[level-membership-for-basic-science-category]
Chapter 20 Psychotropic drugs
Diagnostic issues
Also falling within the scope of modern psychiatric diagnostic systems are organic mental disorders (e.g. dementia in Alzheimer’s disease), disorders due to substance misuse (e.g. alcohol and opiate dependence; see Ch. 11), personality disorders, disorders of childhood and adolescence (e.g. attention deficit/hyperactivity disorder, Tourette’s syndrome), and mental retardation (learning disabilities).
Antidepressant drugs
Antidepressants can be broadly divided into four main classes (Table 20.1), tricyclics (TCA, named after their three-ringed structure), selective serotonin reuptake inhibitors (SSRIs), monoamine oxidase inhibitors (MAOIs) and novel compounds, some of which are related to TCAs or SSRIs. Clinicians who wish to have a working knowledge of antidepressants would be advised to be familiar with the use of at least one drug from each of the four main categories tabulated. A more thorough knowledge-base would demand awareness of the distinct characteristics of the subgroups of novel compounds (e.g. serotonin and noradrenaline/norepinephrine reuptake inhibitors (SNRIs), mirtazapine, reboxetine and agomelatine) and differences between individual SSRIs and TCAs. As antidepressants are largely similar in their therapeutic efficacy, awareness of profiles of unwanted effects is of particular importance.
| Tricyclics | Selective serotonin reuptake inhibitors | Monoamine oxidase inhibitors |
|---|---|---|
| Dosulepin Amitriptyline Lofepramine Clomipramine Imipramine Trimipramine Doxepin Nortriptyline Protriptyline Desipramine |
Fluoxetine Paroxetine Sertraline Citaloprama Escitaloprama Fluvoxamine |
Phenelzine Isocarboxazid Tranylcypromine Moclobemide (RIMA) |
| Novel compounds | ||
|---|---|---|
| Mainly noradrenergic | Mainly serotonergic | |
| Reboxetine (NaRI) | Trazodoneb Nefazodoneb,c |
|
| Mixed |
|---|
| Venlafaxine (SNRI) Mirtazapine (nassa)b Duloxetine (SNRI) Milnacipran (SNRI)d Agomelatine |
RIMA, reversible inhibitor of monoamine oxidase; NaRI, noradrenaline/norepinephrine reuptake inhibitor; SNRI, serotonin and noradrenaline/norepinephrine reuptake inhibitor; NaSSA, noradrenaline/norepinephrine and specific serotonergic antidepressant.
a Escitalopram is the active S-enantiomer of citalopram.
b Trazodone, nefazodone and mirtazapine have been classed as ‘receptor blocking’ antidepressants based on their antagonism of postsynaptic serotonin receptors (trazodone, nefazodone, mirtazapine) and presynaptic α2-receptors (trazodone, mirtazapine).
c Nefazodone has additional weak SSRI activity but has now been withdrawn due to risk of hepatitis.
An alternative categorisation of antidepressants is based solely on mechanism of action (Fig. 20.1). The majority of antidepressants, including SSRIs, TCAs and related compounds, are reuptake inhibitors. Certain novel agents, including trazodone and mirtazapine, are receptor blockers, whereas MAOIs are enzyme inhibitors.
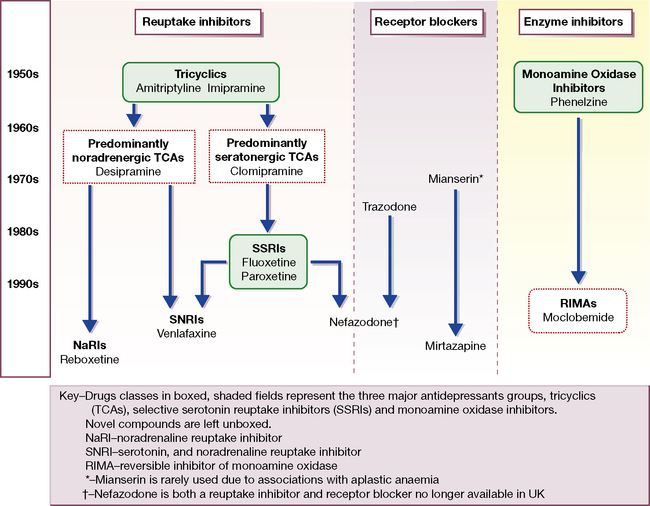
Fig. 20.1 Flow chart of the evolution of antidepressant drugs, and classification by mechanism of action.
The first TCAs (imipramine and amitriptyline) and MAOIs appeared between 1957 and 1961 (see Fig. 20.1). The MAOIs were developed from antituberculous agents that unexpectedly improved mood. Imipramine was a chlorpromazine derivative that showed antidepressant rather than antipsychotic properties. Over the next 25 years the TCA class enlarged to more than 10 agents with heterogeneous pharmacological profiles, and further modifications of the original three-ringed structure gave rise to the related (but pharmacologically distinct) antidepressant trazodone.
Mechanism of action
The monoamine hypothesis proposes that, in depression, there is deficiency of the neurotransmitters noradrenaline/norepinephrine and serotonin in the brain which can be restored by antidepressants. Drugs that alleviate depression also enhance monoamine availability and release (Fig. 20.2), increasing activity at postsynaptic receptors. It is relevant that (older) antihypertensive agents, e.g. reserpine, which reduced the availability of noradrenaline/norepinephrine, caused depression.
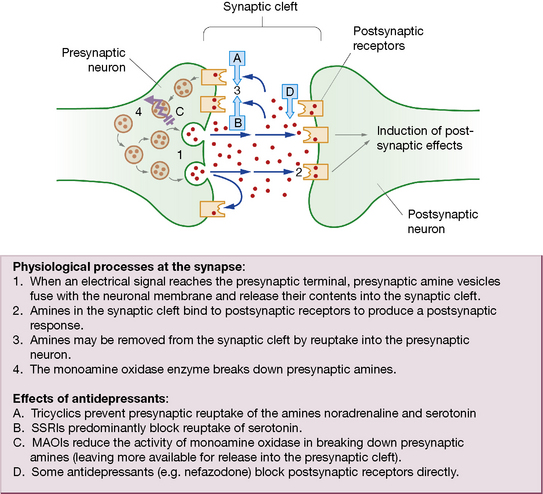
MAOIs increase the availability of noradrenaline/norepinephrine and serotonin by preventing their destruction by the monoamine oxidase type A enzyme in the presynaptic terminal (see Ch. 21, Table 21.3). The older MAOIs, phenelzine, tranylcypromine and isocarboxazid, bind irreversibly to monamine oxidase by forming strong (covalent) bonds. The enzyme is thus rendered permanently ineffective such that amine metabolising activity can be restored only by production of fresh enzyme, which takes weeks. These MAOIs are thus called ‘hit and run’ drugs as their effects greatly outlast their detectable presence in the blood.
Drugs with similar modes of action to antidepressants find other uses in medicine. Bupropion (amfebutamone) inhibits reuptake of both dopamine and noradrenaline/norepinephrine. It was originally developed and used as an antidepressant but is now more frequently used to assist smoking cessation (see p. 321). It is also prescribed in attention deficit hyperactivity disorder (see p. 345). Sibutramine, licensed as an anorectic agent, is a serotonin and noradrenaline/norepinephrine reuptake inhibitor (SNRI).
Pharmacokinetics
The antidepressants listed in Table 20.1 are generally well absorbed after oral administration. Steady-state plasma concentrations of TCAs show great individual variation but correlate with therapeutic effect. Where there is a failure of response, measurement of plasma concentration can be useful as the failure may be attributable to low plasma levels due to ultra-rapid metabolism (though it is often not available). Antidepressants in general are metabolised principally by hepatic cytochrome P450 enzymes. Of the many isoenzymes identified, the most important in antidepressant metabolism are CYP P450 2D6 (Table 20.2A) and CYP 3A4 (Table 20.2B). Other important P450 enzymes are CYP 1A2 (inhibited by the SSRI fluvoxamine, induced by cigarette smoking; substrates include caffeine and the atypical antipsychotics clozapine and olanzapine) and the CYP 2 C group (inhibition by fluvoxamine and fluoxetine, involved in breakdown of escitalopram and moclobemide). Sometimes several CYP enzymes are capable of mediating the same metabolic step. For example, at least six isoenzymes, including CYP 2D6, 3A4 and 2 C9, can mediate the desmethylation of the SSRI sertraline to its major metabolite.
Table 20.2A Psychotropic (and selected other) drugs known to be CYP 2D6 substrates, inhibitors and inducers
| CYP 2D6 inhibitors |
|---|
| Antidepressants |
| Paroxetine Fluoxetine |
| CYP 2D6 substrates | ||
|---|---|---|
| Antidepressants | Antipsychotics | Miscellaneous |
| Paroxetine Fluoxetine Citalopram Sertraline Venlafaxinea Duloxetine Amitriptyline Clomipramine Desipramine Imipramine Nortriptyline Reboxetine |
Chlorpromazine Haloperidol Zuclopenthixol Perphenazine Risperidone |
Dexfenfluramine Donepezil Opioids Codeine Hydrocodone Dihydrocodeine Tramadol Ethyl morphine MDMA (ecstasy) β-Blockers Propranolol Metoprolol Timolol Bufaralol Carvidelol |
A substrate is a substance that is acted upon and changed by an enzyme. Where two substrates of the same enzyme are prescribed together, they will compete and, if present in sufficient quantities, the metabolism of one or other, or both, drugs may also be inhibited, resulting in increased plasma concentration and possibly in enhanced therapeutic or adverse effects. An enzyme inducer accelerates the metabolism of co-prescribed drugs that are substrates of the same enzyme, reducing their effects. An enzyme inhibitor retards metabolism of co-prescribed drugs, increasing their effects.
a CYP 2D6 is involved only in the breakdown of venlafaxine to its active metabolite and therefore implications of 2D6 interactions for efficacy are of limited significance.
