Pulmonary drug delivery
Kevin M.G. Taylor
Chapter contents
Inhalation aerosols and the importance of size distribution
Particle deposition in the airways
Formulating and delivering therapeutic inhalation aerosols
Pressurized metered-dose inhalers
Methods of aerosol size analysis
Key points
• Pulmonary delivery may be used for drugs having local or systemic activity.
• Nebulizers deliver relatively large doses of drugs, as either aqueous solutions or suspensions.
Inhaled drug delivery
Therapeutic agents for the treatment or prophylaxis of airways diseases, such as bronchial asthma, chronic obstructive pulmonary disease (COPD) and cystic fibrosis are usually delivered directly to the respiratory tract. The administration of a drug at its site of action can result in a rapid onset of activity, which may be highly desirable, for instance when delivering bronchodilating drugs for the treatment of asthma. Additionally, smaller doses can be administered locally compared to delivery by the oral or parenteral routes, thereby reducing the potential incidence of adverse systemic effects and reducing drug costs. The pulmonary route is also useful where a drug is poorly absorbed orally, e.g. sodium cromoglicate, or where it is rapidly metabolized orally, e.g. isoprenaline. The avoidance of first-pass metabolism in the liver may also be advantageous, although the lung itself has some metabolic capability.
The lung may also be used as a route for delivering drugs having systemic activity, because of its large surface area, the abundance of capillaries and the thinness of the air–blood barrier. This has been exploited in the treatment of migraine with ergotamine, and the potential for delivering biopharmaceuticals, such as insulin, vaccines and growth hormone via the airways is now well established.
Lung anatomy
The lung is the organ of external respiration, in which oxygen and carbon dioxide are exchanged between blood and inhaled air. The structure of the airways also efficiently prevents the entry, and promotes removal of airborne foreign particles, including microorganisms.
The respiratory tract can be considered as comprising conducting (central) regions (trachea, bronchi, bronchioles, terminal and respiratory bronchioles) and respiratory (peripheral) regions (respiratory bronchioles and alveolar regions), although there is no clear demarcation between them (Fig. 37.1). The upper respiratory tract comprises the nose, throat, pharynx and larynx; the lower tract comprises the trachea, bronchi, bronchioles and the alveolar regions. Simplistically, the airways can be described by a symmetrical model in which each airway divides into two equivalent branches or generations. In fact, the trachea (generation 0) branches into two main bronchi (generation 1), of which the right bronchus is wider and leaves the trachea at a smaller angle than the left, and hence is more likely to receive inhaled material. Further branching of the airways ultimately results in terminal bronchioles. These divide to produce respiratory bronchioles, which connect with alveolar ducts leading to the alveolar sacs (generation 23). These contain approximately 2–6 × 108 alveoli, producing a surface area of 100–140 m2 in an adult male.
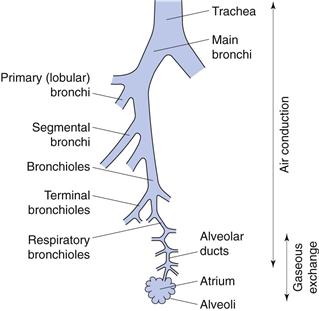
The conducting airways are lined with ciliated epithelial cells. Insoluble particles deposited on the airways walls in this region are trapped by the mucus, swept upwards from the lungs by the beating cilia to the throat, and are swallowed.
Inhalation aerosols and the importance of size distribution
To deliver a drug into the airways, it must be presented as an aerosol (with the exception of medical gases). In pharmacy, an aerosol is defined as a two-phase system of solid particles or liquid droplets dispersed in air or other gaseous phase, having sufficiently small size to display considerable stability as a suspension.
The deposition of a drug/aerosol in the airways is dependent on four factors: the physicochemical properties of the drug, the formulation, the delivery/liberating device, and the patient (breathing patterns and clinical status).
The most fundamentally important physical property of an aerosol for inhalation is its size. The particle size of an aerosol is usually standardized by calculation of its aerodynamic diameter, da, which is the physical diameter of a unit density sphere which settles through air with a velocity equal to the particle in question. Therapeutic aerosols are heterodispersed (polydispersed) and the distribution of sizes is generally represented by the geometric standard deviation (GSD or σg), when the size is log-normally distributed.
For approximately spherical particles:
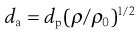 (37.1)
(37.1)
where dp is physical diameter, ρ is particle density and ρ0 is unit density, i.e. 1 g/cm3.
When dp is the mass median diameter (MMD), da is termed the mass median aerodynamic diameter (MMAD).
Large porous particles, with large physical diameters of the order of 20 µm are efficiently delivered to and deposited in the lungs. Their low density, due to the porous or hollow nature of their structure means such particles have a small aerodynamic diameter and are thus carried in the inspired air, deep into the lungs. Additionally, large particles are less prone to aggregation than smaller ones (see below) offering formulation advantages and the particles are too large to be cleared from the airways by alveolar macrophages.
Influence of environmental humidity on particle size
As a particle enters the respiratory tract, the change from ambient to high relative humidity (approximately 99%) results in condensation of water on to the particle surface, which continues until the vapour pressure of the water equals that of the surrounding atmosphere. For water-insoluble materials, this results in a negligibly thin film of water; however, with water-soluble materials a solution is formed on the particle surface. As the vapour pressure of the solution is lower than that of pure solvent at the same temperature, water will continue to condense until equilibrium between vapour pressures is reached, i.e. the particle will increase in size. The final equilibrium diameter is constrained by the Kelvin effect, i.e. the vapour pressure of a droplet solution is higher than that for a planar surface, and is a function of the particle’s original diameter. Hygroscopic growth will affect the deposition of particles, resulting in deposition higher in the respiratory tract than would have been predicted from measurements of their initial size.
Particle deposition in the airways
The efficacy of a therapeutic aerosol is dependent on its ability to penetrate the respiratory tract and be deposited. To penetrate to the peripheral (respiratory) regions, aerosols require a size less than about 5 or 6 µm, with less than 2 µm being preferable for alveolar deposition. Literature values for ‘respirable’ size vary and must be considered alongside the environmental changes in size described above and the heterodispersed nature of inhalation aerosol size distributions. Larger particles or droplets are deposited in the upper respiratory tract and are rapidly removed from the lung by the mucociliary clearance process. As a consequence, the drug becomes available for systemic absorption and may potentially cause adverse effects. Steroid aerosols of sufficiently large size may deposit in the mouth and throat, with the potential to cause adverse effects, including oral candidiasis. The size of aerosolized drug may be especially important in the treatment of certain conditions where penetration to the peripheral airways is particularly desirable, for instance the treatment and prophylaxis of the alveolar infection Pneumocystis carinii pneumonia.
There are three main mechanisms responsible for particulate deposition in the lung: gravitational sedimentation, impaction and diffusion.
