11 Pulmonary Hypertension
Biopsy for evaluation of pulmonary hypertension is relatively uncommon, in part because of the dangers of fatal arrhythmias in such patients, and has sometimes been viewed as offering little therapeutic benefit. As argued by Wagenvoort,1 however, biopsy in patients with pulmonary hypertension serves three purposes:
This information is important in deciding whether to perform corrective surgery in congenital heart disease1 and appears to be of value in predicting response to vasodilator therapy.3
Morphologic Features of the Pulmonary Vasculature
Pulmonary Arteries
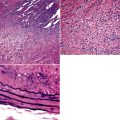
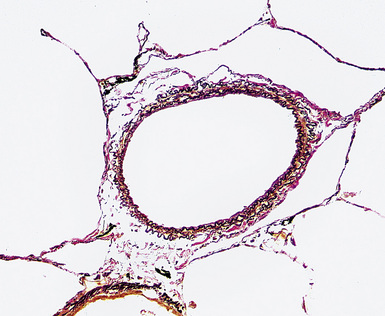
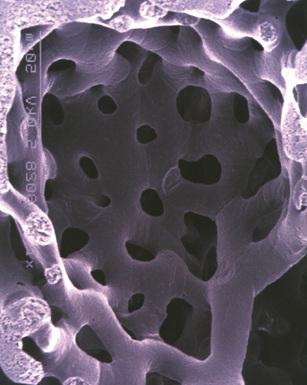
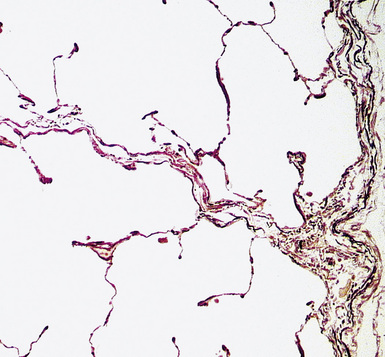
Any diagnosis of pulmonary hypertension requires recognition of the different types of vessels seen in the lung. This is aided by the use of elastic tissue stains, which should be a routine approach when a biopsy or larger specimen shows potential vascular disease. Knowledge of the structure of the normal pulmonary vascular bed is important in assessing biopsy material.4,5 Branches of the pulmonary artery run with the bronchi and then the bronchioles. Arteries associated with the bronchi are typically larger than 1 mm in diameter and have a fairly extensive elastic fiber meshwork in their walls. Muscular pulmonary arteries (Fig. 11-1) are usually associated with bronchioles and measure between 100 and 1000 μm. They are frequently abnormal in pulmonary hypertension. Elastic stain (Fig. 11-2) shows that they have both an internal and an external elastic lamina. In the normal lung the diameter of a muscular pulmonary artery and its accompanying airway should be about the same. Below a diameter of about 100 μm, the pulmonary artery branches lose the internal elastica and are termed arterioles. Arterioles run adjacent to the alveolar ducts and can be found as a corner vessel by the alveolar saccules, but should not be found in alveolar walls. A well-defined mesh of capillaries arranged in a single layer of rings and spokes forms the gas exchange system in the alveoli6 (Fig. 11-3).
Pulmonary Veins
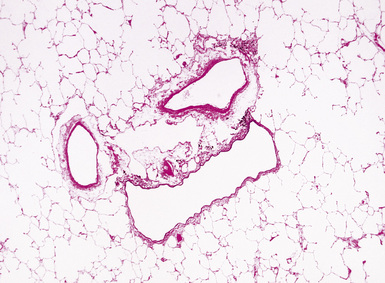
Normal pulmonary veins have only a single elastica and a thin layer of muscle. Veins are best identified by anatomic location. Larger pulmonary veins run in the interlobular septa (Fig. 11-4). Smaller veins are found associated with the alveolar saccules and are indistinguishable by morphology from pulmonary arterioles; thus, the identification of a small vessel as a vein often requires tracing it back through several sections until it joins a definite vein in an interlobular septum. Of note, in pulmonary venous hypertension, the larger veins may acquire both a double elastica and additional muscle and resemble muscular arteries, but the location in the septa indicates their true nature.
Recognition of Right Ventricular Hypertrophy
Significant degrees of pulmonary hypertension are usually associated with right ventricular hypertrophy. A quick, but relatively inaccurate, determination of ventricular hypertrophy can be made by simple measurement of the right ventricular wall muscle thickness (Fig. 11-5). Wall thickness in the normal adult population should be approximately 2 to 3 mm, with measurements greater than 5 mm thought to represent hypertrophy.7
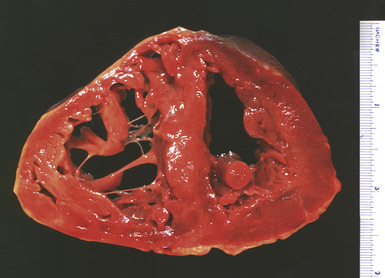
Partitioning of the heart into right ventricle and left ventricle plus septum8 provides a sensitive estimation of ventricular hypertrophy, with a right ventricular weight of 65 g or greater considered abnormal.7 Although a portion of the septum will enlarge with the right ventricle, a ratio of left ventricular weight to right ventricular weight of less than 1.9 is considered to represent right ventricular hypertrophy. Obviously, such ratios are only useful if there is no enlargement of the left ventricle.
Microscopic examination of the right ventricle does not show the generalized increase of fibrosis that can be found in left ventricular hypertrophy. Detailed measurement of the myocardiocytes will demonstrate enlarged fiber diameters,9 but this finding may be too subtle to recognize visually.
Classification of Pulmonary Hypertension
The normal pressure in the pulmonary artery is 20/12 mm Hg (mean, 15 mm Hg) at sea level and 38/14 mm Hg (mean, 25 mm Hg) at approximately 15,000-feet altitude. In general, a mean arterial pressure of 20 mm Hg at sea level is considered abnormal, whereas at 15,000 feet, a pressure of 25 mm Hg is considered abnormal. Pulmonary hypertension is defined clinically as a mean pulmonary artery pressure at rest of greater than 25 mm Hg, or a mean pressure greater than 30 mm Hg during exercise.10 Pulmonary hypertension may be a manifestation of a primary pulmonary vascular disease, or may be secondary to other (nonvascular) diseases in the lung, but the morphologic patterns seen in the vessels in pulmonary hypertension are fairly limited, and, thus, clinical correlation is required for a specific diagnosis.