Table 20.2B Psychotropic (and selected other) drugs known to be CYP 3A4 substrates, inhibitors and inducers
| CYP 3A4 inhibitors | |
|---|---|
| Antidepressants | Other drugs |
| Fluoxetine Nefazodone |
Cimetidine Erythromycin Ketoconazole (grapefruit juice) |
| CYP 3A4 substrates | ||
|---|---|---|
| Antidepressants | Anxiolytics, hypnotics and antipsychotics | Miscellaneous |
| Fluoxetine Sertraline Amitriptyline Imipramine Nortriptyline Trazodonea |
Alpraxolam Aripiprazole Buspirone Diazepam Midazolam Triazolam Zoplicone Haloperidol Zuclopethixol Quetiapine Sertindole |
Buprenorphine Carbamazepine Cortisol Dexamethasone Methadone Testosterone Calcium channel blockers Diltiazem Nifedipine Amlodipine Other drugs Amiodarone Omeprazole Oral contraceptives Simvastatin |
| CYP 3A4 inducers | |
|---|---|
| Antidepressants | Miscellaneous |
| St John’s wort | Carbamazepine Phenobarbital Phenytoin |
A substrate is a substance that is acted upon and changed by an enzyme. Where two substrates of the same enzyme are prescribed together, they will compete and, if present in sufficient quantities, the metabolism of one or other, or both, drugs may also be inhibited, resulting in increased plasma concentration and possibly in enhanced therapeutic or adverse effects. An enzyme inducer accelerates the metabolism of co-prescribed drugs that are substrates of the same enzyme, reducing their effects. An enzyme inhibitor retards metabolism of co-prescribed drugs, increasing their effects.
a mCPP (meta-chlorophenylpiperazine), the active metabolite of trazodone, is a CYP 2D6 substrate; observe for unwanted effects when trazodone is co-administered with the 2D6 inhibitors fluoxetine or paroxetine.
Therapeutic efficacy
• Venlafaxine, in high dose (> 150 mg/day) and Escitalopram may have greater efficacy than other antidepressants.
• Amitriptyline appears to be slightly more effective than other TCAs and also SSRIs for severe depression, but this advantage is compromised by its poor tolerability and lack of safety in overdose relative to more modern agents.
• The older MAOIs (e.g. phenelzine) may be more effective than other classes in ‘atypical’ depression, a form of depressive illness where mood reactivity is preserved, lack of energy may be extreme and biological features are the opposite of the normal syndrome, i.e. excess sleep and appetite with weight gain.
Changing and stopping antidepressants
When changing between antidepressant doses, a conservative approach would be to reduce the first antidepressant progressively over 2 or more weeks before starting the new drug. The gradual reduction is particularly important with paroxetine and venlafaxine which are known to cause ‘discontinuation syndromes’ if stopped abruptly, and less important with fluoxetine due to its long half-life active metabolite which offers ‘built-in’ protection against withdrawal problems. A more proactive approach would involve ‘cross-tapering’ the second antidepressant – i.e. starting it while the first antidepressant is being reduced and gradually titrating the dose up. However, an important exception concerns changes to or from MAOIs, which must be handled with great caution due to the dangers of interactions between antidepressants (see below). Therefore MAOIs cannot safely be introduced within 2 weeks of stopping most antidepressants (3 weeks for imipramine and clomipramine; combination of the latter with tranylcypromine is particularly dangerous), and not until 5 weeks after stopping fluoxetine, due to its long half-life active metabolite. Similarly, other antidepressants should not be introduced until 2–3 weeks have elapsed from discontinuation of MAOI (as these are irreversible inhibitors; see p. 313). No washout period is required when using the reversible MAOI, moclobemide.
Augmentation
Another important augmentation strategy employs the mood stabiliser lithium carbonate. Controlled trials suggest that up to 50% of patients who have not responded to standard antidepressants can respond after lithium augmentation but the evidence is stronger for augmenting tricyclics than for augmenting SSRIs. Addition of lithium requires careful titration of the plasma concentration up to the therapeutic range, with periodic checks thereafter and monitoring for toxicity (see p. 331).
More recently, augmentation of SSRIs with atypical antipsychotics has been effective in clinical trials. Trial evidence is strongest using olanzapine, and also exists for quetiapine, risperidone and aripiprazole. Antipsychotics also have important potential for side-effects which must be taken into account before their introduction (see p. 322).
Other indications for antidepressants
Antidepressants may benefit most forms of anxiety disorder, including panic disorder, generalised anxiety disorder, post-traumatic stress disorder, obsessive–compulsive disorder and social phobia (see p. 331). SSRIs are recognised as first-line drug treatment for all five of these anxiety disorders.
Adverse effects
Interactions
Pharmacodynamic interactions
• Most antidepressants (including SSRIs and tricylics) may cause central nervous system (CNS) toxicity if co-prescribed with the dopaminergic drugs entacapone and selegiline (for Parkinson’s disease). SSRIs increase the risk of the serotonin syndrome when combined with drugs that enhance serotonin transmission, e.g. the antimigraine triptan drugs which are 5HT1-receptor antagonists, and the antiobesity drug sibutramine.
• Most antidepressants lower the convulsion threshold, complicate the drug control of epilepsy and lengthen seizure time in electroconvulsive therapy (ECT). The situation is made more complex by the capacity of carbamazepine to induce the metabolism of antidepressants and of certain antidepressants to inhibit carbamazepine metabolism (see below).
• SSRIs are known to interfere with platelet aggregation and may increase the risk of gastrointestinal bleeding, especially in those with existing risk factors.
• Trazodone and many tricyclics cause sedation and therefore co-prescription with other sedative agents such as opioid analgesics, H1-receptor antihistamines, anxiolytics, hypnotics and alcohol may lead to excessive drowsiness and daytime somnolence.
• The majority of tricyclics have undesirable cardiovascular effects, in particular prolongation of the QTc interval. Numerous other drugs also prolong the QTc interval, e.g. amiodarone, disopyramide, procainamide, propafenone, quinidine, terfenadine, and psychotropic agents such as pimozide and sertindole. Their use in combination with TCAs that prolong QTc enhances the risk of ventricular arrhythmias.
• Tricylics potentiate the effects of catecholamines and other sympathomimetics, but not those of β2-receptor agonists used in asthma. Even the small amounts of adrenaline/epinephrine or noradrenaline/norepinephrine in dental local anaesthetics may produce a serious rise in blood pressure.
Pharmacokinetic interactions
Metabolism by cytochrome P450 enzymes provides ample opportunity for interaction of antidepressants with other drugs by inhibition of, competition for, or induction of enzymes. Tables 20.2A and 20.2B indicate examples of mechanisms by which interaction that may occur when relevant drugs are added to, altered in dose or discontinued from regimens that include antidepressants.
In depressive psychosis, antidepressants are commonly prescribed with antipsychotics and there is potential for enhanced drug effects with paroxetine + perphenazine (CYP 2D6), fluoxetine + sertindole (3A4) and fluvoxamine + olanzapine (1A2). Rapid tranquillisation with zuclopenthixol acetate (see p. 325) of an agitated patient who is also taking fluoxetine or paroxetine can result in toxic plasma concentrations with excessive sedation and respiratory depression due to inhibition of zuclopenthixol metabolism by CYP 2D6 and CYP 3A4. P450 enzyme inhibition by fluoxetine or paroxetine may also augment effects of alcohol, tramadol (danger of serotonin syndrome) methadone, terfenadine (danger of cardiac arrhythmia), -caine anaesthetics and theophylline.
e.g. carbamazepine and several other antiepilepsy drugs, accelerate the metabolism of antidepressants, reducing their therapeutic efficacy and requiring adjustment of dose. Epilepsy is a particularly common co-morbid illness in patients who have both psychiatric illness and learning disabilities, and the combination of an anticonvulsant and an antidepressant or major psychotropic drug is to be anticipated. Depression and hypertension are both such common conditions that some co-morbidity is inevitable. Panic disorder (an indication for an antidepressant drug) is associated with hypertension.1 An antidepressant that is an enzyme inhibitor may exaggerate antihypertensive effects of metoprolol (metabolised by CYP 2D6), or diltiazem or amlodipine (3A4).
Monoamine oxidase inhibitors
Foods likely to produce hypertensive effects in patients taking MAOIs include:
• Cheese, especially if well matured.
• Red wines (especially Chianti) and some white wines; some beers (non- or low-alcohol varieties contain variable but generally low amounts of tyramine).
• Yeast extracts (Marmite, Oxo, Bovril).
• Broad bean pods (contain dopa, a precursor of adrenaline/epinephrine).
• Over-ripe bananas, avocados, figs.
• Fermented bean curds including soy sauce.
MAOI interactions with other drugs
MAOI–SSRI combinations may provoke the life-threatening ‘serotonin syndrome’ (see above). Strict rules apply regarding washout periods when switching between MAOIs and other drugs (see above, Changing antidepressants, p. 316). Very occasionally, MAOIs are co-prescribed with other antidepressants, but as many combinations are highly dangerous such practice should be reserved for specialists only and then as a last resort.