Inertial impaction
The air stream changes direction in the throat, or where a bifurcation occurs in the respiratory tract. Particles within the air stream, having sufficiently high momentum, will impact on the airways’ walls rather than following the changing air stream. This deposition mechanism is particularly important for large particles having a diameter greater than 5 µm, and particularly greater than 10 µm, and is common in the upper airways, being the principal mechanism for deposition in the nose, mouth, pharynx and larynx and the large conducting airways. With the continuous branching of the conducting airways, the velocity of the air stream decreases and impaction becomes a less important mechanism for deposition.
The probability of impaction is proportional to:
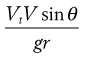 (37.2)
(37.2)
where θ is the change in airways direction, V is air stream velocity and r is the airway’s radius. Vt is the terminal settling velocity (see Eqn 37.3).
Gravitational sedimentation
From Stokes’ Law, particles settling under gravity will attain a constant terminal settling velocity, Vt:
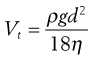 (37.3)
(37.3)
where ρ is particle density, g is the gravitational constant, d is particle diameter and η is air viscosity.
Thus, gravitational sedimentation of an inhaled particle is dependent on its size and density, in addition to its residence time in the airways. Sedimentation is an important deposition mechanism for particles in the size range 0.5–3 µm, in the small airways and alveoli, for particles that have escaped deposition by impaction.
Brownian diffusion
Collision and bombardment of small particles by molecules in the respiratory tract produce Brownian motion. The resultant movement of particles from high to low concentrations causes them to move from the aerosol cloud to the airways’ walls. Diffusion is inversely proportional to particle size. It is the predominant mechanism for particles smaller than 0.5 µm, with the rate of diffusion, given by the Stokes-Einstein equation:
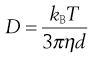 (37.4)
(37.4)
where D is the diffusion coefficient, kB is the Boltzmann’s constant, T is the absolute temperature, η is viscosity and d is particle diameter.
Other mechanisms of deposition
Although impaction, sedimentation and diffusion are the most important mechanisms for drug deposition in the respiratory tract, other mechanisms may occur. These include interception, whereby particles having extreme shapes, such as fibres, physically catch on to the airways’ walls as they pass through the respiratory tract, and electrostatic attraction, whereby an electrostatic charge on a particle induces an opposing charge on the walls of the respiratory tract, resulting in attraction between particle and walls.
Effect of particle size on deposition mechanism
Different deposition mechanisms are important for different sized particles. Those greater than 5 µm will deposit predominantly by inertial impaction in the upper airways. Particles sized between 1 and 5 µm deposit predominantly by gravitational sedimentation in the lower airways, especially during slow, deep breathing and particles less than 1 µm deposit by Brownian diffusion in the stagnant air of the lower airways. Particles of approximately 0.5 µm are inefficiently deposited, being too large for effective deposition by Brownian diffusion and too small for effective impaction or sedimentation, and they are often quickly exhaled. This size of minimum deposition should thus be considered during formulation, although for the reasons of environmental humidity discussed previously, the equilibrium diameter in the airways may be significantly larger than the original particle size in the formulation.
Breathing patterns
Patient-dependent factors, such as breathing patterns, lung physiology and the presence of pulmonary disease also affect particle deposition. For instance, the larger the inhaled volume, the greater the peripheral distribution of particles in the lung, whilst increasing inhalation flow rate enhances deposition in the larger airways by inertial impaction. Breath-holding after inhalation increases the deposition of particles by sedimentation and diffusion. Optimal aerosol deposition occurs with slow, deep inhalations to total lung capacity, followed by breath-holding prior to exhalation. It should be noted that changes in the airways resulting from disease states, for instance airways’ obstruction, may affect the deposition profile of an inhaled aerosol.
Clearance of inhaled particles and drug absorption
Particles deposited in the ciliated conducting airways are cleared by mucociliary clearance within 24 hours and are ultimately swallowed. The composition of mucus and the process of mucociliary clearance are discussed in Chapter 38. Insoluble particles penetrating to the alveolar regions, and which are not solubilized in situ, are removed more slowly. Alveolar macrophages engulf such particles and may then migrate to the bottom of the mucociliary escalator, or alternatively may be removed via the lymphatics. The clearance of particle-loaded macrophages occurs over a period of days or weeks.
Hydrophobic compounds are usually absorbed at a rate dependent on their oil/water partition coefficients, whereas hydrophilic materials are poorly absorbed through membrane pores at rates inversely proportional to molecular size. Thus, the airways’ membrane, like the gastrointestinal tract, is preferably permeable to the unionized form of a drug. Some drugs, such as sodium cromoglicate, are partly absorbed by a saturable active transport mechanism, whilst large macromolecules may be absorbed by transcytosis. The rate of drug absorption, and consequently drug action, can be influenced by the formulation. Rapid drug action can generally be achieved using solutions or powders of aqueous soluble salts, whereas slower or prolonged absorption may be achieved using suspension formulations, powders of less soluble salts or novel drug delivery systems such as liposomes and microspheres.
Formulating and delivering therapeutic inhalation aerosols
There are currently three main types of aerosol-generating device for use in inhaled drug therapy: pressurized metered-dose inhalers, dry powder inhalers and nebulizers.
Pressurized metered-dose inhalers
Pressurized metered-dose inhalers (pMDIs), also referred to as metered-dose inhalers (MDIs), were introduced in the mid-1950s and are the most commonly used inhalation drug delivery devices. In pMDIs, drug is either dissolved or suspended in liquid propellant(s) together with other excipients, including surfactants, and presented in a pressurized canister fitted with a metering valve (Fig. 37.2). A predetermined dose is released as a spray on actuation of the metering valve. When released from the canister, the formulation undergoes volume expansion in the passage within the valve and forms a mixture of gas and liquid before discharge from the orifice. The high-speed gas flow helps to break up the liquid into a fine spray of droplets.
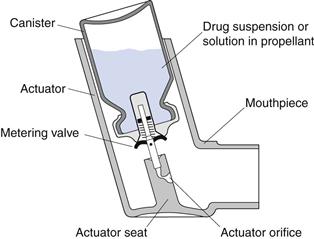
Fig. 37.2 The pressurized metered-dose inhaler.
Containers
Pharmaceutical aerosols may be packaged in tin-plated steel, plastic-coated glass or aluminium containers. In practice, pMDIs are generally presented in aluminium canisters, produced by extrusion to give seamless containers with a capacity of 10–30 mL. Aluminium is relatively inert and may be used uncoated where there is no chemical instability between container and contents. Alternatively, aluminium containers with an internal coating of a chemically resistant organic material, such as an epoxy resin or polytetrafluoroethylene (PTFE), can be used.
