Chapter 4 Evaluation of drugs in humans
Experimental therapeutics
Modern medicine is sometimes accused of callous application of science to human problems and of subordinating the interest of the individual to those of the group (society).1 Official regulatory bodies rightly require scientific evaluation of drugs. Drug developers need to satisfy the official regulators and they also seek to persuade the medical profession to prescribe their products. Patients, too, are more aware of the comparative advantages and limitations of their medicines than they used to be. To some extent, this helps encourage patients to participate in trials so that future patients can benefit, as they do now, from the knowledge gained from such trials. An ethical framework is required to ensure that the interests of the individual participant take precedence over those of society (and, more obviously, those of an individual or corporate investigator).
Research involving human subjects
The definition of research continues to present difficulties. The distinction between medical research and innovative medical practice derives from the intent. In medical practice the sole intention is to benefit the individual patient consulting the clinician, not to gain knowledge of general benefit, though such knowledge may incidentally emerge from the clinical experience gained. In medical research the primary intention is to advance knowledge so that patients in general may benefit; the individual patient may or may not benefit directly.2
Consider also the process of audit, which is used extensively to assess performance, e.g. by individual health-care workers, by departments within hospitals or between hospitals. Audit is a systematic examination designed to determine the degree to which an action or set of actions achieves predetermined standards. Whereas research seeks to address ‘known unknowns’ and often discovers ‘unknown unknowns,3 audit is limited to the monitoring of ‘known knowns’: maybe important, but clearly limited.
Ethics of research in humans4
Some dislike the word ‘experiment’ in relation to humans, thinking that its mere use implies a degree of impropriety in what is done. It is better that all should recognise from the true meaning of the word, ‘to ascertain or establish by trial’,5 that the benefits of modern medicine derive almost wholly from experimentation and that some risk is inseparable from much medical advance.
The moral obligation of all doctors lies in ensuring that in their desire to help patients (the ethical principle of beneficence) they should never allow themselves to put the individual who has sought their aid at any disadvantage (the ethical principle of non-maleficence) for ‘the scientist or physician has no right to choose martyrs for society’.6
In principle, it may be thought proper to perform a therapeutic trial only when doctors (and patients) have genuine uncertainty as to which treatment is best.7 Not all trials are comparisons of different treatments. Some, especially early phase trials of new drugs, are comparisons of different doses. Comparisons of new with old should usually offer patients the chance of receiving current best treatment with one which might be better. Since this is often rather more than is offered in resource-constrained routine care, the obligatory patient information sheet mantra that ‘the decision whether to take part has no bearing on your usual care’ may be economical with the truth. But it is also simplistic to view the main purpose of all trials with medicines as comparative.
The ethics of the randomised and placebo-controlled trial
Providing that ethical surveillance is rooted in the ethical principles of justice,8 there should be no difficulty in clinical research adapting to current needs. And even if the nature of early phase research is changing, the randomised controlled trial will remain the cornerstone of how cause and effect is proven in clinical practice, and how drugs demonstrate the required degree of efficacy and safety to obtain a licence for their prescription.
The use of a placebo (or dummy) raises both ethical and scientific issues (see placebo medicines and the placebo effect, Ch. 2). There are clear-cut cases when placebo use would be ethically unacceptable and scientifically unnecessary, e.g. drug trials in epilepsy and tuberculosis, when the control groups comprise patients receiving the best available therapy.
The pharmacologically inert (placebo) treatment arm of a trial is useful:
• To distinguish the pharmacodynamic effects of a drug from the psychological effects of the act of medication and the circumstances surrounding it, e.g. increased interest by the doctor, more frequent visits, for these latter may have their placebo effect. Placebo responses have been reported in 30–50% of patients with depression and in 30–80% with chronic stable angina pectoris.
• To distinguish drug effects from natural fluctuations in disease that occur with time, e.g. with asthma or hay fever, and other external factors, provided active treatment, if any, can be ethically withheld. This is also called the ‘assay sensitivity’ of the trial.
• To avoid false conclusions. The use of placebos is valuable in Phase 1 healthy volunteer studies of novel drugs to help determine whether minor but frequently reported adverse events are drug related or not. Although a placebo treatment can pose ethical problems, it is often preferable to the continued use of treatments of unproven efficacy or safety. The ethical dilemma of subjects suffering as a result of receiving a placebo (or ineffective drug) can be overcome by designing clinical trials that provide mechanisms to allow them to be withdrawn (‘escape’) when defined criteria are reached, e.g. blood pressure above levels that represent treatment failure. Similarly, placebo (or new drug) can be added against a background of established therapy; this is called the ‘add on’ design.
• To provide a result using fewer research subjects. The difference in response when a test drug is compared with a placebo is likely to be greater than that when a test drug is compared with the best current, i.e. active, therapy (see later).
• The severity of the disease.
• The effectiveness of standard therapy.
• Whether the novel drug under test aims to give only symptomatic relief, or has the potential to prevent or slow up an irreversible event, e.g. stroke or myocardial infarction.
