[level-membership-for-basic-science-category]
Rectal and vaginal drug delivery
Sanjay Garg and Josef J. Tukker
Chapter contents
Anatomy and physiology of the rectum
Absorption of drugs from the rectum
Anatomy and physiology of the vagina
Absorption of drugs from the vagina
Manufacture of rectal and vaginal dosage forms
Quality control of vaginal and rectal dosage forms
Key points
• Both routes have a use in systemic drug delivery when the oral route is not available.
• These routes are relatively less popular because of their anogenital nature and privacy aspects.
Introduction
Although the oral route is the most commonly used route for drug administration, alternatives, such as parenteral, nasal, ophthalmic, rectal and vaginal may be used because they offer specific advantages. Arguments for choosing the rectal or vaginal route for drug administration include the following:
Besides these apparent advantages, the rectal and vaginal routes also have several drawbacks. These routes are generally disliked by patients because of their anogenital and sexual associations, and hence alternative routes are preferred. Depending on culture and tradition, there are strong feelings of aversion in certain countries, such as the UK and the USA, to rectal administration of drugs, whereas there is complete acceptance in continental and eastern Europe. More rational points include the slow and sometimes incomplete drug absorption and the considerable inter- and intra-subject variations. Moreover, the development of proctitis has been reported with long-term rectal delivery. There are also problems with the large-scale production of suppositories, and of achievement of a suitable shelf-life.
Thus, the rectum and vagina are not routes of first choice. Consequently, these routes of drug delivery are amongst the less desirable, resulting in the market size of the rectal and vaginal formulations being less than 1% of the total pharmaceuticals’ market.
Rectal drug delivery
Introduction
The rectal route is used in many different therapies, intended either for local or for systemic effect.
Local action.
This is desired for the local treatment of pain and itching, mostly due to the occurrence of haemorrhoids (painful, swollen veins in the lower part of the rectum and anus). Locally active drugs include astringents, antiseptics, local anaesthetics, vasoconstrictors, anti-inflammatory compounds and soothing and protective agents. Some laxatives also fall into this category.
Systemic action.
All drugs which are orally administered can be given by this route, and many are in spite of the limitations discussed above. Anti-asthmatic, anti-inflammatory and analgesic drugs are widely administered by the rectal route. Rectal preparations may also be used for diagnostic purposes.
Anatomy and physiology of the rectum
Rectal dosage forms are introduced into the body through the anus and are thus brought into contact with the most caudal part of the gastrointestinal tract, i.e. the rectum. Anatomically, the rectum is part of the colon, forming the final 150–200 mm of the gastrointestinal tract.
The rectum can be subdivided into the anal canal and the ampulla, the latter forming approximately 80% of the organ. It is separated from the outside world by a circular muscle, the anus. The rectum can be considered as a hollow organ with a relatively flat wall surface, without villi and with only three major folds – the rectal valves. The rectal wall is composed of epithelium, which is one cell layer thick, and contains cylindrical cells and goblet cells that secrete mucus. A part of the rectal wall and the rectum’s venous drainage are shown in Figure 42.1.
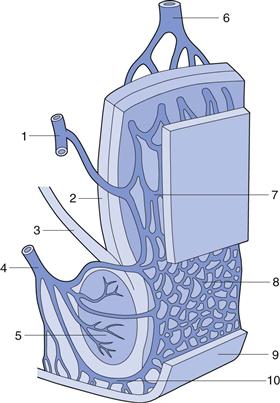
Fig. 42.1 Venous drainage of the human rectum: 1 middle haemorrhoidal (middle rectal) vein; 2 tunica muscularis; stratum longitudinale; 3 muscularis levator ani; 4 inferior haemorrhoidal (lower rectal) vein; 5 muscularis sphincter ani externus; 6 superior haemorrhoidal (upper rectal) vein; 7 and 8 plexus venosus rectalis (submucosus); 9 skin; 10 venosus marginalis. (Adapted from Tondury, 1981, with permission.)
The total volume of mucus is estimated at approximately 3 mL, spread over a total surface area of approximately 300 cm2. The pH of the mucous layer is reported as approximately 7.5 (i.e. close to neutral) in adults, and slightly alkaline in most children. Furthermore, there seems to be little buffering capacity. This point is discussed in relation to drug absorption later in this chapter.
Under normal circumstances the rectum is empty and filling provokes a defaecation reflex under voluntary control. Data comparing drug absorption from freshly prepared and aged (and therefore more viscous) suppositories suggest that there is sufficient motility to provoke spreading even of relatively viscous suppositories.
Absorption of drugs from the rectum
Absorption of drug from the rectum is primarily by passive diffusion. Because of inter-individual variations and the venous drainage of the rectum, the bioavailability of drugs following rectal administration is very unpredictable. In general, the rate and extent of drug absorption is lower than the oral route, mainly due to the small surface area for absorption.
Knowledge of the venous drainage from the rectum is important for the understanding of drug absorption. As can be seen from Figure 42.1, there are three separate veins. The lower and middle rectal (inferior and middle haemorrhoidal) veins drain into the interior vena cava, hence this blood goes directly to the heart and into the general circulation. In contrast, the upper rectal (superior haemorrhoidal) vein drains into the portal vein and therefore this blood passes through the liver before reaching the heart.
This means that drug molecules from the rectum can enter the general circulation either directly or by passing through the highly metabolizing liver. Drug absorbed in the middle and lower part of the rectum will pass directly to the general circulation and avoid first-pass metabolism in the liver. Bioavailability from the upper part of the rectum will be low for certain drugs, as much will be metabolized by the liver during its ‘first pass’ and only a proportion of the drug molecules (if they are of the high clearance type) will enter the general circulation intact.
Investigations have shown that avoiding the first-passage through the liver is possible by keeping the dosage form, and thus the released drug, in the lower part of the rectum. Compared to the small intestine, this situation is very favorable as most gastrointestinal veins drain into the portal vein.
Insertion of a suppository into the rectum results in a chain of events leading to the absorption of the drug. This is represented in a simplified scheme in Figure 42.2. Depending on the character of its vehicle (see later in this chapter), a suppository will either dissolve in the rectal fluid or melt on the mucous layer. Since the volume of rectal fluid is so small, dissolution of the complete vehicle will be difficult and requires extra water. Due to osmotic effects (of the dissolving vehicle) water is attracted, with a resultant unpleasant sensation for the patient. Independent of the vehicle type, drugs dissolved in the suppository will diffuse out towards the rectal membranes. Suspended drugs will first need to leave the vehicle (if it is water immiscible) under the influence of either gravity or motility movements and then begin to dissolve in the rectal fluid.
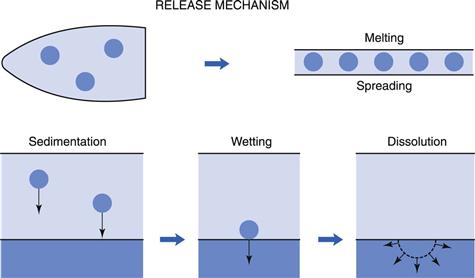
Dissolved drug molecules will have to diffuse through the mucous layer and then into and through the epithelium forming the rectal wall. The process of absorption will be by passive diffusion, as it is throughout the whole gastrointestinal tract for almost all drugs. Active transport processes, as found in the upper regions of the gastrointestinal tract, have not been shown to be present in the rectal area.
For a generalized discussion on drug absorption, the reader is referred to Part 4 of this book. However, some specific points concerning rectal absorption will be discussed here. Box 42.1 provides a summary of the physiological factors that are important in rectal absorption.
The quantity of fluid available for drug dissolution is very small (approximately 3 mL) spread in a layer of approximately 100 µm thick over the organ. Only under non-physiological circumstances is this volume enlarged, e.g. by osmotic attraction by water-soluble vehicles or during diarrhoea. Thus, the dissolution of poorly water-soluble drugs can easily be the slowest step in the absorptive process.
Properties of the rectal fluid, such as composition, viscosity and surface tension, are essentially unknown and have to be estimated from data available for other parts of the gastrointestinal tract. The pH and the buffering capacity of the rectum have been considered earlier in this chapter. The rectum is usually empty, except temporarily when faecal matter arrives from the colon. This material is either expelled or transported back into the colon, depending on voluntary control of the anal sphincter.
The rectal wall may exert pressure on a suppository present in the lumen by two distinct mechanisms. First, the abdominal organs may simply press on to the rectum, especially when the body is in an upright position. This may stimulate spreading and thus promote absorption. The second source of pressure is the motility of the muscles of the rectal wall, which may originate from the normally occurring colonic motor complexes. These are waves of contractions running over the wall of the colon in a caudal direction and are associated with the presence of food residues in the colon.