Propellants
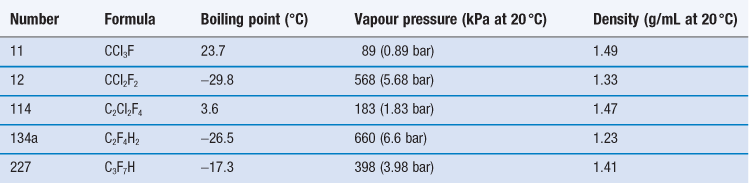
The propellants used in pMDI formulations are liquefied gases, traditionally chlorofluorocarbons (CFCs) which are now largely replaced by hydrofluoroalkanes (HFAs). At room temperature and pressure, these are gases but they are readily liquefied by decreasing temperature or increasing pressure. The head space of the aerosol canister is filled with propellant vapour, producing the saturation vapour pressure at that temperature. On spraying, medicament and propellant are expelled and the head volume increases. To reestablish the equilibrium, more propellant evaporates and so a constant pressure system with consistent spray characteristics is produced. The CFCs currently employed in pMDI formulations are trichlorofluoromethane (CFC-11), dichlorodifluoromethane (CFC-12) and dichlorotetrafluoroethane (CFC-114). Formulations generally comprise blends of CFC-11 and CFC-12 or CFC-11, CFC-12 and CFC-114 (Table 37.1), together with a surfactant such as a sorbitan ester, oleic acid or lecithin, which acts as a suspending agent and lubricates the valve.
CFCs and HFAs are numbered using a universal system. The first digit is the number of carbon atoms minus 1 (omitted if zero), the second is the number of hydrogen atoms plus 1, and the third is the number of fluorine atoms. Chlorine fills any remaining valencies, given the total number of atoms required to saturate the compound. If asymmetry is possible, this is designated by a letter. The symmetrical isomer is assigned the number described above; of the asymmetrical isomers, that designated the letter a is the most symmetrical, b the next most symmetrical, and so on. The CFCs are perfectly miscible with each other and suitable blends give a useful intermediate vapour pressure, usually about 450 kPa. The vapour pressure of the mixture of propellants is given by Raoult’s Law, i.e. the vapour pressure of a mixed system is equal to the sum of the mole fraction of each component multiplied by its vapour pressure:
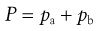 (37.5)
(37.5)
where P is the total vapour pressure of the system and pa and pb are the partial vapour pressures of the components, a and b:
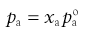 (37.6)
(37.6)
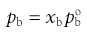 (37.7)
(37.7)
where xa and xb are the mole fractions and 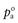 and
and 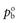 are the partial vapour pressures of components a and b, respectively.
are the partial vapour pressures of components a and b, respectively.
The reaction of CFCs with the ozone in the earth’s stratosphere, which absorbs ultraviolet radiation at 300 nm, and their contribution to global warming are major environmental concerns. CFCs pass to the stratosphere, where in the presence of UV they liberate chlorine, which reacts with ozone. The depletion of stratospheric ozone results in increased exposure to the UV-B part of the UV spectrum, resulting in a number of adverse effects, in particular an increased incidence of skin cancer. The Montreal Protocol of 1987 was a global ban on the production of the five worst ozone-depleting CFCs by the year 2000. This was amended in 1992, so that production of CFCs in developed countries was phased out by January 1996. In the European Union and USA, all ozone-depleting CFCs were banned by the end of 1995, except for certain limited uses. The inclusion of CFCs in pMDIs currently has an ‘essential use exemption’, which will remain until medically acceptable non-ozone depleting alternatives to the remaining CFC-based pMDIs are available. This exemption is reviewed regularly and very few CFC-based pMDIs are now marketed. In household and cosmetic aerosols, CFCs have been replaced by hydrocarbons, such as propane and butane. Alternatively, non-toxic compressed gases such as nitrogen dioxide, nitrogen and carbon dioxide may be used, for instance in food products. However, compressed gases do not maintain a constant pressure within the canister throughout its use, as the internal pressure is inversely proportionate to the head volume, and so product performance changes with usage. For reasons of toxicity and inflammability, hydrocarbons are not considered appropriate alternatives to CFCs for inhalation products and so non-ozone depleting alternatives to CFCs have been developed.
Propellants HFA-134a (trifluoromonofluoroethane) and HFA-227 (heptafluoropropane) are non-ozone depleting, non-flammable HFAs, also called hydrofluorocarbons (HFCs), which are now used as alternatives to CFC-12 (see Table 37.1). However, these gases contribute to global warming and further replacements may be sought in the future.
HFA-134a and HFA-227 have some physical properties, including density, which are similar to those of CFC-12 and, to a lesser extent, CFC-114. However, they have presented major formulation challenges; in particular, they are poor solvents for the surfactants commonly used in pMDI formulation and no alternative to CFC-11 is currently available. Ethanol is approved for use in formulations containing HFAs to allow dissolution of surfactants, and is included in marketed HFA pMDI products. However, ethanol has low volatility and its inclusion may consequently increase the droplet size of the emitted aerosols.
Metering valve
The metering valve of a pMDI permits the reproducible delivery of small volumes (25–100 µL) of product. Compared to the non-metering continuous-spray valve of conventional pressurized aerosols, the metering valve in pMDIs is used in the inverted position (Fig. 37.3). Depression of the valve stem allows the contents of the metering chamber to be discharged through the orifice in the valve stem and made available to the patient. After actuation, the metering chamber refills with liquid from the bulk and is ready to dispense the next dose. A corollary of this is that the pMDI needs to be primed, i.e. the metering chamber must be filled, prior to the first use by a patient. pMDI valves are complex in design and must protect the product from the environment, while also protecting against product loss during repeated use. The introduction of HFA propellants with different solvent properties has necessitated the development of new valve elastomers. The valve stem fits into the actuator, which is made of polyethylene or polypropylene. The dimensions of the orifice in the actuator play a crucial role, along with the propellant vapour pressure, in determining the shape and speed of the emitted aerosol plume.
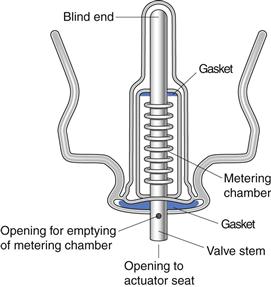
Fig. 37.3 The metering valve.
Formulating pressurized metered-dose inhalers
Pressurized aerosols may be formulated as either solutions or suspensions of drug in the liquefied propellant. Solution preparations are two-phase systems. However, the propellants are poor solvents for most drugs. Cosolvents such as ethanol or isopropanol may be used, although their low volatility retards propellant evaporation. In practice, pressurized inhaler formulations have traditionally been almost exclusively suspensions. These three-phase systems are harder to formulate and all the problems of conventional suspension formulation, such as caking, agglomeration, particle growth, etc., need to be considered. Careful consideration must be given to the particle size of the solid (usually micronized to between 2 and 5 µm), valve clogging, moisture content, the solubility of active pharmaceutical ingredient in propellant (a salt may be desirable), the relative densities of propellant and drug, and the use of surfactants as suspending agents, e.g. lecithin, oleic acid and sorbitan trioleate (usually included at concentrations between 0.1 and 2.0% w/w). These surfactants are very poorly soluble (<0.02% w/w) in HFAs and so ethanol is usually employed as a cosolvent, though alternative surfactants are being developed. Solution formulations of some drugs, such as beclometasone dipropionate are now available. Evaporation of HFA propellant following actuation of these formulations results in smaller particle sizes than with conventional suspension formulations of the same drug, with consequent changes in its pulmonary distribution and bioavailability. The dose may be adjusted accordingly or the volatility of the product modified by the addition of a less volatile component, such as glycerol.
