Pseudohypoparathyroidism, Albright’s Hereditary Osteodystrophy, and Progressive Osseous HeteroplasiA
Disorders Caused by Inactivating GNAS Mutations
In 1942, Albright et al.1 described a group of patients who displayed certain physical features—including obesity, short stature, brachydactyly, and cognitive impairment—combined with hypocalcemia and hyperphosphatemia. In these patients, exogenous, biologically active parathyroid hormone (PTH), extracted from parathyroid glands, failed to result in a full phosphaturic response; hence, the term pseudohypoparathyroidism (PHP) was introduced, indicating that hypocalcemia and hyperphosphatemia in these patients resulted from target organ resistance to, rather than deficiency of, PTH. Consistent with resistance to the actions of this hormone, it was later shown that patients affected by PHP have elevated concentrations of immunoreactive PTH.2 Subsequently, it was shown that some patients affected by PHP have resistance toward additional hormones; however, PTH resistance is the most prominent feature of the disease.
The primary site of PTH resistance in PHP is the renal proximal tubule, as the actions of PTH in bone and in the distal tubule appear normal.3–5 Patients with PHP have reduced serum concentrations of 1,25-dihydroxyvitamin D3 [1,25(OH)2D3], which is the main cause of hypocalcemia.6–9 Low serum 1,25(OH)2D3 and hyperphosphatemia are the direct results of PTH resistance at the proximal tubule. Hyperphosphatemia typically is worsened by the elevation of PTH in the circulation and the absence of resistance to bone resorptive actions of this hormone; on the other hand, serum PTH increase can prevent symptomatic hypocalcemia in some PHP patients largely by its unimpaired actions on the bone and the renal distal tubule.10–14 However, at some point in their lives, most of these patients manifest hypocalcemia and therefore present with associated clinical findings.
Diagnosis, Progression, and Treatment of PTH Resistance
PTH exerts its actions by binding to a seven-transmembrane, G protein–coupled receptor (the PTH/PTH-related protein receptor, PTHR1).15 Although PTHR1 can couple to several different G proteins,16 most PTH actions are mediated primarily through the stimulatory G protein, which acts on adenylyl cyclases and thereby increases the formation of intracellular second messenger cyclic adenosine monophosphate (cAMP).15,17 PTH-induced cAMP formation is used as an important indicator of renal tubular PTH function, because most PHP patients display an inadequate or absent increase in urinary cAMP in response to exogenous, biologically active PTH (Fig. 10-1).18 In fact, the nephrogenous response to exogenously administered PTH is utilized as a test for establishing the diagnosis of PHP (Ellsworth-Howard test), although currently used high-sensitivity PTH assays often suffice to make the diagnosis when serum PTH is elevated in the presence of hypocalcemia and hyperphosphatemia. Nonetheless, depending on the nature of the nephrogenous response to exogenous PTH, PHP is subdivided into two main types. PHP type I is defined by blunted urinary excretion of both cAMP and phosphate, and PHP type II by blunted urinary excretion of phosphate only.1,18,19
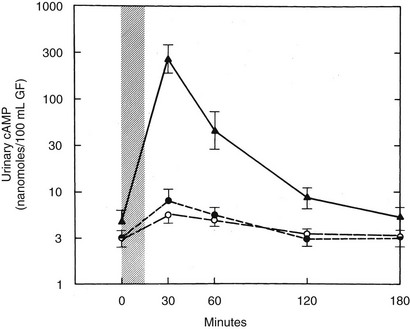
FIGURE 10-1 Urinary cyclic adenosine monophosphate (cAMP) excretion in response to an infusion of bovine parathyroid extract (300 USP units). The peak response in normal subjects (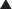 ) and those with pseudopseudohypoparathyroidism (PPHP) (not shown) is 50- to 100-fold times basal. Subjects with PHP type Ia (o) or PHP type Ib (•) show only a two- to fivefold increase. Urinary cAMP is expressed as nanomoles per 100 mL of glomerular filtrate (GF), UcAMP (nanomoles per 100 mL GF) = UcAMP (nanomoles/dL) × SCre (mg/dL)/UCre (mg/dL). (Data from Levine et al., 1986.65)
) and those with pseudopseudohypoparathyroidism (PPHP) (not shown) is 50- to 100-fold times basal. Subjects with PHP type Ia (o) or PHP type Ib (•) show only a two- to fivefold increase. Urinary cAMP is expressed as nanomoles per 100 mL of glomerular filtrate (GF), UcAMP (nanomoles per 100 mL GF) = UcAMP (nanomoles/dL) × SCre (mg/dL)/UCre (mg/dL). (Data from Levine et al., 1986.65)
Signs and symptoms of decreased serum calcium often reflect increased neuromuscular excitability. Although the most common manifestations of hypocalcemia include muscle tetany and spasms, findings vary markedly among patients. In more severe cases, patients present with seizures. Other neurologic symptoms can arise from hypocalcemia, and some patients with PHP initially have been diagnosed with movement disorders.20–24 In one report,25 two sisters with PHP Ib (see below) presented with paroxysmal kinesigenic choreoathetosis, and the diagnosis of PTH resistance in one sister was made through genetic testing and biochemical evaluation only after approximately 4 years of antiepileptic oral treatment.25 Some patients presenting with seizures demonstrate epileptiform activity on electroencephalogram (EEG), and, because this activity typically responds to antiepileptic treatment, the diagnosis of PHP can be delayed.26,27 As another complication of changes in serum calcium and phosphorus, brain imaging studies in PHP patients frequently show intracranial calcifications.20,28–34
PHP is a congenital disorder, and few reports describe findings consistent with PTH resistance during the neonatal period.35,36 However, clinical manifestations of hypocalcemia typically occur only later in childhood. PTH resistance and resultant changes in serum calcium and phosphorus develop gradually.37–40 In a longitudinal study of a child with PHP Ia (see below), cAMP response to PTH was found to be normal at age 3 months, although it was blunted at age 2.6 years.38 In another PHP Ia case, a gradual decline in serum calcium, preceded by increasing serum phosphorus and PTH levels, was demonstrated.37 In addition, a PHP Ib patient diagnosed by genetic analysis (see below) at birth was shown to have normal serum PTH levels until age 18 months, when an elevation in PTH was first detected despite normal serum calcium and phosphorus.39 It thus appears that PTH responses are intact during the early postnatal period despite the existence of the molecular defect underlying PHP. The mechanisms that allow normal PTH signaling during this developmental stage remain unknown.
The primary goal of treatment entails correction of abnormal serum biochemistries that result from PTH and, in some cases, other hormone resistance. Regarding hypocalcemia, treatment involves oral calcium supplements and 1,25(OH)2D (calcitriol) preparations. Note that the active form of vitamin D is required because of the lowered capacity of the proximal tubule to convert 25(OH)D into the biologically active 1,25(OH)2D. In addition, treatment for patients with PTH resistance should be aimed at keeping the serum PTH level within or close to the normal range, rather than simply avoiding symptomatic hypocalcemia, because persistent elevation of serum PTH will increase bone resorption and eventually may lead to hyperparathyroid bone disease, including osteitis fibrosa cystica.41 PTH actions in the distal tubule, which are not impaired, provide sufficient calcium reabsorption from the glomerular filtrate; therefore, urinary calcium levels are usually low, and affected individuals do not have an increased risk of developing kidney stones and nephrocalcinosis. In fact, during the course of treatment, urinary calcium elevation typically does not occur. Nevertheless, blood chemistries and urinary calcium excretion in patients undergoing treatment should be monitored annually, but more frequently during pubertal development and once skeletal growth is completed, as the requirements for treatment with calcium and 1,25(OH)2D may need adjustment.
Pseudohypoparathyroidism Type Ia
Among the two main PHP types, PHP type I is much more common. Clinical variants of PHP type I have been described, based on the presence or absence of clinical manifestations that coexist with PTH resistance, diminished stimulatory G protein activity in easily accessible cells, and imprinting abnormalities of the GNAS gene encoding the α subunit of the stimulatory G protein (Gsα) (Table 10-1).
Table 10-1
Clinical and Molecular Features of PHP Forms and Variants
*POH-like severe heterotopic-ossifications have been reported in some patients with homone resistance.
†Some recent studies have demonstrated co-existence of AHO features and GNAS imprinting defects.
As was described originally by Albright et al.,1 some PHP patients exhibit characteristic physical features that may include obesity, round facies, short stature, brachydactyly, ectopic ossification, and mental impairment (Fig. 10-2). These features are now termed Albright’s hereditary osteodystrophy (AHO), and PHP patients who present with these features are classified as having pseudohypoparathyroidism type Ia (PHP Ia). The brachydactyly observed in patients with AHO typically involves the metacarpal and/or metatarsal bones; thus, the pattern of shortening of hand bones differs from that seen in other disorders with brachydactyly, such as familial brachydactyly and Turner’s syndrome.42 Because of shortened metacarpals, dimpling over the knuckles of a clenched fist (Archibald sign) is often observed.43 Shortening of the distal phalanx of the thumb, however, is the most common skeletal abnormality (called “murderer’s” or “potter’s” thumb), and some patients have shortening of all digits.44 Mental impairment is mild, often presenting as cognitive defects. It is possible that the cause of mental impairment is the deficiency of Gsα signaling in the brain. Although hypocalcemia and/or hypothyroidism may play a role in this phenotype, correction of these biochemical defects does not prevent mental impairment in all cases. Remarkable patient-to-patient variability is seen in AHO, even among patients who carry the same genetic mutation and belong to the same family (see below for discussion of the underlying genetic defect). Some patients may exhibit a single AHO feature only, such as obesity; others may present with multiple different AHO features. Furthermore, the severity of each feature differs vastly among patients. In addition, individual AHO features are not unique to PHP, as they can be observed in other unrelated disorders. The variable expressivity and the lack of specificity of individual features can make the AHO diagnosis challenging. Although the coexistence of hormone resistance in PHP Ia patients is often helpful, it also can be misleading. This is particularly important in differential diagnosis of different PHP forms characterized by the presence of AHO features alone or hormone resistance alone (see below).
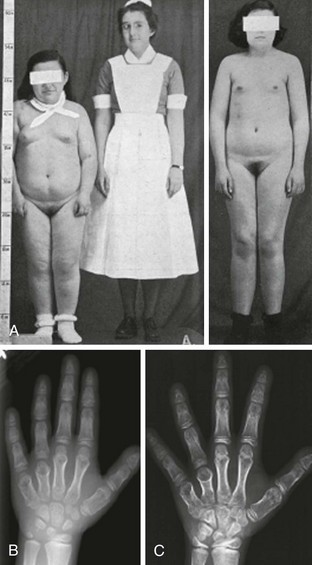
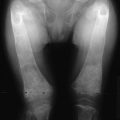
FIGURE 10-2 Albright’s hereditary osteodystrophy (AHO). A, Short stature and obesity are among the typical features of AHO.
(A Adapted from Albright et al: Pseudohypoparathyroidism—an example of “Seabright-Bantam syndrome,” Endocrinology 30:922–932, 1942).
B, Hand radiograph of a child with AHO demonstrating short fourth and fifth metacarpals at age 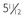 years. C, The same patient’s hand radiograph at age
years. C, The same patient’s hand radiograph at age 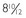 years.
years.
