitive surgery is performed. Tumors with extension into the inferior vena cava or invasion through the renal capsule are easier to resect after tumor shrinkage produced by chemotherapy.1,2,18
Imaging: Radiologic evaluation of Wilms tumor is focused on identifying the site(s) of involvement, extension, and metastases to assist in surgical planning. Preoperative imaging may include conventional chest radiography, abdominal and pelvic sonography, and thoracic, abdominal, and pelvic CT, MRI, and potentially fluorine deoxyglucose positron emission tomography (FDG-PET) fused with CT (PET-CT).1,2,19,20
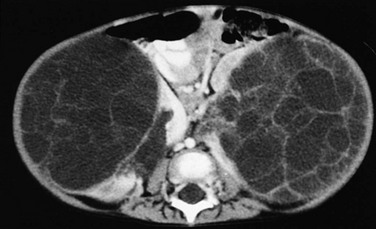
On sonography, a Wilms tumor is an intrarenal mass of heterogeneous echogenicity. Some Wilms tumors may contain cystic components, portions of obstructed and entrapped pelvicalyceal systems, or hemorrhagic and necrotic tumor. Extension into the inferior vena cava and the right atrium are characteristic routes of tumor growth and can be well visualized on ultrasound (e-Fig. 116-5), CT (Fig. 116-6), and MRI. The tumor typically forms a pseudocapsule but may invade the renal capsule, seed the peritoneal space, or grow directly into the mesentery and omentum. Hepatic metastases also are possible. The contralateral kidney may contain a smaller Wilms tumor or nephroblastomatosis. Synchronous or metachronous bilateral Wilms tumor may occur in up to 10% of patients.1,2,6
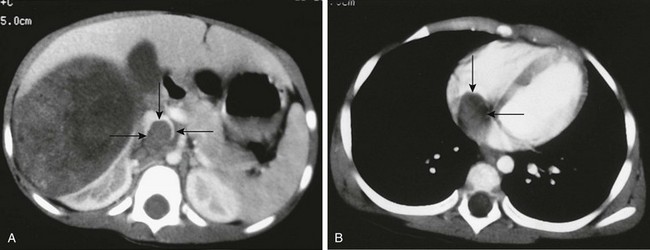
Figure 116-6 A Wilms tumor extending into the inferior vena cava and right atrium.
A, A contrast-enhanced computed tomography scan reveals a large, round, heterogeneous mass in the right kidney that enhances to a lesser extent than normal renal parenchyma. Contrast material opacifies the lateral and anterior margins of the inferior vena cava that contain hypoattenuating tumor (arrows). B, The tumor (arrows) extends into the right atrium.
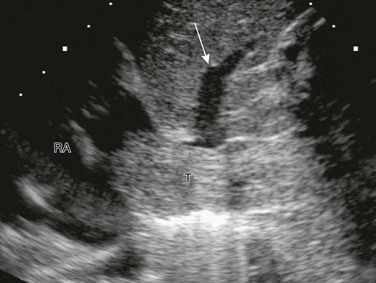
e-Figure 116-5 A Wilms tumor extending into the inferior vena cava and the right atrium (RA).
A longitudinal sonogram shows tumor thrombus (T) expanding the inferior vena cava and extending into the RA. The tumor partially occludes drainage from one of the hepatic veins (arrow).
On CT, Wilms tumor is generally spherical and intrarenal. It may contain small amounts of fat or fine calcification (Fig. 116-7 and e-Fig. 116-8) because the metanephric blastema cell is a pluripotential embryonal cell. Dystrophic calcifications are seen in about 9% of Wilms tumors. The tumor enhances to a lesser extent than does renal parenchyma. In patients who have been screened ultrasonographically because of genetic or syndromic conditions, the tumor is usually smaller than 4 cm in diameter. Children who are evaluated because of physical examination abnormalities generally have tumors larger than 10 cm in diameter. CT and conventional radiographs of the chest are used to identify pulmonary metastases.1,2,6,21,22
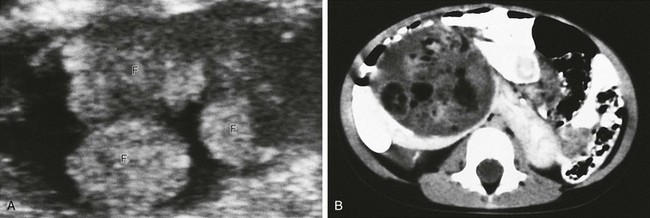
Figure 116-7 A Wilms tumor containing fat in a horseshoe kidney.
A, An axial sonogram of part of the Wilms tumor reveals hyperechoic lobules containing fat (F). B, A contrast-enhanced computed tomography scan shows areas of fat within the mass. The isthmus of the renal parenchyma extends across the midline.
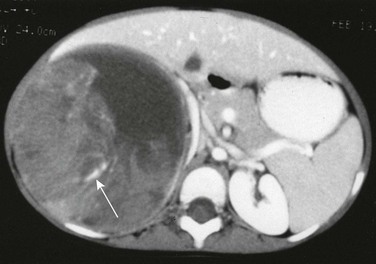
e-Figure 116-8 A Wilms tumor containing calcification.
A contrast-enhanced computed tomography scan reveals fluid-attenuating material anteromedially, most likely hemorrhage, within the large heterogeneous mass. Linear calcification is present posteriorly (arrow).
On MRI, a Wilms tumor is isointense with respect to normal renal parenchyma on T1-weighted sequences and hyperintense with respect to normal renal parenchyma on T2-weighted sequences. After administration of contrast material, a Wilms tumor is hypointense relative to normal renal parenchyma and has inhomogeneous signal intensity (Fig. 116-9). An effectively treated Wilms tumor may be hypointense on T2-weighted sequences.1,2,7,8,23
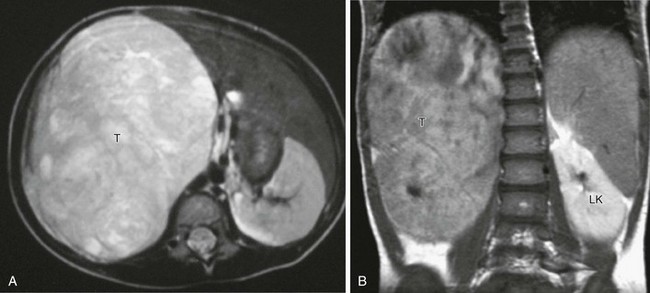
Figure 116-9 A Wilms tumor.
A, Axial T2-weighted magnetic resonance imaging (MRI) shows a large tumor (T) replacing the right kidney, which is hyperintense to other soft tissue structures. B, Coronal T1-weighted MRI after gadolinium in the same patient shows that the large tumor (T) is hypointense relative to the normally enhancing left kidney (LK).
FDG-PET fused with CT is a rapidly developing modality for imaging accelerated metabolism in malignant tissue. FDG-PET has better accuracy than MRI and bone scanning for staging. Wilms tumors are presumed to be FDG-avid, but the role of FDG in Wilms evaluation is as yet unclear. FDG-PET can help with a targeted biopsy of a viable Wilms tumor and biologically aggressive elements (e.g., anaplastic Wilms). Sensitivity for lung metastases is dependent on nodule size and respiratory motion. Treatment response of the primary tumor can be monitored. Potential benefits of FDG-PET will require ongoing investigation.2
Treatment: Surgery for a Wilms tumor typically begins with a complete abdominal exploration before attention is given to the kidneys. The unaffected kidney initially is visualized and palpated to assess for masses or areas of superficial nephroblastomatosis before en bloc resection of the affected kidney is performed. An excisional biopsy of lung nodules is performed to ensure that the stage of the tumor is correctly assigned. Patients with bilateral disease undergo staging surgery as well. Results of the surgical staging and the histologic examination determine the selection of therapy. In patients with bilateral disease, treatment is provided according to the tumor histology of the higher stage side, along with later nephron-sparing surgery. When the tumor is very large or when the tumor extends into the right atrium via the inferior vena cava, the surgeon may defer surgery until several courses of chemotherapy have reduced the tumor size or extension. Substantial progress in therapy for persons with a Wilms tumor over recent decades has resulted in a greater than 90% long-term survival for localized disease and greater than 70% survival for metastatic disease.1,2,24–30
Clear Cell Sarcoma of the Kidney
Overview: Clear cell sarcoma of the kidney accounts for approximately 5% of primary renal tumors in childhood and was considered a sarcomatous subtype of Wilms tumor before 1978. It was reclassified as a separate entity from Wilms tumor because of the tumor’s histologically and biologically unique features. No familial or syndrome association has been identified. Clear cell sarcoma occurs in an age group similar to that affected by Wilms tumor (1 to 4 years of age), and has a male predominance. Immunohistochemical staining has shown no characteristic marker pattern, but negativity to the WT1 gene is important.
