[level-membership-for-neurosurgery-category]
CHAPTER 73 Rationale for Surgical Interventions in Movement Disorders
Neuroscience research has led to major insights into the structure and function of the basal ganglia and the pathophysiologic basis of basal ganglia disorders such as Parkinson’s disease (PD).1–4 Several factors have contributed to progress in this field, specifically, the availability of effective animal models, improved methods of physiologic and pharmacologic investigation, and the development of powerful brain imaging methods for studies of patients and animal models of these disorders.
Overview of Anatomy and Function of the Basal Ganglia
PD and other movement disorders are increasingly being recognized as “circuit disorders” that involve not only the basal ganglia but also related nodes in the thalamus and the cerebral cortex. This chapter focuses on a description of these larger cortical-subcortical circuits. The anatomic organization of the basal ganglia is discussed in detail in Chapter 74.
Anatomic and physiologic studies have demonstrated that the basal ganglia are components of a family of parallel reentrant brain circuits in which cortical information is sent to the basal ganglia, processed, sent to the thalamus, and then returned to the cerebral cortex.5–9 The different circuits are segregated but share anatomic features, thus supporting the view that the basal ganglia and cortex interact in a modular fashion in which similar processing steps are carried out in each module even though the different modules are engaged in different functions. Depending on the presumed function of the cortical region that is involved in these different circuits, the circuits are commonly designated as “motor,” “oculomotor,” “prefrontal,” and “limbic” (Fig. 73-1). As shown in Figure 73-1, dysfunction within the individual circuits may be associated with specific disease states.
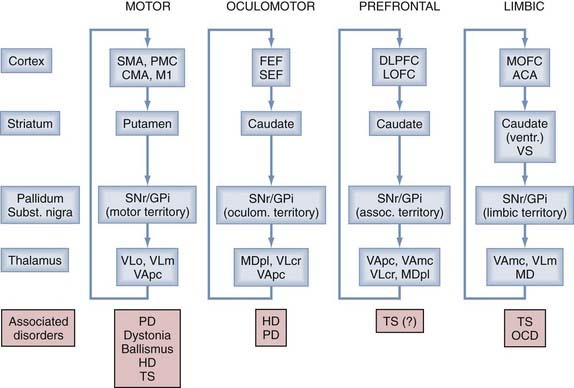
Movement disorders prominently affect the “motor” circuit, which takes origin in the frontal cortical precentral and postcentral sensorimotor areas, including the primary motor cortex (M1), the supplementary motor area (SMA), the premotor cortex (PMC), the cingulate motor area (CMA), and interconnected sensory cortical areas, and involves specific “motor” portions in each of the basal ganglia structures and thalamus. Similar to the other transbasal ganglia circuits, the motor circuit is at least partially closed, with thalamocortical projections terminating in the same frontal cortical regions from which the circuit originates. The anatomy of the motor circuit is shown in greater detail in Figure 73-2A.
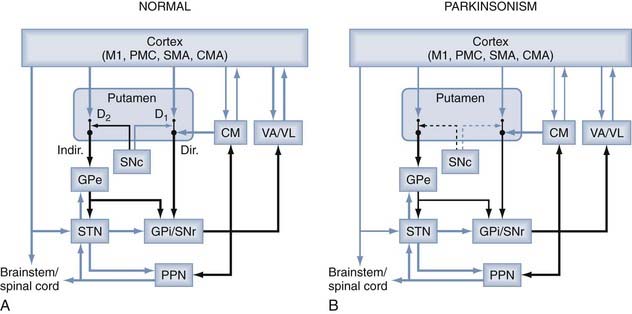
FIGURE 73-2 Parkinsonism-related changes in overall activity in the basal ganglia–thalamocortical motor circuit. Black arrows indicate inhibitory connections; blue arrows indicate excitatory connections. The thickness of the arrows corresponds to their presumed activity. CM, centromedian nucleus; Dir., direct pathway; D1, D2, dopamine receptor subtypes; Indir., indirect pathway; PPN, pedunculopontine nucleus; SNc, substantia nigra pars compacta; STN, subthalamic nucleus. For other abbreviations, see the legend to Figure 73-1.
(From Galvan A, Wichmann T. Pathophysiology of parkinsonism. Clin Neurophysiol. 2008;119:1459.)
The striatum and STN are the main entry points for cortical input, whereas the GPi and SNr provide basal ganglia output to the thalamus. The corticostriatal projections are topographically organized.5–7,10,11 Movement-related cortical input terminates in the postcommissural putamen, and nonmotor projections terminate in other areas of the striatum. Thus, prefrontal cortical areas project to the caudate nucleus and the precommissural putamen, and projections from limbic cortices, amygdala, and hippocampus terminate preferentially in the ventral striatum. The cortical-subthalamic projections are also topographically arranged.12,13 Afferents from M1 reach the dorsolateral STN, whereas afferents from the PMC and SMA innervate mainly the mediodorsal third of the nucleus.14
The striatum is linked to the basal ganglia output structures, GPi and SNr, via two distinct pathways, the so-called direct and indirect pathways (Fig. 73-2).1,5,7 The direct pathway arises from sets of striatal medium spiny neurons (MSNs) that project monosynaptically to neurons in the GPi and SNr. These neurons also contain the neuropeptides substance P and dynorphin and preferentially express dopamine D1-like receptors. It is thought that MSNs in the direct pathway receive a larger share of the thalamostriatal pathway than do neurons in the indirect pathway. The indirect pathway arises from a set of striatal MSNs that project to the GPe.15,16 The striatal neurons that give rise to the indirect pathway preferentially express enkephalin and dopamine D2-like receptors.17,18 Although most authors emphasize the separation of these pathways, single-cell labeling of striatal MSNs has shown that at least some send collaterals to both segments of the globus pallidus (thus participating anatomically in both the direct and indirect pathways).19,20
Similar to the corticostriatal projections, the direct and indirect pathways are topographically organized. For instance, populations of GPe neurons within the sensorimotor, associative, or limbic territory are reciprocally connected with populations of neurons in the same functional territories of the STN, and neurons in each of these regions, in turn, innervate the same functional territory of the GPi.15,16
In terms of basal ganglia output, the caudolateral “motor” territory of the GPi projects almost exclusively to the posterior part of the ventrolateral (VL) nucleus, which sends projections toward the SMA,21,22 M1, and PMC.23 The outflow from pallidal motor areas directed at M1, PMC, and SMA appears to arise from separate populations of pallidothalamic neurons.23 These findings indicate that the larger “motor circuit” is composed of segregated subcircuits centered on the individual cortical motor areas.24,25 The more rostromedial associative areas of the GPi project preferentially to the parvocellular part of the ventral anterior (VA) and the dorsal VL nucleus (VLc in macaques),26,27 specifically to thalamic areas preferentially connected to the prefrontal cortex.28,29
Collaterals from the pallidofugal and nigrofugal projections also reach the intralaminar nuclei of the thalamus, the centromedian (CM) and parafascicular (PF) nuclei. These projections are part of a system of segregated basal ganglia–thalamostriatal feedback projections.27,30 In primates, the CM nucleus receives input from motor areas in the GPi and projects to the motor portions of the putamen and STN, whereas PF input and output are related to the associative and limbic territories of the basal ganglia.31,32 CM terminations are found mostly along the shafts of striatal direct pathway MSNs,33,34 separate from the sites of termination of cortical input and of dopaminergic synapses.30,35–41 Basal ganglia output also reaches the pedunculopontine nucleus (PPN),42,43 which in turn gives rise to ascending projections to the basal ganglia, thalamus, and basal forebrain and to descending projections to the pons, medulla, and spinal cord.44,45
Electrophysiologic and metabolic mapping studies in animals and functional imaging data in humans support the view that the anatomically and physiologically defined basal ganglia “motor” areas are indeed involved in movement.25,46–48 The model of the basal ganglia–thalamocortical motor circuit shown in Figure 73-2 is often used as the basis for speculations regarding the function of the basal ganglia in movement. The model poses that activation of MSNs that give rise to the direct pathway reduces inhibitory basal ganglia output from targeted neurons with subsequent disinhibition of related thalamocortical neurons.5,49,50 The net effect of this process is increased activity in appropriate cortical neurons that results in facilitation of the movement. By contrast, activation of MSNs that give rise to the indirect pathway would lead to increased (inhibitory) basal ganglia output on thalamocortical neurons and to suppression of movement. The balance between direct and indirect pathway activity may regulate the overall amount of movement, whereas specific activation patterns (for instance, a center-surround type of activation involving the direct and indirect pathways) may limit the extent or duration of ongoing movements.
Other views of basal ganglia function are currently evolving.51–55 Thus, synaptic processing and modification of synaptic strength in the striatum, or alteration of the level of interneuronal synchrony, may give the basal ganglia a role in the regulation of habit formation or procedural learning.51–5355
In models of basal ganglia function, the modulatory effects of dopamine on striatal transmission are highly important. Dopamine is released in the striatum from terminals of projections from the SNc and modulates the activity of the basal ganglia output neurons in the GPi and SNr by facilitating corticostriatal transmission on the direct pathway and inhibiting corticostriatal transmission on MSNs that give rise to the indirect pathway. These opposing actions are probably mediated by the two different sets of dopamine receptors (D1-like and D2-like receptors) that are differentially expressed in these pathways.56–59 Through the different effects of activation of the direct and indirect pathways, the net effect of striatal dopamine release appears to be reduction of basal ganglia output to the thalamus and other targets. Evidence indicates that dopamine also acts directly on receptors in the STN and pallidum to influence discharge patterns and rates in these structures.