St John’s wort
The herbal remedy St John’s wort (Hypericum perforatum) has found favour in some patients with mild to moderate depression. The active ingredients in the hypericum extract have yet to be identified, and their mode of action is unclear. Several of the known mechanisms of action of existing antidepressants are postulated, including inhibition of monoamine reuptake and the MAO enzyme, as well as a stimulation of GABA receptors. Much of the original research into the efficacy of St John’s wort was performed in Germany, where its use is well established. Several direct comparisons with tricyclic antidepressants have shown equivalent rates of response, but the interpretation of these studies is complicated by the fact that many failed to use standardised ratings for depressive symptoms, patients tended to receive TCAs below the minimum therapeutic dose, and sometimes received St John’s wort in doses above the maximum recommended in commercially available preparations. Use of St John’s wort is further complicated by the lack of standardisation of the ingredients. A large multi-centre trial found only limited evidence of benefit for St John’s wort over placebo in significant major depression.2
Antipsychotics
Classification
Originally tested as an antihistamine, chlorpromazine serendipitously emerged as an effective treatment for psychotic illness in the 1950s. Chlorpromazine-like drugs were originally termed ‘neuroleptics’ or ‘major tranquillisers’, but the preferred usage now is ‘antipsychotics’. Classification is by chemical structure, e.g. phenothiazines, butyrophenones. Within the large phenothiazine group, compounds are divided into three types on the basis of the side-chain, as this tends to predict adverse effect profiles (Table 20.3). The continuing search for greater efficacy and better tolerability led researchers and clinicians to reinvestigate clozapine, a drug that was originally licensed in the 1960s but subsequently withdrawn because of toxic haematological effects. Clozapine appeared to offer greater effectiveness in treatment-resistant schizophrenia, to have efficacy against ‘negative’ in addition to ‘positive’ psychiatric symptoms (see Table 20.4), and to be less likely to cause extrapyramidal motor symptoms. It regained its licence in the early 1990s with strict requirements on dose titration and haematological monitoring. The renewed interest in clozapine and its unusual efficacy and tolerability stimulated researchers to examine other ‘atypical’ antipsychotic drugs.
| Atypical antipsychoticsa | Classical antipsychotics | |
|---|---|---|
| Clozapine | Phenothiazines | |
| Olanzapine | Type 1 | Chlorpromazine |
| Quetiapine | Promazine | |
| Risperidone | Type 2 | Pericyazine |
| Ziprasidone | Type 3 | Trifluoperazine |
| Amisulprideb | Prochlorperazine | |
| Zotepine | Fluphenazine | |
| Sertindolec | Butyrophenones | Haloperidol |
| Aripiprazole | Benperidol | |
| Paliperidone | Substituted benzamide | Sulpirideb,c |
| Thioxanthines | Flupentixol Zuclopenthixol |
|
| Other | Pimozide Loxapine |
a No recognised classification system exists for atypical antipsychotics. Tentative terms based on receptor binding profiles have been applied to certain drug groupings, e.g. ‘broad-spectrum atypicals’ for clozapine, olanzapine and quetiapine, whereas risperidone and ziprasidone have been described as ‘high-affinity serotonin–dopamine antagonists’.
b Amisulpride and sulpiride are structurally related.
c Sertindole is available only on a named-patient basis when at least one previous antipsychotic has failed owing to lack of efficacy or adverse effects.
Table 20.4 Symptoms of schizophrenia
| Positive symptoms | Negative symptoms |
|---|---|
| Hallucinations: most commonly auditory (i.e. voices) in the third person, which patients may find threatening. The voices may also give commands. Visual hallucinations are rare | Affective flattening manifest by unchanging facial expression with lack of communication through expression, poor eye contact, lack of responsiveness, psychomotor slowing |
| Delusions: most commonly persecutory. ‘Passivity phenomena’ include delusions of thought broadcasting, thought insertion or thought withdrawal, made actions, impulses or feelings | Alogia (literally‘absence of words’), manifesting clinically as a lack of spontaneous speech (poverty of speech) |
| Bizarre behaviours including agitation, sexual disinhibition, repetitive behaviour, wearing of striking but inappropriate clothing | Anhedonia (inability to derive pleasure from any activity) and associality (narrowing of repertoire of interests and impaired relationships) |
| Thought disorder manifest by failure in the organisation of speech such that it drifts away from the point (tangentiality), never reaches the point (circumstantiality), moves from one topic to the next illogically (loosened associations, knight’s move thinking), breaks off abruptly only to continue on an unrelated topic (derailment) or moves from one topic to the next on the basis of a pun or words that sound similar (clang association) | Apathy/avolution involving lack of energy, lack of motivation to work, participate in activities or initiate any goal-directed behaviour, and poor personal hygiene Attention problems involving an inability to focus on any one issue or engage fully with communication |
Mechanism of action
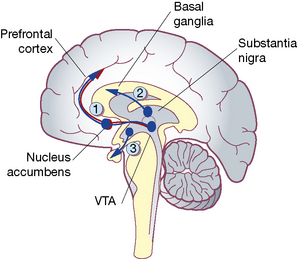
The common action of conventional antipsychotics is to decrease brain dopamine function by blocking the dopamine D2 receptors (Fig. 20.3). However, the atypical drugs act on numerous receptors and modulate several interacting transmitter systems. All atypicals (except amisulpride) exhibit greater affinity for 5HT2 receptors than D2 receptors, unlike the classical agents. Atypical drugs that do antagonise dopamine D2 receptors appear to have some selectivity of effect for those in the mesolimbic system (producing antipsychotic effect) rather than the nigrostriatal system (associated with unwanted motor effects). Clozapine and risperidone exert substantial antagonism of α2-adrenoceptors, a property that may explain their benefits against negative symptoms. Blockade of muscarinic acetylcholine receptors as with chlorpromazine and clozapine reduces the occurrence of extrapyramidal effects. Aripiprazole is a unique drug because it is a partial dopamine D2-receptor agonist that acts conversely as an antagonist in regions where dopamine is overactive, such as the limbic system. It increases dopamine function where this is low (such as in the frontal cortex) and has little motor effect.
Pharmacokinetics
Antipsychotics are well absorbed after oral administration and distribute widely. They are metabolised mainly by hepatic cytochrome P450 isoenzymes, e.g. CYP 2D6 (risperidone, perphenazine; see Table 20.2A), CYP 3A4 (sertindole; see Table 20.2B), CYP 1A2 (olanzapine, clozapine). Metabolism of some compounds is complex, e.g. zuclopenthixol and haloperidol are metabolised by both CYP 2D6 and CYP 3A4, which means that co-prescription of inhibitors of either enzyme can cause raised plasma concentrations of these antipsychotics. Amisulpride is an exception to the general rule as it is mainly eliminated unchanged by the kidneys with little hepatic metabolism. Elimination t½ values range from quetiapine 7 h, clozapine 12 h, haloperidol 18 h to olazapine 33 h. Depot preparations usefully release drug over 2–4 weeks after intramuscular injection (see below).
Efficacy
Symptoms in schizophrenia are defined as positive and negative (Table 20.4). Although a classical antipsychotic drug should provide adequate treatment of positive symptoms including hallucinations and delusions, at least 60% of patients may have unresolved negative symptoms such as apathy, flattening of affect and alogia. Evidence suggests that clozapine may have a significant advantage against negative symptoms. This drug has a further advantage over all other antipsychotics, whether classical or atypical, in that it is the most effective agent for ‘resistant’ schizophrenia, i.e. where other antipsychotics prescribed at adequate doses fail to produce improvement or are not tolerated.
Mode of use
The longer a psychosis is left untreated, the less favourable is the outcome, and drug treatment should be instigated as soon as an adequate period of assessment has allowed a provisional diagnosis to be established. Patients who are ‘neuroleptic naïve’, i.e. have never previously taken any antipsychotic agent, should start at the lowest available dose (see Table 20.5, below). For most atypical agents a period of dose titration by protocol from the starting dose up to a stated lowest therapeutic dose is usual, e.g. risperidone 4 mg/day, quetiapine 300 mg/day. Dose increases are indicated when there is no response until the desired effect on psychotic symptoms or calming of disturbed behaviour is achieved, the urgency of the situation determining the interval between increments until the maximum licensed dose is achieved. Conservative dose titration is advisable for the elderly and patients with learning disabilities (who may require antipsychotics for psychosis or severe behavioural disturbance).
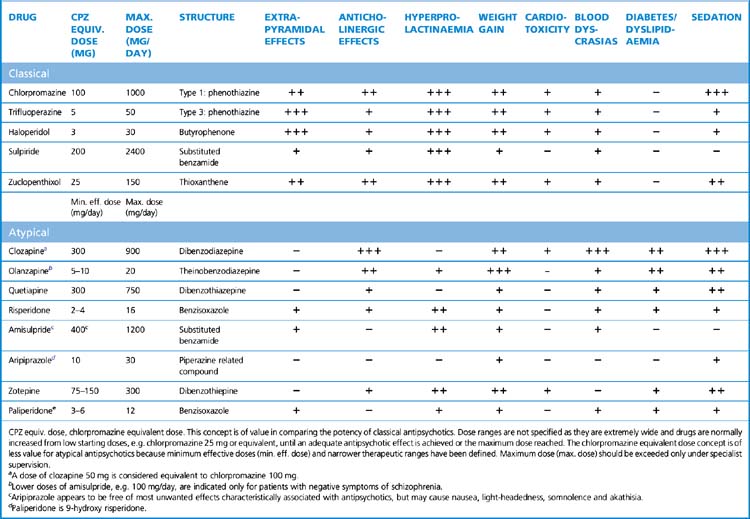
Prescription of conventional antipsychotics follows similar rules to those for atypical drugs, starting at low doses in neuroleptic-naïve patients, e.g. haloperidol 0.5 mg/day in case the patient is particularly susceptible to adverse, especially extrapyramidal, motor side-effects. There is a wide range of effective doses for many classical agents (e.g. chlorpromazine, once the most prescribed antipsychotic but now rarely used, has a dose range from 25 to 1000 mg/day). As the potency (therapeutic efficacy in relation to dose) of antipsychotic agents varies markedly between compounds, it is useful to think of the effective antipsychotic dose of classical agents in terms of ‘chlorpromazine equivalents’ (see Table 20.5). For example, haloperidol has a relatively high antipsychotic potency, such that 2–3 mg is equivalent to chlorpromazine 100 mg, whereas 200 mg sulpiride (low potency) is required for equivalent antipsychotic effect. Prescribing in excess of this requires specialist involvement. With co-prescribed antipsychotics, their total maximum antipsychotic dose should not exceed 1000 mg chlorpromazine equivalents per day, except under specialist supervision.
Adverse effects (Table 20.5)
Classical antipsychotics
Acute extrapyramidal symptoms. Dystonias are manifest as abnormal movements of the tongue and facial muscles with fixed postures and spasm, including torticollis and bizarre eye movements (‘oculogyric crisis’). Parkinsonian symptoms result in the classical triad of bradykinesia, rigidity and tremor. Both dystonias and parkinsonian symptoms are believed to result from a shift in favour of cholinergic rather than dopaminergic neurotransmission in the nigrostriatal pathway (see p. 323). Anticholinergic (antimuscarinic) agents, e.g. procyclidine, orphenadrine or benztropine, act to restore the balance in favour of dopaminergic transmission but are liable to provoke antimuscarinic effects (dry mouth, urinary retention, constipation, exacerbation of glaucoma and confusion) and they offer no relief for tardive dyskinesia, which may even worsen. They should be used only to treat established symptoms and not for prophylaxis. Benzodiazepines are an alternative.