A variety of schemes for classification of pulmonary hypertension have been proposed.2,11–17Box 11-1 shows a recent clinical classification, commonly called the 2003 Venice classification.17 This classification has replaced the older term primary pulmonary hypertension with the terms idiopathic and familial to recognize the occurrence of genetic mutations in the bone morphogenetic protein receptor 2 (BMPR2) gene in many cases. A problematic aspect of this classification is the inclusion of pulmonary veno-occlusive disease (PVOD) and pulmonary capillary hemangiomatosis (PCH) in the general category of pulmonary arterial hypertension on the basis of dubious claims that these entities can show all the changes of classic idiopathic pulmonary arterial hypertension, including plexiform lesions (see the sections “Pulmonary Veno-Occlusive Disease” and “Pulmonary Capillary Hemangiomatosis” later in the chapter), and on the basis of a handful of reports of cases with BMPR2 mutations.18 Since PVOD and PCH patients often develop pulmonary edema after vasodilator therapy and have a worse outcome than patients in Venice categories 1.1 to 1.3,18 we believe there is considerable logic in maintaining PVOD and PCH in a separate group (see the section “Treatment of Pulmonary Hypertension” later in the chapter).
Box 11-1 The Venice 2003 Clinical Classification of Pulmonary Hypertension
From Simonneau G, Galiè N, Rubin LJ, et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2004;43(12 suppl):5S–12S.
Partly for these reasons, we have not adopted the new pathologic classification of pulmonary hypertension arising from the 2003 Venice meeting,19 but prefer the older and more generally accepted scheme shown in Box 11-2. Some authors have argued that there is no real difference between thrombotic and plexogenic arteriopathy, largely because thrombotic lesions may be found in both.14 However, we believe, as Wagenvoort and Mulder20 have argued, there are generally clear clinical and morphologic differences among the entities shown in Box 11-2, and that thrombosis is a secondary phenomenon in most types of pulmonary hypertension (see later on).
Plexogenic Arteriopathy
Clinical Features
In pulmonary hypertension associated with anorectic agents, the pulmonary vascular lesions are associated with cardiac valvular lesions, predominately on the left side of the heart.21,22 In those patients who develop their pulmonary hypertension in association with human immunodeficiency virus (HIV) disease, the majority can be directly related to HIV infection; other cofactors include liver disease and coagulation abnormalities. There is a wide age range of the affected population. The interval between HIV infection and clinical presentation of pulmonary hypertension may be up to 3 years, but after presentation the prognosis is poor.23,24
Radiologic Features
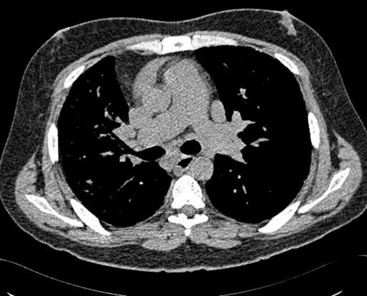
Plain chest film early in the disease may be totally normal in appearance; with more advanced disease, enlarged pulmonary arteries become apparent, and with the development of cor pulmonale, the right ventricle may be visibly enlarged. With long-standing pulmonary hypertension, calcification of the large arteries, presumably representing atherosclerosis, can be seen. Computed tomography (CT) scanning allows measurements of the diameters of the main pulmonary artery; in general, if the diameter of the pulmonary artery is larger than that of the ascending aorta—strictly speaking, if the diameter of the main pulmonary artery at the level of its bifurcation is greater than 29 mm (Fig. 11-6)—there is a high probability of pulmonary hypertension.25 Angiography classically demonstrates vascular “pruning,” in which the vessels have a simplified branching pattern.
Morphologic Features
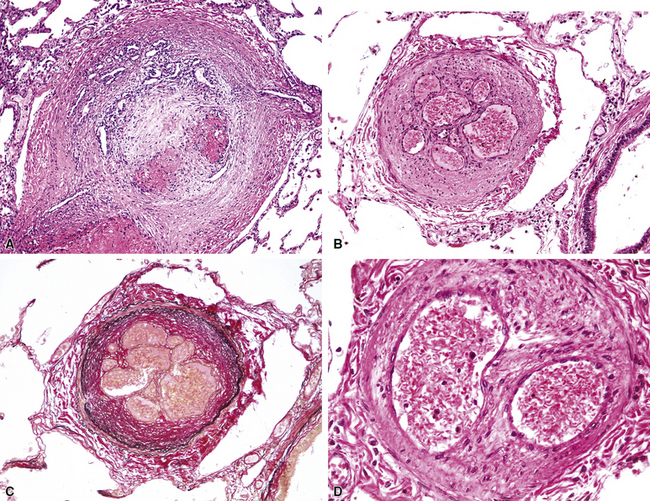
Plexiform lesions were first clearly characterized by Heath and Edwards.16 The term plexogenic pulmonary arteriopathy was coined by Wagenvoort11 to describe a morphologic response pattern that sometimes, but not always, is characterized by the formation of peculiar thrombi with multiple small channels—“plexiform lesions.” Such lesions are the end result of a series of vascular changes, however, and a given case of plexogenic arteriopathy may show only the lower-grade changes without formation of plexiform lesions.
Plexogenic arteriopathy primarily affects muscular arteries and arterioles, but larger arteries may demonstrate increased atherosclerosis, a finding that may be seen in pulmonary hypertension of any cause or in the absence of hypertension. Statistically, however, the most common cause of atherosclerosis in the pulmonary artery is severe systemic atherosclerosis.26
The vascular changes in the muscular arteries and arterioles in plexogenic arteriopathy appear to reflect, in general, the level of pulmonary artery pressure and, to a lesser extent, the length of time hypertension has been present; thus, in a broad sense, higher-grade lesions (see later on) are found in individuals with higher pulmonary artery pressures. The correlations are not exact, however, and only lower-grade lesions may be found in some patients with quite marked pulmonary hypertension. There is also some controversy about the order in which different lesions develop.2,13,14,16 It is our belief that the original Heath and Edwards classification16 is incorrect and that the actual sequence of changes is that proposed by Wagenvoort and Wagenvoort,11 as follows.
Grade I: Muscular Hypertrophy
Muscular hypertrophy appears as thickening of the walls of muscular arteries, often with obvious narrowing of the lumina (Fig. 11-7). Elastic stain shows that the space between the internal and external elastica has become widened by the new muscle. Normal preacinar muscular pulmonary arteries should have, in the fully distended state, a medial thickness that is 1% to 2% of the vessel diameter, although in the smaller muscular arteries (30–100 μm in external diameter), the medial thickness may be up to 5%.24 However, these values are based on arteries fixed by inflation, and in ordinary specimens, some allowance needs to be made for the fact that uninflated vessels will normally have thicker-appearing walls than inflated vessels.