Imaging and Treatment: Clear cell sarcoma imaging features are not distinct from those of the Wilms tumor (Fig. 116-10). Clear cell sarcoma has a predilection for bone metastases (formerly known as the bone metastasizing renal tumor of childhood) that may occur at presentation or upon relapse. A pathologic diagnosis of clear cell sarcoma necessitates an evaluation of the skeletal system. Bone scintigraphy or a conventional radiographic skeletal survey may be used. However, metastases to lymph nodes, lung, and liver still are seen more frequently than bone metastases. Because of the aggressive behavior of clear cell sarcoma, it is associated with a higher rate or relapse and mortality than is Wilms tumor, with a reported long-term survival of 60% to 70%. Treatment consists of nephrectomy and aggressive chemotherapy.1,31–38
Rhabdoid Tumor of the Kidney
Overview: Rhabdoid tumor of the kidney accounts for 2% of pediatric renal malignancies and was considered a sarcomatous variant of Wilms tumor before 1978, when it was reclassified as a separate entity. The name is derived from its monomorphous histologic appearance resembling that of skeletal muscle, although a myogenic origin has not been proved. Rhabdoid tumors occur in a younger age group than do Wilms tumors (80% occur in children younger than 2 years of age). The most common clinical presentation is hematuria secondary invasion of the renal pelvis. Fever, hypertension, hypercalcemia (elevated parathormone levels), and cutaneous nodules (“blueberry muffin baby”) also are reported.
Imaging: As is seen with clear cell sarcoma of the kidney, rhabdoid tumors of the kidney have imaging features similar to those of Wilms tumors. Imaging features that may suggest the diagnosis include subcapsular fluid collections, medial location within the kidney, and tumor lobules separated by low-density hemorrhage or necrosis and outlined by calcifications. Rhabdoid tumor of the kidney is highly aggressive, metastasizes early, and presents with advanced disease. A distinct association with synchronous or metachronous primary or metastatic central nervous system lesions is seen, often in the posterior fossa. Primitive neuroectodermal tumors, medulloblastomas, ependymomas, and cerebellar and brainstem astrocytomas all have been reported. After tissue diagnosis, MRI of the brain is recommended.
Congenital Mesoblastic Nephroma
Overview: Congenital mesoblastic nephroma (CMN) is the most common solid renal tumor of infancy. Originally thought to represent a congenital Wilms tumor, it is recognized as a distinct neoplasm and also is referred to as a fetal renal hamartoma. It usually is identified in the first 3 months of life. Clinical presentation is that of a palpable abdominal mass and, less frequently, hematuria. Histologically, the tumor is composed of monomorphic infiltrating spindle-shaped mesenchymal cells and embryonal metaplasia of entrapped renal tissue. CMN is subtyped into classic, cellular, and mixed forms. On gross sectioning, the tumor resembles a uterine leiomyoma.
Imaging: A mesoblastic nephroma tends to be a large infiltrative mass; it has poorly defined margins and lacks a capsule. Ultrasound should be the initial imaging study performed. A mesoblastic nephroma is predominantly solid but may contain cystic components (Fig. 116-11). A concentric hyperechoic and hypoechoic ring pattern may be seen reflecting dilated vasculature and entrapped nephrons. The mass may distort and displace the pelvicalyceal system. On CT, CMN may be homogeneous or heterogeneous, is unilateral and unifocal, displaces adjacent vasculature without invasion, and shows no calcification. Low-density foci representing necrosis, seroma, fluid-filled cysts, or hemorrhage may be seen. Enhancement patterns are variable and can be related to entrapped functioning nephrons. On MRI, the tumor typically has a low signal on T1-weighted images both before and after administration of contrast material and has a variable signal on T2-weighted sequences. T1 shortening is associated with hemorrhage.
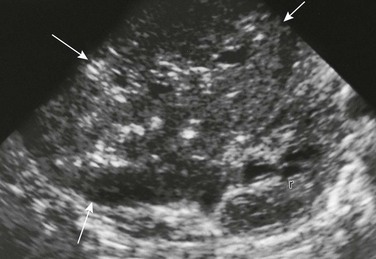
Treatment: CMN usually is associated with a favorable outcome after complete surgical excision. However, the cellular variant (resembling congenital infantile fibrosarcoma) is associated with local recurrences (10%) and metastases. Adjuvant chemotherapy is not recommended if complete resection is feasible in either classic or cellular variants of CMN.1,47–53
Multilocular Cystic Renal Tumor
Overview and Pathophysiology: Multilocular cystic renal tumor occurs predominantly in boys during infancy and toddler stages (3 months to 4 years) and in women during their seventh and eighth decades. Multilocular cystic tumors with fibrous septa that contain mature tubules are called cystic nephromas. When the cystic mass has blastemal elements within its septa, it is called a cystic partially differentiated nephroblastoma. A cystic Wilms tumor is present if solid masses of nephroblastomatous tissue are present. A potential theory suggests that cystic nephromas, cystic partially differentiated nephroblastomas, and Wilms tumors represent benign and malignant ends of a spectrum, analogous to the continuum of ganglioneuromas, ganglioneuroblastomas, and neuroblastomas. Partially differentiated cystic nephroblastomas present in childhood, whereas cystic nephromas are more commonly identified in adult women. Lesions are nonfamilial, are sporadically associated with congenital anomalies, and present clinically as painless abdominal masses.
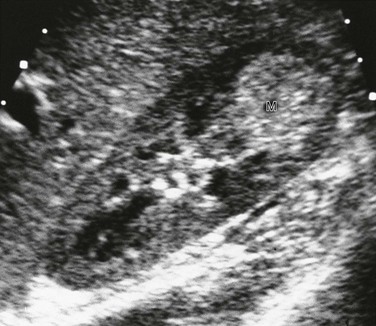
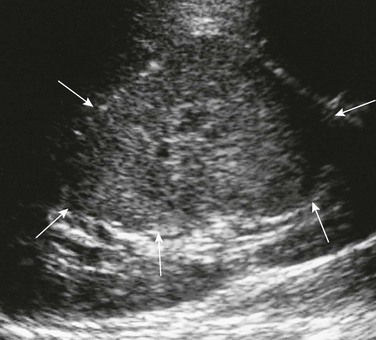
Imaging: Partially differentiated cystic nephroblastomas and cystic nephromas cannot be distinguished on the basis of imaging features. The multilocular cystic renal tumor may involve the entire kidney or only a small portion of it. Ultrasound is more sensitive than CT for identification of septa (Fig. 116-12). The lesions are well-circumscribed, encapsulated masses of multiple anechoic cysts varying in size from a few millimeters to 4 cm. When the cystic spaces are small, the lesion may appear solid. The septa may be thin or thick. The pelvicalyceal system may be distorted and displaced. On CT, the septa enhance but the locules do not enhance, and contrast material does not accumulate within individual locules (Fig. 116-13). Small curvilinear calcifications occasionally are identified. Herniated tumoral cysts may project within the renal pelvis. MRI demonstrates a low T1 and variable T2 signal, depending on the cyst’s contents (hemorrhage or protein). After administration of gadolinium, the cyst septa enhance.