Classical antipsychotics may also be associated with:
• Weight gain (a problem with almost all classical antipsychotics with the exception of loxapine; most pronounced with fluphenazine and flupentixol).
• Seizures (chlorpromazine is especially likely to lower the convulsion threshold).
• Interference with temperature regulation (hypothermia or hyperthermia, especially in the elderly).
• Skin problems (phenothiazines, particularly chlorpromazine, may provoke photosensitivity necessitating advice about limiting exposure to sunlight); rashes and urticaria may also occur.
• Sexual dysfunction (ejaculatory problems through α-adrenoceptor blockade).
• Retinal pigmentation (chlorpromazine can cause visual impairment if the dose is prolonged and high).
• Blood dyscrasias (agranulocytosis and leucopenia).
• Osteoporosis (associated with increased prolactin levels).
Neuroleptic malignant syndrome
Raised plasma creatine kinase concentration and white cell count are suggestive (but not conclusive) of neuroleptic malignant syndrome. There is some clinical overlap with the ‘serotonin syndrome’ (see p. 318) and concomitant use of SSRI antidepressants (or possibly TCAs) with antipsychotics may increase the risk.
Comparison of conventional and atypical antipsychotics
Atypical antipsychotics may have advantages in two areas:
1. Tolerability, and thus compliance, appears to be better, in particular with less likelihood of inducing extrapyramidal effects and hyperprolactinaemia (although the latter remains common with risperidone and amisulpride).
2. Regarding efficacy it was originally thought that all atypicals had an advantage over conventional agents at least for negative symptoms. More recently, evidence has emerged that for overall efficacy in schizophrenia, olanzapine, risperidone and amisulpride, in addition to clozapine, have an advantage over the other atypicals and the conventional antipsychotics. Note that clozapine is normally only used when at least two atypical antipsychotics have been tried without adequate response.
Mood stabilisers
Lithium
Lithium salts were known anecdotally to have beneficial psychotropic effects as long ago as the middle of the 19th century, but scientific evidence of their efficacy was not obtained until 1949, when lithium carbonate was tried in manic patients; it was found to be effective in the acute state and, later, to prevent recurrent attacks.3
Lithium is also used to augment the action of antidepressants in treatment-resistant depression (see p. 318).
are encountered in three general categories:
• Those occurring at plasma concentrations within the therapeutic range (0.4–1 mmol/L), which include fine tremor (especially involving the fingers; a β-blocker may benefit), constipation, polyuria, polydipsia, metallic taste in the mouth, weight gain, oedema, goitre, hypothyroidism, acne, rash, diabetes insipidus and cardiac arrhythmias. Mild cognitive and memory impairment also occur.
• Signs of intoxication, associated with plasma concentrations greater than 1.5 mmol/L, are mainly gastrointestinal (diarrhoea, anorexia, vomiting) and neurological (blurred vision, muscle weakness, drowsiness, sluggishness and coarse tremor, leading on to giddiness, ataxia and dysarthria).
• Frank toxicity, due to severe overdosage or rapid reduction in renal clearance, usually associated with plasma concentrations over 2 mmol/L, constitutes a medical emergency. Hyperreflexia, hyperextension of limbs, convulsions, hyperthermia, toxic psychoses, syncope, oliguria, coma and even death may result if treatment is not instigated urgently.
Carbamazepine
Carbamazepine is licensed as an alternative to lithium for prophylaxis of bipolar affective disorder, although clinical trial evidence is actually stronger to support its use in the treatment of acute mania. While lithium monotherapy is more effective than carbamazepine for prophylaxis of classical bipolar affective disorder, carbamazepine appears to be more effective than lithium for rapidly cycling bipolar disorders, i.e. with recurrent swift transitions from mania to depression. It is also effective in combination with lithium. (See also Epilepsy, p. 349.)
Drugs used in anxiety and sleep disorders
Classification of anxiety disorders
The diagnostic criteria of DSM-IV (Diagnostic and Statistical Manual) or ICD-10 (International Classification of Disease) are generally used. Both divide anxiety into a series of subsyndromes with clear operational criteria to assist in distinguishing them. At any one time many patients may have symptoms of more than one syndrome, but making the primary diagnosis is important as this can markedly influence the choice of treatment (Table 20.6).
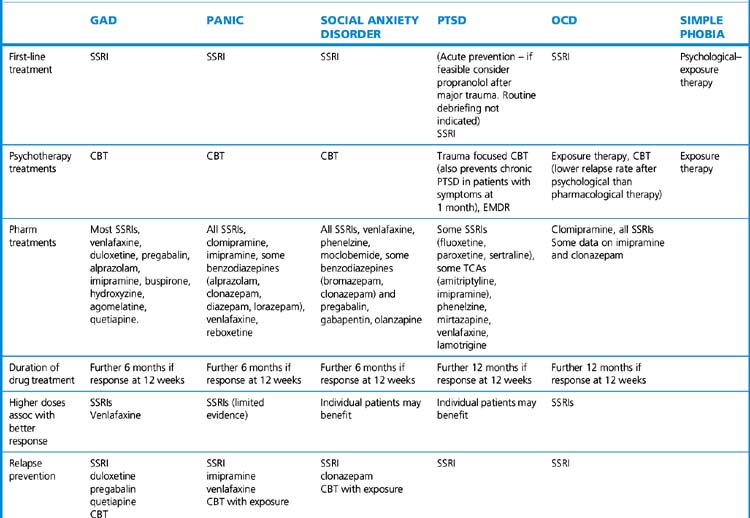
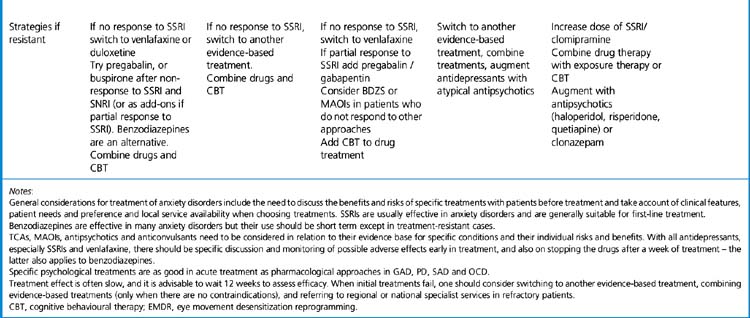
Panic disorder (PD)
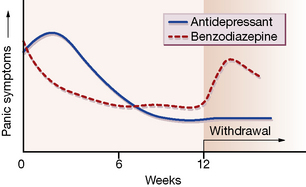
The choice lies between a fast-acting benzodiazepine, such as lorazepam or alprazolam, and one with delayed efficacy but fewer problems of withdrawal, such as a tricyclic antidepressant (TCA), e.g. clomipramine or imipramine, or an SSRI, e.g. paroxetine. The different time course of these two classes of agent in panic disorder is depicted in Figure 20.4 (see also Table 20.6).
Social anxiety disorder
Obsessive–compulsive disorder (OCD)
Obsessive–compulsive disorder has two main components:
• The repetition of acts or thoughts which are involuntary, recognised by the sufferer to be generated by their own brain but unwanted, irrational, and out of keeping with their morals or values, and so very distressing.
• Anxiety provoked by the occurrence of such thoughts, or by prevention of the compulsive acts.
OCD on its own often starts in adolescence and has a chronic and pervasive course unless treated. OCD starting later on in life is often associated with affective or anxiety disorders. Symptoms often abate briefly if the individual is taken to a new environment.
General comments about treating anxiety disorders (Table 20.7)
• There is need to discuss the benefits and risks of specific treatments with patients before treatment and take account of their clinical features, needs, preferences and availability of local services when choosing treatments.
• SSRIs are usually effective in anxiety disorders and are generally suitable for first-line treatment.
• Benzodiazepines are effective in many anxiety disorders but their use should be short term except in treatment-resistant cases.
• SNRIs, TCAs, MAOIs, pregabalin and antipsychotics need to be considered in relation to their evidence base for specific conditions and their individual risks and benefits.
• With all antidepressants, especially SSRIs and SNRIs, there should be specific discussion and monitoring of possible adverse effects early in treatment, and also on stopping the drugs after a week of treatment; this latter also applies to benzodiazepines.
• SSRI/SNRI treatment is often slow to produce the therapeutic effect, and it is advisable to wait 12 weeks to assess efficacy.
• In a first episode, patients may need medication for at least 6 months, withdrawing over a further 4–8 weeks if they are well. Those with recurrent illness may need treatment for 1–2 years to enable them to learn and put into place psychological approaches to their problems. In many cases the illness is life-long and chronic maintenance treatment is justified if it significantly improves their well-being and function.
• Specific psychological treatments are also effective in treatment.
• When initial treatments fail, consider switching to another evidence-based treatment, combining evidence-based treatments (only when there are no contraindications), and referring to regional or national specialist services in refractory patients.
Sleep disorders
Types of sleep disorder
Several types of sleep disorder are recognised, and their differentiation is important; a simplified summary is given below but reference to DSM-IV, ICD-10 and ICSD4 will clarify the exact diagnostic criteria:







Insomnia
A summary of precipitating factors for insomnia is shown in Box 20.1.
Box 20.1 Precipitating factors for insomnia
Psychiatric
• Patients with depressive illnesses often have difficulty falling asleep at night and complain of restless, disturbed and unrefreshing sleep, and early morning waking. When their sleep is analysed by polysomnography, time to sleep onset is indeed prolonged, and there is a tendency for more REM sleep to occur in the first part of the night, with reduced deep quiet sleep in the first hour or so after sleep onset, and increased awakenings during the night. They may wake early in the morning and fail to get back to sleep again.
• Anxiety disorders may cause patients to complain about their sleep, either because there is a reduction in sleep continuity or because normal periods of nocturnal waking are somehow less well tolerated. Nocturnal panic attacks can make patients fearful of going off to sleep.
• Bipolar patients in the hypomanic or manic phase will sleep less than usual, and often changes in sleep pattern give early warning that an episode is imminent.