In addition to having PTH resistance and AHO, patients with PHP Ia show clinical evidence that is consistent with target organ resistance to other hormones. The most common additional hormone resistance involves the actions of thyroid-stimulating hormone (TSH) that lead to hypothyroidism.45,46 In fact, unlike PTH resistance, which typically develops later in life, TSH resistance can be present at birth.47–50 Resistance toward gonadotropins and growth hormone–releasing factor has been reported,51–53 whereas resistance toward other peptide hormones that also mediate their actions through Gsα-coupled receptors, such as vasopressin or adrenocorticotropic hormone (ACTH), does not appear to become clinically overt.52,54–58
The genetic mutation that causes PHP Ia is located within Gsα-coding GNAS exons.59,60 A protein that is essential for the actions of many hormones, Gsα primarily mediates agonist-induced generation of intracellular cAMP. Activation of a stimulatory G protein–coupled receptor by its agonist, such as PTH, leads to a guanosine diphosphate (GDP)-guanosine triphosphate (GTP) exchange on Gsα, causing dissociation of the latter from Gβγ subunits (Fig. 10-3). This allows both Gsα and Gβγ to stimulate their respective effectors. In its GTP-bound state, Gsα can directly activate several different effectors, such as Src tyrosine kinase,61 and certain Ca channels.62,63 Apart from these effectors, however, adenylyl cyclase is by far the most ubiquitous and the most extensively investigated effector molecule stimulated by Gsα. An integral membrane protein, adenylyl cyclase catalyzes the synthesis of the ubiquitous second messenger cAMP, which then triggers various cell-specific responses. Activation of adenylyl cyclase and other effectors by Gsα is regulated by the intrinsic GTP hydrolase (GTPase) activity of Gsα. Conversion of GTP into GDP results in reassembly of the G protein heterotrimer and thereby prevents further effector stimulation (see Fig. 10-3).
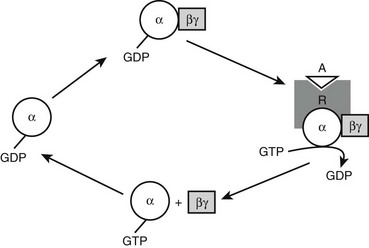
FIGURE 10-3 Heterotrimeric G protein activation cycle. The heterotrimeric complex is assembled at the basal state, with the α subunit associated with a guanosine diphosphate (GDP) molecule. Upon binding of an agonist (A) to its Gs-coupled receptor (R), the GDP molecule bound to the α subunit is replaced with a guanosine triphosphate (GTP) molecule, that is, the activated receptor acts as a guanine nucleotide exchange factor for the α subunit. The GTP-bound form of the α subunit dissociates from the βγ subunits and thereby stimulates its downstream effectors, which, in the case of Gsα, include adenylyl cyclase. Note that the free Gβγ dimer can also stimulate different downstream effectors. The intrinsic GTP hydrolase activity of the α subunit converts GTP into GDP, resulting in reassociation of the heterotrimer and, thus, termination of effector stimulation.
Mutations identified in PHP-Ia patients are heterozygous and scattered throughout all 13 GNAS exons that encode Gsα, including missense and nonsense amino acid changes, insertions/deletions that cause frameshift, and nucleotide alterations that disrupt pre-mRNA splicing (an extensive list of these mutations can be found at OMIM entry #139320 at http://www.ncbi.nlm.nih.gov). Constitutional deletions of the chromosomal arm containing GNAS have also been identified.64 Consistent with heterozygous mutations, Gsα level/activity is reduced by approximately 50% in easily accessible tissues from PHP Ia patients, such as erythrocytes, skin fibroblasts, and platelets.45,65–78 Deficiency of Gsα has been demonstrated in renal membranes from a patient with PHP.79 A complementation assay is typically used to examine Gsα activity, involving patient-derived erythrocyte membranes and membranes from turkey erythrocyte that lack endogenous Gsα (Fig. 10-4). The detection of reduced Gsα activity by this means is important for the establishment of PHP Ia diagnosis, particularly in cases where genetic analysis fails to reveal a GNAS mutation. Reduction of Gsα activity in PHP Ia is consistent with the fact that PTH and the other hormones with impaired actions in this disorder act via cAMP-mediated signaling pathways.
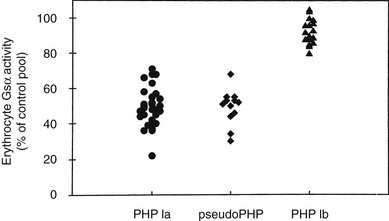
FIGURE 10-4 Gsα activity of erythrocyte membranes. Gsα is quantified in complementation assays with S49 cyc− membranes, which genetically lack Gsα but retain all other components necessary for hormone responsive adenylyl cyclase activity. Activity is reduced by approximately 50% in patients with Albright’s hereditary osteodystrophy (AHO) with pseudohypothyroidism (PHP) type Ia or pseudoPHP but is normal in patients with PHP type Ib.
Among the many different inactivating GNAS mutations, a 4 bp deletion in exon 7 has been identified in numerous families, representing a genetic “hot spot.” In addition, two different mutations are associated with additional phenotypes. A missense mutation in exon 13 (A366S) was identified in two unrelated boys who presented with PHP Ia and precocious puberty.80 This mutant Gsα protein is temperature sensitive and thus rapidly degrades at normal body temperature. The amino acid substitution, however, renders the protein constitutively active, resulting in elevated cAMP signaling in the cooler temperature of the testis. Recently, another mutant Gsα protein was described in a unique case of familial PHP Ia and transient neonatal diarrhea.81 The mutation, which entails a repeated AVDT sequence at residues 366 to 369, generates an unstable but constitutively active Gsα mutant as the result of enhanced GDP-GTP exchange and reduced GTPase activity. Although hormone resistance results from the instability of the Gsα-AVDT mutant, diarrhea is attributed to increased plasma membrane localization of the mutant protein in the small intestinal epithelium.
PHP Ic has been described as a distinct variant of PHP Ia,68 but the clinical features of patients with PHP Ic are indistinguishable from those with PHP Ia, in that they have both AHO and multihormone resistance. In contrast to PHP Ia, however, biochemical assays demonstrate no reduction in Gsα activity in erythrocytes obtained from PHP Ic patients, suggesting the absence of mutations within the Gsα gene. Nevertheless, recent molecular characterizations have revealed Gsα mutations in at least some PHP Ic patients. However, the Gsα mutants show impaired coupling to different G protein–coupled receptors, yet show normal Gsα activity in complementation assays that use nonhydrolyzable GTP analogues rather than a receptor agonist for stimulation of Gsα activity.44,82 Thus, the mutant Gsα protein that causes PHP Ic allows basal cAMP formation but is unable to trigger an increase in response to hormones. Hence, at least some PHP Ic cases are allelic variants of PHP Ia. Although it remains possible that some patients who match the description of PHP Ic develop hormone resistance and AHO as the result of a novel gene mutation that affects cAMP production without functionally impairing Gsα itself (e.g., inactivating mutations that affect one of the adenylyl cyclases, or activating mutations in the phosphodiesterase), it appears unlikely that such putative mutations affect only a few G protein–coupled receptors and thus result in limited hormonal resistance as observed in PHP Ia and PHP Ic.
The Complex GNAS Locus
GNAS is a complex locus giving rise to multiple different coding and noncoding transcripts that show monoallelic, parent-of-origin specific expression profiles (Fig. 10-5). GNAS maps to the telomeric end of the long arm of chromosome 20 (20q13.2-20q13.3).83–85 Gsα is encoded by 13 exons,86 but because of alternative pre-mRNA splicing, the Gsα transcript has several variants. The long and short Gsα variants (Gsα-L and Gsα-S, respectively) differ from each other by the inclusion or exclusion of exon 386–88; these Gsα variants are typically detected as 52 and 45 kDa protein bands on Western blots. Showing further complexity, each Gsα form either includes or excludes a CAG trinucleotide (encoding serine) at the start of exon 4. Small but potentially important differences have been reported between the activities of Gsα-L and Gsα-S. For example, Gsα-L has been shown to display a greater ability to mediate receptor signaling than Gsα-S when partially purified proteins from rabbit liver were examined,89 although the opposite finding was reported upon the use of cultured pancreatic islet cells.90 Furthermore, the GDP release rate from Gsα-L appears to be approximately twofold higher than that of Gsα-S,91 and, accordingly, fusion proteins involving the β2-adrenergic receptor and Gsα-L have shown higher constitutive activity than those involving the receptor and Gsα-S.92 Moreover, differences have been reported in the subcellular targeting of these two Gsα variants.93–95 Currently, it remains unclear whether these differences translate into biologically significant effects, such as divergence in the variety of effectors and/or the efficiency of effector activation. Nonetheless, a mutation in exon 3 has been identified recently in two siblings affected by a mild form of PHP Ia.96 The mildness of the phenotype is consistent with the disruption of only one of the two main Gsα variants—that is, Gsα-L. It remains unknown whether this exon 3 mutation impairs agonist responses in an effector- and tissue-specific manner; this possibility depends on the putative effector selectivity and relative expression levels of Gsα-L and Gsα-S in different tissues.
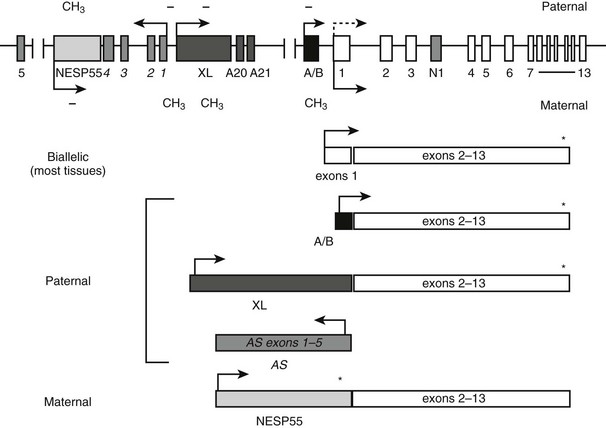
FIGURE 10-5 The GNAS locus. The complex GNAS locus gives rise to multiple transcripts. Boxes and connecting lines indicate exons and introns, respectively. Arrows show the direction of transcription for each transcript. The main transcriptions derived from GNAS and the utilized exons are depicted as rectangles below the gene schematic. Gsα is biallelically expressed, except in a small number of tissues, including renal proximal tubules, thyroid, gonads, and pituitary, in which expression from the paternal allele is silenced (dashed arrow). XLαs, A/B and antisense (AS) transcripts are derived from the paternal allele, and the NESP55 transcript from the maternal allele. Promoters of XLαs, A/B, antisense, and NESP55 transcripts are methylated on the silenced allele, as indicated by CH3 (methylated CpGs) and − (unmethylated CpGs).