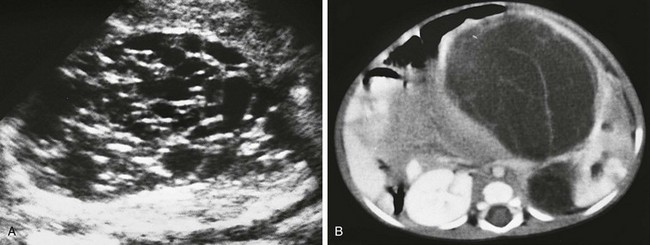
Treatment: Surgical excision generally is curative, and the prognosis is excellent. Imaging on a regular interval is recommended for follow-up. Local recurrence may occur if the tumor is incompletely excised. Recurrence is treated with radiation therapy and chemotherapy. Metastases have not been reported.1,54–66
Renal Cell Carcinoma
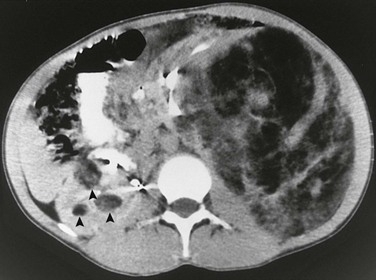
Overview: Renal cell carcinoma (RCC) is rare in the first two decades of life and accounts for approximately 5% of all renal tumors (median age at diagnosis is 9 to 12 years). Increasing evidence indicates that RCC in children differs from adult RCC in biology and behavior. Translocation RCC accounts for most instances of childhood RCC and generally involves the TFE3 gene on chromosome Xp11.2 and less commonly the TEFB gene on chromosome 6p21. Prior chemotherapy currently is the only known risk factor for the development of Xp11 translocation RCC. Children with tuberous sclerosis complex, von Hippel–Lindau syndrome, and neuroblastoma appear to be at increased risk for the development of RCC. Histologically, nearly 80% of RCCs are predominately papillary in architecture. Wilms tumors outnumber RCCs in the first decade of life by a ratio of 30 : 1. During the second decade of life, a solid renal mass is equally likely to be an RCC or a Wilms tumor. Clinical presentation includes gross hematuria, flank pain, and a palpable abdominal mass. Fever, hypercalcemia, polycythemia, and hypertension are uncommon.
Imaging: Although an RCC tends to be smaller than a Wilms tumor, its gross morphology is similar, and the two can be indistinguishable preoperatively. The tumor forms an infiltrative mass with variable necrosis, hemorrhage, calcification, and cystic degeneration. The tumor invades locally and spreads to retroperitoneal nodes. Metastases to the lungs, bones, liver, or brain are found in 20% of patients at diagnosis. Compared with Wilms tumors, RCCs are more likely to be bilateral and have bone metastases. Well-defined intrarenal lesions have been described on ultrasound (Fig. 116-14). On CT and MRI, an RCC appears as a nonspecific solid intrarenal mass with little enhancement. Regions of hemorrhage and necrosis may be present. In some case reports, the tumor has been of increased attenuation relative to renal parenchyma. About 25% of renal cell carcinomas contain calcifications. RCC enhances to a lesser extent than does normal renal parenchyma.
Treatment: Staging uses the Tumor, Node, Metastasis system. The standard therapy for RCC in children and adolescents remains radical nephrectomy; RCC is among the tumors most resistant to systemic chemotherapy or radiotherapy. Nephron-sparing surgery currently is recommended only in adults. Retroperitoneal lymph node dissection remains controversial in children. Overall survival rates are approximately 50% to 60%, with the best prognosis for localized disease confined to the kidney. Xp11 translocation RCC has the potential to metastasize late, as many as 20 or 30 years after diagnosis.1,67–78
Medullary Carcinoma of the Kidney
Overview: Medullary carcinoma of the kidney was described as a distinct entity in 1995. It is a highly aggressive malignant tumor of epithelial origin that appears almost exclusively in patients of African descent with the sickle cell (SC) trait or hemoglobin SC disease. The tissue of origin appears to be within the caliceal epithelium, a common region of papillary necrosis in the SC population. The age at presentation is 20 years (range, 10 to 39 years). A male-to-female ratio of 3 : 1 is seen in patients younger than 25 years. Presenting symptoms include gross hematuria, abdominal or flank pain, a palpable mass, and weight loss. A medullary carcinoma metastasizes rapidly to regional lymph nodes and the lungs.
Imaging: Medullary carcinomas develop in the renal medulla, infiltrate the cortex, encase the renal pelvis, invade the lymphatics and vasculature, and have a predilection for the right kidney. Ultrasound (Fig. 116-15) and CT show a centrally located, infiltrative lesion with peripheral calyectasis, reniform enlargement, and peripheral satellite nodules. The tumor has a heterogeneous echotexture on ultrasound and shows heterogeneous enhancement on CT, which is related to hemorrhage and extensive necrosis. MRI is superior for revealing the extent of parenchymal and nodal invasion, as well as intratumoral hemorrhage and liver metastases.
Renal Angiomyolipoma
Overview: Angiomyolipomas are lesions consisting of a disordered arrangement of vascular, smooth muscle, and fatty elements. Histologic composition suggests a hamartoma, but currently angiomyolipomas are thought to represent a true neoplasm. In children, angiomyolipomas are rare in the absence of tuberous sclerosis. In most children with tuberous sclerosis, angiomyolipomas develop by 10 years of age. Angiomyolipomas also are associated with neurofibromatosis and von Hippel–Lindau syndrome. Lesions that are smaller than 4 cm typically are asymptomatic. Larger lesions are susceptible to spontaneous hemorrhage, resulting in flank or abdominal pain, hematuria, and severe retroperitoneal hemorrhage (Wunderlich syndrome).
Imaging: The imaging appearance of angiomyolipomas is variable depending on the histologic contents. CT (Fig. 116-16) and MRI generally are diagnostic when fat density or signal is identified within the lesion, remembering that fat may be seen in Wilms tumors and RCCs. Ultrasound will identify highly echogenic nonshadowing lesions. Bilateral lesions often are seen in persons with tuberous sclerosis. On angiography, angiomyolipomas demonstrate characteristic tortuous, dilated vessels with aneurysm formation. Rarely are lesions hypovascular.
Renal Lymphoma
Overview: Lymphoma of the kidney is due to hematogenous spread or extension from retroperitoneal sites; the kidney normally does not contain lymphoid tissue, and for this reason, primary renal lymphoma is rare. In children, non-Hodgkin lymphoma (especially Burkitt lymphoma) is most common. Lymphomatous involvement of the kidney typically does not produce symptoms until late in its course. Flank pain, abdominal pain, hematuria, anemia, weight loss, and a palpable mass are most common.
Imaging: The most common radiographic pattern is multiple parenchymal masses distorting the collecting system; less common findings are a solitary renal mass, diffuse infiltration, and isolated perinephric disease. Both CT and MRI are better than sonography for locating renal lesions and identifying extension into adjacent structures. On CT, multiple round homogeneous and hypoattenuating masses are seen on precontrast and postcontrast images (Fig. 116-17). The CT findings of perinephric disease include thickening of Gerota fascia, soft tissue attenuating nodules, or a hypoechoic plaque that is hyperattenuating on nonenhanced images and hypoattenuating on contrast-enhanced images. MRI demonstrates that lymphomatous masses are isointense or slightly hypointense on T1-weighted images and hypointense on T2-weighted images relative to the renal cortex. Enhancement of the lesions after administration of gadolinium usually is less intense than surrounding renal parenchyma, particularly on postcontrast images. On sonography, the mass(es) typically are hypoechoic, with frequent through transmission. Hypoechoic, isoechoic, and hyperechoic subcortical masses all have been described (e-Fig. 116-18). Lymphomatous lesions, however, may be occult or may present as nephromegaly because of diffuse tumor infiltration as the only finding.1,93–99
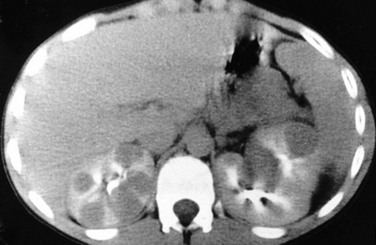
Figure 116-17 Non-Hodgkin lymphoma of the kidneys.