Dopamine receptor activation may not only act to modulate the activity of the direct and indirect pathways but might also have a role in the proposed learning functions of the basal ganglia. Activation of dopamine receptors on MSNs has been shown to be involved in the induction of long-term potentiation and depression at glutamatergic (presumably corticostriatal) synapses.60–64 Recent research has elucidated complex interactions between dopaminergic transmission and transmission involving adenosine, glutamate receptors, and endocannabinoid receptors in this process.61,63–66
Parkinsonism
The cardinal features of PD—the triad of akinesia/bradykinesia, tremor at rest, and muscular rigidity—result from decreased dopaminergic transmission in the motor portions of the basal ganglia, in particular, the putamen, as a result of progressive loss of dopaminergic neurons in the SNc. Dopaminergic replacement therapies are highly effective in reversing these features of the disorder. These dopamine-responsive features are often accompanied by other issues that are poorly or entirely unresponsive to dopaminergic medications, such as depression, autonomic dysfunction, sleep disorders, cognitive impairment, and gait/balance problems. Although some of these problems may result, in part, from decreased dopamine within the nonmotor portions of the basal ganglia, widespread progressive pathologic changes in the brainstem, thalamus, and eventually the cerebral cortex appear to play a major role.67
Pathophysiology of Parkinsonism
The following paragraphs are focused on the motor aspects of PD that result from dopamine deficiency. Study of these changes has been facilitated by the development of animal models of dopamine depletion in which changes in the basal ganglia, thalamus, and cerebral cortex can be conveniently studied, including the 6-hydroxydopamine (6-OHDA)-treated rat and primates treated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP).68,69
In early studies of MPTP-treated primates, alterations in activity of the striatopallidal pathways were strongly emphasized. Such changes were suggested by studies of metabolic activity in the basal ganglia that indicated increased synaptic activity in the GPe and GPi.70,71 Possible interpretations of this finding were that the activity of the striatal-GPe connection and the STN-GPi pathway is increased in parkinsonism or that the STN projections to both pallidal segments were overactive. Subsequent microelectrode recording studies in MPTP-treated primates showed a reduction in neuronal discharge in the GPe and increased firing in the STN, GPi, and SNr in comparison to normal controls,72–75 as well as high neuronal discharge rates in the GPi in PD patients undergoing functional neurosurgical procedures.76–78 These changes may be related to one another in that striatal dopamine depletion may lead to increased activity of striatal neurons in the indirect pathway and result in inhibition of the GPe and subsequent disinhibition of the STN and GPi/SNr (Fig 73-2B). A role of local dopamine loss in the extrastriatal structures (specifically in the STN, GPi, and SNr) may play a role as well, specifically with regard to the emergence of abnormal firing patterns in these nuclei (see later).
Other structures and feedback loops, such as those involving the PPN and CM, may contribute to the abnormalities in discharge that are found in the basal ganglia output nuclei. A series of studies have demonstrated that brainstem areas such as the PPN may also be involved in the development of parkinsonian signs. Lesioning of the PPN in normal monkeys was shown to lead to akinesia.79,80 At the cortical level, positron emission tomography (PET) studies in parkinsonian patients have consistently shown reduced activation of motor and premotor areas.46,81
The notion that rate changes in the basal ganglia underlie the development of parkinsonism (the so-called rate model, depicted in Fig. 73-2) is generally supported by studies that have shown that inactivation of the sensorimotor portions of the STN or GPi increases metabolic activity in the cortical motor areas and improves bradykinesia and tremor in parkinsonian subjects. Thus, lesions of the STN, GPi, or SNr in MPTP-treated primates reverse some or all signs of parkinsonism, presumably by reducing basal ganglia output,19,82,83 and GPi or STN lesions are highly effective antiparkinsonian treatments in patients with PD.84–90 However, the “rate model” has largely been rejected because several crucial observations contradict a purely rate-based view of basal ganglia–processing abnormalities. For instance, lesions of the VA/VL nuclei of the thalamus (which completely removes thalamic output) do not lead to parkinsonism and are, in fact, beneficial in the treatment of tremor and rigidity.91,92 Similarly, lesions of the GPi in the setting of parkinsonism improve the cardinal features of PD without producing dyskinesias or other obvious detrimental effects. In fact, they are highly effective in reducing drug-induced dyskinesias.86,87,93
Because of these discrepancies, the rate model has given way to the view that abnormalities in basal ganglia activity other than rate changes are more relevant to the pathophysiology of parkinsonism. Among the most discussed changes in discharge patterns in the basal ganglia of parkinsonian subjects (Fig. 73-3) is the development of oscillatory phenomena.94,95 Abnormal oscillations have been identified in the activity of single neurons in the GPi, SNr, and STN in animals and patients and, more recently, in recordings of local field potentials (LFPs) in patients, and these oscillations may reflect the activity of more extensive networks of connections. The LFP recordings were made by using implanted DBS macroelectrodes as recording probes during the time immediately after the implantation surgery. Recordings in the STN and other basal ganglia areas with this method have demonstrated the presence of LFP oscillations in the 10- to 25-Hz (beta) range in the STN, GPi, and cortex in unmedicated PD patients. The studies have also shown that beta-range oscillations give way to oscillations in the 60- to 80-Hz (gamma) range when the patients are treated with levodopa or when the STN is stimulated at high frequencies.95,96 The prominence of beta-band activity is apparent throughout the basal ganglia–thalamocortical circuitry. For example, the desynchronization of electroencephalography (EEG) oscillations that normally precedes movement was shown to be abnormally small in parkinsonian patients, perhaps interfering with the initiation of movement.97–99
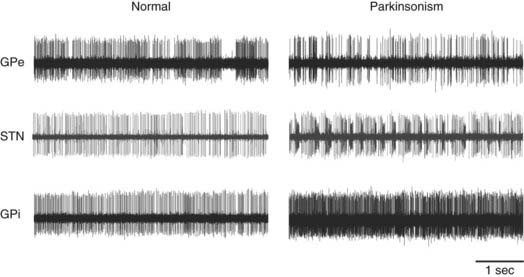
Another parkinsonism-related abnormality in spontaneous discharge is the emergence of abnormal synchrony between neurons. Under physiologic conditions, basal ganglia activity is highly specific in its relation to movement parameters and body part and appears to be segregated even at the cellular level, where neighboring neurons are rarely found to fire in synchrony.100 In parkinsonism, this level of segregation is lost, and the discharge of neighboring neurons is often found to be correlated and abnormally synchronized.95,101
Finally, the proportion of cells in the STN, GPi, and SNr that discharge in bursts (oscillatory or nonoscillatory) is greatly increased in parkinsonism.73,95,101–103 Oscillatory burst discharge patterns are also seen in conjunction with tremor, which may reflect tremor-related proprioceptive input or more active participation of basal ganglia in the generation of tremor.
Although spontaneous basal ganglia firing has been extensively studied in MPTP-treated monkeys, much less is known about the way information processing is altered in the basal ganglia. Studies of changes in neuronal firing patterns preceding or following active movements are difficult to perform and interpret in akinetic or bradykinetic animals. However, studies of neuronal responses to passive limb manipulation have been carried out and showed that such responses in the STN, GPi, and thalamus occur more often, are more pronounced, and have widened receptive fields after treatment with MPTP.72,73,101,104
The importance of specific electrophysiologic features in basal ganglia, thalamic, or cortical activity for development of the behavioral signs of parkinsonism remains unclear. Although burst-like, synchronized oscillatory activity in the basal ganglia–thalamocortical circuits is associated with parkinsonism,105 direct links between oscillatory activity and specific parkinsonian deficits have not been established. In fact, recent studies in monkeys that underwent a gradual MPTP treatment protocol in which parkinsonism was slowly induced have cast doubt on the notion that synchronous oscillatory firing contributes strongly to (early) parkinsonism.106 In these single-neuron recording studies, synchrony and oscillations in neuronal spiking activity were detected only after the development of bradykinesia and akinesia. Similarly, recent rodent experiments have suggested that abnormal oscillations in the basal ganglia do not result simply from (acute) lack of dopaminergic stimulation but may rather be due to the chronic absence of dopamine.107 Such “late” changes could be related to anatomic alterations secondary to dopamine depletion, such as loss of the dendritic spines of MSNs,108–110 which has been described as homeostatic pruning in response to altered glutamatergic transmission in the striatum.108,109
Rationale for Treating Parkinsonism by Surgical Interventions
Two general types of procedures are used to surgically treat patients with parkinsonism: lesioning and DBS. Unilateral lesioning of the sensorimotor territory of the GPi results in significant contralateral antiparkinsonian effects and significantly reduces drug-induced involuntary movements and motor fluctuations. For DBS, a stimulation electrode is introduced into the same targets in the GPi and STN, and high-frequency stimulation is applied by way of an implanted, externally programmable pulse generator. DBS alleviates parkinsonian motor signs and reduces the severity of “off” periods and the dyskinesias, dystonia, and motor fluctuations that result from long-term drug administration.111–113
The mechanism of action of these interventions remains unclear. The therapeutic benefits of GPi and STN lesions suggest that a total lack of basal ganglia output is more tolerable in PD patients than disruptive abnormal output on the brainstem and thalamocortical systems. Although it was initially believed that DBS, like lesioning, functionally inactivates the target because both procedures lead to almost identical outcomes, considerable evidence now suggests more complex mechanisms of action of DBS. The electrophysiologic effects of GPi or STN DBS appear to depend on the distance of the stimulated elements from the electrode and may differ between the elements themselves. Stimulation in the STN has been demonstrated to evoke complex excitatory effects in the GPi, one of the primary recipients of STN efferents,114 and may alter the oscillatory resonance characteristics of the STN-GPi network.115 Modeling studies have shown that STN DBS may inhibit the somata of STN cells through activation of local release of γ-aminobutyric acid from GPe afferents while simultaneously directly activating STN axons116 and thereby activating synaptically innervated GPi cells. Anterograde activation of efferents plus retrograde activation of afferents by DBS is likely to secondarily alter firing patterns in the associated corticosubthalamic and thalamocortical circuitry. Recent modeling studies have suggested that this may strongly affect (normalize) thalamic bursting activity.117 STN DBS has also been shown to normalize intracortical inhibitory mechanisms in studies of the effects of transcranial magnetic stimulation,118 and functional imaging studies have shown that pallidotomy or DBS of the STN or GPi acts to restore relatively normal levels in the frontal motor areas.87,119,120
DBS at other nodes along the basal ganglia–thalamocortical motor circuitry has also been shown to be useful. For instance, parkinsonian tremor is effectively treated by thalamic DBS at the border between the thalamic nucleus ventralis oralis and nucleus ventralis intermedius.121,122 Stimulation of the PPN may also be beneficial.123–126
Dystonia
Dystonia may arise from a variety of disease processes, and many of these processes involve the basal ganglia. Dystonia is categorized as “primary” if no clear cause is identified and as “secondary” if an underlying structural or biochemical defect is present. One of the main forms of generalized dystonia, idiopathic torsion dystonia, is caused by a genetic defect in the DYT1 gene on chromosome 9.127,128 Secondary dystonia may occur in the setting of PD, after exposure to drugs (tardive dystonia), as a sequela of focal damage to the striatum (particularly the putamen) or the thalamus, or even after peripheral injuries. The onset of dystonia is typically delayed for weeks or months after the inciting lesion.
