6 Radiotherapy and Chemotherapy
Exploitable Strategies in Combining Chemotherapy With Radiation
The primary objective of combining chemotherapy with radiation is to achieve an improved therapeutic result, which can be evaluated as a function of enhanced tumor response or reduced normal tissue toxicity. In the late 1970s, Steel and Peckham developed a conceptual framework for analyzing drug-radiation interactions.1 In this seminal work, four mechanisms were described in which combined modality therapy could improve therapeutic outcome: spatial cooperation, toxicity independence, protection of normal tissue, and enhancement of tumor response. For more than 20 years, these mechanisms provided the backbone for evaluating chemoradiation combinations clinically. Based on lessons learned from these clinical investigations, coupled with the rapid emergence of molecularly targeted agents, Bentzen and colleagues proposed an updated conceptual framework to evaluate drug-radiation combinations, which are summarized in the following text and in Fig. 6-1.2
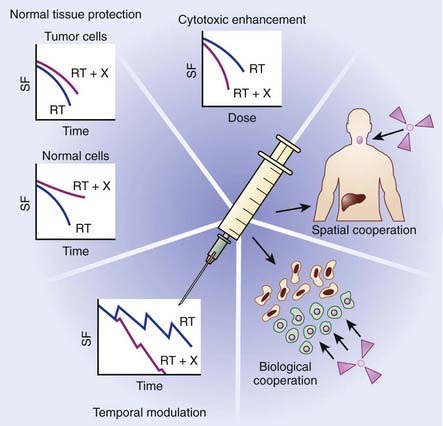
Spatial cooperation refers to combining a drug that is efficacious against systemic disease with radiation, which is effective against locoregional disease. Because a full dose of radiotherapy and chemotherapy is required, and spatial cooperation does not require an interaction at the cellular level, these modalities are typically administered sequentially in an effort to reduce toxicity. Examples of spatial cooperation include adjuvant chemotherapy and radiation therapy in breast cancer and the combination of hormonal therapy and radiation in patients with high-risk prostate cancer. Although this approach is advantageous in terms of toxicity, the issue of overall treatment must also be considered in the design of these schedules. By integrating a chemotherapeutic agent either before or after definitive radiation therapy, the overall treatment time would consequently increase, potentially contributing toward accelerated cellular repopulation and diminished response.3,4 For example, a recent overview of randomized controlled trials of combined chemoradiation for small cell lung cancer concluded that shorter time from the start of any cytotoxic chemotherapy to the end of radiotherapy was significantly correlated with improved treatment outcome.5 Similar findings have been identified for head and neck cancer.
Cytotoxic enhancement refers to the capacity of chemotherapy to interact with radiation and produce a greater effect on the local tumor than would be expected from simple additivity of cell killing. A drug with additive cell killing properties might be seen as a borderline case of cytotoxic enhancement, but unless it possesses active biologic targeting, it will not provide an advantage over a simple radiation dose escalation. With the rapid development of molecular oncology, which has identified numerous molecularly targeted agents that target tumor-specific biologic targets, the identification of tumor-specific agents is being actively investigated. Interestingly, preclinical models suggest that many of these agents also appear to enhance radiation response. Numerous trials are currently underway in multiple tumor types to determine the capacity of these agents to serve as radiation sensitizers. The mechanism that underlies this favorable interaction likely involves induction or repair of radiation-induced cellular deoxyribonucleic acid (DNA) damage. For example, incorporation of halogenated pyrimidines such as 5-fluorouracil into DNA seems to enhance the initial induction of DNA damage by radiation.6 The diverse and complex biologic processes that may be targeted by chemotherapy occurring during the interval between fractionated radiotherapy, including tumor-cell repopulation, reoxygenation, and cellular redistribution, have been collectively termed temporal modulation. For example, combining epidermal growth factor receptor (EGFR) blockade with fractionated radiation may reduce cellular proliferation during therapy, thereby attenuating radiation-induced accelerated cellular proliferation.7
Both toxicity independence and normal tissue protection were initially described by Steel and Peckham as exploitable strategies of chemoradiation; however, lessons learned from clinical investigations over the years suggest these mechanisms may no longer have direct clinical relevance. Toxicity independence refers to the concept of combining a drug that caused systemic toxicity with radiation, in which toxicity is expressed locally. It was suggested that this combination could allow for treatment intensification without unacceptable toxicity, even if the anticancer effects of the two modalities were simply additive. Although this approach was eagerly pursued, a number of studies have shown that effective radiochemotherapy combinations did, in fact, increase radiation-related side effects.8–10 Normal tissue protection was initially based on the potential of some agents, such as cyclophosphamide and methotrexate, given before radiation, to reduce the effect of a subsequent irradiation in some normal tissues, including bone marrow and intestinal epithelium. This effect of dosing chemotherapy before radiotherapy is now understood to be a result of induced cellular repopulation after the first cytotoxic insult. Unfortunately, attempts to exploit this mechanism clinically have been unsuccessful. However, normal tissue protection is continuing to be actively investigated using agents specifically designed to provide cytoprotection or modulate the cytotoxic response of normal tissue. A number of strategies in preclinical or early clinical testing fall under this category, such as the stimulation of stem-cell proliferation in early responding normal tissues,11,12 for example the use of keratinocyte growth factors to ameliorate mucositis,13 or cytoprotection using the thiol-based free radical scavenger amifostine.14,15
Advances in molecular oncology have fostered the development of the mechanism termed biologic cooperation, which has been proposed by Bentzen and colleagues to refer to strategies that target distinct cell populations, or employ different mechanisms for cell killing or delaying tumor regrowth.2 An example could be a drug that targets hypoxic tumor cells, thereby complementing the effect of radiation, which has greater response in well-oxygenated cells. This approach has been applied with such agents as misonidazole, nimorazole, tirapazamine, and mitomycin C, which target hypoxic tumor cells either as radiation sensitizers or bioreductive cytotoxins.16 More recently, clinical interest has focused on agents that affect hypoxia indirectly by targeting the tumor microenvironment. For example, vascular-targeted antiangiogenic agents such as combretastatin cause a shutdown of tumor vasculature, leading to tumor cell death via hemorrhagic necrosis. Agents such as combretastatin are mainly effective in the central regions of the tumor where hypoxic radioresistance may be a therapeutic challenge. Biologic cooperation arises because radiation is particularly effective against the well-oxygenated cells in the periphery of the tumor where vascular shut-down is less effective. Biologic cooperation can even be applied when combining angiogenic agents with radiation. For example, Jain recently presented the case of antiangiogenic agents inducing a normalization of tumor blood vessels, which in turn reduced tumor hypoxia, creating potential biologic cooperation between this class of drugs and radiation.17
Methods for Assessing and Defining Drug–Radiation Interactions
Preclinical studies are typically initiated in vitro, using cell-culture systems. With respect to assays that may be used, the clonogenic assay, which detects all forms of radiation-induced cell death, is considered the “gold standard” for radiosensitivity analysis.18 The underlying biologic end-point evaluated in this assay involves reproductive integrity. Following irradiation, a cell may be physically present and apparently intact, and may even progress through one to two cycles of mitosis; however, if it has lost the capacity to divide indefinitely and produce a large number of progeny, it loses its reproductive integrity. In the context of radiation treatment, losing reproductive integrity prevents continued growth and metastases; therefore, from a practical perspective, the tumor is eradicated. A surviving cell, however, retains its reproductive integrity and is able to proliferate indefinitely to produce a large clone or colony, referred to as clonogenic. In the clonogenic assay, cell survival is determined as a function of radiation dose, with the surviving fraction of cells (colonies) plotted on a logarithmic scale and the dose of radiation plotted linearly. To assess the effect of a drug on cell radiosensitivity, the combined drug–radiation curve is commonly plotted after the cytotoxicity produced by the drug alone is excluded, referred to as normalization. The radiation cell-survival curve is not changed if the drug does not influence cell radiosensitivity, regardless of whether the drug is cytotoxic on it own. In this case, the cytotoxicity of the drug contributes only to the overall cell killing by the combined treatment of both agents, referred to as an additive effect. Investigational agents may interact with radiation by altering cell radiosensitivity such that the combination results in a supra-additive or subadditive effect, depending on whether the cell killing is greater or smaller than the sum of cell killings produced by individual agents. This type of interaction often changes the shape of the cell-survival curve. For instance, a modification of the shoulder region indicates an interaction affecting the repair of radiation-induced DNA damage. An example of a clonogenic survival experiment demonstrating the capacity of an investigational agent to enhance radiation response is depicted in Fig. 6-2A.