• The objective of the trial (equivalence, superiority or non-inferiority; see p. 45). Thus it may be quite ethical to compare a novel analgesic against placebo for 2 weeks in the treatment of osteoarthritis of the hip (with escape analgesics available). It would not be ethical to use a placebo alone as comparator in a 6-month trial of a novel drug in active rheumatoid arthritis, even with escape analgesia.
The precise use of the placebo will depend on the study design, e.g. whether crossover, when all patients receive placebo at some point in the trial, or parallel group, when only one cohort receives placebo. Generally, patients easily understand the concept of distinguishing between the imagined effects of treatment and those due to a direct action on the body. Provided research subjects are properly informed and give consent freely, they are not the subject of deception in any ethical sense; but a patient given a placebo in the absence of consent is deceived and research ethics committees will, rightly, decline to agree to this. (See also: Lewis et al (2002) in Guide to further reading, at the end of this chapter.)
Injury to research subjects9
The question of compensation for accidental (physical) injury due to participation in research is a vexed one. Plainly there are substantial differences between the position of healthy volunteers (whether or not they are paid) and that of patients who may benefit and, in some cases, who may be prepared to accept even serious risk for the chance of gain. There is no simple answer. But the topic must always be addressed in any research carrying risk, including the risk of withholding known effective treatment. The CIOMS/WHO Guidelines4 state:
Payment of subjects in clinical trials
There is an intuitive abreaction by physicians to pay patients (compared with healthy volunteers), because they feel the accusation of inducement or persuasion could be levelled at them, and because they assuage any feeling of taking advantage of the doctor–patient relationship by the hope that the medicines under test may be of benefit to the individual. This is not an entirely comfortable position.10
Rational introduction of a new drug to humans
Phases of clinical development
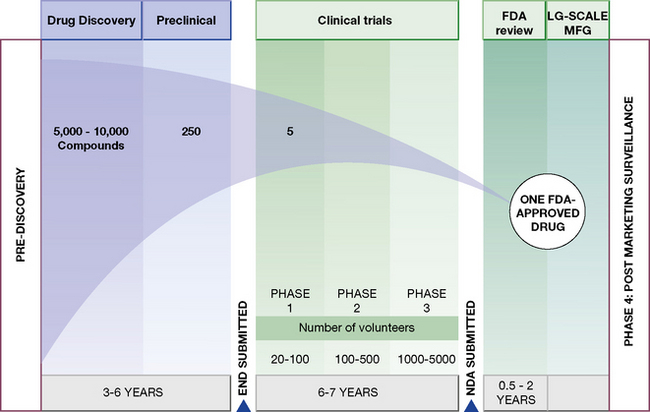
Human experiments progress in a commonsense manner that is conventionally divided into four phases (Fig. 4.1). These phases are divisions of convenience in what is a continuous expanding process. It begins with a small number of subjects (healthy subjects and volunteer patients) closely observed in laboratory settings, and proceeds through hundreds of patients, to thousands before the drug is agreed to be a medicine by a national or international regulatory authority. It is then licensed for general prescribing (though this is by no means the end of the evaluation). The process may be abandoned at any stage for a variety of reasons, including poor tolerability or safety, inadequate efficacy and commercial pressures. The phases are:
• Phase 1. Human pharmacology (20–50 subjects):



• Phase 2. Therapeutic exploration (50–300 subjects):

 pharmacokinetics and pharmacodynamic dose ranging, in carefully controlled studies for efficacy and safety,11 which may involve comparison with placebo.
pharmacokinetics and pharmacodynamic dose ranging, in carefully controlled studies for efficacy and safety,11 which may involve comparison with placebo.• Phase 3. Therapeutic confirmation (randomised controlled trials; 250–1000 + subjects):


• Phase 4. Therapeutic use (pharmacovigilance, post-licensing studies) (2000–10 000 + subjects):

Official regulatory guidelines and requirements12
For studies in humans (see also Ch. 6) these ordinarily include:
• Studies of pharmacokinetics and bioavailability and, in the case of generics, bioequivalence (equal bioavailability) with respect to the reference product.
• Therapeutic trials (reported in detail) that substantiate the safety and efficacy of the drug under likely conditions of use, e.g. a drug for long-term use in a common condition will require a total of at least 1000 patients (preferably more), depending on the therapeutic class, of which (for chronic diseases) at least 100 have been treated continuously for about 1 year.
• Special groups. If the drug will be used in, for example, the elderly or children, then these populations should be studied. New drugs are not normally studied in pregnant women. Studies in patients having disease that affects drug metabolism and elimination may be needed, such as patients with impaired liver or kidney function.
• Fixed-dose combination products will require explicit justification for each component.
• Interaction studies with other drugs likely to be taken simultaneously. Plainly, all possible combinations cannot be evaluated; a rational choice, based on knowledge of pharmacodynamics and pharmacokinetics, is made.