In contrast with the upper part of the gastrointestinal tract, no esterase or peptidase activity is present in the rectum, resulting in a much greater stability of peptide-like drugs (thus allowing attempts at their delivery by this route). Administration of these compounds using the rectal or vaginal routes has been satisfactory but only if absorption enhancers, such as surfactants are used concomitantly. All types of surfactants seem to be effective, but polyoxyethylene lauryl alcohol ether seems to have the greatest effect. One major drawback of these enhancers, however, is their irritation of the rectal mucosa in the long term. Less irritating enhancers are thus needed to explore this interesting area of drug delivery in greater depth.
Rectal dosage forms
The advantages and limitations of rectal drug administration are outlined in Box 42.2. Several categories of rectal preparations for drug delivery are available: suppositories, rectal capsules, rectal solutions, emulsions and suspensions, powders and tablets for rectal solutions and suspensions, semi-solid rectal preparations, rectal foams and rectal tampons. Of these, suppositories are the most commonly used and these are discussed first, followed by a briefer description of some of the other rectal dosage forms.
Formulation of suppositories
Suppositories are the primary, but not exclusive, dosage form used for the administration of drugs via the rectal route. Suppositories are single-dose preparations with a shape, volume and consistency appropriate for rectal administration. Administration of other suppository-type products (bougies) through other body orifices, e.g. ear, nose and urethra, is rarely undertaken and is not discussed here. Alternative dosage forms for the rectal route are discussed later.
Rectal suppositories are formulated in different shapes and sizes (usually 1–4 g). They contain one or more active substances dispersed or dissolved in a suitable base that may be soluble or dispersible in water or may melt at body temperature. Excipients such as diluents, adsorbents, surface-active agents, lubricants, antimicrobial preservatives and colouring matter may be added if necessary. Their drug content varies widely from less than 0.1% up to almost 40%.
Vehicle (suppository base)
An ideal suppository vehicle or base should melt, dissolve or disperse at body temperature. It should be non-irritating, physically and chemically stable, and pharmacologically inert. Compatibility with a range of drugs is a desirable feature. It should also be convenient to handle during manufacturing and storage. Leakage following administration is likely to be less problematic if the viscosity of the vehicle after melting or dispersion is high.
There are two main classes of vehicles in use: glyceride-type fatty bases and water-soluble bases. Although the ideal vehicle has not been found, the large variety of bases which are available enables a well-considered choice for every drug that has to be formulated into a suppository.
Choosing the optimum base requires much practical experience and can at present only partly be guided by scientifically sound data. However, some general guidelines can be given.
Requirements of the vehicle. There is no doubt that a suppository should either melt after insertion in the body or dissolve in (and mix with) the available volume of rectal fluid. For fatty bases, this means a melting range lower than approximately 37 °C (formulators should be aware that the body temperature might be as low as 36 °C at night). The melting range should be small enough to give rapid solidification after preparation, thus preventing agglomeration or sedimentation of suspended, especially high-density, drug particles. When the solidification rate is high, for example when rapid cooling is applied, this may result in fissures in the suppository. The melting temperature range, on the other hand, should be sufficiently wide to permit easy preparation, which may take a considerable length of time on an industrial scale. During solidification, a suppository should exhibit enough volume contraction to permit removal from the mould or plastic former.
The viscosity of the molten base plays an important role from both a technological and a biopharmaceutical viewpoint. During preparation, the viscosity determines not only the flow into the moulds but also the separation of drug particles. Clearly a compromise has to be found. During and after melting in the rectal cavity, the suppository mass is forced to spread over the absorbing surface. The rate of spreading is determined partly by the viscosity of the suppository at body temperature. The rate of transportation of drug particles from within the base to the interface with rectal fluid, in order to be released and absorbed, will also be affected by the viscosity.
A good suppository base should be chemically and physically stable during storage as a bulk product and after preparation into a suppository. It should have no incompatibility with, and should permit optimal release of, the drug it contains.
Clearly, this list of requirements cannot always be completely fulfilled and often an acceptable compromise is the best that can be expected.
Fatty vehicles.
The fatty vehicles in use nowadays are almost exclusively semi- or fully synthetic. Theobroma oil (also known as cocoa butter) – once a very commonly used base – is no longer used because of its many disadvantages in practice, such as its polymorphic behaviour, insufficient contraction during cooling, low softening point, chemical instability, poor water-absorptive power and its price.
A number of substitutes have been developed, which are becoming increasingly popular. The newer semi-synthetic fatty vehicles have few or none of the problems mentioned above. Commercial examples include: Cotmar, Dehydag, Fattibase, Suppocire and Witepsol. These are mixtures of natural or synthetic vegetable oils, consisting of mixed triglycerides of C12-C18 saturated fatty acids, waxes and fatty alcohols. By using a combination of components, they can be designed to have an adjustable range of melting points, e.g. different grades of Witepsol have melting points ranging from 29 to 44 °C.
The ‘hydroxyl number’ of these bases is a parameter that refers directly to the amount of mono- and di-glycerides present in the fatty base. A high number means that the base is less hydrophobic and its power to absorb water is high. This may lead to an increased rate of decomposition for drugs that are easily hydrolysed. This capacity could also lead to the formation of a w/o emulsion in the rectum. This is generally to be avoided because of a very low drug release rate. An advantage of a high hydroxyl number is the larger melting and solidifying ranges that permit easier manufacture.
Water-soluble vehicles.
Hydrophilic water-soluble (or miscible) vehicles are much less frequently used. They comprise the classic glycerinated gelatin (glycerol-gelatin) and polyethylene glycol (macrogol) bases. Glycerol-gelatin bases are mostly used for laxative purposes and in vaginal dosage forms (see below).
Glycerinated gelatin is a mixture of glycerol, gelatin and water. The mixture forms a translucent, gelatinous mixture, dispersible in the rectum. The ratio of glycerol, gelatin and water can affect the dispersion time and thus the duration of action. A higher proportion of gelatin in the mixture makes it more rigid and longer acting. The following is an example composition of the base:
Preservatives such as methyl and/or propyl parabens may be added. Formulations made with gelatin and glycerol tend to be hygroscopic and require well-closed containers for packaging.
Polyethylene glycols (PEGs) are very versatile polymers in their properties and applications. They consist of mixtures of polyethylene glycols of different molecular weight. Lower molecular weight PEGs (PEG 400 and 600) are liquid, those around 1000 are semi-solid and those above 4000 are waxy solids. Different molecular weights of PEG can be combined to produce the desired properties, as seen in Table 42.1.
Table 42.1
Compositions of PEG bases with different physical characteristics
| Base A | Base B | |
| PEG 1000 | 95% | 75% |
| PEG 4000 | 5% | 25% |
| Properties | Low melting temperature, immediate drug release | Higher melting temperature, sustained drug release |
The melting point of these vehicles is well above body temperature, which means that they need to mix with the rectal fluid. PEGs of all molecular weights are miscible with water and rectal fluids, thereby releasing drug by dispersion; the available volume of rectal fluid is too small for true dissolution. Because of their high melting point, PEG-based formulations are especially suited for application in tropical climates but several disadvantages have to be considered.
They are hygroscopic and therefore attract water after administration, resulting in an uncomfortable sensation for the patient. Incorporation of at least 20% water in the base and moistening before insertion can help to reduce this problem. A considerable number of incompatibilities with various drugs have been reported. Due to the solubilizing character of this base (which has a low dielectric constant) drugs may tend to remain in the base, and drug release may be slow.
PEG bases can develop peroxides on storage, therefore airtight packaging is recommended and the formulation should be monitored for peroxides during stability studies when ascertaining shelf-life.
Choice of vehicle.
A summary of the points which are important for the choice of a suppository base is given in Box 42.3. The parameters outlined are evidently not independent of each other. One additional parameter is the volume of the suppository. Usually suppositories for adults are 2 mL and for children 1 mL. It has been suggested that the larger volume may provoke a reaction of the rectal wall, thus helping to spread the melt over a larger area. Indeed, an increase in volume of, for example, paracetamol suppositories results in faster and more complete absorption of the drug.
Drug
Box 42.4 lists the factors relating to the drug substance that may have possible consequences for the quality of suppositories.
Drug type.