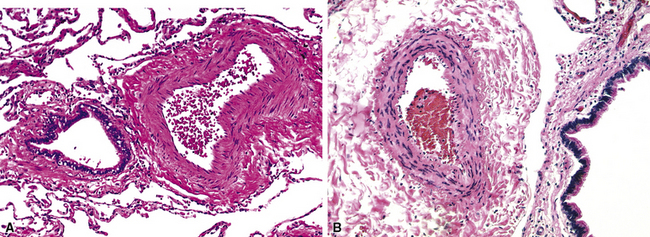
Muscular hypertrophy in small arteries is often accompanied by muscularization of arterioles, such that the arteriole acquires both a double elastica and muscle between the elasticas (Fig. 11-8). Thus, muscularized arterioles come to resemble ordinary muscular arteries but are found in the lung parenchyma; this finding is a clue to the correct diagnosis because arteries are normally present only next to accompanying airways.
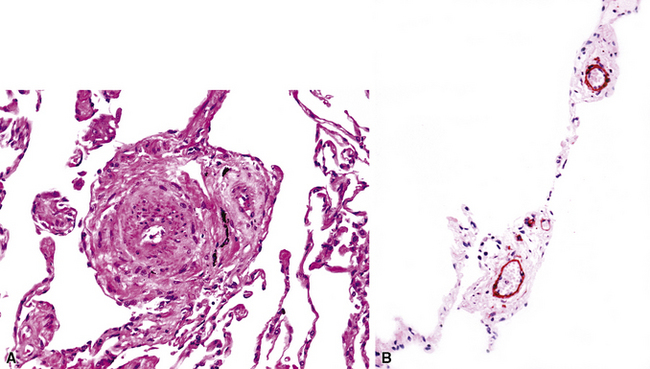
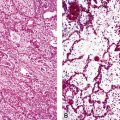
Grade II: Intimal Proliferation
In this stage, proliferation of intimal cells leads to a thickened intima superimposed on a thickened muscular media (Fig. 11-9). The intimal cells do not show any special organization.
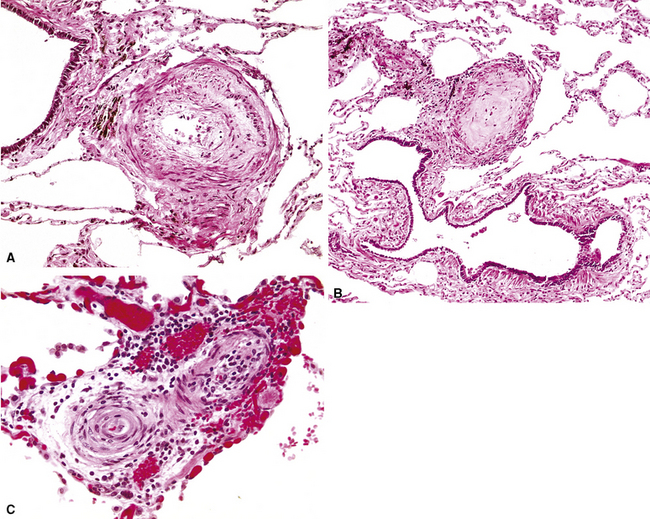
Grade III: Concentric Laminar Intimal Fibrosis
In this stage, the intima is markedly thickened and organized in a series of concentric bands of collagen and spindle-shaped cells, which lend a whorled appearance (Fig. 11-10). The lumen is often dramatically narrowed.
Grade IV: Necrotizing Vasculitis
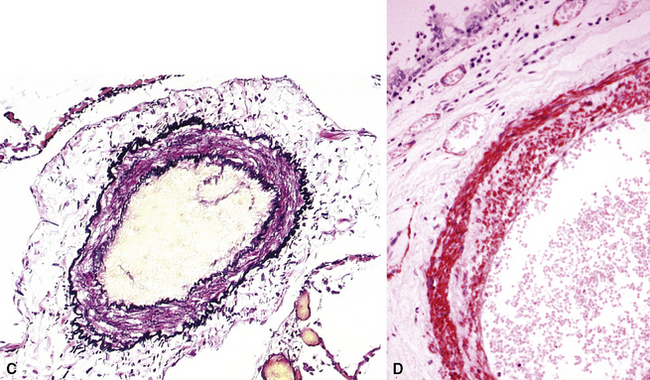
As a result of markedly increased pressure, the arterial wall may become necrotic. The typical pattern is that of fibrinoid necrosis with eosinophilic granular necrotic material replacing the normal arterial wall (Fig. 11-11). Inflammatory cells, usually polymorphonuclear leukocytes but sometimes eosinophils, may be present. Elastic stains show that, typically, the internal elastic is destroyed.
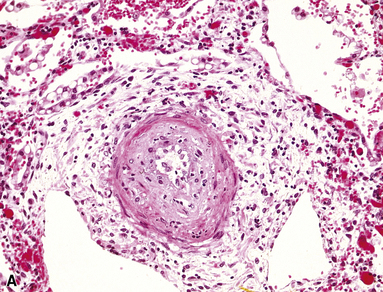
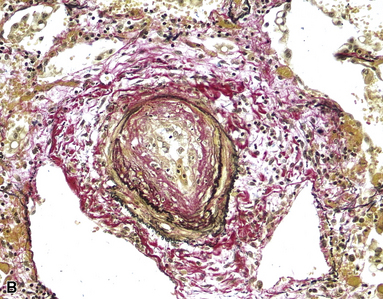
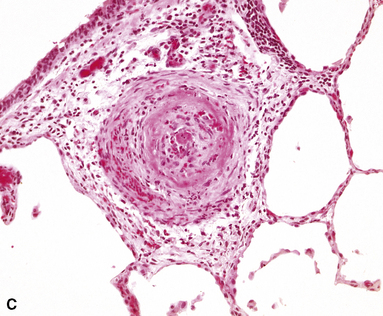
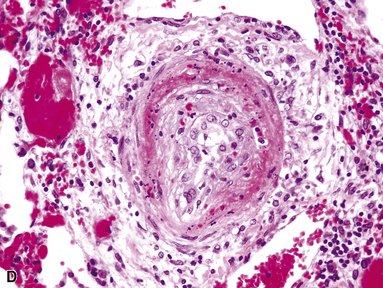
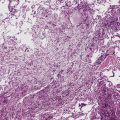
Grade V: Plexiform Lesions
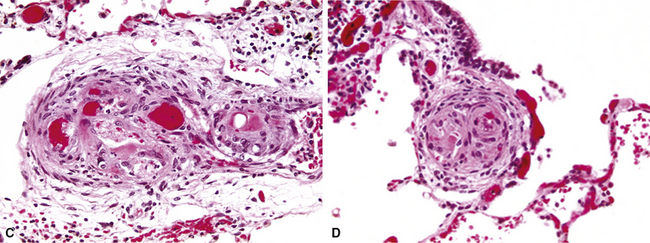
Plexiform lesions are typically found in small muscular arteries at branch points. The artery immediately proximal to the plexiform lesion often shows marked muscular hypertrophy and intimal hyperplasia. In the plexiform lesion itself the vessel is often dilated and the lumen is characteristically filled with capillary channels that very much resemble a fairly cellular organizing thrombus (Fig. 11-12). However, in contradistinction to most thrombi, where the elastic laminae are intact,2 elastic stains show that the inner elastica is typically destroyed in the region of the plexiform lesion (see Fig. 11-12B), and this feature is useful when a question of thrombotic versus plexogenic arteriopathy arises. This set of findings reflects the fact that plexiform lesions are actually the result of fibrinoid necrosis of the vessels, with subsequent thrombosis and organization. The acute plexiform lesions may show small fibrin thrombi and small numbers of inflammatory cells in the capillary channels; as the lesions age, they scar and become paucicellular. Plexiform lesions are usually not very numerous and can be widely scattered within the lung parenchyma; thus, a certain amount of hunting may be required to demonstrate them. It has been suggested that in plexogenic arteriopathy associated with congenital left-to-right shunts, the plexiform lesions occur in arteries 100 to 200 µM in external diameter; whereas in idiopathic (primary) pulmonary hypertension, the lesions occur in arteries smaller than 100 µM.19
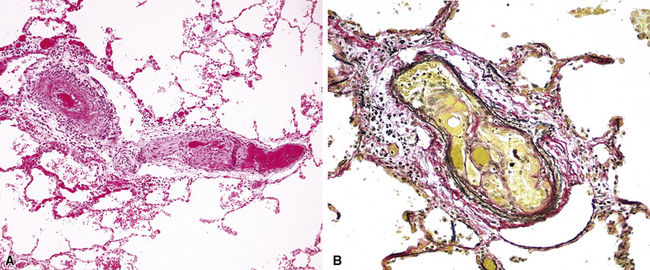
Grade VI: Dilatation and Angiomatoid Lesions
These arterial lesions are located distal to plexiform lesions and probably are related to changes in flow produced by the plexiform lesions. They consist of thin-walled, often dilated and tortuous, channels with a single elastica; these channels are not of obvious arterial structure, but their origin can be proved by tracing back through serial sections (Fig. 11-13). Dilatation and angiomatoid lesions may rupture with resulting pulmonary hemorrhage, and in some instances these lesions appear to anastomose with the bronchial circulation, thus exposing these relatively weak structures to systemic arterial pressures.