A delayed contrast-enhanced computed tomography image reveals multiple, round hypoattenuating cortical masses.
Ossifying Renal Tumor of Infancy
Overview: An ossifying renal tumor of infancy is an exceedingly rare benign renal mass presenting in children; only 13 cases have been reported in the literature. The age at presentation ranges from 6 days to 14 months. Boys are affected more commonly, with a lesion predilection for the left kidney and upper pole calices. Hematuria is the presenting symptom. Histologically, the lesion consists of an osteoid core, osteoblasts, and spindle cells.
Imaging: The mass arises from the urothelium at the papillary region of the renal pyramids, is polypoid in configuration, and is 2 to 3 cm in size. At imaging, filling defects are seen in the collecting system with partial obstruction. On ultrasound, the mass is echogenic with acoustic shadowing. On CT, a well-defined calcified mass is seen with poor contrast enhancement.
Treatment: The treatment is resection—either complete or partial nephrectomy was performed in the reported cases. The biologic behavior appears to be benign, with no case of malignant spread or postsurgical disease at follow-up.1,100–102
 What the Clinician Needs to Know
What the Clinician Needs to Know
• Renal involvement (unilateral or bilateral)
• Size and imaging characteristics (a solid, cystic, or mixed lesion and the presence of fat)
• Extent (confined to the kidney or extrarenal extension)
• Vascular invasion (inferior vena cava) and extent and distribution of metastatic disease
• Imaging recommendations for further diagnostic evaluation and follow-up
Argani, P, Ladanyi, M. Recent advances in pediatric renal neoplasia. Adv Anat Pathol. 2003;10(5):243–260.
Broecker, B. Non-Wilms’ renal tumors in children. Urol Clin North Am. 2000;27(3):463–469. ix
Cohen, MM, Jr. Beckwith-Wiedemann syndrome: historical, clinicopathological, and etiopathogenetic perspectives. Pediatr Dev Pathol. 2005;8(3):287–304.
Geller, E, Kochan, PS. Renal neoplasms of childhood. Radiol Clin North Am. 2011;49(4):689–709. vi
Glick, RD, Hicks, MJ, Nuchtern, JG, et al. Renal tumors in infants less than 6 months of age. J Pediatr Surg. 2004;39(4):522–525.
Lowe, LH, Isuani, BH, Heller, RM, et al. Pediatric renal masses: Wilms tumor and beyond. Radiographics. 2000;20(6):1585–1603.
Miniati, D, Gay, AN, Parks, KV, et al. Imaging accuracy and incidence of Wilms’ and non-Wilms’ renal tumors in children. J Pediatr Surg. 2008;43(7):1301–1307.
Powis, M. Neonatal renal tumours. Early Hum Dev. 2010;86(10):607–612.
Sarhan, OM, El-Baz, M, Sarhan, MM, et al. Bilateral Wilms’ tumors: single-center experience with 22 cases and literature review. Urology. 2010;76(4):946–951.
Sebire, NJ, Vujanic, GM. Paediatric renal tumours: recent developments, new entities and pathological features. Histopathology. 2009;54(5):516–528.
Shet, T, Viswanathan, S. The cytological diagnosis of paediatric renal tumours. J Clin Pathol. 2009;62(11):961–969.
Zhang, J, Israel, GM, Krinsky, GA, et al. Masses and pseudomasses of the kidney: imaging spectrum on MR. J Comput Assist Tomogr. 2004;28(5):588–595.
References
1. Lowe, LH, Isuani, BH, Heller, RM, et al. Pediatric renal masses: Wilms tumor and beyond. Radiographics. 2000;20(6):1585–1603.
2. Owens, CM, Brisse, HJ, Olsen ØE, et al. Bilateral disease and new trends in Wilms tumour. Pediatr Radiol. 2008;38(1):30–39.
3. Lonergan, GJ, Martínez-León, MI, Agrons, GA, et al. Nephrogenic rests, nephroblastomatosis, and associated lesions of the kidney. Radiographics. 1998;18(4):947–968.
4. Hennigar, RA, O’Shea, PA, Grattan-Smith, JD. Clinicopathologic features of nephrogenic rests and nephroblastomatosis. Adv Anat Pathol. 2001;8(5):276–289.
5. Hoyme, HE, Seaver, LH, Jones, KL, et al. Isolated hemihyperplasia (hemihypertrophy): report of a prospective multicenter study of the incidence of neoplasia and review. Am J Med Genet. 1998;79:274.
6. Rohrschneider, WK, Weirich, A, Rieden, K, et al. US, CT, and MR imaging characteristics of nephroblastomatosis. Pediatr Radiol. 1998;28:435.
7. Hoffer, FA. Magnetic resonance imaging of abdominal masses in the pediatric patient. Semin Ultrasound CT MR. 2005;26(4):212–223.
8. Gylys-Morin, V, Hoffer, FA, Kozakewich, H, et al. Wilms’ tumor and nephroblastomatosis: imaging characteristics at gadolinium-enhanced MR imaging. Radiology. 1993;188:517.
9. Cozzi, DA, Zani, A. Nephron-sparing surgery in children with primary renal tumor: indications and results. Semin Pediatr Surg. 2006;15(1):3–9.
10. Beckwith, JB. Questions and answers: Wilms’ tumor screening. AJR Am J Roentgenol. 1995;164:1294.
11. Choyke, PL, Siegel, MJ, Craft, AW, et al. Screening for Wilms’ tumor in children with Beckwith-Wiedemann syndrome or idiopathic hemihypertrophy. Med Pediatr Oncol. 1999;32:196.
12. Borer, JG, Kaefer, M, Barnewolt, CE, et al. Renal findings on radiological follow-up of patients with Beckwith-Wiedemann syndrome. J Urol. 1999;161:235.
13. Perlman, EJ, Faria, P, Soares, A, et al. Hyperplastic perilobar nephroblastomatosis: long-term survival of 52 patients. Pediatr Blood Cancer. 2006;46:203.
14. Applegate, KE, Ghei, M, Perez-Atayde, AR. Prenatal detection of a Wilms’ tumor. Pediatr Radiol. 1999;29:64.
15. Md Zin, R, Murch, A, Charles, A. Pathology, genetics and cytogenetics of Wilms’ tumour. Pathology. 2011;43(4):302–312.
16. Kullendorff, C-M, Wiebe, T. Wilms’ tumour in infancy. Acta Paediatr. 1998;87:747.
17. Suzuki, K, Miyake, H, Tashiro, M, et al. Extrarenal Wilms’ tumor. Pediatr Radiol. 1993;23:149.
18. Sultan, I, Ajlouni, F, Al-Jumaily, U, et al. Distinct features of teratoid Wilms tumor. J Pediatr Surg. 2010;45(10):e13–e19.
19. Cohen, MD. Commentary: imaging and staging of Wilms’ tumors: problems and controversies. Pediatr Radiol. 1996;26:307.