• The application for a licence for general use (marketing application) should include a draft Summary of Product Characteristics for prescribers. A Patient Information Leaflet must be submitted. These should include information on the form of the product (e.g. tablet, capsule, sustained-release, liquid), its uses, dosage (adults, children, elderly where appropriate), contraindications, warnings and precautions (less strong), side-effects/adverse reactions, overdose and how to treat it.
The emerging discipline of pharmacogenomics seeks to identify patients who will respond beneficially or adversely to a new drug by defining certain genotypic profiles. Individualised dosing regimens may be evolved as a result (see p. 101). This tailoring of drugs to individuals is consuming huge resources from drug developers but has yet to establish a place in routine drug development.
Therapeutic investigations
• The therapeutic effect itself (sleep, eradication of infection), i.e. the outcome.
• A surrogate effect, a short-term effect that can be reliably correlated with long-term therapeutic benefit, e.g. blood lipids or glucose or blood pressure. A surrogate endpoint might also be a pharmacokinetic parameter, if it is indicative of the therapeutic effect, e.g. plasma concentration of an antiepileptic drug.
Therapeutic evaluation
The aims of therapeutic evaluation are three-fold:
1. To assess the efficacy, safety and quality of new drugs to meet unmet clinical needs.
2. To expand the indications for the use of current drugs (or generic drugs13) in clinical and marketing terms.
3. To protect public health over the lifetime of a given drug.
The process of therapeutic evaluation may be divided into pre- and post-registration phases (Table 4.1), the purposes of which are set out below.

If the drug is found useful in these trials, it becomes desirable next to find out how closely the ideal may be approached in the rough and tumble of routine medical practice: in patients of all ages, at all stages of disease, with complications, taking other drugs and relatively unsupervised. Interest continues in all patients from the moment they are entered into the trial and it is maintained if they fail to complete, or even to start, the treatment; the need is to know the outcome in all patients deemed suitable for therapy, not only in those who successfully complete therapy.14
Need for statistics
In order truly to know whether patients treated in one way are benefited more than those treated in another, it is essential to use numbers. Statistics has been defined as ‘a body of methods for making wise decisions in the face of uncertainty’.15 Used properly, they are tools of great value for promoting efficient therapy. More than 100 years ago Francis Galton saw this clearly:
The human mind is … a most imperfect apparatus for the elaboration of general ideas … In our general impressions far too great weight is attached to what is marvellous … Experience warns us against it, and the scientific man takes care to base his conclusions upon actual numbers … to devise tests by which the value of beliefs may be ascertained.16
Concepts and terms
Hypothesis of no difference
Confidence intervals
The problem with the P value is that it conveys no information on the amount of the differences observed or on the range of possible differences between treatments. A result that a drug produces a uniform 2% reduction in heart rate may well be statistically significant but it is clinically meaningless. What doctors are interested to know is the size of the difference, and what degree of assurance (confidence) they may have in the precision (reproducibility) of this estimate. To obtain this it is necessary to calculate a confidence interval (see Figs 4.2 and 4.3).18
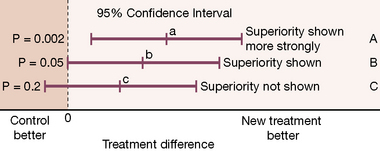
A confidence interval expresses a range of values that contains the true value with 95% (or other chosen percentage) certainty. The range may be broad, indicating uncertainty, or narrow, indicating (relative) certainty. A wide confidence interval occurs when numbers are small or differences observed are variable and points to a lack of information, whether the difference is statistically significant or not; it is a warning against placing much weight on (or confidence in) the results of small or variable studies. Confidence intervals are extremely helpful in interpretation, particularly of small studies, as they show the degree of uncertainty related to a result. Their use in conjunction with non-significant results may be especially enlightening.19
Types of therapeutic trial
a carefully, and ethically, designed experiment with the aim of answering some precisely framed question. In its most rigorous form it demands equivalent groups of patients concurrently treated in different ways or in randomised sequential order in crossover designs. These groups are constructed by the random allocation of patients to one or other treatment … In principle the method has application with any disease and any treatment. It may also be applied on any scale; it does not necessarily demand large numbers of patients.21
Other factors to consider when designing or critically appraising a trial are:
• The characteristics of the patients.
• The general applicability of the results.
• Whether a treatment is effective.
• The magnitude of that effect (compared with other remedies – or doses, or placebo).
• The types of patients in whom it is effective.
• The best method of applying the treatment (how often, and in what dosage if it is a drug).
Dose–response trials
• Confirmation of efficacy (hence a therapeutic trial).
• Investigation of the shape and location of the dose–response curve.
• The estimation of an appropriate starting dose.
• The identification of optimal strategies for individual dose adjustments.
• The determination of a maximal dose beyond which additional benefit is unlikely to occur.
Superiority, equivalence and non-inferiority in clinical trials
In some cases the purpose of a comparison is to show not necessarily superiority, but either equivalence or non-inferiority. Such trials avoid the use of placebo, explore possible advantages of safety, dosing convenience and cost, and present an alternative or ‘second-line’ therapy. Examples of a possible outcome in a ‘head to head’ comparison of two active treatments appear in Figure 4.2.