Pharmacological
• Non-prescription drugs such as caffeine or alcohol. Alcohol reduces the time to onset of sleep, but disrupts sleep later in the night. Regular and excessive consumption disrupts sleep continuity; insomnia is a key feature of alcohol withdrawal. Excessive intake of caffeine and theophylline, in tea, coffee or cola drinks, also contributes to sleeplessness, probably because caffeine acts as an antagonist to adenosine, the endogenous sleep-promoting neurotransmitter.
• Starting treatment with certain antidepressants, especially serotonin reuptake inhibitors (e.g. fluoxetine, fluvoxamine), or monoamine uptake inhibitors; sleep disruption is likely to resolve after 3–4weeks.
• Other drugs that increase central noradrenergic and serotonergic activity include stimulants, such as amfetamine, cocaine and methylphenidate, and sympathomimetics, such as the β-adrenergic agonist salbutamol and associated substances.
• Withdrawal from hypnotic drugs; this is usually short lived.
• Treatment with β-adrenoceptor blockers may disrupt sleep, perhaps because of their serotonergic action; a β-blocking drug that crosses blood–brain barrier less readily is preferred, e.g. atenolol.
Physical
• Pain, in which case adequate analgesia will improve sleep.
• Coughing or wheezing: adequate control of asthma with stimulating drugs as above, may paradoxically improve sleep by reducing waking due to breathlessness.
• Respiratory and cardiovascular disorders.
• Need to urinate; this may be affected by timing of diuretic medication.
• Neurological disorders, e.g. stroke, movement disorders.
• Periodic leg movements of sleep (frequent jerks or twitches during the descent into deeper sleep); rarely reduce subjective sleep quality but are more likely to cause them in the subject’s sleeping partner.
• Restless legs syndrome (irresistible desire to move the legs), which is usually worse in the evening and early night, and can prevent sleep.







Drugs for insomnia
The promotion of sleep is regulated by a number of other neurotransmitters; primary among these is γ-aminobutyric acid (GABA), the major inhibitory neurotransmitter in the brain. The majority of brain cells are inhibited by GABA so increasing its function reduces arousal and produces sleep, and eventually anaesthesia. There are many subsets of GABA neurones distributed throughout the brain but a particular cluster in the hypothalamus (ventrolateral preoptic nucleus) can be considered to be the sleep ‘switch’.5 These neurones switch off brain arousal systems at the level of the cell bodies and therefore promote sleep. GABA receptors in the cortex can also promote sedation and sleep by inhibiting the target neurones of the arousal system. Most drugs used in insomnia act by increasing the effects of GABA at the GABAA receptor.
The GABAA–benzodiazepine receptor complex
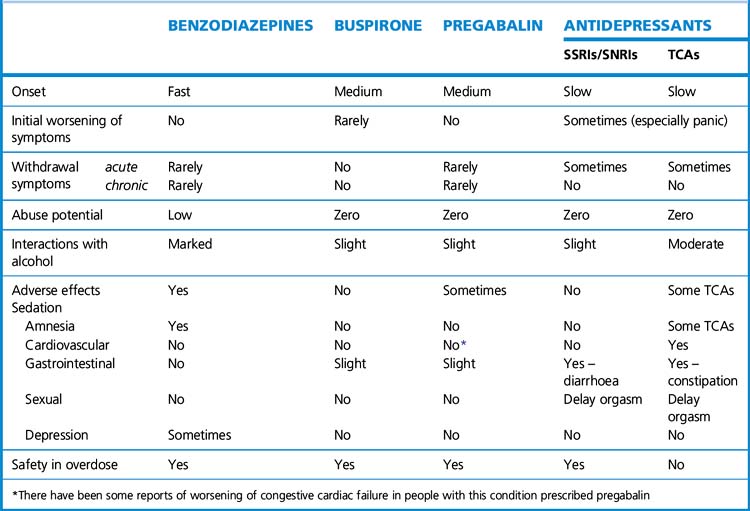
When GABA binds with the GABAAreceptor, the permeability of the central pore of the receptor complex opens, so allowing more chloride ions into the neurone and decreasing excitability. Classical benzodiazepines (BZDs) in clinical use bind to another receptor on the complex (the benzodiazepine receptor) and enhance the effectiveness of GABA, producing a larger inhibitory effect (Fig. 20.5). These drugs are agonists at the receptor and an antagonist, flumazenil, prevents agonists from binding at the receptor site; it is used clinically to reverse benzodiazepine actions.
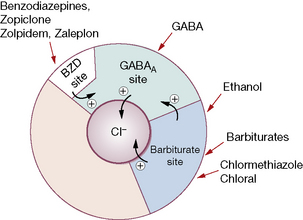
Benzodiazepines
Benzodiazepines are effective after administration by mouth but enter the circulation at very different rates that are reflected in the speed of onset of action, e.g. alprazolam is rapid, oxazepam is slow (Table 20.8). Hepatic breakdown produces metabolites, some with long t½ which greatly extend drug action, e.g. chlordiazepoxide, clorazepate and diazepam all form demethyldiazepam (t½ 80 h).
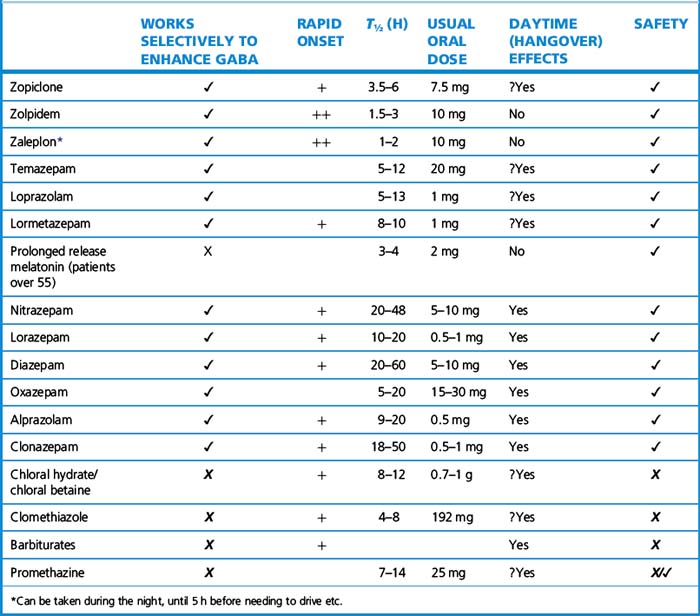
Oral benzodiazepines are used for: insomnia, anxiety, alcohol withdrawal states, muscle spasm due to a variety of causes, including tetanus and cerebral spasticity. Injectable preparations are used for rapid tranquillisation in psychosis (see previous section), anaesthesia and sedation for minor surgery and invasive investigations (see Index). The choice of drug as hypnotic and anxiolytic is determined by pharmacokinetic properties (see before, and Table 20.7).
Clinical uses include reversal of benzodiazepine sedation after endoscopy, dentistry and in intensive care. Heavily sedated patients become alert within 5 minutes. The t½ of 1 h is much shorter than that of most benzodiazepines (see Table 20.7), so that repeated i.v. administration may be needed to maintain the effect. Thus the recovery period needs supervision lest sedation recurs; if used in day surgery it is important to tell patients that they may not drive a car home. The dose is 200 micrograms by i.v. injection given over 15 s, followed by 100 micrograms over 60 s if necessary, to a maximum of 300–600 micrograms. Flumazenil is useful for diagnosis of self-poisoning, and also for treatment, when 100–400 micrograms are given by continuous i.v. infusion and adjusted to the degree of wakefulness.
Non-benzodiazepine hypnotics that act at the GABAA-benzodiazepine receptor
Zopiclone, a cyclopyrrolone, has an onset of action that is relatively rapid (about 1 h) and that lasts for 6–8 h, making it suitable for both initial and maintenance treatment of insomnia. It may cause fewer problems on withdrawal than benzodiazepines. The duration of action is prolonged in the elderly, and in hepatic insufficiency. About 40% of patients experience a metallic aftertaste (genetically determined). People who take zopiclone have been shown to be at increased risk of road traffic accidents. Care should be taken with concomitant medication that affects its metabolic pathway (see Table 20.2A).
Other drugs used in insomnia
Antihistamines. Most proprietary (over-the-counter) sleep remedies contain H1-receptor antihistamines with sedative action (see Ch. 9). Promethazine (Phenergan) has a slow (1–2 h) onset and long duration of action (t½ 12 h). It reduces sleep onset latency and awakenings during the night after a single dose, but there have been no studies showing enduring action. It is sometimes used as a hypnotic in children. There are no controlled studies showing improvements in sleep after other antihistamines. Alimemazine (trimeprazine) is used for short-term sedation in children. Most antihistamine sedatives have a relatively long action and may cause daytime sedation.
Antidepressants. In the depressed patient, improvement in mood is almost always accompanied by improvement in subjective sleep, and therefore choice of antidepressant should not usually involve additional consideration of sleep effects. Nevertheless, some patients are more likely to continue with medication if there is a short-term improvement, in which case mirtazapine or trazodone may provide effective antidepressant together with sleep-promoting effects. Antidepressant drugs, particularly those with 5HT2-blocking effects, may occasionally be effective in long-term insomnia (but see Table 20.7). Antipsychotics have been used to promote sleep in resistant insomnia occurring as part of another psychiatric disorder, probably because of the combination of 5HT2-antagonism, α1-adrenoceptor antagonism and histamine H1-receptor antihistamine effects in addition to their primary dopamine antagonist effects. Their long action leads to daytime sedation, and extrapyramidal movement disorders may result from their blockade of dopamine receptors (see above, Antipsychotics). They therefore should be used with great care in the context of insomnia. Nevertheless, modern antipsychotics, e.g. quetiapine, are being used for intractable insomnia, usually at a dose well below the one required to treat psychosis, e.g. 25–50 mg/day.
Summary of pharmacotherapy for insomnia
• Drug treatment is usually effective for a short period (2–4 weeks).
• Some patients may need long-term medication.
• Intermittent medication which is taken only on nights that symptoms occur is desirable and may often be possible with modern, short-acting, compounds.