Pathophysiology of Dystonia
The pathophysiology of dystonia is far less clear than that of PD and potentially varies substantially between different forms of dystonia. Research in this field is hampered by the fact that no fully convincing mouse or primate model of dystonia exists129 and, of course, by the fact that clear pathologic changes do not appear to be present in most cases of dystonia.
There is some evidence that a disturbance in the dopamine system may be involved in DYT1 dystonia130 and other forms of dystonia. For instance, dystonia may develop acutely in normal individuals treated with dopamine receptor blocking agents or appear after long-term treatment with these drugs (tardive dystonia). In PD, dystonia usually develops in patients who have been exposed to dopaminergic drugs, but it may also occur as an early sign of the disease itself, independent of medications. Dystonia is also present in a group of patients with familial dystonia and parkinsonian features who respond dramatically to treatment with low-dose levodopa (dopamine-responsive dystonia [DRD]).131,132 Most of these patients suffer from a genetic defect in dopamine synthesis caused by reduced activity of guanosine triphosphate cyclohydrolase,133–135 the rate-limiting enzyme in the biosynthesis of a cofactor of the dopamine-synthesizing enzyme tyrosine hydroxylase, tetrahydrobiopterin.
When dystonia results from lesions affecting the striatum or its dopaminergic supply,136 these lesions may affect the affinity or number of dopamine receptors in the nonlesioned portion of the striatum or may lead to reorganization of striatal topography, which eventually results in altered activity in the basal ganglia output structures. Metabolic studies in primates have suggested that dystonia may be associated with a reduction of activity along the putamen-GPe connection and increased inhibition of the STN and GPi by GPe efferents.137,138 Other PET and single-cell recording studies in human patients with dystonia have emphasized changes in activity in the direct pathway instead (see later).
Involvement of the direct and indirect pathways in the pathophysiology of dystonia is also supported by pharmacologic studies. For instance, it has been shown that D2-like receptor antagonists may induce dystonia, presumably by increasing striatal outflow to the GPe via the indirect pathway, whereas D1-like receptor antagonists may be beneficial in this regard, presumably by reducing striatal outflow to the GPi along the direct pathway.139,140 These data suggest that a relative increase in activity along the direct pathway (versus that along the indirect pathway) contributes to dystonia. Single-cell recording studies in human patients undergoing functional neurosurgical procedures are in partial agreement with this concept. These studies have demonstrated that discharge rates in the GPe and GPi are lower than expected,141–146 although such changes in firing rate were not found by Hutchison and colleagues.147
Similar to parkinsonism, oscillatory activity in the basal ganglia may also be involved in the pathophysiology of dystonia. Low-frequency coherence between the discharge of neurons in the basal ganglia142 or thalamus146,148 and the electromyographic activity of dystonic muscles has been demonstrated. LFP recordings from DBS electrodes implanted in the GPi have shown increased power in the 4- to 10-Hz band149,150 and high correlation with simultaneously recorded electromyographic activity.149,151
True to the concept that dystonia is a circuit disorder similar to PD, there is substantial evidence for altered cortical functioning in dystonia, specifically from imaging and electrophysiologic studies that have used transcranial magnetic stimulation and other methods. These studies are strongly suggestive of abnormal plasticity in dystonia. For instance, PET studies in dystonic patients have demonstrated widespread changes in activity of the prefrontal areas,152–155 specifically, changes involving the SMA, anterior cingulate, and dorsolateral prefrontal motor areas.152,153,155 In focal dystonia, abnormal somatotopic maps were demonstrated in M1,156,157 with increased intracortical excitability in the motor areas158 and a decreased Bereitschaftspotential/contingent negative variation.159–162 Patients with writer’s cramp were also shown to have an abnormally small degree of beta-band EEG desynchronization.163
Sensory abnormalities may also play a role in dystonia. There is convincing evidence of reduced corticocortical inhibition in the sensory system164–167 and increased and improperly modulated precentral sensory evoked potentials (N30).168–174 In addition, single-cell recording and imaging studies have suggested altered somatotopic representation at the cortical,175–177 putamen,178 and thalamic levels,148,179 although such changes were not seen in recent GPi recording studies.143,180
Rationale for Treating Dystonia by Surgical Interventions
Despite the lack of an overarching pathophysiologic model of dystonia, the disease has been treated for decades by inducing pallidal or thalamic lesions and, more recently, by DBS. Specifically, GPi DBS is currently used in many cases of advanced intractable dystonia. This procedure is highly effective in cases of generalized dystonia181–183 but less so for secondary dystonia.184 Other emerging indications for GPi DBS are cervical and tardive dystonia.182,185,186 Recent case reports suggest that STN DBS may also be effective.20,187
As is the case with ablative or DBS surgeries for PD, selection of the surgical target for treatment of dystonia is primarily driven by our knowledge of the functional anatomy of the basal ganglia. The effects of surgical interventions for dystonia remain unclear. It has been shown that GPi DBS, administered to motor portions of the GPi, modulates activity in the basal ganglia–thalamocortical motor circuit. PET activation studies have shown that GPi DBS reverses the overactivity of the motor cortical areas present in dystonia,188 and electrophysiologic studies have demonstrated that GPi DBS enhances motor cortex excitability through modulation of thalamocortical projections.189 In distinction to the use of neurosurgical treatments in PD, the clinical benefits of DBS or pallidotomy in dystonia are usually delayed, often by weeks or months,190 thus suggesting that anatomic or functional remodeling of neuronal interactions, reflecting abnormal plasticity within the basal ganglia–thalamocortical circuitry, may be necessary for the beneficial effects to occur.
Tourette’s Syndrome
Rationale for Treating Tourette’s Syndrome by Surgical Interventions
In the small group of patients in whom incapacitating tics persist despite conventional treatments, surgical procedures may be useful.191 Given the paucity of knowledge regarding the pathophysiology of the disease, target selection and other aspects of these procedures remain entirely empirical. Several targets are currently being explored, including the medial thalamus CM/PF nuclei,192–194 the motor and limbic portions of the GPi,192 and the anterior limb of the internal capsule.195 In small open-label studies, these procedures are reported to substantially reduce vocal and motor tics.
Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12:366.
Alexander GE, DeLong MR, Strick PL. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu Rev Neurosci. 1986;9:357.
Braak H, Del Tredici K, Rub U, et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197.
Breakefield XO, Blood AJ, Li Y, et al. The pathophysiological basis of dystonias. Nat Rev Neurosci. 2008;9:222.
Brown P. Oscillatory nature of human basal ganglia activity: relationship to the pathophysiology of Parkinson’s disease. Mov Disord. 2003;18:357.
Butefisch CM, Boroojerdi B, Chen R, et al. Task-dependent intracortical inhibition is impaired in focal hand dystonia. Mov Disord. 2005;20:545.
Ceballos-Baumann AO, Brooks DJ. Basal ganglia function and dysfunction revealed by PET activation studies. Adv Neurol. 1997;74:127.
Coubes P, Cif L, El Fertit H, et al. Electrical stimulation of the globus pallidus internus in patients with primary generalized dystonia: long-term results. J Neurosurg. 2004;101:189.
DeLong MR. Primate models of movement disorders of basal ganglia origin. Trends Neurosci. 1990;13:281.
Doyon J. Motor sequence learning and movement disorders. Curr Opin Neurol. 2008;21:478.
Eidelberg D, Moeller JR, Ishikawa T, et al. The metabolic topography of idiopathic torsion dystonia. Brain. 1995;118:1473.
Gerfen CR, Engber TM, Mahan LC, et al. D1 and D2 dopamine receptor–regulated gene expression of striatonigral and striatopallidal neurons. Science. 1990;250:1429.
Grafton ST, Turner RS, Desmurget M, et al. Normalizing motor-related brain activity: subthalamic nucleus stimulation in Parkinson disease. Neurology. 2006;66:1192.
Graybiel AM. Habits, rituals, and the evaluative brain. Annu Rev Neurosci. 2008;31:359.
Hammond C, Bergman H, Brown P. Pathological synchronization in Parkinson’s disease: networks, models and treatments. Trends Neurosci. 2007;30:357.
Hashimoto T, Elder CM, Okun MS, et al. Stimulation of the subthalamic nucleus changes the firing pattern of pallidal neurons. J Neurosci. 2003;23:1916.
McIntyre CC, Savasta M, Walter BL, et al. How does deep brain stimulation work? Present understanding and future questions. J Clin Neurophysiol. 2004;21:40.
Mena-Segovia J, Bolam JP, Magill PJ. Pedunculopontine nucleus and basal ganglia: distant relatives or part of the same family? Trends Neurosci. 2004;27:585.
Middleton FA, Strick PL. Basal ganglia and cerebellar loops: motor and cognitive circuits. Brain Res Brain Res Rev. 2000;31:236.
Mink JW. Neurobiology of basal ganglia and Tourette syndrome: basal ganglia circuits and thalamocortical outputs. Adv Neurol. 2006;99:89.
Smith Y, Raju DV, Pare JF, et al. The thalamostriatal system: a highly specific network of the basal ganglia circuitry. Trends Neurosci. 2004;27:520.
Temel Y, Visser-Vandewalle V. Surgery in Tourette syndrome. Mov Disord. 2004;19:3.
Turner RS, Grafton ST, Votaw JR, et al. Motor subcircuits mediating the control of movement velocity: a PET study. J Neurophysiol. 1998;80:2162.
1 Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12:366.
2 DeLong MR. Primate models of movement disorders of basal ganglia origin. Trends Neurosci. 1990;13:281.
3 DeLong MR, Wichmann T. Circuits and circuit disorders of the basal ganglia. Arch Neurol. 2007;64:20.
4 Wichmann T, Delong MR. Deep brain stimulation for neurologic and neuropsychiatric disorders. Neuron. 2006;52:197.
5 Alexander GE, Crutcher MD. Functional architecture of basal ganglia circuits: neural substrates of parallel processing. Trends Neurosci. 1990;13:266.