Design of trials
Techniques to avoid bias
The two most important techniques are:
Randomisation
Randomisation may be accomplished in simple or more complex ways, such as:
• Sequential assignments of treatments (or sequences in crossover trials).
• Randomising subjects in blocks. This helps to increase comparability of the treatment groups when subject characteristics change over time or there is a change in recruitment policy. It also gives a better guarantee that the treatment groups will be of nearly equal size.
• By dynamic allocation, in which treatment allocation is influenced by the current balance of allocated treatments.23
Blinding
The fact that both doctors and patients are subject to bias due to their beliefs and feelings has led to the invention of the double-blind technique, which is a control device to prevent bias from influencing results. On the one hand, it rules out the effects of hopes and anxieties of the patient by giving both the drug under investigation and a placebo (dummy) of identical appearance in such a way that the subject (the first ‘blind’ person) does not know which he or she is receiving. On the other hand, it also rules out the influence of preconceived hopes of, and unconscious communication by, the investigator or observer by keeping him or her (the second ‘blind’ person) ignorant of whether he or she is prescribing a placebo or an active drug. At the same time, the technique provides another control, a means of comparison with the magnitude of placebo effects. The device is both philosophically and practically sound.24
Some common design configurations
N-of-1 trials
Patients give varied treatment responses and the average effect derived from a population sample may not be helpful in expressing the size of benefit or harm for an individual. In the future, pharmacogenomics may provide an answer, but in the meantime the best way to settle doubt as to whether a test drug is effective for an individual patient is the N-of-1 trial. This is a crossover design in which each patient receives two or more administrations of drug or placebo in random manner; the results from individuals can then be displayed. Two conditions apply. First, the disease in which the drug is being tested must be chronic and stable. Second, the treatment effect must wear off rapidly. N-of-1 trials are not used routinely in drug development and, if so, only at the Phase 3 stage.25,26
Size of trials
1. The magnitude of the difference sought or expected on the primary efficacy endpoint (the target difference). For between-group studies, the focus of interest is the mean difference that constitutes a clinically significant effect.
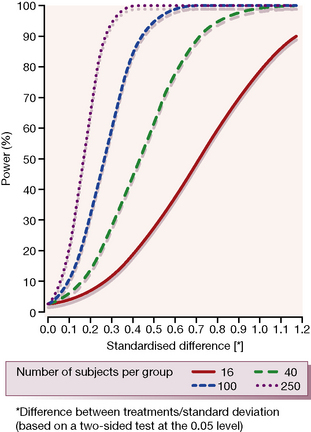
2. The variability of the measurement of the primary endpoint as reflected by the standard deviation of this primary outcome measure. The magnitude of the expected difference (above) divided by the standard deviation of the difference gives the standardised difference (Fig. 4.3).
3. The defined significance level, i.e. the level of chance for accepting a Type I (α) error. Levels of 0.05 (5%) and 0.01 (1%) are common targets.
4. The power or desired probability of detecting the required mean treatment difference, i.e. the level of chance for accepting a Type II (β) error. For most controlled trials, a power of 80–90% (0.8–0.9) is frequently chosen as adequate, although higher power is chosen for some studies.
It will be intuitively obvious that a small difference in the effect that can be detected between two treatment groups, or a large variability in the measurement of the primary endpoint, or a high significance level (low P value) or a large power requirement, all act to increase the required sample size. Figure 4.3 gives a graphical representation of how the power of a clinical trial relates to values of clinically relevant standardised difference for varying numbers of trial subjects (shown by the individual curves). It is clear that the larger the number of subjects in a trial, the smaller is the difference that can be detected for any given power value.
Fixed sample size and sequential designs
An alternative (or addition) to repeating the fixed sample size trial is to use a sequential design in which the trial is run until a useful result is reached.27 These adaptive designs, in which decisions are taken on the basis of results to date, can assess results on a continuous basis as data for each subject become available or, more commonly, on groups of subjects (group sequential design). The essential feature of these designs is that the trial is terminated when a predetermined result is attained and not when the investigator looking at the results thinks it appropriate. Reviewing results in a continuous or interim basis requires formal interim analysis and there are specific statistical methods for handling the data, which need to be agreed in advance. Group sequential designs are especially successful in large long-term trials of mortality or major non-fatal endpoints when safety must be monitored closely.
Meta-analysis
The collecting together of a number of trials with the same objective in a systematic review28and analysing the accumulated results using appropriate statistical methods is termed meta-analysis. The principles of a meta-analysis are that:
• It should be comprehensive, i.e. include data from all trials, published and unpublished.
• Only randomised controlled trials should be analysed, with patients entered on the basis of ‘intention to treat’.29
• The results should be determined using clearly defined, disease-specific endpoints (this may involve a re-analysis of original trials).
• An effect of reasonable size ought to be demonstrable in a single trial.
• Different study designs cannot be pooled.
• Lack of accessibility of all relevant studies.