Filling pressurized metered-dose inhaler canisters
Canisters are filled either by liquefying the propellant by reducing its temperature (cold filling) or by filling the vapour at elevated pressure (pressure filling).
In cold filling, active compound, excipients and propellant are chilled and filled at about −60 °C. Additional propellant is then added at the same temperature and the canister sealed with the valve. In pressure filling, a drug/propellant (CFC-11) concentrate is produced and filled at effectively room temperature and pressure (in fact, usually slightly chilled to below 20 °C). The valve is crimped on to the canister and additional propellant (e.g. CFC-12) is filled at elevated pressure through the valve, in a process known as gassing. Pressure filling is most frequently employed for inhalation aerosols. However, no HFAs have the properties (high boiling point: 23.7 °C) of CFC-11 so a single-stage pressure filling process has been developed for HFA-based formulations whereby a concentrated solution or suspension of drug in propellant, under pressure, is filled into canisters through the valve, followed by addition of further propellant.
Once filled, the canisters are leak tested by placing them in a water bath at elevated temperature, usually 50–60 °C. Following storage to allow equilibration of the formulation and valve components, the containers are weighed to check for further leakage, prior to spray testing and insertion into actuators.
Advantages and disadvantages of pressurized metered-dose inhalers
The major advantages of pMDIs are their portability, low cost and disposability. Many doses (up to 200) are stored in the small canister and dose delivery is reproducible. The inert conditions created by the propellant vapour, together with the hermetically sealed container, protect drugs from oxidative degradation and microbiological contamination. However, pMDIs have disadvantages. They are inefficient at drug delivery. On actuation, the first propellant droplets exit at a high velocity which may exceed 30 m/s. Consequently, much of the drug is lost through impaction of these droplets in the oropharyngeal areas. The mean emitted droplet size typically exceeds 40 µm, and propellants may not evaporate sufficiently rapidly for their size to decrease to that suitable for deep lung deposition. Vaporization of the droplets is hindered by the low volatility of CFC-11, which is present in concentrations of at least 25% in most CFC-based formulations. Evaporation, such that the aerodynamic diameter of the particles is close to that of the original micronized drug, may not occur until 5 seconds after actuation.
An additional problem with pMDIs, which is beyond the control of the formulator and manufacturer, is their incorrect use by patients. Reported problems include:
• failure to remove the protective cap covering the mouthpiece
• the inhaler being used in an inverted position
• failure to shake the canister
• failure to inhale slowly and deeply
Correct use by patients is vital for effective drug deposition and therapeutic action. Ideally, the pMDI should be actuated during the course of a slow, deep inhalation, followed by a period of breath-holding. Many patients find this difficult, especially children and the elderly. The misuse of pMDIs through poor inhalation/actuation coordination can be significantly reduced with appropriate instruction and counselling. However, it should be noted that even using the correct inhalation technique, only 10–20% of the stated emitted dose may be delivered to the site of action.
Spacers and breath-actuated metered-dose inhalers
Some of the disadvantages of pMDIs, namely inhalation/actuation coordination and the premature deposition of large droplets high in the airways, can be overcome by using extension devices or ‘spacers’ positioned between the pMDI and the patient (Fig. 37.4), and thus these are frequently employed with a pMDI, for administering aerosol medications to young children (see also Chapter 43). The dose from a pMDI is discharged directly into the reservoir prior to inhalation. This reduces the initial droplet velocity, large droplets may be removed by impaction, efficient propellant evaporation occurs and the need for actuation/inhalation coordination is removed. The disadvantage of traditional spacers, though effective, is that they may be cumbersome because of their large volume, e.g. Volumatic® (GlaxoSmithKline), though smaller, medium-volume spacers are now available, e.g. AeroChamber Plus® (GlaxoSmithKline). Alternatively, extension tubes may be built into the design of the pMDI itself as an extended mouthpiece, e.g. Syncroner® (Sanofi-Aventis) and Spacer Inhalers® (AstraZeneca). Breath-actuated pMDIs do not release drug until inspiration occurs. In the Autohaler® (3M), an inspiratory demand valve triggers a spring mechanism to release drug, whilst in the Easi-breathe® (Teva), a vacuum in the device is released on inspiration to trigger the actuation. Breath-actuated devices overcome the coordination problems of conventional pMDIs and are easy to use without adding bulk to the device.
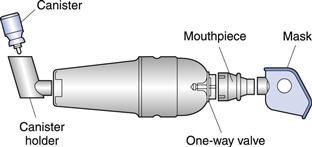
Dry powder inhalers
In dry powder inhaler (DPI) systems, drug is inhaled as a cloud of fine particles. The drug is either preloaded in an inhalation device or filled into hard gelatin capsules or foil blister discs which are loaded into a device prior to use.
DPIs have several advantages over pMDIs. DPI formulations are propellant-free and usually do not contain any excipient, other than a carrier (see below), which is usually lactose. They are breath-actuated, avoiding the problems of inhalation/actuation coordination encountered with pMDIs. DPIs can also deliver larger drug doses than pMDIs, which are limited by the volume of the metering valve and the maximum suspension concentration that can be employed without causing valve clogging. However, DPIs have several disadvantages. Liberation of powders from the device and the deaggregation of particles are limited by the patient’s ability to inhale, which in the case of respiratory disease may be impaired. An increase in turbulent air flow created by an increase in inhaled air velocity increases the deaggregation of the emerging particles but also increases the potential for inertial impaction in the upper airways and throat, and so a compromise has to be found. Further, DPIs are exposed to ambient atmospheric conditions, which may reduce formulation stability. For instance, elevated humidity may cause powders to aggregate. Finally, DPIs are generally less efficient at drug delivery than pMDIs, such that twice the dose is often required for delivery from a DPI compared to the equivalent pMDI.
Formulating dry powder inhalers
To produce particles of a suitable size (preferably less than 5 µm), drug powders for use in inhalation systems are usually micronized. Alternatives are spray drying, spray freeze drying and supercritical fluid technology. The high-energy powders produced by micronization have poor flow properties because of their static, cohesive and adhesive nature. The flowability of a powder is affected by physical properties, including particle size and shape, density, surface roughness, hardness, moisture content and bulk density.
To improve their flow properties, poorly flowing drug particles are generally mixed with larger ‘carrier’ particles (median size usually 30–150 µm) of an inert excipient, usually lactose (α-lactose monohydrate). Drug and carrier particles are mixed to produce an ordered mix in which the small drug particles attach to the surface of the larger carrier particles. This not only improves liberation of the drug from the inhalation device by improving powder flow, but also improves the uniformity of capsule or device filling. Once liberated from the device, the turbulent air flow generated within the inhalation device should be sufficient for the deaggregation of the drug/carrier aggregates. The larger carrier particles impact in the throat, whereas smaller drug particles are carried in the inhaled air deeper into the respiratory tract.