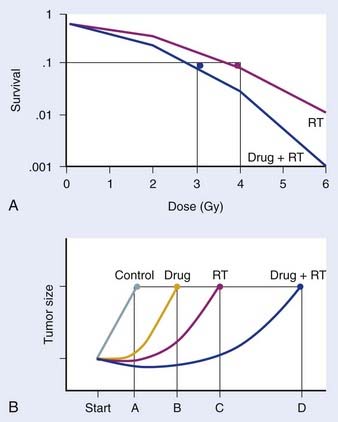
Favorable findings in vitro are often followed by in vivo exploration of drug–radiation interactions. A commonly applied technique involves a mouse xenograft model, in which human tumor cells are grown in culture and inoculated in the flank region of immunodeficient mice. The efficacy of treatment is then determined by the extent of tumor growth delay19 or the rate of tumor cure.20 When the treatment endpoint is delay in tumor growth in drug and radiation combinations, typically four treatment arms are required (control, radiation alone, drug alone, and drug and radiation combined), and tumors are followed until they meet predetermined size criteria. As in the clonogenic assay, growth delays are normalized by subtracting delays of independent treatments (radiation alone and drug alone) from the difference between the drug–radiation combination and the untreated control arm. An example of a growth delay experiment is depicted in Figure 6-2B. The 50% tumor control dose (TCD50; i.e., the dose of radiation that achieves 50% tumor control) assay is used when the treatment endpoint is rate of tumor cure. In this experiment, tumor cure is measured as a function of radiation dose, and the dose that is required to cure or control 50% of the tumors are compared between mice treated with radiation alone and those treated with combined treatment. If combined treatment displaces the curve to the left (i.e., a lower TCD50), then addition of the drug improves tumor curability21; however, this assay does not discriminate between independent drug activity and a direct influence on radiation response. The TCD50 assay has been suggested to be a more relevant model to assess the curative potential of radiation when combined with an investigational agent, as its primary endpoint involves clonogenic cell death.22
Although these in vivo methodologies certainly have their limitations, their potential advantage over traditional in vitro studies is that they also provide insight into the exploitable strategy of biologic cooperation, because both fractionation and tumor microenvironment are evaluated. In an effort to better replicate tumor environment for a specific tumor type, this assay may be of further value if performed orthotopically (e.g., brain tumor grown intracranially, breast tumor grown in mammary fat pads, etc.).23–25 It has been historically suggested that this model may be applied to address normal tissue toxicity; however, its overall clinical application has been limited thus far. Therefore, it is of great importance to identify novel model systems that may be used in this context, because this would provide insight into an improved therapeutic ratio, rather than solely cytotoxic enhancement, which would be the necessary endpoint to further clinical gains.
Initial Radiation Damage
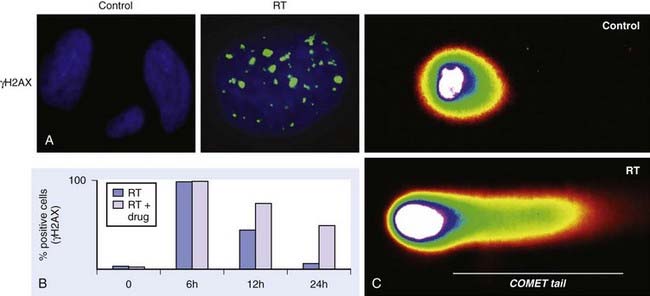
The primary methodology employed to assess radiation damage preclinically is the neutral comet or single-cell gel electrophoresis assay, which is a rapid and sensitive method for detecting DNA damage at the level of individual cells.26 It is based on the ability of negatively charged loops or fragments of DNA to be drawn through an agarose gel in response to an electric field. The extent of DNA migration is directly influenced by the amount of DNA damage present in the cell. In this assay, a suspension of cells are mixed with agarose and spread onto a microscope glass slide. DNA unwinding and electrophoresis is carried out at a specific pH. Unwinding of the DNA and electrophoresis at neutral pH (termed neutral comet) predominantly facilitates the detection of DSBs and crosslinks, whereas, when performed at pH levels greater than 12 (termed alkaline comet), the test facilitates the detection of SSBs and DSBs, incomplete excision repair sites, and crosslinks. When subjected to an electric field, the DNA migrates out of the cell in the direction of the anode, appearing like a “comet” with a distinct head composed of intact DNA, and a tail consisting of damaged or broken pieces of DNA. The size and shape of the comet and the distribution of DNA within the comet correlate with the extent of DNA damage.27
DNA Damage Repair
Immediately following radiation-induced DNA damage, a dynamic and well-orchestrated repair process is initiated, which includes DNA damage recognition, chromatin relaxation, formation of multiunit repair protein complexes at sites of DNA damage, DNA DSB repair, and finally repair protein dissolution and chromatin restoration. Despite its complexity, the entire repair process of DNA DSBs is rapid; a majority of base pairs are repaired within 6 hours. Several modalities have been used preclinically to evaluate DNA damage repair, including constant or pulsed-field gel electrophoresis, but these have been criticized because of the need for large radiation doses. The neutral comet assay, which, as described previously, determines initial damage repair, can also be applied to measure DNA DSB repair when evaluated in a time course manner. A methodology that has gained significant popularity in recent years to assess DNA damage repair involves immunofluorescent cytochemistry to assess phosphorylation of H2AX. At DSB sites, the histone H2AX becomes rapidly phosphorylated (γH2AX), forming readily visible foci. The dephosphorylation and dispersal of γH2AX in irradiated cells correlates with repair of the DNA DSBs. Therefore, prolonged expression of γH2AX, for example, when an investigational agent is combined with radiation, suggests abrogation of DNA repair processes. In addition, the γH2AX assay is sensitive enough to detect damage and repair at very low doses in the clinical and subclinical range.28–31 Common methods used to assess DNA damage repair and initial DNA damage are depicted in Fig. 6-3.
Cell Cycle Kinetics
The mechanistic interplay between radiation and chemotherapy through cell cycle kinetics may be appreciated at multiple levels. One potential interaction involves cell cycle redistribution. Focused radiobiologic studies have determined differential radiosensitivity based on cell cycle phase. In general, cells in the G2 or M phases are the most sensitive, and cells in the S phase are the most resistant cells to radiation. This variation in radiosensitivity during the cell cycle can be potentially exploited when designing effective chemoradiation therapy strategies. One example involves the mitotic-spindle poison taxane. These agents act to stabilize microtubules, and thereby prevent chromosome separation at M phase, leading to cell cycle arrest in the radiosensitive G2 and M phases. Another approach by which cell cycle kinetics may be exploited in radiation and chemotherapy interactions involves differential cytotoxicity. Although the S phase of the cell cycle appears to render cells more resistant to the cytotoxic effects radiation, this phase is particularly sensitive to nucleoside analogs that become incorporated in cellular DNA, such as fludarabine or gemcitabine. Therefore, the preferential targeting of cells in radioresistant phases of the cell cycle represents a form of biologic cooperation underlying the observed radioenhancement. An important point to remember is that although chemotherapeutic agents may have a specific cell cycle phase during which they inhibit cell cycle progression, it is often cells in another phase that might be more sensitive. Classic examples are the vinca-alkaloids, which arrest cells in mitosis but are most toxic when cells are exposed in S phase. Fig. 6-4 summarizes these relationships for a series of chemotherapeutic agents.