• Publication bias (‘positive’ trials are more likely to be published).
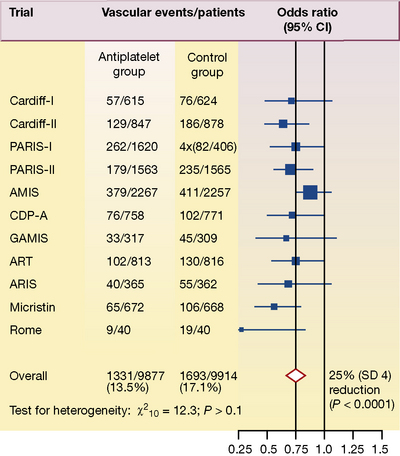
Figure 4.4 shows detailed results from 11 trials in which antiplatelet therapy after myocardial infarction was compared with a control group. The number of vascular events per treatment group is shown in the second and third columns, and the odds ratios with the point estimates (the value most likely to have resulted from the study) are represented by black squares and their 95% confidence intervals (CI) in the fourth column.
Results: implementation
Relative and absolute risk
To make clinical decisions, readers of therapeutic studies need to know: how many patients must be treated30 (and for how long) to obtain one desired result (number needed to treat). This is the inverse (or reciprocal) of absolute risk reduction.
Relative risk reductions can remain high (and thus make treatments seem attractive) even when susceptibility to the events being prevented is low (and the corresponding numbers needed to be treated are large). As a result, restricting the reporting of efficacy to just relative risk reductions can lead to great – and at times excessive – zeal in decisions about treatment for patients with low susceptibilities.31
In secondary prevention of myocardial infarction, 50 patients need to be treated for 2 years, while in primary prevention 200 patients need to be treated for 5 years, for one non-fatal myocardial infarction to be prevented. In other words, it takes 100 patient-years of treatment in primary prevention to produce the same beneficial outcome of one fewer non-fatal myocardial infarction.32
Pharmacoepidemiology
Pharmacoepidemiology is the study of the use and effects of drugs in large numbers of people. Some of the principles of pharmacoepidemiology are used to gain further insight into the efficacy, and especially the safety, of new drugs once they have passed from limited exposure in controlled therapeutic pre-registration trials to the looser conditions of their use in the community. Trials in this setting are described as observational because the groups to be compared are assembled from subjects who are, or who are not (the controls), taking the treatment in the ordinary way of medical care. These (Phase 4) trials are subject to greater risk of selection bias33 and confounding34 than experimental studies (randomised controlled trials) where entry and allocation of treatment are strictly controlled (increasing internal validity). Observational studies, nevertheless, come into their own when sufficiently large randomised trials are logistically and financially impracticable. The following approaches are used.
Observational cohort35 studies
Investigation of the question of thromboembolism and the combined oestrogen–progestogen contraceptive pill by means of an observational cohort study required enormous numbers of subjects36 (the adverse effect is, fortunately, uncommon) followed over years. An investigation into cancer and the contraceptive pill by an observational cohort would require follow-up for 10–15 years. Happily, epidemiologists have devised a partial alternative: the case–control study.
Case–control studies
This reverses the direction of scientific logic from a forward-looking, ‘what happens next’ (prospective) to a backward-looking, ‘what has happened in the past’ (retrospective)37 investigation. The case–control study requires a definite hypothesis or suspicion of causality, such as an adverse reaction to a drug. The investigator assembles a group of patients who have the condition. A control group of people who have not had the reaction is then assembled (matched, e.g. for sex, age, smoking habits) from hospital admissions for other reasons, primary care records or electoral rolls. A complete drug history is taken from each group, i.e. the two groups are ‘followed up’ backwards to determine the proportion in each group that has taken the suspect agent. Case–control studies do not prove causation.38 They reveal associations and it is up to investigators and critical readers to decide the most plausible explanation.
Surveillance systems: pharmacovigilance
Four kinds of logic can be applied to drug safety monitoring:
• To gain experience from regular reporting of suspected adverse drug reactions from health professionals during the regular clinical use of the drug.
• To attempt to follow a complete cohort of (new) drug users for as long as it is deemed necessary to have adequate information.
• To perform special studies in areas which may be predicted to give useful information.
Voluntary reporting
Prescription event monitoring
This is a form of observational cohort study. Prescriptions for a drug (say, 20 000) are collected (in the UK this is made practicable by the existence of a National Health Service in which prescriptions are sent to a single central authority for pricing and payment of the pharmacist). The prescriber is sent a questionnaire and asked to report all events that have occurred (not only suspected adverse reactions) with no judgement regarding causality. Thus ‘a broken leg is an event. If more fractures were associated with this drug they could have been due to hypotension, CNS effects or metabolic disease’.40 By linking general practice and hospital records and death certificates, both prospective and retrospective studies can be done and unsuspected effects detected. Prescription event monitoring can be used routinely on newly licensed drugs, especially those likely to be widely prescribed in general practice, and it can also be implemented quickly in response to a suspicion raised, e.g. by spontaneous reports.