The success of DPI formulations depends on the adhesion of drug and carrier during mixing and filling of devices or hard gelatin capsules, followed by the ability of the drug to detach from the carrier during inhalation, such that free drug is available to penetrate to the peripheral airways. Adhesion and detachment will depend on the morphology of the particle surfaces and surface energies, which may be influenced by the chemical nature of the materials involved and the nature of powder processing. Additionally, a ternary mix may be employed. Thus, fine particle size lactose can be added to conventional carrier lactose, to occupy the high energy sites on the larger carrier particles. Only low energy sites remain for drug-carrier interaction, enhancing the detachment of drug particles during inhalation of the formulation. Likewise, materials such as leucine and magnesium stearate may be included in formulations to modify the adhesion properties between drug and carrier particles. The interactions between micronized drugs, fine carrier particles and large carrier particles in DPI formulations are discussed in Chapter 8.
The performance of DPI systems is thus strongly dependent on formulation factors. Other factors affecting aerosol delivery and deposition include the design of the delivery device and a patient’s inhalation technique.
Unit-dose devices with drug in hard gelatin capsules
The first DPI device developed was the Spinhaler® (Rhône-Poulenc Rorer) for the delivery of sodium cromoglicate. Each dose, contained in a hard gelatin capsule, was placed individually into the device, in a loose-fitting rotor. The capsule was pierced by two metal needles on either side of the capsule, and inhaled air flow though the device caused a turbovibratory air pattern as the rotor rotated rapidly, resulting in the powder being dispersed to the capsule walls and out through the perforations into the inspired air. Although the tendency nowadays is to develop multiple-dosing devices, other hard gelatin capsule-based devices, working on similar principles are still available, e.g. the HandiHaler® (Boehringer Ingelheim/Pfizer) and the Aerolizer/Cyclohaler® (Novartis/Teva, Fig. 37.5).

Multidose devices with drug in foil blisters
The main disadvantage of hard gelatin capsule-based devices, namely the individual loading of each dose, has been overcome with the development of the Diskhaler® (GlaxoSmithKline). In this system, drug is mixed with a coarse lactose carrier and filled into an aluminium foil blister disc which is loaded, by the patient, into the device on a support wheel (Fig. 37.6). Each disc contains four or eight doses of drug and the blisters are pierced with a needle as a result of mechanical leverage of the lid. Air flow through the blister causes the powder to disperse as the patient inhales through the mouthpiece. The foil blisters are numbered, so that the patient knows the number of doses remaining.

Fig. 37.6 The Diskhaler® dry powder inhaler.
Multidose devices with drug preloaded in inhaler
The evolution of the Diskhaler led to the Accuhaler® or Diskus® Inhaler (GlaxoSmithKline), in which drug/carrier mix is preloaded into the device in foil-covered blister pockets containing 60 doses (Fig. 37.7). The foil lid is peeled off the drug-containing pockets as each dose is advanced, with the blisters and lids being wound up separately within the device, which is discarded at the end of operation. As each dose is packaged separately and only momentarily exposed to ambient conditions prior to inhalation, the Diskhaler and Accuhaler are relatively insensitive to humidity compared to hard gelatin capsule-based systems.
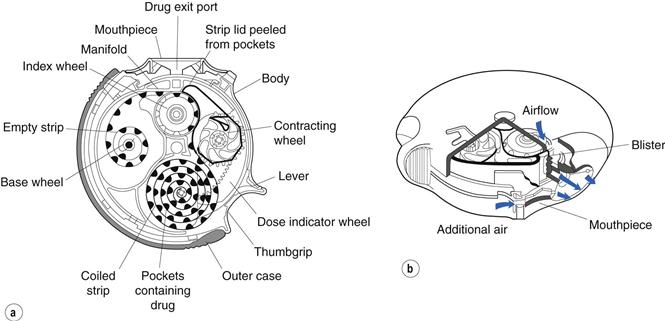
Fig. 37.7 The Accuhaler/Diskus® dry powder inhaler, showing (a) a schematic diagram and (b) a cross-sectional representation of the device. (Courtesy of Prime et al, 1996, with permission.)
An alternative approach is a reservoir type of device, in which a dose is accurately measured and delivered from a drug reservoir. In the Clickhaler® DPI (Innovata Biomed), a drug–lactose blend is stored in a reservoir. Metering cups are filled by gravity from this reservoir and delivered to an inhalation passage, from which the dose is inhaled. The device is shaken before use. The device is capable of holding up to 200 doses and incorporates a dose counter which indicates the number of metered doses and informs patients when the device, which is discarded after use, is nearly empty. After the final dose has been dispensed, the push button locks to prevent further use.
The Turbohaler® (AstraZeneca) has overcome the need for both a carrier and the loading of individual doses (Fig. 37.8). The device contains a large number of doses (up to 200) of undiluted, loosely aggregated micronized drug, which is stored in a reservoir from which it flows on to a rotating disc in the dosing unit. The fine holes in the disc are filled and excess drug is removed by scrapers. As the rotating disc is turned, by moving a turning grip back and forth, one metered dose is presented to the inhalation channel and this is inhaled by the patient, with the turbulent air flow created within the device breaking up any drug aggregates. A dose indicator is incorporated. The Turbohaler requires a higher inspiratory effort than the Diskhaler, owing to its higher internal resistance, and is more sensitive to humidity if not closed quickly after each use.
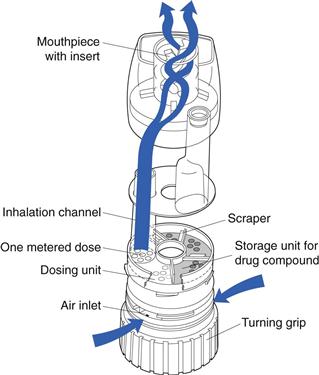
Breath-assisted devices
Several devices have been developed which reduce or eliminate the reliance on the patient’s inspiratory effort to disperse the drug. This is advantageous as inspiratory effort may be affected by the patient’s age and/or clinical condition. For instance, Nektar Therapeutics produced a device for the delivery of insulin as a fine powder, in which compressed air was used to disperse drug from a unit-dose package into a large holding chamber, from which it was inhaled by the patient. The device was marketed briefly by Pfizer for the delivery of insulin by inhalation, but was withdrawn for many reasons including; cost, the cumbersome design of the delivery device and the need for injections of insulin to supplement inhaled drug. Another novel device, the Spiros® DPI (Dura) (Fig. 37.9), is a breath-actuated, effort-assisted device which uses a battery-powered impeller to deaggregate and aerosolize the drug powder, which is held in a rotating cassette. The device functions relatively independently of the patient’s inspiratory flow rate and can be used by those patients who can only achieve low inspiratory flow rates.