• Discontinuing hypnotic drugs is usually not a problem if the patient knows what to expect. There will be a short period (usually 1–2 nights) of rebound insomnia on stopping hypnotic drugs, which can be ameliorated by phased withdrawal.
Drugs for Alzheimer’s6 disease (dementia)
Dementia is described as a syndrome ‘due to disease of the brain, usually of chronic or progressive nature, in which there is disturbance of multiple higher cortical functions, including memory, thinking, orientation, comprehension, calculation, learning capacity, language and judgement, without clouding of consciousness’.7 Deterioration in emotional control, social behaviour or motivation may accompany or precede cognitive impairment. Alzheimer’s and vascular (multi-infarct) disease are the two most common forms of dementia, accounting for about 80% of presentations. Alzheimer’s disease is associated with deposition of β-amyloid in brain tissue and abnormal phosphorylation of the intracellular tau (τ) proteins causing abnormalities of microtubule assembly and collapse of the cytoskeleton. Pyramidal cells of the cortex and subcortex are particularly affected.
Evidence indicates that cholinergic transmission is diminished in Alzheimer’s disease. The first three drugs available for use in Alzheimer’s each act to enhance acetylcholine activity by inhibiting the enzyme acetylcholinesterase which metabolises and inactivates synaptically released acetylcholine. Consequently acetylcholine remains usable for longer. Individual drugs are categorised by the type of enzyme inhibition they cause. Donepezil is classed as a ‘reversible’ agent, as binding to the acetylcholinesterase enzymes lasts only minutes, whereas rivastigmine is considered ‘pseudo-irreversible’ as inhibition lasts for several hours. Galantamine is associated both with reversible inhibition and with enhanced acetylcholine action on nicotinic receptors.8 Clinical trials show that these agents produce an initial increase in patients’ cognitive ability. There also may be associated global benefits, including improvements in non-cognitive aspects such as depressive symptoms. But the drugs do not alter the underlying process, and the relentless progress of the disease is paralleled by a reduction in acetylcholine production with a decline in cognition.
The beneficial effects of drugs are therefore to:
• stabilise the condition initially and sometimes improve cognitive function
• delay the overall pace of decline (and therefore the escalating levels of support required)
The severity of cognitive deficits in patients suffering from, or suspected of having, dementia can be quantified by a simple 30-point schedule, the mini mental state examination (MMSE) of Folstein. A score of 21–26 denotes mild, 10–20 moderate and less than 10 severe Alzheimer’s disease. The MMSE can also be used to monitor progress.
• Alzheimer’s disease must be diagnosed and assessed in a specialist clinic; the clinic should also assess cognitive, global and behavioural functioning, activities of daily living, and the likelihood of compliance with treatment.
• Treatment should be initiated by specialists but may be continued by family practitioners under a shared-care protocol.
• The carers’ views of the condition should be sought before and during drug treatment.
• The patient should be assessed every 6 months and drug treatment should continue only if the MMSE score remains at or above 10 points and if treatment has had a worthwhile effect on global, behavioural and functional parameters.
• Donepezil 5–10 mg nocte increasing to 10 mg nocte after 1 month.
• Galantamine 4 mg twice daily increasing to 8–12 mg twice daily at 4-week intervals.
• Rivastigmine 1.5 mg twice daily increasing to 3–6 mg twice daily at 2-week intervals.
• Memantine 5 mg daily increasing weekly by 5 mg to a maximum of 20 mg daily.
Drugs in attention deficit/hyperactivity disorder
Stimulant drugs are the first-choice treatment for ADHD in both children and adults (Table 20.9). Methylphenidate and dexamfetamine increase synaptic dopamine and noradrenaline/norepinephrine; dexamfetamine also blocks the reuptake of both neurotransmitters. Methylphenidate is the first-line treatment in the UK for ADHD; it is available both in conventional and extended release formulations which can be combined to obtain maximum therapeutic effect. Treatment in children is frequently restricted to school terms, giving the child a ‘drug holiday’ when out of school. Drug holidays are necessary to determine efficacy of treatment, adjust or change dosage and avoid tolerance effects. Treatment in adults is more complex as attentional requirements are often continuous, so drug holidays may not be possible. Dosage tends to be slightly higher and combinations of different psychotropic drugs are more common. Dexamfetamine is an alternative that has similar efficacy to methylphenidate in ADHD and is the preferred drug in children with epilepsy. It has a greater potential for abuse than methylphenidate.
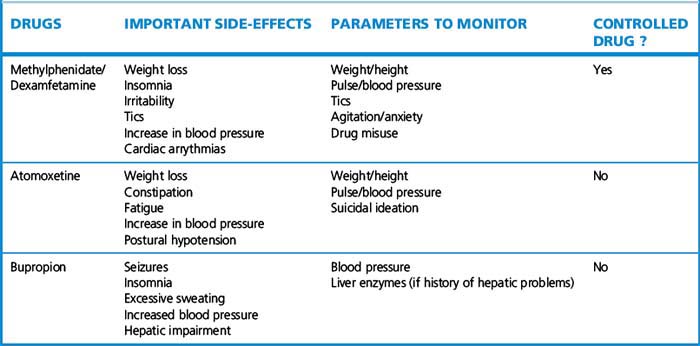
Drugs for psychiatric illness in childhood
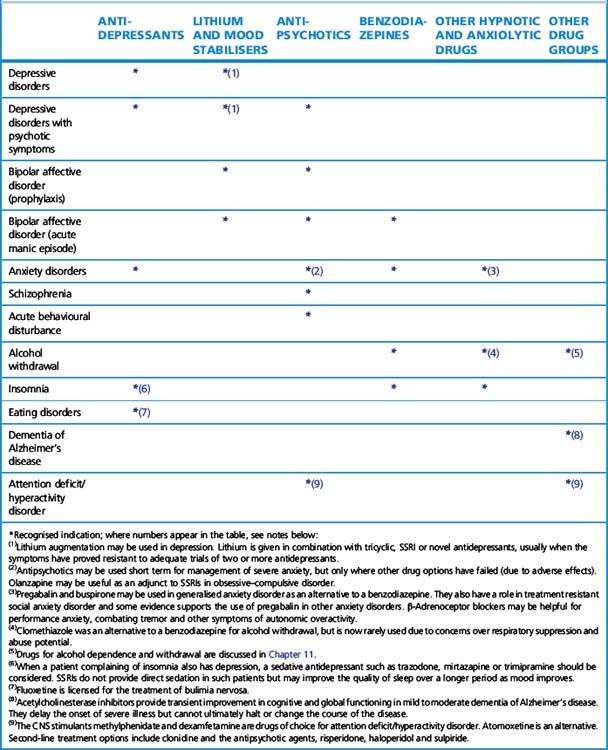
Table 20.10 summarises indications of the major groups of psychotropic drugs. Remember that psychiatric illnesses are often associated with co-morbid conditions, which may themselves require treatment; for example, schizophrenia may be associated with depression, so both antipsychotics and antidepressants may be required.
Anderson I.M., Ferrier I.N., Baldwin R.C., et al. Evidence-based guidelines for treating depressive disorders with antidepressants: a revision of the 2000 British Association for Psychopharmacology guidelines. J. Psychopharmacol.. 2008;22:343–396.
Baldwin D.S., Anderson I.M., Nutt D.J., et al. Evidence-based guidelines for the pharmacological treatment of anxiety disorders: recommendations from the British Association for Psychopharmacology. J. Psychopharmacol.. 2005;19(6):567–596.
Ballenger J.C., Davidson J.R.T., Lecrubier Y., et al. Consensus statement on panic disorder from the International Consensus Group on Depression and Anxiety. J. Clin. Psychiatry. 1998;59:47–54.
Ballenger J.C., Davidson J.R.T., Lecrubier Y., et al. Consensus statement on social anxiety disorder from the International Consensus Group on Depression and Anxiety. J. Clin. Psychiatry. 1998;59:54–60.
Ballenger J.C., Davidson J.R.T., Lecrubier Y., et al. Consensus statement on posttraumatic stress disorder from the International Consensus Group on Depression and Anxiety. J. Clin. Psychiatry. 2000;61:60–66.
Biederman J., Faraone S.V. Attention-deficit hyperactivity disorder. Lancet. 2005;366:237–248.
Blennow K., de Leon M.J., Zetterberg H. Alzheimer’s disease. Lancet. 2006;368:387–403.
Fink M. Convulsive therapy: a review of the first 55 years. J. Affect. Disord.. 2001;63:1–15.
Goodwin G.M. Evidence-based guidelines for treating bipolar disorder: revised second edition – recommendations from the British Association for Psychopharmacology. J. Psychopharmacol.. 2009;23(4):346–388.
Heyman I., Mataix-Cols D., Fineberg N.A. Obsessive–compulsive disorder. Br. Med. J.. 2006;333:424–429.
Leucht S., Corves C., Arbter D., et al. Second-generation versus first-generation antipsychotic drugs for schizophrenia: a meta-analysis. Lancet. 2009;373(9657):31–41.
Lieberman J.A., Mataix-Cols D., Fineberg N.A., for the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N. Engl. J. Med. 2005;353(12):1209–1223.
Mann J.J. The medical management of depression. N. Engl. J. Med.. 2005;353(17):1819–1834.
Ryan N.D. Treatment of depression in children and adolescents. Lancet. 2005;366:933–940.
Taylor C.B. Panic disorder. Br. Med. J.. 2006;332:951–955.
Wong I.C.K., Besag F.M.C., Santosh P.J., Murray M.L. Use of selective serotonin reuptake inhibitors in children and adolescents. Drug Saf.. 2004;27(13):991–1000.
1 Davies S J C, Ghahramani P, Jackson P R et al 1999 Association of panic disorder and panic attacks with hypertension. American Journal of Medicine 107:310–316.
2 Shelton R C, Keller M B, Gelenberg A et al 2001 Effectiveness of St John’s wort in major depression. A randomised control trial. Journal of the American Medical Association 285:1978–1986.