6 Alexander GE, Crutcher MD, DeLong MR. Basal ganglia–thalamocortical circuits: parallel substrates for motor, oculomotor, ‘prefrontal’ and ‘limbic’ functions. Prog Brain Res. 1990;85:119.
7 Alexander GE, DeLong MR, Strick PL. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu Rev Neurosci. 1986;9:357.
8 Middleton FA, Strick PL. Basal ganglia and cerebellar loops: motor and cognitive circuits. Brain Res Brain Res Rev. 2000;31:236.
9 Parent A, Hazrati LN. Functional anatomy of the basal ganglia. I. The cortico–basal ganglia–thalamo-cortical loop. Brain Res Brain Res Rev. 1995;20:91.
10 Haber SN, Kunishio K, Mizobuchi M, et al. The orbital and medial prefrontal circuit through the primate basal ganglia. J Neurosci. 1995;15:4851.
11 Parent A. Extrinsic connections of the basal ganglia. Trends Neurosci. 1990;13:254.
12 Hartmann-von Monakow K, Akert K, Kunzle H. Projections of the precentral motor cortex and other cortical areas of the frontal lobe to the subthalamic nucleus in the monkey. Exp Brain Res. 1978;33:395.
13 Nambu A, Takada M, Inase M, et al. Dual somatotopical representations in the primate subthalamic nucleus: evidence for ordered but reversed body-map transformations from the primary motor cortex and the supplementary motor area. J Neurosci. 1996;16:2671.
14 Takada M, Tokuno H, Hamada I, et al. Organization of inputs from cingulate motor areas to basal ganglia in macaque monkey. Eur J Neurosci. 2001;14:1633.
15 Shink E, Bevan MD, Bolam JP, et al. The subthalamic nucleus and the external pallidum: two tightly interconnected structures that control the output of the basal ganglia in the monkey. Neuroscience. 1996;73:335-357.
16 Smith Y, Bevan MD, Shink E, et al. Microcircuitry of the direct and indirect pathways of the basal ganglia. Neuroscience. 1998;86:353.
17 Gerfen CR, Engber TM, Mahan LC, et al. D1 and D2 dopamine receptor–regulated gene expression of striatonigral and striatopallidal neurons. Science. 1990;250:1429.
18 Surmeier DJ, Song WJ, Yan Z. Coordinated expression of dopamine receptors in neostriatal medium spiny neurons. J Neurosci. 1996;16:6579.
19 Lieberman DM, Corthesy ME, Cummins A, et al. Reversal of experimental parkinsonism by using selective chemical ablation of the medial globus pallidus. J Neurosurg. 1999;90:928.
20 Pastor-Gomez J, Hernando-Requejo V, Luengo-Dos Santos A, et al. [Treatment of a case of generalised dystonia using subthalamic stimulation.]. Rev Neurol. 2003;37:529.
21 Inase M, Tanji J. Thalamic distribution of projection neurons to the primary motor cortex relative to afferent terminal fields from the globus pallidus in the macaque monkey. J Comp Neurol. 1995;353:415.
22 Schell GR, Strick PL. The origin of thalamic inputs to the arcuate premotor and supplementary motor areas. J Neurosci. 1984;4:539.
23 Hoover JE, Strick PL. Multiple output channels in the basal ganglia. Science. 1993;259:819.
24 Turner RS, Grafton ST, McIntosh AR, et al. The functional anatomy of parkinsonian bradykinesia. Neuroimage. 2003;19:163.
25 Turner RS, Grafton ST, Votaw JR, et al. Motor subcircuits mediating the control of movement velocity: a PET study. J Neurophysiol. 1998;80:2162.
26 DeVito JL, Anderson ME. An autoradiographic study of efferent connections of the globus pallidus in Macaca mulatta. Exp Brain Res. 1982;46:107.
27 Sidibe M, Bevan MD, Bolam JP, et al. Efferent connections of the internal globus pallidus in the squirrel monkey: I. Topography and synaptic organization of the pallidothalamic projection. J Comp Neurol. 1997;382:323.
28 Goldman-Rakic PS, Porrino LJ. The primate mediodorsal (MD) nucleus and its projection to the frontal lobe. J Comp Neurol. 1985;242:535.
29 Middleton FA, Strick PL. Anatomical evidence for cerebellar and basal ganglia involvement in higher cognitive function. Science. 1994;266:458.
30 Smith Y, Raju DV, Pare JF, et al. The thalamostriatal system: a highly specific network of the basal ganglia circuitry. Trends Neurosci. 2004;27:520.
31 Sadikot AF, Parent A, Francois C. Efferent connections of the centromedian and parafascicular thalamic nuclei in the squirrel monkey: a PHA-L study of subcortical projections. J Comp Neurol. 1992;315:137.
32 Smith Y, Parent A. Differential connections of caudate nucleus and putamen in the squirrel monkey (Saimiri sciureus). Neuroscience. 1986;18:347.
33 Parthasarathy HB, Graybiel AM. Cortically driven immediate-early gene expression reflects influence of sensorimotor cortex on identified striatal neurons in the squirrel monkey. J Neurosci. 1997;17:2477.
34 Sidibe M, Smith Y. Differential synaptic innervation of striatofugal neurones projecting to the internal or external segments of the globus pallidus by thalamic afferents in the squirrel monkey. J Comp Neurol. 1996;365:445.
35 Lacey CJ, Bolam JP, Magill PJ. Novel and distinct operational principles of intralaminar thalamic neurons and their striatal projections. J Neurosci. 2007;27:4374.
36 Magill PJ. Bursting with information: how the intralaminar thalamus communicates with the striatum. In Abstracts of the International Basal Ganglia Society IX. Egmond aan Zee, Netherlands: IBAGS; 2007;45
37 Moss J, Bolam JP. The spatial relationships between cortical, thalamic and nigral terminals in the striatum. In Abstracts of the International Basal Ganglia Society IX. Egmond aan Zee, Netherlands: IBAGS; 2007;76
38 Raju DV, Shah DJ, Wright TM, et al. Differential synaptology of vGluT2-containing thalamostriatal afferents between the patch and matrix compartments in rats. J Comp Neurol. 2006;499:231.
39 Raju DV, Smith Y. Differential localization of vesicular glutamate transporters 1 and 2 in the rat striatum. In: Bolam JP, Ingham CA, Magill PJ, editors. The Basal Ganglia VIII. Singapore: Springer; 2005:601.
40 Smith AD, Bolam JP. The neural network of the basal ganglia as revealed by the study of synaptic connections of identified neurones. Trends Neurosci. 1990;13:259.
41 Smith Y, Bennett BD, Bolam JP, et al. Synaptic relationships between dopaminergic afferents and cortical or thalamic input in the sensorimotor territory of the striatum in monkey. J Comp Neurol. 1994;344:1.
42 Harnois C, Filion M. Pallidofugal projections to thalamus and midbrain: a quantitative antidromic activation study in monkeys and cats. Exp Brain Res. 1982;47:277.
43 Rye DB, Lee HJ, Saper CB, et al. Medullary and spinal efferents of the pedunculopontine tegmental nucleus and adjacent mesopontine tegmentum in the rat. J Comp Neurol. 1988;269:315.
44 Inglis WL, Winn P. The pedunculopontine tegmental nucleus: where the striatum meets the reticular formation. Prog Neurobiol. 1995;47:1.
45 Mena-Segovia J, Bolam JP, Magill PJ. Pedunculopontine nucleus and basal ganglia: distant relatives or part of the same family? Trends Neurosci. 2004;27:585.
46 Eidelberg D, Edwards C. Functional brain imaging of movement disorders. Neurolo Res. 2000;22:305.
47 Ghilardi M, Ghez C, Dhawan V, et al. Patterns of regional brain activation associated with different forms of motor learning. Brain Res. 2000;871:127.
48 Grafton ST, DeLong M. Tracing the brain’s circuitry with functional imaging. Nat Med. 1997;3:602.
49 Chevalier G, Deniau JM. Disinhibition as a basic process in the expression of striatal functions. Trends Neurosci. 1990;13:277.
50 Inase M, Buford JA, Anderson ME. Changes in the control of arm position, movement, and thalamic discharge during local inactivation in the globus pallidus of the monkey. J Neurophysiol. 1996;75:1087.
51 Bar-Gad I, Bergman H. Stepping out of the box: information processing in the neural networks of the basal ganglia. Curr Opin Neurobiol. 2001;11:689.
52 Doyon J. Motor sequence learning and movement disorders. Curr Opin Neurol. 2008;21:478.
53 Graybiel AM. Habits, rituals, and the evaluative brain. Annu Rev Neurosci. 2008;31:359.
54 Mink JW, Thach WT. Basal ganglia intrinsic circuits and their role in behavior. Curr Opin Neurobiol. 1993;3:950.
55 Moustafa AA, Sherman SJ, Frank MJ. A dopaminergic basis for working memory, learning and attentional shifting in Parkinsonism. Neuropsychologia. 2008;46:3144-3156.
56 Gerfen CR. Dopamine receptor function in the basal ganglia. Clin Neuropharmacol. 1995;18:S162.
57 Murer MG, Tseng KY, Kasanetz F, et al. Brain oscillations, medium spiny neurons, and dopamine. [erratum appears in Cell Mol Neurobiol. 2003 Jun;23(3):449]. Cell Mol Neurobiol. 2002;22:611.
58 Tseng KY, Kasanetz F, Kargieman L, et al. Cortical slow oscillatory activity is reflected in the membrane potential and spike trains of striatal neurons in rats with chronic nigrostriatal lesions. J Neurosci. 2001;21:6430.
59 Wilson CJ, Kawaguchi Y. The origins of two-state spontaneous membrane potential fluctuations of neostriatal spiny neurons. J Neurosci. 1996;16:2397.
60 Calabresi P, Picconi B, Tozzi A, et al. Dopamine-mediated regulation of corticostriatal synaptic plasticity. Trends Neurosci. 2007;30:211.
61 Kreitzer AC, Malenka RC. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson’s disease models. Nature. 2007;445:643.
62 Reynolds JN, Hyland BI, Wickens JR. A cellular mechanism of reward-related learning. Nature. 2001;413:67.
63 Shen W, Flajolet M, Greengard P, et al. Dichotomous dopaminergic control of striatal synaptic plasticity. Science. 2008;321:848.
64 Singla S, Kreitzer AC, Malenka RC. Mechanisms for synapse specificity during striatal long-term depression. J Neurosci. 2007;27:5260.