Clinical Correlations
As noted earlier, assessment of reversibility or potential for response to treatment is an important reason for performing lung biopsies in patients with pulmonary hypertension. However, the question of what features actually predict reversibility is controversial. As a first approximation, lesions can be separated as shown in Box 11-3.
Wagenvoort12,13 and Palevsky and associates3 have more recently suggested that simple qualitative assessment of the types of lesions present is inadequate by itself for predicting response, and that quantitative measurements are required, particularly measurements of intimal proliferation. For example, Palevsky’s group3 found that an average intimal area of more than 18% of the vascular cross section predicted a poor response to therapy. Interested readers should consult the appropriate references.2,3,13,27
The long-term outlook for patients with pulmonary hypertension associated with plexogenic arteriopathy tends to be poor, with deaths from cor pulmonale or sudden arrhythmias (see the section “Treatment of Pulmonary Hypertension” later in the chapter). In some forms of congenital heart disease, hypertension may be reversed by repair of the cardiac defect.
Differential Diagnosis
It should be remembered that some degree of muscular arterial hypertrophy is seen in virtually all forms of pulmonary hypertension, including veno-occlusive disease, pulmonary capillary hemangiomatosis, and venous hypertension secondary to mitral stenosis or other cardiac lesions.2,28,29 Thus, the finding of muscular hypertrophy as the only vascular abnormality does not necessarily mean that the patient has plexogenic arteriopathy. Equally important, low-grade morphologic changes, especially isolated muscular hypertrophy, are not necessarily predictors of the degree of pulmonary hypertension; some patients with only muscular hypertrophy, nonetheless, can have quite high pulmonary artery pressures.
A further problem in interpretation is that thrombotic lesions, presumably reflecting in situ thromboses caused by abnormal flow, are now recognized as a finding in many different morphologic types of pulmonary hypertension2,3,20 (see morphologic description in the next section) and certainly may be found in plexogenic arteriopathy. This does not invalidate the notion that plexogenic arteriopathy is morphologically separate from thrombotic hypertension.20
Thrombotic and Embolic Hypertension
Pathologic Findings
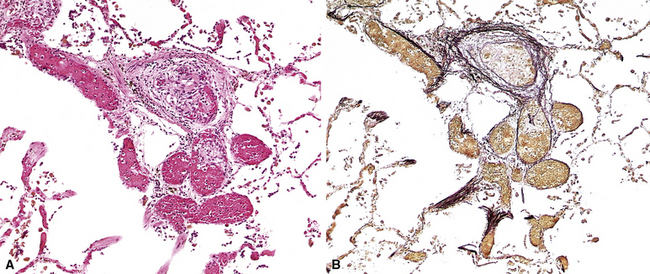
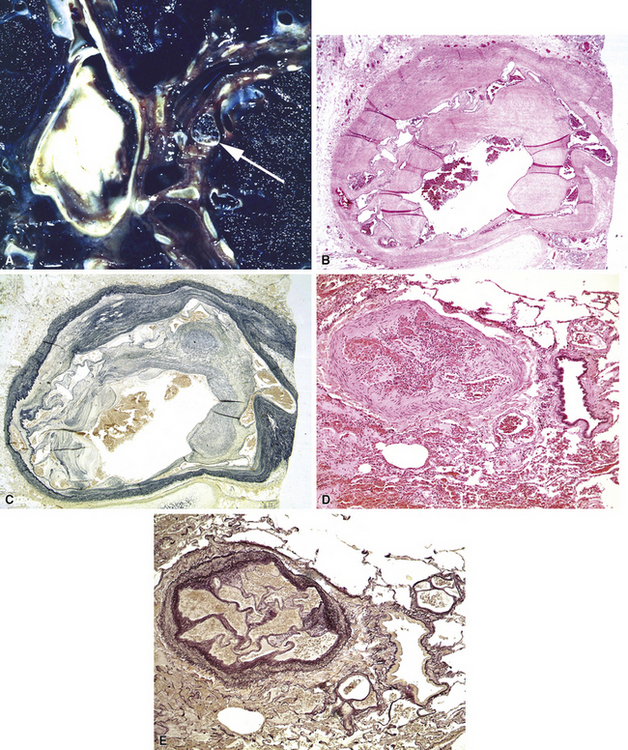
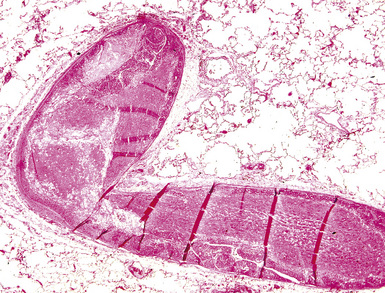
Pathologic findings vary with the type of underlying lesion. In classic thrombotic or thromboembolic hypertension, thrombi in various stages of organization, mostly old, are seen in branches of the small muscular pulmonary arteries. Of note, both elastic laminae are usually intact in thrombotic disease, as opposed to the destruction of the internal elastic in plexiform lesions.2 Larger arteries may show “webs” (Fig. 11-14), which are simply organized thrombi with channels large enough to be seen grossly. In some cases of thrombotic or embolic hypertension, thrombi are only found in the main branches of the pulmonary artery, sometimes with webs as well; these patients often have underlying (nonhypertensive) lung disease or left-sided cardiac disease, as well as peripheral vascular thromboses.29
A helpful feature that should alert the pathologist to the presence of thrombi and emboli is the finding of eccentric intimal proliferation or intimal fibrosis in arterial vessels (Fig. 11-15); these lesions sometimes represent old small organizing thrombi. Both eccentric lesions and recanalized thrombi may be seen in pulmonary hypertension of other causes.20
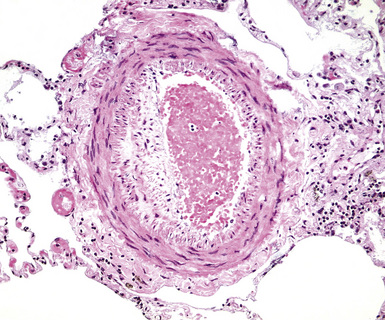
Figure 11-15 Eccentric intimal proliferation representing an old thrombus. Compare with the concentric intimal proliferation of plexogenic arteriopathy in Figures 11-10C and 11-12A.