20. Goske, MJ, Mitchell, C, Reslan, WA. Imaging of patients with Wilms’ tumor. Semin Urol Oncol. 1999;17:11.
21. Slasky, BS, Bar-Ziv, J, Freeman, AI, et al. CT appearance of involvement of the peritoneum, mesentery and omentum in Wilms’ tumor. Pediatr Radiol. 1997;27:14.
22. Wootton-Gorges, SL, Albano, EA, Riggs, JM, et al. Chest radiography versus chest CT in the evaluation for pulmonary metastases in patients with Wilms’ tumor: a retrospective review. Pediatr Radiol. 2000;30:533.
23. Linam, LE, Yu, X, Calvo-Garcia, MA, et al. Contribution of magnetic resonance imaging to prenatal differential diagnosis of renal tumors: report of two cases and review of the literature. Fetal Diagn Ther. 2010;28(2):100–108.
24. Gratias, EJ, Dome, JS. Current and emerging chemotherapy treatment strategies for Wilms tumor in North America. Paediatr Drugs. 2008;10(2):115–124.
25. Grundy, P, Perlman, E, Rosen, NS, et al. Current issues in Wilms tumor management. Curr Probl Cancer. 2005;29(5):221–260.
26. Ko, EY, Ritchey, ML. Current management of Wilms’ tumor in children. J Pediatr Urol. 2009;5(1):56–65.
27. Ehrlich, PF, Hamilton, TE, Grundy, P, et al. The value of surgery in directing therapy for patients with Wilms’ tumor with pulmonary disease: a report from the National Wilms’ Tumor Study Group (National Wilms’ Tumor Study 5). J Pediatr Surg. 2006;41:162.
28. Olsen, OE, Jeanes, AC, Sebire, NJ, et al. Changes in computed tomography features following preoperative chemotherapy for nephroblastoma: relation to histopathological classification. Eur Radiol. 2004;14:990.
29. Meisel, JA, Guthrie, KA, Breslow, NE, et al. Significance and management of computed tomography detected pulmonary nodules: a report from the National Wilms’ Tumor Study Group. Int J Radiat Oncol Biol Physics. 1999;44:579.
30. Porteus, MH, Narkool, P, Neuberg, D, et al. Characteristics and outcome of children with Beckwith-Wiedemann syndrome and Wilms’ tumor: a report from the National Wilms’ Tumor Study Group. J Clin Oncol. 2000;18:2026.
31. Hadley, GP, Sheik-Gafoor, MH. Clear cell sarcoma of the kidney in children: experience in a developing country. Pediatr Surg Int. 2010;26(4):345–348.
32. Boo, YJ, Fisher, JC, Haley, MJ, et al. Vascular characterization of clear cell sarcoma of the kidney in a child: a case report and review. J Pediatr Surg. 2009;44(10):2031–2036.
33. Hung, NA. Congenital “clear cell sarcoma of the kidney. Virchows Arch. 2005;446(5):566–568.
34. Ahmed, HU, Arya, M, Levitt, G, et al. Part I: Primary malignant non-Wilms’ renal tumours in children. Lancet Oncol. 2007;8(8):730–737.
35. Ahmed, HU, Arya, M, Levitt, G, et al. Part II: Treatment of primary malignant non-Wilms’ renal tumours in children. Lancet Oncol. 2007;8(9):842–848.
36. Portugal, R, Barroca, H. Clear cell sarcoma, cellular mesoblastic nephroma and metanephric adenoma: cytological features and differential diagnosis with Wilms tumour. Cytopathology. 2008;19(2):80–85.
37. Argani, P, Perlman, EJ, Breslow, NE, et al. Clear cell sarcoma of the kidney: a review of 351 cases from the National Wilms’ Tumor Study Group Pathology Center. Am J Surg Pathol. 2000;24:4.
38. Glass, RBJ, Davidson, AJ, Fernbach, SK. Clear cell sarcoma of the kidney: CT, sonographic, and pathological correlation. Radiology. 1991;180:715.
39. Chang, CJ, Yeh, ML, Chen, CC. Rhabdoid tumor of the kidney with spontaneous rupture: case report and review of literature. Pediatr Surg Int. 2008;24(4):451–453.
40. Winger, DI, Buyuk, A, Bohrer, S, et al. Radiology-Pathology Conference: rhabdoid tumor of the kidney. Clin Imaging. 2006;30(2):132–136.
41. Luo, CC, Lin, JN, Jaing, TH, et al. Malignant rhabdoid tumour of the kidney occurring simultaneously with a brain tumour: a report of two cases and review of the literature. Eur J Pediatr. 2002;161(6):340–342.
42. Amar, AM, Tomlinson, G, Green, DM, et al. Clinical presentation of rhabdoid tumors of the kidney. J Pediatr Hematol Oncol. 2001;23(2):105–108.
43. Isaacs, H, Jr. Fetal and neonatal rhabdoid tumor. J Pediatr Surg. 2010;45(3):619–626.
44. Agrons, GA, Kingsman, KD, Wagner, BJ, et al. Rhabdoid tumor of the kidney in children: a comparative study of 21 cases. AJR Am J Roentgenol. 1997;168:447.
45. Chung, CJ, Lorenzo, R, Rayder, S, et al. Rhabdoid tumors of the kidney in children: CT findings. AJR Am J Roentgenol. 1995;164:697.
46. Tomlinson, GE, Breslow, NE, Dome, J, et al. Rhabdoid tumor of the kidney in The National Wilms’ tumor study: age at diagnosis as a prognostic factor. J Clin Oncol. 2005;23:7641.
47. Bayindir, P, Guillerman, RP, Hicks, MJ, et al. Cellular mesoblastic nephroma (infantile renal fibrosarcoma): institutional review of the clinical, diagnostic imaging, and pathologic features of a distinctive neoplasm of infancy. Pediatr Radiol. 2009;39(10):1066–1074.
48. England, RJ, Haider, N, Vujanic, GM, et al. Mesoblastic nephroma: a report of the United Kingdom Children’s Cancer and Leukaemia Group (CCLG). Pediatr Blood Cancer. 2011;56(5):744–748.
49. Chaudry, G, Perez-Atayde, AR, Ngan, BY, et al. Imaging of congenital mesoblastic nephroma with pathological correlation. Pediatr Radiol. 2009;39(10):1080–1086.
50. Goldberg, J, Liu, P, Smith, C. Congenital mesoblastic nephroma presenting with hemoperitoneum and shock. Pediatr Radiol. 1994;24:54.
51. Hartman, DS, Lim Lesar, MS, Madewell, JE, et al. Mesoblastic nephroma: radiologic-pathologic correlation of 20 cases. AJR Am J Roentgenol. 1981;136:69.
52. Schlesinger, AE, Rosenfield, NS, Castle, VP, et al. Congenital mesoblastic nephroma metastatic to the brain: a report of two cases. Pediatr Radiol. 1995;25:S73.
53. Willert, JR, Feuser, J, Beckwith, JB. Congenital mesoblastic nephroma: a rare cause of perinatal anemia. J Pediatr. 1999;134:248.
54. Agrons, GA, Wagner, BJ, Davidson, AJ, et al. Multilocular cystic renal tumor in children: radiologic-pathologic correlation. Radiographics. 1995;15:653.
55. Deeg, KH, Gerdemann, C, Weingärtner, K, et al. Sonographic diagnosis of an unusual case of multilocular cystic nephroma mimicking polycystic kidney disease. Ultraschall Med. 2008;29(suppl 5):264–267.
56. Bouhafs, A, Cherradi, N, Lamaalmi, N, et al. An unusual case of multilocular cystic nephroma with prominent renal pelvis involvement. Int J Urol. 2006;13(4):436–438.
57. Sodhi, KS, Suri, S, Samujh, R, et al. Case report: bilateral multilocular cystic nephromas: a rare occurrence. Br J Radiol. 2005;78(929):450–452.