Strength of evidence
A number of types of clinical investigation are described in this chapter, and elsewhere in the book. When making clinical decisions about a course of therapeutic action, it is obviously relevant to judge the strength of evidence generated by different types of study. This has been summarised as follows, in rank order:41
1. Systematic reviews and meta-analyses.
2. Randomised controlled trials with definitive results (confidence intervals that do not overlap the threshold of the clinically significant effect).42
3. Randomised controlled trials with non-definitive results (a difference that suggests a clinically significant effect but with confidence intervals overlapping the threshold of this effect).
Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin. Pharmacol. Ther.. 2001;69(3):89–95.
Bland J.M., Altman D.G. Statistical notes: the odds ratio. Br. Med. J.. 2000;320:1468.
Bracken M.B. Why animal studies are often poor indicators of human reactions to exposure. J. R. Soc. Med.. 2008;101:120–122.
Chatellier G., Zapletal E., Lemaitre D., et al. The number needed to treat: a clinically useful nomogram in its proper context. Br. Med. J.. 1996;312:426–429.
Doll R. Controlled trials: the 1948 watershed. Br. Med. J.. 1998;317:1217–1220. (and following articles)
Egger M., Smith G.D., Phillips A.N. Meta-analysis: principles and procedures. Br. Med. J. 1997;315:1533–1537. (see also other articles in the series entitled ‘Meta-analysis’)
Emanuel E.J., Miller F.G. The ethics of placebo-controlled trials – a middle ground. N. Engl. J. Med.. 2001;345:915–919.
Garattini S., Chalmers I. Patients and the public deserve big changes in the evaluation of drugs. Br. Med. J.. 2009;338:804–806.
GRADE Working Group. GRADE: what is ‘quality of evidence’ and why is it important to clinicians?. Br. Med. J.. 2008;336:924–929. (and the other papers of this series)
Greenhalgh T. Papers that report drug trials. Br. Med. J. 1997;315:480–483. (see also other articles in the series entitled ‘How to read a paper’)
Kaptchuk T.J. Powerful placebo: the dark side of the randomised controlled trial. Lancet. 1998;351:1722–1725.
Khan K.S., Kunz R., Kleijnen J., Antes G. Five steps to conducting a systematic review. J. R. Soc. Med.. 2003;96:118–121.
Lewis J.A., Jonsson B., Kreutz G., et al. Placebo-controlled trials and the Declaration of Helsinki. Lancet. 2002;359:1337–1340.
Miller F.G., Rosenstein D.L. The therapeutic orientation to clinical trials. N. Engl. J. Med.. 2003;348:1383–1386.
Rochon P.A., Gurwitx J.H., Sykora K., et al. Reader’s guide to critical appraisal of cohort studies: 1. Role and design. Br. Med. J.. 2005;330:895–897.
Rothwell P.M. External validity of randomised controlled trials: ‘to whom do the results of this trial apply? Lancet. 2005;365:82–93.
Rothwell P.M. Treating individuals 2. Subgroup analysis in randomised controlled trials: importance, indications, and interpretation. Lancet. 2005;365:176–186.
Sackett D., Rosenberg W., Gray J., et al. Evidence based medicine: what it is and what it isn’t [editorial]. Can. Med. Assoc. J.. 2009;312:1–8.
Silverman W.A., Altman D.G. Patients’ preferences and randomised trials. Lancet. 1996;347:171–174.
Vlahakes G.J. Editorial. The value of phase 4 clinical testing. N. Engl. J. Med. 2006;354(4):413–415.
Waller P.C., Jackson P.R., Tucker G.T., Ramsay L.E. Clinical pharmacology with confidence [intervals]. Br. J. Clin. Pharmacol.. 1994;37(4):309.
Williams R.L., Chen M.L., Hauck W.W. Equivalence approaches. Clin. Pharmacol. Ther.. 2002;72:229–237.
Zwarenstein M., Treweek S., Gagnier J.J., et al. CONSORT group: Pragmatic Trials in Healthcare (Practihc) group. Improving the reporting of pragmatic trials: an extension of the CONSORT statement. Br. Med. J. 2008;337:a2390.
1 Guidance to researchers in this matter is clear. The World Medical Association Declaration of Helsinki (Edinburgh revision 2000) states that ‘considerations related to the well-being of the human subject should take precedence over the interests of science and society’. The General Assembly of the United Nations adopted in 1966 the International Covenant on Civil and Political Rights, of which Article 7 states, ‘In particular, no one shall be subjected without his free consent to medical or scientific experimentation’. This means that subjects are entitled to know that they are being entered into research even though the research be thought ‘harmless’. But there are people who cannot give (informed) consent, e.g. the demented. The need for special procedures for such is now recognised, for there is a consensus that, without research, they and the diseases from which they suffer will become therapeutic ‘orphans’.
2 Report: Royal College of Physicians of London 1996 Guidelines on the Practice of Ethics Committees in Medical Research Involving Human Subjects. Royal College of Physicians, London.