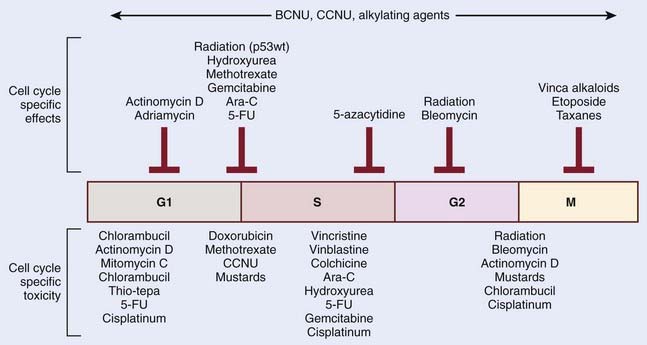
In addition to cell cycle redistribution, the modulation of checkpoint response represents another approach by which chemotherapeutic agents may influence radiosensitization. The activation of the G2 checkpoint allows for DNA repair before progression into mitosis and is considered to protect against radiation-induced cell death.32 Cell cycle kinetics is typically assessed in vitro by way of flow cytometry. By using a DNA-specific stain (typically propidium iodide or Hoechst) the DNA profile can be determined based on the relative amount of DNA. When relative fluorescence (i.e., the amount of DNA per cell) is plotted as a function of cell number, cells in G1 have the least amount of DNA per cell (2n) and therefore represent the first spike on this plot. Cells accumulate DNA during the S phase, representing the intermediate region, and finally, cells in G2 and M have the most quantitative DNA per cell (4n), and therefore represent the final spike on this plot. However, this approach, which assesses DNA quantity per cell, is unable to differentiate cells in G2 and M phases. Therefore, to distinguish between G2 and mitotic cells, in addition to labeling cellular DNA, cells are also labeled with an antibody specific to phosphorylated histone H3, which is specifically expressed in mitotic cells. Done as a function of time after irradiation, this analysis provides a measure of the progression of G2 cells into M phase and thus the activation of the G2 checkpoint.33
Tumor Microenvironment/Hypoxia
In addition to the aberrant genetic alterations driving uncontrolled tumor growth, the surrounding microenvironment of a tumor plays a critical role in its continued growth and may also influence therapeutic response. An important way in which the microenvironment influences tumor growth is by way of its supporting vasculature. The continued growth of a tumor requires the formation of new blood vessels to facilitate the delivery of nutrients and oxygen, a process called angiogenesis. A critical mediator of tumor angiogenesis is the vascular endothelial growth factor, and targeting this protein using a variety of molecular agents represents a dominant theme in anticancer therapy in nearly all solid tumor types. Despite a robust angiogenic response, the oxygenation concentrations are quite heterogeneous within a tumor, with a gradient of intratumoral oxygenation governed by the diffusion capacity of host vessels. Therefore, regions within a tumor distant from the supporting vasculature develop subpopulations of hypoxic cells. In addition to distance from vasculature, hypoxic cells are also induced from defective vascularization within a tumor, both in the number of blood vessels and vessel function. For example, tumor blood vessels are commonly irregular and tortuous, and have blind ends, arteriovenous shunts, incomplete endothelial linings, and basement membranes, leading to areas of poor oxygenation within a tumor. It has long been suggested that hypoxia contributes to radiation resistance,34 a phenomenon that was initially interpreted as reflecting the requirement for oxygen as a source of radiation-induced free radicals that mediate tumor cell killing. Although mechanisms still remain unclear, a more generally accepted principle is that hypoxia influences radiation response at a molecular level, modulating tumor phenotype and angiogenesis through upregulation of key mediators, including hypoxia-inducible factor (HIF-1).35
Although the evaluation of angiogenesis inhibitors as anticancer agents is apparent, their application when combined with radiation is not as intuitive. For example, based on these proposed mechanisms, although angiogenesis inhibitors purportedly prevent continued growth of the tumor, it may also contribute to inducing hypoxia and theoretically minimizing therapeutic efficacy when combined with radiation. However, tumor vasculature is often functionally and structurally abnormal and contributes to hypoxia through spatial and temporal heterogeneity in tumor blood flow. Recent findings suggest that certain angiogenic agents may normalize the abnormal structure and function of tumor vasculature to abrogate hypoxia and potentially even increase the efficacy of conventional therapies.17
Cell Repopulation
Both radiation therapy and chemotherapy are typically administered in multiple, temporally spaced, doses. With respect to radiation treatments, this is to allow for recovery from sublethal radiation-induced damage and allow repopulation of normal tissues between treatments. However, repopulation of surviving tumor cells can also occur during this interval, increasing tumor burden. In addition, the rate of repopulation may increase during a protracted course of therapy, a phenomenon termed accelerated repopulation, which may further limit the effectiveness of therapy.4 Although studies aimed to quantify accelerated repopulation are often confounded by multiple factors, often being derived from retrospective data with a heterogeneous dose of radiation per fraction, their findings are striking. In an important paper, Withers and colleagues36 analyzed pooled clinical data for TCD50 in squamous cell carcinoma of the head and neck. A marked increase in TCD50 was demonstrated if the treatment lasted more than 4 weeks, which was attributed to accelerated repopulation. Further, the added radiation dose required to overcome repopulation has been estimated to range from 0.5 to 1 Gy per day of prolonged treatment.4 The critical importance of minimizing treatment time in cancer therapy has been corroborated by a large prospective randomized trial that demonstrated clinical gains in head and neck cancer with accelerated fractionated radiotherapy (delivered for 6 weeks) when compared with a conventional fractionated radiotherapy (delivered for 7 weeks).37
Commonly Applied Chemotherapies and Mechanisms
Cisplatin and Analogs
Cisplatin is probably the most widely used anticancer agent in combination with radiation. Cisplatin is a water-soluble, coplanar complex that is converted to its active form by replacing its chloride ions with hydroxyl groups.38 The active metabolite reacts with cellular DNA to form interstrand and intrastrand crosslinks that impair DNA replication and ribonucleic acid (RNA) transcription. This leads to DNA breaks and miscoding that may be repaired, mutagenic, or lethal, causing activation of an irreversible apoptotic program.
The cytotoxicity of cisplatin is primarily ascribed to its interaction with nucleophilic N7-sites of purine bases in DNA39 to form both DNA–protein and DNA–DNA interstrand and intrastrand crosslinks. Although there is controversy involving which lesion is dominant in cisplatin toxicity,40 evidence favors the intrastrand adducts. The intrastrand cis-Pt(NH3)2-d(GpG) and cis-Pt(NH3)2-d(ApG) crosslinks represent approximately 65% and 25%, respectively, of the total lesions present in DNA. Binding of the drug causes physical distortions in DNA that attract the attention of a myriad of proteins. More than 20 candidate proteins have been suggested, including components of the mismatch repair complex and the nonhistone chromosomal high-mobility group 1 and 2 proteins.41 It has been suggested that each of the recognition proteins may initiate one or more specific events resulting in several seemingly unrelated biologic effects. This concept is consistent with the cisplatin-induced disruption of replication and transcription, simultaneously initiating responses that may be both survival and death signals.41,42
The consequence of cisplatin adduct formation is a cascade of cellular events involving signaling pathways, checkpoint activation, DNA repair activity, and apoptosis. Previously, the ataxia-telangiectasia RAD3-related protein has been implicated as the main kinase activated by cisplatin,43,44 resulting in phosphorylation of CHK1 kinase, although recent evidence suggests that cisplatin may activate CHK2 in an ataxia-telangiectasia mutated–dependent manner.45 CHK1 and CHK2 orchestrate a complex pathway of cell cycle checkpoint activation involved in G1, S, and G2 arrest.46 In particular, activation of p53 and its downstream mediators47 and the mitogen-activated protein kinase cascade48 are considered key events determining the ultimate fate of the cell.
Cisplatin and Radiation
In vitro studies have shown that the most effective combinations between the two agents tend to be achieved with lower doses of the two modalities. Myint and colleagues49 showed increased radiosensitivity of murine embryonic fibroblast cells at low levels of cisplatin, but when concentrations were increased, instead of observing an increase in radiosensitivity, the data revealed an increasing radioresistance. Gorodetsky and colleagues50 showed that when human ovarian carcinoma (OV-1063) and murine mammary adenocarcinoma (EMT-6) cell lines were pre-irradiated at a low dose of 2 Gy, a clear additional effect of the drug was observed, but this was almost totally eliminated when cells were irradiated with a higher dose (6 Gy). This dose sensitivity argues against a simple model of physical interaction because of proximity between the lesions caused by the two modalities.
The most plausible explanation, supported by the timing of optimal exposure to the agents, is that synergy is a consequence of cisplatin inhibition of repair of radiation-induced DNA damage.50–52 In two cell lines, the EMT-6 and OV-1063 cells, a 2-hour postradiation drug exposure resulted in a supra-additive combined effect, whereas a 24-hour pre-irradiation exposure or protracted postirradiation exposure yielded an additive or slightly subadditive response.50 In experimental tumors, the greatest dose-enhancement factors were observed when cisplatin was administered immediately before a daily fraction of radiation.52
The concept of sublethal damage repair was developed to explain the recovery of cells to full mitotic potential after exposure to a radiation dose insufficient to cause cell death. Early studies suggested that the addition of cisplatin to radiation inhibited this process through modification of two critical pathways involved in DNA DSB repair, homologous recombination53 and nonhomologous end-joining (NHEJ).49,54 The first support for the involvement of NHEJ in the mechanism of cisplatin and radiation interaction came from Frit and colleagues,55 who showed that cross-resistance to ionizing radiation and cisplatin was associated with increased KU80 activity. It was later found that cisplatin-induced DNA damage reduced the ability of the DNA-dependent protein kinase to interact with duplex DNA molecules in vitro56 and that cisplatin radiosensitization was not evident in KU80-deficient cells compared with their wild-type counterparts.49
The involvement of homologous recombination in the sensitization mechanism has been suggested from studies in repair-proficient wild-type and recombinational repair-deficient (RAD52) strains of the yeast Saccharomyces cerevisiae.53 Cisplatin exposure sensitized wild-type yeast cells with a competent recombinational repair mechanism, but could not sensitize cells defective in recombinational repair, indicating that the radiosensitizing effect of cisplatin involved inhibition of RAD52-dependent recombinational repair of DNA. This study also suggested that excision repair was not involved in the sensitization mechanism because strains homozygous for RAD3-2 showed synergy between radiation and cisplatin.