65 Gerdeman GL, Ronesi J, Lovinger DM. Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci. 2002;5:446.
66 Tozzi A, Tscherter A, Belcastro V, et al. Interaction of A2A adenosine and D2 dopamine receptors modulates corticostriatal glutamatergic transmission. Neuropharmacology. 2007;53:783.
67 Braak H, Del Tredici K, Rub U, et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197.
68 Burns RS, Chiueh CC, Markey SP, et al. A primate model of parkinsonism: selective destruction of dopaminergic neurons in the pars compacta of the substantia nigra by N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Proc Natl Acad Sci U S A. 1983;80:4546.
69 Forno LS, DeLanney LE, Irwin I, et al. Similarities and differences between MPTP-induced parkinsonism and Parkinson’s disease. Neuropathologic considerations. Adv Neurol. 1993;60:600.
70 Crossman AR, Mitchell IJ, Sambrook MA. Regional brain uptake of 2-deoxyglucose in N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism in the macaque monkey. Neuropharmacology. 1985;24:587.
71 Schwartzman RJ, Alexander GM. Changes in the local cerebral metabolic rate for glucose in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) primate model of Parkinson’s disease. Brain Res. 1985;358:137.
72 Filion M, Tremblay L, Bedard PJ. Abnormal influences of passive limb movement on the activity of globus pallidus neurons in parkinsonian monkeys. Brain Res. 1988;444:165.
73 Miller WC, DeLong MR. Altered tonic activity of neurons in the globus pallidus and subthalamic nucleus in the primate MPTP model of parkinsonism. In: Carpenter MB, Jayaraman A, editors. The Basal Ganglia II. New York: Plenum Press; 1987:415.
74 Wichmann T, Bergman H, DeLong MR. The primate subthalamic nucleus. III. Changes in motor behavior and neuronal activity in the internal pallidum induced by subthalamic inactivation in the MPTP model of parkinsonism. J Neurophysiol. 1994;72:521.
75 Wichmann T, Bergman H, Starr PA, et al. Comparison of MPTP-induced changes in spontaneous neuronal discharge in the internal pallidal segment and in the substantia nigra pars reticulata in primates. Exp Brain Res. 1999;125:397.
76 Dogali M, Beric A, Sterio D, et al. Anatomic and physiological considerations in pallidotomy for Parkinson’s disease. Stereotact Funct Neurosurg. 1994;62:53.
77 Lozano A, Hutchison W, Kiss Z, et al. Methods for microelectrode-guided posteroventral pallidotomy. J Neurosurg. 1996;84:194.
78 Vitek JL, Kaneoke Y, Turner R, et al. Neuronal activity in the internal (GPi) and external (GPe) segments of the globus pallidus (GP) of parkinsonian patients is similar to that in the MPTP-treated primate model of parkinsonism. Soc Neurosci Abstr. 1993;19:1584.
79 Kojima J, Yamaji Y, Matsumura M, et al. Excitotoxic lesions of the pedunculopontine tegmental nucleus produce contralateral hemiparkinsonism in the monkey. Neurosci Lett. 1997;226:111.
80 Munro-Davies LE, Winter J, Aziz TZ, et al. The role of the pedunculopontine region in basal-ganglia mechanisms of akinesia. Exp Brain Res. 1999;129:511.
81 Ceballos-Baumann AO, Brooks DJ. Basal ganglia function and dysfunction revealed by PET activation studies. Adv Neurol. 1997;74:127.
82 Bergman H, Wichmann T, DeLong MR. Reversal of experimental parkinsonism by lesions of the subthalamic nucleus. Science. 1990;249:1436.
83 Wichmann T, Kliem MA, DeLong MR. Antiparkinsonian and behavioral effects of inactivation of the substantia nigra pars reticulata in hemiparkinsonian primates. Exp Neurol. 2001;167:410.
84 Alvarez L, Macias R, Guridi J, et al. Dorsal subthalamotomy for Parkinson’s disease. Mov Disord. 2001;16:72.
85 Alvarez L, Macias R, Lopez G, et al. Bilateral subthalamotomy in Parkinson’s disease: initial and long-term response. Brain. 2005;128:570.
86 Baron MS, Vitek JL, Bakay RA, et al. Treatment of advanced Parkinson’s disease by posterior GPi pallidotomy: 1-year results of a pilot study. Ann Neurol. 1996;40:355.
87 Dogali M, Fazzini E, Kolodny E, et al. Stereotactic ventral pallidotomy for Parkinson’s disease. Neurology. 1995;45:753.
88 Gill SS, Heywood P. Bilateral subthalamic nucleotomy can be accomplished safely. Mov Disord. 1998;13:201.
89 Laitinen LV, Bergenheim AT, Hariz MI. Leksell’s posteroventral pallidotomy in the treatment of Parkinson’s disease. J Neurosurg. 1992;76:53.
90 Lozano AM, Lang AE, Galvez-Jimenez N, et al. Effect of GPi pallidotomy on motor function in Parkinson’s disease. Lancet. 1995;346:1383.
91 Giller CA, Dewey RB, Ginsburg MI, et al. Stereotactic pallidotomy and thalamotomy using individual variations of anatomic landmarks for localization. Neurosurgery. 1998;42:56.
92 Tasker RR, Lang AE, Lozano AM. Pallidal and thalamic surgery for Parkinson’s disease. Exp Neurol. 1997;144:35.
93 Rabey JM, Orlov E, Spiegelman R. Levodopa-induced dyskinesias are the main feature improved by contralateral pallidotomy in Parkinson’s disease. Neurology. 1995;45:A377.
94 Gatev P, Darbin O, Wichmann T. Oscillations in the basal ganglia under normal conditions and in movement disorders. Mov Disord. 2006;21:1566.
95 Hammond C, Bergman H, Brown P. Pathological synchronization in Parkinson’s disease: networks, models and treatments. Trends Neurosci. 2007;30:357.
96 Brown P, Williams D. Basal ganglia local field potential activity: character and functional significance in the human. Clin Neurophysiol. 2005;116:2510.
97 Klostermann F, Nikulin VV, Kuhn AA, et al. Task-related differential dynamics of EEG alpha- and beta-band synchronization in cortico-basal motor structures. Eur J Neurosci. 2007;25:1604.
98 Wang HC, Lees AJ, Brown P. Impairment of EEG desynchronisation before and during movement and its relation to bradykinesia in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1999;66:442.
99 Williams D, Tijssen M, Van Bruggen G, et al. Dopamine-dependent changes in the functional connectivity between basal ganglia and cerebral cortex in humans. Brain Cogn. 2002;125:1558.
100 Wichmann T, Bergman H, DeLong MR. The primate subthalamic nucleus. I. Functional properties in intact animals. J Neurophysiol. 1994;72:494.
101 Bergman H, Wichmann T, Karmon B, et al. The primate subthalamic nucleus. II. Neuronal activity in the MPTP model of parkinsonism. J Neurophysiol. 1994;72:507.
102 Filion M, Tremblay L. Abnormal spontaneous activity of globus pallidus neurons in monkeys with MPTP-induced parkinsonism. Brain Res. 1991;547:142.
103 Wichmann T, Soares J. Neuronal firing before and after burst discharges in the monkey basal ganglia is predictably patterned in the normal state and altered in parkinsonism. J Neurophysiol. 2006;95:2120.
104 Boraud T, Bezard E, Bioulac B, et al. Ratio of inhibited-to-activated pallidal neurons decreases dramatically during passive limb movement in the MPTP-treated monkey. J Neurophysiol. 2000;83:1760.
105 Brown P. Oscillatory nature of human basal ganglia activity: relationship to the pathophysiology of Parkinson’s disease. Mov Disord. 2003;18:357.
106 Leblois A, Meissner W, Bioulac B, et al. Late emergence of synchronized oscillatory activity in the pallidum during progressive parkinsonism. Eur J Neurosci. 2007;26:1701.
107 Mallet N, Pogosyan A, Sharott A, et al. Disrupted dopamine transmission and the emergence of exaggerated beta oscillations in subthalamic nucleus and cerebral cortex. J Neurosci. 2008;28:4795.
108 Chan CS, Guzman JN, Ilijic E, et al. ‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature. 2007;447:1081.
109 Peterson JD, Surmeier DJ. Dopamine depletion results in differential alterations in corticostriatal and thalamostriatal glutamatergic synapses onto striatonigral and striatopallidal neurons. Soc Neurosci Abstr. 2007;590:16.
110 Villalba R, Lee H, Smith Y. Dopaminergic denervation and spine loss in the striatum of MPTP-treated monkeys. Experimental Neurology. 2009;215:200-207.
111 Anderson VC, Burchiel KJ, Hogarth P, et al. Pallidal vs subthalamic nucleus deep brain stimulation in Parkinson disease. Arch Neurol. 2005;62:554.
112 Rodriguez-Oroz MC, Zamarbide I, Guridi J, et al. Efficacy of deep brain stimulation of the subthalamic nucleus in Parkinson’s disease 4 years after surgery: double blind and open label evaluation. J Neurol Neurosurg Psychiatry. 2004;75:1382.
113 Weaver F, Follett K, Hur K, et al. Deep brain stimulation in Parkinson disease: a metaanalysis of patient outcomes. J Neurosurg. 2005;103:956.
114 Hashimoto T, Elder CM, Okun MS, et al. Stimulation of the subthalamic nucleus changes the firing pattern of pallidal neurons. J Neurosci. 2003;23:1916.
115 Brown P, Mazzone P, Oliviero A, et al. Effects of stimulation of the subthalamic area on oscillatory pallidal activity in Parkinson’s disease. Exp Neurol. 2004;188:480.
116 McIntyre CC, Savasta M, Walter BL, et al. How does deep brain stimulation work? Present understanding and future questions. J Clin Neurophysiol. 2004;21:40.
117 Guo Y, Rubin JE, McIntyre CC, et al. Thalamocortical relay fidelity varies across subthalamic nucleus deep brain stimulation protocols in a data-driven computational model. J Neurophysiol. 2008;99:1477.
118 Pierantozzi M, Palmieri MG, Mazzone P, et al. Deep brain stimulation of both subthalamic nucleus and internal globus pallidus restores intracortical inhibition in Parkinson’s disease paralleling apomorphine effects: a paired magnetic stimulation study. Clin Neurophysiol. 2002;113:108.