3 American Defence Secretary Donald Rumsfeld, on 12 February 2002, at a press briefing where he addressed the absence of evidence linking the government of Iraq with the supply of weapons of mass destruction to terrorist groups.
4 For extensive practical detail, see Council for International Organisations of Medical Sciences (CIOMS) in collaboration with the World Health Organization (WHO) 2002 International Ethical Guidelines for Biomedical Research Involving Human Subjects. CIOMS, Geneva. (WHO publications are available in all UN member countries.) See also: Guideline for Good Clinical Practice, International Conference on Harmonisation Tripartite Guideline. EU Committee on Proprietary Medicinal Products (CPMP/ICH/135/95). Smith T 1999 Ethics in Medical Research: A Handbook of Good Practice. Cambridge University Press, Cambridge.
5 Oxford English Dictionary. See also: Edwards M 2004 Historical keywords: Tria. Lancet 364:1659.
6 Kety S. Quoted by Beecher H K 1959 Journal of the American Medical Association 169:461.
7 This is the uncertainty principle: the concept that patients entering a randomised therapeutic trial will have equal potential for benefit and risk is referred to as equipoise.
8 The ‘four principles’ approach (above) is widely utilised in biomedical ethics. A full description and an analysis of the contribution of this and other ethical theories to decision-making in clinical, including research, practice can be found in: Beauchamp T L, Childress J F 2001 Principles of Biomedical Ethics, 5th edn. Oxford University Press, Oxford.
9 Injury to participants in clinical trials is uncommon and serious injury is rare. In March 2006, eight healthy young men entered a trial of a humanised monoclonal antibody designed to be an agonist of a particular receptor on T lymphocytes that stimulates their production and activation. This was the first administration to humans; preclinical testing in rabbits and monkeys at doses up to 500 times those received by the volunteers apparently showed no ill effect. Six of the volunteers quickly became seriously ill and required admission to an intensive care facility with multi-organ failure due to a ‘cytokine release syndrome’, in effect a massive attack on the body’s own tissues. All the volunteers recovered but some with disability. This toxicity in humans, despite apparent safety in animals, may be due to the specifically humanised nature of the monoclonal antibody. Testing of perceived high-risk new medicines is likely to be subject to particularly stringent regulation in future. See Wood A J J, Darbyshire J 2006 Injury to research volunteers – the clinical research nightmare. New England Journal of Medicine 354:1869–1871.
10 Freedman B 1987 Equipoise and the ethics of clinical research. New England Journal of Medicine 317:141–145.
11 Moderate to severe adverse events have occurred in about 0.5% of healthy subjects. See Orme M, Harry J, Routledge P, Hobson S 1989 British Journal of Clinical Pharmacology 27:125; Sibille M et al 1992 European Journal of Clinical Pharmacology 42:393.
12 Guidelines for the conduct and analysis of a range of clinical trials in different therapeutic categories are released from time to time by the Committee on Medicinal Products for Human Use (CHMP) of the European Commission. These guidelines apply to drug development in the European Union. Other regulatory authorities issue guidance, e.g. the Food and Drug Administration in the USA, the Ministry of Health, Labour and Welfare in Japan. There has been considerable success in aligning different guidelines across the world through the International Conferences on Harmonisation (ICH). The source for CHMP guidelines is info@mhra.gsi.gov.uk
13 A drug for which the original patent has expired, so that any pharmaceutical company may market it in competition with the inventor. The term ‘generic’ has come to be synonymous with the non-proprietary or approved name (see Ch. 7).
14 Information on both categories (method effectiveness and use effectiveness) is valuable (Sheiner L B, Rubin D B 1995 Intention-to-treat analysis and the goals of clinical trials. Clinical Pharmacology and Therapeutics 57(1):6–15).
15 Wallis W A, Roberts H V 1957 Statistics, A New Approach. Methuen, London.
16 Galton F 1879 Generic images. Proceedings of the Royal Institution.
17 Altman D G, Gore S M, Gardner M J, Pocock S J 1983 Statistical guidelines for contributors to medical journals. British Medical Journal 286:1489–1493.
18 Gardner M J, Altman D G 1986 Confidence intervals rather than P values: estimation rather than hypothesis testing. British Medical Journal 292:746–750.
19 Altman D G, Gore S M, Gardner M J, Pocock S J 1983 Statistical guidelines for contributors to medical journals. British Medical Journal 286:1489–1493.
20 The target difference. Differences in trial outcomes fall into three grades: (1) that the doctor will ignore, (2) that will make the doctor wonder what to do (more research needed), and (3) that will make the doctor act, i.e. change prescribing practice.
21 Bradford Hill A 1977 Principles of Medical Statistics. Hodder and Stoughton, London. If there is a ‘father’ of the modern scientific therapeutic trial, it is he.
22 Particularly in large-scale outcome trials, an independent data monitoring committee is given access to the results as these are accumulated; the committee is empowered to discontinue a trial if the results show significant advantage or disadvantage to one or other treatment.