Patients with sickle cell disease may develop thrombi in their distal pulmonary arterial branches during sickle crises, and recurrent episodes can lead to a form of thrombotic pulmonary hypertension. The lesions look like organizing thrombi, but close examination reveals the presence of sickled cells (Fig. 11-16).
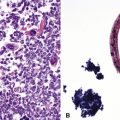
Intravenous injection of licit drugs intended for oral use, or sometimes of illicit drugs such as heroin or cocaine,30,31 tends to produce deposits of insoluble filler material from the drug in the small muscular arteries. In mild disease, small numbers of birefringent particles are seen in the lumina or in the arterial walls (Fig. 11-17), presumably having been incorporated into organizing thrombi. With injection of large amounts of drug, the inflammatory and thrombotic reaction to the particles leads to formation of thrombus-like formations with capillary channels called angiothrombotic lesions.31 These are probably just peculiar in situ thrombi caused by large numbers of particles. They more or less completely obstruct the arterial branch, and accumulation of such lesions leads to pulmonary hypertension. Polarized light examination is often useful in demonstrating the particulate matter. Under polarizing light, starch appears as Maltese cross–like images; talc, as brightly birefringent plates; and microcrystalline cellulose, as periodic acid/Schiff–positive rectangular crystals that are also birefringent. For illicit drugs, exact identification of the filler may not be possible.
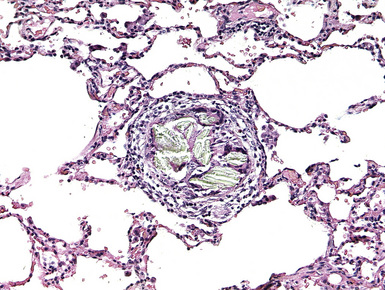
Figure 11-17 Organized thrombus and birefringent particles in a muscular pulmonary artery from an intravenous drug abuser.
Pulmonary hypertension may develop as a result of filling of the small arterial branches with tumor emboli (Fig. 11-18). Radiographically or pathologically visible metastases may or may not be present. Pulmonary hypertension in this setting has been most commonly reported with lung, breast, stomach, ovarian, and hepatocellular carcinomas.29 An unusual form of tumor-associated pulmonary hypertension is pulmonary tumor thrombotic microangiopathy32 in which the tumor elicits a marked intimal proliferation with only small numbers of tumor cells present.
Differential Diagnosis
As noted, scattered thrombi are fairly common in other types of pulmonary hypertension, such as plexogenic arteriopathy, and the presence of thrombi or eccentric intimal lesions does not automatically indicate a diagnosis of thrombotic and embolic hypertension. Although it has been claimed that plexiform lesions can, rarely, be seen in thrombotic and embolic hypertension, including sickle cell disease,17,33 we believe that most such cases, in fact, represent plexogenic arteriopathy with more than the usual number of thrombi. Thus, it is important to be sure that typical plexogenic lesions are not present and to demonstrate the presence of multiple thrombi or emboli when making a diagnosis of thrombi or embolic hypertension.
Pulmonary Veno-Occlusive Disease and Other Types of Pulmonary Venous Hypertension
Radiologic Features
On plain films, patients with advanced disease show pulmonary artery enlargement and, with cor pulmonale, right ventricular hypertrophy. A helpful finding in PVOD is the presence of prominent Kerley B lines and mild to moderate interstitial infiltrates. The CT scan shows distinctly thickened interlobular septa.34
Pathologic Findings
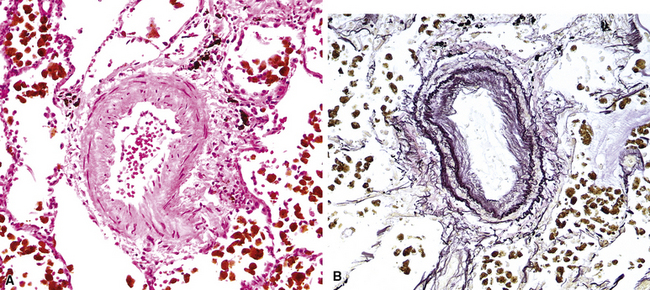
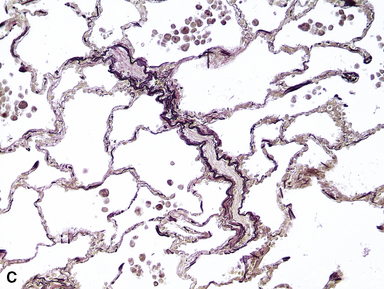
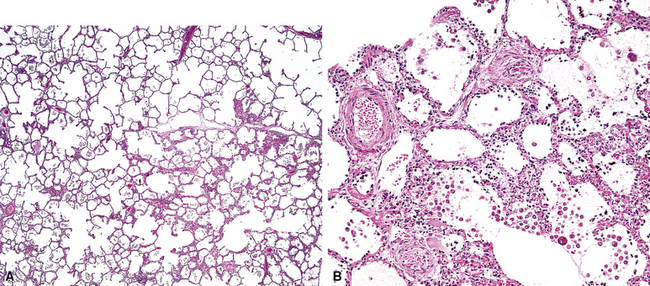
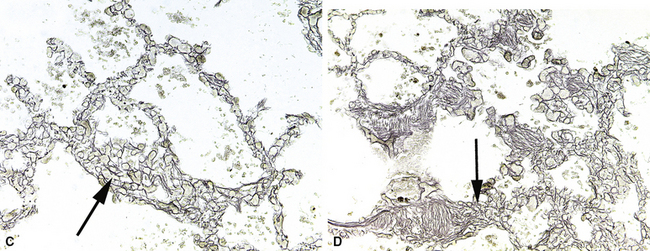
The fundamental lesion in PVOD is thrombosis, typically old thrombosis, of small pulmonary veins and venules, although a recent report documented venulitis in a small proportion of cases.35 Thrombosis is most easily seen in veins in the interlobular septa (Fig. 11-19) where the location ensures that the structure is indeed a vein. With increasing pressure, such veins may become arterialized, that is, they develop a double elastic lamina and a distinct layer of muscle (see Fig. 11-19C), and are, thus, distinguishable from arteries only by their location.