Recent studies have revealed that GNAS gives rise to multiple additional gene products that show parent-of-origin specific, monoallelic expression. Besides Gsα, at least two translated GNAS transcripts exist, using distinct upstream promoters and alternative first exons that splice onto exons 2 to 13 encoding Gsα. The most upstream promoter relative to the Gsα promoter drives expression of a neuroendocrine secretory protein with an apparent molecular mass of 55,000 (NESP55).97 The paternal NESP55 promoter is methylated, and transcription occurs from the maternal allele.98,99 In humans, the NESP55 protein is encoded by a single exon, so that Gsα exons 2 to 13 make up the 3′ untranslated region.98,99 Expressed in neuroendocrine tissues, the peripheral and central nervous system, and some endocrine tissues,97,100–103 NESP55 is a chromogranin-like protein that is associated with the constitutive secretory pathway.104 Loss of NESP55 expression does not seem to have an overt clinical outcome in humans, as evidenced from patients with PHP type Ib (see below). However, its disruption in mice results in a subtle behavioral phenotype characterized by increased reactivity to novel environments.105
Another GNAS product is XLαs, which is expressed from the paternal allele.99,106,107 XLαs also uses a distinct upstream promoter and a unique first exon that splices onto Gsα exons 2 to 13.99,107 Unlike in NESP55 transcript, however, the latter portion of the XLαs transcript is included in the translated product, resulting in a protein that is partially identical to Gsα.106 Consistent with its structural similarity to Gsα, XLαs can mediate receptor-activated adenylyl cyclase stimulation in transfected cells.44,108,109 However, the phenotype of mice with targeted disruption of the XL exon suggests that XLαs has unique, albeit as yet undefined, cellular functions. These mice show high early postnatal mortality as the result of poor adaptation to feeding and impairment in glucose and energy metabolism,110 in contrast to Gsα knockout mice, which to a large extent recapitulate the findings in patients with PHP Ia.111,112
The paternal GNAS gives rise to two additional transcripts. From the sense strand originates the A/B transcript (also termed 1A or 1′), which, similar to NESP55 and XLαs, utilizes an upstream promoter and an alternative first exon (exon A/B) that splices onto Gsα exons 2 to 13.113,114 Exon A/B does not comprise a translation initiation codon, but, as demonstrated in vitro, translation can be started through the use of an AUG located within exon 2, thereby giving rise to an N-terminally truncated protein that localizes to the plasma membrane.114 However, no evidence currently supports the existence of endogenous A/B protein. It is thought instead that the A/B transcript is a noncoding RNA. Another paternal GNAS transcript is derived from the antisense strand.115,116 The GNAS antisense transcript, which is formed in humans by several primary exons that show multiple alternative splicing patterns,115,117 is also considered to be noncoding. The promoter of GNAS antisense transcript is immediately upstream of the promoter of XLαs. Although the promoters of XLαs and antisense transcript are located together within a large maternally methylated region, the female germline-specific imprint is established at the antisense promoter only.118 The A/B promoter likewise is methylated in the female, but not in the male, germline.119 Thus, the two germline imprint marks at the GNAS locus include promoters of the antisense and A/B transcripts. Accordingly, data from different genetically manipulated mouse models show that these noncoding transcripts are essential for the regulation of imprinted gene expression from GNAS.120–122 Imprinting of the A/B transcript is particularly important for the development of hormone resistance seen in patients with PHP Ib (see below).
Unlike the promoters of NESP55, antisense, XLαs, and A/B transcripts, the promoter of Gsα is not differentially methylated and, accordingly, Gsα expression is biallelic in most tissues.98,107,123,124 Biallelic Gsα expression has been shown specifically in human bone and adipose tissue.125 However, paternal Gsα expression is silenced in a small subset of tissues through as yet undefined mechanisms, so that the maternal allele is the predominant source of Gsα expression. These tissues include the renal proximal tubule, thyroid, pituitary, and gonads.112,126–128 Although devoid of differential methylation, the Gsα promoter exhibits parent-of-origin specific histone modifications in those tissues in which it is monoallelic. The active maternal Gsα promoter shows a greater ratio of trimethylated to dimethylated histone-3 Lys4 compared with the silenced paternal promoter in the proximal tubule, whereas the quantity of methylated histones is similar in maternal and paternal Gsα promoters in liver—a tissue in which Gsα is biallelic.129 As is discussed below, tissue-specific, paternal Gsα silencing has a key role in the development of PTH resistance in patients with PHP Ia and PHP Ib.
Clinically Distinct, Genetically Related PHP Ia Variants
pseudopseudohypoparathyroidism
Physical abnormalities similar to those observed in patients with PHP Ia but without evidence of an abnormal regulation of calcium and phosphate homeostasis were first reported in 1952.130 Because of the lack of abnormal regulation of mineral ion homeostasis, the name pseudopseudohypoparathyroidism (PPHP) was coined to describe this disorder.130 It is interesting to note that patients with PPHP also carry GNAS mutations that lead to diminished Gsα function, and these mutations can be found in the same kindred as those with PHP Ia. However, both disorders are never seen in the same sibling kinship, and careful analysis of multiple families has revealed that the mode of inheritance of each disorder depends on the gender of the parent who transmits the Gsα mutations.131 Thus, an inactivating Gsα mutation causes PHP Ia (i.e., hormone resistance and AHO) after maternal inheritance, whereas the same mutation results in PPHP (AHO only). Because AHO appears to develop regardless of the parent of origin, it is primarily hormone resistance that displays an imprinted mode of inheritance. Recent findings regarding tissue-specific imprinting of Gsα expression (see above) now can explain the parent-of-origin specific inheritance of hormone resistance. In those tissues where Gsα expression is paternally silenced (i.e., Gsα is expressed exclusively or predominantly from the maternal allele), an inactivating mutation located on the paternal allele is not predicted to alter Gsα function, whereas the same mutation located on the maternal allele is predicted to abolish Gsα function completely (Fig. 10-6). Tissue-specific imprinting of Gsα expression also explains why the target organ resistance involves only a small subset of tissues despite the involvement of Gsα signaling in a multitude of physiologic responses. Hormone resistance is observed in those tissues where Gsα is imprinted, such as the proximal renal tubule and the thyroid; hormone responses are unimpaired in those tissues where Gsα is biallelic, such as the distal renal tubules. The role of tissue-specific Gsα imprinting in the development of PTH resistance has been demonstrated through the generation of mice heterozygous for maternal or paternal disruption of Gnas.126
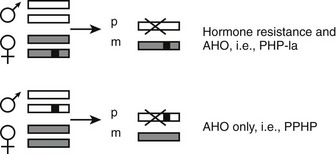
FIGURE 10-6 The effect of paternal Gsα silencing in the development of hormone resistance. Gsα is biallelic in most tissues; a heterozygous inactivating Gsα mutation therefore causes an approximately 50% reduction in Gsα activity/expression regardless of the parent of origin of the mutation. However, in some tissues, such as the proximal tubule and thyroid, the paternally inherited Gsα transcript is silenced (X). Thus, if a Gsα mutation (black square) is inherited from a female individual, this mutation nearly abolishes the expression or activity of Gsα in those tissues, thus leading to hormone resistance (PHP Ia). In contrast, upon paternal inheritance, the same mutation Gsα does not lead to a significant change in expression/activity, and thus, hormone responses are unimpaired. Paternal and maternal Gsα alleles are depicted by white or gray rectangles.
Most AHO features develop regardless of the parent transmitting a Gsα mutation; this observation has led to the hypothesis that inheritance of AHO is due to Gsα haploinsufficiency in various tissues, which appears to be true in certain settings. PTH-related protein-induced cAMP generation is critical for proper control of hypertrophic differentiation of growth plate chondrocytes,132 and Gsα haploinsufficiency has been demonstrated in this tissue through the study of mice chimeric for wild-type cells and mutant cells heterozygous for disruption of Gnas exon 2.133 Regardless of the parental origin of the Gnas exon 2 disruption, mutant cells displayed premature hypertrophy compared with their wild-type neighbors, although paternal disruption (i.e., loss of one Gsα allele combined with a complete loss of XLαs) resulted in significantly more premature hypertrophy than did maternal disruption (loss of one Gsα allele only). Thus, the brachydactyly and/or short stature observed in the context of AHO likely results from diminished Gsα signaling in growth plate chondrocytes. Although these data correlate well with the notion that AHO develops after both maternal and paternal inheritance of a Gsα mutation, recent evidence suggests that individual AHO features can be subject to imprinting. Careful analysis of multiple patients with PHP Ia and PPHP revealed that obesity is primarily a feature of PHP Ia patients, which develops after maternal inheritance.134 Given that Gsα is biallelic in white adipose tissue,125 it was proposed that Gsα may also be imprinted—predominantly by maternal expression—in areas of the central nervous system that control satiety and body weight.134 Recent reports have demonstrated that cognitive impairment is more prevalent in PHP Ia than PPHP, thus indicating that tissue-specific Gsα imprinting may involve additional brain regions.135 On the other hand, imprinted inheritance has not been reported for short stature, despite the finding that Gsα is imprinted in the pituitary,127,128 and that PHP Ia patients display growth hormone-releasing hormone (GHRH) resistance and growth hormone (GH) deficiency.52,53 Future analyses of patients with PHP Ia and PPHP will be helpful in determining the relative roles of genomic imprinting and haploinsufficiency in the development of individual AHO features.
Progressive Osseous Heteroplasia
A disorder termed progressive osseous heteroplasia (POH) has been described in patients with severe extraskeletal ossifications that involve deep connective tissue and skeletal muscle (Fig. 10-7).136,137 In POH, ectopic bone is primarily intramembranous, as opposed to a similar disease, termed fibrodysplasia ossificans progressiva (FOP), in which extraskeletal bone formation occurs via endochondral mechanisms and is accompanied by skeletal malformations.138,139 Few patients with POH demonstrate AHO features and, consistent with the occasional coexistence of these two sets of clinical defects, heterozygous inactivating Gsα mutations have been identified as a cause of POH.140–142 Several of the identified mutations are identical to those identified in PHP Ia/PPHP kindreds.141,142 Gsα activity and downstream signaling have been implicated in the control of osteogenic differentiation. Patients who are mosaic for heterozygous GNAS mutations that result in constitutive Gsα activity develop fibrous dyplasia of bone characterized by irregular woven bone disrupted by fibrous tissue.143 Moreover, in human mesenchymal stem cells, reduction of Gsα protein levels has been shown to cause osteogenic differentiation, while inhibiting the formation of adipocytes.144,145 In addition, Runx2, a key regulator of osteoblast-specific gene expression, appears to suppress Gsα expression.146 Thus, osteoprogenitor formation and early stages of osteoblastic differentiation require reduced levels of cAMP signaling, consistent with the association of inactivating Gsα mutations with the severe ectopic bone formation observed in POH.
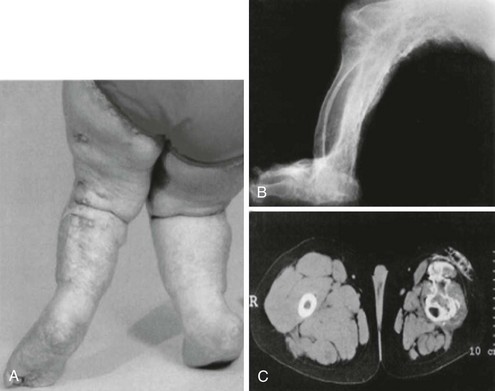
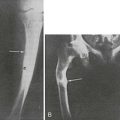
FIGURE 10-7 Clinical and radiographic appearance of progressive osseous heteroplasia (POH). A, Posterior view of the legs and feet of a 5-year-old girl with POH, showing severe maculopapular lesions caused by extensive dermal and subcutaneous ossification. B, A lateral radiogram of the right leg of an 11-year-old girl with POH demonstrating severe heterotopic ossification of the soft tissues. C, Computed tomographic image of the thighs of a 10-year-old boy with POH, showing atrophied soft tissues in the left leg and extensive ossification of the skin, subcutaneous fat, and quadriceps muscles. (Adapted from Shore EM, et al.142)
Because of the presence of GNAS mutations in both AHO and POH, it appears that POH is an extreme manifestation of AHO, and that additional factors, such as genetic background, epigenetic events, or environmental factors, may determine the penetrance and severity of ectopic ossifications in these patients that show approximately 50% loss of Gsα activity. Nevertheless, clinical and genetic data demonstrate several important differences between patients with AHO and those with POH. First, the ectopic bone in AHO is limited to subcutaneous tissue. In addition, in nearly all patients with POH, severe ectopic bone formation is isolated (i.e., other typical AHO features are not manifest).142,147 Moreover, mutations leading to isolated POH are inherited from male obligate gene carriers only (i.e., the inheritance pattern is exclusively paternal).142 In fact, in one large kindred, paternal inheritance of a GNAS mutation caused POH, and maternal inheritance of the same mutation caused typical AHO findings (without severe heterotopic ossification).142 These findings suggest that the disease mechanism underlying POH is significantly different from that underlying AHO, and that a GNAS product with exclusive paternal expression, such as XLαs, contributes to the molecular pathogenesis of POH.