58. Broecker, B. Non-Wilms’ renal tumors in children. Urol Clin North Am. 2000;27(3):463–469. [ix].
59. MacLennan, GT, Ross, J, Cheng, L. Cystic partially differentiated nephroblastoma. J Urol. 2010;183(4):1585–1586.
60. van den Hoek, J, de Krijger, R, van de Ven, K, et al. Cystic nephroma, cystic partially differentiated nephroblastoma and cystic Wilms’ tumor in children: a spectrum with therapeutic dilemmas. Urol Int. 2009;82(1):65–70.
61. Baker, JM, Viero, S, Kim, PC, et al. Stage III cystic partially differentiated nephroblastoma recurring after nephrectomy and chemotherapy. Pediatr Blood Cancer. 2008;50(1):129–131.
62. Puvaneswary, M, Macintosh, J, Cassey, J. Cystic partially differentiated nephroblastoma. Australas Radiol. 2006;50(3):255–257.
63. Blakely, ML, Shamberger, RC, Norkool, P, et al. Outcome of children with cystic partially differentiated nephroblastoma treated with or without chemotherapy. J Pediatr Surg. 2003;38(6):897–900.
64. Rajangam, K, Narasimhan, KL, Trehan, A, et al. Partial nephrectomy in cystic partially differentiated nephroblastoma. J Pediatr Surg. 2000;35(3):510–512.
65. Hartman, DS, Davis, CJ, Sanders, RC, et al. The multiloculated renal mass: considerations and differential features. Radiographics. 1987;7:29.
66. Madewell, JE, Goldman, SM, Davis, CJ, et al. Multilocular cystic nephroma: a radiographic-pathologic correlation of 58 patients. Radiology. 1983;146:309.
67. Spreafico, F, Collini, P, Terenziani, M, et al. Renal cell carcinoma in children and adolescents. Expert Rev Anticancer Ther. 2010;10(12):1967–1978.
68. Ross, H, Argani, P. Xp11 translocation renal cell carcinoma. Pathology. 2010;42(4):369–373.
69. Armah, HB, Parwani, AV. Xp11.2 translocation renal cell carcinoma. Arch Pathol Lab Med. 2010;134(1):124–129.
70. Sausville, JE, Hernandez, DJ, Argani, P, et al. Pediatric renal cell carcinoma. J Pediatr Urol. 2009;5(4):308–314.
71. Selle, B, Furtwängler, R, Graf, N, et al. Population-based study of renal cell carcinoma in children in Germany, 1980-2005: more frequently localized tumors and underlying disorders compared with adult counterparts. Cancer. 2006;107(12):2906–2914.
72. Silberstein, J, Grabowski, J, Saltzstein, SL, et al. Renal cell carcinoma in the pediatric population: results from the California Cancer Registry. Pediatr Blood Cancer. 2009;52(2):237–241.
73. Schultz, TD, Sergi, C, Grundy, P, et al. Papillary renal cell carcinoma: report of a rare entity in childhood with review of the clinical management. J Pediatr Surg. 2011;46(6):e31–e34.
74. Allison, JW, James, CA, Figarola, MS. Pediatric case of the day: renal cell carcinoma in a child with tuberous sclerosis. Radiographics. 1999;19:1388.
75. Donnelly, LF, Rencken, IO, Shardell, K, et al. Renal cell carcinoma after therapy for neuroblastoma. AJR Am J Roentgenol. 1996;167:915.
76. Fenton, DS, Taub, JW, Amundson, GM, et al. Renal cell carcinoma occurring in a child 2 years after chemotherapy for neuroblastoma. AJR Am J Roentgenol. 1993;161:165.
77. Kabala, JE, Shield, J, Duncan, A. Renal cell carcinoma in childhood. Pediatr Radiol. 1992;22:203.
78. Cook, A, Lorenzo, AJ, Salle, JL, et al. Pediatric renal cell carcinoma: single institution 25-year case series and initial experience with partial nephrectomy. J Urol. 2006;175(4):1456–1460. [discussion 1460].
79. Davidson, AJ, Choyke, PL, Hartman, DS, et al. Renal medullary carcinoma associated with sickle cell trait: radiologic findings. Radiology. 1995;195:83.
80. Davis, CJ, Mostofi, FK, Sesterhenn, IA. Renal medullary carcinoma: the seventh sickle cell nephropathy. Am J Surg Pathol. 1995;19:1.
81. Kalyanpur, A, Schwartz, DS, Fields, JM, et al. Renal medulla carcinoma in a white adolescent. AJR Am J Roentgenol. 1997;169:1037.
82. Pickhardt, PJ. Renal medullary carcinoma: an aggressive neoplasm in patients with sickle cell trait. Abdom Imaging. 1998;23:531.
83. Kim, JY, Kim, JK, Kim, N, et al. CT histogram analysis: differentiation of angiomyolipoma without visible fat from renal cell carcinoma at CT imaging. Radiology. 2008;246(2):472–479.
84. Silverman, SG, Mortele, KJ, Tuncali, K, et al. Hyperattenuating renal masses: etiologies, pathogenesis, and imaging evaluation. Radiographics. 2007;27(4):1131–1143.
85. Kim, JK, Kim, SH, Jang, YJ, et al. Renal angiomyolipoma with minimal fat: differentiation from other neoplasms at double-echo chemical shift FLASH MR imaging. Radiology. 2006;239(1):174–180.
86. Winterkorn, EB, Daouk, GH, Anupindi, S, et al. Tuberous sclerosis complex and renal angiomyolipoma: case report and review of the literature. Pediatr Nephrol. 2006;21(8):1189–1193.
87. Casper, KA, Donnelly, LF, Chen, B, et al. Tuberous sclerosis complex: renal imaging findings. Radiology. 2002;225(2):451–456.
88. Carter, TC, Angtuaco, TL, Shah, HR. Ultrasound case of the day: large, bilateral angiomyolipomas of the kidneys with tuberous sclerosis. Radiographics. 1999;19:555.
89. Ewalt, DH, Sheffield, E, Sparagana, SP, et al. Renal lesion growth in children with tuberous sclerosis complex. J Urol. 1998;160:141.
90. Henske, EP. Tuberous sclerosis and the kidney: from mesenchyme to epithelium, and beyond. Pediatr Nephrol. 2005;20:854.
91. Kim, JK, Park, SY, Shon, JH, et al. Angiomyolipoma with minimal fat: differentiation from renal cell carcinoma at biphasic helical CT. Radiology. 2004;230:677.
92. Tchaprassian, Z, Mognato, G, Paradias, G, et al. Renal angiomyolipoma in children: diagnostic difficulty in 3 patients. J Urol. 1998;159:1654.
93. Pedrosa, I, Sun, MR, Spencer, M, et al. MR imaging of renal masses: correlation with findings at surgery and pathologic analysis. Radiographics. 2008;28(4):985–1003.
94. Zhang, J, Pedrosa, I, Rofsky, NM. MR techniques for renal imaging. Radiol Clin North Am. 2003;41(5):877–907.
95. Weinberger, E, Rosenbaum, DM, Pendergrass, TW. Renal involvement in children with lymphoma: comparison of CT with sonography. AJR Am J Roentgenol. 1990;155:347.
96. Sheth, S, Ali, S, Fishman, E. Imaging of renal lymphoma: patterns of disease with pathologic correlation. Radiographics. 2006;26(4):1151–1168.
97. Urban, BA, Fishman, EK. Renal lymphoma: CT patterns with emphasis on helical CT. Radiographics. 2000;20(1):197–212.
98. Fernbach, SK, Glass, RBJ. Uroradiographic manifestations of Burkitt’s lymphoma in children. J Urol. 1986;135:986.
99. Hartman, DS, Davis, CJ, Goldman, SM, et al. Renal lymphoma: radiologic-pathologic correlation of 21 cases. Radiology. 1982;144:759.