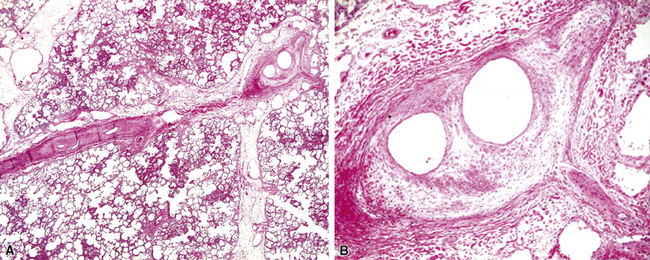
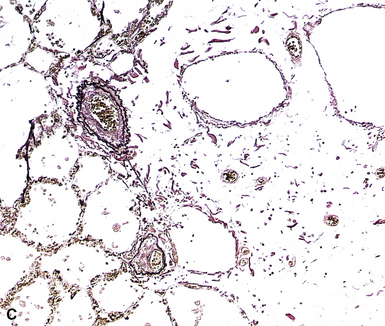
The venules in PVOD are often thrombosed as well, but this may be difficult to document. Venous hypertension of any cause is associated with intimal fibrosis and luminal narrowing of the veins and venules, but in PVOD the lumen of small venules may simply be obliterated by fibrous tissue (Fig. 11-20). Use of elastic stains is mandatory to find such vessels, and once small vessels with apparent luminal obliteration are found, they may need to be traced back through several sections until they connect with a vein in an interlobular septum, thus proving their nature. The diagnosis of PVOD can be exceedingly difficult when only small venules are affected.
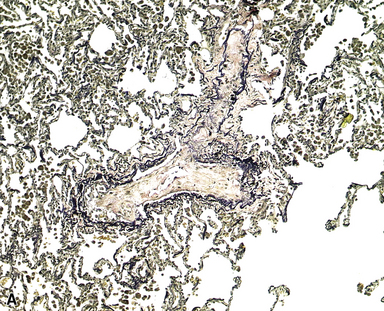
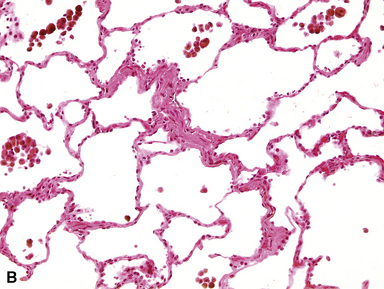
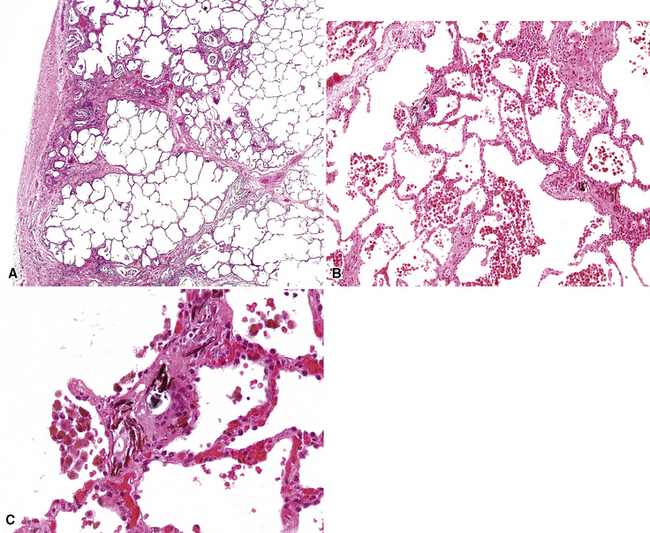
The vascular changes in PVOD do not occur in isolation. The interlobular septa are generally edematous and the lymphatics prominently dilated (see Fig. 11-19). A peculiar, usually mild, form of interstitial fibrosis that tends to be more marked in the very periphery of the lung under the pleura (Fig. 11-21) is common in PVOD (47% of cases in a recent large series35). The fibrosis is fairly homogeneous, typically paucicellular, and raises the morphologic question of a chronic interstitial pneumonia. Accompanying the fibrosis are usually very small foci of acute or old hemorrhage with hemosiderin-laden macrophages (see Fig. 11-21B). The combination of mild, homogeneous fibrosis and small hemorrhages should bring the diagnosis of PVOD to mind and prompt examination of the veins in the interlobular septa.
Arterial changes may be present in PVOD and consist of muscular hypertrophy and sometimes mild intimal fibrosis. Although it has been claimed that plexiform lesions can be seen in PVOD,19 in our experience this is not true, and plexiform lesions were not seen in 30 patients reported by Lantuéjoul.35
Another change that may be seen in PVOD, but also in any disease characterized by repeated pulmonary hemorrhage, is so-called endogenous pneumoconiosis, in which elastic fibers are coated by iron and calcium, with a resulting appearance that resembles a ferruginous body (see Fig. 11-21C) of the type seen in asbestosis. Foci of dystrophic ossification also may be present but are a fairly nonspecific marker of venous hypertension (see later discussion).
Clinical Correlations
In most cases, PVOD has no known cause, but it has been reported in a small number of patients given chemotherapeutic agents36 and patients with autoantibodies against platelets or anticardiolipin antibodies.35 In general, the prognosis is poor, and, at present, transplantation offers the only hope of cure. Vasodilator therapy must be approached with caution because it may induce severe pulmonary edema.