119 Ceballos-Bauman AO, Obeso JA, Vitek JL, et al. Restoration of thalamocortical activity after posteroventrolateral pallidotomy in Parkinson’s disease. Lancet. 1994;344:814.
120 Grafton ST, Turner RS, Desmurget M, et al. Normalizing motor-related brain activity: subthalamic nucleus stimulation in Parkinson disease. Neurology. 2006;66:1192.
121 Kumar R, Lozano AM, Sime E, et al. Long-term follow-up of thalamic deep brain stimulation for essential and parkinsonian tremor. Neurology. 2003;61:1601.
122 Ondo W, Jankovic J, Schwartz K, et al. Unilateral thalamic deep brain stimulation for refractory essential tremor and Parkinson’s disease tremor. Neurology. 1998;51:1063.
123 Mazzone P, Lozano A, Stanzione P, et al. Implantation of human pedunculopontine nucleus: a safe and clinically relevant target in Parkinson’s disease. Neuroreport. 2005;16:1877.
125 Nandi D, Liu X, Winter JL, et al. Deep brain stimulation of the pedunculopontine region in the normal non-human primate. J Clin Neurosci. 2002;9:170.
125 Plaha P, Gill SS. Bilateral deep brain stimulation of the pedunculopontine nucleus for Parkinson’s disease. Neuroreport. 2005;16:1883.
126 Stefani A, Lozano AM, Peppe A, et al. Bilateral deep brain stimulation of the pedunculopontine and subthalamic nuclei in severe Parkinson’s disease. Brain. 2007;130:1596.
127 Ozelius LJ, Hewett J, Kramer P, et al. Fine localization of the torsion dystonia gene (DYT1) on human chromosome 9q34: YAC map and linkage disequilibrium. Genome Res. 1997;7:483.
128 Risch N, deLeon D, Ozelius L, et al. Genetic analysis of idiopathic torsion dystonia in Ashkenazi Jews and their recent descent from a small founder population. Nat Genet. 1995;9:152.
129 Raike RS, Jinnah HA, Hess EJ. Animal models of generalized dystonia. NeuroRx. 2005;2:504.
130 Wichmann T. Commentary: Dopaminergic dysfunction in DYT1 dystonia. Exp Neurol. 2008;212:719-730.
131 Nygaard TG. Dopa-responsive dystonia. Curr Opin Neurol. 1995;8:310.
132 Patel K, Roskrow T, Davis JS, et al. Dopa responsive dystonia. Arch Dis Child. 1995;73:256.
133 Ichinose H, Nagatsu T. Molecular genetics of hereditary dystonia—mutations in the GTP cyclohydrolase I gene. Brain Res Bull. 1997;43:35.
134 Ichinose H, Ohye T, Takayashi E, et al. Hereditary progressive dystonia with marked diurnal fluctuation caused by mutations in the GTP cyclohydrolase I gene. Nat Genet. 1994;8:236.
135 Nagatsu T, Ichinose H. GTP cyclohydrolase I gene, dystonia, juvenile parkinsonism, and Parkinson’s disease. J Neural Transm Suppl. 1997;49:203.
136 Perlmutter JS, Tempel LW, Black KJ, et al. MPTP induces dystonia and parkinsonism. Clues to the pathophysiology of dystonia. Neurology. 1997;49:1432.
137 Hantraye P, Riche D, Maziere M, et al. A primate model of Huntington’s disease: behavioral and anatomical studies of unilateral excitotoxic lesions of the caudate-putamen in the baboon. Exp Neurol. 1990;108:91.
138 Mitchell IJ, Luquin R, Boyce S, et al. Neural mechanisms of dystonia: evidence from a 2-deoxyglucose uptake study in a primate model of dopamine agonist-induced dystonia. Mov Disord. 1990;5:49.
139 Casey DE. Dopamine D1 (SCH 23390) and D2 (haloperidol) antagonists in drug-naive monkeys. Psychopharmacology (Berl). 1992;107:18.
140 Gerlach J, Hansen L. Clozapine and D1/D2 antagonism in extrapyramidal functions. Br J Pychiatry. 1997;17(Suppl):34.
141 Lenz FA, Suarez JI, Metman LV, et al. Pallidal activity during dystonia: somatosensory reorganisation and changes with severity. J Neurol Neurosurg Psychiatry. 1998;65:767.
142 Starr PA, Rau GM, Davis V, et al. Spontaneous pallidal neuronal activity in human dystonia: comparison with Parkinson’s disease and normal macaque. J Neurophysiol. 2005;93:3165.
143 Tang JK, Moro E, Mahant N, et al. Neuronal firing rates and patterns in the globus pallidus internus of patients with cervical dystonia differ from those with Parkinson’s disease. J Neurophysiol. 2007;98:720.
144 Vitek JL. Pathophysiology of dystonia: a neuronal model. Mov Disord. 2002;17(Suppl 3):S49.
145 Vitek JL, Chockkan V, Zhang JY, et al. Neuronal activity in the basal ganglia in patients with generalized dystonia and hemiballismus. Ann Neurol. 1999;46:22.
146 Zhuang P, Li Y, Hallett M. Neuronal activity in the basal ganglia and thalamus in patients with dystonia. Clin Neurophysiol. 2004;115:2542.
147 Hutchison WD, Lang AE, Dostrovsky JO, et al. Pallidal neuronal activity: implications for models of dystonia. Ann Neurol. 2003;53:480.
148 Lenz FA, Jaeger CJ, Seike MS, et al. Thalamic single neuron activity in patients with dystonia: dystonia-related activity and somatic sensory reorganization. J Neurophysiol. 1999;82:2372.
149 Chen CC, Kuhn AA, Hoffmann KT, et al. Oscillatory pallidal local field potential activity correlates with involuntary EMG in dystonia. Neurology. 2006;66:418.
150 Silberstein P, Kuhn AA, Kupsch A, et al. Patterning of globus pallidus local field potentials differs between Parkinson’s disease and dystonia. Brain. 2003;126:2597.
151 Liu X, Yianni J, Wang S, et al. Different mechanisms may generate sustained hypertonic and rhythmic bursting muscle activity in idiopathic dystonia. Exp Neurol. 2006;198:204.
152 Eidelberg D, Moeller JR, Ishikawa T, et al. The metabolic topography of idiopathic torsion dystonia. Brain. 1995;118:1473.
153 Galardi G, Perani D, Grassi F, et al. Basal ganglia and thalamo-cortical hypermetabolism in patients with spasmodic torticollis. Acta Neurol Scand. 1996;94:172.
154 Karbe H, Holthoff VA, Rudolf J, et al. Positron emission tomography demonstrates frontal cortex and basal ganglia hypometabolism in dystonia. Neurology. 1992;42:1540.
155 Playford ED, Passingham RE, Marsden CD, et al. Increased activation of frontal areas during arm movement in idiopathic torsion dystonia. Mov Disord. 1998;13:309.
156 Byrnes ML, Thickbroom GW, Wilson SA, et al. The corticomotor representation of upper limb muscles in writer’s cramp and changes following botulinum toxin injection. Brain. 1998;121:977.
157 Rona S, Berardelli A, Vacca L, et al. Alterations of motor cortical inhibition in patients with dystonia. Mov Disord. 1998;13:118.
158 Sommer M, Ruge D, Tergau F, et al. Intracortical excitability in the hand motor representation in hand dystonia and blepharospasm. Mov Disord. 2002;17:1017.
159 Deuschl G, Toro C, Matsumoto J, et al. Movement-related cortical potentials in writer’s cramp. Ann Neurol. 1995;38:862.
160 Hamano T, Kaji R, Katayama M, et al. Abnormal contingent negative variation in writer’s cramp. Clin Neurophysiol. 1999;110:508.
161 Ikeda A, Shibasaki H, Kaji R, et al. Abnormal sensorimotor integration in writer’s cramp: study of contingent negative variation. Mov Disord. 1996;11:683.
162 Kaji R, Ikeda A, Ikeda T, et al. Physiological study of cervical dystonia. Task-specific abnormality in contingent negative variation. Brain. 1995;118:511.
163 Toro C, Deuschl G, Hallett M. Movement-related electroencephalographic desynchronization in patients with hand cramps: evidence for motor cortical involvement in focal dystonia. Ann Neurol. 2000;47:456.
164 Butefisch CM, Boroojerdi B, Chen R, et al. Task-dependent intracortical inhibition is impaired in focal hand dystonia. Mov Disord. 2005;20:545.
165 Chen R, Wassermann EM, Canos M, et al. Impaired inhibition in writer’s cramp during voluntary muscle activation. Neurology. 1997;49:1054.
166 Filipovic SR, Ljubisavljevic M, Svetel M, et al. Impairment of cortical inhibition in writer’s cramp as revealed by changes in electromyographic silent period after transcranial magnetic stimulation. Neurosci Lett. 1997;222:167.
167 Ridding MC, Sheean G, Rothwell JC, et al. Changes in the balance between motor cortical excitation and inhibition in focal, task specific dystonia. J Neurol Neurosurg Psychiatry. 1995;59:493.
168 Berardelli A, Rothwell JC, Hallett M, et al. The pathophysiology of primary dystonia. Brain. 1998;121:1195.
169 Hallett M. The neurophysiology of dystonia. Arch Neurol. 1998;55:601.
170 Kanovsky P, Streitova H, Dufek J, et al. Lateralization of the P22/N30 component of somatosensory evoked potentials of the median nerve in patients with cervical dystonia. Mov Disord. 1997;12:553.
171 Kanovsky P, Streitova H, Dufek J, et al. Lateralization of the P22/N30 precentral cortical component of the median nerve somatosensory evoked potentials is different in patients with a tonic or tremulous form of cervical dystonia. Mov Disord. 1999;14:642.
172 Murase N, Kaji R, Shimazu H, et al. Abnormal premovement gating of somatosensory input in writer’s cramp. Brain. 2000;123:1813.
173 Reilly JA, Hallett M, Cohen LG, et al. The N30 component of somatosensory evoked potentials in patients with dystonia. Electroencephalogr Clin Neurophysiol. 1992;84:243.
174 Tinazzi M, Frasson E, Polo A, et al. Evidence for an abnormal cortical sensory processing in dystonia: selective enhancement of lower limb P37-N50 somatosensory evoked potential. Mov Disord. 1999;14:473.