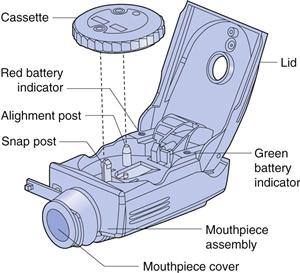
Nebulizers
Nebulizers deliver relatively large volumes of drug solutions and suspensions and are frequently used for drugs that cannot be conveniently formulated into pMDIs or DPIs, or where the therapeutic dose is too large for delivery with these alternative systems. Nebulizers also have the advantage over pMDI and DPI systems in that drug may be inhaled during normal tidal breathing through a mouthpiece or facemask, and thus they are useful for patients such as children, the elderly and patients with arthritis, who experience difficulties with pMDIs.
There are three categories of commercially available nebulizer: jet, ultrasonic and mesh.
Jet nebulizers
Jet nebulizers (also called air-jet or air-blast nebulizers) use compressed gas (air or oxygen) from a compressed gas cylinder, hospital air-line or electrical compressor to convert a liquid (usually an aqueous solution) into a spray. The jet of high-velocity gas is passed either tangentially or coaxially through a narrow Venturi nozzle, typically 0.3–0.7 mm in diameter. An area of negative pressure, where the air jet emerges, causes liquid to be drawn up a feed tube from a fluid reservoir by the Bernoulli effect (Fig. 37.10). Liquid emerges as fine filaments, which collapse into droplets as a result of surface tension. A proportion of the resultant (primary) aerosol leaves the nebulizer directly; the remaining large, non-respirable droplets impact on baffles or the walls of the nebulizer chamber and are recycled into the reservoir fluid.
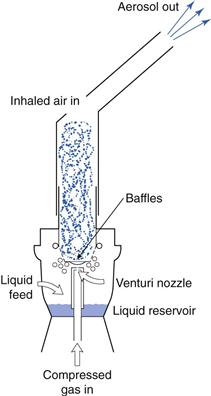
Nebulizers are operated continuously and because the inspiratory phase of breathing constitutes approximately one-third of the breathing cycle, a large proportion of the emitted aerosol is not inhaled but is released into the environment. Open-vent nebulizers, incorporating inhalation and exhalation valves, e.g. the Pari LC® nebulizer (Pari), have been developed in which the patient’s own breath boosts nebulizer performance, with aerosol production matching the patient’s tidal volume and greatly enhancing drug delivery. On exhalation, the aerosol being produced is generated only from the compressor gas source, thereby minimizing drug wastage.
The rate of gas flow driving atomization is the major determinant of the aerosol droplet size and rate of drug delivery for jet nebulizers; for instance, there may be up to a 50% reduction in the mass median aerodynamic diameter when the flow rate is increased from 4 to 8 L/min, with a linear increase in the proportion of droplets less than 5 µm.
Ultrasonic nebulizers
In ultrasonic nebulizers the energy necessary to atomize liquids comes from a piezoelectric crystal vibrating at high frequency. At sufficiently high ultrasonic intensities, a fountain of liquid is formed in the nebulizer chamber. Large droplets are emitted from the apex and a ‘fog’ of small droplets is emitted from the lower part (Fig. 37.11). Some models have a fan to blow the respirable droplets out of the device, whereas in others the aerosol only becomes available to the patient during inhalation.
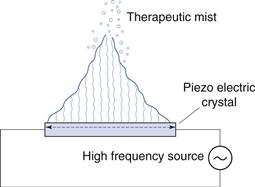
Mesh nebulizers
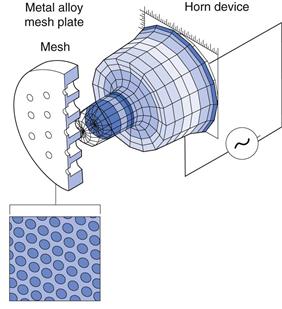
Recently, mesh nebulizers have come commercially available, in which aerosols are generated by passing liquids through a vibrating mesh or plate with multiple apertures. The energy of vibration comes from a vibrating piezoelectric crystal attached to a horn transducer which transmits vibrations to a perforated plate with up to 6000 tapered holes (Fig. 37.12) or from an aerosol generator comprising a domed aperture plate, with up to 10 000 tapered holes and a vibrational element which contracts and expands to generate the aerosol. These devices generate aerosols with a high fine particle fraction, deliver fluids very rapidly and have very small residual volumes (see below) compared to jet and ultrasonic nebulizers. A recent development, has been the ‘Adaptive Aerosol Delivery’ (AAD) system employed in the I-neb AAD® mesh nebulizer (Philips Respironics), which analyse the patient’s breathing pattern and emits aerosol only during inhalation, thus eliminating wastage during exhalation.
Formulating nebulizer fluids
Nebulizer fluids are formulated in water, occasionally with the addition of a cosolvent such as ethanol, and with the addition of surfactants for suspension formulations. Because hypoosmotic and hyperosmotic solutions may cause bronchoconstriction, as may high hydrogen ion concentrations, isoosmotic solutions of pH greater than 5 are usually employed. Stabilizers such as antioxidants and preservatives may also be included, although these may also cause bronchospasm, and for this reason sulfites in particular are generally avoided as antioxidants in such formulations. Although chemically preserved multidose preparations are commercially available, nebulizer formulations are generally presented as sterile, isotonic unit doses (usually 1–2.5 mL) without a preservative.
Whilst most nebulizer formulations are solutions, suspensions of micronized drug are also available for delivery from nebulizers. In general, suspensions are poorly delivered from ultrasonic nebulizers, whilst mesh nebulizers are considered suitable for delivering suspensions. With jet nebulizers the efficiency of drug delivery increases as the size of suspended drug is decreased, with little or no delivery of particles when they exceed the droplet size of the nebulized aerosol.
As the formulation of fluids for delivery by nebulizers is relatively simple, these devices are frequently the first to be employed when investigating the delivery of new entities to the human lung. Recently, they have been used for the delivery of biopharmaceuticals and drug delivery systems, such as liposomes. In general, ultrasonic nebulizers have not been successful for delivering either biopharmaceuticals or liposomes, because of denaturation resulting from the elevated temperatures produced. Consequently, ultrasonic nebulizers are expressly excluded for the delivery of recombinant human deoxyribonuclease in the management of cystic fibrosis. Mesh and jet nebulizers have been successfully used to deliver some peptides, nucleic acids and liposome formulations, although the shearing forces that occur in jet nebulizers may produce time-dependent damage to some materials.
Physicochemical properties of nebulizer fluids
The viscosity and surface tension of a liquid being nebulized may affect the output of nebulizers, as energy is required to overcome viscous forces and to create a new surface. However, the size selectivity of the nebulizer design and dimensions, with more than 99% of the primary aerosol mass being recycled into the reservoir liquid, means that changes in the size distribution of the primary aerosol resulting from changes in the properties of the solution being atomized may not be reflected in the size distribution of the emitted aerosol. In general, the size of aerosol droplets is inversely proportional to viscosity for jet and mesh nebulizers and directly proportional to viscosity for ultrasonic nebulizers, with more viscous solutions requiring longer to nebulize to dryness and leaving larger residual volumes in the nebulizer following atomization. Surface tension effects are more complex, but usually a decrease in surface tension is associated with a reduction in mean aerosol size.