5-Fluoruracil and Analogs
5-Fluoruracil (5-FU) is used widely in the treatment of solid tumors and belongs to a class of anticancer agents termed antimetabolites. It is an analogue of uracil with a fluorine atom at the C5 position in place of hydrogen. The mechanism of cytotoxicity of 5-FU has been ascribed to the misincorporation of fluoronucleotides into RNA and DNA and to the inhibition of the nucleotide synthetic enzyme thymidylate synthase (TS). 5-FU rapidly enters the cell using the same facilitated transport mechanism as uracil, and is converted intracellularly to several active metabolites that disrupt RNA synthesis and the action of TS.57,58
Another metabolite of 5-FU, fluoridine triphosphate (FUTP), is extensively incorporated into RNA, disrupting normal RNA processing and function. The toxicity of 5-FU–induced toxicity to RNA is present at several levels.57 This includes inhibiting the processing of pre-rRNA into mature rRNA,59,60 disrupting posttranslational modifications of tRNAs,61,62 and the assembly and activity of snRNA/protein complexes, thereby inhibiting splicing of pre-mRNA.63,64 In addition to their effect on cellular RNA, 5-FU may also be misincorporated into cellular DNA. This is accomplished through another one of its active metabolites, fluorodeoxyuridine triphosphate (FdUTP). Based on this mechanism, 5-FU has particular activity in cells in S phase through incorporation into DNA, leading to both SSBs and DSBs.
5-FU and Radiation
There are a number of mechanisms by which 5-FU could increase radiation sensitivity at the cellular level. As described previously, the toxicity of 5-FU appears to be S phase–specific, a phase in the cell cycle that is particularly resistant to radiation. However, this mechanism is not sufficient to account for all of the increase in radiation sensitivity produced by the drug because noncytotoxic concentrations of 5-FU have also been demonstrated to increase radiation sensitivity.65,67 Recent data suggest that increased radiation sensitivity occurs in cells that have inappropriate progression though S phase in the presence of drug, suggesting a disordered S-phase checkpoint response.67
Preclinical studies suggest that protracted infusions of 5-FU during a course of fractionated radiotherapy would be required to achieve optimal radiosensitization. This is logical if one considers the mechanism of 5-FU inhibition of TS and the short half-life of 5-FU and its metabolites.67 Indeed, protracted venous infusion of 5-FU has become a standard therapy in several tumor types. The introduction of oral forms of 5-FU, such as the prodrug capecitabine, allow for added convenience for protracted treatment. In addition to ease of use, this agent may also offer further advantages when combined with radiation. Capecitabine is converted to its active form in a multistep process that relies on the enzyme thymidine phosphorylase (TP). TP has higher concentrations in many tumor types compared with matched normal tissue, and therefore may serve as a potent prodrug.68,69 However, over and above this favorable activation profile, interest in integrating capecitabine in combination with radiation has been heightened by the observation that radiation itself can induce TP activity.70,71 Local irradiation with a single dose of 5 Gy increased TP levels by up to 13-fold at 9 days after irradiation. Therefore, there seems to be great potential for synergy between radiation and capecitabine through its capacity to act as a prodrug and enhanced drug activation during irradiation, in addition to the established interaction between the two modalities through cell-cycle effects.
Gemcitabine
Gemcitabine is another nucleoside analogue that acts as a very potent radiation sensitizer.72,73 Early studies in leukemic cells noted a dramatic effect of gemcitabine on DNA metabolism, with notable decreases in cellular deoxynucleotide triphosphates, which are required for DNA synthesis and repair.74 It was later shown than once inside the cell, gemcitabine is rapidly phosphorylated by deoxycytidine kinase, the rate-limiting enzyme for the formation of the active metabolites gemcitabine diphosphate (dFdCDP) and gemcitabine triphosphate (dFdCTP). Ribonucleotide reductase is inhibited by dFdCDP, which is responsible for producing the deoxynucleotides. In addition, the subsequent decrease in cellular deoxynucleotides (particularly dCTP) favors dFdCTP in its competition with dCTP for incorporation into DNA. Therefore, this reduction in cellular dCTP represents an important self-potentiating mechanism resulting in increased gemcitabine nucleotide incorporation into DNA. Once the gemcitabine nucleotide is incorporated on the end of the elongating DNA strand, one more deoxynucleotide is added, following which the DNA polymerases are unable to proceed. This action, termed masked chain termination, appears to lock the drug into DNA, and is strongly correlated with the inhibition of further DNA synthesis, which is likely attributed to the inability of proof-reading exonucleases to remove the gemcitabine nucleotide.75
Gemcitabine and Radiation
Gemcitabine demonstrates a significant enhancement of radiation-induced cell killing at both nontoxic and cytotoxic concentrations.76 There appears to be no radiosensitization when cells are irradiated before gemcitabine, whereas the greatest enhancement of radiation was observed when cells were incubated for 24 hours before irradiation. Subsequent studies explained these results by revealing that maximum sensitization was produced under conditions in which cells were depleted of phosphorylated deoxynucleotides, which, as previously described, is one of their primary mechanisms of action through inhibition of ribonucleotide reductase.72 This mechanism is further supported by the finding that ribonucleotide reductase overexpressing cells were resistant to gemcitabine-mediated radiosensitization.77 Another key mechanism that likely underlies gemcitabine-mediated radiosensitization involves cell cycle distribution. Similar to 5-FU, incorporation of gemcitabine’s active metabolites into dividing DNA renders cells more susceptible in the radiation resistant S phase. It has therefore been suggested that maximum sensitization requires simultaneous redistribution into S phase along with deoxyadenosine triphosphate pool depletion.78 Early clinical trials combining gemcitabine with radiation has indeed demonstrated potent radiosensitization; however, its continued development has been hindered by normal tissue toxicity.67
Temozolomide
Temozolomide is an imidazotetrazine derivative of the alkylating agent dacarbazine. It undergoes rapid chemical conversion in the systemic circulation at physiologic pH to the active compound monomethyl triazeno imidazole carboxamide (MTIC). In contrast, MTIC is formed from dacarbazine only after metabolism by the liver. Because hepatic metabolism can be influenced by agents commonly taken by brain-tumor patients such as anticonvulsant drugs and corticosteroids, it is thought that bioavailability of MTIC may be more consistent with temozolomide than with dacarbazine. In addition, temozolomide is administered orally and has strong capacity to enter the cerebrospinal fluid without accumulation with repeat dosing, further contributing to its rapidly developing clinical interest and applications.79–81
The antitumor activity of temozolomide has largely been attributed to methylation of DNA, specifically at the O6 position of guanine, which has been found to be especially mutagenic and cytotoxic. Methyl adducts at the O6 guanine in DNA are repaired by the cytoprotective DNA repair protein methyl guanine methyl transferase (MGMT; also referred to as arginine glycine amidinotransferase), which transfers the methyl group to an internal cysteine acceptor residue. This reaction results in an irreversible inactivation of MGMT, requiring increased de novo protein synthesis to restore repair activity. Although methylation at the O6 position of guanine is attributed to temozolomide toxicity, it makes up only a small proportion (≈5%) of the DNA that is methylated. For example, nearly 70% of total DNA methylation by temozolomide occurs at the N7 position of guanine and approximately 9% of adducts formed at the N3 position at adenine.80,81 However, these sites are typically repaired rapidly, using the highly conserved and efficient base excision repair pathway. Although methylation at the O6 position of guanine is also readily repaired by MGMT, a subset of tumors has been identified with epigenetic silencing of this repair protein through promoter methylation. In a seminal work reported by Hegi and colleagues, it was demonstrated that promoter methylation of MGMT in glioblastoma could serve as a biomarker for predicting response to temozolomide.82 Although these findings are currently being validated prospectively, this is perhaps the first study that has identified a predictive marker for a cytotoxic agent, and therefore serves as an important step in the direction of individualized therapy based on tumor biology. However, despite representing progress, MGMT methylation is clearly not an exclusive factor, as nearly half of these patients still do not benefit from therapy, and a subset of patients who do not carry this methylation demonstrate long-term survival; therefore, more specific biomarkers are still required.