Pseudohypoparathyroidism Type Ib
Another form of PHP was described by Peterman and Garvey148 and by Reynolds et al.149 Now known as pseudohypoparathyroidism type Ib (PHP Ib), this PHP form is characterized by the presence of PTH-resistant hypocalcemia and hyperphosphatemia, but without evidence of AHO. In addition to increased serum PTH, patients with PHP Ib can demonstrate elevated serum alkaline phosphatase activity, which suggests normal PTH-dependent bone turnover.150 In fact, hyperparathyroid bone disease sometimes is observed in association with PHP Ib, leading to the introduction of the term “pseudohypo-hyperparathyroidism” (PHP-HPT).151–153 The intact PTH response in the bone is consistent with the lack of Gsα imprinting in bone.125 Moreover, because the genetic defect underlying PHP Ib does not involve a coding Gsα mutation (see below), this intact response appears more likely to result in increased bone resorption as the result of elevated serum PTH in this PHP subtype compared with that in PHP Ia.
The hormone resistance observed in PHP Ib patients develops only after maternal inheritance of the genetic defect154 (i.e., the mode of inheritance is identical to hormone resistance in PHP Ia). PTH resistance and related changes in calcium and phosphorous homeostasis are the major laboratory findings in PHP Ib, but recent reports have demonstrated that some PHP Ib patients also display mild hypothyroidism with slightly elevated TSH levels,26,155–157 as well as some elevation in calcitonin level.155 Hypothyroidism, as in patients with PHP Ia, likely results from mild TSH resistance and is consistent with the predominantly maternal expression of Gsα in the thyroid.128,156 Nevertheless, evidence for resistance to other hormones, such as gonadotropins, whose actions also involve tissues in which Gsα is imprinted, has not been reported for PHP Ib patients. A recent study assessed growth hormone responsiveness to GHRH plus arginine stimulation in PHP Ib, revealing a normal response in 9 of 10 patients.157 On the other hand, in addition to PTH and mild thyroid-stimulating hormone (TSH) resistance, hypouricemia due to increased fractional excretion of uric acid has been reported in the affected individuals of two unrelated PHP Ib kindreds.27,159 Although this finding implicates impaired PTH actions in the development of hypouricemia in these patients, and this interpretation is consistent with two previous reports describing hyperuricemia inassociation with hyperparathyroidism,160,161 the hypouricemia reported in one of the PHP Ib kindreds seemed to have resolved following treatment with calcium and calcitriol.27
Patients with PHP Ib display normal Gsα bioactivity/levels in easily accessible tissues. Accordingly, coding Gsα mutations are ruled out in these patients. In one family, however, a mutation located in exon 13 (in-frame deletion of Ile382) was reported, leading to the uncoupling of Gsα from PTHR1 but not other receptors, including TSHR, LHR, and β-adrenergic receptor.162 Analyses were performed in LLCPK cells, which are of renal origin and express endogenous Gsα. These findings are consistent with isolated PTH resistance seen in PHP Ib, and this mutation may represent a rare cause of PHP Ib. However, this conclusion has been questioned, as the use of transfected mouse embryonic fibroblasts null for endogenous Gsα showed that del382Ile leads to uncoupling from not only PTHR1 but also the β-adrenergic receptor.44 Because of a lack of Gsα mutations, and because Gsα activity/levels in easily accessible tissues are normal in PHP Ib patients, inactivating mutations that affect the gene encoding PTHR1 were considered in the past.163 However, several different studies have excluded such mutations as the cause of this disease.164–167 Instead, inactivating mutations of PTHR1 have been revealed as the cause of Bloomstrand’s chondrodysplasia, an embryonic lethal disorder with severe skeletal abnormalities.168
Based on genome-wide linkage analysis, the genetic defect underlying PHP Ib maps to a region of chromosome 20q13 that comprises the GNAS locus,154 but the critical interval excludes all the coding GNAS exons, including those that encode Gsα.26 On the other hand, patients with PHP Ib display epigenetic abnormalities within the GNAS locus.26,169 The most consistent epigenetic defect is a loss of imprinting at exon A/B (also termed exon 1A), which is found primarily as an isolated defect in familial PHP Ib cases.26,169 In addition, many sporadic and some familial PHP Ib cases show additional loss of imprinting at the DMR comprising the XLαs and antisense promoters and gain of imprinting at the differentially methylated region (DMR) comprising exon NESP55.13,169 These abnormalities are associated with biallelic expression of A/B, XLαs, and antisense transcripts and the silencing of NESP55 transcript. Together with the genetic linkage data, these imprinting defects suggest that the mutation causing PHP Ib disrupts a regulatory element that controls GNAS imprinting. However, evidence for incomplete penetrance regarding GNAS imprinting defects has been reported in one kindred, in whom some individuals lacked loss of imprinting and were healthy despite maternally inheriting the disease-associated haplotype.170 Thus, imprinting abnormalities of GNAS appear to be required for the development of PHP Ib. Consistent with the importance of imprinting in the disease mechanism, a patient with this disorder has been reported to have paternal uniparental isodisomy of chromosome 20q.155 In addition to having a paternal-exclusive imprinting profile at the GNAS locus and PTH resistance, the patient presented with mild developmental delay and craniosynostosis, which may reflect unmasking of recessive mutations inherited from the father or disruption of gene expression at other imprinted loci in this genomic region, such as the gene encoding neuronatin (NNAT).
In multiple familial PHP Ib cases with isolated exon A/B loss of imprinting, a unique 3 kb microdeletion at the neighboring STX16 locus has been identified (Fig. 10-8).171 The deleted region comprises STX16 exons 4 to 6 and is flanked by two direct repeats, which may underlie the mechanism whereby this deletion occurs. This is consistent with the presence of the same microdeletion in many unrelated families with different ethnic and racial origin.* In a single kindred, a different microdeletion within STX16 has been reported, removing exons 2 through 4 and overlapping with the 3 kb microdeletion by approximately 1.3 kb.39 Thus, disruption of STX16 appears to be the common genetic defect in cases with isolated loss of exon A/B imprinting. The parental origin of these STX16 deletions correlates well with the mode of inheritance of PHP Ib. It is inherited maternally in affected individuals and is inherited paternally in unaffected carriers. This gene encodes syntaxin-16, a member of the SNARE family proteins. However, STX16 does not appear to be imprinted,39,171 and it is therefore unlikely that the loss of one STX16 copy leads to PHP Ib. Instead, because maternal inheritance is associated with loss of exon A/B imprinting on the same allele, these deletions are presumed to disrupt a cis-acting element that controls the establishment or maintenance of exon A/B imprinting. Other than genetic evidence, however, no currently available data corroborate this prediction. A mouse model carrying a deletion equivalent to the 3-kb STX16 deletion in humans has been generated, but neither maternal nor paternal inheritance of this genetic alteration causes PTH resistance or any alterations in GNAS imprinting174; animals with the homozygous Stx16 deletion are also healthy. It thus appears plausible that the imprinting control element of GNAS located within STX16 in the human is not precisely at the same location in the mouse. Nonetheless, the absence of a phenotype in the Stx16 deletion mice argues against a model in which syntaxin 16, the product of this gene, is required in the oocyte for proper exon A/B imprinting.
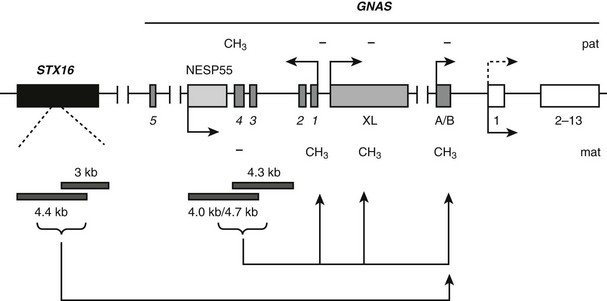
FIGURE 10-8 Mutations identified in patients with autosomal dominant PHP Ib (AD-PHP-Ib) and their effects on GNAS imprinting. The most frequent mutation is a 3 kb deletion within STX16, a gene located more than 200 kb upstream of GNAS. This deletion and another overlapping deletion in the same gene are predicted to disrupt a cis-acting control element of GNAS that is required for the imprint mark located at exon A/B. Deletions of the NESP55 DMR, including exons 3 and 4 of the antisense transcript and a recently identified deletion that includes only antisense transcript exons 3 and 4, have been identified in some AD-PHP-Ib kindreds. These reveal a cis-acting element that controls imprinting of the entire maternal GNAS allele. Boxes and connecting lines indicate exons and introns, respectively. STX16 exons and GNAS exons 2 through 13 are shown as single rectangles for simplicity. Paternal (pat) and maternal (mat) methylation (CH3) and parental origin of transcription (arrows) are indicated. Tissue-specific silencing of the paternal Gsα transcription is depicted by a dotted arrow. Identified deletions are shown by gray horizontal bars.
In two unrelated familial cases of PHP Ib in which the affected individuals carried broad GNAS imprinting defects, maternally inherited deletions of the entire NESP55 DMR, including exons 3 and 4 of the antisense transcript, have been identified (see Fig. 10-8).117 The deletions are 4 kb and 4.7 kb large and have breakpoints located in similar locations. Unaffected carriers in these families display an apparent loss of NESP55 methylation as the result of the loss of this region from the normally methylated paternal allele, but they do not show other imprinting GNAS defects. Affected individuals show loss of imprinting in the entire maternal allele. The presence of similarly large deletions at the NESP55 DMR has been excluded in a number of sporadic PHP Ib cases.13,117 However, a different 4.2 kb deletion has been identified recently in the affected individuals of a different PHP Ib kindred who displayed broad GNAS imprinting defects175 (see Fig. 10-8). This new deletion also includes antisense exons 3 and 4 but spares exon NESP55, overlapping with the previously identified two deletions by about 1.5 kb. Thus, these identified deletions reveal the putative location of another control element required for the imprinting of the entire maternal GNAS allele. The study of a mouse model in which Nesp55 transcription was prematurely truncated revealed loss of imprinting at exon 1A and, less consistently, the antisense promoter and exon XL.176 Taken together with the genetic findings in patients with PHP Ib, it appears that the establishment of imprinting on the maternal GNAS allele, which allows expression of Gsα in the proximal renal tubule and other tissues in which this GNAS product is monoallelic, such as thyroid, requires transcription from the NESP55 promoter. It is thus possible that even small mutations that prevent the generation of NESP55 pre-mRNA can lead to PHP Ib.