Temperature effects during nebulization
The aerosol output from a jet nebulizer comprises drug solution and solvent vapour, which saturates the outgoing air. This causes solute concentration to increase with time and results in a rapid decrease in the temperature of the liquid being nebulized by approximately 10–15 °C. This temperature decrease may be important clinically, as some asthma sufferers experience bronchoconstriction on inhalation of cold solutions. Further, the cooling effect within the reservoir fluid will reduce drug solubility and result in increased liquid surface tension and viscosity. Precipitation is uncommon with bronchodilators, which have high aqueous solubility, but problems may arise with less soluble drugs. In such instances the use of an ultrasonic nebulizer may be appropriate, as the operation of such devices may increase solution temperature by up to 10–15 °C. As indicated above, this temperature increase may have detrimental effects on heat-sensitive materials intended for nebulization. Mesh nebulizers have a negligible effect on fluid temperature.
Duration of nebulization and residual volume
Clinically, liquids may be nebulized for a specified period of time or, more commonly, they may be nebulized to ‘dryness’, which may be interpreted as sputtering time, which is the time when air is drawn up the feed tube and nebulization becomes erratic, although agitation of the nebulizer permits treatment to be continued; clinical time, which is the time at which therapy is ceased following sputtering; or total time, which is the time at which the production of aerosol ceases.
Regardless of the duration of nebulization, not all the fluid in the nebulizer can be atomized. Some liquid, usually about 1 mL for jet and ultrasonic nebulizers, remains as the ‘dead’ or ‘residual’ volume, associated with the baffles, internal structures and walls of the nebulizer. The proportion of drug retained as residual volume is more marked for smaller fill volumes; hence for a 2 mL fill volume, approximately 50% of fluid will remain associated with the nebulizer and be unavailable for delivery to the patient. This reduces to approximately 25% with a 4 mL fill volume, although there is a commensurate increase in the time necessary to nebulize to dryness. For this reason small-volume nebulizer fluids may be diluted to a larger volume by addition of a suitable diluent, usually sterile 0.9% sodium chloride solution. Vibrating-mesh nebulizers have a much smaller residual volume than jet or ultrasonic devices, which may mean that the nominal dose, or dose volume used with a mesh nebulizer should be reduced compared to that employed with a conventional nebulizer.
Variability between nebulizers
Many different models of nebulizer and compressor are commercially available, and the size of aerosols produced and the dose delivered can vary enormously. Variability may not only exist between different nebulizers but also between individual nebulizers of the same type, and repeated use of a single nebulizer may cause variability due to baffle wear and non-uniformity of assembly. Nebulizers, unlike the DPI and pMDI devices, are not manufactured by the producers of nebulizer solutions and suspension. The choice of nebulizer employed for their delivery is thus usually beyond the influence of the pharmaceutical manufacturer.
Novel delivery devices
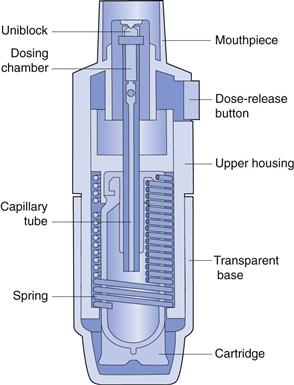
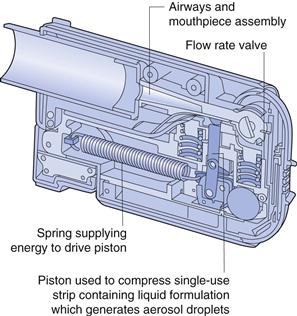
A number of new inhalation devices are currently being developed, which do not fit neatly into the traditional categories of pMDI, DPI and nebulizers, since they operate on alternative principles. Two metered-dose liquid inhalers are the Respimat® (Boehringer Ingelheim, Fig. 37.13) and the AERx Pulmonary Delivery System® (Aradigm, Fig. 37.14). The Respimat Soft Mist® Inhaler is a handheld device with a multidose reservoir releasing a cloud of 1.5 seconds duration. This is much slower than for conventional pMDIs, allowing more time for coordination between actuation and inhalation. The device is mechanically actuated without employing a propellant. The aerosol generated by the device has lower velocity and smaller particle size than that generated with a conventional pMDI, resulting in superior peripheral lung deposition. The AERx Pulmonary Delivery System® has drug contained in unit-dose blister packs. Drug delivery is computer controlled and involves extrusion of liquid through a single-use nozzle containing numerous spherical holes, with exits approximately 1 µm in diameter. This produces very fine aerosols, suitable for delivery of biopharmaceuticals, such as insulin, to the peripheral airways. The device electronically monitors the patient’s inspiratory flow rate, and releases drug at the inspiratory flow rate determined to be optimal for drug delivery.

Methods of aerosol size analysis
The regional distribution of aerosols in the airways can be measured directly using γ-scintigraphy, by radiolabelling droplets or particles, usually with the short half-life γ-emitter technetium-99m (99mTc). However, more commonly, in vitro measurements of aerosol size are used to predict clinical performance. The principal methods that have been employed for size characterization of aerosols are microscopy, laser diffraction and cascade impaction.
Optical methods of measuring the physical size of deposited aerosols using microscopy are laborious and do not give an indication of their likely deposition within the humid airways while being carried in an air stream. With methods of analysis based on laser diffraction, aerosolized droplets or particles are sized as they traverse a laser beam to give a volume median diameter. Again, the aerodynamic properties of an aerosol are not being measured. In addition, spraying droplets into a beam exposes them to ambient conditions of temperature and humidity, which may result in solvent evaporation.
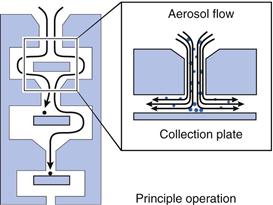
Cascade impactors and impingers
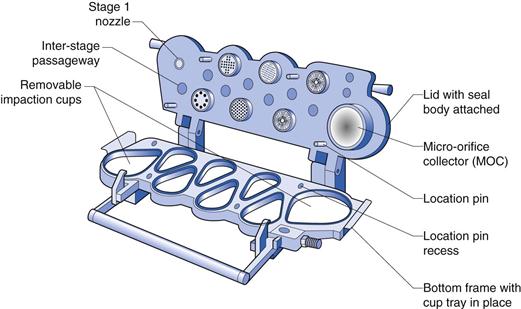
Cascade impactors comprise a series of progressively finer jets and collection plates, allowing fractionation of aerosols according to their aerodynamic size distribution as the aerosol is drawn through the device at a known flow rate. Large, dense particles will deposit higher in the impactor, whereas smaller, less dense particles will follow the air flow and only deposit when they have been given sufficient momentum as they are accelerated through the finer jets lower in the impactor (Fig. 37.15). The first stage of the impactor is usually preceded by a 90° bend of metal or glass to mimic the human throat. Traditional cascade impactors are constructed from metal. The two most commonly used are 1) the Andersen Cascade Impactor (ACI) which comprises eight impaction stages, with metal collection plates followed by a terminal filter and 2) the Next Generation Impactor (NGI) which consists of seven impaction stages followed by a micro-orifice collector, which can be used in place of a terminal filter in most cases. The NGI uses collection cups rather than plates and can be used with flow rates from 15 to 100 L/min (Fig. 37.16). The cut-off diameters for each stage at a particular air-flow rate can be determined using monodisperse aerosols or calculated using calibration curves. When determining the size of an aerosol, cumulative percentage undersize plots of deposited aerosol on each stage are plotted against the cut-off diameter for that stage to allow calculation of the MMAD. Cascade impactors are used for aerodynamic assessment of fine particles in pMDIs and DPIs (USP and PhEur).