Temozolomide and Radiation
The addition of temozolomide with definitive radiation has doubled 2-year survival among patients with glioblastoma, an achievement that had not been accomplished in the prior 30 years of clinical investigations of this tumor.83 Clearly, temozolomide has independent activity in glioblastoma, although preclinical investigations suggest it may also enhance radiation response. Kil and colleagues demonstrated modest in vitro enhancement of radiation response in a glioma cell line and a breast cancer cell line with a proclivity to form brain metastases, although in vivo sensitization was more striking, with a dose enhancement factor of 2.8 in tumor growth delay.84 Of the numerous mechanisms studied, it appeared that mitotic catastrophe played a dominant role in the observed sensitization. Chakravarti and colleagues demonstrated similar findings and went on to suggest that sensitization was most effective in MGMT-negative tumors.85
Timing of Drug Administration
Depending on the principal aim, chemotherapeutic agents may be administered before (neoadjuvant), during (concurrent), or following (adjuvant) the course of radiation therapy. In general, when a chemotherapeutic agent is delivered alone, using either a neoadjuvant or adjuvant approach, the primary goal is for the treatment of disseminated micrometastatic disease. However, activity at the primary tumor site is also important. For example, a potential application of neoadjuvant chemotherapy is for tumor down-sizing, which would attempt to convert patients with locally advanced tumors, previously unresectable, into surgical candidates. By tumor down-sizing, neoadjuvant chemotherapy can also potentially be used to decrease radiation field sizes, thereby reducing toxicity. Although both of these approaches clearly have their respective roles, there is likely little interaction between the chemotherapeutic agent and the biologic processes underlying radiation response. In addition, one must judiciously choose such an approach, as it may have negative effects on clinical outcome. For example, neoadjuvant chemotherapy would actually increase overall treatment time, which has been shown to negatively correlate with prognosis and contribute to accelerated cellular repopulation.4
Normal Tissue Effects
Integrating agents that may serve as normal tissue radioprotectors also represents an active area of investigation.86 A majority of work has focused on the thiol-containing prodrug amifostine, the primary mechanism of which involves free-radical scavenging. A phase III randomized trial was conducted from 1995 to 1997 to assess the ability of this drug to reduce the incidence of grade ≥ 2 acute and late xerostomia and grade ≥ 3 acute mucositis in locally advanced head and neck cancer. Amifostine did not reduce the incidence of mucositis, although it did appear to reduce both acute and late xerostomia.14 Although amifostine has an approved xerostomia indication in the setting of radiation alone, its activity when combined with chemoradiation or modern radiation techniques that significantly minimizes dose to salivary glands, including IMRT, has yet to be defined. A similar study has been performed in NSCLC to determine the capacity of amifostine to mitigate radiation-induced esophagitis.15 Although no reduction in esophagitis was observed, less swallowing dysfunction was demonstrated in patients who received amifostine. Although it has been a challenge to identify compounds that could selectively protect normal tissue (and not protect tumor) and integrate them in the clinic, success in such an approach would demonstrate a clear method of improving the therapeutic ratio.
Strategies for Improvement
Despite clear clinical gains demonstrated with the integration of chemotherapeutic agents into definitive radiation treatment regimens, investigators are actively seeking novel approaches to continue these improvements. One sought-after strategy for improvement in current treatment regimens has been to profile tumors using gene microarrays in an effort to identify patients most likely to respond to a particular chemotherapeutic agent, and tailor treatment accordingly. However, because of complex mechanisms of action and the multitiered biologic processes driving tumorigenesis and response and resistance, modest progress has been made to date using this approach. One promising example has involved predicting response to temozolomide in glioblastoma patients. Although findings are currently being prospectively validated, the promoter-methylation status of repair protein MGMT, which is involved in the repair of temozolomide-induced DNA damage, may serve as an important biomarker for temozolomide response.82
Another strategy that has generated enthusiasm in furthering clinical gains involves integrating molecularly targeted agents into the chemoradiation platform. Such investigations are supported by preclinical investigations demonstrating the capacity of many of these agents to enhance radiation response. When developing these early phase clinical trials, it is important to recognize that these targeted agents are not benign and clearly have their own toxicity profile. However, as their toxicities are typically distinct from those commonly attributed to radiation and standard chemotherapeutics, this may allow for an improved therapeutic ratio. Targeting the tumor microenvironment with angiogenesis inhibitors represents another actively investigated approach to improve current chemoradiation regimens. In addition to having independent activity, these agents may have added benefits by inducing vascular normalization to alleviate hypoxia and increase intratumoral drug concentrations.17
Clinical Results
One of the first modern chemoradiation platforms involved the use of the antimetabolite 5-FU in the late 1950s,87 which interestingly still plays a significant role in the treatment of gastrointestinal malignancies. This was followed by one of the Radiation Therapy Oncology Group’s (RTOG’s) first studies, which was a prospective randomized trial evaluating methotrexate combined with radiation in head and neck cancer.88 Since these initial investigations, combining chemotherapy with radiation has been an active area of investigation, contributing to improved clinical outcomes in a majority of solid tumors. Key clinical investigations leading to current chemoradiation platforms are summarized in the following text.
Lung
The Cancer and Leukemia Group B was one of the earliest groups evaluating the role of combining chemotherapy with radiation in locally advanced NSCLC.89 This study used induction chemotherapy consisting of 5 weeks of cisplatin and vinblastine, which was subsequently followed by radiotherapy. Results demonstrated a statistically significant improvement in median survival of 4.1 months in patients receiving induction chemotherapy, which was also demonstrated by a similar trial performed by the RTOG and the Eastern Cooperative Oncology Group (ECOG).90 Following these studies, the next critical question was to determine if chemotherapy delivered concurrently with radiation could further clinical gains. Both the RTOG and the West Japan Lung Cancer Group91 identified concurrent chemoradiation to be superior, using cisplatin/vinblastine and mitomycin/vindesine/cisplatin platforms, respectively. Another regimen that is commonly used is concurrent paclitaxel and carboplatin, which has shown promising results in phase II studies.92
As the natural history of small cell carcinoma (SCC) involves early seeding of distant metastases, chemotherapy has been the mainstay of therapy. However, in limited-stage small cell, radiation has demonstrated both an improvement in local control and survival. The use of concurrent cisplatin and etoposide with radiation has been generally accepted, largely based on results presented by ECOG (in collaboration with RTOG and the Southwest Oncology Group [SWOG]), using this platform with a hyperfractionated radiation regimen.93
Head and Neck
In a report from the journal Lancet in 2000, the Meta-Analysis of Chemotherapy in Head and Neck Cancer Group identified that the addition of chemotherapy to locoregional treatment provides a modest overall improvement in survival (4% at 5 years) for patients with locoregionally advanced head and neck cancer.94 The most promising benefit (8%) was evident with the use of concomitant chemoradiation with no significant benefit for the use of induction or adjuvant chemotherapy. These results were recently updated by incorporating data from randomized trials performed between 1994 and 2000.95 Twenty-four new trials, most of them involving concomitant chemotherapy, were included, totaling 87 trials with more than 17,000 total patients. This updated analysis confirmed the small overall survival benefit of chemotherapy of 4% at 5 years. Similarly, analysis of the 50 concomitant chemoradiation trials confirmed the previous results, demonstrating an absolute survival benefit of 8% at 5 years. As with the prior meta-analysis, there was no significant survival benefit identified for the use of induction or adjuvant chemotherapy for patients with head and neck cancer, only in the concurrent chemoradiation setting.
Key clinical trials demonstrating benefit of concurrent chemotherapy in head and neck cancer include results presented by the Groupe d’Oncology Radiothérapie Téte et Cou, which used a carboplatin and 5-FU platform in SCC of the oropharynx.96 Improved local control and survival came at a cost of increased acute mucositis and bone marrow toxicity, although it did not lead to an increase in late toxicities. In nasopharyngeal cancer, cisplatin delivered with radiation, followed by adjuvant cisplatin and 5-FU, was also shown to improve local control and survival.97 In addition to the potential of improved survival, another key rationale for concurrent chemoradiation is organ preservation. Improved local control offered by concurrent chemoradiation in locally advanced laryngeal cancer, when compared with both radiation alone and induction chemotherapy, led to an improvement in larynx preservation.98
Molecularly targeted agents have also been actively investigated in head and neck cancer. Results from an international, randomized phase III clinical trial of 424 locoregionally advanced head and neck cancer patients treated with curative intent using either high dose radiotherapy alone or high-dose radiotherapy plus the anti-EGFR monoclonal antibody cetuximab demonstrated a near doubling of the median survival for patients treated with radiotherapy plus cetuximab over radiotherapy alone, 49 months versus 29 months.99 The percentage of patients who achieved locoregional control at 1 year and at 2 years following treatment was 69% and 56% in the cetuximab-treated patients, compared with 59% and 48% for those treated with radiotherapy alone. There was a statistically significant improvement (p = 0.02) in locoregional disease control (9% at 2 years) and overall survival (10% at 3 years) favoring the cetuximab arm. Because there has been a shift in the management of locally advanced head and neck cancer since the initiation of this study, with concurrent cisplatin now being accepted as a standard regimen with radiation, current trials are examining whether cetuximab combined with concurrent cisplatin and radiation may lead to further clinical gains.
Cervical Cancer
In 1999 the National Cancer Institute advised that cisplatin-based chemotherapy administered concurrently with radiotherapy exhibited a marked superiority over standard radiotherapy alone in cervical cancer and therefore was the new standard of care for this disease.100 This was largely based on five of six randomized prospective studies that demonstrated a survival advantage of concurrent chemotherapy in the definitive treatment of cervical cancer.101 In addition, concurrent chemotherapy also demonstrated benefit in the postoperative setting for patients exhibiting high-risk features, including positive margins, parametrial invasion, or positive lymph node.102 The RTOG is now evaluating the benefit of integrating the angiogenesis inhibitor bevacizumab to this platform.
