Chapter 427 Pulmonary Hypertension
427.1 Primary Pulmonary Hypertension
Pathophysiology
Primary pulmonary hypertension is characterized by pulmonary vascular obstructive disease and right-sided heart failure. It occurs at any age, although in pediatric patients the diagnosis is usually made in adolescence. In older patients, females outnumber males 1.7 : 1; in younger patients, both genders are represented equally. Some patients have evidence of either an immunologic disorder or a hypercoagulable state. Mutations in the gene for bone morphogenetic protein receptor-2 (BMPR-2, a member of the transforming growth factor-β receptor family) on chromosome 2q33 have been identified in patients with autosomal dominant familial primary pulmonary hypertension (known as PPH1). This genetic variant demonstrates a female preponderance and is found in 60% of patients with a family history and up to 25% of patients with sporadic disease. Other potential disease causing genes have been identified, including chromosome 2q31 (PPH2) and 12q13 (ALK1). Viral infection, such as with the vasculotropic human herpesvirus 8 (HHV8), has been suggested as a trigger factor in many patients. Diet pills, particularly fenfluramine, have also been implicated. Pulmonary hypertension is a common complication of sickle cell anemia and other hemolytic anemias. Pulmonary hypertension is associated with precapillary obstruction of the pulmonary vascular bed as a result of hyperplasia of the muscular and elastic tissues and a thickened intima of the small pulmonary arteries and arterioles. Atherosclerotic changes may be found in the larger pulmonary arteries as well. In children, pulmonary veno-occlusive disease may account for some cases of primary pulmonary hypertension. Before a diagnosis of primary pulmonary hypertension can be made, other causes of elevated pulmonary arterial pressure must be eliminated (chronic pulmonary parenchymal disease, persistent obstruction of the upper airway, congenital cardiac malformations, recurrent pulmonary emboli, alveolar capillary dysplasia, liver disease, autoimmune disease, and moyamoya disease). A classification system is noted in Table 427-1. Pulmonary hypertension places an afterload burden on the right ventricle, which results in right ventricular hypertrophy. Dilatation of the pulmonary artery is present, and pulmonary valve insufficiency may occur. In the later stages of the disease, the right ventricle dilates, tricuspid insufficiency develops, and cardiac output is decreased. Arrhythmias, syncope, and sudden death are common.
Table 427-1 REVISED WHO CLASSIFICATION OF PH
From ACCF/AHA: 2009 Expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association, J Am Coll Cardiol 53:1573–1619, 2009.
Diagnosis
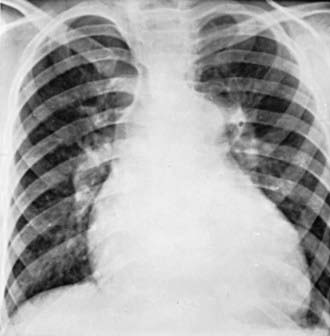
Chest roentgenograms reveal a prominent pulmonary artery and right ventricle (Fig. 427-1). The pulmonary vascularity in the hilar areas may be prominent, in contrast to the peripheral lung fields in which pulmonary markings are decreased. The electrocardiogram shows right ventricular hypertrophy, often with spiked P waves. Echocardiography is used to screen for any congenital cardiac malformations. Doppler evaluation of the tricuspid valve, if insufficiency is present, will allow estimation of the right ventricular (and hence pulmonary arterial) systolic pressure.
At cardiac catheterization, the presence of left-sided obstructive lesions (pulmonary venous stenosis, mitral stenosis, restrictive cardiomyopathy) that result in pulmonary venous hypertension can be evaluated (Chapters 421.9, 425.7, and 433.3). The presence of pulmonary arterial hypertension with a normal pulmonary capillary wedge pressure is diagnostic of primary pulmonary hypertension. If the wedge pressure is elevated and left ventricular end-diastolic pressure is normal, obstruction at the level of the pulmonary veins, left atrium, or mitral valve should be suspected. If left ventricular end-diastolic pressure is also elevated, the diagnosis of restrictive cardiomyopathy should be entertained. The risks associated with cardiac catheterization are increased in severely ill patients with primary pulmonary hypertension.