Due to the limited rectal area for absorption and the small amount of water available, the rectum is thought to be unsuitable for absorption of very hydrophilic or very low water-soluble compounds. For example, after rectal dosing, tamoxifen (which has an aqueous solubility of 400 mg/L, but a solubility in saline of approximately 40 mg/L) shows a marked decrease to 10% of its oral availability. This example illustrates that switching from oral to rectal dosing without proper information is not always a simple exercise.
Drug solubility in vehicle.
The drug solubility in the vehicle is of particular interest from the biopharmaceutical point of view. It determines directly the type of product, i.e. solution or suspension suppository. The drug solubility in the rectal fluid determines the maximum attainable concentration and thus the driving force for absorption. When a drug has a high vehicle-to-water partition coefficient, it is likely to be in solution to an appreciable extent (or completely) in the suppository. This generally means that the tendency to leave the dosage form will be low and thus the release rate into the rectal fluid will be slow. This is obviously unfavourable for rapid absorption. On the other hand, some lipid solubility is required for penetration through the rectal membranes (see above).
The optimal balance between these two requirements is usually found using the rules listed in Table 42.2. This table assumes that the release from the dosage form is considered as the rate-limiting step. Thus, the tendency to remain in the base should be lowered as much as possible (rules 1 and 2). When the drug solubility in fat and water are both low, no definite rule can be given. It may be that the dissolution rate of the drug will become the controlling step, and thus it seems advisable to use micronized drug particles.
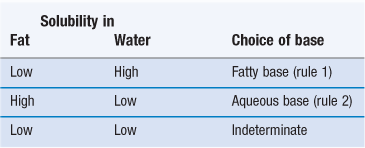
It should be stated as a general rule that w/o emulsion-type suppositories are strongly discouraged. The transfer of drug molecules present in a dissolved state in the inner phase will be very slow and thus drug absorption will be very much retarded.
It seems logical therefore that the first choice of a formulation would be a readily water-soluble form of the drug dispersed in a fatty base. This lays special emphasis on the water solubility of drugs and the methods to improve this. For a detailed discussion of these points, the reader may refer to Chapters 2, 3, 20 and 23.
Surface properties.
The surface properties of drug particles are also important as these particles will be transferred from one phase to another (see Fig. 42.2). This happens first when the drug is brought into contact with the vehicle (i.e. during compounding or manufacture) and air has to be displaced from its surface. When this is not achieved efficiently particles may form agglomerates. This adversely affects final content uniformity by an increased tendency for the solid particles to separate out by sedimentation prior to setting. If wetting by the vehicle has taken place, displacement by rectal fluid will be required to let the drug move into solution – a prerequisite for absorption. This is the underlying reason why formulators add surfactants to formulations (see below).
Particle size.
The particle size of the drug is an important parameter, both technologically and biopharmaceutically. However, as occurs many times during formulation design, there are conflicting requirements and a compromise is often required.
To prevent undue sedimentation during or after preparation, the particle size should be limited. Data available from the literature do not allow definition of an exact limit; however, the use of particles smaller than approximately 150 µm is recommended. It is, of course, assumed that no agglomeration will take place.
The smaller the particles the less the possible mechanical irritation to the patient (especially if they are < 50 µm).
The smaller the particles the higher will be the dissolution rate. Therefore drugs with low water solubility should be dispersed as small, preferably micronized, particles.
One should be aware of the increased tendency of these small particles to agglomerate due to strongly increased van der Waals forces as particle size is reduced.
Unnecessary manufacturing operations, such as size reduction, should be avoided when possible.
There are indications that size reduction is not necessarily a good option for all drugs. It has been shown, especially for readily water-soluble drugs, that large particles give blood levels which are higher than, or at least equivalent to, smaller particles. This would lead to the suggestion to use particles in the size range 50–100 µm in that case. The lower limit of 50 µm to increase transport through the molten vehicle (see Fig. 42.2) and the upper limit of 100 µm is a safe protection against undue sedimentation during preparation. There is no clear-cut recommendation, however, for which solubility class this would be the rule to follow. For example, paracetamol (solubility in water approximately 15 mg/mL) gave the best blood levels when the particle size was smaller than 45 µm.
It should also be borne in mind that the spreading suppository mass should take with it the suspended particles, so as to maximize the surface area for absorption. For dense particles it has become clear that this is a problem, but there is no evidence that organic drugs (density usually 1.2–1.4 g cm–3) suffer from this disadvantage when dispersed as, for example, 150 µm size particles. A decision on particle size is an important part of the development plan of a new suppository formulation.
Amount of drug.
An additional complicating factor is the amount (proportion) of drug present in a suppository. If the number of particles increases, this would also increase the rate of agglomerate formation. This will depend on the particle size and on the presence of additives. The theory describing the agglomeration behaviour of dispersed systems (DLVO theory, see Chapter 5) can be applied to the non-aqueous systems described here, but certain refinements are necessary. Another consequence of the presence of suspended particles is the increased viscosity of the molten base. Also, in this case, the formulator has to rely largely on empirical data rather than on theory.
Other additives
For several widely varying reasons, formulators of suppositories make use of additives to improve their product. The dispensing aspects include formulations for specific drugs which affect the melting point of the suppository; it may become depressed (by a soluble liquid compound) or increased (by a high amount of soluble high melting active compound). The important point to consider in these situations is the possible influence of formulation changes on the release characteristics.
The inclusion of viscosity-increasing additives (e.g. colloidal silicon oxide or aluminium monostearate, both at approximately 1–2%) will create a gel-like system with, in general, a slower release rate of the drug. In vitro relationships can be easily established, but whether the actual release in vivo will also be depressed cannot be easily predicted, since rectal motility will in certain cases be able to overcome this problem.
The addition of lecithin is a worthwhile possibility when high amounts of solid drug are used. It is thought that the lecithin decreases the attraction between the drug particles, enhancing the flow properties of the dispersion.
The addition of surface-active agents has been practiced extensively but still remains a source of great uncertainty. When these compounds are used to create a w/o emulsion system, this has to be discouraged since the release will be unacceptably slow. It may well be, however, that surfactants act as wetting agents. This can influence the release rate in a positive sense, but there is little evidence to show that wetting (i.e. displacement of base by rectal fluid) is a real problem. Surfactants may also act as ‘deglomerators’, which may prevent the formation of a cake in the melting suppository, which in turn would slow down the rate of drug release. The role of surfactants as spreading enhancers has not been clarified and this factor is strongly interrelated with the occurrence of rectal motility. There is evidence that the presence of surfactants in a concentration higher than the critical micelle concentration can retard the release of some drugs from suppositories.
Other rectal preparations
Other rectal preparations include rectal capsules, rectal tablets, rectal solutions (including enemas), suspensions and emulsions, powders and tablets for rectal solutions and suspensions, semi-solids (ointments and creams), rectal foams and rectal tampons.
For the treatment of local conditions, like haemorrhoids, fatty ointments are used widely. For the systemic administration of drugs, delivery forms such as tablets, capsules and microenemas are employed.
Rectal preparations may contain a number of excipients, such as viscosity enhancers, buffers, solubilizing agents, antimicrobial agents and antioxidants. Addition of absorption enhancers, such as surfactants can be used to promote bioavailability. As in all drug delivery, the safety of excipients should be evaluated carefully. Local irritation with rectal formulations is frequently reported.
Rectal capsules
Rectal capsules (shell suppositories) are solid, single-dose preparations generally similar to soft capsules (Chapter 34). They are of elongated shape and have a smooth external appearance that may be lubricated. Capsules used to achieve a systemic effect are usually filled with a solution or suspension of the drug in vegetable oil or liquid paraffin. There is limited experience with this dosage form, but it seems that no striking differences exist between the bioavailability from rectal capsules and fatty suppositories.
Rectal tablets
Tablets are not an ideal dosage form because they cannot disintegrate rapidly, due to the low amount of water present in the rectum. Rectal tablets releasing CO2 after insertion can be used, stimulating defaecation.
Rectal solutions, emulsions and suspensions
Rectal solutions, emulsions and suspensions (also historically referred to as enemas) are liquid preparations intended for rectal use. They are used for systemic or local effect and for diagnostic purposes. These liquid formulations are applied rectally to evacuate, cleanse or treat the lower parts of the gastrointestinal tract. In the treatment of rectocolitis, enemas are used having a relatively large volume, e.g. 100 mL. This enables the drug to reach the upper part of the rectum and the sigmoid colon. Larger volumes are used for the process known as colon cleansing or colonic irrigation.