In some sporadic PHP Ib cases that show GNAS methylation defects, the maternal allele is shared between affected and unaffected siblings, suggesting that these cases may carry small de novo mutations in this region. Alternatively, it is possible that some of the sporadic PHP Ib cases carry mutations in an entirely different genomic location with a putative autosomal recessive mode of inheritance. It has also been suggested that the broad GNAS imprinting defects observed in sporadic PHP Ib patients result from stochastic defects in the regulation of imprinting.172
Despite having distinct epigenetic abnormalities at the GNAS locus (i.e., isolated A/B loss of imprinting vs. broad imprinting defects that involve exon A/B and at least one other GNAS DMR), PHP Ib patients seem to have similar clinical findings with respect to serum calcium, phosphate, and PTH levels.13 Analysis of 20 families in which the affected individuals show an isolated loss of A/B imprinting reveals that a significant portion of such familial cases are asymptomatic at the time of diagnosis. In these cases, the diagnosis frequently is made only on the basis of elevated serum PTH. Comparison of male and female patients among sporadic PHP Ib cases that exhibit GNAS imprinting defects at two or more GNAS DMRs reveals that female patients have significantly higher serum PTH levels than male patients, suggesting that hormone resistance is more severe in females.13
By definition, PHP Ib patients do not show AHO features. However, some recent reports identified patients who carry genetic and epigenetic defects associated with PHP Ib, yet present with mild AHO features, particularly shortness of metacarpal bones.27,173,177 This may suggest that Gsα imprinting occurs in more tissues than is currently recognized, although alternative explanations have been put forth. Given that individual AHO features can be observed in other disorders, the presence of AHO features may be unrelated to the molecular genetic defects underlying PHP Ib in these cases. In one case, the mother of two affected siblings with short metacarpals and loss of A/B imprinting also exhibited short metacarpals despite lacking any GNAS epigenetic abnormalities,173 suggesting that the finding of short metacarpals is unrelated to the epigenetic defect in that family. In addition, the observed coexistence of GNAS imprinting defects and AHO can result from a large genomic deletion comprising at least the promoter of Gsα and one or more differentially methylated regions. Such deletions have been ruled out in some but not all cases.27,173,177
Pseudohypoparathyroidism Type II
Dissociation regarding the impairment of PTH-induced nephrogenous cAMP formation and phosphaturia (i.e., PHP II) appears to be the least common form of PHP. Although typically sporadic, a case with a familial form of PHP II type has been reported,178 and several reports describe evidence of a self-limited form of this disease in newborns, which could indicate that it is transient in nature.35,179–181 The molecular defect and pathophysiologic mechanisms underlying this PHP variant remains to be discovered. Because the defect underlying PHP II is associated with normal cAMP generation in response to exogenous PTH, it is possible that it is caused by molecular defects that involve downstream cAMP generation, such as protein kinase A.19 Alternatively, the PTH signaling pathways that utilize G proteins other than Gs, such as Gq, may be defective in patients with PHP II. Signaling mediated by Gq involves activation of phopholipase C, which, in turn, leads to the formation of second messengers inositol 1,4,5-triphophate (IP3) and diacylglycerol (DAG). This signaling pathway results in stimulation of protein kinase C (PKC) and an increase in intracellular calcium ions. Serum calcium levels, which may affect the efficient utilization of intracellular calcium signaling pathways, appear to be important for restoring PTH responsiveness in PHP II, as shown in some patients who normalized their phosphaturic response to PTH following normalization of serum calcium.182 It is also possible that sodium-phosphate transporters in the proximal renal tubule are nonresponsive to PTH, thus leading to a defective phosphaturic, but not cAMP, response to exogenous PTH. Such a defect, however, should preserve the action of PTH on 25(OH)D-1α-hydroxylase, leading to normal serum 1,25(OH)2D, unless it is combined with vitamin D deficiency. Hypocalcemia as a result of vitamin D deficiency has been associated with PTH resistance that entailed the phosphaturic effect of this hormone without altering its potential to raise urinary cAMP,183 suggesting that some PHP II cases may in fact reflect vitamin D deficiency.184,185
References
1. Albright, F, Burnett, CH, Smith, PH, et al. Pseudohypoparathyroidism—an example of “Seabright-Bantam syndrome,”. Endocrinology. 1942;30:922–932.
2. Tashjian, AH, Jr., Frantz, AG, Lee, JB. Pseudohypoparathyroidism: assays of parathyroid hormone and thyrocalcitonin. Proc Natl Acad Sci U S A. 1966;56:1138–1142.
3. Ish-Shalom, S, Rao, LG, Levine, MA, et al. Normal parathyroid hormone responsiveness of bone-derived cells from a patient with pseudohypoparathyroidism. J Bone Miner Res. 1996;11:8–14.
4. Murray, T, Gomez Rao, E, Wong, MM, et al. Pseudohypoparathyroidism with osteitis fibrosa cystica: direct demonstration of skeletal responsiveness to parathyroid hormone in cells cultured from bone. J Bone Miner Res. 1993;8:83–91.
5. Stone, M, Hosking, D, Garcia-Himmelstine, C, et al. The renal response to exogenous parathyroid hormone in treated pseudohypoparathyroidism. Bone. 1993;14:727–735.
6. Breslau, NA, Weinstock, RS. Regulation of 1,25(OH)2D synthesis in hypoparathyroidism and pseudohypoparathyroidism. Am J Physiol. 1988;255:E730–E736.
7. Drezner, MK, Neelon, FA, Haussler, M, et al. 1,25-Dihydroxycholecalciferol deficiency: the probable cause of hypocalcemia and metabolic bone disease in pseudohypoparathyroidism. J Clin Endocrinol Metab. 1976;42:621–628.
8. Braun, JJ, Birkenhager, JC, Visser, TJ, et al. Lack of response of 1,25-dihydroxycholecalciferol to exogenous parathyroid hormone in a patient with treated pseudohypoparathyroidism. Clin Endocrinol (Oxf). 1981;14:403–407.
9. Yamaoka, K, Seino, Y, Ishida, M, et al. Effect of dibutyryl adenosine 3′,5′-monophosphate administration on plasma concentrations of 1,25-dihydroxyvitamin D in pseudohypoparathyroidism type I. J Clin Endocrinol Metab. 1981;53:1096–1100.
10. Drezner, MK, Haussler, MR. Normocalcemic pseudohypoparathyroidism, Association with normal vitamin D3 metabolism. Am J Med. 1979;66:503–508.
11. Balachandar, V, Pahuja, J, Maddaiah, VT, et al. Pseudohypoparathyroidism with normal serum calcium level. Am J Dis Child. 1975;129:1092–1095.
12. Breslau, NA, Notman, DD, Canterbury, JM, et al. Studies on the attainment of normocalcemia in patients with pseudohypoparathyroidism. Am J Med. 1980;68:856–860.
13. Linglart, A, Bastepe, M, Jüppner, H. Similar clinical and laboratory findings in patients with symptomatic autosomal dominant and sporadic pseudohypoparathyroidism type Ib despite different epigenetic changes at the GNAS locus. Clin Endocrinol (Oxf). 2007;67:822–831.
14. Tamada, Y, Kanda, S, Suzuki, H, et al. A pseudohypoparathyroidism type Ia patient with normocalcemia. Endocr J. 2008;55:169–173.
15. Jüppner, H, Abou-Samra, AB, Freeman, MW, et al. A G protein-linked receptor for parathyroid hormone and parathyroid hormone-related peptide. Science. 1991;254:1024–1026.
16. Gardella, TJ, Jüppner, H. Molecular properties of the PTH/PTHrP receptor. Trends Endocrinol Metab. 2001;12:210–217.
17. Abou-Samra, AB, Jüppner, H, Force, T, et al. Expression cloning of a common receptor for parathyroid hormone and parathyroid hormone-related peptide from rat osteoblast-like cells: a single receptor stimulates intracellular accumulation of both cAMP and inositol triphosphates and increases intracellular free calcium. Proc Natl Acad Sci U S A. 1992;89:2732–2736.
18. Chase, LR, Melson, GL, Aurbach, GD. Pseudohypoparathyroidism: defective excretion of 3′,5′-AMP in response to parathyroid hormone. J Clin Invest. 1969;48:1832–1844.
19. Drezner, M, Neelon, FA, Lebovitz, HE. Pseudohypoparathyroidism type II: a possible defect in the reception of the cyclic AMP signal. N Engl J Med. 1973;289:1056–1060.
20. Siejka, SJ, Knezevic, WV, Pullan, PT. Dystonia and intracerebral calcification: pseudohypoparathyroidism presenting in an eleven-year-old girl. Aust N Z J Med. 1988;18:607–609.
21. Dure, L, St., Mussell, HG. Paroxysmal dyskinesia in a patient with pseudohypoparathyroidism. Mov Disord. 1998;13:746–748.
22. Huang, CW, Chen, YC, Tsai, JJ. Paroxysmal dyskinesia with secondary generalization of tonic-clonic seizures in pseudohypoparathyroidism. Epilepsia. 2005;46:164–165.
23. Prashantha, DK, Pal, PK. Pseudohypoparathyroidism manifesting with paroxysmal dyskinesias and seizures. Mov Disord. 2009;24:623–624.
24. Kinoshita, M, Komori, T, Ohtake, T, et al. Abnormal calcium metabolism in myotonic dystrophy as shown by the Ellsworth-Howard test and its relation to CTG triplet repeat length. J Neurol. 1997;244:613–622.
25. Mahmud, FH, Linglart, A, Bastepe, M, et al. Molecular diagnosis of pseudohypoparathyroidism type Ib in a family with presumed paroxysmal dyskinesia. Pediatrics. 2005;115:e242–e244.
26. Bastepe, M, Pincus, JE, Sugimoto, T, et al. Positional dissociation between the genetic mutation responsible for pseudohypoparathyroidism type Ib and the associated methylation defect at exon A/B: evidence for a long-range regulatory element within the imprinted GNAS1 locus. Hum Mol Genet. 2001;10:1231–1241.
27. Unluturk, U, Harmanci, A, Babaoglu, M, et al. Molecular diagnosis and clinical characterization of pseudohypoparathyroidism type-Ib in a patient with mild Albright’s hereditary osteodystrophy-like features, epileptic seizures, and defective renal handling of uric acid. Am J Med Sci. 2008;336:84–90.
28. Windeck, R, Menken, U, Benker, G, et al. Basal ganglia calcification in pseudohypoparathyroidism type II. Clin Endocrinol (Oxf). 1981;15:57–63.
29. Chen, H, Tseng, F, Su, D, et al. Multiple intracranial calcifications and spinal compressions: rare complications of type la pseudohypoparathyroidism. J Endocrinol Invest. 2005;28:646–650.
30. Illum, F, Dupont, E. Prevalences of CT-detected calcification in the basal ganglia in idiopathic hypoparathyroidism and pseudohypoparathyroidism. Neuroradiology. 1985;27:32–37.
31. Manabe, Y, Araki, M, Takeda, K, et al. Pseudohypoparathyroidism with striopallidodentate calcification—a case report and review of the literature. Jpn J Med. 1989;28:391–395.
32. Pearson, DW, Durward, WF, Fogelman, I, et al. Pseudohypoparathyroidism presenting as severe Parkinsonism. Postgrad Med J. 1981;57:445–447.
33. Saito, H, Saito, M, Saito, K, et al. Subclinical pseudohypoparathyroidism type II: evidence for failure of physiologic adjustment in calcium metabolism during pregnancy. Am J Med Sci. 1989;297:247–250.
34. Zachariah, SB, Zachariah, B, Antonios, N, et al. Pseudohypoparathyroidism and cerebrovascular disease with dural calcification. J Fla Med Assoc. 1991;78:26–28.