175 Bara-Jimenez W, Catalan MJ, Hallett M, et al. Abnormal somatosensory homunculus in dystonia of the hand. Ann Neurol. 1998;44:828.
176 Byl NN, Merzenich MM, Jenkins WM. A primate genesis model of focal dystonia and repetitive strain injury: I. Learning-induced dedifferentiation of the representation of the hand in the primary somatosensory cortex in adult monkeys. Neurology. 1996;47:508.
177 Elbert T, Candia V, Altenmuller E, et al. Alteration of digital representations in somatosensory cortex in focal hand dystonia. Neuroreport. 1998;9:3571.
178 Delmaire C, Krainik A, Tezenas du Montcel S, et al. Disorganized somatotopy in the putamen of patients with focal hand dystonia. Neurology. 2005;64:1391.
179 Lenz FA, Byl NN. Reorganization in the cutaneous core of the human thalamic principal somatic sensory nucleus (ventral caudal) in patients with dystonia. J Neurophysiol. 1999;82:3204.
180 Chang EF, Turner RS, Ostrem JL, et al. Neuronal responses to passive movement in the globus pallidus internus in primary dystonia. J Neurophysiol. 2007;98:3696.
181 Coubes P, Cif L, El Fertit H, et al. Electrical stimulation of the globus pallidus internus in patients with primary generalized dystonia: long-term results. J Neurosurg. 2004;101:189.
182 Eltahawy HA, Saint-Cyr J, Poon YY, et al. Pallidal deep brain stimulation in cervical dystonia: clinical outcome in four cases. Can J Neurol Sci. 2004;31:328.
183 Vidailhet M, Vercueil L, Houeto JL, et al. Bilateral deep-brain stimulation of the globus pallidus in primary generalized dystonia. N Engl J Med. 2005;352:459.
184 Cif L, El Fertit H, Vayssiere N, et al. Treatment of dystonic syndromes by chronic electrical stimulation of the internal globus pallidus. J Neurosurg Sci. 2003;47:52.
185 Loher TJ, Hasdemir MG, Burgunder JM, et al. Long-term follow-up study of chronic globus pallidus internus stimulation for posttraumatic hemidystonia. J Neurosurg. 2000;92:457.
186 Trottenberg T, Volkmann J, Deuschl G, et al. Treatment of severe tardive dystonia with pallidal deep brain stimulation. Neurology. 2005;64:344.
187 Sun B, Chen S, Zhan S, et al. Subthalamic nucleus stimulation for primary dystonia and tardive dystonia. Acta Neurochir Suppl. 2007;97:207.
188 Detante O, Vercueil L, Thobois S, et al. Globus pallidus internus stimulation in primary generalized dystonia: a H215O PET study. Brain. 2004;127:1899.
189 Kuhn AA, Meyer BU, Trottenberg T, et al. Modulation of motor cortex excitability by pallidal stimulation in patients with severe dystonia. Neurology. 2003;60:768.
190 Krauss JK, Loher TJ, Pohle T, et al. Pallidal deep brain stimulation in patients with cervical dystonia and severe cervical dyskinesias with cervical myelopathy. J Neurol Neurosurg Psychiatry. 2002;72:249.
191 Mink JW. Neurobiology of basal ganglia and Tourette syndrome: basal ganglia circuits and thalamocortical outputs. Adv Neurol. 2006;99:89.
192 Houeto JL, Karachi C, Mallet L, et al. Tourette’s syndrome and deep brain stimulation. J Neurol Neurosurg Psychiatry. 2005;76:992.
193 Temel Y, Visser-Vandewalle V. Surgery in Tourette syndrome. Mov Disord. 2004;19:3.
194 Visser-Vandewalle V, Temel Y, van der Linden C, et al. Deep brain stimulation in movement disorders. The applications reconsidered. Acta Neurol Belg. 2004;104:33.
195 Flaherty AW, Williams ZM, Amirnovin R, et al. Deep brain stimulation of the anterior internal capsule for the treatment of Tourette syndrome: technical case report. Neurosurgery. 2005;57(4 Suppl):E403.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
CHAPTER 73 Rationale for Surgical Interventions in Movement Disorders
Neuroscience research has led to major insights into the structure and function of the basal ganglia and the pathophysiologic basis of basal ganglia disorders such as Parkinson’s disease (PD).1–4 Several factors have contributed to progress in this field, specifically, the availability of effective animal models, improved methods of physiologic and pharmacologic investigation, and the development of powerful brain imaging methods for studies of patients and animal models of these disorders.
Overview of Anatomy and Function of the Basal Ganglia
PD and other movement disorders are increasingly being recognized as “circuit disorders” that involve not only the basal ganglia but also related nodes in the thalamus and the cerebral cortex. This chapter focuses on a description of these larger cortical-subcortical circuits. The anatomic organization of the basal ganglia is discussed in detail in Chapter 74.
Anatomic and physiologic studies have demonstrated that the basal ganglia are components of a family of parallel reentrant brain circuits in which cortical information is sent to the basal ganglia, processed, sent to the thalamus, and then returned to the cerebral cortex.5–9 The different circuits are segregated but share anatomic features, thus supporting the view that the basal ganglia and cortex interact in a modular fashion in which similar processing steps are carried out in each module even though the different modules are engaged in different functions. Depending on the presumed function of the cortical region that is involved in these different circuits, the circuits are commonly designated as “motor,” “oculomotor,” “prefrontal,” and “limbic” (Fig. 73-1). As shown in Figure 73-1, dysfunction within the individual circuits may be associated with specific disease states.
Movement disorders prominently affect the “motor” circuit, which takes origin in the frontal cortical precentral and postcentral sensorimotor areas, including the primary motor cortex (M1), the supplementary motor area (SMA), the premotor cortex (PMC), the cingulate motor area (CMA), and interconnected sensory cortical areas, and involves specific “motor” portions in each of the basal ganglia structures and thalamus. Similar to the other transbasal ganglia circuits, the motor circuit is at least partially closed, with thalamocortical projections terminating in the same frontal cortical regions from which the circuit originates. The anatomy of the motor circuit is shown in greater detail in Figure 73-2A.
FIGURE 73-2 Parkinsonism-related changes in overall activity in the basal ganglia–thalamocortical motor circuit. Black arrows indicate inhibitory connections; blue arrows indicate excitatory connections. The thickness of the arrows corresponds to their presumed activity. CM, centromedian nucleus; Dir., direct pathway; D1, D2, dopamine receptor subtypes; Indir., indirect pathway; PPN, pedunculopontine nucleus; SNc, substantia nigra pars compacta; STN, subthalamic nucleus. For other abbreviations, see the legend to Figure 73-1.
(From Galvan A, Wichmann T. Pathophysiology of parkinsonism. Clin Neurophysiol. 2008;119:1459.)
The striatum and STN are the main entry points for cortical input, whereas the GPi and SNr provide basal ganglia output to the thalamus. The corticostriatal projections are topographically organized.5–7,10,11 Movement-related cortical input terminates in the postcommissural putamen, and nonmotor projections terminate in other areas of the striatum. Thus, prefrontal cortical areas project to the caudate nucleus and the precommissural putamen, and projections from limbic cortices, amygdala, and hippocampus terminate preferentially in the ventral striatum. The cortical-subthalamic projections are also topographically arranged.12,13 Afferents from M1 reach the dorsolateral STN, whereas afferents from the PMC and SMA innervate mainly the mediodorsal third of the nucleus.14
The striatum is linked to the basal ganglia output structures, GPi and SNr, via two distinct pathways, the so-called direct and indirect pathways (Fig. 73-2).1,5,7 The direct pathway arises from sets of striatal medium spiny neurons (MSNs) that project monosynaptically to neurons in the GPi and SNr. These neurons also contain the neuropeptides substance P and dynorphin and preferentially express dopamine D1-like receptors. It is thought that MSNs in the direct pathway receive a larger share of the thalamostriatal pathway than do neurons in the indirect pathway. The indirect pathway arises from a set of striatal MSNs that project to the GPe.15,16 The striatal neurons that give rise to the indirect pathway preferentially express enkephalin and dopamine D2-like receptors.17,18 Although most authors emphasize the separation of these pathways, single-cell labeling of striatal MSNs has shown that at least some send collaterals to both segments of the globus pallidus (thus participating anatomically in both the direct and indirect pathways).19,20
Similar to the corticostriatal projections, the direct and indirect pathways are topographically organized. For instance, populations of GPe neurons within the sensorimotor, associative, or limbic territory are reciprocally connected with populations of neurons in the same functional territories of the STN, and neurons in each of these regions, in turn, innervate the same functional territory of the GPi.15,16
In terms of basal ganglia output, the caudolateral “motor” territory of the GPi projects almost exclusively to the posterior part of the ventrolateral (VL) nucleus, which sends projections toward the SMA,21,22 M1, and PMC.23 The outflow from pallidal motor areas directed at M1, PMC, and SMA appears to arise from separate populations of pallidothalamic neurons.23 These findings indicate that the larger “motor circuit” is composed of segregated subcircuits centered on the individual cortical motor areas.24,25 The more rostromedial associative areas of the GPi project preferentially to the parvocellular part of the ventral anterior (VA) and the dorsal VL nucleus (VLc in macaques),26,27 specifically to thalamic areas preferentially connected to the prefrontal cortex.28,29
Collaterals from the pallidofugal and nigrofugal projections also reach the intralaminar nuclei of the thalamus, the centromedian (CM) and parafascicular (PF) nuclei. These projections are part of a system of segregated basal ganglia–thalamostriatal feedback projections.27,30 In primates, the CM nucleus receives input from motor areas in the GPi and projects to the motor portions of the putamen and STN, whereas PF input and output are related to the associative and limbic territories of the basal ganglia.31,32 CM terminations are found mostly along the shafts of striatal direct pathway MSNs,33,34 separate from the sites of termination of cortical input and of dopaminergic synapses.30,35–41 Basal ganglia output also reaches the pedunculopontine nucleus (PPN),42,43 which in turn gives rise to ascending projections to the basal ganglia, thalamus, and basal forebrain and to descending projections to the pons, medulla, and spinal cord.44,45
Electrophysiologic and metabolic mapping studies in animals and functional imaging data in humans support the view that the anatomically and physiologically defined basal ganglia “motor” areas are indeed involved in movement.25,46–48 The model of the basal ganglia–thalamocortical motor circuit shown in Figure 73-2 is often used as the basis for speculations regarding the function of the basal ganglia in movement. The model poses that activation of MSNs that give rise to the direct pathway reduces inhibitory basal ganglia output from targeted neurons with subsequent disinhibition of related thalamocortical neurons.5,49,50 The net effect of this process is increased activity in appropriate cortical neurons that results in facilitation of the movement. By contrast, activation of MSNs that give rise to the indirect pathway would lead to increased (inhibitory) basal ganglia output on thalamocortical neurons and to suppression of movement. The balance between direct and indirect pathway activity may regulate the overall amount of movement, whereas specific activation patterns (for instance, a center-surround type of activation involving the direct and indirect pathways) may limit the extent or duration of ongoing movements.