100. Höglund, HH, Kellner, MW, Körber, F, et al. Ossifying renal tumor of infancy (ORTI)—a rare diagnosis. Klin Padiatr. 2011;223(3):178–179.
101. Vazquez, JL, Barnewolt, CE, Shamberger, RC, et al. Ossifying renal tumor of infancy presenting as a palpable abdominal mass. Pediatr Radiol. 1998;28(6):454–457.
102. Chatten, J, Cromie, WJ, Duckett, JW. Ossifying tumor of infantile kidney: report of two cases. Cancer. 1980;45(3):609–612.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]Chapter 116
Renal Neoplasms
Nephroblastomatosis Complex
Overview and Pathophysiology: Nephrogenesis is completed by the 36th gestational week. A focus of fetal metanephric blastema or embryonal renal tissue that persists beyond 36 weeks gestation is called a nephrogenic rest. Multiple foci or diffuse nephrogenic rests are termed nephroblastomatosis. Nephrogenic rests are identified in about 1% of neonatal autopsies but usually are no longer found after 4 months of age. Malignant transformation of the fetal metanephric blastema may occur, along with development of a Wilms tumor (nephroblastoma), or nephroblastomatosis may resolve spontaneously.1–3
Nephrogenic rests can occur anywhere in the kidney, depending on when nephrogenesis is interrupted. These rests may be located in the renal lobe (intralobar) or in the cortex that envelops the renal lobe (perilobar). The two types of nephrogenic rests have different appearances, malignant potential, and associated genetic abnormalities. Nephrogenic rests also are classified according to histologic features of development; hyperplastic and neoplastic rests are thought to be active and to have malignant potential, whereas dormant or sclerosing nephrogenic rests are considered inactive.1–4
Intralobar nephrogenic rests are less common than perilobar nephrogenic rests and are more likely to degenerate into a Wilms tumor. Intralobar nephrogenic rests tend to be few and are located randomly in the renal lobe; they are seen in patients with sporadic aniridia, Drash syndrome (i.e., male pseudohermaphrodism and nephritis), and WAGR syndrome (i.e., Wilms tumor, aniridia, genital anomalies, and mental retardation). Patients with sporadic aniridia have a 30% to 40% risk of having a Wilms tumor develop, which represents the greatest likelihood of all of the genetic and syndromic abnormalities associated with nephroblastomatosis.1–4
Perilobar nephrogenic rests are multiple and are located at the corticomedullary junction or in the cortex. Also called diffuse perilobar nephrogenic rests or diffuse perilobar nephroblastomatosis, they are found in persons with hemihypertrophy and in persons with Beckwith-Wiedemann syndrome (i.e., macroglossia, macrosomia, and omphalocele), Perlman syndrome (i.e., fetal gigantism and multiple congenital anomalies), and trisomy 18 syndrome. Patients with hemihypertrophy and Beckwith-Wiedemann syndrome have about a 5% risk of having a Wilms tumor develop.1,3–5
Imaging: Microscopic nephrogenic rests cannot be identified radiologically. Diffuse perilobar nephroblastomatosis and multifocal nephroblastomatosis can be evaluated through ultrasonography, computed tomography (CT), and magnetic resonance imaging (MRI). In diffuse perilobar nephroblastomatosis, the affected kidney may be enlarged. On sonography, corticomedullary differentiation is absent. Regions of nephroblastomatosis may be hypoechoic or isoechoic with respect to normal renal cortex (Fig. 116-1). Multifocal nephroblastomatosis is more difficult to identify on sonography.2,3,6
Figure 116-1 Nephroblastomatosis.
A, Diffuse perilobar form. A longitudinal sonogram shows an enlarged right kidney with lobulated contour and hypoechoic cortical masses (arrows). B, Focal intralobar form. A longitudinal sonogram shows a lower pole mass (arrows) that is isoechoic with renal cortex.
CT is more sensitive than ultrasound for the evaluation of nephroblastomatosis. On contrast-enhanced CT, areas of nephroblastomatosis are well defined because they enhance to a lesser extent than does normal renal cortex (Fig. 116-2). Bulky masses of nephroblastomatosis may distort the pelvicalyceal system. Involvement may be symmetric or asymmetric (e-Fig. 116-3). In diffuse perilobar nephroblastomatosis, a thick rind of lower attenuation tissue encases normally enhancing but architecturally distorted parenchyma. In persons with multifocal nephroblastomatosis, multiple round masses of low attenuation are present. Flat or plaquelike areas of involvement may be difficult to identify on CT.2,3,6
Figure 116-2 Diffuse perilobar nephroblastomatosis with symmetric involvement.
On a contrast-enhanced computed tomography scan, the kidneys are enlarged and multiple, round, peripheral masses of low attenuation are present. Architectural distortion at this level is pronounced; in the medial portion of the right kidney, only a peripheral area of normally enhancing kidney (arrows) is present.
e-Figure 116-3 Diffuse perilobar nephroblastomatosis with asymmetric involvement.
On a contrast-enhanced computed tomography scan, the left kidney is much larger than the right. The architecture of the kidneys is distorted. A portion (arrowheads) of the normally enhancing right kidney consists of parenchyma and calyces.
On MRI, nephroblastomatosis cannot be distinguished from normal renal parenchyma on T1-weighted sequences and is isointense or hyperintense on T2-weighted sequences. Nephroblastomatosis appears hypointense relative to normal renal parenchyma after administration of contrast material on T1-weighted sequences, which are the most sensitive for detection. Active areas of nephroblastomatosis typically are hyperintense on T2-weighted sequences, and inactive areas are hypointense. The signal intensity of nephroblastomatosis is homogeneous, whereas foci of Wilms tumor tend to be heterogeneous.2,3,6–8
Treatment and Follow-up: Although the subject is somewhat controversial, presently no specific treatment is advocated for nephrogenic rests/nephroblastomatosis. Close radiologic follow-up is recommended for children with genetic abnormalities or syndromes associated with nephroblastomatosis. Patients should be monitored to detect Wilms tumor development, because the prognosis for Wilms tumor is best with small lesions. Children with hemihypertrophy or Beckwith-Wiedemann syndrome are at risk for the development of other embryonal tumors, such as hepatoblastoma and adrenal cell carcinoma. No large studies have been performed to establish the optimal screening interval for Wilms tumor surveillance; however, large tumors with metastases that develop within a 6-month period are reported. A baseline CT of nephroblastomatosis-related genetic abnormalities or syndromes at 6 months of age (or at diagnosis if the patient is older than 6 months), followed by ultrasound examinations every 3 to 4 months until the child is 8 years of age, is recommended on the basis of results from the National Wilms Tumor Studies. An area of nephroblastomatosis that grows larger and rounder is suspicious for malignant degeneration.2,3,9–13
Wilms Tumor
Overview, Pathophysiology, and Staging: Wilms tumor is the most common abdominal malignancy of childhood and accounts for 87% of renal masses. Its peak incidence is at 3 to 4 years of age (80% occur in children <5 years), but it has been described in the fetus, neonate, teenager, and adult. Clinical presentation includes a palpable mass, abdominal pain, hematuria, and occasional hypertension (from tumor renin production). As discussed in the Nephroblastomatosis Complex section of this chapter, certain syndromes and genetic abnormalities predispose to development of a Wilms tumor (e-Fig. 116-4). Two loci on chromosome 11 have been implicated in the genesis of Wilms tumors: 11p13 (WT1 gene—WAGR or Drash syndrome) and 11p15 (WT2 gene—Beckwith-Wiedemann syndrome or hemihypertrophy). Bilateral Wilms tumors occur almost exclusively in patients with nephroblastomatosis. Most Wilms tumors arise from the renal parenchyma; however, extrarenal Wilms tumors rarely may develop in the abdomen or at distant sites.1,14–17
e-Figure 116-4 Diffuse perilobar nephroblastomatosis and a right Wilms tumor.