Differential Diagnosis
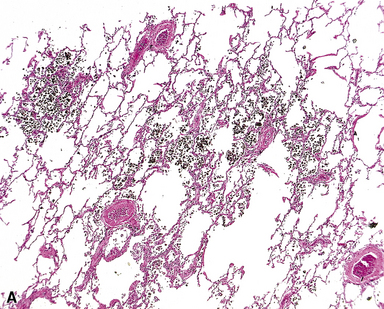
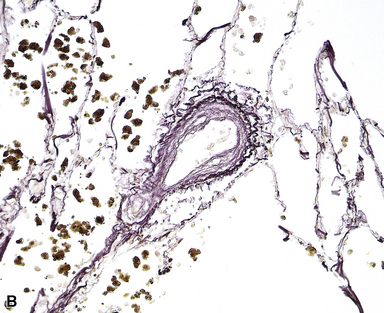
PVOD is the most dramatic and severe form of pulmonary venous hypertension. Venous hypertension may also be seen in patients with left-sided heart disease, especially mitral stenosis, and, rarely, atrial myxomas; in conditions such as sclerosing mediastinitis, in which the pulmonary veins are obliterated or narrowed in the mediastinum; in some types of congenital cardiac malformations, in which the major pulmonary veins are abnormal or absent; and in some patients with collagen vascular diseases.37 Intimal fibrosis in the small pulmonary veins and venules, mild interstitial inflammation and edema, small hemorrhages, “endogenous pneumoconiosis,” and dystrophic ossification are common in all these conditions, and muscular hypertrophy in the small pulmonary arterial branches may be present as well (Fig. 11-22). However, true venous obliteration, the hallmark of PVOD, is absent. Idiopathic interstitial pneumonias and even real pneumoconioses come into the differential diagnosis, as indicated previously.
Pulmonary Capillary Hemangiomatosis
Clinical and Radiologic Features
PCH is a very rare disease, mostly seen in adults. The usual nonspecific signs of pulmonary hypertension are present and small hemoptyses may occur. Chest radiography demonstrates interstitial infiltrates with thickened interlobular septa.38
Pathologic Findings
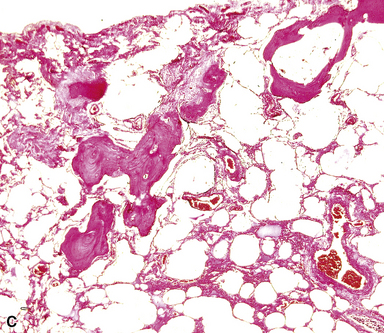
PCH is characterized by proliferation of dilated capillary-sized channels along and in the alveolar walls (Fig. 11-23). In this it resembles a very severe form of passive congestion, but careful examination shows that there appears to be duplicate or multiple capillary channels in an alveolar wall, something not present in passive congestion.39 The proliferating capillary channels extend into arterioles and venules, producing a peculiar pattern of capillary proliferation within the walls of the larger vessel with resulting luminal narrowing or obliteration; it is this involvement of larger vessels that is thought to produce pulmonary hypertension. Often there is an admixture of very abnormal areas of lung with extensive capillary proliferation combined with perfectly normal-appearing lung, again a helpful finding in separating capillary hemangiomatosis from passive congestion (the latter should be more homogeneous). Small acute and old hemorrhages may be present, as may muscular hypertrophy in small pulmonary arteries.
It has recently been suggested that most cases of PCH are really examples of PVOD.35 Thrombotic occlusion of small veins can be found in PCH, along with small foci of hemorrhage and areas of interstitial fibrosis; conversely, PCH-like foci can be demonstrated in many cases of PVOD as well as other forms of pulmonary venous hypertension and also in patients who do not have pulmonary hypertension.35
Pulmonary Hypertension Secondary to Other Forms of Intrinsic Lung Disease
Pulmonary hypertension is commonly found associated with different forms of nonvascular intrinsic lung disease, including emphysema, bronchiectasis, usual interstitial pneumonia, and any other conditions that produce extensive scarring of the parenchyma or that produce chronic hypoxia. In emphysema, claims have been made that hypertension is secondary to loss of capillary bed, although this may not be correct, and vascular changes may be caused by direct effects of cigarette smoke on the vasculature or by local hypoxic vasoconstriction.40 Pulmonary hypertension is increasingly being recognized as an important complication of usual interstitial pneumonia; it was seen in 46% of a large series of patients with usual interstitial pneumonia awaiting transplantation.41 With pulmonary hypertension, the development of cor pulmonale is common in all of these diseases and may be the cause of death.
Morphologic Mimics of Pulmonary Hypertension
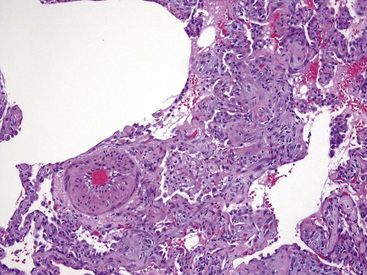
In our experience, biopsies from patients with interstitial lung disease frequently show muscular arterial hypertrophy, even when cardiac catheterization shows normal pressures (Fig. 11-24). Intimal fibrosis also increases as a normal function of age.35 Thus, considerable caution should be exercised in the individual case when interpreting what appear to be low-grade hypertensive changes when the morphologic changes occur in a clinical or pathologic setting not suggestive of pulmonary hypertension.
Treatment of Pulmonary Hypertension
The pathophysiology of pulmonary hypertension is complex and involves multiple pathways, including the endothelin (vasoconstriction), protacyclin, and nitric oxide (vasodilation) cascades. Various therapies have been directed toward vasodilation by antagonism or enhancement of the above pathways, in addition to the supportive therapies of diuretics and oxygen; these have shown some benefit.42,43
1 Wagenvoort C.A. Lung biopsy specimens in the evaluation of pulmonary vascular disease. Chest. 1980;77:614-625.
2 Pietra G.G., Edwards W.D., Kay J.M., et al. Histopathology of primary pulmonary hypertension. Circulation. 1989;80:1198-1206.
3 Palevsky H.I., Schloo B.L., Pietra G.G., et al. Primary pulmonary hypertension. Vascular structure, morphometry, and responsiveness to vasodilator agents. Circulation. 1989;80:1207-1220.
4 Wagenvoort C.A., Wagenvoort N. Pulmonary vascular bed: normal anatomy and responses to disease. In: Moser K.M., editor. Pulmonary Vascular Diseases: Lung Biology in Health and Disease. New York: Marcel Dekker; 1979:1-110.
5 Rabinovitch M. Morphology of the developing pulmonary bed: pharmacologic implications. Pediatr Pharm. 1985;5:31-48.
6 Schraufnagel D.E. Corrosion casting of the lung for scanning electron microscopy. In: Lenfant C., editor. Electron Microscopy of the Lung. New York: Marcel Dekker; 1990:257-297.