3 Cade J F 1970 The story of lithium. In: Ayd F J, Blackwell B (eds) Biological Psychiatry. Lippincott, Philadelphia.
4 DSM-IV American Psychiatric Association 1994 Diagnostic and Statistical Manual of Mental Disorders (DSM IV). American Psychiatric Association, Washington DC.
ICSD American Sleep Disorders Association 1992 International Classification of Sleep Disorders: Diagnostic and Coding Manual. ASDA, Rochester.
ICD-10 WHO 1994 Classification of Mental and Behavioural Disorders. World Health Organization, Geneva.
5 Saper CB, Scammell TE, LuJ (2005) Hypothalamic regulation of sleep and circadian rhythms. Nature 437:1257–1263.
6 Alois Alzheimer (1864–1915), German psychiatrist who studied the brains of demented and senile patients and correlated histological findings with clinical features.
7 International Classification of Diseases, 10th edition, Diagnostic System.
8 Irreversible antagonists exist but, not surprisingly, are not used in therapeutics (sarin nerve gas is an example).
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Chapter 20 Psychotropic drugs
Diagnostic issues
Also falling within the scope of modern psychiatric diagnostic systems are organic mental disorders (e.g. dementia in Alzheimer’s disease), disorders due to substance misuse (e.g. alcohol and opiate dependence; see Ch. 11), personality disorders, disorders of childhood and adolescence (e.g. attention deficit/hyperactivity disorder, Tourette’s syndrome), and mental retardation (learning disabilities).
Antidepressant drugs
Antidepressants can be broadly divided into four main classes (Table 20.1), tricyclics (TCA, named after their three-ringed structure), selective serotonin reuptake inhibitors (SSRIs), monoamine oxidase inhibitors (MAOIs) and novel compounds, some of which are related to TCAs or SSRIs. Clinicians who wish to have a working knowledge of antidepressants would be advised to be familiar with the use of at least one drug from each of the four main categories tabulated. A more thorough knowledge-base would demand awareness of the distinct characteristics of the subgroups of novel compounds (e.g. serotonin and noradrenaline/norepinephrine reuptake inhibitors (SNRIs), mirtazapine, reboxetine and agomelatine) and differences between individual SSRIs and TCAs. As antidepressants are largely similar in their therapeutic efficacy, awareness of profiles of unwanted effects is of particular importance.
| Tricyclics | Selective serotonin reuptake inhibitors | Monoamine oxidase inhibitors |
|---|---|---|
| Dosulepin Amitriptyline Lofepramine Clomipramine Imipramine Trimipramine Doxepin Nortriptyline Protriptyline Desipramine |
Fluoxetine Paroxetine Sertraline Citaloprama Escitaloprama Fluvoxamine |
Phenelzine Isocarboxazid Tranylcypromine Moclobemide (RIMA) |
| Novel compounds | ||
|---|---|---|
| Mainly noradrenergic | Mainly serotonergic | |
| Reboxetine (NaRI) | Trazodoneb Nefazodoneb,c |
|
| Mixed |
|---|
| Venlafaxine (SNRI) Mirtazapine (nassa)b Duloxetine (SNRI) Milnacipran (SNRI)d Agomelatine |
RIMA, reversible inhibitor of monoamine oxidase; NaRI, noradrenaline/norepinephrine reuptake inhibitor; SNRI, serotonin and noradrenaline/norepinephrine reuptake inhibitor; NaSSA, noradrenaline/norepinephrine and specific serotonergic antidepressant.
a Escitalopram is the active S-enantiomer of citalopram.
b Trazodone, nefazodone and mirtazapine have been classed as ‘receptor blocking’ antidepressants based on their antagonism of postsynaptic serotonin receptors (trazodone, nefazodone, mirtazapine) and presynaptic α2-receptors (trazodone, mirtazapine).
c Nefazodone has additional weak SSRI activity but has now been withdrawn due to risk of hepatitis.
An alternative categorisation of antidepressants is based solely on mechanism of action (Fig. 20.1). The majority of antidepressants, including SSRIs, TCAs and related compounds, are reuptake inhibitors. Certain novel agents, including trazodone and mirtazapine, are receptor blockers, whereas MAOIs are enzyme inhibitors.
Fig. 20.1 Flow chart of the evolution of antidepressant drugs, and classification by mechanism of action.
The first TCAs (imipramine and amitriptyline) and MAOIs appeared between 1957 and 1961 (see Fig. 20.1). The MAOIs were developed from antituberculous agents that unexpectedly improved mood. Imipramine was a chlorpromazine derivative that showed antidepressant rather than antipsychotic properties. Over the next 25 years the TCA class enlarged to more than 10 agents with heterogeneous pharmacological profiles, and further modifications of the original three-ringed structure gave rise to the related (but pharmacologically distinct) antidepressant trazodone.
Mechanism of action
The monoamine hypothesis proposes that, in depression, there is deficiency of the neurotransmitters noradrenaline/norepinephrine and serotonin in the brain which can be restored by antidepressants. Drugs that alleviate depression also enhance monoamine availability and release (Fig. 20.2), increasing activity at postsynaptic receptors. It is relevant that (older) antihypertensive agents, e.g. reserpine, which reduced the availability of noradrenaline/norepinephrine, caused depression.
MAOIs increase the availability of noradrenaline/norepinephrine and serotonin by preventing their destruction by the monoamine oxidase type A enzyme in the presynaptic terminal (see Ch. 21, Table 21.3). The older MAOIs, phenelzine, tranylcypromine and isocarboxazid, bind irreversibly to monamine oxidase by forming strong (covalent) bonds. The enzyme is thus rendered permanently ineffective such that amine metabolising activity can be restored only by production of fresh enzyme, which takes weeks. These MAOIs are thus called ‘hit and run’ drugs as their effects greatly outlast their detectable presence in the blood.
Drugs with similar modes of action to antidepressants find other uses in medicine. Bupropion (amfebutamone) inhibits reuptake of both dopamine and noradrenaline/norepinephrine. It was originally developed and used as an antidepressant but is now more frequently used to assist smoking cessation (see p. 321). It is also prescribed in attention deficit hyperactivity disorder (see p. 345). Sibutramine, licensed as an anorectic agent, is a serotonin and noradrenaline/norepinephrine reuptake inhibitor (SNRI).
Pharmacokinetics
The antidepressants listed in Table 20.1 are generally well absorbed after oral administration. Steady-state plasma concentrations of TCAs show great individual variation but correlate with therapeutic effect. Where there is a failure of response, measurement of plasma concentration can be useful as the failure may be attributable to low plasma levels due to ultra-rapid metabolism (though it is often not available). Antidepressants in general are metabolised principally by hepatic cytochrome P450 enzymes. Of the many isoenzymes identified, the most important in antidepressant metabolism are CYP P450 2D6 (Table 20.2A) and CYP 3A4 (Table 20.2B). Other important P450 enzymes are CYP 1A2 (inhibited by the SSRI fluvoxamine, induced by cigarette smoking; substrates include caffeine and the atypical antipsychotics clozapine and olanzapine) and the CYP 2 C group (inhibition by fluvoxamine and fluoxetine, involved in breakdown of escitalopram and moclobemide). Sometimes several CYP enzymes are capable of mediating the same metabolic step. For example, at least six isoenzymes, including CYP 2D6, 3A4 and 2 C9, can mediate the desmethylation of the SSRI sertraline to its major metabolite.
Table 20.2A Psychotropic (and selected other) drugs known to be CYP 2D6 substrates, inhibitors and inducers
| CYP 2D6 inhibitors |
|---|
| Antidepressants |
| Paroxetine Fluoxetine |
| CYP 2D6 substrates | ||
|---|---|---|
| Antidepressants | Antipsychotics | Miscellaneous |
| Paroxetine Fluoxetine Citalopram Sertraline Venlafaxinea Duloxetine Amitriptyline Clomipramine Desipramine Imipramine Nortriptyline Reboxetine |
Chlorpromazine Haloperidol Zuclopenthixol Perphenazine Risperidone |
Dexfenfluramine Donepezil Opioids Codeine Hydrocodone Dihydrocodeine Tramadol Ethyl morphine MDMA (ecstasy) β-Blockers Propranolol Metoprolol Timolol Bufaralol Carvidelol |
A substrate is a substance that is acted upon and changed by an enzyme. Where two substrates of the same enzyme are prescribed together, they will compete and, if present in sufficient quantities, the metabolism of one or other, or both, drugs may also be inhibited, resulting in increased plasma concentration and possibly in enhanced therapeutic or adverse effects. An enzyme inducer accelerates the metabolism of co-prescribed drugs that are substrates of the same enzyme, reducing their effects. An enzyme inhibitor retards metabolism of co-prescribed drugs, increasing their effects.
a CYP 2D6 is involved only in the breakdown of venlafaxine to its active metabolite and therefore implications of 2D6 interactions for efficacy are of limited significance.
Table 20.2B Psychotropic (and selected other) drugs known to be CYP 3A4 substrates, inhibitors and inducers
| CYP 3A4 inhibitors | |
|---|---|
| Antidepressants | Other drugs |
| Fluoxetine Nefazodone |
Cimetidine Erythromycin Ketoconazole (grapefruit juice) |
| CYP 3A4 substrates | ||
|---|---|---|
| Antidepressants | Anxiolytics, hypnotics and antipsychotics | Miscellaneous |
| Fluoxetine Sertraline Amitriptyline Imipramine Nortriptyline Trazodonea |
Alpraxolam Aripiprazole Buspirone Diazepam Midazolam Triazolam Zoplicone Haloperidol Zuclopethixol Quetiapine Sertindole |
Buprenorphine Carbamazepine Cortisol Dexamethasone Methadone Testosterone Calcium channel blockers Diltiazem Nifedipine Amlodipine Other drugs Amiodarone Omeprazole Oral contraceptives Simvastatin |
| CYP 3A4 inducers | |
|---|---|
| Antidepressants | Miscellaneous |
| St John’s wort | Carbamazepine Phenobarbital Phenytoin |
A substrate is a substance that is acted upon and changed by an enzyme. Where two substrates of the same enzyme are prescribed together, they will compete and, if present in sufficient quantities, the metabolism of one or other, or both, drugs may also be inhibited, resulting in increased plasma concentration and possibly in enhanced therapeutic or adverse effects. An enzyme inducer accelerates the metabolism of co-prescribed drugs that are substrates of the same enzyme, reducing their effects. An enzyme inhibitor retards metabolism of co-prescribed drugs, increasing their effects.
a mCPP (meta-chlorophenylpiperazine), the active metabolite of trazodone, is a CYP 2D6 substrate; observe for unwanted effects when trazodone is co-administered with the 2D6 inhibitors fluoxetine or paroxetine.