Other views of basal ganglia function are currently evolving.51–55 Thus, synaptic processing and modification of synaptic strength in the striatum, or alteration of the level of interneuronal synchrony, may give the basal ganglia a role in the regulation of habit formation or procedural learning.51–5355
In models of basal ganglia function, the modulatory effects of dopamine on striatal transmission are highly important. Dopamine is released in the striatum from terminals of projections from the SNc and modulates the activity of the basal ganglia output neurons in the GPi and SNr by facilitating corticostriatal transmission on the direct pathway and inhibiting corticostriatal transmission on MSNs that give rise to the indirect pathway. These opposing actions are probably mediated by the two different sets of dopamine receptors (D1-like and D2-like receptors) that are differentially expressed in these pathways.56–59 Through the different effects of activation of the direct and indirect pathways, the net effect of striatal dopamine release appears to be reduction of basal ganglia output to the thalamus and other targets. Evidence indicates that dopamine also acts directly on receptors in the STN and pallidum to influence discharge patterns and rates in these structures.
Dopamine receptor activation may not only act to modulate the activity of the direct and indirect pathways but might also have a role in the proposed learning functions of the basal ganglia. Activation of dopamine receptors on MSNs has been shown to be involved in the induction of long-term potentiation and depression at glutamatergic (presumably corticostriatal) synapses.60–64 Recent research has elucidated complex interactions between dopaminergic transmission and transmission involving adenosine, glutamate receptors, and endocannabinoid receptors in this process.61,63–66
Parkinsonism
The cardinal features of PD—the triad of akinesia/bradykinesia, tremor at rest, and muscular rigidity—result from decreased dopaminergic transmission in the motor portions of the basal ganglia, in particular, the putamen, as a result of progressive loss of dopaminergic neurons in the SNc. Dopaminergic replacement therapies are highly effective in reversing these features of the disorder. These dopamine-responsive features are often accompanied by other issues that are poorly or entirely unresponsive to dopaminergic medications, such as depression, autonomic dysfunction, sleep disorders, cognitive impairment, and gait/balance problems. Although some of these problems may result, in part, from decreased dopamine within the nonmotor portions of the basal ganglia, widespread progressive pathologic changes in the brainstem, thalamus, and eventually the cerebral cortex appear to play a major role.67
Pathophysiology of Parkinsonism
The following paragraphs are focused on the motor aspects of PD that result from dopamine deficiency. Study of these changes has been facilitated by the development of animal models of dopamine depletion in which changes in the basal ganglia, thalamus, and cerebral cortex can be conveniently studied, including the 6-hydroxydopamine (6-OHDA)-treated rat and primates treated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP).68,69
In early studies of MPTP-treated primates, alterations in activity of the striatopallidal pathways were strongly emphasized. Such changes were suggested by studies of metabolic activity in the basal ganglia that indicated increased synaptic activity in the GPe and GPi.70,71 Possible interpretations of this finding were that the activity of the striatal-GPe connection and the STN-GPi pathway is increased in parkinsonism or that the STN projections to both pallidal segments were overactive. Subsequent microelectrode recording studies in MPTP-treated primates showed a reduction in neuronal discharge in the GPe and increased firing in the STN, GPi, and SNr in comparison to normal controls,72–75 as well as high neuronal discharge rates in the GPi in PD patients undergoing functional neurosurgical procedures.76–78 These changes may be related to one another in that striatal dopamine depletion may lead to increased activity of striatal neurons in the indirect pathway and result in inhibition of the GPe and subsequent disinhibition of the STN and GPi/SNr (Fig 73-2B). A role of local dopamine loss in the extrastriatal structures (specifically in the STN, GPi, and SNr) may play a role as well, specifically with regard to the emergence of abnormal firing patterns in these nuclei (see later).
Other structures and feedback loops, such as those involving the PPN and CM, may contribute to the abnormalities in discharge that are found in the basal ganglia output nuclei. A series of studies have demonstrated that brainstem areas such as the PPN may also be involved in the development of parkinsonian signs. Lesioning of the PPN in normal monkeys was shown to lead to akinesia.79,80 At the cortical level, positron emission tomography (PET) studies in parkinsonian patients have consistently shown reduced activation of motor and premotor areas.46,81
The notion that rate changes in the basal ganglia underlie the development of parkinsonism (the so-called rate model, depicted in Fig. 73-2) is generally supported by studies that have shown that inactivation of the sensorimotor portions of the STN or GPi increases metabolic activity in the cortical motor areas and improves bradykinesia and tremor in parkinsonian subjects. Thus, lesions of the STN, GPi, or SNr in MPTP-treated primates reverse some or all signs of parkinsonism, presumably by reducing basal ganglia output,19,82,83 and GPi or STN lesions are highly effective antiparkinsonian treatments in patients with PD.84–90 However, the “rate model” has largely been rejected because several crucial observations contradict a purely rate-based view of basal ganglia–processing abnormalities. For instance, lesions of the VA/VL nuclei of the thalamus (which completely removes thalamic output) do not lead to parkinsonism and are, in fact, beneficial in the treatment of tremor and rigidity.91,92 Similarly, lesions of the GPi in the setting of parkinsonism improve the cardinal features of PD without producing dyskinesias or other obvious detrimental effects. In fact, they are highly effective in reducing drug-induced dyskinesias.86,87,93
Because of these discrepancies, the rate model has given way to the view that abnormalities in basal ganglia activity other than rate changes are more relevant to the pathophysiology of parkinsonism. Among the most discussed changes in discharge patterns in the basal ganglia of parkinsonian subjects (Fig. 73-3) is the development of oscillatory phenomena.94,95 Abnormal oscillations have been identified in the activity of single neurons in the GPi, SNr, and STN in animals and patients and, more recently, in recordings of local field potentials (LFPs) in patients, and these oscillations may reflect the activity of more extensive networks of connections. The LFP recordings were made by using implanted DBS macroelectrodes as recording probes during the time immediately after the implantation surgery. Recordings in the STN and other basal ganglia areas with this method have demonstrated the presence of LFP oscillations in the 10- to 25-Hz (beta) range in the STN, GPi, and cortex in unmedicated PD patients. The studies have also shown that beta-range oscillations give way to oscillations in the 60- to 80-Hz (gamma) range when the patients are treated with levodopa or when the STN is stimulated at high frequencies.95,96 The prominence of beta-band activity is apparent throughout the basal ganglia–thalamocortical circuitry. For example, the desynchronization of electroencephalography (EEG) oscillations that normally precedes movement was shown to be abnormally small in parkinsonian patients, perhaps interfering with the initiation of movement.97–99
Another parkinsonism-related abnormality in spontaneous discharge is the emergence of abnormal synchrony between neurons. Under physiologic conditions, basal ganglia activity is highly specific in its relation to movement parameters and body part and appears to be segregated even at the cellular level, where neighboring neurons are rarely found to fire in synchrony.100 In parkinsonism, this level of segregation is lost, and the discharge of neighboring neurons is often found to be correlated and abnormally synchronized.95,101
Finally, the proportion of cells in the STN, GPi, and SNr that discharge in bursts (oscillatory or nonoscillatory) is greatly increased in parkinsonism.73,95,101–103 Oscillatory burst discharge patterns are also seen in conjunction with tremor, which may reflect tremor-related proprioceptive input or more active participation of basal ganglia in the generation of tremor.
Although spontaneous basal ganglia firing has been extensively studied in MPTP-treated monkeys, much less is known about the way information processing is altered in the basal ganglia. Studies of changes in neuronal firing patterns preceding or following active movements are difficult to perform and interpret in akinetic or bradykinetic animals. However, studies of neuronal responses to passive limb manipulation have been carried out and showed that such responses in the STN, GPi, and thalamus occur more often, are more pronounced, and have widened receptive fields after treatment with MPTP.72,73,101,104
The importance of specific electrophysiologic features in basal ganglia, thalamic, or cortical activity for development of the behavioral signs of parkinsonism remains unclear. Although burst-like, synchronized oscillatory activity in the basal ganglia–thalamocortical circuits is associated with parkinsonism,105 direct links between oscillatory activity and specific parkinsonian deficits have not been established. In fact, recent studies in monkeys that underwent a gradual MPTP treatment protocol in which parkinsonism was slowly induced have cast doubt on the notion that synchronous oscillatory firing contributes strongly to (early) parkinsonism.106 In these single-neuron recording studies, synchrony and oscillations in neuronal spiking activity were detected only after the development of bradykinesia and akinesia. Similarly, recent rodent experiments have suggested that abnormal oscillations in the basal ganglia do not result simply from (acute) lack of dopaminergic stimulation but may rather be due to the chronic absence of dopamine.107 Such “late” changes could be related to anatomic alterations secondary to dopamine depletion, such as loss of the dendritic spines of MSNs,108–110 which has been described as homeostatic pruning in response to altered glutamatergic transmission in the striatum.108,109
Rationale for Treating Parkinsonism by Surgical Interventions
Two general types of procedures are used to surgically treat patients with parkinsonism: lesioning and DBS. Unilateral lesioning of the sensorimotor territory of the GPi results in significant contralateral antiparkinsonian effects and significantly reduces drug-induced involuntary movements and motor fluctuations. For DBS, a stimulation electrode is introduced into the same targets in the GPi and STN, and high-frequency stimulation is applied by way of an implanted, externally programmable pulse generator. DBS alleviates parkinsonian motor signs and reduces the severity of “off” periods and the dyskinesias, dystonia, and motor fluctuations that result from long-term drug administration.111–113
[/not-level-membership-for-neurosurgery-category]