Gastrointestinal Disease
Nearly all subtypes of tumor in the gastrointestinal track have demonstrated clear benefit when chemotherapy is administered concurrently with radiation. Many of these regimens are based on a 5-FU platform, which was one of the first chemotherapeutic agents combined with radiation nearly 50 years ago.87 In esophageal cancer, the definitive trial was performed by the RTOG, demonstrating a 5-year survival rate of 26% when cisplatin and 5-FU was combined with radiation, compared with 0% in the radiation-alone arm in unresectable disease.103 In gastric cancer, an Intergroup study using a 5-FU and leucovorin platform demonstrated an improved local control and overall survival in patients with T3, T4, or N+ disease when delivered in the postoperative setting.104
In rectal cancer, the Gastrointestinal Tumor Study Group performed one of the initial studies demonstrating benefit with combined modality therapy.105 In this study, following surgery, patients were randomized to fours arms: no further therapy, radiation alone, chemotherapy alone (consisting of 5-FU and methyl-CCNU), and combined 5-FU and radiotherapy followed by adjuvant chemotherapy. Results demonstrated an improved local control with combined modality therapy. The North Central Cancer Treatment Group went on to demonstrate that protracted venous infusion of 5-FU improved recurrence rate and overall survival when compared with bolus 5-FU when delivered concurrently with radiation, suggesting its direct influence on radiation response.106 With the established role of chemoradiation in rectal cancer, another important question that needed to be answered was the most effective time of delivery, either pre- or postoperative. This was addressed by the German Rectal Cancer Study group, which showed that preoperative concurrent 5-FU and radiation led to improvements in local control, colostomy-free survival, and an improved toxicity profile.107 This regimen has now been established as standard therapy for locally advanced rectal cancer.
The initial rationale for combining chemotherapy with radiation in anal cancer was to increase resectability. However, in 1974, Nigro and coworkers observed complete tumor regression in its initial patients treated with a 5-FU and mitomycin C preoperative regimen, suggesting the potential for cure without surgery and colostomy.108 A subsequent report described the outcome for 45 patients treated with the same regimen with a complete response rate of 84% on biopsy 6 weeks after completion of treatment.109 The benefit of a concurrent chemoradiation regimen over radiation alone was demonstrated in two randomized phase III trials by the European Organization for Research and Treatment of Cancer (EORTC) and the United Kingdom Coordinating Committee on Cancer Research, both demonstrating increased local control and colostomy-free survival.110,111 The potential of replacing mitomycin C with cisplatin was investigated in the randomized Intergroup RTOG trial that showed no difference between the two regimens in disease-free survival, its primary endpoint; however, the colostomy-free survival rate was significantly higher in patients receiving mitomycin C.112 Therefore, the 5-FU and mitomycin regimen remains standard concurrent chemotherapy in anal cancer.
Glioblastoma
For nearly 30 years, surgical resection to the extent that is safely feasible, followed by radiotherapy has been the standard of care in glioblastoma. Because an overwhelming majority of patients experience local recurrence and eventually succumb to uncontrolled disease progression despite therapy, numerous attempts had been made at integrating chemotherapy to this regimen. Although individual trials did not show benefit, a meta-analysis demonstrated approximately a 5% improvement in overall survival with the addition of chemotherapy.113 Despite the discouraging historical context involving glioblastoma management, novel therapeutic strategies offering clinical gains have emerged. A survival benefit in glioblastoma has recently been published by the EORTC.83 This regimen, which is now the current standard of care in newly diagnosed glioblastoma patients, consists of concomitant low-dose temozolomide with radiation, followed by high-dose adjuvant temozolomide. Determining ways to integrate novel agents upon this platform to further clinical gains in this disease remains an active area of investigation.
1 Steel GG, Peckham MJ. Exploitable mechanisms in combined radiotherapy-chemotherapy: the concept of additivity. Int J Radiat Oncol Biol Phys. 1979;5(1):85-91.
2 Bentzen SM, Harari PM, Bernier J. Exploitable mechanisms for combining drugs with radiation: concepts, achievements and future directions. Nat Clin Pract Oncol. 2007;4(3):172-180.
3 Bentzen SM. Repopulation in radiation oncology: perspectives of clinical research. Int J Radiat Biol. 2003;79(7):581-585.
4 Kim JJ, Tannock IF. Repopulation of cancer cells during therapy: an important cause of treatment failure. Nat Rev Cancer. 2005;5(7):516-525.
5 De Ruysscher D, Pijls-Johannesma M, Bentzen SM, et al. Time between the first day of chemotherapy and the last day of chest radiation is the most important predictor of survival in limited-disease small-cell lung cancer. J Clin Oncol. 2006;24(7):1057-1063.
6 Nishimura Y. Rationale for chemoradiotherapy. Int J Clin Oncol. 2004;9(6):414-420.
7 Harari PM, Huang SM. Head and neck cancer as a clinical model for molecular targeting of therapy: combining EGFR blockade with radiation. Int J Radiat Oncol Biol Phys. 2001;49(2):427-433.
8 Bentzen SM, Overgaard M, Thames HD, et al. Early and late normal-tissue injury after postmastectomy radiotherapy alone or combined with chemotherapy. Int J Radiat Biol. 1989;56(5):711-715.
9 Denis F, Garaud P, Bardet E, et al. Late toxicity results of the GORTEC 94-01 randomized trial comparing radiotherapy with concomitant radiochemotherapy for advanced-stage oropharynx carcinoma: comparison of LENT/SOMA, RTOG/EORTC, and NCI-CTC scoring systems. Int J Radiat Oncol Biol Phys. 2003;55(1):93-98.
10 Trotti A, Bentzen SM. The need for adverse effects reporting standards in oncology clinical trials. J Clin Oncol. 2004;22(1):19-22.
11 Bentzen SM. Preventing or reducing late side effects of radiation therapy: radiobiology meets molecular pathology. Nat Rev Cancer. 2006;6(9):702-713.
12 Trotti A. Toxicity antagonists in head and neck cancer. Semin Radiat Oncol. 1998;8(4):282-291.
13 Spielberger R, Stiff P, Bensinger W, et al. Palifermin for oral mucositis after intensive therapy for hematologic cancers. N Engl J Med. 2004;351(25):2590-2598.
14 Brizel DM, Wasserman TH, Henke M, et al. Phase III randomized trial of amifostine as a radioprotector in head and neck cancer. J Clin Oncol. 2000;18(19):3339-3345.
15 Movsas B, Scott C, Langer C, et al. Randomized trial of amifostine in locally advanced non-small-cell lung cancer patients receiving chemotherapy and hyperfractionated radiation: radiation therapy oncology group trial 98-01. J Clin Oncol. 2005;23(10):2145-2154.
16 Peters KB, Brown JM. Tirapazamine: a hypoxia-activated topoisomerase II poison. Cancer Res. 2002;62(18):5248-5253.
17 Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307(5706):58-62.
18 Hall EJ. Radiobiology for the radiologist. Philadelphia: Lippincott Williams & Wilkins; 1993.
19 Begg AC. Rodent Tumor Models in Experimental Cancer Therapy. Principles and Practices of the Tumor Growth Delay Assay. 1987:114-121.
20 Suit HD, Sedlacek R, Thames HDJr. Radiation dose-response assays of tumor control. principles and practices of the tumor growth delay assay. 1987:138-148.
21 Mason KA, Hunter NR, Milas M, et al. Docetaxel enhances tumor radioresponse in vivo. Clin Cancer Res. 1997;3(12 Pt 1):2431-2438.
22 Krause M, Zips D, Thames HD, et al. Preclinical evaluation of molecular-targeted anticancer agents for radiotherapy. Radiother Oncol. 2006;80(2):112-122.
23 Morikawa K, Walker SM, Nakajima M, et al. Influence of organ environment on the growth, selection, and metastasis of human colon carcinoma cells in nude mice. Cancer Res. 1988;48(23):6863-6871.
24 Nakajima M, Morikawa K, Fabra A, et al. Influence of organ environment on extracellular matrix degradative activity and metastasis of human colon carcinoma cells. J Nat Cancer Inst. 1990;82(24):1890-1898.
25 Yokoi K, Sasaki T, Bucana CD, et al. Simultaneous inhibition of EGFR, VEGFR, and platelet-derived growth factor receptor signaling combined with gemcitabine produces therapy of human pancreatic carcinoma and prolongs survival in an orthotopic nude mouse model. Cancer Res. 2005;65(22):10371-10380.
26 Singh NP, McCoy MT, Tice RR, et al. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res. 1988;175(1):184-191.
27 Olive PL. DNA damage and repair in individual cells: applications of the comet assay in radiobiology. Int J Radiat Biol. 1999;75(4):395-405.