Therapeutic efficacy
• Venlafaxine, in high dose (> 150 mg/day) and Escitalopram may have greater efficacy than other antidepressants.
• Amitriptyline appears to be slightly more effective than other TCAs and also SSRIs for severe depression, but this advantage is compromised by its poor tolerability and lack of safety in overdose relative to more modern agents.
• The older MAOIs (e.g. phenelzine) may be more effective than other classes in ‘atypical’ depression, a form of depressive illness where mood reactivity is preserved, lack of energy may be extreme and biological features are the opposite of the normal syndrome, i.e. excess sleep and appetite with weight gain.
Changing and stopping antidepressants
When changing between antidepressant doses, a conservative approach would be to reduce the first antidepressant progressively over 2 or more weeks before starting the new drug. The gradual reduction is particularly important with paroxetine and venlafaxine which are known to cause ‘discontinuation syndromes’ if stopped abruptly, and less important with fluoxetine due to its long half-life active metabolite which offers ‘built-in’ protection against withdrawal problems. A more proactive approach would involve ‘cross-tapering’ the second antidepressant – i.e. starting it while the first antidepressant is being reduced and gradually titrating the dose up. However, an important exception concerns changes to or from MAOIs, which must be handled with great caution due to the dangers of interactions between antidepressants (see below). Therefore MAOIs cannot safely be introduced within 2 weeks of stopping most antidepressants (3 weeks for imipramine and clomipramine; combination of the latter with tranylcypromine is particularly dangerous), and not until 5 weeks after stopping fluoxetine, due to its long half-life active metabolite. Similarly, other antidepressants should not be introduced until 2–3 weeks have elapsed from discontinuation of MAOI (as these are irreversible inhibitors; see p. 313). No washout period is required when using the reversible MAOI, moclobemide.
Augmentation
Another important augmentation strategy employs the mood stabiliser lithium carbonate. Controlled trials suggest that up to 50% of patients who have not responded to standard antidepressants can respond after lithium augmentation but the evidence is stronger for augmenting tricyclics than for augmenting SSRIs. Addition of lithium requires careful titration of the plasma concentration up to the therapeutic range, with periodic checks thereafter and monitoring for toxicity (see p. 331).
More recently, augmentation of SSRIs with atypical antipsychotics has been effective in clinical trials. Trial evidence is strongest using olanzapine, and also exists for quetiapine, risperidone and aripiprazole. Antipsychotics also have important potential for side-effects which must be taken into account before their introduction (see p. 322).
Other indications for antidepressants
Antidepressants may benefit most forms of anxiety disorder, including panic disorder, generalised anxiety disorder, post-traumatic stress disorder, obsessive–compulsive disorder and social phobia (see p. 331). SSRIs are recognised as first-line drug treatment for all five of these anxiety disorders.
Adverse effects
Interactions
Pharmacodynamic interactions
• Most antidepressants (including SSRIs and tricylics) may cause central nervous system (CNS) toxicity if co-prescribed with the dopaminergic drugs entacapone and selegiline (for Parkinson’s disease). SSRIs increase the risk of the serotonin syndrome when combined with drugs that enhance serotonin transmission, e.g. the antimigraine triptan drugs which are 5HT1-receptor antagonists, and the antiobesity drug sibutramine.
• Most antidepressants lower the convulsion threshold, complicate the drug control of epilepsy and lengthen seizure time in electroconvulsive therapy (ECT). The situation is made more complex by the capacity of carbamazepine to induce the metabolism of antidepressants and of certain antidepressants to inhibit carbamazepine metabolism (see below).
• SSRIs are known to interfere with platelet aggregation and may increase the risk of gastrointestinal bleeding, especially in those with existing risk factors.
• Trazodone and many tricyclics cause sedation and therefore co-prescription with other sedative agents such as opioid analgesics, H1-receptor antihistamines, anxiolytics, hypnotics and alcohol may lead to excessive drowsiness and daytime somnolence.
• The majority of tricyclics have undesirable cardiovascular effects, in particular prolongation of the QTc interval. Numerous other drugs also prolong the QTc interval, e.g. amiodarone, disopyramide, procainamide, propafenone, quinidine, terfenadine, and psychotropic agents such as pimozide and sertindole. Their use in combination with TCAs that prolong QTc enhances the risk of ventricular arrhythmias.
• Tricylics potentiate the effects of catecholamines and other sympathomimetics, but not those of β2-receptor agonists used in asthma. Even the small amounts of adrenaline/epinephrine or noradrenaline/norepinephrine in dental local anaesthetics may produce a serious rise in blood pressure.
Pharmacokinetic interactions
Metabolism by cytochrome P450 enzymes provides ample opportunity for interaction of antidepressants with other drugs by inhibition of, competition for, or induction of enzymes. Tables 20.2A and 20.2B indicate examples of mechanisms by which interaction that may occur when relevant drugs are added to, altered in dose or discontinued from regimens that include antidepressants.
In depressive psychosis, antidepressants are commonly prescribed with antipsychotics and there is potential for enhanced drug effects with paroxetine + perphenazine (CYP 2D6), fluoxetine + sertindole (3A4) and fluvoxamine + olanzapine (1A2). Rapid tranquillisation with zuclopenthixol acetate (see p. 325) of an agitated patient who is also taking fluoxetine or paroxetine can result in toxic plasma concentrations with excessive sedation and respiratory depression due to inhibition of zuclopenthixol metabolism by CYP 2D6 and CYP 3A4. P450 enzyme inhibition by fluoxetine or paroxetine may also augment effects of alcohol, tramadol (danger of serotonin syndrome) methadone, terfenadine (danger of cardiac arrhythmia), -caine anaesthetics and theophylline.
e.g. carbamazepine and several other antiepilepsy drugs, accelerate the metabolism of antidepressants, reducing their therapeutic efficacy and requiring adjustment of dose. Epilepsy is a particularly common co-morbid illness in patients who have both psychiatric illness and learning disabilities, and the combination of an anticonvulsant and an antidepressant or major psychotropic drug is to be anticipated. Depression and hypertension are both such common conditions that some co-morbidity is inevitable. Panic disorder (an indication for an antidepressant drug) is associated with hypertension.1 An antidepressant that is an enzyme inhibitor may exaggerate antihypertensive effects of metoprolol (metabolised by CYP 2D6), or diltiazem or amlodipine (3A4).
Monoamine oxidase inhibitors
Foods likely to produce hypertensive effects in patients taking MAOIs include:
• Cheese, especially if well matured.
• Red wines (especially Chianti) and some white wines; some beers (non- or low-alcohol varieties contain variable but generally low amounts of tyramine).
• Yeast extracts (Marmite, Oxo, Bovril).
• Broad bean pods (contain dopa, a precursor of adrenaline/epinephrine).
• Over-ripe bananas, avocados, figs.
• Fermented bean curds including soy sauce.
MAOI interactions with other drugs
MAOI–SSRI combinations may provoke the life-threatening ‘serotonin syndrome’ (see above). Strict rules apply regarding washout periods when switching between MAOIs and other drugs (see above, Changing antidepressants, p. 316). Very occasionally, MAOIs are co-prescribed with other antidepressants, but as many combinations are highly dangerous such practice should be reserved for specialists only and then as a last resort.
St John’s wort
The herbal remedy St John’s wort (Hypericum perforatum) has found favour in some patients with mild to moderate depression. The active ingredients in the hypericum extract have yet to be identified, and their mode of action is unclear. Several of the known mechanisms of action of existing antidepressants are postulated, including inhibition of monoamine reuptake and the MAO enzyme, as well as a stimulation of GABA receptors. Much of the original research into the efficacy of St John’s wort was performed in Germany, where its use is well established. Several direct comparisons with tricyclic antidepressants have shown equivalent rates of response, but the interpretation of these studies is complicated by the fact that many failed to use standardised ratings for depressive symptoms, patients tended to receive TCAs below the minimum therapeutic dose, and sometimes received St John’s wort in doses above the maximum recommended in commercially available preparations. Use of St John’s wort is further complicated by the lack of standardisation of the ingredients. A large multi-centre trial found only limited evidence of benefit for St John’s wort over placebo in significant major depression.2
Antipsychotics
Classification
Originally tested as an antihistamine, chlorpromazine serendipitously emerged as an effective treatment for psychotic illness in the 1950s. Chlorpromazine-like drugs were originally termed ‘neuroleptics’ or ‘major tranquillisers’, but the preferred usage now is ‘antipsychotics’. Classification is by chemical structure, e.g. phenothiazines, butyrophenones. Within the large phenothiazine group, compounds are divided into three types on the basis of the side-chain, as this tends to predict adverse effect profiles (Table 20.3). The continuing search for greater efficacy and better tolerability led researchers and clinicians to reinvestigate clozapine, a drug that was originally licensed in the 1960s but subsequently withdrawn because of toxic haematological effects. Clozapine appeared to offer greater effectiveness in treatment-resistant schizophrenia, to have efficacy against ‘negative’ in addition to ‘positive’ psychiatric symptoms (see Table 20.4), and to be less likely to cause extrapyramidal motor symptoms. It regained its licence in the early 1990s with strict requirements on dose titration and haematological monitoring. The renewed interest in clozapine and its unusual efficacy and tolerability stimulated researchers to examine other ‘atypical’ antipsychotic drugs.
| Atypical antipsychoticsa | Classical antipsychotics | |
|---|---|---|
| Clozapine | Phenothiazines | |
| Olanzapine | Type 1 | Chlorpromazine |
| Quetiapine | Promazine | |
| Risperidone | Type 2 | Pericyazine |
| Ziprasidone | Type 3 | Trifluoperazine |
| Amisulprideb | Prochlorperazine | |
| Zotepine | Fluphenazine |