7 Murphy M.L., Bone R.C. Cor Pulmonale in Chronic Bronchitis and Emphysema. New York: Future Publishing, 1984.
8 Fulton R.M., Hutchinson E.C., Jones A.M. Ventricular weight in cardiac hypertrophy. Br Heart J. 1952;14:413-420.
9 Ishikawa S., Fattal G.A. Functional morphometry of myocardial fibres in cor pulmonale. Am Rev Respir Dis. 1972;105:358-367.
10 Fishman A.P. Pulmonary hypertension and cor pulmonale. In Fishman A.P., editor: Pulmonary Diseases and Disorders, 2nd ed, New York: McGraw-Hill, 1988.
11 Wagenvoort C.A., Wagenvoort N. Pathology of Pulmonary Hypertension. New York: John Wiley & Sons, 1977.
12 Wagenvoort C.A. Lung biopsies in the differential diagnosis of thromboembolic versus primary pulmonary hypertension. Prog Resp Res. 1980;13:16-21.
13 Wagenvoort C.A. Grading of pulmonary vascular lesions—a reappraisal. Histopathology. 1981;5:595-598.
14 Pietra G.G., Ruttner J.R. Specificity of pulmonary vascular lesions in primary pulmonary hypertension. A reappraisal. Respiration. 1982;52:81-85.
15 Burke A.P., Farb A., Virmani R. The pathology of primary pulmonary hypertension. Mod Pathol. 1991;4:269-277.
16 Heath D., Edwards J.E. The pathology of hypertensive pulmonary vascular disease. Circulation. 1958;18:533-543.
17 Simonneau G., Galiè N., Rubin L.J., et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol.. 2004;43(12 Suppl S):5S-12S.
18 Montani D., Achouh L., Dorfmüller P., et al. Pulmonary veno occlusive disease: clinical, functional, radiologic, and hemodynamic characteristics and outcome of 24 cases confirmed by histology. Medicine (Baltimore).. 2008;87:220-233.
19 Pietra G.G., Capron F., Stewart S., et al. Pathologic assessment of vasculopathies in pulmonary hypertension. J Am Coll Cardiol.. 2004;43(12 Suppl S):25S-32S.
20 Wagenvoort C.A., Mulder R.G.H. Thrombotic lesion in primary plexogenic arteriopathy: similar pathogenesis or complication? Chest. 1993;103:844-849.
21 Abenhaim L., Moride U.Y., Brenot F., et al. Appetite-suppressant drugs and the risk of primary pulmonary hypertension. N Engl J Med. 1996;335:609-616.
22 Fishman A.P. Aminorex to fen/phen: an epidemic foretold. Circulation. 1999;99:156-161.
23 Mehta N.J., Khan L.A., Mehta R.N., Sepkowitz D.A. HIV-related pulmonary hypertension: analytic review of 131 cases. Chest. 2000;118:1133-1141.
24 Pellicelli A.M., Barbaro G., Palmieri F., et al. Primary pulmonary hypertension in HIV patients: a systematic review. Angiology. 2001;51:31-41.
25 Ng C.S., Wells A.U., Padley S.P. A CT sign of chronic pulmonary arterial hypertension: the ratio of main pulmonary artery to aortic diameter. J Thorac Imaging. 1999;14:270-278.
26 Moore G.W., Smith R.R., Hutchins G.M. Pulmonary artery atherosclerosis: correlation with systemic atherosclerosis and hypertensive pulmonary vascular disease. Arch Pathol Lab Med. 1982;106:378-380.
27 Yamaki S., Wagenvoort C.A. Plexogenic pulmonary arteriopathy: significance of medial thickness with respect to advanced pulmonary vascular lesions. Am J Pathol. 1981;105:70-75.
28 Fernie J.M., Lamb D. Effects of age and smoking on intima of muscular pulmonary arteries. J Clin Pathol. 1986;39:1204-1208.
29 Katzenstein A.B.L. Pulmonary hypertension and other vascular disorders. In: Katzenstein A.B.L., editor. Katzenstein and Askin’s Surgical Pathology of Non-Neoplastic Lung Disease. 3rd ed. Philadelphia: WB Saunders; 1997:322-360.
30 Yakel D.L., Eisenberg M.J. Pulmonary artery hypertension in chronic intravenous cocaine users. Am Heart J. 1995;130:398-399.
31 Tomashefski J.F., Hirsch C.S. The pulmonary vascular lesions of intravenous drug abuse. Hum Pathol. 1980;11:133-145.
32 von Herbay A., Illes A., Waldherr R., Otto H.F. Pulmonary tumor thrombotic microangiopathy with pulmonary hypertension. Cancer. 1990;66:587-592.
33 Moser K.M., Bloor C.M. Pulmonary vascular lesions occurring in patients with chronic major vessel thromboembolic pulmonary hypertension. Circulation. 1981;210:507-511.
34 Holcomb B.S., Loyd J.E., Ely W., et al. Pulmonary veno-occlusive disease. Chest. 2000;118:1671-1679.
35 Lantuéjoul S., Sheppard M.N., Corrin B., et al. Pulmonary veno-occlusive disease and pulmonary capillary hemangiomatosis: a clinicopathologic study of 35 cases. Am J Surg Pathol.. 2006;30:850-857.
36 Lombard C., Churg A., Winokur A. Pulmonary veno-occlusive disease following therapy for malignant neoplasms. Chest. 1987;92:871-876.
37 Dorfmüller P., Humbert M., Perros F., et al. Fibrous remodeling of the pulmonary venous system in pulmonary arterial hypertension associated with connective tissue diseases. Hum Pathol.. 2007;38:893-902.
38 Al-Fawaz I.M., Al Mobiareek K.F., Al-Suhaibani M., Ashour M. Pulmonary capillary hemangiomatosis. Pediatr Pulmonol. 1995;19:243-248.
39 Tron V., Magee F., Wright J., et al. Pulmonary capillary hemangiomatosis. Hum Pathol. 1986;17:1144-1150.
40 Wright J.L., Levy R.D., Churg A. Pulmonary hypertension in chronic obstructive pulmonary disease: current theories of pathogenesis and their implications for treatment. Thorax.. 2005;60:605-609.
41 Shorr A.F., Wainright J.L., Cors C.S., et al. Pulmonary hypertension in patients with pulmonary fibrosis awaiting lung transplant. Eur Respir J.. 2007;30:715-721.
42 Badesch D.B., Abman S.H., Simonneau G., et al. Medical therapy for pulmonary arterial hypertension: updated ACCP evidence based clinical practice guidelines. Chest.. 2007;131:1917-1928.
43 Humbert M., Sitbon O., Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med.. 2004;351:1425-1436.