28 Banath JP, Macphail SH, Olive PL. Radiation sensitivity, H2AX phosphorylation, and kinetics of repair of DNA strand breaks in irradiated cervical cancer cell lines. Cancer Res. 2004;64(19):7144-7149.
29 Olive PL, Banath JP. Phosphorylation of histone H2AX as a measure of radiosensitivity. Int J Radiat Oncol Biol Phys. 2004;58(2):331-335.
30 Taneja N, Davis M, Choy JS, et al. Histone H2AX phosphorylation as a predictor of radiosensitivity and target for radiotherapy. J Biol Chem. 2004;279(3):2273-2280.
31 Rothkamm K, Lobrich M. Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc Natl Acad Sci U S A. 2003;100(9):5057-5062.
32 Carson CT, Schwartz RA, Stracker TH, et al. The Mre11 complex is required for ATM activation and the G2/M checkpoint. EMBO J. 2003;22(24):6610-6620.
33 Xu B, Kim ST, Lim DS, Kastan MB. Two molecularly distinct G(2)/M checkpoints are induced by ionizing irradiation. Mol Cell Biol. 2002;22(4):1049-1059.
34 Gray LH, Conger AD, Ebert M, et al. The concentration of oxygen dissolved in tissues at the time of irradiation as a factor in radiotherapy. Br J Radiol. 1953;26(312):638-648.
35 Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3(10):721-732.
36 Withers HR, Taylor JM, Maciejewski B. The hazard of accelerated tumor clonogen repopulation during radiotherapy. Acta Oncol. 1988;27(2):131-146.
37 Fu KK, Pajak TF, Trotti A, et al. A Radiation Therapy Oncology Group (RTOG) phase III randomized study to compare hyperfractionation and two variants of accelerated fractionation to standard fractionation radiotherapy for head and neck squamous cell carcinomas: first report of RTOG 9003. Int J Radiat Oncol Biol Phys. 2000;48(1):7-16.
38 Rosenberg B, VanCamp L, Trosko JE, et al. Platinum compounds: a new class of potent antitumour agents. Nature. 1969;222(5191):385-386.
39 Eastman A, Barry MA. Interaction of trans-diamminedichloroplatinum(II) with DNA: formation of monofunctional adducts and their reaction with glutathione. Biochemistry. 1987;26(12):3303-3307.
40 Jones JC, Zhen WP, Reed E, et al. Gene-specific formation and repair of cisplatin intrastrand adducts and interstrand cross-links in Chinese hamster ovary cells. J Biol Chem. 1991;266(11):7101-7107.
41 Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22(47):7265-7279.
42 Jordan P, Carmo-Fonseca M. Molecular mechanisms involved in cisplatin cytotoxicity. Cell Mol Life Sci. 2000;57(8–9):1229-1235.
43 Damia G, Filiberti L, Vikhanskaya F, et al. Cisplatinum and Taxol induce different patterns of p53 phosphorylation. Neoplasia. 2001;3(1):10-16.
44 Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001;21(13):4129-4139.
45 Agner J, Falck J, Lukas J, et al. Differential impact of diverse anticancer chemotherapeutics on the Cdc25A-degradation checkpoint pathway. Exp Cell Res. 2005;302(2):162-169.
46 Wilson GD. Radiation and the cell cycle, revisited. Cancer Metastasis Rev. 2004;23(3–4):209-225.
47 Appella E, Anderson CW. Post-translational modifications and activation of p53 by genotoxic stresses. Eur J Biochem. 2001;268(10):2764-2772.
48 Losa JH, Parada Cobo C, Viniegra JG, et al. Role of the p38 MAPK pathway in cisplatin-based therapy. Oncogene. 2003;22(26):3998-4006.
49 Myint WK, Ng C, Raaphorst GP. Examining the non-homologous repair process following cisplatin and radiation treatments. Int J Radiat Biol. 2002;78(5):417-424.
50 Gorodetsky R, Levy-Agababa F, Mou X, et al. Combination of cisplatin and radiation in cell culture: effect of duration of exposure to drug and timing of irradiation. Int J Cancer. 1998;75(4):635-642.
51 Bartelink H, Kallman RF, Rapacchietta D, et al. Therapeutic enhancement in mice by clinically relevant dose and fractionation schedules of cis-diamminedichloroplatinum (II) and irradiation. Radiother Oncol. 1986;6(1):61-74.
52 Kanazawa H, Rapacchietta D, Kallman RF. Schedule-dependent therapeutic gain from the combination of fractionated irradiation and cis-diamminedichloroplatinum (II) in C3H/Km mouse model systems. Cancer Res. 1988;48(11):3158-3164.
53 Dolling JA, Boreham DR, Brown DL, et al. Cisplatin-modification of DNA repair and ionizing radiation lethality in yeast, Saccharomyces cerevisiae. Mutat Res. 1999;433(2):127-136.
54 Haveman J, Castro Kreder N, Rodermond HM, et al. Cellular response of x-ray sensitive hamster mutant cell lines to gemcitabine, cisplatin and 5-fluorouracil. Oncol Rep. 2004;12(1):187-192.
55 Frit P, Canitrot Y, Muller C, et al. Cross-resistance to ionizing radiation in a murine leukemic cell line resistant to cis-dichlorodiammineplatinum(II): role of Ku autoantigen. Mol Pharmacol. 1999;56(1):141-146.
56 Turchi JJ, Henkels KM, Zhou Y. Cisplatin-DNA adducts inhibit translocation of the Ku subunits of DNA-PK. Nucleic Acids Res. 2000;28(23):4634-4641.
57 Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3(5):330-338.
58 Shewach DS, Lawrence TS. Antimetabolite radiosensitizers. J Clin Oncol. 2007;25(26):4043-4050.
59 Ghoshal K, Jacob ST. Specific inhibition of pre-ribosomal RNA processing in extracts from the lymphosarcoma cells treated with 5-fluorouracil. Cancer Res. 1994;54(3):632-636.
60 Kanamaru R, Kakuta H, Sato T, et al. The inhibitory effects of 5-fluorouracil on the metabolism of preribosomal and ribosomal RNA in L-1210 cells in vitro. Cancer Chemother Pharmacol. 1986;17(1):43-46.
61 Randerath K, Tseng WC, Harris JS, et al. Specific effects of 5-fluoropyrimidines and 5-azapyrimidines on modification of the 5 position of pyrimidines, in particular the synthesis of 5-methyluracil and 5-methylcytosine in nucleic acids. Recent Results Cancer Res. 1983;84:283-297.
62 Santi DV, Hardy LW. Catalytic mechanism and inhibition of tRNA (uracil-5-)methyltransferase: evidence for covalent catalysis. Biochemistry. 1987;26(26):8599-8606.
63 Doong SL, Dolnick BJ. 5-Fluorouracil substitution alters pre-mRNA splicing in vitro. J Biol Chem. 1988;263(9):4467-4473.
64 Patton JR. Ribonucleoprotein particle assembly and modification of U2 small nuclear RNA containing 5-fluorouridine. Biochemistry. 1993;32(34):8939-8944.
65 Byfield JE. 5-Fluorouracil radiation sensitization—a brief review. InvestNew Drugs. 1989;7(1):111-116.
66 Wilson GD, Bentzen SM, Harari PM. Biologic basis for combining drugs with radiation. Semin Radiat Oncol. 2006;16(1):2-9.
67 Lawrence TS, Blackstock AW, McGinn C. The mechanism of action of radiosensitization of conventional chemotherapeutic agents. Semin Radiat Oncol. 2003;13(1):13-21.
68 Hotta T, Taniguchi K, Kobayashi Y, et al. Increased expression of thymidine phosphorylase in tumor tissue in proportion to TP-expression in primary normal tissue. Oncol Rep. 2004;12(3):539-541.
69 Nishimura G, Terada I, Kobayashi T, et al. Thymidine phosphorylase and dihydropyrimidine dehydrogenase levels in primary colorectal cancer show a relationship to clinical effects of 5′-deoxy-5-fluorouridine as adjuvant chemotherapy. Oncol Rep. 2002;9(3):479-482.
70 Barton-Burke M. Gemcitabine: a pharmacologic and clinical overview. Cancer Nurs. 1999;22(2):176-183.
71 Sawada N, Ishikawa T, Sekiguchi F, et al. X-ray irradiation induces thymidine phosphorylase and enhances the efficacy of capecitabine (Xeloda) in human cancer xenografts. Clin Cancer Res. 1999;5(10):2948-2953.
72 Lawrence TS, Chang EY, Hahn TM, et al. Delayed radiosensitization of human colon carcinoma cells after a brief exposure to 2′,2′-difluoro-2′-deoxycytidine (Gemcitabine). Clin Cancer Res. 1997;3(5):777-782.