These preparations contain vehicle only (e.g. arachis oil enema) or one or more active substances dissolved or dispersed in water, glycerol, macrogols (PEGs) or other suitable solvents. Rectal solutions, emulsions and suspensions may contain excipients, for example to adjust the viscosity of the preparation, to adjust or stabilize the pH, to increase the solubility of the active substance(s) or to chemically stabilize the drug in the preparation. These substances should not adversely affect the intended clinical effect or, at the concentrations used, cause undue local irritation. Rectal solutions, emulsions and suspensions are supplied in single-dose containers containing a volume in the range of 2.5 mL up to a few hundred mL. The container design is adapted to deliver the preparation to the rectum or is accompanied by a suitable applicator.
Microenemas.
These are solutions or dispersions of the drug in a small volume (approximately 3 mL) of water or vegetable oil. This dosage form is supplied in a small plastic container, equipped with an application tube. After insertion of the tube, the container is emptied by compression of the bulb. The advantage of this delivery system, if water is used as a vehicle, is that no melting and dissolution is necessary before drug release can begin. Microenemas have been shown to perform well, but they are still of limited applicability because of their relatively high cost compared to, for example, suppositories. Moreover, administration cannot be performed easily by patients themselves, and it is rather difficult to deliver the total content of the plastic container.
Powders and tablets for rectal solutions and suspensions
Powders and tablets intended for the preparation of rectal solutions or suspensions are single-dose solid preparations that are dissolved or dispersed in water or other suitable solvents at the time of administration. They may contain excipients to facilitate dissolution or dispersion, or to prevent aggregation of the particles. Such powders are often produced by freeze drying (lyophilization). This results in a porous product that has a large surface are and dissolves rapidly (Chapters 28 and 29).
Semi-solid rectal preparations
Semi-solid rectal preparations are ointments, creams or gels. They are often supplied as single-dose preparations in containers provided with a suitable applicator.
Rectal tampons
Rectal tampons are solid, single-dose preparations intended to be inserted into the lower part of the rectum for a limited time and then removed.
Vaginal drug delivery
Introduction
Administration of drugs via the vagina is less common than rectal administration. Vaginal delivery is used for both local and systemic effects, though applications for local effects are far more common.
Local action
For drugs targeted for local action, vaginal administration permits use of smaller doses with reduced absorption and systemic distribution and toxicity. Such drugs include anti-infectives, for instance: clotrimazole, miconazole, clindamycin. With antifungal drugs, such as miconazole, treatment of vaginal infections can be achieved using a much lower dose applied vaginally, as compared to oral administration. Spermicides, such as nonoxynol-9 have been delivered by the vaginal route for contraceptive activity. In the over-the-counter category, a number of formulations are available for hygiene, lubrication or sexual pleasure enhancement.
A new category of drugs currently under development are ‘microbicides’. These are compounds and formulations that can prevent transmission of HIV and/or sexually transmitted infections, such as chlamydia and trichomonas. Microbicides may be used by women to protect themselves during sexual activity, if their male partner is infected. Currently, there are no approved products in clinical use; however some compounds such as dapivirine and tenofovir are in advanced stages of clinical development.
Systemic action
Some drugs are administered vaginally to achieve systemic effects. In some cases, drugs given by the intravaginal route have a higher bioavailability compared to the oral route, because the drug enters the systemic circulation without passing through the metabolizing liver (as explained below).
Only a few systemically acting drugs are currently administered by this route. Among these are oestrogens, progesterone and prostaglandin analogues that are usually administered as vaginal creams or hydrogels. Some drugs (e.g. progesterone) are poorly absorbed after oral administration and undergo extensive first-pass metabolism. Vaginal application of progesterone cream offers significant advantages for the development of the uterine lining. A number of other drugs have been evaluated for vaginal delivery.
Anatomy and physiology of the vagina
The human vagina has a fibro-muscular tube structure that connects the uterus to the external environment. The vaginal wall is coated with mucus or cervico-vaginal fluids, which provides some protection and necessary lubrication during sexual activity. The fluid comprises mucin, salts, acids, proteins and water.
In healthy adult women, vaginal fluid is slightly acidic (with a pH range between 3.5 and 4.5) because of the presence of microflora, consisting primarily of lactobacillus. The bacteria convert glycogen from epithelial cells into lactic acid. The vaginal ecology is dynamic in nature and responds to changes in hormone levels, administration of contraceptives, use of vaginal formulations, age, menstrual cycle and a number of other factors.
The composition and volume of cervico-vaginal fluids and environment varies significantly with age, stage of menstrual cycle, pregnancy, sexual activity and infections. The pH tends to rise during local infections and in the post-menopausal stage. The acidic pH provides natural protection from infections, including those which are sexually transmitted. Human semen on the other hand, is highly buffered and slightly alkaline in pH. After intercourse, deposition of semen leads to a transient change in pH of the cervico-vaginal fluid, sufficient for survival of sperm and subsequent fertilization.
Vaginitis is a common clinical condition caused by local infections and characterized by itching, discharge and change of odour. A range of sexually transmitted infections such as chlamydia, gonorrhoea, trichomonas and candida also affect the vagina. Herpes simplex virus (HSV) and Herpes papilloma virus (HPV) infections are also frequently reported.
These conditions may, in turn, affect the efficiency of action of vaginally administered drugs and their systemic absorption.
Absorption of drugs from the vagina
Drugs administered for local action in the cervico-vaginal area are not required to be absorbed. In fact, non-absorption of these drugs is an advantage since it avoids any systemic toxic effects.
Drugs designed for systemic action are absorbed by passive transport. The vaginal wall is very well suited for the absorption of drugs for systemic use, since it contains a vast network of blood vessels. Blood drainage from the vagina is via the vaginal vein. From here the blood moves into the inferior vena cava and thus avoids the hepatic portal system, i.e. the drugs absorbed through vaginal wall by-passes first-pass metabolism in liver.
A number of physiological factors, such as the volume, viscosity and pH of vaginal fluid, the stage of menstrual cycle and sexual activity can influence drug absorption. Drug-related factors that influence absorption include drug solubility, ionization behaviour, molecular weight, release characteristics from the formulation and the dosage form.
Vaginal dosage forms
The advantages and limitations of drug delivery by vaginal administration are shown in Box 42.5. Formulation design and other considerations for the vaginal route are similar in many respects to rectal preparations. A range of vaginal dosage forms are currently available and used in practice. These include pessaries, liquids (vaginal solutions, emulsions, suspensions), semi-solids (creams, gels, ointments), solids (vaginal tablets, films, capsules and rings), tablets for vaginal solutions and suspensions, gaseous preparations (sprays and foams) and medicated vaginal tampons. Not all will be medicated, but may also exist without an active ingredient, e.g. formulations designed for lubricant activity. Table 42.3 summarizes the types of vaginal dosage forms and commonly used excipients.
Table 42.3
| Dosage form | Excipients |
| Creams and ointments | Oily and aqueous bases, emulsifying agents, water, preservatives, antioxidants |
| Gels | Gelling agent, humectant, water, preservative |
| Pessaries | Aqueous/oily/emulsifying base, antioxidant, preservative |
| Foams and sprays | Propellant blend, emulsifier, solvents, water, vehicle |
| Suspensions, solutions and emulsions | Suspending or emulsifying agent, thickening agent, water, fatty acids, waxes, preservative, and antioxidant |
| Tablets or capsules | Diluent, binders, lubricating agent, capsule shell, filler, disintegrating agent |
| Films | Polymers, plasticizers, water, solvents |
| Rings | Polymers, plasticizers, solvents |
Worldwide, there are variations in the preferred types of vaginal dosage forms used. In some countries, such as the USA, creams and foams are popular; pessaries are more commonly seen in France, while in other parts of the world, such as India, tablets tend to be more popular because of their better stability in the tropical climatic conditions. Semi-solid formulations such as creams, gels and ointments can serve dual purposes of providing medication as well as lubrication, which is more difficult with tablets.
Depending upon the dosage form, a number of excipients, including diluents, surfactants, emulsifying agents, lubricants, preservatives, antioxidants, colouring agents and humectants may need to be included in the formulation. As with all dosage forms, these excipients should be pharmaceutically safe and of acceptable quality.
Measuring accurate doses of semi-solids with an applicator tends to be relatively difficult, while tablets, capsules, and films offer the advantage of unit-dose delivery.
Ideal vaginal dosage form
Some of the important characteristics of an ideal vaginal dosage form are:
• should be long acting, reducing the frequency of administration
• should be stable in a range of climatic conditions
• should not lead to any irritation, burning or itching
• should not cause any leakage
• should not cause staining or discolouring of under garments
• the formulation should be colourless and odourless
• the formulation should not adversely affect sexual activity
• women should be able to use it without the knowledge of a male partner
• should be easy to insert and/or apply, without the need for an applicator.