A contrast-enhanced computed tomography scan shows round, peripheral masses that enhance to a lesser extent than normal renal parenchyma in the upper pole of the left kidney. The right kidney has a thick rind of lobulated, hypoattenuating tissue without a discreet mass. A biopsy showed Wilms tumor on the right, and a nephrectomy was performed.
In the United States, tumor stage (Box 116-1) is determined operatively, and the grade is established on pathologic examination. The classic triphasic Wilms tumor arises from mesodermal precursors of the renal parenchyma (metanephros) and contains blastemal, stromal, and epithelial elements. The “teratoid Wilms tumor” contains tissue not normally found in the kidney (e.g., bone, cartilage, and muscle). Tumors with a favorable histology do not contain any anaplastic changes. The prognosis for tumors with a favorable histology is excellent, even for those at higher stages. In Europe, the staging system is based completely on radiologic findings. Tumors are classified on the basis of their imaging appearance, and chemotherapy is given before definitive surgery is performed. Tumors with extension into the inferior vena cava or invasion through the renal capsule are easier to resect after tumor shrinkage produced by chemotherapy.1,2,18
Imaging: Radiologic evaluation of Wilms tumor is focused on identifying the site(s) of involvement, extension, and metastases to assist in surgical planning. Preoperative imaging may include conventional chest radiography, abdominal and pelvic sonography, and thoracic, abdominal, and pelvic CT, MRI, and potentially fluorine deoxyglucose positron emission tomography (FDG-PET) fused with CT (PET-CT).1,2,19,20
On sonography, a Wilms tumor is an intrarenal mass of heterogeneous echogenicity. Some Wilms tumors may contain cystic components, portions of obstructed and entrapped pelvicalyceal systems, or hemorrhagic and necrotic tumor. Extension into the inferior vena cava and the right atrium are characteristic routes of tumor growth and can be well visualized on ultrasound (e-Fig. 116-5), CT (Fig. 116-6), and MRI. The tumor typically forms a pseudocapsule but may invade the renal capsule, seed the peritoneal space, or grow directly into the mesentery and omentum. Hepatic metastases also are possible. The contralateral kidney may contain a smaller Wilms tumor or nephroblastomatosis. Synchronous or metachronous bilateral Wilms tumor may occur in up to 10% of patients.1,2,6
Figure 116-6 A Wilms tumor extending into the inferior vena cava and right atrium.
A, A contrast-enhanced computed tomography scan reveals a large, round, heterogeneous mass in the right kidney that enhances to a lesser extent than normal renal parenchyma. Contrast material opacifies the lateral and anterior margins of the inferior vena cava that contain hypoattenuating tumor (arrows). B, The tumor (arrows) extends into the right atrium.
e-Figure 116-5 A Wilms tumor extending into the inferior vena cava and the right atrium (RA).
A longitudinal sonogram shows tumor thrombus (T) expanding the inferior vena cava and extending into the RA. The tumor partially occludes drainage from one of the hepatic veins (arrow).
On CT, Wilms tumor is generally spherical and intrarenal. It may contain small amounts of fat or fine calcification (Fig. 116-7 and e-Fig. 116-8) because the metanephric blastema cell is a pluripotential embryonal cell. Dystrophic calcifications are seen in about 9% of Wilms tumors. The tumor enhances to a lesser extent than does renal parenchyma. In patients who have been screened ultrasonographically because of genetic or syndromic conditions, the tumor is usually smaller than 4 cm in diameter. Children who are evaluated because of physical examination abnormalities generally have tumors larger than 10 cm in diameter. CT and conventional radiographs of the chest are used to identify pulmonary metastases.1,2,6,21,22
Figure 116-7 A Wilms tumor containing fat in a horseshoe kidney.
A, An axial sonogram of part of the Wilms tumor reveals hyperechoic lobules containing fat (F). B, A contrast-enhanced computed tomography scan shows areas of fat within the mass. The isthmus of the renal parenchyma extends across the midline.
e-Figure 116-8 A Wilms tumor containing calcification.
A contrast-enhanced computed tomography scan reveals fluid-attenuating material anteromedially, most likely hemorrhage, within the large heterogeneous mass. Linear calcification is present posteriorly (arrow).
On MRI, a Wilms tumor is isointense with respect to normal renal parenchyma on T1-weighted sequences and hyperintense with respect to normal renal parenchyma on T2-weighted sequences. After administration of contrast material, a Wilms tumor is hypointense relative to normal renal parenchyma and has inhomogeneous signal intensity (Fig. 116-9). An effectively treated Wilms tumor may be hypointense on T2-weighted sequences.1,2,7,8,23
Figure 116-9 A Wilms tumor.
A, Axial T2-weighted magnetic resonance imaging (MRI) shows a large tumor (T) replacing the right kidney, which is hyperintense to other soft tissue structures. B, Coronal T1-weighted MRI after gadolinium in the same patient shows that the large tumor (T) is hypointense relative to the normally enhancing left kidney (LK).
FDG-PET fused with CT is a rapidly developing modality for imaging accelerated metabolism in malignant tissue. FDG-PET has better accuracy than MRI and bone scanning for staging. Wilms tumors are presumed to be FDG-avid, but the role of FDG in Wilms evaluation is as yet unclear. FDG-PET can help with a targeted biopsy of a viable Wilms tumor and biologically aggressive elements (e.g., anaplastic Wilms). Sensitivity for lung metastases is dependent on nodule size and respiratory motion. Treatment response of the primary tumor can be monitored. Potential benefits of FDG-PET will require ongoing investigation.2
Treatment: Surgery for a Wilms tumor typically begins with a complete abdominal exploration before attention is given to the kidneys. The unaffected kidney initially is visualized and palpated to assess for masses or areas of superficial nephroblastomatosis before en bloc resection of the affected kidney is performed. An excisional biopsy of lung nodules is performed to ensure that the stage of the tumor is correctly assigned. Patients with bilateral disease undergo staging surgery as well. Results of the surgical staging and the histologic examination determine the selection of therapy. In patients with bilateral disease, treatment is provided according to the tumor histology of the higher stage side, along with later nephron-sparing surgery. When the tumor is very large or when the tumor extends into the right atrium via the inferior vena cava, the surgeon may defer surgery until several courses of chemotherapy have reduced the tumor size or extension. Substantial progress in therapy for persons with a Wilms tumor over recent decades has resulted in a greater than 90% long-term survival for localized disease and greater than 70% survival for metastatic disease.1,2,24–30
Clear Cell Sarcoma of the Kidney
Overview: Clear cell sarcoma of the kidney accounts for approximately 5% of primary renal tumors in childhood and was considered a sarcomatous subtype of Wilms tumor before 1978. It was reclassified as a separate entity from Wilms tumor because of the tumor’s histologically and biologically unique features. No familial or syndrome association has been identified. Clear cell sarcoma occurs in an age group similar to that affected by Wilms tumor (1 to 4 years of age), and has a male predominance. Immunohistochemical staining has shown no characteristic marker pattern, but negativity to the WT1 gene is important.
Imaging and Treatment: Clear cell sarcoma imaging features are not distinct from those of the Wilms tumor (Fig. 116-10). Clear cell sarcoma has a predilection for bone metastases (formerly known as the bone metastasizing renal tumor of childhood) that may occur at presentation or upon relapse. A pathologic diagnosis of clear cell sarcoma necessitates an evaluation of the skeletal system. Bone scintigraphy or a conventional radiographic skeletal survey may be used. However, metastases to lymph nodes, lung, and liver still are seen more frequently than bone metastases. Because of the aggressive behavior of clear cell sarcoma, it is associated with a higher rate or relapse and mortality than is Wilms tumor, with a reported long-term survival of 60% to 70%. Treatment consists of nephrectomy and aggressive chemotherapy.1,31–38
[/not-level-membership-for-pediatrics-category]