73 Shewach DS, Lawrence TS. Radiosensitization of human tumor cells by gemcitabine in vitro. Semin Oncol. 1995;22(4 Suppl 11):68-71.
74 Huang P, Chubb S, Hertel LW, et al. Action of 2′,2′-difluorodeoxycytidine on DNA synthesis. Cancer Res. 1991;51(22):6110-6117.
75 Plunkett W, Huang P, Xu YZ, et al. Gemcitabine: metabolism, mechanisms of action, and self-potentiation. Semin Oncol. 1995;22(4 Suppl 11):3-10.
76 Shewach DS, Hahn TM, Chang E, et al. Metabolism of 2′,2′-difluoro-2′-deoxycytidine and radiation sensitization of human colon carcinoma cells. Cancer Res. 1994;54(12):3218-3223.
77 Zhou BS, Hsu NY, Pan BC, et al. Overexpression of ribonucleotide reductase in transfected human KB cells increases their resistance to hydroxyurea: M2 but not M1 is sufficient to increase resistance to hydroxyurea in transfected cells. Cancer Res. 1995;55(6):1328-1333.
78 Ostruszka LJ, Shewach DS. The role of cell cycle progression in radiosensitization by 2′,2′-difluoro-2′-deoxycytidine. Cancer Res. 2000;60(21):6080-6088.
79 Denny BJ, Wheelhouse RT, Stevens MF, et al. NMR and molecular modeling investigation of the mechanism of activation of the antitumor drug temozolomide and its interaction with DNA. Biochemistry. 1994;33(31):9045-9051.
80 Danson SJ, Middleton MR. Temozolomide: a novel oral alkylating agent. Expert Rev Anticancer Ther. 2001;1(1):13-19.
81 Ostermann S, Csajka C, Buclin T, et al. Plasma and cerebrospinal fluid population pharmacokinetics of temozolomide in malignant glioma patients. Clin Cancer Res. 2004;10(11):3728-3736.
82 Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997-1003.
83 Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987-996.
84 Kil WJ, Cerna D, Burgan WE, et al. In vitro and in vivo radiosensitization induced by the DNA methylating agent temozolomide. Clin Cancer Res. 2008;14(3):931-938.
85 Chakravarti A, Erkkinen MG, Nestler U, et al. Temozolomide-mediated radiation enhancement in glioblastoma: a report on underlying mechanisms. Clin Cancer Res. 2006;12(15):4738-4746.
86 Brizel DM. Pharmacologic approaches to radiation protection. J Clin Oncol. 2007;25(26):4084-4089.
87 Heidelberger C, Chaudhuri NK, Danneberg P, et al. Fluorinated pyrimidines, a new class of tumour-inhibitory compounds. Nature. 1957;179(4561):663-666.
88 Kramer S. Use of methotrexate and radiation therapy for advanced cancer of the head and neck. Front Radiat Ther Oncol. 4(116), 1969.
89 Dillman RO, Herndon J, Seagren SL, et al. Improved survival in stage III non-small-cell lung cancer: seven-year follow-up of cancer and leukemia group B (CALGB) 8433 trial. J Natl Cancer Inst. 1996;88(17):1210-1215.
90 Sause WT, Scott C, Taylor S, et al. Radiation Therapy Oncology Group (RTOG) 88-08 and Eastern Cooperative Oncology Group (ECOG) 4588: preliminary results of a phase III trial in regionally advanced, unresectable non-small-cell lung cancer. J Natl Cancer Inst. 1995;87(3):198-205.
91 Furuse K, Fukuoka M, Kawahara M, et al. Phase III study of concurrent versus sequential thoracic radiotherapy in combination with mitomycin, vindesine, and cisplatin in unresectable stage III non-small-cell lung cancer. J Clin Oncol. 1999;17(9):2692-2699.
92 Choy H, Akerley W, Safran H, et al. Multiinstitutional phase II trial of paclitaxel, carboplatin, and concurrent radiation therapy for locally advanced non-small-cell lung cancer. J Clin Oncol. 1998;16(10):3316-3322.
93 Turrisi AT, Kim K, Blum R, et al. Twice-daily compared with once-daily thoracic radiotherapy in limited small-cell lung cancer treated concurrently with cisplatin and etoposide. N Engl J Med. 1999;340(4):265-271.
94 Pignon JP, Bourhis J, Domenge C, et al. Chemotherapy added to locoregional treatment for head and neck squamous-cell carcinoma: three meta-analyses of updated individual data. MACH-NC Collaborative Group. Meta-Analysis of Chemotherapy on Head and Neck Cancer. Lancet. 2000;355(9208):949-955.
95 Pignon JP, le Maitre A, Bourhis J. Meta-Analyses of Chemotherapy in Head and Neck Cancer (MACH-NC): an update. Int J Radiat Oncol Biol Phys. 2007;69(2 Suppl):S112-114.
96 Denis F, Garaud P, Bardet E, et al. Final results of the 94-01 French Head and Neck Oncology and Radiotherapy Group randomized trial comparing radiotherapy alone with concomitant radiochemotherapy in advanced-stage oropharynx carcinoma. J Clin Oncol. 2004;22(1):69-76.
97 Al-Sarraf M, LeBlanc M, Giri PG, et al. Chemoradiotherapy versus radiotherapy in patients with advanced nasopharyngeal cancer: phase III randomized Intergroup study 0099. J Clin Oncol. 1998;16(4):1310-1317.
98 Forastiere AA, Goepfert H, Maor M, et al. Concurrent chemotherapy and radiotherapy for organ preservation in advanced laryngeal cancer. N Engl J Med. 2003;349(22):2091-2098.
99 Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354(6):567-578.
100 Moore DH. Chemotherapy for advanced, recurrent, and metastatic cervical cancer. J Natl Compr Canc Netw. 2008;6(1):53-57.
101 Monk BJ, Tewari KS, Koh WJ. Multimodality therapy for locally advanced cervical carcinoma: state of the art and future directions. J Clin Oncol. 2007;25(20):2952-2965.
102 Peters WA, Liu PY, Barrett RJ, et al. Concurrent chemotherapy and pelvic radiation therapy compared with pelvic radiation therapy alone as adjuvant therapy after radical surgery in high-risk early-stage cancer of the cervix. J Clin Oncol. 2000;18(8):1606-1613.
103 Cooper JS, Guo MD, Herskovic A, et al. Chemoradiotherapy of locally advanced esophageal cancer: long-term follow-up of a prospective randomized trial (RTOG 85-01). Radiation Therapy Oncology Group. JAMA. 1999;281(17):1623-1627.
104 Macdonald JS, Smalley SR, Benedetti J, et al. Chemoradiotherapy after surgery compared with surgery alone for adenocarcinoma of the stomach or gastroesophageal junction. N Engl J Med. 2001;345(10):725-730.
105 Gastrointestinal Tumor Study Group. Prolongation of the disease-free interval in surgically treated rectal carcinoma. Gastrointestinal Tumor Study Group. N Engl J Med. 1985;312(23):1465-1472.
106 O’Connell MJ, Martenson JA, Wieand HS, et al. Improving adjuvant therapy for rectal cancer by combining protracted-infusion fluorouracil with radiation therapy after curative surgery. N Engl J Med. 1994;331(8):502-507.
107 Sauer R, Becker H, Hohenberger W, et al. Preoperative versus postoperative chemoradiotherapy for rectal cancer. N Engl J Med. 2004;351(17):1731-1740.
108 Nigro ND, Vaitkevicius VK, Considine BJr. Combined therapy for cancer of the anal canal: a preliminary report. Dis Colon Rectum. 1974;17(3):354-356.
109 Leichman L, Nigro N, Vaitkevicius VK, et al. Cancer of the anal canal. Model for preoperative adjuvant combined modality therapy. Am J Med. 1985;78(2):211-215.
110 Epidermoid anal cancer: results from the UKCCCR randomised trial of radiotherapy alone versus radiotherapy, 5-fluorouracil, and mitomycin. UKCCCR Anal Cancer Trial Working Party. UK Co-ordinating Committee on Cancer Research. Lancet. 1996;348(9034):1049-1054.
111 Bartelink H, Roelofsen F, Eschwege F, et al. Concomitant radiotherapy and chemotherapy is superior to radiotherapy alone in the treatment of locally advanced anal cancer: results of a phase III randomized trial of the European Organization for Research and Treatment of Cancer Radiotherapy and Gastrointestinal Cooperative Groups. J Clin Oncol. 1997;15(5):2040-2049.
112 Ajani JA, Winter KA, Gunderson LL, et al. Fluorouracil, mitomycin, and radiotherapy vs fluorouracil, cisplatin, and radiotherapy for carcinoma of the anal canal: a randomized controlled trial. JAMA. 2008;299(16):1914-1921.
113 Stewart LA. Chemotherapy in adult high-grade glioma: a systematic review and meta-analysis of individual patient data from 12 randomised trials. Lancet. 2002;359(9311):1011-1018.