Drug characteristics
The physicochemical properties of active ingredient(s) influence the formulation design and performance. The following is a brief outline of the most important properties. Many of these factors are similar to those for rectal administration discussed above.
Particle size.
Insoluble particles larger than 50 µm are likely to cause mechanical irritation after administration. At the same time, particles greater than 150 µm have the potential for rapid sedimentation during manufacturing, i.e. during molten mixture stage, before setting.
Solubility.
The aqueous solubility of the drug is a critical preformulation parameter that dictates formulation options, for example, a lipophilic drug will solubilize in fatty bases, while a hydrophilic drug will be suspended.
Dose or drug loading.
It can be difficult to incorporate high unit doses in formulations, e.g. fatty pessaries are difficult to optimize when they contain greater than 10% of oily drugs, while maintaining the required mechanical strength and the melting point.
Bulk density.
Drugs insoluble in the vehicle with low bulk density, especially those with high doses, can be difficult to incorporate into the formulation.
Surface characteristics.
Addition of surfactant may be required to help with some drugs to permit rapid wetting after administration.
Pessaries
Pessaries are solid, single-dose preparations for vaginal insertion. Pessaries can be prepared in various shapes, usually ovoid, with a volume and consistency suitable for insertion into the vagina. Typically, they weigh around one gram. Pessaries contain one or more active substances dissolved or dispersed in a suitable base that may be soluble or dispersible in water, or may melt at body temperature. Additionally, they may contain diluents, adsorbents, surface-active agents, lubricants, antimicrobial preservatives, colouring matter and other stabilizers if necessary. The particle size of the active and excipients, if suspended in the vehicle, is controlled to avoid any irritation.
Selection of a pessary base
A number of factors influence the selection of a base for pessaries. Many of these considerations are similar to those for rectal suppositories discussed above.
Vaginal suppositories (pessaries) are most often prepared with glycerol-gelatin bases, since this mixture is well tolerated. Polyethylene glycols are less common, since they are said to promote irritation. Fatty excipients are little used. Most drug delivery forms for vaginal application demand an auxiliary device, in order to obtain deep insertion of the dosage form.
The following are some of the important factors that should be critically evaluated during the development phase for large-scale manufacture of pessaries.
Drug solubility and release.
The solubility of a drug in the vehicle and body fluids affects the release profile from the formulation. Because of partitioning, the drug tends to prefer the phase where it is more soluble. Additionally, the miscibility of a base with body fluids affects the drug release profile. If the drug is highly lipophilic and poorly hydrophilic, a water-soluble base is preferred. Similarly, if the drug is more hydrophilic, a lipid base will be desirable. However, use of a highly lipid base will hinder the release of lipophilic drugs. A desirable combination is a water-soluble form of drug, dispersed in lipidic base, which will ensure rapid release of drug and miscibility with vaginal fluids.
Desired target.
Consideration must be given as to whether local or systemic activity is required.
Chemical stability.
Synthetic bases such as Witepsol and Fattibase offer better stability than theobroma oil.
Melting point.
Certain drugs can lower the melting point of the base, making it necessary to re-optimize the formulation to achieve the melting point close to body temperature.
Semi-solid vaginal preparations
Semi-solid vaginal preparations are ointments, creams or gels. They are relatively commonly used for vaginal application, for local as well as systemic delivery. Ideally, these should be developed by optimizing the rheological properties of the semi-solid, its spreading behaviour, volume, pH, osmolarity, ease of insertion and retention and patient acceptability. However, in most cases these formulations are developed on the basis of empirical knowledge of topical semi-solid formulations. Because of the aqueous environment in semi-solids, stability may be an issue with drugs susceptible to hydrolysis.
Vaginal semi-solids may be supplied in multidose, collapsible tubes, provided with a suitable applicator. Delivery of an accurate dose is an issue with vaginal semi-solids; the applicators tend to be messy in use and add to the cost of the product. Single-dose containers are also available that usually administer a 3 to 5 mL dose per application. Prefilled syringe-type applicators are being evaluated clinically. From the large-scale manufacturing perspective, filling a highly viscous gel or cream into an applicator at high speed is a difficult process.
Vaginal films
Vaginal films are thin layers of a polymeric material, which are designed to dissolve in the vaginal fluids and release the drug. These are single-dose preparations and can be applied without the need for an applicator. In terms of composition, the films usually contain polymer, plasticizer and solvent. Other components can include disintegrating agents, colourants, antimicrobial agents and other stabilizers. A vaginal contraceptive film (VCF), containing nonoxynol-9 as an active ingredient, is an example of a marketed product.
Vaginal tablets
Vaginal tablets are solid, single-dose preparations. They are similar to oral tablets with respect to their formulation and manufacture; they may be coated or uncoated. Vaginal tablets can be prepared in various shapes, ovoid being the most popular. Tablet formulations offer the advantages of ease of storage, ease of use, low cost and well-controlled large-scale manufacturing. These are especially suitable for drugs susceptible to degradation in the presence of moisture. In tropical countries, vaginal tablets are the more commonly used vaginal dosage form.
An ideal vaginal tablet formulation should rapidly disintegrate in a small volume of vaginal fluids and release the drug. Excipients are used to improve the disintegration of vaginal tablets, e.g. a bicarbonate together with an organic acid, which results in CO2 release. A good filler for vaginal tablets is lactose, since this is a natural substrate for the vaginal microflora that converts lactose into lactic acid, resulting in a pH value of 4–4.5.
Both vaginal tablets and films can be designed to include bioadhesive polymers such as Carbopol and xanthan gum, which result in a thick bioadhesive dispersion after dispersion of the formulation contents, minimizing leakage and improving retention.
Vaginal capsules
Vaginal capsules (shell pessaries) are solid, single-dose preparations. They are generally similar to oral soft capsules (Chapter 34) but their shape is often (but not universally) an elongated ovoid, and they can be of larger size. They should have a smooth surface.
Vaginal rings
Vaginal rings offer the flexibility of controlled release of the drug over a prolonged period of time. Rings are a flexible, circular system containing the drug entrapped in a polymer network. An example of vaginal ring is NuvaRing®, containing estrogen and progestin, used as a contraceptive. After administration, NuvaRing® is left in the vagina for three weeks, during which time it releases the drugs over that time scale.
Vaginal solutions, emulsions and suspensions
Vaginal solutions, emulsions and suspensions are liquid preparations intended for a local effect, for irrigation or for diagnostic purposes. Excipients may be added, for example to adjust the viscosity of the preparation, to adjust or stabilize the pH, to increase the solubility of the active substance(s) or to stabilize the preparation. The excipients should not adversely affect the intended medical action or, at the concentrations used, cause undue local irritation. Vaginal emulsions may show evidence of phase separation but are readily redispersed on shaking. Vaginal suspensions may show sediment that is readily dispersed on shaking to give a suspension that remains sufficiently stable to enable a homogeneous preparation to be delivered. They are supplied in single-dose containers. The container is designed to deliver the preparation to the vagina, or it is accompanied by a suitable applicator.
Tablets for vaginal solutions and suspensions
Tablets intended for the preparation of vaginal solutions and suspensions are single-dose preparations that are dissolved or dispersed in water at the time of administration. They may contain excipients to facilitate dissolution or dispersion, or to prevent caking.
Medicated vaginal tampons
Medicated vaginal tampons are solid, single-dose preparations intended to be inserted in the vagina for a limited time and then removed.
Drug-release mechanisms
There are two major drug-release mechanisms from solid vaginal dosage forms and these are similar to those described for rectal dosage forms, i.e.:
Melting.
Formulations, such as pessaries, may be designed to melt at body temperature releasing the drug when the vehicle melts and the molten mass disperses in the available space.
Disintegration.
Solid vaginal dosage forms typically release the drug by dissolution or disintegration, followed by dispersion of the contents in the vaginal fluids.
The drug release from semi-solid dosage forms, such as creams or gels, is by diffusion or dispersion of contents from the formulation to vaginal fluids.
Manufacture of rectal and vaginal dosage forms
Suppositories and pessaries
Suppositories and pessaries are manufactured by hand on a small scale, in batches of 6–50, and on a (semi) automatic scale in batches of up to 20 000 per hour. Essentially the mode of manufacture is similar in both cases and involves:
Most suppositories are packed individually in a plastic (PVC) or aluminium foil strip pack. Requirements for good protection against moisture and oxygen can be deduced from the individual needs of the drug and the properties of the packaging material. The requirement to follow current good manufacturing practices (GMP) is the same as for other pharmaceutical dosage forms.