Prognosis and Treatment
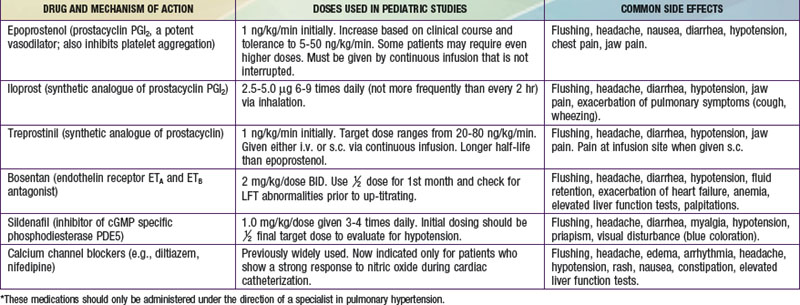
Primary pulmonary hypertension is progressive, and no cure is currently available. Some success has been reported with oral calcium channel blocking agents such as nifedipine in children who demonstrate pulmonary vasoreactivity when these agents are administered during catheterization. Continuous intravenous infusion of the arachidonic acid metabolite, prostacyclin (epoprostenol), provides relief as long as the infusion is continued. Despite the success of prostacyclin in reducing symptoms and improving quality of life, it slows but does not stop the progression of the disease. Treprostinil, a prostacyclin analogue with a longer half-life has also been shown to be effective. Continuous administration of nitric oxide via nasal cannula, nebulized forms of prostacyclin (iloprost), and orally administered pulmonary vasodilators (bosentan, an antagonist of endothelin receptors; or sildenafil, a phosphodiesterase type 5 inhibitor) have been used with success in adults, although clinical studies in children are of small numbers (Table 427-2). Anticoagulation may be of value in patients with previous pulmonary thromboemboli, and some of these patients will respond to balloon angioplasty of narrowed pulmonary artery segments. Despite many advances, definitive therapy is still heart-lung or lung transplantation (Chapter 437.2). In patients with severe pulmonary hypertension and low cardiac output, the terminal event is often sudden and related to a lethal arrhythmia. Patients with primary pulmonary hypertension diagnosed in infancy often have rapid progression and high mortality.
Table 427-2 SUMMARY OF DRUGS USED TO TREAT PULMONARY HYPERTENSION*
427.2 Pulmonary Vascular Disease (Eisenmenger Syndrome)
Pathology and Pathophysiology
Pulmonary vascular disease occurs more rapidly in patients with trisomy 21 who have left-to-right shunts. It also complicates the natural history of patients with elevated pulmonary venous pressure secondary to mitral stenosis or left ventricular dysfunction, especially in those patients with restrictive cardiomyopathy (Chapter 439.3). Pulmonary vascular disease can also occur in any patient with transmission of systemic pressure to the pulmonary circulation via a shunt at the interventricular or great vessel level, and in patients chronically exposed to low PO2 (because of high altitude). Patients with cyanotic congenital heart lesions associated with unrestricted pulmonary blood flow are at particularly high risk.
Diagnosis
Roentgenographically, the heart varies in size from normal to greatly enlarged; the latter usually occurs late in the course of the disease. The main pulmonary artery is generally prominent, similar to primary pulmonary hypertension (see Fig. 427-1). The pulmonary vessels are enlarged in the hilar areas and taper rapidly in caliber in the peripheral branches. The right ventricle and atrium are prominent. The electrocardiogram shows marked right ventricular hypertrophy. The P wave may be tall and spiked. Cyanotic patients have various degrees of polycythemia that depend on the severity and duration of hypoxemia.
Treatment
Medical treatment of Eisenmenger syndrome is primarily symptomatic. Many patients benefit substantially from either oral (calcium channel blocker, endothelin antagonist, phosphodiesterase inhibitors) or chronic intravenous (prostacyclin) therapy. Combined heart-lung or bilateral lung transplantation is the only surgical option for many of these patients (Chapter 437.2).