35. Narang, M, Salota, R, Sachdev, SS. Neonatal pseudohypoparathyroidism. Indian J Pediatr. 2006;73:97–98.
36. Sajitha, S, Krishnamoorthy, PN, Shenoy, UV. Pseudohypoparathyroidism in newborn—a rare presentation. Indian J Pediatr. 2003;40:47–49.
37. Tsang, R, Venkataraman, P, Ho, M, et al. The development of pseudohypoparathyroidism, Involvement of progressively increasing serum parathyroid hormone concentrations, increased 1,25-dihydroxyvitamin D concentrations, and “migratory” subcutaneous calcifications. Am J Dis Child. 1984;138:654–658.
38. Barr, DG, Stirling, HF, Darling, JA. Evolution of pseudohypoparathyroidism: an informative family study. Arch Dis Child. 1994;70:337–338.
39. Linglart, A, Gensure, RC, Olney, RC, et al. A novel STX16 deletion in autosomal dominant pseudohypoparathyroidism type Ib redefines the boundaries of a cis-acting imprinting control element of GNAS. Am J Hum Genet. 2005;76:804–814.
40. Gelfand, IM, Eugster, EA, Dimeglio, LA. Presentation and clinical progression of pseudohypoparathyroidism with multi-hormone resistance and Albright hereditary osteodystrophy: a case series. J Pediatr. 2006;149:877–880.
41. Farfel, Z. Pseudohypohyperparathyroidism-pseudohypoparathyroidism type Ib. J Bone Miner Res. 1999;14:1016.
42. Poznanski, AK, Werder, EA, Giedion, A, et al. The pattern of shortening of the bones of the hand in PHP and PPHP—a comparison with brachydactyly E, Turner syndrome, and acrodysostosis. Radiology. 1977;123:707–718.
43. Archibald, RM, Finby, N, De Vito, F. Endocrine significance of short metacarpals. J Clin Endocrinol Metab. 1959;19:1312–1322.
44. Linglart, A, Mahon, MJ, Kerachian, MA, et al. Coding GNAS mutations leading to hormone resistance impair in vitro agonist- and cholera toxin-induced adenosine cyclic 3′,5′-monophosphate formation mediated by human XLαs. Endocrinology. 2006;147:2253–2262.
45. Levine, MA, Downs, RW, Jr., Moses, AM, et al. Resistance to multiple hormones in patients with pseudohypoparathyroidism, Association with deficient activity of guanine nucleotide regulatory protein. Am J Med. 1983;74:545–556.
46. Mallet, E, Carayon, P, Amr, S, et al. Coupling defect of thyrotropin receptor and adenylate cyclase in a pseudohypoparathyroid patient. J Clin Endocrinol Metab. 1982;54:1028–1032.
47. Yu, D, Yu, S, Schuster, V, et al. Identification of two novel deletion mutations within the Gs alpha gene (GNAS1) in Albright hereditary osteodystrophy. J Clin Endocrinol Metab. 1999;84:3254–3259.
48. Levine, MA, Jap, TS, Hung, W. Infantile hypothyroidism in two sibs: an unusual presentation of pseudohypoparathyroidism type Ia. J Pediatr. 1985;107:919–922.
49. Weisman, Y, Golander, A, Spirer, Z, et al. Pseudohypoparathyroidism type 1a presenting as congenital hypothyroidism. J Pediatr. 1985;107:413–415.
50. Yokoro, S, Matsuo, M, Ohtsuka, T, et al. Hyperthyrotropinemia in a neonate with normal thyroid hormone levels: the earliest diagnostic clue for pseudohypoparathyroidism. Biol Neonate. 1990;58:69–72.
51. Wolfsdorf, JI, Rosenfield, RL, Fang, VS, et al. Partial gonadotrophin-resistance in pseudohypoparathyroidism. Acta Endocrinol (Copenh). 1978;88:321–328.
52. Mantovani, G, Maghnie, M, Weber, G, et al. Growth hormone-releasing hormone resistance in pseudohypoparathyroidism type Ia: new evidence for imprinting of the Gs alpha gene. J Clin Endocrinol Metab. 2003;88:4070–4074.
53. Germain-Lee, EL, Groman, J, Crane, JL, et al. Growth hormone deficiency in pseudohypoparathyroidism type 1a: another manifestation of multihormone resistance. J Clin Endocrinol Metab. 2003;88:4059–4069.
54. Moses, AM, Weinstock, RS, Levine, MA, et al. Evidence for normal antidiuretic responses to endogenous and exogenous arginine vasopressin in patients with guanine nucleotide-binding stimulatory protein-deficient pseudohypoparathyroidism. J Clin Endocrinol Metab. 1986;62:221–224.
55. Stirling, HF, Barr, DGD, Kelnar, CJH. Familial growth hormone releasing factor deficiency in pseudohypoparathyroidism. Arch Dis Child. 1991;66:533–535.
56. Namnoum, AB, Merriam, GR, Moses, AM, et al. Reproductive dysfunction in women with Albright’s hereditary osteodystrophy. J Clin Endocrinol Metab. 1998;83:824–829.
57. Weinstein, LS. Albright hereditary osteodystrophy, pseudohypoparathyroidism, and Gs deficiency. In: Spiegel AM, ed. G Proteins, Receptors, and Disease. Totowa, NJ: Humana Press; 1998:23–56.
58. Levine, MA. Pseudohypoparathyroidism: from bedside to bench and back. J Bone Miner Res. 1999;14:1255–1260.
59. Weinstein, LS, Gejman, PV, Friedman, E, et al. Mutations of the Gs alpha-subunit gene in Albright hereditary osteodystrophy detected by denaturing gradient gel electrophoresis. Proc Natl Acad Sci U S A. 1990;87:8287–8290.
60. Patten, JL, Johns, DR, Valle, D, et al. Mutation in the gene encoding the stimulatory G protein of adenylate cyclase in Albright’s hereditary osteodystrophy. N Engl J Med. 1990;322:1412–1419.
61. Ma, YC, Huang, J, Ali, S, et al. Src tyrosine kinase is a novel direct effector of G proteins. Cell. 2000;102:635–646.
62. Yatani, A, Imoto, Y, Codina, J, et al. The stimulatory G protein of adenylyl cyclase, Gs, also stimulates dihydropyridine-sensitive Ca2+ channels. Evidence for direct regulation independent of phosphorylation by cAMP-dependent protein kinase or stimulation by a dihydropyridine agonist. J Biol Chem. 1988;263:9887–9895.
63. Mattera, R, Graziano, MP, Yatani, A, et al. Splice variants of the alpha subunit of the G protein Gs activate both adenylyl cyclase and calcium channels. Science. 1989;243:804–807.
64. Aldred, MA, Aftimos, S, Hall, C, et al. Constitutional deletion of chromosome 20q in two patients affected with Albright hereditary osteodystrophy. Am J Med Genet. 2002;113:167–172.
65. Levine, MA, Jap, TS, Mauseth, RS, et al. Activity of the stimulatory guanine nucleotide-binding protein is reduced in erythrocytes from patients with pseudohypoparathyroidism and pseudohypoparathyroidism: biochemical, endocrine, and genetic analysis of Albright’s hereditary osteodystrophy in six kindreds. J Clin Endocrinol Metab. 1986;62:497–502.
66. Miric, A, Vechio, JD, Levine, MA. Heterogeneous mutations in the gene encoding the alpha-subunit of the stimulatory G protein of adenylyl cyclase in Albright hereditary osteodystrophy. J Clin Endocrinol Metab. 1993;76:1560–1568.
67. Farfel, Z, Bourne, HR. Deficient activity of receptor-cyclase coupling protein in platelets of patients with pseudohypoparathyroidism. J Clin Endocrinol Metab. 1980;51:1202–1204.
68. Farfel, Z, Brothers, VM, Brickman, AS, et al. Pseudohypoparathyroidism: inheritance of deficient receptor-cyclase coupling activity. Proc Natl Acad Sci U S A. 1981;78:3098–3102.
69. Bourne, HR, Kaslow, HR, Brickman, AS, et al. Fibroblast defect in pseudohypoparathyroidism, type I: reduced activity of receptor-cyclase coupling protein. J Clin Endocrinol Metab. 1981;53:636–640.
70. Spiegel, AM, Levine, MA, Aurbach, GD, et al. Deficiency of hormone receptor-adenylate cyclase coupling protein: basis for hormone resistance in pseudohypoparathyroidism. Am J Physiol. 1982;243:E37–E42.
71. Motulsky, HJ, Hughes, RJ, Brickman, AS, et al. Platelets of pseudohypoparathyroid patients: evidence that distinct receptor-cyclase coupling proteins mediate stimulation and inhibition of adenylate cyclase. Proc Natl Acad Sci U S A. 1982;79:4193–4197.
72. Farfel, Z, Abood, ME, Brickman, AS, et al. Deficient activity of receptor-cyclase coupling protein is transformed lymphoblasts of patients with pseudohypoparathyroidism, type I. J Clin Endocrinol Metab. 1982;55:113–117.
73. Levine, MA, Eil, C, Downs, RW, Jr., et al. Deficient guanine nucleotide regulatory unit activity in cultured fibroblast membranes from patients with pseudohypoparathyroidism type I. A cause of impaired synthesis of 3′,5′-cyclic AMP by intact and broken cells. J Clin Invest. 1983;72:316–324.
74. Levine, MA, Ahn, TG, Klupt, SF, et al. Genetic deficiency of the alpha subunit of the guanine nucleotide-binding protein Gs as the molecular basis for Albright hereditary osteodystrophy. Proc Natl Acad Sci U S A. 1988;85:617–621.
75. Patten, JL, Levine, MA. Immunochemical analysis of the α-subunit of the stimulatory G-protein of adenylyl cyclase in patients with Albright’s hereditary osteodystrophy. J Clin Endocrinol Metab. 1990;71:1208–1214.
76. Carter, A, Bardin, C, Collins, R, et al. Reduced expression of multiple forms of the α subunit of the stimulatory GTP-binding protein in pseudohypoparathyroidism type Ia. Proc Natl Acad Sci U S A. 1987;84:7266–7269.
77. Farfel, Z, Brickman, AS, Kaslow, HR, et al. Defect of receptor-cyclase coupling protein in pseudohypoparathyroidism. N Engl J Med. 1980;303:237–242.
78. Levine, MA, Downs, RW, Jr., Singer, M, et al. Deficient activity of guanine nucleotide regulatory protein in erythrocytes from patients with pseudohypoparathyroidism. Biochem Biophys Res Commun. 1980;94:1319–1324.
79. Downs, RW, Jr., Levine, MA, Drezner, MK, et al. Deficient adenylate cyclase regulatory protein in renal membranes from a patient with pseudohypoparathyroidism. J Clin Invest. 1983;71:231–235.
80. Iiri, T, Herzmark, P, Nakamoto, JM, et al. Rapid GDP release from Gs in patients with gain and loss of function. Nature. 1994;371:164–168.
81. Makita, N, Sato, J, Rondard, P, et al. Human G(salpha) mutant causes pseudohypoparathyroidism type Ia/neonatal diarrhea, a potential cell-specific role of the palmitoylation cycle. Proc Natl Acad Sci U S A. 2007;104:17424–17429.
82. Linglart, A, Carel, JC, Garabedian, M, et al. GNAS1 lesions in pseudohypoparathyroidism Ia and Ic: genotype phenotype relationship and evidence of the maternal transmission of the hormonal resistance. J Clin Endocrinol Metab. 2002;87:189–197.
83. Gejman, PV, Weinstein, LS, Martinez, M, et al. Genetic mapping of the Gs-α subunit gene (GNAS1) to the distal long arm of chromosome 20 using a polymorphism detected by denaturing gradient gel electrophoresis. Genomics. 1991;9:782–783.