The level of automation and scale of manufacturing varies significantly in machines from different manufacturers. The steps in the commercial manufacturing process are as follows:
Vaginal films
To manufacture films, the active ingredient is dissolved or dispersed in a concentrated polymer solution in an appropriate solvent. Plasticizer is added and the viscous solution is spread on a glass surface and dried. Films are cut to the required size and packaged in blister packs or sachets. Parameters to optimize during development of films include dissolution and release behaviour in small volumes of fluids, mechanical strength, thickness, content uniformity, texture, and process parameters such as drying time. Because of their light weight, films are suitable for small-dose drugs only.
Rectal and vaginal tablets
Compaction of vaginal tablets and tablets for vaginal solutions and suspensions is similar to the manufacturing of oral tablets by using punch and die to produce suitable shapes (see Chapter 30).
Other rectal and vaginal dosage forms
The industrial manufacture of many other types of rectal and vaginal dosage forms (solutions, emulsions, suspensions, creams, gels, ointments, etc.) differ little, or not at all, from those described elsewhere in this book.
Quality control of vaginal and rectal dosage forms
Rectal and vaginal dosage forms are evaluated by a set of in-vitro and in-vivo tests for quality and safety, as well as effectiveness. A list of properties that should be controlled is given in Box 42.6. These include organoleptic evaluation (for colour, odour, shape and surface), release characteristics, melting and solubility, stability, pH, viscosity, spreading, bioadhesion and mechanical strength. Some of these are pharmacopoeial requirements and others are carried out during the development phase. A number of these tests will form part of the release and expiry specifications of the dosage forms. Formulations are also required to comply with the monograph for the particular dosage form. For example, medicated vaginal tampons must comply with the specific monograph of medicated tampons.
Quality assessments
The following are common pharmacopoeial and non-pharmacopoeial in-vitro tests for rectal and vaginal preparations. Clearly not all these tests are appropriate for every dosage form. Local pharmacopoeias will provide information on national requirements.
1. Uniformity of weight (mass) of the dosage form.
3. Uniformity of content of active ingredient in individual dosage units.
5. Disintegration time could be important for rectal and vaginal tablets and capsules.
6. Dissolution and drug release. This is considered further below.
Assessment of drug release from suppositories and pessaries
In-vitro testing
Since there are few ways of obtaining in-vivo drug release information, this will usually have to be interpreted from in-vitro release data. Present knowledge does not permit the choice of an in-vitro method with a high predictive power for in-vivo performance for these dosage forms. Some aspects can be discussed, however, to give helpful pointers in this respect. Box 42.7 lists the parameters to be examined when testing drug release from suppositories and pessaries in vitro.
The temperature to be used for testing rectal and vaginal dosage forms is easily defined as the body temperature. Although for most practical purposes this temperature can be set at 37 °C, this is not so for, especially, fatty-based suppository and pessary testing. Since the body temperature may be as low as 36 °C at night, this implies that the release rate measured at 37 °C may be an overestimate. Similarly comparing bases at 37 °C may lead to erroneous conclusions. The temperature at which testing is performed might be crucial, especially when ageing has occurred following storage. Special attention should therefore be given to the actual testing temperature.
In the set-up shown in Figure 42.3, the temperature at the surface of the water layer inside the tube, where molten suppository material is gathered, may be a few degrees lower than the bulk temperature. By choosing the right dimension and closing the tube at the upper side, this problem is eliminated.
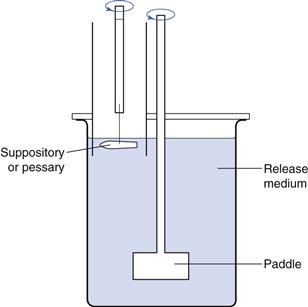
Fig. 42.3 Tube apparatus for testing of sedimentation-controlled release from suppositories and pessaries.
The contact area in the rectum or vagina over which spreading occurs cannot be standardized without introducing either an overestimate or an underestimate. In Figure 42.3 the contact area is relatively small (i.e. approximately 10 cm2) compared to the total surface area of the rectum or vagina. This type of apparatus is therefore intended to be used for comparative studies only and not for a complete in-vivo simulation. At present no method is available which closely mimics the in-vivo situation.
Another parameter to be considered is the release medium. Insufficient information is available on the actual composition and structure of rectal and vaginal fluids to allow the design of precise simulation fluids. A relatively large volume of water or buffer solution is usually chosen. Since the rate-limiting step in the bioavailability from fatty suppositories and pessaries is very often the release from the suppository, it seems reasonable to include mucins which control the viscosity in vivo. The large volume in most in-vitro methods would then also not be so important. More difficult is the choice of buffer and especially its strength, since little is known about this factor in the in-vivo situation. For water-soluble vehicles the problem is even greater, and essentially no ideal volume and composition of release medium has yet been found.
Interest has been directed towards ways of incorporating a pressure feature in release testing of rectal products. It is clear that rectal motility has an effect on spreading and may influence bioavailability. Attempts have been made to incorporate this feature in the design of a release testers but no successful design is currently available.
Often membranes have been used in apparatus for assessing drug release from suppositories and pessaries. They are used to envelop the suppository in a smaller volume of release medium. This has the enormous drawback in that the release, as measured in the outer compartment, is not equal to the actual release taking place in the inner compartment. Most results published do not take into account that the membrane itself may form a resistance to passing drug molecules and that the actual release may be underestimated. It seems advisable therefore to avoid membranes in a release test, whenever possible.
In-vivo considerations
Studies to evaluate release of an active ingredient from the formulation in a dissolution system acts as a surrogate for in-vivo drug release and performance of a formulation. The data obtained from in-vitro tests is extrapolated or correlated with in-vivo behaviour. The test systems and in-vitro/in-vivo correlation for most oral preparations are well established; however, the information available for rectal and vaginal preparations is limited. Some of the reasons for this gap in knowledge are the small volume of research being carried out, biological complexities, lack of appropriate animal models and the differences in practices in use of these routes of drug delivery around the world. A number of different systems have been explored by scientists, but there is a lack of universally accepted or compendial models.
In-vivo tests depend upon the drug and biological activity. Tests for assessing distribution, spreading, retention, and efficacy are carried out in animal models, such as rabbits, monkeys and sheep. A test for vaginal irritation is usually carried out in rabbits and is normally the first screening test in developing a new drug, excipient or formulation. Whenever possible, data should be obtained in humans, since at present no animal model is available which is sufficiently reliable.
For a more detailed discussion on the general aspects of bioavailability testing, and in-vitro/in-vivo correlations, Chapter 21 can be consulted.
Reference
1. Tondury G. Angewandte and Topographische Anatomie. 5th edn Stuttgart, Germany: Georg Thieme Verlag; 1981.
Bibliography
1. de Blaey CJ, Polderman J. Rationales in the design of rectal and vaginal delivery forms of drugs. In: Drug Design. New York: Academic Press; 1989;Ariëns EJ, ed. Drug Design vol. 9.
2. de Boer AG, Molenaar F, de Leede LGJ, Breimer DD. Rectal drug administration: clinical pharmacokinetic considerations. Clinical Pharmacokinetics. 1982;7:285–311.
3. British Pharmacopoeia (2011) British Pharmacopoeia Commission Secretariat, UK.
4. Garg S, Kandarapu R, Vermani K, et al. Development pharmaceutics of microbicide formulations Part I: preformulation considerations and challenges. AIDS Patient Care and STDs. 2003;17(1):17–32.
5. Garg S, Kandarapu R, Vermani K, et al. Development pharmaceutics of microbicide formulations: Part II, Formulation and evaluation challenges. AIDS Patient Care and STDs. 2003;17(8):377–399.
6. Garg S, Vermani K, Kohli G, Kandarapu R, Zaneveld LJ, et al. Survey of vaginal formulations available on the Indian market: physicochemical characterization of selected products. International Journal of Pharmaceutical Medicine. 2002;16(3):141–152.
7. Garg S, Anderson RA, Chany CJ, Waller DP, Diao XH, Vermani K. Properties of a new acid-buffering bioadhesive vaginal formulation (ACIDFORM). Contraception. 2001;64(1):67–75.
8. Gupta J, Tao JQ, Garg S, Al-Kassas R. Design and development of an in-vitro assay for evaluation of solid vaginal dosage forms. Pharmacology and Pharmacy. 2011;2:289–298.