23 Note also patient preference trials. Conventionally, patients are invited to participate in a clinical trial, give consent and are then randomised to a particular treatment group. In special circumstances, randomisation takes place first, the patients are informed of the treatment to be offered and are allowed to opt for this or another treatment. This is called pre-consent randomisation or ‘pre-randomisation’. In a trial of simple mastectomy versus lumpectomy with or without radiotherapy for early breast cancer, recruitment was slow because of the disfiguring nature of the mastectomy option. A policy of pre-randomisation was then adopted, letting women know the group to which they would be allocated should they consent. Recruitment increased sixfold and the trial was completed, providing sound evidence that survival was as long with the less disfiguring option (Fisher B, Bauer M, Margolese R et al 1985 Five-year results of a randomised clinical trial comparing total mastectomy and segmental mastectomy with and without radiotherapy in the treatment of breast cancer. New England Journal of Medicine 312:665–673). However, the benefit of enhanced recruitment may be limited by potential for introducing bias.
24 Modell W, Houde R W 1958 Factors influencing clinical evaluation of drugs; with special reference to the double-blind technique. Journal of the American Medical Association 167:2190–2199.
25 Senn S 1997 N-of-1 Trials: Statistical Issues in Drug Development. John Wiley, Chichester, pp. 249–255.
26 Jull A, Bennet D 2005 Do N-of-1 trials really tailor treatment? Lancet 365:1992–1994.
27 Whitehead J 1992 The Design Analysis of Sequential Clinical Trials, 2nd edn. Ellis Horwood, Chester.
28 A review that strives comprehensively to identify and synthesise all the literature on a given subject (sometimes called an overview). The unit of analysis is the primary study, and the same scientific principles and rigour apply as for any study. If a review does not state clearly whether and how all relevant studies were identified and synthesised, it is not a systematic review (Cochrane Library 1998).
29 Reports of therapeutic trials should contain an analysis of all patients entered, regardless of whether they dropped out or failed to complete, or even started the treatment for any reason. Omission of these subjects can lead to serious bias (Laurence D R, Carpenter J 1998 A Dictionary of Pharmacological and Allied Topics. Elsevier, Amsterdam).
30 See Cooke R J, Sackett D L 1995 The number needed to treat: a clinically useful treatment effect. British Medical Journal 310:452.
31 Sackett D L, Cooke R J 1994 Understanding clinical trials: what measures of efficacy should journal articles provide busy clinicians? British Medical Journal 309:755.
32 For example, drug therapy for high blood pressure carries risks, but the risks of the disease vary enormously according to severity of disease: ‘Depending on the initial absolute risk, the benefits of lowering blood pressure range from preventing one cardiovascular event a year for about every 20 people treated, to preventing one event for about every 5000–10 000 people treated. The level of risk at which treatment should be started is debatable’ (Jackson R, Barham P, Bills J et al 1993 Management of raised blood pressure in New Zealand: a discussion document. British Medical Journal 307:107–110).
33 A systematic error in the selection or randomisation of patients on admission to a trial such that they differ in prognosis, i.e. the outcome is weighted one way or another by the selection, not by the trial.
34 When the interpretation of an observed association between two variables may be affected by a strong influence from a third variable (which may be hidden or unknown). Examples of confounders would be concomitant drug therapy or differences in known risk factors, e.g. smoking, age, sex.
35 Used here for a group of people having a common attribute, e.g. they have all taken the same drug.
36 The Royal College of General Practitioners (UK) recruited 23 000 women takers of the pill and 23 000 controls in 1968 and issued a report in 1973. It found an approximately doubled incidence of venous thrombosis in combined-pill takers (the dose of oestrogen was reduced because of this study).
37 For this reason such studies have been named trohoc (cohort spelled backwards) studies (Feinstein A 1981 Journal of Chronic Diseases 34:375).
38 Experimental cohort studies (i.e. randomised controlled trials) are on firmer ground with regard to causation as there should be only one systematic difference between the groups (i.e. the treatment being studied). In case–control studies the groups may differ systematically in several ways.
39 Edwards I R 1998 A perspective on drug safety. In: Edwards I R (ed) Drug Safety. Adis International, Auckland, p. xii.
40 Inman W H W, Rawson N S B, Wilton L V 1986 Prescription-event monitoring. In: Inman W H W (ed) Monitoring for Drug Safety, 2nd edn. MTP, Lancaster, p. 217.
41 Guyatt G H, Sackett D L, Sinclair J C et al 1995 Users’ guides to the medical literature. IX. A method for grading health care recommendations. Evidence-Based Medicine Working Group. Journal of the American Medical Association 274:1800–1804.
42 The reporting of randomised controlled trials has been systemised so that only high-quality studies will be considered. See Moher D, Schulz K F, Altman D G 2001 CONSORT Group. The CONSORT statement: revised recommendations for improving the quality of reports of parallel group randomised trials. Lancet 357:1191–1194.
43 ‘Quick, let us prescribe this new drug while it remains effective’. Richard Asher.