ACCF/AHA. 2009 Expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association. J Am Coll Cardiol. 2009;53:1573-1619.
Archer SL, Michelakis ED. Phosphodiesterase type 5 inhibitors for pulmonary arterial hypertension. N Engl J Med. 2009;361:1864-1870.
Barnett CF, Hsue PY, Machado RF. Pulmonary hypertension: An increasingly recognized complication of hereditary hemolytic anemias and HIV infection. JAMA. 2008;299:324-331.
Beghetti M, Galiè N. Eisenmenger syndrome a clinical perspective in a new therapeutic era of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;53:733-740.
Beghetti M, Hoeper MM, Kiely DG, et al. Safety experience with Bosentan in 146 children 2–11 years old with pulmonary arterial hypertension: results from the European postmarketing surveillance program. Pediatr Res. 2008;64:200-204.
Berger S, Konduri GG. Pulmonary hypertension in children: the twenty-first century. Pediatr Clin North Am. 2006;53:961-987.
Davies RJ, Morrell NW. Molecular mechanisms of pulmonary arterial hypertension: role of mutations in the bone morphogenetic protein type II receptor. Chest. 2008;134:1271-1277.
Dimopoulos K, Giannakoulas G, Wort SJ, et al. Pulmonary arterial hypertension in adults with congenital heart disease: distinct differences from other causes of pulmonary arterial hypertension and management implications. Curr Opin Cardiol. 2008;23:545-554.
Farber HW, Loscalzo J. Pulmonary arterial hypertension. N Engl J Med. 2004;351:1655-1666.
Feinstein JA. Evaluation, risk stratification, and management of pulmonary hypertension in patients with congenital heart disease. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu. 2009:106-111.
Feinstein JA, Goldhaber SZ, Lock JE, et al. Balloon pulmonary angioplasty for treatment of chronic thromboembolic pulmonary hypertension. Circulation. 2001;103:10-13.
Grünig E, Koehler R, Miltenberger-Miltenyi G, et al. Primary pulmonary hypertension in children may have a different genetic background than in adults. Pediatr Res. 2004;56:571-578.
Li X, Zhang X, Leathers R, et al. Notch3 signaling promotes the development of pulmonary arterial hypertension. Nat Med. 2009;15:1289-1297.
McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension. Circulation. 2009;119:2250-2294.
McLaughlin VV, Sitbon O, Badesch DB, et al. Survival with first-line bosentan in patients with primary pulmonary hypertension. Eur Respir J. 2005;25:244-249.
The Medical Letter. Tadalafil (Adcirca) for pulmonary arterial hypertension. Med Lett. 2009;51:87-88.
Michelakis ED, Wilkins MR, Rabinovitch M. Emerging concepts and translational priorities in pulmonary arterial hypertension. Circulation. 2008 Sep 30;118:1486-1495.
Newman JH, Wheeler L, Lane KB, et al. Mutation in the gene for bone morphogenetic protein receptor II as a cause of primary pulmonary hypertension in a large kindred. N Engl J Med. 2001;345:319-324.
Rosenzweig EB, Barst RJ. Pulmonary arterial hypertension in children: a medical update. Curr Opin Pediatr. 2008;20:288-293.
Simonneau G, Rubin LJ, Galie N, et al. Addition of sildenafil to long-term intravenous epoprostenol therapy in patients with pulmonary arterial hypertension. Ann Intern Med. 2008;149:521-530.
Tissot C, Ivy DD, Beghetti M: Medical therapy for pediatric pulmonary arterial hypertension, J Pediatr 157(4):528–532, 2010.
Van Albada ME, Loot FG, Fokkema R, et al. Biological serum markers in the management of pediatric pulmonary arterial hypertension. Pediatr Res. 2008;63:321-327.
Van Loon RL, Roofthooft MTR, van Osch-Gevers M, et al. Clinical characterization of pediatric pulmonary hypertension: complex presentation and diagnosis. J Pediatr. 2009;155:176-182.