9. Müller B, ed. Suppositorien. Stuttgart, Germany: Wissenschaftliche Verlagsgesellschaft mbH; 1986.
10. Romano J, Malcolm K, Garg S, Rohan LR, Kaptur P. Microbicide delivery: formulation technologies and strategies. Current Opinions in HIV and AIDS. 2008;3:558–566.
11. Rowe RC, Sheskey PJ, Cook WG, Fenton ME, eds. Handbook of Pharmaceutical Excipients. 7th edn London: Pharmaceutical Press; 2012.
12. Rytting JH. Rectal route of peptide and protein delivery. In: Lee VH, ed. Peptide and Protein Drug Delivery. New York: Marcel Dekker; 1990.
13. Shitut NR, Rastogi SK, Singh S, Kang F, Singh J. Rectal and vaginal routes of drug delivery. In: Ghosh TK, Jasti BR, eds. Theory and Practice of Contemporary Pharmaceutics. Boca Raton: CRC Press; 2005;455–478.
14. Thoma K. Arzneiformen zur rectalen and vaginalen Applikation. Frankfurt am Main, Germany: Werbe-und Vertriebsgesellschaft Deutscher Apotheker mbH; 1980.
15. Tukker JJ, Blankenstein MA, Nortier JWR. Comparison of bioavailability of tamoxifen after oral and rectal administration as measured by plasma levels of tamoxifen and its major metabolite N-desmethyltamoxifen. Journal of Pharmacy and Pharmacology. 1986;38:888–892.
16. Vermani K, Garg S. The scope and potential of vaginal drug delivery systems. Pharmaceutical Science and Technology Today. 2000;3:359–364.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Rectal and vaginal drug delivery
Sanjay Garg and Josef J. Tukker
Chapter contents
Anatomy and physiology of the rectum
Absorption of drugs from the rectum
Anatomy and physiology of the vagina
Absorption of drugs from the vagina
Manufacture of rectal and vaginal dosage forms
Quality control of vaginal and rectal dosage forms
Key points
• Both routes have a use in systemic drug delivery when the oral route is not available.
• These routes are relatively less popular because of their anogenital nature and privacy aspects.
Introduction
Although the oral route is the most commonly used route for drug administration, alternatives, such as parenteral, nasal, ophthalmic, rectal and vaginal may be used because they offer specific advantages. Arguments for choosing the rectal or vaginal route for drug administration include the following:
Besides these apparent advantages, the rectal and vaginal routes also have several drawbacks. These routes are generally disliked by patients because of their anogenital and sexual associations, and hence alternative routes are preferred. Depending on culture and tradition, there are strong feelings of aversion in certain countries, such as the UK and the USA, to rectal administration of drugs, whereas there is complete acceptance in continental and eastern Europe. More rational points include the slow and sometimes incomplete drug absorption and the considerable inter- and intra-subject variations. Moreover, the development of proctitis has been reported with long-term rectal delivery. There are also problems with the large-scale production of suppositories, and of achievement of a suitable shelf-life.
Thus, the rectum and vagina are not routes of first choice. Consequently, these routes of drug delivery are amongst the less desirable, resulting in the market size of the rectal and vaginal formulations being less than 1% of the total pharmaceuticals’ market.
Rectal drug delivery
Introduction
The rectal route is used in many different therapies, intended either for local or for systemic effect.
Local action.
This is desired for the local treatment of pain and itching, mostly due to the occurrence of haemorrhoids (painful, swollen veins in the lower part of the rectum and anus). Locally active drugs include astringents, antiseptics, local anaesthetics, vasoconstrictors, anti-inflammatory compounds and soothing and protective agents. Some laxatives also fall into this category.
Systemic action.
All drugs which are orally administered can be given by this route, and many are in spite of the limitations discussed above. Anti-asthmatic, anti-inflammatory and analgesic drugs are widely administered by the rectal route. Rectal preparations may also be used for diagnostic purposes.
Anatomy and physiology of the rectum
Rectal dosage forms are introduced into the body through the anus and are thus brought into contact with the most caudal part of the gastrointestinal tract, i.e. the rectum. Anatomically, the rectum is part of the colon, forming the final 150–200 mm of the gastrointestinal tract.
The rectum can be subdivided into the anal canal and the ampulla, the latter forming approximately 80% of the organ. It is separated from the outside world by a circular muscle, the anus. The rectum can be considered as a hollow organ with a relatively flat wall surface, without villi and with only three major folds – the rectal valves. The rectal wall is composed of epithelium, which is one cell layer thick, and contains cylindrical cells and goblet cells that secrete mucus. A part of the rectal wall and the rectum’s venous drainage are shown in Figure 42.1.
Fig. 42.1 Venous drainage of the human rectum: 1 middle haemorrhoidal (middle rectal) vein; 2 tunica muscularis; stratum longitudinale; 3 muscularis levator ani; 4 inferior haemorrhoidal (lower rectal) vein; 5 muscularis sphincter ani externus; 6 superior haemorrhoidal (upper rectal) vein; 7 and 8 plexus venosus rectalis (submucosus); 9 skin; 10 venosus marginalis. (Adapted from Tondury, 1981, with permission.)
The total volume of mucus is estimated at approximately 3 mL, spread over a total surface area of approximately 300 cm2. The pH of the mucous layer is reported as approximately 7.5 (i.e. close to neutral) in adults, and slightly alkaline in most children. Furthermore, there seems to be little buffering capacity. This point is discussed in relation to drug absorption later in this chapter.
Under normal circumstances the rectum is empty and filling provokes a defaecation reflex under voluntary control. Data comparing drug absorption from freshly prepared and aged (and therefore more viscous) suppositories suggest that there is sufficient motility to provoke spreading even of relatively viscous suppositories.
Absorption of drugs from the rectum
Absorption of drug from the rectum is primarily by passive diffusion. Because of inter-individual variations and the venous drainage of the rectum, the bioavailability of drugs following rectal administration is very unpredictable. In general, the rate and extent of drug absorption is lower than the oral route, mainly due to the small surface area for absorption.
Knowledge of the venous drainage from the rectum is important for the understanding of drug absorption. As can be seen from Figure 42.1, there are three separate veins. The lower and middle rectal (inferior and middle haemorrhoidal) veins drain into the interior vena cava, hence this blood goes directly to the heart and into the general circulation. In contrast, the upper rectal (superior haemorrhoidal) vein drains into the portal vein and therefore this blood passes through the liver before reaching the heart.
This means that drug molecules from the rectum can enter the general circulation either directly or by passing through the highly metabolizing liver. Drug absorbed in the middle and lower part of the rectum will pass directly to the general circulation and avoid first-pass metabolism in the liver. Bioavailability from the upper part of the rectum will be low for certain drugs, as much will be metabolized by the liver during its ‘first pass’ and only a proportion of the drug molecules (if they are of the high clearance type) will enter the general circulation intact.
Investigations have shown that avoiding the first-passage through the liver is possible by keeping the dosage form, and thus the released drug, in the lower part of the rectum. Compared to the small intestine, this situation is very favorable as most gastrointestinal veins drain into the portal vein.
Insertion of a suppository into the rectum results in a chain of events leading to the absorption of the drug. This is represented in a simplified scheme in Figure 42.2. Depending on the character of its vehicle (see later in this chapter), a suppository will either dissolve in the rectal fluid or melt on the mucous layer. Since the volume of rectal fluid is so small, dissolution of the complete vehicle will be difficult and requires extra water. Due to osmotic effects (of the dissolving vehicle) water is attracted, with a resultant unpleasant sensation for the patient. Independent of the vehicle type, drugs dissolved in the suppository will diffuse out towards the rectal membranes. Suspended drugs will first need to leave the vehicle (if it is water immiscible) under the influence of either gravity or motility movements and then begin to dissolve in the rectal fluid.
Dissolved drug molecules will have to diffuse through the mucous layer and then into and through the epithelium forming the rectal wall. The process of absorption will be by passive diffusion, as it is throughout the whole gastrointestinal tract for almost all drugs. Active transport processes, as found in the upper regions of the gastrointestinal tract, have not been shown to be present in the rectal area.
For a generalized discussion on drug absorption, the reader is referred to Part 4 of this book. However, some specific points concerning rectal absorption will be discussed here. Box 42.1 provides a summary of the physiological factors that are important in rectal absorption.
The quantity of fluid available for drug dissolution is very small (approximately 3 mL) spread in a layer of approximately 100 µm thick over the organ. Only under non-physiological circumstances is this volume enlarged, e.g. by osmotic attraction by water-soluble vehicles or during diarrhoea. Thus, the dissolution of poorly water-soluble drugs can easily be the slowest step in the absorptive process.