84. Rao, VV, Schnittger, S, Hansmann, I. G protein Gs alpha (GNAS 1), the probable candidate gene for Albright hereditary osteodystrophy, is assigned to human chromosome 20q12-q13.2. Genomics. 1991;10:257–261.
85. Levine, MA, Modi, WS, O’Brien, SJ. Mapping of the gene encoding the alpha subunit of the stimulatory G protein of adenylyl cyclase (GNAS1) to 20q13.2-q13.3 in human by in situ hybridization. Genomics. 1991;11:478–479.
86. Kozasa, T, Itoh, H, Tsukamoto, T, et al. Isolation and characterization of the human Gsα gene. Proc Natl Acad Sci U S A. 1988;85:2081–2085.
87. Bray, P, Carter, A, Simons, C, et al. Human cDNA clones for four species of G alpha s signal transduction protein. Proc Natl Acad Sci U S A. 1986;83:8893–8897.
88. Robishaw, JD, Smigel, MD, Gilman, AG. Molecular basis for two forms of the G protein that stimulates adenylate cyclase. J Biol Chem. 1986;261:9587–9590.
89. Sternweis, PC, Northup, JK, Smigel, MD, et al. The regulatory component of adenylate cyclase: purification and properties. J Biol Chem. 1981;256:11517–11526.
90. Walseth, TF, Zhang, HJ, Olson, LK, et al. Increase in Gs and cyclic AMP generation in HIT cells: evidence that the 45-kDa alpha-subunit of Gs has greater functional activity than the 52-kDa alpha-subunit. J Biol Chem. 1989;264:21106–21111.
91. Graziano, MP, Freissmuth, M, Gilman, AG. Expression of Gs alpha in Escherichia coli: purification and properties of two forms of the protein. J Biol Chem. 1989;264:409–418.
92. Seifert, R, Wenzel-Seifert, K, Lee, TW, et al. Different effects of Gsalpha splice variants on beta2-adrenoreceptor-mediated signaling: the beta2-adrenoreceptor coupled to the long splice variant of Gsalpha has properties of a constitutively active receptor. J Biol Chem. 1998;273:5109–5116.
93. Kvapil, P, Novotny, J, Svoboda, P, et al. The short and long forms of the alpha subunit of the stimulatory guanine-nucleotide-binding protein are unequally redistributed during (−)-isoproterenol-mediated desensitization of intact S49 lymphoma cells. Eur J Biochem. 1994;226:193–199.
94. el Jamali, A, Rachdaoui, N, Jacquemin, C, et al. Long-term effect of forskolin on the activation of adenylyl cyclase in astrocytes. J Neurochem. 1996;67:2532–2539.
95. Bourgeois, C, Duc-Goiran, P, Robert, B, et al. G protein expression in human fetoplacental vascularization: functional evidence for Gs alpha and Gi alpha subunits. J Mol Cell Cardiol. 1996;28:1009–1021.
96. Thiele, S, Werner, R, Ahrens, W, et al. A disruptive mutation in exon 3 of the GNAS gene with Albright hereditary osteodystrophy, normocalcemic pseudohypoparathyroidism, and selective long transcript variant Gsalpha-L deficiency. J Clin Endocrinol Metab. 2007;92:1764–1768.
97. Ischia, R, Lovisetti-Scamihorn, P, Hogue-Angeletti, R, et al. Molecular cloning and characterization of NESP55, a novel chromogranin-like precursor of a peptide with 5-HT1B receptor antagonist activity. J Biol Chem. 1997;272:11657–11662.
98. Hayward, BE, Moran, V, Strain, L, et al. Bidirectional imprinting of a single gene: GNAS1 encodes maternally, paternally, and biallelically derived proteins. Proc Natl Acad Sci U S A. 1998;95:15475–15480.
99. Peters, J, Wroe, SF, Wells, CA, et al. A cluster of oppositely imprinted transcripts at the GNAS locus in the distal imprinting region of mouse chromosome 2. Proc Natl Acad Sci U S A. 1999;96:3830–3835.
100. Lovisetti-Scamihorn, P, Fischer-Colbrie, R, Leitner, B, et al. Relative amounts and molecular forms of NESP55 in various bovine tissues. Brain Res. 1999;829:99–106.
101. Weiss, U, Ischia, R, Eder, S, et al. Neuroendocrine secretory protein 55 (NESP55): alternative splicing onto transcripts of the GNAS gene and posttranslational processing of a maternally expressed protein. Neuroendocrinology. 2000;71:177–186.
102. Bauer, R, Weiss, C, Marksteiner, J, et al. The new chromogranin-like protein NESP55 is preferentially localized in adrenaline-synthesizing cells of the bovine and rat adrenal medulla. Neurosci Lett. 1999;263:13–16.
103. Li, JY, Lovisetti-Scamihorn, P, Fischer-Colbrie, R, et al. Distribution and intraneuronal trafficking of a novel member of the chromogranin family, NESP55, in the rat peripheral nervous system. Neuroscience. 2002;110:731–745.
104. Fischer-Colbrie, R, Eder, S, Lovisetti-Scamihorn, P, et al. Neuroendocrine secretory protein 55: a novel marker for the constitutive secretory pathway. Ann N Y Acad Sci. 2002;971:317–322.
105. Plagge, A, Isles, AR, Gordon, E, et al. Imprinted Nesp55 influences behavioral reactivity to novel environments. Mol Cell Biol. 2005;25:3019–3026.
106. Kehlenbach, RH, Matthey, J, Huttner, WB. XLαs is a new type of G protein (Erratum in Nature 375:253, 1995). Nature. 1994;372:804–809.
107. Hayward, B, Kamiya, M, Strain, L, et al. The human GNAS1 gene is imprinted and encodes distinct paternally and biallelically expressed G proteins. Proc Natl Acad Sci U S A. 1998;95:10038–10043.
108. Klemke, M, Pasolli, H, Kehlenbach, R, et al. Characterization of the extra-large G protein alpha-subunit XLalphas, II, Signal transduction properties. J Biol Chem. 2000;275:33633–33640.
109. Bastepe, M, Gunes, Y, Perez-Villamil, B, et al. Receptor-mediated adenylyl cyclase activation through XLalphas, the extra-large variant of the stimulatory G protein alpha-subunit. Mol Endocrinol. 2002;16:1912–1919.
110. Plagge, A, Gordon, E, Dean, W, et al. The imprinted signaling protein XLalphas is required for postnatal adaptation to feeding. Nat Genet. 2004;36:818–826.
111. Chen, M, Gavrilova, O, Liu, J, et al. Alternative Gnas gene products have opposite effects on glucose and lipid metabolism. Proc Natl Acad Sci U S A. 2005;102:7386–7391.
112. Germain-Lee, EL, Schwindinger, W, Crane, JL, et al. A mouse model of Albright hereditary osteodystrophy generated by targeted disruption of exon 1 of the Gnas gene. Endocrinology. 2005;146:4697–4709.
113. Swaroop, A, Agarwal, N, Gruen, JR, et al. Differential expression of novel Gs alpha signal transduction protein cDNA species. Nucleic Acids Res. 1991;19:4725–4729.
114. Ishikawa, Y, Bianchi, C, Nadal-Ginard, B, et al. Alternative promoter and 5′ exon generate a novel Gsα mRNA. J Biol Chem. 1990;265:8458–8462.
115. Hayward, B, Bonthron, D. An imprinted antisense transcript at the human GNAS1 locus. Hum Mol Genet. 2000;9:835–841.
116. Wroe, SF, Kelsey, G, Skinner, JA, et al. An imprinted transcript, antisense to Nesp, adds complexity to the cluster of imprinted genes at the mouse GNAS locus. Proc Natl Acad Sci U S A. 2000;97:3342–3346.
117. Bastepe, M, Fröhlich, LF, Linglart, A, et al. Deletion of the NESP55 differentially methylated region causes loss of maternal GNAS imprints and pseudohypoparathyroidism type-Ib. Nat Genet. 2005;37:25–37.
118. Coombes, C, Arnaud, P, Gordon, E, et al. Epigenetic properties and identification of an imprint mark in the Nesp-Gnasxl domain of the mouse GNAS imprinted locus. Mol Cell Biol. 2003;23:5475–5488.
119. Liu, J, Yu, S, Litman, D, et al. Identification of a methylation imprint mark within the mouse GNAS locus. Mol Cell Biol. 2000;20:5808–5817.
120. Williamson, CM, Ball, ST, Nottingham, WT, et al. A cis-acting control region is required exclusively for the tissue-specific imprinting of GNAS. Nat Genet. 2004;36:894–899.
121. Williamson, CM, Turner, MD, Ball, ST, et al. Identification of an imprinting control region affecting the expression of all transcripts in the GNAS cluster. Nat Genet. 2006;38:350–355.
122. Liu, J, Chen, M, Deng, C, et al. Identification of the control region for tissue-specific imprinting of the stimulatory G protein alpha-subunit. Proc Natl Acad Sci U S A. 2005;102:5513–5518.
123. Zheng, H, Radeva, G, McCann, JA, et al. Gαs transcripts are biallelically expressed in the human kidney cortex: implications for pseudohypoparathyroidism type Ib. J Clin Endocrinol Metab. 2001;86:4627–4629.
124. Campbell, R, Gosden, CM, Bonthron, DT. Parental origin of transcription from the human GNAS1 gene. J Med Genet. 1994;31:607–614.
125. Mantovani, G, Bondioni, S, Locatelli, M, et al. Biallelic expression of the Gsalpha gene in human bone and adipose tissue. J Clin Endocrinol Metab. 2004;89:6316–6319.
126. Yu, S, Yu, D, Lee, E, et al. Variable and tissue-specific hormone resistance in heterotrimeric Gs protein α-subunit (Gsα) knockout mice is due to tissue-specific imprinting of the Gsα gene. Proc Natl Acad Sci U S A. 1998;95:8715–8720.
127. Hayward, B, Barlier, A, Korbonits, M, et al. Imprinting of the G(s)alpha gene GNAS1 in the pathogenesis of acromegaly. J Clin Invest. 2001;107:R31–R36.
128. Mantovani, G, Ballare, E, Giammona, E, et al. The Gsalpha gene: predominant maternal origin of transcription in human thyroid gland and gonads. J Clin Endocrinol Metab. 2002;87:4736–4740.
129. Sakamoto, A, Liu, J, Greene, A, et al. Tissue-specific imprinting of the G protein Gsalpha is associated with tissue-specific differences in histone methylation. Hum Mol Genet. 2004;13:819–828.
130. Albright, F, Forbes, AP, Henneman, PH. Pseudo-pseudohypoparathyroidism. Trans Assoc Am Physicians. 1952;65:337–350.
131. Davies, AJ, Hughes, HE. Imprinting in Albright’s hereditary osteodystrophy. J Med Genet. 1993;30:101–103.
132. Kronenberg, HM. Developmental regulation of the growth plate. Nature. 2003;423:332–336.
133. Bastepe, M, Weinstein, LS, Ogata, N, et al. Stimulatory G protein directly regulates hypertrophic differentiation of growth plate cartilage in vivo. Proc Natl Acad Sci U S A. 2004;101:14794–14799.
134. Long, DN, McGuire, S, Levine, MA, et al. Body mass index differences in pseudohypoparathyroidism type 1a versus pseudopseudohypoparathyroidism may implicate paternal imprinting of Galpha(s) in the development of human obesity. J Clin Endocrinol Metab. 2007;92:1073–1079.