Multistage liquid impingers (MSLIs), work on the same principle of cascade impaction. They are constructed from glass or glass and metal and have three, four or five stages, with wet sintered glass collection plates followed by a terminal filter. The five-stage liquid impinger (multistage liquid impinger, MSLI) (Fig. 37.17), with an appropriate induction port and mouthpiece adapter, is used to determine the aerodynamic size of DPIs and pMDIs. The MSLI may be operated at a flow rate between 30 and 100 L/min. At 60 L/min (i.e. 1 L/s), the effective cut-off diameters of stages 1, 2, 3 and 4 are 13.0, 6.8, 3.1 and 1.7 µm, respectively. The fifth stage comprises an integral filter which captures particles smaller than 1.7 µm.
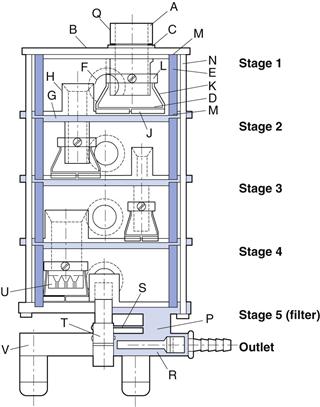
When testing DPIs to USP requirements, an air-flow rate (Q) calculated to produce a pressure drop of 4.0 kPa over the inhaler is employed. If this exceeds 100 L/min, then 100 L/min is used. The cut-off diameters of each stage at flow rate (Q) can be calculated from:
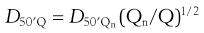 (37.8)
(37.8)
where D50′Q is the cut-off diameter at the flow rate Q and D50′Qn refers to the nominal cut-off values determined when Qn is 60 L/min (values given above).
The use of cascade impaction methods to determine the size of aerosols has a number of disadvantages. The high flow rates employed (typically 28.3–100 L/min) result in rapid solvent evaporation and droplets may be re-entrained in the air stream, whilst particles may ‘bounce off’ metal collection plates, although this latter effect may be reduced by coating the collection surface, for instance, with a silicone fluid or glycerol. These effects can result in a significant decrease in the measured aerosol size. Also, these measuring devices are operated at a constant air-flow rate. However, the dispersion of dry powder formulations and the deposition profile of inhaled aerosols will vary considerably with flow rate. To overcome the limitations of measurement at a single flow rate, an ‘electronic lung’ can be used, whereby a computer-controlled piston draws air through the inhaler and into an impaction sizer, following a predetermined inhalation profile.
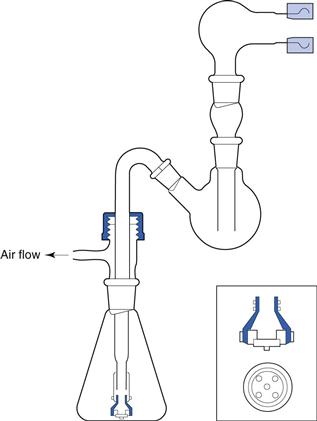
Cascade impactor methods are invasive, laborious and time-consuming but necessary to derive information about median aerodynamic size and the polydispersity of the aerosol. To ensure that inhalation products are likely to be clinically effective, in addition to size properties, the emitted dose is calculated together with the ‘fine particle fraction’ (that fraction of the emitted dose less than a stated size, usually 5 µm or sometimes 6.4 µm), which are combined to give a ‘therapeutically useful’ or ‘respirable’ dose or mass (fine particle dose). For routine analysis, a simplified glass two-stage (twin) impinger may be employed (Fig. 37.18). Aerosol collected in the throat and the upper stage (stage 1) is considered ‘non-respirable’, whereas that collected in the lower stage (stage 2) is considered ‘respirable’. For this glass device, the cut-off diameter for stage 2 is 6.4 µm, i.e. aerosols collected in this stage have an aerodynamic diameter less than 6.4 µm and for this measurement technique constitute the fine particle fraction. This two-stage impinger may be used for fine particle assessment of aerosols generated from pMDIs, DPIs and nebulizers (PhEur), but gives much less information about aerosol particle size distribution than a multi-stage impactor or impinger.
References
1. Atkins PJ, Barker NP, Mathisen D. The design and development of inhalation drug delivery systems. In: Hickey ASJ, ed. Pharmaceutical Inhalation Aerosol Technology. New York: Marcel Dekker; 1992.
2. Nelson H, Kemp JP, Bieler S, Vaughan LM, Hill MR. Comparative efficacy and safety of albutarol sulfate Spiros inhaler and albuterol metered-dose inhaler. Chest. 1999;115:329–335.
3. Prime D, Slater AL, Haywood PA, Smith IJ. Assessing dose delivery from the Flixotide Diskus Inhaler – a multidose powder inhaler. Pharmaceutical Technology Europe. 1996;8(3):23–34.
Bibliography
1. Campen LV, Venthoye G. Inhalation: dry powder. In: Swarbrick J, ed. Encyclopedia of Pharmaceutical Technology. 3rd edn New York: Informa Healthcare; 2006.
2. Carvalho TC, Peters JI, Williams III RO. Influence of particle size on regional lung deposition – what evidence is there? International Journal of Pharmaceutics. 2011;406:1–10.
3. Hickey ASJ, ed. Pharmaceutical Inhalation Aerosol Technology. 2nd edn New York: Marcel Dekker; 2003.
4. Munro SJM, Cripps AL. Metered dose inhalers. In: Swarbrick J, ed. Encyclopedia of Pharmaceutical Technology. 3rd edn New York: Informa Healthcare; 2006.
5. Placke ME, Ding J, Zimlich WC. Inhalation, liquids. In: Swarbrick J, ed. Encyclopedia of Pharmaceutical Technology. 3rd edn New York: Informa Healthcare; 2006.
6. Taylor G, Kellaway I. Pulmonary drug delivery. In: Hillery AM, Lloyd AW, Swarbrick J, eds. Drug Delivery and Targeting for Pharmaceutical Scientists. London: Taylor and Francis; 2001.
7. Taylor KMG, McCallion ONM. Ultrasonic nebulizers. In: Swarbrick J, ed. Encyclopedia of Pharmaceutical Technology. 3rd edn New York: Informa Healthcare; 2006.