Properties of the rectal fluid, such as composition, viscosity and surface tension, are essentially unknown and have to be estimated from data available for other parts of the gastrointestinal tract. The pH and the buffering capacity of the rectum have been considered earlier in this chapter. The rectum is usually empty, except temporarily when faecal matter arrives from the colon. This material is either expelled or transported back into the colon, depending on voluntary control of the anal sphincter.
The rectal wall may exert pressure on a suppository present in the lumen by two distinct mechanisms. First, the abdominal organs may simply press on to the rectum, especially when the body is in an upright position. This may stimulate spreading and thus promote absorption. The second source of pressure is the motility of the muscles of the rectal wall, which may originate from the normally occurring colonic motor complexes. These are waves of contractions running over the wall of the colon in a caudal direction and are associated with the presence of food residues in the colon.
In contrast with the upper part of the gastrointestinal tract, no esterase or peptidase activity is present in the rectum, resulting in a much greater stability of peptide-like drugs (thus allowing attempts at their delivery by this route). Administration of these compounds using the rectal or vaginal routes has been satisfactory but only if absorption enhancers, such as surfactants are used concomitantly. All types of surfactants seem to be effective, but polyoxyethylene lauryl alcohol ether seems to have the greatest effect. One major drawback of these enhancers, however, is their irritation of the rectal mucosa in the long term. Less irritating enhancers are thus needed to explore this interesting area of drug delivery in greater depth.
Rectal dosage forms
The advantages and limitations of rectal drug administration are outlined in Box 42.2. Several categories of rectal preparations for drug delivery are available: suppositories, rectal capsules, rectal solutions, emulsions and suspensions, powders and tablets for rectal solutions and suspensions, semi-solid rectal preparations, rectal foams and rectal tampons. Of these, suppositories are the most commonly used and these are discussed first, followed by a briefer description of some of the other rectal dosage forms.
Formulation of suppositories
Suppositories are the primary, but not exclusive, dosage form used for the administration of drugs via the rectal route. Suppositories are single-dose preparations with a shape, volume and consistency appropriate for rectal administration. Administration of other suppository-type products (bougies) through other body orifices, e.g. ear, nose and urethra, is rarely undertaken and is not discussed here. Alternative dosage forms for the rectal route are discussed later.
Rectal suppositories are formulated in different shapes and sizes (usually 1–4 g). They contain one or more active substances dispersed or dissolved in a suitable base that may be soluble or dispersible in water or may melt at body temperature. Excipients such as diluents, adsorbents, surface-active agents, lubricants, antimicrobial preservatives and colouring matter may be added if necessary. Their drug content varies widely from less than 0.1% up to almost 40%.
Vehicle (suppository base)
An ideal suppository vehicle or base should melt, dissolve or disperse at body temperature. It should be non-irritating, physically and chemically stable, and pharmacologically inert. Compatibility with a range of drugs is a desirable feature. It should also be convenient to handle during manufacturing and storage. Leakage following administration is likely to be less problematic if the viscosity of the vehicle after melting or dispersion is high.
There are two main classes of vehicles in use: glyceride-type fatty bases and water-soluble bases. Although the ideal vehicle has not been found, the large variety of bases which are available enables a well-considered choice for every drug that has to be formulated into a suppository.
Choosing the optimum base requires much practical experience and can at present only partly be guided by scientifically sound data. However, some general guidelines can be given.
Requirements of the vehicle. There is no doubt that a suppository should either melt after insertion in the body or dissolve in (and mix with) the available volume of rectal fluid. For fatty bases, this means a melting range lower than approximately 37 °C (formulators should be aware that the body temperature might be as low as 36 °C at night). The melting range should be small enough to give rapid solidification after preparation, thus preventing agglomeration or sedimentation of suspended, especially high-density, drug particles. When the solidification rate is high, for example when rapid cooling is applied, this may result in fissures in the suppository. The melting temperature range, on the other hand, should be sufficiently wide to permit easy preparation, which may take a considerable length of time on an industrial scale. During solidification, a suppository should exhibit enough volume contraction to permit removal from the mould or plastic former.
The viscosity of the molten base plays an important role from both a technological and a biopharmaceutical viewpoint. During preparation, the viscosity determines not only the flow into the moulds but also the separation of drug particles. Clearly a compromise has to be found. During and after melting in the rectal cavity, the suppository mass is forced to spread over the absorbing surface. The rate of spreading is determined partly by the viscosity of the suppository at body temperature. The rate of transportation of drug particles from within the base to the interface with rectal fluid, in order to be released and absorbed, will also be affected by the viscosity.
A good suppository base should be chemically and physically stable during storage as a bulk product and after preparation into a suppository. It should have no incompatibility with, and should permit optimal release of, the drug it contains.
Clearly, this list of requirements cannot always be completely fulfilled and often an acceptable compromise is the best that can be expected.
Fatty vehicles.
The fatty vehicles in use nowadays are almost exclusively semi- or fully synthetic. Theobroma oil (also known as cocoa butter) – once a very commonly used base – is no longer used because of its many disadvantages in practice, such as its polymorphic behaviour, insufficient contraction during cooling, low softening point, chemical instability, poor water-absorptive power and its price.
A number of substitutes have been developed, which are becoming increasingly popular. The newer semi-synthetic fatty vehicles have few or none of the problems mentioned above. Commercial examples include: Cotmar, Dehydag, Fattibase, Suppocire and Witepsol. These are mixtures of natural or synthetic vegetable oils, consisting of mixed triglycerides of C12-C18 saturated fatty acids, waxes and fatty alcohols. By using a combination of components, they can be designed to have an adjustable range of melting points, e.g. different grades of Witepsol have melting points ranging from 29 to 44 °C.
The ‘hydroxyl number’ of these bases is a parameter that refers directly to the amount of mono- and di-glycerides present in the fatty base. A high number means that the base is less hydrophobic and its power to absorb water is high. This may lead to an increased rate of decomposition for drugs that are easily hydrolysed. This capacity could also lead to the formation of a w/o emulsion in the rectum. This is generally to be avoided because of a very low drug release rate. An advantage of a high hydroxyl number is the larger melting and solidifying ranges that permit easier manufacture.
Water-soluble vehicles.
Hydrophilic water-soluble (or miscible) vehicles are much less frequently used. They comprise the classic glycerinated gelatin (glycerol-gelatin) and polyethylene glycol (macrogol) bases. Glycerol-gelatin bases are mostly used for laxative purposes and in vaginal dosage forms (see below).
Glycerinated gelatin is a mixture of glycerol, gelatin and water. The mixture forms a translucent, gelatinous mixture, dispersible in the rectum. The ratio of glycerol, gelatin and water can affect the dispersion time and thus the duration of action. A higher proportion of gelatin in the mixture makes it more rigid and longer acting. The following is an example composition of the base:
Preservatives such as methyl and/or propyl parabens may be added. Formulations made with gelatin and glycerol tend to be hygroscopic and require well-closed containers for packaging.
Polyethylene glycols (PEGs) are very versatile polymers in their properties and applications. They consist of mixtures of polyethylene glycols of different molecular weight. Lower molecular weight PEGs (PEG 400 and 600) are liquid, those around 1000 are semi-solid and those above 4000 are waxy solids. Different molecular weights of PEG can be combined to produce the desired properties, as seen in Table 42.1.
Table 42.1
Compositions of PEG bases with different physical characteristics
| Base A | Base B | |
| PEG 1000 | 95% | 75% |
| PEG 4000 | 5% | 25% |
| Properties | Low melting temperature, immediate drug release | Higher melting temperature, sustained drug release |
The melting point of these vehicles is well above body temperature, which means that they need to mix with the rectal fluid. PEGs of all molecular weights are miscible with water and rectal fluids, thereby releasing drug by dispersion; the available volume of rectal fluid is too small for true dissolution. Because of their high melting point, PEG-based formulations are especially suited for application in tropical climates but several disadvantages have to be considered.
They are hygroscopic and therefore attract water after administration, resulting in an uncomfortable sensation for the patient. Incorporation of at least 20% water in the base and moistening before insertion can help to reduce this problem. A considerable number of incompatibilities with various drugs have been reported. Due to the solubilizing character of this base (which has a low dielectric constant) drugs may tend to remain in the base, and drug release may be slow.
PEG bases can develop peroxides on storage, therefore airtight packaging is recommended and the formulation should be monitored for peroxides during stability studies when ascertaining shelf-life.
Choice of vehicle.
A summary of the points which are important for the choice of a suppository base is given in Box 42.3
[/not-level-membership-for-basic-science-category]