135. Mouallem, M, Shaharabany, M, Weintrob, N, et al. Cognitive impairment is prevalent in pseudohypoparathyroidism type Ia, but not in pseudopseudohypoparathyroidism: possible cerebral imprinting of Gsalpha. Clin Endocrinol (Oxf). 2008;68:233–239.
136. Kaplan, FS, Craver, R, MacEwen, GD, et al. Progressive osseous heteroplasia: a distinct developmental disorder of heterotopic ossification, Two new case reports and follow-up of three previously reported cases. J Bone Joint Surg Am. 1994;76:425–436.
137. Kaplan, FS, Shore, EM. Progressive osseous heteroplasia. J Bone Miner Res. 2000;15:2084–2094.
138. Buyse, G, Silberstein, J, Goemans, N, et al. Fibrodysplasia ossificans progressiva: still turning into wood after 300 years? Eur J Pediatr. 1995;154:694–699.
139. Shore, EM, Kaplan, FS. Insights from a rare genetic disorder of extra-skeletal bone formation, fibrodysplasia ossificans progressiva (FOP). Bone. 2008;43:427–433.
140. Eddy, MC, De Beur, SM, Yandow, SM, et al. Deficiency of the alpha-subunit of the stimulatory G protein and severe extraskeletal ossification. J Bone Miner Res. 2000;15:2074–2083.
141. Yeh, GL, Mathur, S, Wivel, A, et al. GNAS1 mutation and Cbfa1 misexpression in a child with severe congenital platelike osteoma cutis. J Bone Miner Res. 2000;15:2063–2073.
142. Shore, EM, Ahn, J, Jan de Beur, S, et al. Paternally inherited inactivating mutations of the GNAS1 gene in progressive osseous heteroplasia. N Engl J Med. 2002;346:99–106.
143. Weinstein, LS, Yu, S, Warner, DR, et al. Endocrine manifestations of stimulatory G protein alpha-subunit mutations and the role of genomic imprinting. Endocr Rev. 2001;22:675–705.
144. Lietman, SA, Ding, C, Cooke, DW, et al. Reduction in Gsalpha induces osteogenic differentiation in human mesenchymal stem cells. Clin Orthop Relat Res. 2005;434:231–238.
145. Zhao, Y, Ding, S. A high-throughput siRNA library screen identifies osteogenic suppressors in human mesenchymal stem cells. Proc Natl Acad Sci U S A. 2007;104:9673–9678.
146. Bertaux, K, Broux, O, Chauveau, C, et al. Runx2 regulates the expression of GNAS on SaOs-2 cells. Bone. 2006;38:943–950.
147. Adegbite, NS, Xu, M, Kaplan, FS, et al. Diagnostic and mutational spectrum of progressive osseous heteroplasia (POH) and other forms of GNAS-based heterotopic ossification. Am J Med Genet. 2008;146A:1788–1796.
148. Peterman, MG, Garvey, JL. Pseudohypoparathyroidism; case report. Pediatrics. 1949;4:790.
149. Reynolds, TB, Jacobson, G, Edmondson, HA, et al. Pseudohypoparathyroidism: report of a case showing bony demineralization. J Clin Endocrinol Metab. 1952;12:560.
150. Elrick, H, Albright, F, Bartter, FC, et al. Further studies on pseudo-hypoparathyroidism: report of four new cases. Acta Endocrinol. 1950;5:199–225.
151. Costello, JM, Dent, CE. Hypo-hyperparathyroidism. Arch Dis Child. 1963;38:397–407.
152. Allen, EH, Millard, FJC, Nassim, JR. Hypo-hyperparathyroidism. Arch Dis Child. 1968;43:295–301.
153. Frame, B, Hanson, CA, Frost, HM, et al. Renal resistance to parathyroid hormone with osteitis fibrosa: “pseudohypohyperparathyroidism.”. Am J Med. 1972;52:311–321.
154. Jüppner, H, Schipani, E, Bastepe, M, et al. The gene responsible for pseudohypoparathyroidism type Ib is paternally imprinted and maps in four unrelated kindreds to chromosome 20q13.3. Proc Natl Acad Sci U S A. 1998;95:11798–11803.
155. Bastepe, M, Lane, AH, Jüppner, H. Paternal uniparental isodisomy of chromosome 20q (patUPD20q)—and the resulting changes in GNAS1 methylation—as a plausible cause of pseudohypoparathyroidism. Am J Hum Genet. 2001;68:1283–1289.
156. Liu, J, Erlichman, B, Weinstein, LS. The stimulatory G protein α-subunit Gsα is imprinted in human thyroid glands: implications for thyroid function in pseudohypoparathyroidism types 1A and 1B. J Clin Endocrinol Metab. 2003;88:4336–4341.
157. Mantovani, G, Bondioni, S, Linglart, A, et al. Genetic analysis and evaluation of resistance to thyrotropin and growth hormone-releasing hormone in pseudohypoparathyroidism type Ib. J Clin Endocrinol Metab. 2007;92:3738–3742.
159. Laspa, E, Bastepe, M, Jüppner, H, et al. Phenotypic and molecular genetic aspects of pseudohypoparathyroidism type Ib in a Greek kindred: evidence for enhanced uric acid excretion due to parathyroid hormone resistance. J Clin Endocrinol Metab. 2004;89:5942–5947.
160. Pepersack, T, Jabbour, N, Fuss, M, et al. Hyperuricemia and renal handling of urate in primary hyperparathyroidism. Nephron. 1989;53:349–352.
161. Westerdahl, J, Valdemarsson, S, Lindblom, P, et al. Urate and arteriosclerosis in primary hyperparathyroidism. Clin Endocrinol (Oxf). 2001;54:805–811.
162. Wu, WI, Schwindinger, WF, Aparicio, LF, et al. Selective resistance to parathyroid hormone caused by a novel uncoupling mutation in the carboxyl terminus of Gαs: a cause of pseudohypoparathyroidism type Ib. J Biol Chem. 2001;276:165–171.
163. Silve, C, Santora, A, Breslau, N, et al. Selective resistance to parathyroid hormone in cultured skin fibroblasts from patients with pseudohypoparathyroidism type Ib. J Clin Endocrinol Metab. 1986;62:640–644.
164. Schipani, E, Weinstein, LS, Bergwitz, C, et al. Pseudohypoparathyroidism type Ib is not caused by mutations in the coding exons of the human parathyroid hormone (PTH)/PTH-related peptide receptor gene. J Clin Endocrinol Metab. 1995;80:1611–1621.
165. Suarez, F, Lebrun, JJ, Lecossier, D, et al. Expression and modulation of the parathyroid hormone (PTH)/PTH-related peptide receptor messenger ribonucleic acid in skin fibroblasts from patients with type Ib pseudohypoparathyroidism. J Clin Endocrinol Metab. 1995;80:965–970.
166. Fukumoto, S, Suzawa, M, Takeuchi, Y, et al. Absence of mutations in parathyroid hormone (PTH)/PTH-related protein receptor complementary deoxyribonucleic acid in patients with pseudohypoparathyroidism type Ib. J Clin Endocrinol Metab. 1996;81:2554–2558.
167. Jan de Beur, S, Ding, C, LaBuda, M, et al. Pseudohypoparathyroidism 1b: exclusion of parathyroid hormone and its receptors as candidate disease genes. J Clin Endocrinol Metab. 2000;85:2239–2246.
168. Blomstrand, S, Claësson, I, Säve-Söderbergh, J. A case of lethal congenital dwarfism with accelerated skeletal maturation. Pediatr Radiol. 1985;15:141–143.
169. Liu, J, Litman, D, Rosenberg, M, et al. A GNAS1 imprinting defect in pseudohypoparathyroidism type IB. J Clin Invest. 2000;106:1167–1174.
170. Jan de Beur, S, Ding, C, Germain-Lee, E, et al. Discordance between genetic and epigenetic defects in pseudohypoparathyroidism type 1b revealed by inconsistent loss of maternal imprinting at GNAS1. Am J Hum Genet. 2003;73:314–322.
171. Bastepe, M, Fröhlich, LF, Hendy, GN, et al. Autosomal dominant pseudohypoparathyroidism type Ib is associated with a heterozygous microdeletion that likely disrupts a putative imprinting control element of GNAS. J Clin Invest. 2003;112:1255–1263.
172. Liu, J, Nealon, JG, Weinstein, LS. Distinct patterns of abnormal GNAS imprinting in familial and sporadic pseudohypoparathyroidism type IB. Hum Mol Genet. 2005;14:95–102.
173. de Nanclares, GP, Fernandez-Rebollo, E, Santin, I, et al. Epigenetic defects of GNAS in patients with pseudohypoparathyroidism and mild features of Albright’s hereditary osteodystrophy. J Clin Endocrinol Metab. 2007;92:2370–2373.
174. Fröhlich, LF, Bastepe, M, Ozturk, D, et al. Lack of GNAS epigenetic changes and pseudohypoparathyroidism type Ib in mice with targeted disruption of syntaxin-16. Endocrinology. 2007;148:2925–2935.
175. Chillambhi S, Turan S, Hwang D-Y, et al: Deletion of the GNAS antisense transcript results in parent-of-origin specific GNAS imprinting defects and phenotypes including PTH-resistance, Presented at: 30th Annual Meeting of the American Society of Bone and Mineral Research, Montreal, September 12–26, 2008. Abstract No, 1052.
176. Chotalia, M, Smallwood, SA, Ruf, N, et al. Transcription is required for establishment of germline methylation marks at imprinted genes. Genes Dev. 2009;23:105–117.
177. Mariot, V, Maupetit-Mehouas, S, Sinding, C, et al. A maternal epimutation of GNAS leads to Albright osteodystrophy and parathyroid hormone resistance. J Clin Endocrinol Metab. 2008;93:661–665.
178. van Dop, C. Pseudohypoparathyroidism: clinical and molecular aspects. Semin Nephrol. 1989;9:168–178.
179. Kruse, K, Kustermann, W. Evidence for transient peripheral resistance to parathyroid hormone in premature infants. Acta Paediatr Scand. 1987;76:115–118.
180. Lee, CT, Tsai, WY, Tung, YC, et al. Transient pseudohypoparathyroidism as a cause of late-onset hypocalcemia in neonates and infants. J Formos Med Assoc. 2008;107:806–810.
181. Manzar, S. Transient pseudohypoparathyroidism and neonatal seizure. J Trop Pediatr. 2001;47:113–114.
182. Kruse, K, Kracht, U, Wohlfart, K, et al. Biochemical markers of bone turnover, intact serum parathyroid hormone and renal calcium excretion in patients with pseudohypoparathyroidism and hypoparathyroidism before and during vitamin D treatment. Eur J Pediatr. 1989;148:535–539.
183. Rao, DS, Parfitt, AM, Kleerekoper, M, et al. Dissociation between the effects of endogenous parathyroid hormone on adenosine 3′,5′-monophosphate generation and phosphate reabsorption in hypocalcemia due to vitamin D depletion: an acquired disorder resembling pseudohypoparathyroidism type II. J Clin Endocrinol Metab. 1985;61:285–290.
184. Srivastava, T, Alon, US. Stage I vitamin D-deficiency rickets mimicking pseudohypoparathyroidism type II. Clin Pediatr (Phila). 2002;41:263–268.
185. Shriraam, M, Bhansali, A, Velayutham, P. Vitamin D deficiency masquerading as pseudohypoparathyroidism type 2. J Assoc Physicians India. 2003;51:619–620.